
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of nickel foam modified by multiwalled hollow spheres of NiCo2O4 as a promising non-enzymatic glucose sensor†
Nada Epriliaa,
Afiten R. Sanjayaa,
Respati K. Pramadewandaru d,
Tiara A. H. Pertiwia,
Yulia M. T. A. Putria,
Isnaini Rahmawatia,
Beti E. Dewic,
Yuni K. Krisnandia,
Hoeil Chung*b and
Tribidasari A. Ivandini*a
d,
Tiara A. H. Pertiwia,
Yulia M. T. A. Putria,
Isnaini Rahmawatia,
Beti E. Dewic,
Yuni K. Krisnandia,
Hoeil Chung*b and
Tribidasari A. Ivandini*a
aDepartment of Chemistry, Faculty of Mathematics and Natural Sciences, Universitas Indonesia, Depok, 16424, Indonesia. E-mail: ivandini.tri@sci.ui.ac.id
bDepartment of Chemistry, College of Natural Sciences, University of Hanyang, Seoul, South Korea. E-mail: hoeil@hanyang.ac.kr
cDepartment of Microbiology, Faculty of Medicine, Universitas Indonesia, Depok, 16424, Indonesia
dDepartement of Materials and Metallurgical Engineering, Institut Teknologi Sepuluh Nopember, Surabaya, Indonesia
First published on 3rd April 2024
Abstract
Nickel foam modified by hollow sphere NiCo2O4 particles was successfully prepared via a hydrothermal method using nanosphere SiO2 particles as the hard template for the hollow structure. Characterisation using SEM-EDX and TEM confirmed the structure as multiwalled hollow spheres with an average size of 270 nm, while characterisation using SEM, XRD, and XPS confirmed that the NiCo2O4 particles were attached on the surface of the nickel foam. BET analysis showed that the surface area of the synthesized NiCo2O4@Ni foam was nearly three times higher compared to that of the unmodified Ni foam. Investigation of the NiCo2O4-modified nickel foam as an electrode for the detection of glucose in sodium hydroxide solution showed high linearity of the anodic currents (R2 = 0.99) in the concentration range of 0–2.5 μM with sensitivity of 0.060 mA μM−1 and an estimated limit of detection of 0.060 μM. Excellent stability of the current response was also obtained with a relative standard deviation of 1.51% (n = 10). Furthermore, the developed sensor demonstrates strong applicability for glucose detection in real samples of human blood plasma, making it highly suitable for practical use. The results indicate that the material is promising for the further development of nickel-based sensors.
Introduction
Nickel is widely used in catalysts due to its excellent performance as an electrocatalyst.1–6 However, nickel has disadvantages due to its high over potential and poor stability.7 Combining nickel with cobalt to form spinel NiCo2O4 for use as an electrocatalyst has received major attention in many electrochemical applications, such as water splitting, supercapacitor, and chemical sensing.8–14 In addition, one of the key factors to obtain a favourable electrocatalyst for use as a working electrode is by modifying its structure and morphology, which could further affect its sensing performance due to the increase of the electrode's surface area.Meanwhile, glucose is a crucial part in human physiological fluids, playing an important role in human metabolism and being the only main fuel for the brain, except in a state of prolonged hunger.15 Strict regulation of glucose metabolism is needed because insufficient glucose intake can disrupt the brain physiology,16 while high glucose concentrations in human blood can trigger diabetes, a disease which can be caused by genetic factors, insensitivity to insulin, and failed insulin secretion.17 To manage blood sugar levels in the body, a healthy lifestyle and regular exercise are necessary. In addition, glucose levels in the body should be monitored periodically. Conventional glucose sensors commonly use an enzymatic reaction to promote glucose oxidation.18 However, loss of biological activity is also often reported due to a change of pH or temperature.19,20 Accordingly, the development of the non-enzymatic glucose sensors attracts considerable attention.21–23 Such sensors commonly use transition metals, in the forms of metal oxides, bimetals, or layered triple hydroxides,24,25 while nickel is the most favourable metal due to its low cost and good reactivity.26,27
Electrochemical detection of glucose based on nickel electrodes has been widely explored. Many types of nickel-based materials have been proposed to be employed as working electrodes for sensors, such as pure nickel metal, oxides, sulfides, and organic frameworks.28–30 In addition, the use of alloys or composites is often found to increase the catalytic activity, while minimizing its high overpotential and instability problems.31,32 On the other hand, the use of porous materials for sensor applications can provide a high surface area, and accordingly accelerate the ion transfer on the electrode's surface, resulting in sensors with high sensitivity.33
In this work, nickel foam was used as the support for the working electrode due to its porous structure, which provides a high surface area to facilitate good electron and electrolyte transfer during the redox process.34 To solve the high over potential problem, the nickel foam was modified with cobalt. Hollow sphere-structured NiCo2O4 was selected for the cobalt source since the multiwalled hollow sphere structure of NiCo2O4 can enhance the sensor's surface area and the sensitivity of glucose oxidation. The NiCo2O4 was synthesized on the surface of the nickel foam using a hydrothermal method with SiO2 nanospheres as the hard template. The holes in the NiCo2O4 structure were produced by etching the template in an alkaline atmosphere. Confirmation of the hollow nanostructure was obtained by using SEM and TEM characterisation, while the advantage of the hollow structure was employed for the detection of glucose.
The prepared NiCo2O4 hollow spheres-modified nickel foam (NiCo2O4@Ni foam) was found to have a high surface area. Furthermore, the modified nickel foam showed good linearity for the anodic current with a low limit of detection and high stability of the current response for glucose detection, indicating that the material is suitable for sensor applications.
Materials and methods
Materials
All reagents, including nickel nitrate hexahydrate (Ni(NO3)2·6H2O), cobalt nitrate hexahydrate (Co(NO3)2·6H2O), sodium hydroxide (NaOH, >98%), urea (CO(NH2)2, 99–100.5%), D-(+) glucose (99.99%), tetraethyl orthosilicate (TEOS, 99.99%), ethanol (99%), and ammonium hydroxide (NH4OH, 28–32%), were purchased from Sigma-Aldrich, while double distilled water with a maximum conductivity of 0.056 μS cm−1 was obtained from a Direct-Q® 3 UV Remote Water Purification System (Direct-Q3 UV, Millipore). All chemical reagents used in this experiment were of analytical grade and were used without further purification.Synthesis of SiO2 nanospheres
SiO2 nanospheres were synthesized by slowly dripping 2 mL of TEOS into a mixture of ethanol (50 mL), ammonia (1.5 mL), and water (5 mL). The mixture was then stirred at 60 °C for 24 h. The SiO2 nanospheres were then obtained by centrifugation and washed with water and ethanol several times, followed by drying under vacuum at 60 °C for 24 h.Synthesis of NiCo2O4 on nickel foam
Prior to use, a piece of nickel foam (dimensions: 10 mm × 10 mm × 1.6 mm) was cleaned with sonication in 3.0 M HCl for 20 min, followed by ethanol and water for 15 min each, then dried at 60 °C for 3 h. Meanwhile, the as-synthesized SiO2 nanospheres (0.1 g) were placed into 50 mL of water and sonicated for 20 min. Ni(NO3)2·6H2O (0.278 g), Co(NO3)2·6H2O (0.476 g), and urea (1.2 g) were added to the mixture, which was then stirred to form a homogenous solution before transferring into a 100 mL Teflon-lined stainless-steel autoclave together with the cleaned nickel foam. The autoclave was then heated in an oven at 100 °C for 48 h. After cooling to room temperature, the treated nickel foam was washed with hot NaOH solution in an ethanol–water solvent (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to remove the SiO2 nanosphere template. The characterisation of the treated nickel foam was performed using Fourier-transform infrared spectrometry (FTIR Bruker, LUMOS), X-ray diffraction analysis (XRD Rigaku, Cu-Kα radiation), scanning electron microscopy with energy dispersive X-ray analysis (SEM-EDX Hitachi S-4800), and X-ray photoelectron spectroscopy (XPS PHI5000 Versa Probe), while transmission electron microscopy (TEM, JEM-2100F) characterisation was performed for the NiCo2O4 particles filtered from the remaining solution in the autoclave.
:1) to remove the SiO2 nanosphere template. The characterisation of the treated nickel foam was performed using Fourier-transform infrared spectrometry (FTIR Bruker, LUMOS), X-ray diffraction analysis (XRD Rigaku, Cu-Kα radiation), scanning electron microscopy with energy dispersive X-ray analysis (SEM-EDX Hitachi S-4800), and X-ray photoelectron spectroscopy (XPS PHI5000 Versa Probe), while transmission electron microscopy (TEM, JEM-2100F) characterisation was performed for the NiCo2O4 particles filtered from the remaining solution in the autoclave.
Electrochemical measurements
The electrochemical measurements were carried out using an eDAQ electrochemical workstation (E-corder 410) with a three-electrode system. The synthesized NiCo2O4@Ni was used as the working electrode, with platinum wire as the counter electrode and an Ag/AgCl (saturated KCl) system as the reference electrode. The electrochemical measurements were conducted in 1.0 M NaOH as the electrolyte. Cyclic voltammetry in the potential range of −0.1–0.6 V with a scan rate of 25 mV s−1 and chronoamperometry at −0.17 V (vs. Ag/AgCl) for 120 s were applied to examine the sensor performance.The sample measurements were carried out using human blood plasma, obtained from the Faculty of Medicine, University of Indonesia, Jakarta, Indonesia. No preparation or purification were required for the samples prior to the measurements. A total volume of 40 μL of sample was diluted in a 100 mL volumetric flask, then 12.5 mL of it was further diluted in another 100 mL volumetric flask with 1.0 M NaOH. The glucose level in the sample was then measured using the nickel foam electrode modified with NiCo2O4 hollow spheres using the CV technique and the results were subsequently validated using a glucose detector (EasyTouch GCU).
Result and discussion
Characterisation of the prepared materials
SiO2 nanospheres were synthesized from TEOS as a template to form the hollow sphere structure of NiCo2O4. Fig. 1 shows the FTIR and XRD spectra of the synthesized SiO2 nanospheres. The FTIR spectrum (Fig. 1a) revealed a band at 797 cm−1, attributed to the Si–O bending vibration, while the peak at 961 cm−1 is due to the Si–O–(H⋯H2O) bending vibration.35,36 The absorption band at 1091 cm−1 is assigned to the asymmetric vibration of Si–OH. The bands at 1624 cm−1 and 3303 cm−1 are attributed to the O–H bending vibration and the O–H stretching vibration of adsorbed molecular water, respectively.35 The results indicated that the SiO2 had been successfully formed.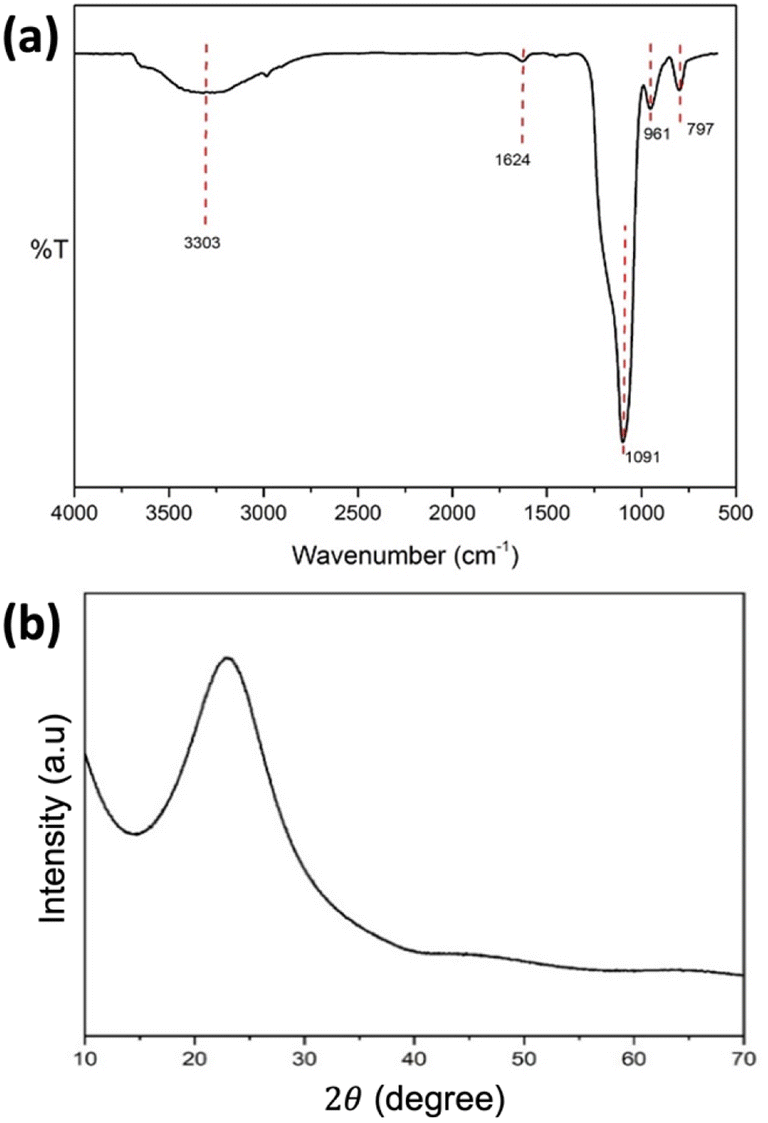 | ||
| Fig. 1 (a) FTIR and (b) XRD spectra of the synthesized SiO2 nanospheres. | ||
Further characterisation with XRD (Fig. 1b) showed a peak at 2θ of 22.75°, which is characteristic of the amorphous SiO2. Meanwhile, the SEM image of the synthesized SiO2 nanospheres (Fig. 2a) shows a spherical shape with the size range from 200 to 300 nm. The EDX characterisation shows ratios of 37.08% and 62.92% for Si and O, respectively, confirming the formation of SiO2.
 | ||
| Fig. 2 Typical SEM images of (a) the SiO2 nanospheres, (b) the NiCo2O4 hollow spheres, (c) the unmodified nickel foam, and (d) the NiCo2O4 hollow spheres deposited on the nickel foam. | ||
Synthesis of the NiCo2O4 hollow spheres was performed using a hydrothermal method in the presence of SiO2 nanospheres as the hard template. Basically, hydrolysis of urea occurs at a temperature of around 90 °C to form ammonia, providing an alkaline solution to further promote the nucleation process of nickel and cobalt ions to form [Ni(NH3)6]2− and [Co(NH3)6]2+. This nucleation process takes place on the surface of the SiO2 nanospheres. During the heating process, reaction between the excess of ammonia molecules with water molecules generates OH− to react with nickel and cobalt to form Ni(OH)2 and Co(OH)2, which then form NiCo2O4 after oxidation, as shown by the following reactions:37
| CO(NH2)2 + 2H2O → H2CO3 + NH3 | (1) |
| Ni2+ + 6NH3 → [Ni(NH3)6]2− | (2) |
| 2CO2 + 6NH3 → [Co(NH3)6]2+ | (3) |
| H2O + NH3 → NH4+ + OH− | (4) |
| Ni2+ + 2Co2+ + 6OH− → NiCo2(OH)6 | (5) |
| 2NiCo2(OH)6 + O2 → 2NiCo2O4 + 6H2O | (6) |
The hollow sphere structure can be seen in the SEM images of the synthesized NiCo2O4 hollow spheres (Fig. 2b), which confirmed the morphology of the synthesized material. Meanwhile, the SEM images for the unmodified Ni foam and the hollow spheres-modified NiCo2O4@Ni foam are shown in Fig. 2c and d, respectively. These figures show that the homogeneous NiCo2O4 hollow spheres were successfully synthesized covering the surface of the nickel foam. Further confirmation using EDX showed that the NiCo2O4 hollow spheres contain 7.92% (w/w) nickel, 16.98% cobalt, 64.37% oxygen, and 10.73% silicon. The nickel:cobalt ratio of 1:2 confirms the formation of NiCo2O4. However, the presence of silicon indicated that the template could not be completely removed, which also explains the high oxygen percentage in the structure.
The structural information of the synthesized NiCo2O4 hollow spheres was confirmed by TEM characterisation of the precipitated NiCo2O4 particles of the remaining solution from the autoclave. Fig. 3 shows the TEM images of the NiCo2O4 particles on three different scales. An average diameter of around 270 nm for the NiCo2O4 hollow spheres was estimated using ImageJ software. Besides, a contrasting colour difference observed between the shells and the inner space confirmed the formation of the hollow structure. The figure also shows that the hollow spheres have multiple walls with thickness of around 5 nm (Fig. 3c).
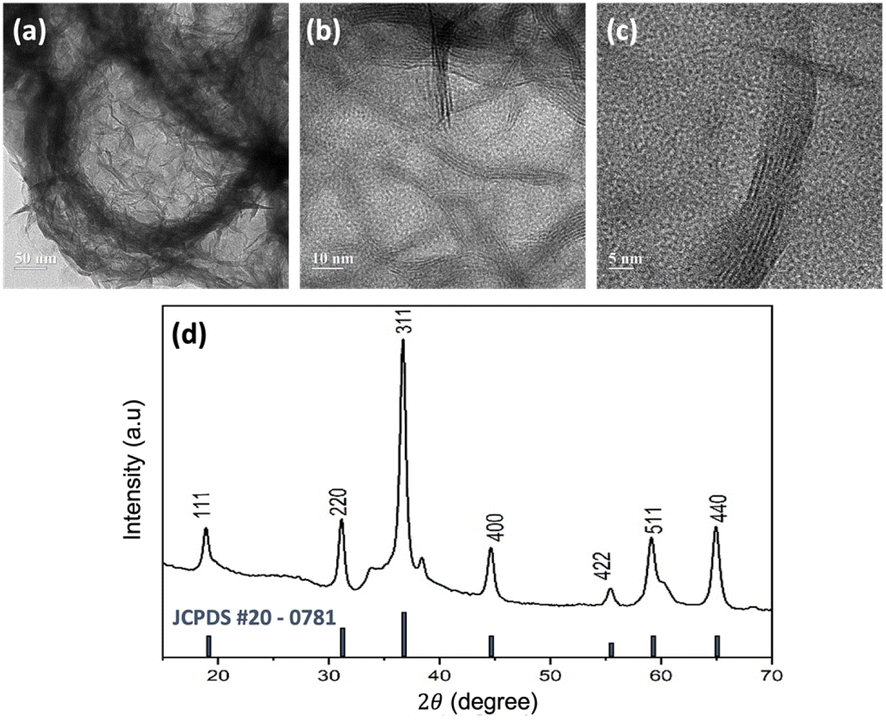 | ||
| Fig. 3 (a–c) Typical TEM images of the NiCo2O4 hollow spheres in different magnifications, and (d) XRD spectra for the NiCo2O4 hollow spheres in comparison with the NiCo2O4 JCPDS card, shown as the black lines, as the reference. | ||
Fig. 3d shows the XRD peak pattern of the synthesized NiCo2O4 hollow spheres. Diffraction peaks were observed at 2θ of 18.95°, 31.15°, 36.7°, 44.65°, 55.45°, 59.15°, and 64.95°, which were attributed to the characteristic peaks of the inverse cubic spinel phase of NiCo2O4.38 This result was confirmed with the JCPDS card no. #20-0781, as shown in the black lines in the figure. The inverse spinel structure of NiCo2O4 consists of two sites, tetrahedral and octahedral. Nickel is distributed at the octahedral site while cobalt is located at both the octahedral and tetrahedral sites. The multi-valency of cobalt makes the electrical conductivity of NiCo2O4 twice as high as those of the monometallic nickel oxide (NiO) or cobalt oxide (Co3O4), which further contributes to accessible electron transfer.39
XPS characterisation was further conducted to determine the composition of the elements present on the surface of the electrodes. The XPS spectrum of the synthesized NiCo2O4@Ni foam in Fig. 4a shows the main peaks of Ni, Co, and O. In addition, the peaks of C and Si are also observed as a result of the remaining template used in the synthesis of the NiCo2O4@Ni foam. The deconvolution of the peak spectra of Ni 2p, Co 2p, and O 1s by using Gaussian fitting is shown in Fig. 4a–c, respectively. The characteristic peaks of Ni 2p1/2 and Ni 2p3/2 were detected at the binding energies of 856.28 eV and 873.88 eV, respectively (Fig. 4b). In addition, two shake-up satellites for Ni 2p1/2 and Ni 2p3/2 were also detected, resulting in a spin energy difference of 17.58 eV, confirming the formation of the nickel hydroxide phase.40
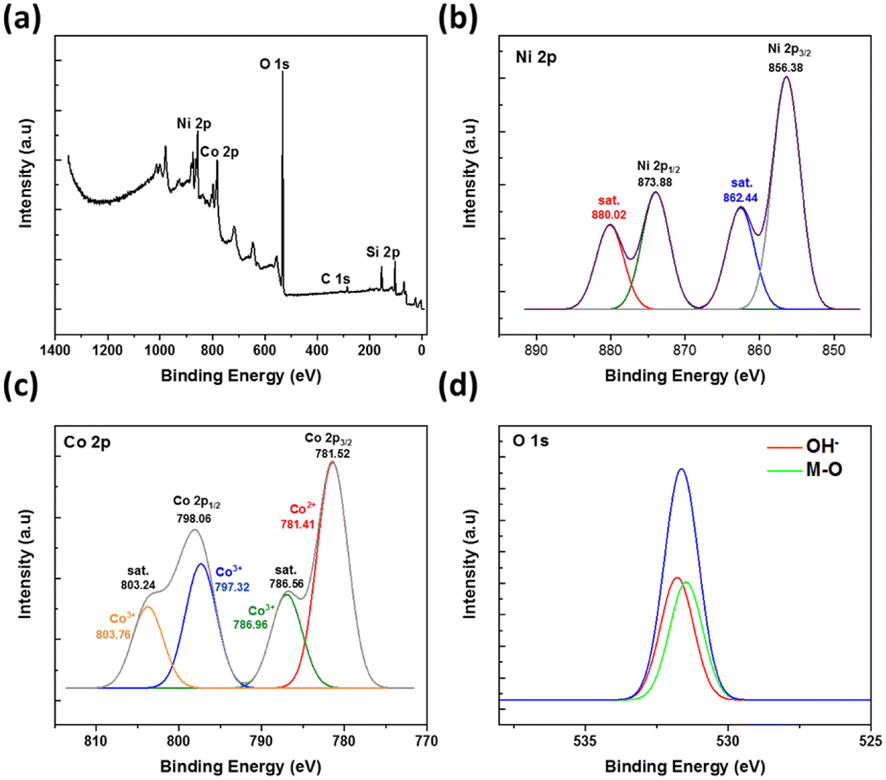 | ||
| Fig. 4 (a) The XPS spectra of the synthesized NiCo2O4@Ni foam together with (b–d, respectively) the related deconvolution peaks of Ni 2p, Co 2p, and O 1s. | ||
Meanwhile, Fig. 4c shows the XPS spectra of Co 2p atoms with two peaks at binding energies of 781.52 and 798.06 eV, which characterise Co 2p3/2 and Co 2p1/2. Typical peaks of Co2+ 2p3/2 and Co2+ 2p1/2 are shown at the binding energies of 781.41 eV and 797.32 eV, while typical peaks of Co3+ 2p3/2 and Co3+ 2p1/2 are shown at the binding energies of 786.96 eV and 803.76 eV, respectively.9 These results confirmed the existence of multi-valence cobalt in the synthesized material.
In the case of the XPS O 1s spectra, two fitting peaks are shown at the binding energy of around 531.66 eV (Fig. 4d), deconvoluted to the metal–oxygen bond at the binding energy of 531.42 eV and the defects, impurities, and a range of surface species, including hydroxyls, chemisorbed oxygen, uncoordinated lattice oxygen, or species intrinsic to the surface of spinel, at the binding energy of 531.77 eV.9
The surface area of the unmodified Ni foam and the synthesized NiCo2O4@Ni foam was examined by BET analysis. Fig. 5a depicts the N2 adsorption/desorption isotherms and Barrett–Joyner–Halenda (BJH) pore-size-distribution plots (Fig. 5b and c) for the electrodes. The isotherms of the unmodified Ni foam and the NiCo2O4@Ni foam sample can be categorized as typical type IV with an H3 hysteresis loop, indicating the existence of a mesoporous structure in the materials.41–44 As shown in Fig. 5, Ni foam exhibits a surface area and a pore volume of 10.294 m2 g−1 and 0.026 cm3 g−1, respectively, while NiCo2O4@Ni foam shows an increase in both the surface area and pore volume to 17.944 m2 g−1 and 0.045 cm3 g−1, respectively. Moreover, the pore size distribution, derived from desorption data and calculated from the BJH model (Fig. 5b and c), shows that the average pore radii of the Ni foam and the NiCo2O4@Ni foam are 2.431 nm and 1.889 nm, respectively.
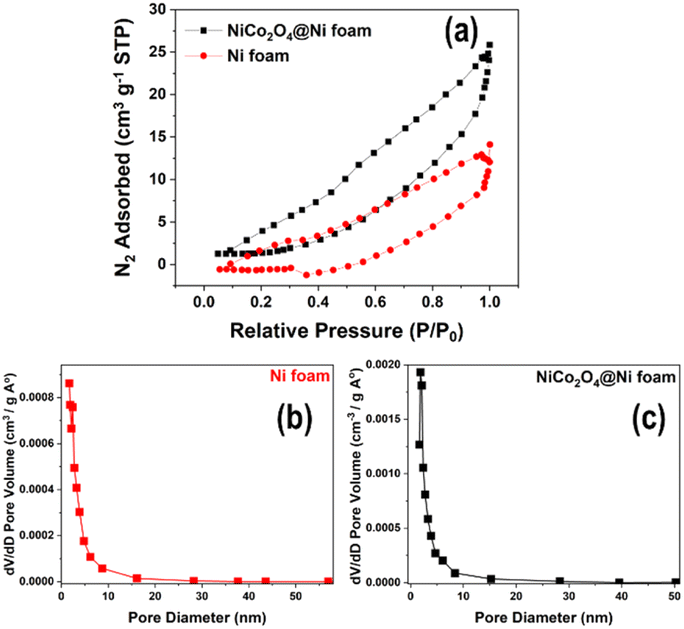 | ||
| Fig. 5 (a) N2 adsorption/desorption isotherms of the unmodified Ni foam and the NiCo2O4@Ni foam; and (b and c) the pore size distributions of the unmodified Ni foam and the NiCo2O4@Ni foam, respectively. | ||
In comparison to the unmodified Ni foam, the synthesized NiCo2O4@Ni foam has a greater surface area and pore volume, as well as smaller pore radius, which is believed to be advantageous for the diffusion of the electrolyte to the electrode's active sites and will improve its electrochemical performance for glucose sensing.
Electrochemical behaviour of glucose at the prepared NiCo2O4@Ni foam
Cyclic voltammetry of a 1.0 M NaOH solution in the presence (solid line) and absence (dashed line) of glucose was performed to study its electrochemical behaviour at the prepared NiCo2O4@Ni foam (red line) compared to the unmodified Ni foam (black line) (Fig. 6a). The prepared NiCo2O4@Ni foam shows a significantly higher current density compared to the unmodified Ni foam, both in the presence and absence of glucose, which is in line with the BET analysis results. Signal-to-background current (S/B) ratios of 0.10 and 0.28 were achieved for the unmodified Ni foam and the NiCo2O4@Ni foam, respectively, which is nearly three times higher than the unmodified Ni foam.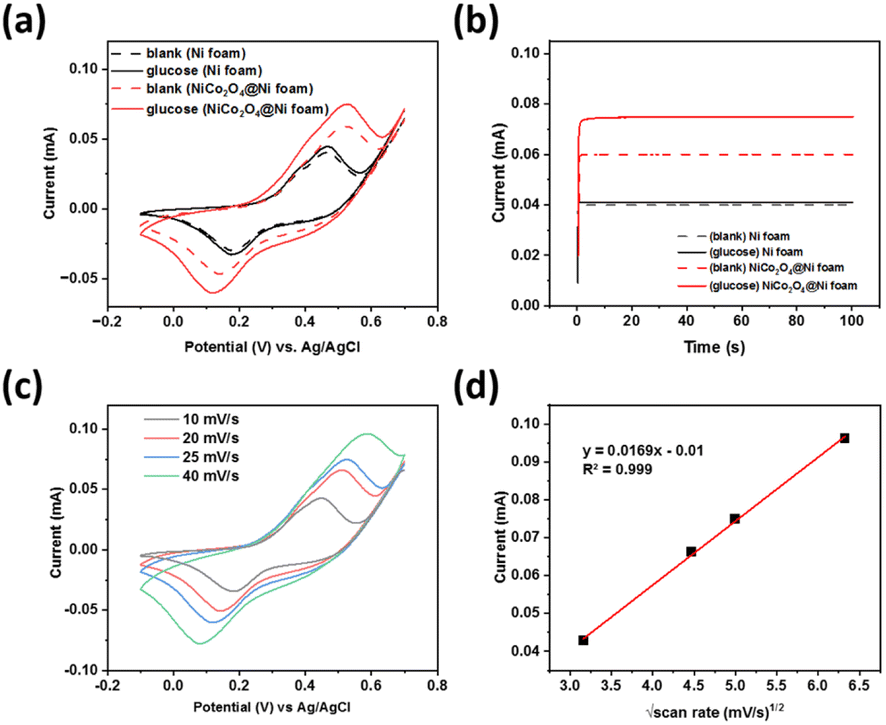 | ||
| Fig. 6 (a) Cyclic voltammograms at a scan rate of 25 mV s−1 and (b) chronoamperograms with the prepared NiCo2O4@Ni foam as the working electrode in comparison with the unmodified Ni foam in 1.0 M NaOH solution with and without the presence of 0.5 μM glucose. (c) Cyclic voltammograms for the prepared NiCo2O4@Ni foam at various scan rates and (d) the related peak currents plots as a function of the square root of the scan rate. | ||
Furthermore, chronoamperometry was conducted to validate the performance of the developed sensors (Fig. 6b), showing a similar trend of the current responses to the CV data. A significant increase of the glucose oxidation currents was observed when using the modified Ni foam. A signal-to-background current (S/B) ratio of 1.025 and 1.25 was achieved for the unmodified Ni foam and NiCo2O4@Ni foam, respectively. The results confirmed that the higher surface area of the NiCo2O4@Ni foam helps to enhance the number of catalytic active sites, which further increase its current density.
Anodic and cathodic peak couple at the potentials of +0.45 V and +0.1 V (vs. Ag/AgCl) were observed, respectively, corresponding to the binary intrinsic-state reduction–oxidation peak couples of Ni3+/Ni2+ and Co3+/Co2+ ions in NiCo2O4.45,46 These peaks were overlapped to form larger shoulder peak due to the similar reduction–oxidation potentials of Ni3+/Ni2+ and Co3+/Co2+.46 When glucose was present in the solution, a significant increase of the oxidation peak currents was observed. This result indicates that NiCo2O4 could be a promising catalyst for glucose electrooxidation, and hence could be an excellent glucose sensor. The possible mechanism of glucose oxidation by NiCo2O4 is through each intrinsic-state reduction–oxidation couple of Ni3+/Ni2+ and Co3+/Co2+ ions, as follows:46,47
| NiCO2O4 + OH− + H2O → NiOOH + 2CoOOH + e− | (7) |
| NiOOH + glucose → Ni(OH)2 + gluconolactone | (8) |
| 2CoOOH + 2glucose → 2Co(OH)2 + 2gluconolactone | (9) |
| NiOOH + 2CoOOH + 3glucose → Ni(OH)2 + 2Co(OH)2 + 3gluconolactone | (10) |
The influence of scan rate was studied to investigate the electron transfer kinetics and optimise the scan rate for glucose detection. Fig. 6b shows the cyclic voltammogram for NiCo2O4@Ni foam in 1.0 M NaOH solution in the presence of glucose at 0.5 μM. A shift in the anodic peak to a more positive potential was observed, whereas the cathodic peak shifted to a more negative potential. This result indicates that a quasi-reversible electron transfer reaction occurs.48 Plots of the square root of the scan rate versus the peak current (Ip) (Fig. 6c), showing a linear fit with the R2 = 0.999, indicate that the electron transfer on the prepared electrode surface is a diffusion-controlled process.49,50 Furthermore, determination of the optimum scan rate is based on the largest signal-to-background ratio [(S − B)/B], where S and B are the anodic peak currents in the presence and absence of glucose, respectively. Based on this calculation, the optimum scan rate of 25 mV s−1 was fixed and used for further experiments.
The performance of the prepared NiCo2O4@Ni foam as the working electrode in the quantitative detection of glucose was then assessed. Fig. 7a reveals that the peak current increases proportionally with the concentration of glucose. The oxidation currents of glucose electrocatalysis by NiCo2O4@Ni foam fell into a linear curve in the concentration range of 0 to 2.5 μM (R2 ≥ 0.99) with the linear equation of y (mA) = 0.0173[glucose (μM)] − 0.0002 (Fig. 2b). From the linear curve, the prepared electrode can provide sensitivity of 0.018 mA μM−1. Furthermore, an estimated limit of detection (LOD) of 0.173 μM could be determined by employing the formula of LOD = 3(Sy/S), where Sy represents the standard deviation of the response curve and S denotes the slope of the calibration curve.
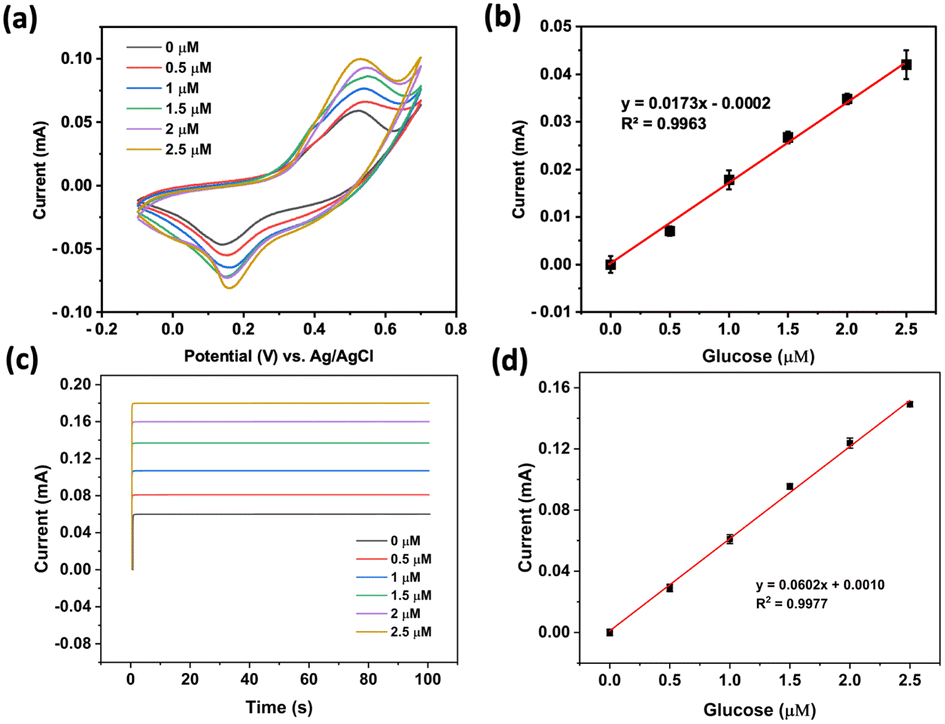 | ||
| Fig. 7 (a and b) Cyclic voltammograms at a scan rate of 25 mV s−1 and the related calibration curve, respectively. (c and d) Chronoamperograms at +0.56 V (vs. Ag/AgCl) and the related calibration curve, respectively. The applied working electrode was NiCo2O4@Ni foam and the electrolytes were various concentrations of glucose in 1.0 M NaOH. | ||
Further investigation using chronoamperometry at an applied potential of +0.56 V (vs. Ag/AgCl) in the same concentration range as that using cyclic voltammetry also showed excellent linearity (R2 ≥ 0.99) with the linear equation of y (mA) = 0.0602[glucose (μM)] − 0.0010 (Fig. 2a and b). A sensitivity of 0.060 mA μM−1 with an estimated LOD of 0.060 μM could be achieved.
The analytical performance of the developed sensor in comparison with other related glucose detection methods is displayed in Table 1, showing that the use of NiCo2O4@Ni foam as the electrode for glucose sensing is favourable among the other reported sensors, with a lower limit of detection than the other electrodes.
A repeatability test was performed to evaluate the precision level of the developed sensor using the prepared electrode. The tests were conducted ten times with cyclic voltammetry technique using a solution of 0.5 μM glucose in 1.0 M NaOH. The developed sensor showed excellent repeatability of the current responses, as confirmed by the relative standard deviation (RSD) of 1.51% (Fig. 8a).
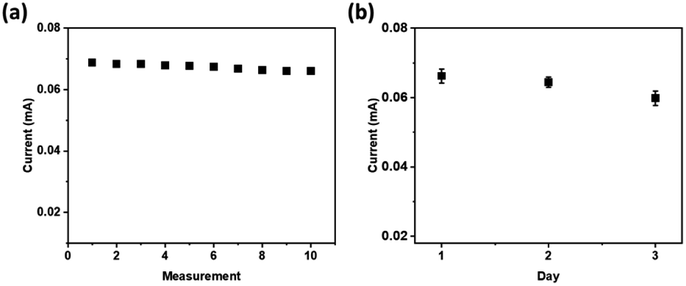 | ||
| Fig. 8 The current peaks of the voltammograms for 0.5 μM glucose in 1.0 M NaOH at NiCo2O4@Ni foam (a) for ten consecutive measurements, and (b) on three different measurement days. | ||
A stability test was carried out for 3 days at one-day intervals to study the stability of the prepared NiCo2O4@Ni foam for glucose oxidation. Good stability of the current responses was obtained with a slight decrease of the current response of around 2.7% after the 3rd day. The results indicated that the hollow spheres of NiCo2O4 were stably attached on the surface of nickel foam, and therefore the prepared NiCo2O4@Ni foam is promising for application in a real glucose sensor.
The selectivity of the developed sensor using the prepared NiCo2O4@Ni foam electrode was investigated by performing an interference study in the presence of possible interferents, including ascorbic acid, uric acid, sucrose, fructose, and albumin. As displayed in Fig. 9, the addition of the compounds at concentrations up to 50 percent analyte did not significantly change the peak currents. The results suggest the high selectivity of the developed sensor towards glucose in the presence of ascorbic acid, uric acid, sucrose, fructose, and albumin.
 | ||
| Fig. 9 Comparison of the signal responses for 0.5 μM glucose in 1.0 M NaOH at NiCo2O4@Ni foam performed in the absence (blank) and in the presence of 0.25 μM ascorbic acid, uric acid, sucrose, fructose, and albumin. The error bars represent the standard deviations from three independent measurements. | ||
The performance of the developed sensor was evaluated by measuring the glucose content in blood plasma sample obtained from the Faculty of Medicine, Universitas Indonesia, Jakarta, Indonesia. Ethical clearance was obtained from the Health Research Ethics Committee, Faculty of Medicine Universitas Indonesia No. 878//UN2.F1/ETIK/PPM.00.03/2020. The diluted blood plasma was electrochemically analysed under the optimised conditions. The current, obtained as an average of three measurements, yielded a value of 0.0664 mA (ESI S1†). Data processing was conducted using the equation derived from the linear calibration curve, indicating a glucose content of 173.28 mg dL−1 in the blood plasma sample (ESI S2†). The electrochemical measurement results were subsequently validated using a glucose detector (EasyTouch GCU), confirming a glucose level of 176 mg dL−1 in the blood plasma sample (ESI S3†). This validates that the detection result using the NiCo2O4@Ni foam electrode is comparable to the result obtained from the glucose detector. Thus, the prepared NiCo2O4@Ni foam electrode demonstrates promising potential as a detection tool (Table 2).
| Methods | Glucose (mg dL−1) |
|---|---|
| CV | 173.28 ± 2 |
| EasyTouch glucose detector | 176 |
Conclusions
Multiwalled hollow spheres of NiCo2O4 were successfully attached on the inner surface of nickel foam with a hydrothermal method with an estimated sphere size of 270 nm. The NiCo2O4 contains 7.92% (w/w) nickel, 16.98% cobalt, 64.37% oxygen, and 10.73% silicon. The nickel-to-cobalt ratio of 1:2 confirms the formation of NiCo2O4, whereas the observed silicon indicates that the template could not be completely removed, which also explains the high oxygen percentage in the structure. Investigation of the modified nickel foam for non-enzymatic-based electrochemical detection of glucose provided a linear calibration curve in the glucose concentration range from 0 to 2.5 μM. A detection limit of 0.060 μM with a sensitivity of 0.060 mA μM−1 could be achieved with excellent repeatability, stability, and selectivity towards glucose in the presence of ascorbic acid, uric acid, sucrose, fructose, and albumin. Furthermore, the developed sensor was effectively utilized for the detection of glucose in a blood plasma sample.
Author contributions
N. E.: data curation, formal analysis, investigation, and writing; T. A. H. P.: data curation, formal analysis, and investigation; A. R. S.: data curation, formal analysis, and investigation; R. K. P.: data curation, formal analysis, and investigation; Y. M. T. A. P.: electrochemical investigation, and writing-review and editing; I. R.: investigation, and writing-review and editing; Y. K. K.: material preparation supervision, validation, and writing-review and editing; H. C.: conceptualization, supervision, and validation; T. A. I.: conceptualization, supervision, validation, funding acquisition, and writing-review and editing. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to sincerely thank Hibah PUTI Q2 UI 2020 Grants No. NKB-1637/UN2.RST/HKP.05.00/2020.References
- Z. Gong, N. Hu, W. Ye, K. Zheng, C. Li, L. Ma, Q. Wei, Z. Yu, K. Zhou, N. Huang, C. te Lin and J. Luo, J. Electroanal. Chem., 2019, 841, 135–141 CrossRef CAS.
- H. A. Ariyanta, T. A. Ivandini and Y. Yulizar, FlatChem, 2021, 29, 100285 CrossRef CAS.
- E. Saepudin, T. Yuliani, N. R. R. Musyarofah, M. A. F. Nasution, J. Gunlazuardi, Y. Einaga and T. A. Ivandini, Sens. Mater., 2021, 33, 1027–1036 CAS.
- P. K. Jiwanti, R. P. Aritonang, I. Abdullah, Y. Einaga and T. A. Ivandini, Makara J. Sci., 2019, 204–209 CrossRef CAS.
- A. R. Sanjaya, S. Amanda, T. A. Ivandini, F. Abnisa, G. T. M. Kadja, U. Pratomo, Y. Alias and M. Khalil, Designs, 2023, 7, 26 CrossRef.
- Y. M. T. A. Putri, J. Gunlazuardi and T. A. Ivandini, Chem. Lett., 2021, 51, 135–138 CrossRef.
- F. Guo, K. Cheng, K. Ye, G. Wang and D. Cao, Electrochim. Acta, 2016, 199, 290–296 CrossRef CAS.
- X. Gao, H. Zhang, Q. Li, X. Yu, Z. Hong, X. Zhang, C. Liang and Z. Lin, Angew. Chem., Int. Ed., 2016, 55, 6290–6294 CrossRef CAS PubMed.
- K. Xu, J. Yang and J. Hu, J. Colloid Interface Sci., 2018, 511, 456–462 CrossRef CAS PubMed.
- Z. Yu, H. Li, X. Zhang, N. Liu, W. Tan, X. Zhang and L. Zhang, Biosens. Bioelectron., 2016, 75, 161–165 CrossRef CAS PubMed.
- Y. M. T. A. Putri, T. W. Chamberlain, V. Degirmenci, J. Gunlazuardi, Y. K. Krisnandi, R. I. Walton and T. A. Ivandini, ACS Appl. Energy Mater., 2023, 6, 2497–2507 CrossRef CAS.
- P. Dhandapani, B. Balan, T. Dinadayalane and S. Angaiah, J. Energy Storage, 2022, 56, 105943 CrossRef.
- P. Dhandapani, D. K. Maurya and S. Angaiah, ChemistrySelect, 2022, 7, e20220100 CrossRef.
- P. Dhandapani, A. Udayakumar, S. P. Rajendra, M. S. AlSalhi, J. Z. Guo and S. Angaiah, Sustainable Energy Fuels, 2023, 7, 2368–2377 RSC.
- Y. Qiao, Q. Liu, S. Lu, G. Chen, S. Gao, W. Lu and X. Sun, J. Mater. Chem. B, 2020, 8, 5411–5415 RSC.
- P. Mergenthaler, U. Lindauer, G. A. Dienel and A. Meisel, Trends Neurosci., 2013, 36, 587–597 CrossRef CAS PubMed.
- P. Bjornstad, E. Nehus, L. el ghormli, F. Bacha, I. M. Libman, S. McKay, S. Willi, L. Laffel, S. Arslanian, K. J. Nadeau, S. McKay, M. Haymond, B. Anderson, C. Bush, S. Gunn, H. Holden, S. M. Jones, G. Jeha, S. McGirk, S. Thamotharan, L. Cuttler, E. Abrams, T. Casey, W. Dahms, C. Ievers-Landis, B. Kaminski, M. Koontz, S. MacLeish, P. McGuigan, S. Narasimhan, M. Geffner, V. Barraza, N. Chang, B. Conrad, D. Dreimane, S. Estrada, L. Fisher, E. Fleury-Milfort, S. Hernandez, B. Hollen, F. Kaufman, E. Law, V. Mansilla, D. Miller, C. Muñoz, R. Ortiz, A. Ward, K. Wexler, Y. K. Xu, P. Yasuda, L. L. Katz, R. Berkowitz, S. Boyd, B. Johnson, J. Kaplan, C. Keating, C. Lassiter, T. Lipman, G. McGinley, H. McKnight, B. Schwartzman, F. Bacha, S. Foster, B. Galvin, T. Hannon, A. Kriska, I. Libman, M. Marcus, K. Porter, T. Songer, E. Venditti, R. Goland, D. Gallagher, P. Kringas, N. Leibel, D. Ng, M. Ovalles, D. Seidman, L. Laffel, A. Goebel-Fabbri, M. Hall, L. Higgins, J. Keady, M. Malloy, K. Milaszewski, L. Rasbach, D. M. Nathan, A. Angelescu, L. Bissett, C. Ciccarelli, L. Delahanty, V. Goldman, O. Hardy, M. Larkin, L. Levitsky, R. McEachern, D. Norman, D. Nwosu, S. Park-Bennett, D. Richards, N. Sherry, B. Steiner, S. Tollefsen, S. Carnes, D. Dempsher, D. Flomo, T. Whelan, B. Wolff, R. Weinstock, D. Bowerman, S. Bristol, J. Bulger, J. Hartsig, R. Izquierdo, J. Kearns, R. Saletsky, P. Trief, P. Zeitler, N. Abramson, A. Bradhurst, N. Celona-Jacobs, J. Higgins, M. Kelsey, G. Klingensmith, K. Nadeau, T. Witten, K. Copeland, E. Boss, R. Brown, J. Chadwick, L. Chalmers, S. Chernausek, A. Hebensperger, C. Macha, R. Newgent, A. Nordyke, D. Olson, T. Poulsen, L. Pratt, J. Preske, J. Schanuel, S. Sternlof, J. Lynch, N. Amodei, R. Barajas, C. Cody, D. Hale, J. Hernandez, C. Ibarra, E. Morales, S. Rivera, G. Rupert, A. Wauters, N. White, A. Arbeláez, D. Flomo, J. Jones, T. Jones, M. Sadler, M. Tanner, A. Timpson, R. Welch, S. Caprio, M. Grey, C. Guandalini, S. Lavietes, P. Rose, A. Syme, W. Tamborlane, K. Hirst, S. Edelstein, P. Feit, N. Grover, C. Long, L. Pyle, B. Linder, S. M. Marcovina, J. Harting, J. Shepherd, B. Fan, L. Marquez, M. Sherman, J. Wang, M. Nichols, E. Mayer-Davis, Y. Liu, J. Lima, S. Gidding, J. Puccella, E. Ricketts, R. Danis, A. Domalpally, A. Goulding, S. Neill, P. Vargo, D. Wilfley, D. Aldrich-Rasche, K. Franklin, C. Massmann, D. O'Brien, J. Patterson, T. Tibbs, D. van Buren, M. Palmert, R. Ratner, D. Dremaine and J. Silverstein, Am. J. Kidney Dis., 2018, 71, 65–74 CrossRef CAS PubMed.
- V. B. Juska and M. E. Pemble, Sensors, 2020, 20, 1–28 CrossRef PubMed.
- D. W. Hwang, S. Lee, M. Seo and T. D. Chung, Anal. Chim. Acta, 2018, 1033, 1–34 CrossRef CAS PubMed.
- J. Monzó, I. Insua, F. Fernandez-Trillo and P. Rodriguez, Analyst, 2015, 140, 7116–7128 RSC.
- N. S. Lopa, M. M. Rahman, F. Ahmed, S. C. Sutradhar, T. Ryu and W. Kim, J. Electroanal. Chem., 2018, 822, 43–49 CrossRef CAS.
- J. Zhu, H. Yin, J. Gong, M. S. H. Al-Furjan and Q. Nie, J. Alloys Compd., 2018, 748, 145–153 CrossRef CAS.
- N. I. Chandrasekaran and M. Manickam, Sens. Actuators, B, 2019, 288, 188–194 CrossRef CAS.
- H. Zhu, L. Li, W. Zhou, Z. Shao and X. Chen, J. Mater. Chem. B, 2016, 4, 7333–7349 RSC.
- M. M. Rahman, A. J. S. Ahammad, J. H. Jin, S. J. Ahn and J. J. Lee, Sensors, 2010, 10, 4855–4886 CrossRef CAS PubMed.
- Y. M. T. A. Putri, P. K. Jiwanti, Irkham, J. Gunlazuardi, Y. Einaga and T. A. Ivandini, Bull. Chem. Soc. Jpn., 2021, 94, 2922–2928 CrossRef CAS.
- Y. M. T. A. Putri, J. Gunlazuardi and T. A. Ivandini, IOP Conf. Ser.: Mater. Sci. Eng., 2019, 496, 012051 CrossRef CAS.
- Z. Cui, H. Yin, Q. Nie, D. Qin, W. Wu and X. He, J. Electroanal. Chem., 2015, 757, 51–57 CrossRef CAS.
- Y. Qiao, R. Zhang, F. He, W. Hu, X. Cao, J. Jia, W. Lu and X. Sun, New J. Chem., 2020, 44, 17849–17853 RSC.
- R. Singh and M. M. Ayyub, ACS Appl. Electron. Mater., 2021, 3, 1912–1919 CrossRef CAS.
- S. N. A. M. Yazid, I. M. Isa, S. A. Bakar, N. Hashim and S. A. Ghani, Anal. Lett., 2014, 47, 1821–1834 CrossRef.
- F. Franceschini and I. Taurino, Physics in Medicine, 2022, 14, 100054 CrossRef.
- M. Li, P. W. Yuan, S. H. Guo, F. Liu and J. P. Cheng, Int. J. Hydrogen Energy, 2017, 42, 28797–28806 CrossRef CAS.
- J. Lu, T. Xiong, W. Zhou, L. Yang, Z. Tang and S. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 5065–5069 CrossRef CAS PubMed.
- A. Sakthisabarimoorthi, S. A. M. B. Dhas and M. Jose, Mater. Sci. Semicond. Process., 2017, 71, 69–75 CrossRef CAS.
- I. A. Rahman, P. Vejayakumaran, C. S. Sipaut, J. Ismail and C. K. Chee, Mater. Chem. Phys., 2009, 114, 328–332 CrossRef CAS.
- N. Garg, M. Basu and A. K. Ganguli, J. Phys. Chem. C, 2014, 118, 17332–17341 CrossRef CAS.
- Z. Wu, X. Pu, Y. Zhu, M. Jing, Q. Chen, X. Jia and X. Ji, J. Alloys Compd., 2015, 632, 208–217 CrossRef CAS.
- D. P. Dubal, P. Gomez-Romero, B. R. Sankapal and R. Holze, Nano Energy, 2015, 11, 377–399 CrossRef CAS.
- H. Liang, J. Lin, H. Jia, S. Chen, J. Qi, J. Cao, T. Lin, W. Fei and J. Feng, J. Power Sources, 2018, 378, 248–254 CrossRef CAS.
- D. R. Kumar, K. R. Prakasha, A. S. Prakash and J. J. Shim, J. Alloys Compd., 2020, 836, 155370 CrossRef CAS.
- L. Li, Y. Cheah, Y. Ko, P. Teh, G. Wee, C. Wong, S. Peng and M. Srinivasan, J. Mater. Chem. A, 2013, 1, 10935–10941 RSC.
- R. Atchudan, T. N. J. I. Edison, D. Chakradhar, N. Karthik, S. Perumal and Y. R. Lee, Ceram. Int., 2018, 44, 2869–2883 CrossRef CAS.
- A. Manalu, K. Tarigan, S. Humaidi, M. Ginting, I. P. Manalu and Ikhwanuddin, Mater. Sci. Energy Technol., 2022, 5, 444–451 CAS.
- J. Wang, Y. Zhang, J. Ye, H. Wei, J. Hao, J. Mu, S. Zhao and S. Hussain, RSC Adv., 2016, 6, 70077–70084 RSC.
- W. Huang, Y. Cao, Y. Chen, J. Peng, X. Lai and J. Tu, Appl. Surf. Sci., 2017, 396, 804–811 CrossRef CAS.
- B. Saravanakumar, T. Priyadharshini, G. Ravi, V. Ganesh, A. Sakunthala and R. Yuvakkumar, J. Sol. Gel Sci. Technol., 2017, 84, 297–305 CrossRef CAS.
- N. F. Shoparwe, M. M. Z. Makhtar, S. A. Sata, M. Mohamad and H. Shukor, IOP Conf. Ser. Earth Environ. Sci., 2021, 765, 012102 CrossRef.
- D. Thomas, Z. Rasheed, J. S. Jagan and K. G. Kumar, J. Food Sci. Technol., 2015, 52, 6719–6726 CrossRef CAS PubMed.
- M. P. Kingsley, P. K. Kalambate and A. K. Srivastava, RSC Adv., 2016, 6, 15101–15111 RSC.
- J. Chen, L. Xu, R. Xing, J. Song, H. Song, D. Liu and J. Zhou, Electrochem. Commun., 2012, 20, 75–78 CrossRef CAS.
- M. Sivakumar, R. Madhu, S. M. Chen, V. Veeramani, A. Manikandan, W. H. Hung, N. Miyamoto and Y. L. Chueh, J. Phys. Chem. C, 2016, 120, 17024–17028 CrossRef CAS.
- J. Yang, M. Cho and Y. Lee, Sens. Actuators, B, 2016, 222, 674–681 CrossRef CAS.
- M. Waqas, L. Wu, H. Tang, C. Liu, Y. Fan, Z. Jiang, X. Wang, J. Zhong and W. Chen, ACS Appl. Nano Mater., 2020, 3, 4788–4798 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra08663a |
| This journal is © The Royal Society of Chemistry 2024 |