DOI:
10.1039/D3RA08609G
(Paper)
RSC Adv., 2024,
14, 4930-4945
Biobran-loaded core/shell nanofibrous scaffold: a promising wound dressing candidate
Received
17th December 2023
, Accepted 25th January 2024
First published on 7th February 2024
Abstract
This research examined the effectiveness of Biobran as a bioactive substance that could potentially improve wound healing. It also looked at how Biobran affects the properties of a nanofibrous scaffold made through coaxial electrospinning. This is the first study exploring the use of Biobran in this context and its interaction with nanofibrous scaffolds. The scaffolds were composed of poly(ε-caprolactone) (PCL) in the shell and various concentrations of Biobran blended with polyvinyl alcohol (PVA) in the core. The properties of the scaffolds were characterized by SEM, TEM, FTIR, XRD, TGA, DSC, stress–strain test, WCA, release test, MTT cytotoxicity assay, wound scratching assay, and the dye exclusion method using trypan blue. The scaffolds loaded with Biobran exhibited a more compact and smooth morphology compared with the scaffold without Biobran. The physical interaction and crystallinity of the polymers in the scaffolds were also affected by Biobran in a concentration-dependent manner. This positively influenced their tensile strength, elongation at break, thermal stability, and hydrophilicity. The porosity, water uptake capacity, and WVTR of the nanofibrous scaffolds are within the optimal ranges for wound healing. The release rate of Biobran, which revealed a biphasic release pattern, decreased with increasing Biobran concentration, resulting in controlled and sustained delivery of Biobran from the nanofiber scaffolds. The cell viability assays showed a dose-dependent effect of Biobran on WISH cells, which might be attributed to the positive effect of Biobran on the physicochemical properties of the nanofibrous scaffolds. These findings suggest that Biobran-loaded core/shell nanofiber scaffolds have a potential application in wound healing as an ideal multifunctional wound dressing.
1. Introduction
Skin injury is one of the most serious problems threatening the global medical system.1 Conventional dressings, such as gauze, have a number of drawbacks, including a high absorbent capacity that can cause wound dehydration and promote bacterial proliferation, along with the potential for additional damage to the newly formed epithelium after gauze removal.2,3 Consequently, the development of multi-functional wound dressings that are more effective in healing has become a global necessity.4 The wound dressing should possess certain characteristics and capabilities to be ideal and versatile in terms of wound therapy. These capabilities include protecting the wound from external environmental influences and microbial infection, keeping the wound site moist to prevent it from drying out, absorbing surplus fluids to avoid exudate pooling on the wound, allowing gas exchange, providing suitable performance and mechanical properties, inability to adhere, facilitating removal or disposability without exposing the wound to trauma, being biocompatible and non-toxic, and inducing biological activity that aids in wound healing.1,2,5–10
Electrospinning (ES) has emerged as an attractive method for producing advanced wound dressings due to its ease of use and ability to generate nanofibers from a diverse range of synthetic and/or natural polymers that mimic the structure of the native extracellular matrix (ECM), which is crucial for maintaining cell adhesion and proliferation processes.11 In addition to other benefits, such as the large surface area-to-volume ratio that provides superior drug loading capabilities.1,12,13 The porous nature of electrospun nanofibers (ENs) allows for the exchange of nutrients and gases, the absorption of excess secretions, and the avoidance of pathogen penetration.14 Blending is a common method for incorporating drugs with ENs.15 However, direct ES of the drug-blended polymer solution can result in rapid and uncontrolled drug release, particularly for hydrophilic drugs.16 To address this issue, coaxial electrospinning (CES) has been developed as an improved technique that can create core–shell nanofibers. In CES, the drug is confined to the core, while the shell prevents degradation and burst release. Additionally, CES can boost drug stability, bioavailability, and efficacy.17 By modifying shell properties such as thickness, composition, and porosity, CES can also regulate drug release.18,19
The degradability, exudate absorption capacity, mechanical properties, morphology, diameter, and porosity of the ENs depend on the type of materials used in the ES process and the way they interact with each other, as well as the ES parameters.20,21 The US Food and Drug Administration (FDA) has approved polycaprolactone (PCL) as a biocompatible polymer. It is characterized by its mechanical stability, slow biodegradability, and ease of processing. Despite being a hydrophobic polymer, when cultivated in vivo, it improves cell adhesion and proliferation due to surface hydrolysis, which leads to the formation of hydrophilic surface functional groups.22 Polyvinyl alcohol (PVA) has excellent chemical resistance, high mechanical characteristics, heat stability, biocompatibility, and the capacity to absorb extra wound fluids.23 Because of its water-soluble nature and superior fiber-forming ability, it can act as a carrier for hydrophilic bioactive agents, allowing for high loading efficiency.24
The skin has the capacity to restore its integrity through the wound healing process.25,26 Bacterial infection is a major impediment to wound healing, as it necessitates more antibiotic regimens and longer recovery times.1,27 Conventional antibiotics have become less effective in managing wound infections due to the emergence and spread of antibiotic-resistant microorganisms, which is partly attributed to antibiotic misuse.27,28 The immune system, comprising the innate and adaptive branches, plays a vital role in resolving infection, clearing necrotic tissue, and facilitating tissue repair, which makes it the ideal remedy for this issue. Various immune cells, such as neutrophils, monocytes, macrophages, and lymphocytes, participate in the wound healing process by secreting factors and cytokines that regulate hemostasis, inflammation, angiogenesis, and skin regeneration. However, immunodeficiency or dysregulation can impair wound healing by causing excessive tissue damage and persistent inflammation.29,30
Biobran is an immunomodulator phytochemical derived from the enzymatic modification of rice bran using carbohydrate hydrolyzing enzymes extracted from shiitake mushrooms.31 Biobran is a hydrophilic, non-toxic and safe bioactive agent, as its 50% lethal dose (LD50) is greater than 36 g kg−1.32 Biobran improves the immune system in various ways. It can make natural killer (NK) cells more active and able to recognize and destroy virus-infected cells and cancer cells.33,34 It can also make macrophages, neutrophils, and monocytes more efficient at engulfing and killing bacteria, as well as producing cytokines that help regulate other immune cells.35 Biobran can also stimulate dendritic cells, which are important for presenting antigens to T and B lymphocytes and activating them.36,37 A reduction in lipid peroxidation, an improvement in the antioxidant defense system, and protection against oxidative stress are signs of Biobran antioxidant activity, according to Ghoneum and El Sayed.32 Additionally, Noaman et al. have revealed that exposure to Biobran increases the activity of the antioxidant enzymes catalase, glutathione peroxidase, superoxide dismutase, and glutathione-S-transferase.38 Biobran has been shown to have antiviral properties against a variety of viral diseases, including human immunodeficiency virus (HIV), hepatitis C virus (HCV), influenza, and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Biobran immunomodulatory effects or direct interactions with viral components may act as a mediator for its antiviral properties.31,36,39,40 Owing to the above-mentioned advantageous features of Biobran, it was hypothesized to be a unique biological agent for wound healing.
This paper presents the first evaluation of Biobran as a potential bioactive agent for wound healing. Moreover, it introduces the first incorporation of Biobran into nanofibers and investigates its effect on their attributes. In this context, we synthesized core–shell nanofibrous scaffolds with different concentrations of Biobran in the core by using CES. We conducted a comprehensive analysis of these scaffolds, assessing their morphology, structure, thermal and mechanical properties, surface wettability, and degradation characteristics. To assess biocompatibility, we employed the MTT cytotoxicity assay in two distinct scenarios: WISH cells directly seeded on the nanofibrous scaffolds and WISH cells seeded with nanofibrous scaffold extracts. Furthermore, we assessed the cell migration and attachment abilities using a wound scratch assay and the dye exclusion method with trypan blue, respectively. This investigation provides valuable insights into the potential application of Biobran in wound healing.
2. Materials and methods
2.1. Materials
PCL pellets (Mw ∼ 80 kDa) were supplied from Acros-Organics (Thermo Fisher Scientific, Waltham, MA, USA). 2,2,2-Trifluoroethanol (TFE, 99.0%) was procured from Panreac Quimica, SA (Barcelona, Spain). PVA (Mw = 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–124000, degree of hydrolysis: 86.0–89.0%) was purchased from S.D. Fine-Chem Limited (SDFCL) (Mumbai, India). Ethanol (EtOH, ≥ 99.8%), dimethyl sulfoxide (DMSO, ≥ 99.7%), 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT), and trypan blue were obtained from Sigma-Aldrich (St. Louis, MO, USA). Human epithelial (WISH, ATCC® CCL-25™) was acquired from the American Type Culture Collection (ATCC, Manassas, VA, USA). Dulbecco's phosphate-buffered saline (DPBS, no calcium, no magnesium, pH = 7.0–7.3), trypsin–EDTA (0.25%, 1×), foetal bovine serum (FBS), Dulbecco's Modified Eagle's Medium (DMEM)/high glucose, and penicillin–streptomycin were purchased from Gibco-BRL (Grand Island, NY, USA). Biobran was provided by Daiwa Pharmaceuticals Co., Ltd. (Tokyo, Japan).
000–124000, degree of hydrolysis: 86.0–89.0%) was purchased from S.D. Fine-Chem Limited (SDFCL) (Mumbai, India). Ethanol (EtOH, ≥ 99.8%), dimethyl sulfoxide (DMSO, ≥ 99.7%), 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT), and trypan blue were obtained from Sigma-Aldrich (St. Louis, MO, USA). Human epithelial (WISH, ATCC® CCL-25™) was acquired from the American Type Culture Collection (ATCC, Manassas, VA, USA). Dulbecco's phosphate-buffered saline (DPBS, no calcium, no magnesium, pH = 7.0–7.3), trypsin–EDTA (0.25%, 1×), foetal bovine serum (FBS), Dulbecco's Modified Eagle's Medium (DMEM)/high glucose, and penicillin–streptomycin were purchased from Gibco-BRL (Grand Island, NY, USA). Biobran was provided by Daiwa Pharmaceuticals Co., Ltd. (Tokyo, Japan).
2.2. Core–shell electrospun nanofiber fabrication
Using CES, fiber mats with a PCL shell and a Biobran-loaded PVA core at different concentrations were produced. Shell solution was prepared by vigorously stirring 1.4 g of PCL pellets in 10 ml TFE overnight at 50 °C on a magnetic stirrer. To prepare the core solutions, firstly 1.0 g of PVA was agitated in 10 ml deionized water for 4 hours at room temperature. Subsequently, different concentrations of Biobran (0, 10, 20, and 30 mg ml−1) were added and magnetically stirred overnight at room temperature. The shell and core solutions, after fully dissolving, were poured into separate syringes, which were then attached to the pumps of the electrospinning apparatus (Nano NC laboratory machine, South Korea Republic, ESR 100), and both syringes were connected to the coaxial needle. At 25 °C and 45% relative humidity (RH), electrospinning was carried out by employing a 20 cm distance from the needle to the stationary aluminum foil collector, a 1.2 ml h−1 shell flow rate, a 0.8 ml h−1 core flow rate, and a 25–28 kV voltage. The nanofibrous scaffolds were subsequently dried for two days in a vacuum oven at 40 °C to remove any leftover TFE. According to the Biobran concentration (0, 10, 20, and 30 mg ml−1) in the core solution, the resultant nanofibrous scaffolds were labelled B0, B10, B20, and B30 respectively.
2.3. Characterizations
2.3.1. Field-emission scanning electron microscopy (FE-SEM). The nanofiber structure and shape were investigated using a FE-SEM (Quattro; Thermo Fisher Scientific, Waltham, MA, USA) at 10 kV voltage. The fibers were coated with a thin layer of gold using a sputter coater (model: S150B, Edwards High Vacuum Ltd., England) prior to imaging. Image analysis software (Image J, National Institute of Health, Bethesda, MD, USA) was used to assess the diameter of selected random fibers (n = 100) in the FE-SEM images.
2.3.2. Transmission electron microscopy (TEM). To validate the formation of core–shell nanofibers, the nanofibrous scaffolds samples were dispersed in ethanol and homogenized for 120 s. A drop of the nanofiber suspension (20 μL) was placed on a 200-mesh carbon-coated copper grid, and the excess solvent was blotted. After that, the grid was left to dry at room temperature before being scanned with TEM (JEOL JEM-2100F, Japan), running at 160 kV of accelerating voltage.
2.3.3. Fourier transform infrared spectroscopy (FT-IR). FT-IR (Shimadzu FTIR-8400 S, Kyoto, Japan) at a scanning resolution of 2 cm−1 in the wavenumber range of 4000–400 cm−1 was used to analyze the composition, molecular interactions, and chemical structure of the nanofibrous scaffolds.
2.3.4. X-ray diffraction analysis (XRD). The nanofibrous scaffolds diffraction patterns were obtained using an X-ray diffractometer (X-ray 7000 Shimadzu, Japan) equipped with a Cu Kα radiation source (λ = 1.5418 Å) operating at 40 kV and 30 mA. The patterns were recorded in a 2θ range of 0–100° at a 4° min−1 scanning rate. The degree of crystallinity was determined using the following relation:41Here, Xc is the degree of crystallinity, Ac is the intensity of the crystallization peak, and Aa is the intensity of the amorphous peak.
2.3.5. Thermal behavior. Thermogravimetric analysis (TGA) and its first derivative (DTG), and differential scanning calorimetry (DSC) were acquired by utilizing Discovery SDT 650 (TA Instruments, New Castle, USA). The measurements were taken from room temperature to 800 °C at a heating rate of 10 °C min−1 under a nitrogen atmosphere of 50 ml min−1. The specific heat capacity (Cp) was established using the following relation:42,43| | |
Cp = heat flow × time/weight × temperature
| (2) |
2.3.6. Mechanical properties. A tensile testing machine (Shimadzu UTM, Kyoto, Japan) was utilized to measure the mechanical properties of the nanofibrous scaffolds. The samples were cut into 5 × 1 cm2 rectangles, and their thicknesses were measured with a 1 μm precision electronic micrometer. The samples were vertically positioned between two mechanical gripping, with a 3 cm gauge length left for mechanical loading. Tensile tests were performed using a cross-head movement at a 5 mm min−1 constant extension rate. From stress (σ)–strain (ε) curves, ultimate tensile strength (UTS) and elongation at break (Eb) were calculated, and Young's modulus (E) was the slope of the first 5% linear portion of the curve. Five samples were tested for each type of nanofibrous scaffolds.44
2.3.7. Water contact angle (WCA). Nanofibrous scaffolds wettability assessment was investigated by WCA measurement (n = 3) using the static sessile drop method (droplet = 6 μL) at room temperature. After 1, 3, 6, and 10 s, the images were recorded and carried out with a CCD camera and analysis software equipped with Attension Theta Optical Tensiometer (Biolin Scientific Company, Finland).
2.3.8. Total porosity determination. The total porosity (P) of nanofibrous scaffolds was measured according to the liquid displacement method using eqn (4).44,45 Briefly, the samples were cut into a square shape from nanofibrous scaffolds sheets, and the volume (V) was calculated by measuring the length, width, and height of the sample with a vernier caliper. The sample was weighed (Wi) and soaked in absolute ethanol at room temperature for up to 6 hours. After soaking, the samples were placed on filter paper to remove residual ethanol and weighed again (Wa).| | |
P = (Wa − Wi) × 100/(ρ × V)
| (4) |
where, ρ represents the density of the ethanol at room temperature (n = 5).
2.3.9. Water uptake capacity. To investigate the exudate absorption ability of nanofibrous scaffolds, the percentage of water uptake (W%) after 1 day was calculated using eqn (5) based on Haixia et al.46 The samples (n = 3) were punched from nanofibrous scaffolds sheets, weighed (Wb), and placed in DPBS at 37 °C for the predefined time. After the time interval, the samples were taken out and filter paper was used to eliminate excess wetness before weighing the samples again (Wa).| | |
W% = (Wa − Wb) × 100/Wb
| (5) |
2.3.10. Water vapor transmission rate (WVTR). The WVTR of nanofibrous scaffolds was measured in accordance with American Society for Testing and Materials (ASTM) standards.47 The sample (n = 3) was cut into a disc shape and inserted into the mouth of a vial containing deionized water. The vial was then tightly packed with the sample and placed in an incubator set at a constant temperature of 37 °C and humidity of 79%. The following formula was used to determine the WVTR:| | |
WVTR = (m1 − m2)/(A × t)
| (6) |
where m1 and m2 are the weights of the sample before and after testing, respectively, A is the surface area of the sample, and t is the time interval of testing.
2.3.11. Degradation of nanofibrous scaffolds. The weight loss method was used to quantify scaffold degradation using the ASTM F1635-04A standard.24 Specimen disks of nanofibrous scaffolds with 10 mm diameter (n = 3) were weighted (Wi). The specimens were placed in a 10 ml DPBS solution in an incubator set to 37 °C. The samples were withdrawn from DPBS at specified intervals, dried in an oven for 48 hours, and weighed again (Wd). The following calculation was used to estimate the weight loss percentage of the specimens:| | |
Weight loss (%) = (Wi − Wd) × 100/Wi
| (7) |
2.3.12. In vitro release performance of Biobran-loaded nanofibrous scaffolds. To investigate the in vitro release profile of Biobran, the calibration curve of Biobran was established by monitoring the absorbance values of serially diluted Biobran concentrations (n = 3) at 362 nm. Subsequently, 20 mg of nanofibrous scaffolds (B10, B20, and B30) were placed in a 6 ml DPBS solution and incubated (n = 3) in a shaker incubator (37 °C, 100 rpm). At predefined time intervals, 4 ml of DPBS was withdrawn and replaced with an equal volume of fresh DPBS. The amount of Biobran released at a wavelength of 362 nm was determined using a UV-spectrophotometer-double beam (T80+, PG Instruments Ltd., England, UK).
2.3.13. Cytocompatibility aspects.
2.3.13.1. Cell seeding and nanofibrous scaffolds sterilization. WISH cells were cultured in DMEM supplemented with 10% FBS and 1% Pen–Strep solution in a humidified incubator (5% CO2, 37 °C). 14 mm discs of nanofibrous scaffolds (n = 3) fastened with stainless steel rings in 24-well plates were washed three times with DPBS after 12 hours of UV light exposure.
2.3.13.2. Direct cytotoxicity assay. The MTT assay was used to quantify the viability of WISH cells on the sterilized nanofibrous scaffolds (n = 3). The nanofibrous scaffolds were submerged in a culture medium overnight to enhance cell adherence prior to seeding cells at a density of 1 × 104 cells per well for 1, 2, and 3 days. Each well received 250 μL of MTT solution (5 mg ml−1) and was incubated at 37 °C after rinsing with DPBS. After 4 h, 750 μL of DMSO was added to each well to dissolve the formazan crystals. The supernatants were transferred to a 96-well plate, and their optical densities at 570 nm were measured with a microplate reader (Multiskan MK3, Thermo Fisher, Waltham, MA, USA).
2.3.13.3. Indirect cytotoxicity assay. To acquire the extracts, sterilized nanofibrous scaffolds (n = 3) were immobilized in 24-well plates by stainless steel rings and immersed in DMEM for 24 hours. The WISH cell line was cultured in 96-well plates for 24 hours with 100 μL of culture medium per well. After this time, the DMEM was replaced by extracts. After 24 hours of cultivation with extracts, cells were rinsed twice by adding 100 μL of DPBS, and an MTT cytotoxicity assay was performed. To obtain a positive control (n = 3), a sterile solution of 0.1% Triton X-100 in DMEM was prepared and incubated for 24 hours. A negative control was created by incubating DMEM for 24 hours (n = 3).
2.3.13.4. Cell attachment. The dye exclusion method using trypan blue was used to evaluate the viable cell count attached to the nanofibrous scaffolds.48,49 WISH cells were seeded on nanofibrous scaffolds (n = 3) into a 24-well plate at a density of 1 × 104 cells per well for 1, 2, and 3 days. The scaffolds were transferred to another well after each time point, rinsed with DPBS, and trypsinized for 3 minutes. Then, 500 μL of DMEM was added to stop the trypsinization process. After centrifuging the cell suspension, DMEM was added to the cell pellet, followed by trypan blue. A hemocytometer was used under a microscope to count the quantity of live and dead cells; dead cells were stained blue. Untreated cells were used as a positive control (100% viable) in the study.
2.3.13.5. Scratch wound assay. To investigate the effect of nanofibrous scaffolds on WISH cell migration, a scratch wound assay was conducted. WISH cells were seeded into a 24-well plate at a density of 4 × 104 cells per well and cultured until they formed a confluent monolayer. The monolayer of cells was then scraped in a straight line using the tip of a sterile pipette, and any cell debris was removed by washing with DPBS. Subsequently, 2 ml of serum-free medium was added to each well, followed by the addition of sterilized samples of nanofibrous scaffolds. The cell migration was recorded using a microscope, and the results were compared to untreated scratched cells. The wound closure area was calculated using ImageJ software.
2.4. Statistical analysis
Statistical analysis of the data was conducted using Costate software. A one-way ANOVA was employed to compare multiple groups, followed by the LSD test. The data were presented as the mean ± standard deviation (SD). In all evaluations, statistical significance was determined by a p-value of less than 0.05.
3. Results and discussion
3.1. Morphology investigation and core–shell demonstration
The morphology of PVA/PCL core/shell nanofiber scaffolds was significantly impacted by Biobran at different concentrations (10, 20, and 30 mg ml−1). Fig. 1 illustrates these morphological changes. The SEM images in Fig. 1A, show that the B0 nanofibrous scaffold has highly interconnected nanofibers and secondary pores in the individual fibers. These features decreased as the Biobran concentration increased. Conversely, fiber homogeneity (i.e., the fiber diameter range becomes less dispersed as in Fig. 1 and 2) and average diameter increased with increasing Biobran concentration up to 20 mg ml−1 (B0 = 213.8 ± 168.6 nm, B10 = 307.5 ± 147.3 nm, B20 = 365.7 ± 119.1 nm). However, at 30 mg ml−1, the average diameter decreased (B30 = 285.8 ± 128.3 nm). The irregularity of the nanofibers in diameter and their high interconnectivity are due to the swelling influence of the PVA extrusion, which affects the shell stretching and consequently the jet bending instability.50–52 The addition of Biobran increases the concentration of the core, which in turn increases viscosity. This reduces the extrudate swell effect of PVA and increases the uniformity and mean diameter of the nanofibers.53,54 However, at 30 mg ml−1, the viscosity increase causes the core solution to drip from the tip of the coaxial needle, hindering the electrospinning process. To overcome this issue, an electrical voltage of 28 kV was used, resulting in a reduction in fiber diameter.6,55,56 The formation of secondary pores in the PVA/PCL core/shell nanofiber scaffolds was achieved based on vapor-induced phase separation (VIPS), non-solvent-induced phase separation (NIPS), and breath figure (BFs) mechanisms. The water present in the PVA core solution creates a highly humid environment for the PCL shell. By increasing the flow rate of the PVA core solution, water molecules in the core rapidly diffuse into the PCL shell before TFE evaporation. This diffusion causes PCL and water to separate into phase separation zones. As the PCL shell solidifies, water evaporation leads to pore development. Due to the initial high-water content and pore formation during the early stages of electrospinning, the pores exhibit folded and long elliptical shapes. Additionally, pores are formed through water vapor condensation on the shell surface, facilitated by a relative humidity of 45% and the high volatility of TFE.57–61 The high hygroscopicity of Biobran reduces water diffusion and evaporation, slowing down the nanofiber drying process. Consequently, both pore size and density decrease as Biobran concentration increases.57 The core–shell morphology of the nanofibrous scaffolds was confirmed by TEM images in Fig. 1, which shows that the PVA and different concentrations of Biobran loaded on the PVA were fully encapsulated by the PCL shell.
 |
| | Fig. 1 Images of (A–D) FE-SEM and (E–H) TEM for nanofibrous scaffolds. B0 (A and E), B10 (B and F), B20 (C and G), and B30 (D and H). Dashed black circles represent the location of secondary pores. | |
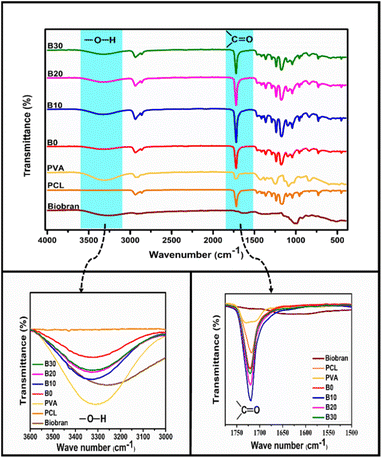 |
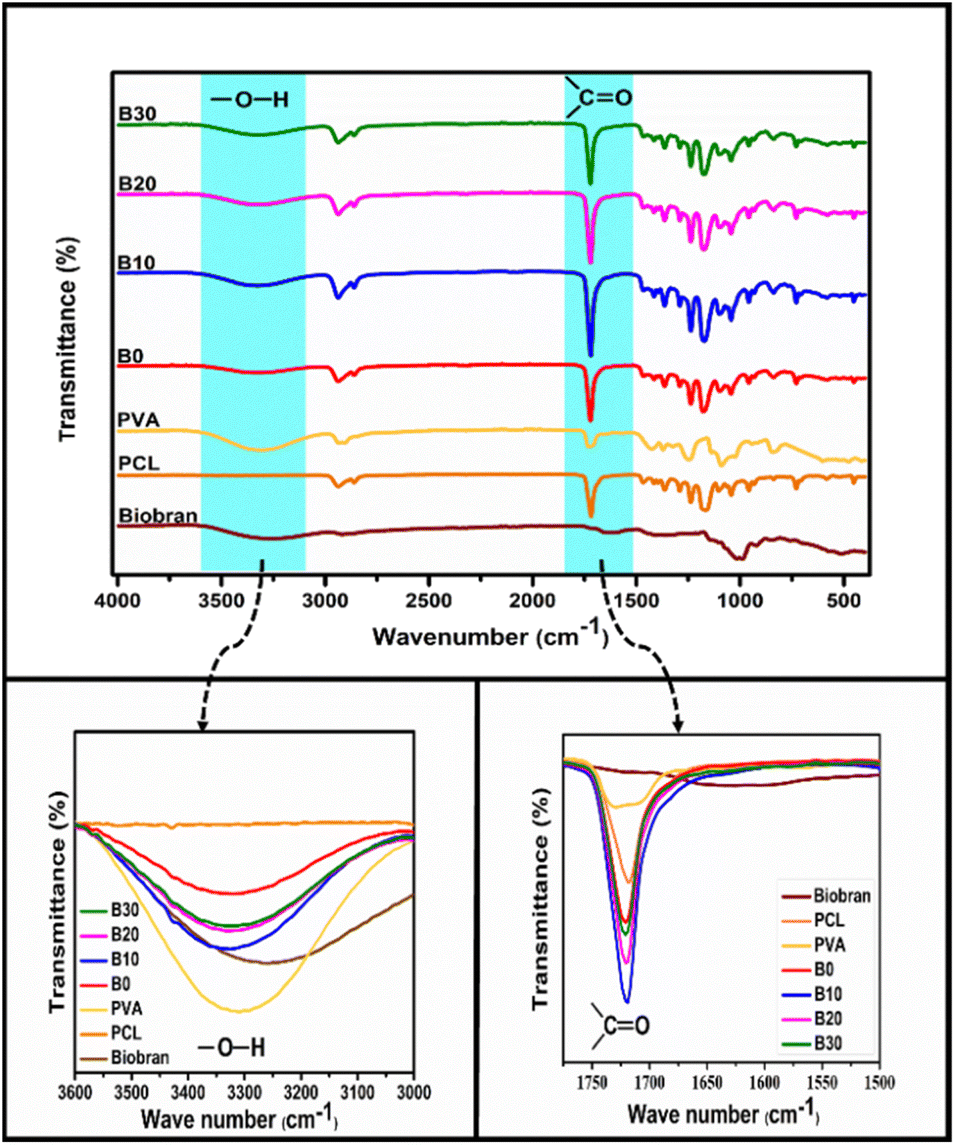
| | Fig. 2 FT-IR spectra of Biobran and nanofibrous scaffolds. | |
3.2. FT-IR spectroscopy
FTIR spectra of PCL, PVA, Biobran, B0, B10, B20, and B30 were utilized, as shown in Fig. 2. The FTIR spectra of Biobran exhibited characteristic peaks at 3280 cm−1 (O–H stretch), 2905 cm−1 (C–H stretch), 1710 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch), 1630 cm−1 (absorbed moisture), 1447 cm−1 (C–H bend), 1360 cm−1 (O–H bend), and 1145 cm−1 (C–O–C stretch). Additionally, peaks at 844 cm−1 and 1072 cm−1 correspond to the glycosidic link and β(1→4) linkages between the monomers.62 The band from 988 cm−1 to 1016 cm−1 is related to the arabinose side chain O-3 on xylose.63 The PCL spectra displays strong bands attributed to CH2 stretching at 2937 cm−1 and 2855 cm−1, CO stretching at 1717 cm−1, C–O–C stretching at 1234 cm−1 and 1165 cm−1 and O–C stretching at 1042 cm−1 and 955 cm−1. The main peaks of PVA were observed at 3310, 2930, 1723, 1420, 1374, 1089, and 838 cm−1. These peaks are assigned to O–H stretch, C–H stretch, CO stretch of the remaining vinyl acetate,64 C–H bend, O–H bend, C–O stretch, and C–C stretching, respectively. In B0 spectrum, no new bands were observed along with the previously identified absorption peaks of PCL and PVA. However, the CO band was detected at 1721 cm−1, and the O–H stretch of PVA shifted to 3320 cm−1, owing to physical interaction between PCL and PVA.65,66 It was observed that the CO band exhibited no shift in the spectra of B10, B20 and B30. Whereas, O–H band was shifted to 3332 cm−1 for B10 and 3323 cm−1 for B20 and B30, which might be due to intermolecular interaction between PCL, PVA, and Biobran along with intramolecular interaction of PVA and Biobran.67,68
O stretch), 1630 cm−1 (absorbed moisture), 1447 cm−1 (C–H bend), 1360 cm−1 (O–H bend), and 1145 cm−1 (C–O–C stretch). Additionally, peaks at 844 cm−1 and 1072 cm−1 correspond to the glycosidic link and β(1→4) linkages between the monomers.62 The band from 988 cm−1 to 1016 cm−1 is related to the arabinose side chain O-3 on xylose.63 The PCL spectra displays strong bands attributed to CH2 stretching at 2937 cm−1 and 2855 cm−1, CO stretching at 1717 cm−1, C–O–C stretching at 1234 cm−1 and 1165 cm−1 and O–C stretching at 1042 cm−1 and 955 cm−1. The main peaks of PVA were observed at 3310, 2930, 1723, 1420, 1374, 1089, and 838 cm−1. These peaks are assigned to O–H stretch, C–H stretch, CO stretch of the remaining vinyl acetate,64 C–H bend, O–H bend, C–O stretch, and C–C stretching, respectively. In B0 spectrum, no new bands were observed along with the previously identified absorption peaks of PCL and PVA. However, the CO band was detected at 1721 cm−1, and the O–H stretch of PVA shifted to 3320 cm−1, owing to physical interaction between PCL and PVA.65,66 It was observed that the CO band exhibited no shift in the spectra of B10, B20 and B30. Whereas, O–H band was shifted to 3332 cm−1 for B10 and 3323 cm−1 for B20 and B30, which might be due to intermolecular interaction between PCL, PVA, and Biobran along with intramolecular interaction of PVA and Biobran.67,68
3.3. XRD analysis and crystallinity investigation
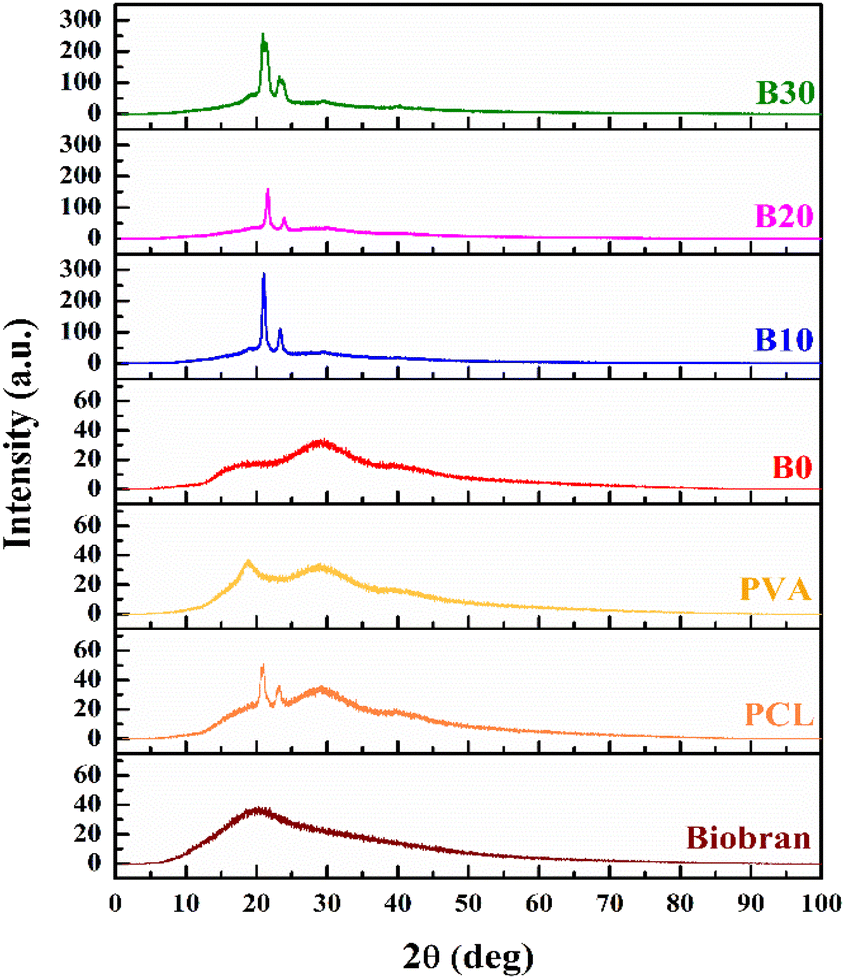
The XRD patterns of nanofibrous scaffolds and Biobran are depicted in the Fig. 3. The XRD pattern of Biobran exhibited a broad peak at 20°, indicating its amorphous structure. For PCL, two sharp peaks were observed at 20.8° and 23.1°, along with two broad peaks at 29.1° and 39.9°, with a low crystallinity of 6.41%. PVA showed two broad peaks at 18.7° and 29.1°, indicating its low crystallinity (8.83%). The low crystallinity of the electrospun polymers is due to the evaporation rate of the solvent used for electrospinning.69 In the case of B0, two broads peaks were observed at 17.7° and 29.1°, and its crystallinity was 7.21%, indicating physical interaction between PCL and PVA, which is consistent with the previously presented FT-IR results.70 B10 showed 19.2% crystallinity along with sharp peaks at 21.0° and 23.3°, in addition to broad peaks at 19.7°, 29.5°, and 39.9°. The XRD pattern for B20 displayed sharp peaks at 21.5° and 23.9° along with broad peaks at 29.4°, and its crystallinity was 12.2%, making the two broad peaks at 19.7° and 39.9° barely visible. For B30, there are sharp peaks at 20.9° and 23.4°, as well as broad peaks at 19.3°, 29.6°, and 40.0°, with 25.7% crystallinity. The change in the intensities and positions of peaks along with crystallinity may be attributed to the physical interaction between Biobran and polymers,71,72 which is in accordance with the FT-IR data and the mean diameter of NFs previously reported.
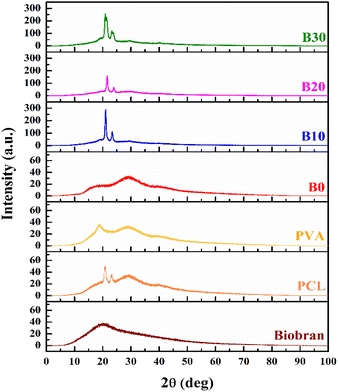 |
| | Fig. 3 XRD patterns of nanofibrous scaffolds and Biobran. | |
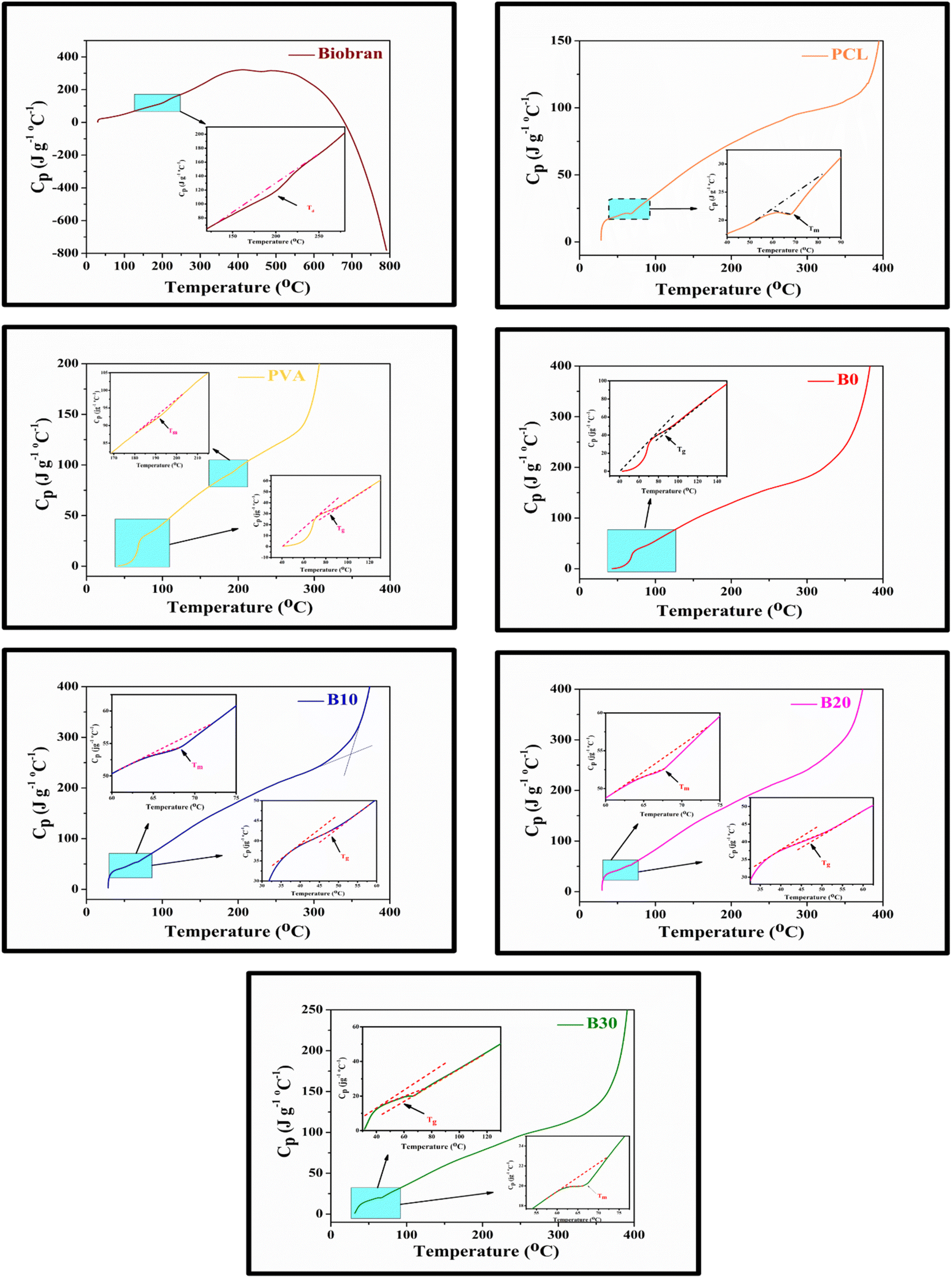
3.4. Thermal characteristics investigation
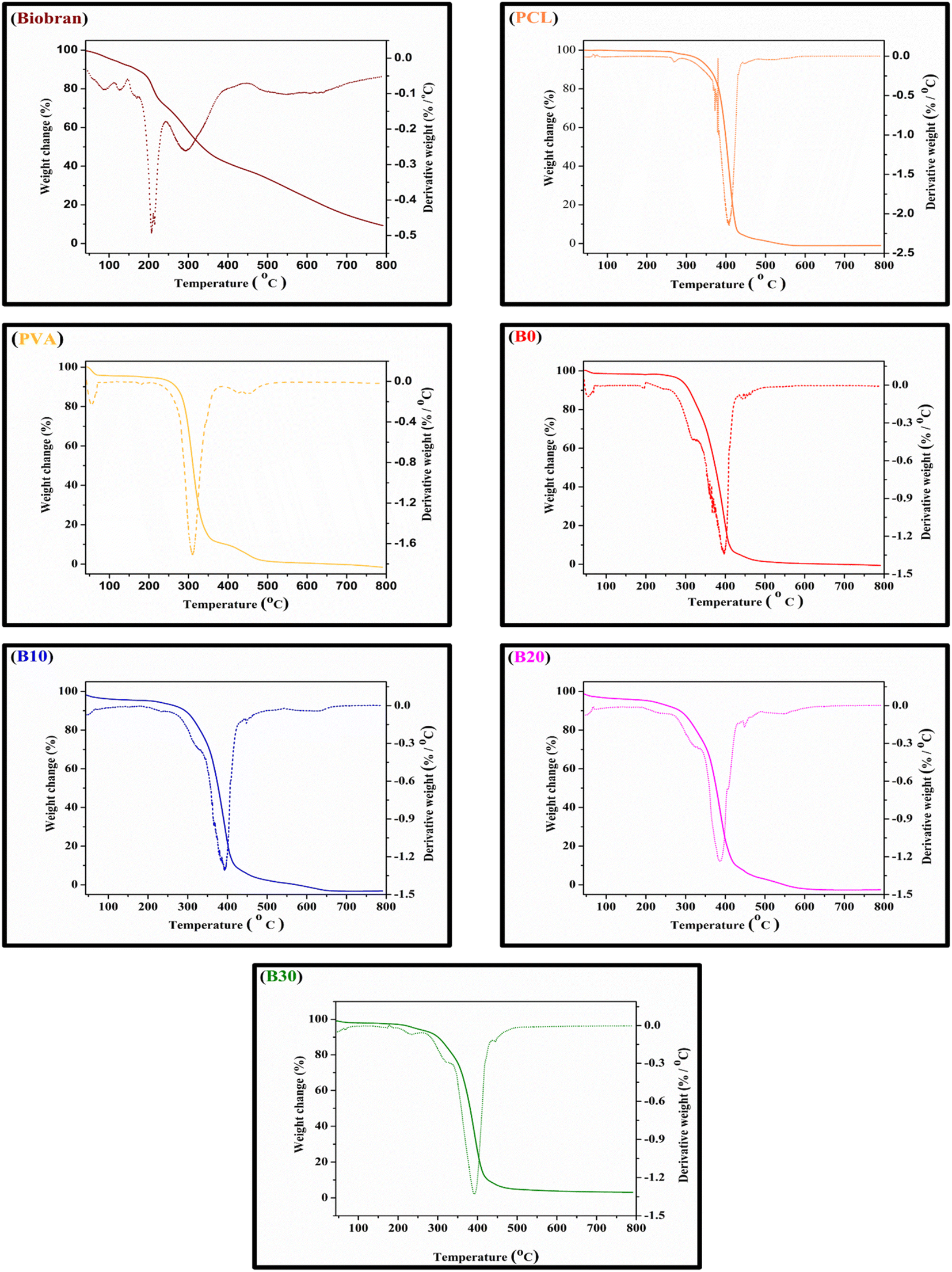
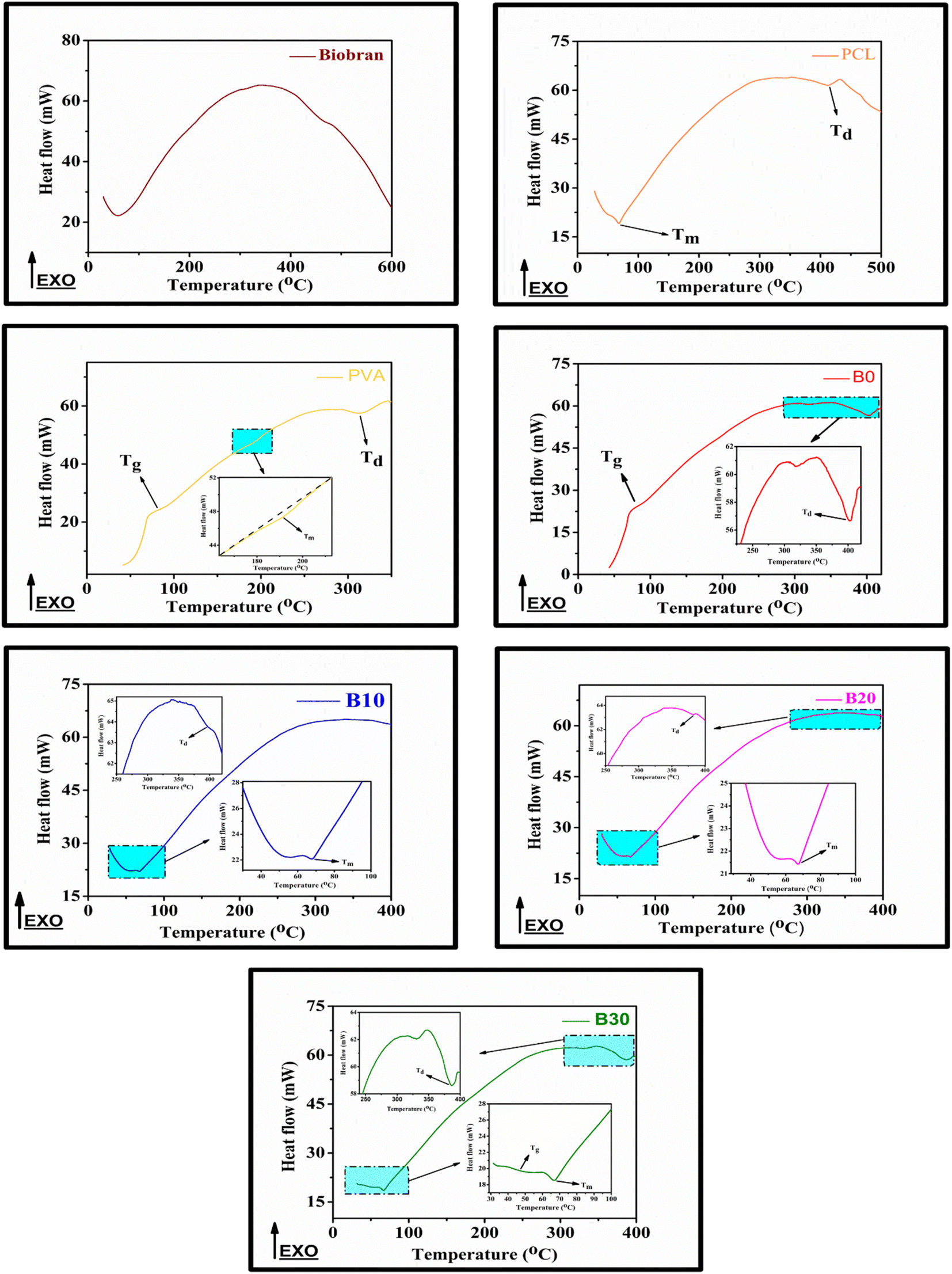
All samples revealed multiple decomposition stages in the representative TGA curves, as illustrated in the Fig. 4–6 and Table 1. The first stage of weight loss might be attributable to absorbed water evaporation.73 The subsequent stages correspond to more intricate processes. As the elimination of H2O and residual acetate groups, as well as chain scission reactions in the case of PVA.74 Furthermore, in the case of PCL, polyester chain rupture and unzipping depolymerization occur.75 Biobran, on the other hand, had a broad mass loss interval and, interestingly, a higher Tmax (temperature at 50% weight loss) than PVA. This could be due to the phenolic compounds in the Biobran structure, such as ferulic acid, which is cross-linked to arabinoxylan.40,76 The increase in Tmax values, shown in Table 1, with increasing Biobran concentration indicates that the thermal breakdown rate of the nanofibrous scaffolds decreased with increasing Biobran content. PVA DSC thermogram exhibited a glass transition temperature (Tg = 82.4 °C), lower than B0 (Tg = 86.0 °C). This shift results from the physical interactions of PCL and PVA, which reduce the mobility of the chain.77 The glass transition temperature decreased significantly with B10 (Tg = 45.1 °C) and then began to increase slightly with increasing Biobran concentration (Tg for B20 = 46.4 °C, Tg for B30 = 55.8 °C). This is because Biobran's side branching introduces defects into the crystal, resulting in free volumes and subsequently increasing chain mobility.78 The decrease in Tm with increasing Biobran concentration, as mentioned in Table 1, supports this interpretation.79,80 The slight increase in Tg is due to the increase in the degree of crystallization previously explained in the XRD results (Fig. 3).81
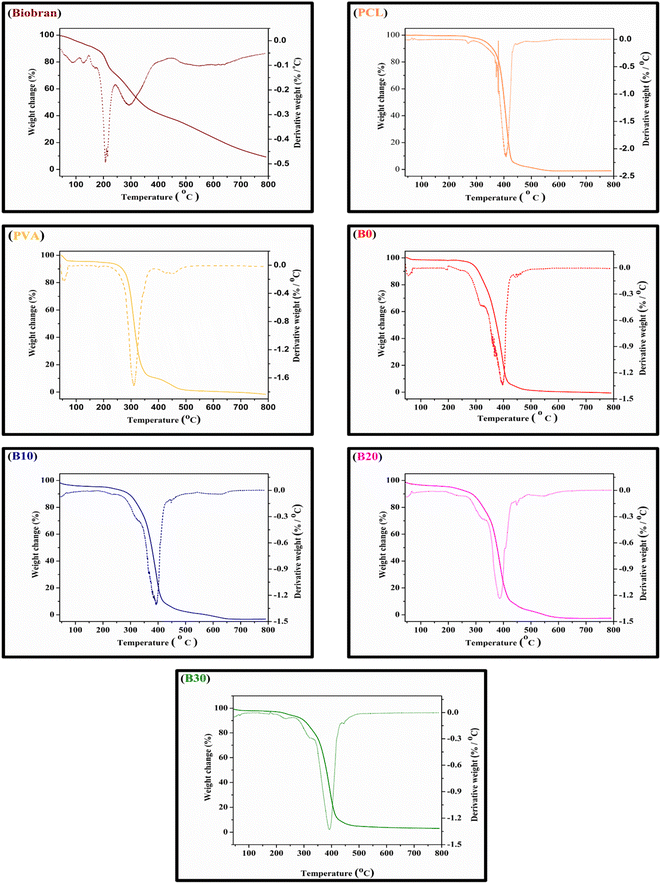 |
| | Fig. 4 Nanofibrous scaffolds and Biobran TGA-DTG thermographs. | |
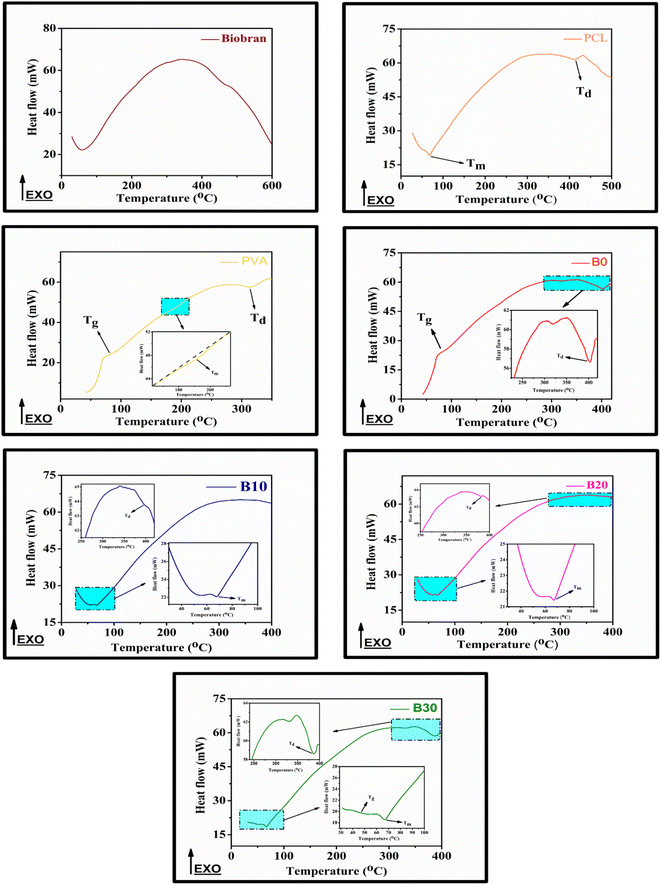 |
| | Fig. 5 Nanofibrous scaffolds and Biobran DSC thermographs. | |
Table 1 Thermal statistics of nanofibrous scaffolds and Biobran, Ti (°C) = the stage onset temperature, W (%) = the weight loss percentage, Cp = specific heat capacity, Tmax = temperature at 50% weight loss, and inflection point (Ip) = the peak point that represents the greatest rate of change on the first derivative weight loss curve
| Thermal characters |
Biobran |
PCL |
PVA |
B0 |
B10 |
B20 |
B30 |
| TGA |
1st stage |
Ti (°C) |
29.8 |
28.2 |
31.5 |
30.9 |
28.7 |
29.6 |
31.5 |
| W (%) |
8.1 |
3.8 |
5.6 |
1.8 |
4.7 |
4.6 |
2.8 |
| 2nd stage |
Ti (°C) |
150.9 |
324.2 |
255.5 |
279.5 |
200.5 |
193.1 |
205.0 |
| W (%) |
18.8 |
91.1 |
83.3 |
92.3 |
2.5 |
4.0 |
4.0 |
| 3rd stage |
Ti (°C) |
231.9 |
437.2 |
371.1 |
425.1 |
290.6 |
285.0 |
295.8 |
| W (%) |
30.8 |
6.0 |
9.5 |
3.7 |
88.7 |
85.4 |
85.2 |
| 4th stage |
Ti (°C) |
391.7 |
— |
506.1 |
479.8 |
465.6 |
456.3 |
476.2 |
| W (%) |
23.7 |
1.3 |
1.7 |
4.0 |
6.0 |
5.11 |
| 5th stage |
Ti (°C) |
653.1 |
— |
— |
— |
— |
— |
— |
| W (%) |
9.0 |
| Tmax (°C) |
333.7 |
402.2 |
312.5 |
375 |
377.5 |
376.8 |
380.9 |
| DTG |
Ip (°C) |
210.6 and 293.9 |
407.3 |
310.0 |
396.1 |
392.3 |
386.2 |
393.6 |
| DSC |
Tg (°C) |
— |
— |
82.4 |
86.0 |
45.1 |
46.4 |
55.8 |
| Cp (J °C−1 g−1) |
32.4 |
44.5 |
41.2 |
40.6 |
18.5 |
| Tm (°C) |
Ti (°C) |
— |
59.5 |
183.1 |
— |
64.2 |
63.1 |
61.9 |
| Cp (J °C−1 g−1) |
21.4 |
89.2 |
— |
53.0 |
50.8 |
19.9 |
| ΔHm (J g−1) |
— |
30.9 |
14.0 |
— |
2.98 |
2.22 |
9.1 |
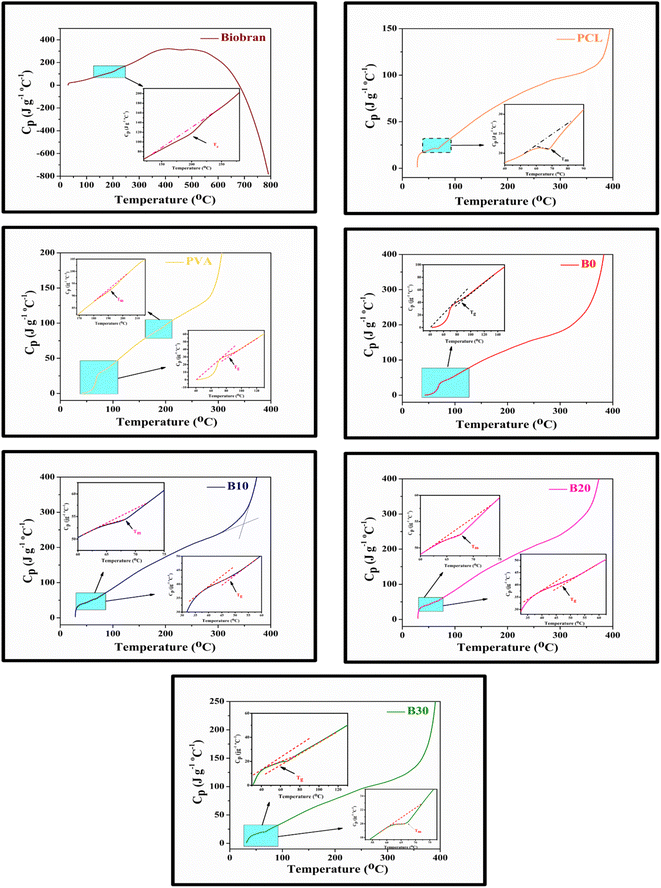 |
| | Fig. 6 Nanofibrous scaffolds and Biobran Cp thermographs. | |
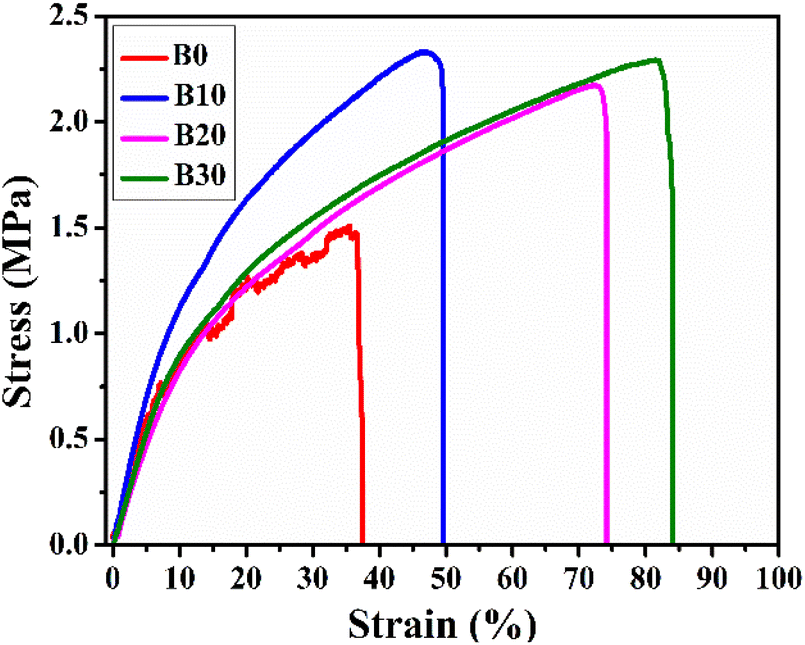
3.5. Mechanical behavior identification
The scaffolds must be sufficiently strong to maintain their structure when placed on the wounds and easily movable without causing damage to the freshly formed tissue.82 In this context, the stress–strain curves of Biobran and nanofibrous scaffolds are represented in Fig. 7, and their mechanical characteristics are quantified in Table 2. As is clear from Table 2, the inclusion of Biobran enhanced nanofibrous scaffolds mechanical properties and resembled those of skin.83,84 Physical interactions and crystallinity are known to influence the mechanical properties of electrospun nanofibers.85,86 As mentioned earlier, the addition of Biobranincreases the physical interaction and crystallinity of the nanofibrous scaffolds in a concentration-dependent manner, which explains the change in tensile strength with the addition of Biobran. The increase in flexibility with Biobran addition corroborates the findings of the previously explained thermal properties, as the decrease in Tm with the increase in Biobran concentration indicates an increase in nanofibrous scaffold flexibility.87–89 The mechanical properties confirmed the suitability and compatibility of Biobran and the polymers for wound healing applications.
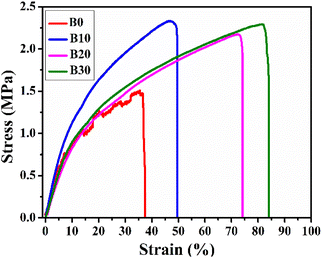 |
| | Fig. 7 Representative stress–strain curves of nanofibrous scaffolds. | |
Table 2 Mechanical parameters clarification
| Samples |
UTS (MPa) |
E (MPa) |
Eb (%) |
| Native skin83,84 |
2.0–16.0 |
4.6–20.0 |
35.0–115.0 |
| B0 |
1.51 ± 0.08 |
12.8 ± 0.44 |
37.55 ± 2.0 |
| B10 |
2.33 ± 0.10 |
14.1 ± 0.73 |
49.67 ± 1.5 |
| B20 |
2.17 ± 0.21 |
9.3 ± 0.28 |
74.13 ± 3.0 |
| B30 |
2.29 ± 0.09 |
10.5 ± 0.48 |
84.06 ± 3.9 |
3.6. Surface wettability
The WCA, which directly reflects the surface wettability, is a critical factor influencing drug delivery, protein absorption, cell adhesion, and cell infiltration.90 As displayed in Fig. 8, the WCA for PCL was found to be 135.58°, 133.48°, 130.22°, and 129.86° at 1, 3, 6, and 10 seconds, respectively. Similarly, the WCA for PVA was measured to be 22.29°, 16.43°, 15.22°, and 9.75° at the same time intervals. The WCA for B0, B10, B20, and B30 were determined to be 113.40°, 73.01°, 48.44°, and 33.18°; 98.86°, 58.89°, 43.93°, and 32.38°; 99.64°, 51.89°, 36.93°, and 11.59°; and 88.29°, 47.50°, 33.04°, and 15.95° at 1, 3, 6, and 10 seconds, respectively. The enhancement in surface wettability due to the addition of Biobran can be ascribed to the alteration in crystallinity, as the degree of crystallinity influences surface characteristics like surface free energy and, consequently, wettability.91–93 Interestingly, the WCA exhibits a remarkable decrease within 10 seconds, indicating rapid water absorption.94 The exceptional water-absorbing capability of PVA,95 combined with the high hygroscopicity of Biobran might be the cause of this result.
 |
| | Fig. 8 WCA images for nanofibrous scaffolds at 1, 3, 6, and 10 s. | |
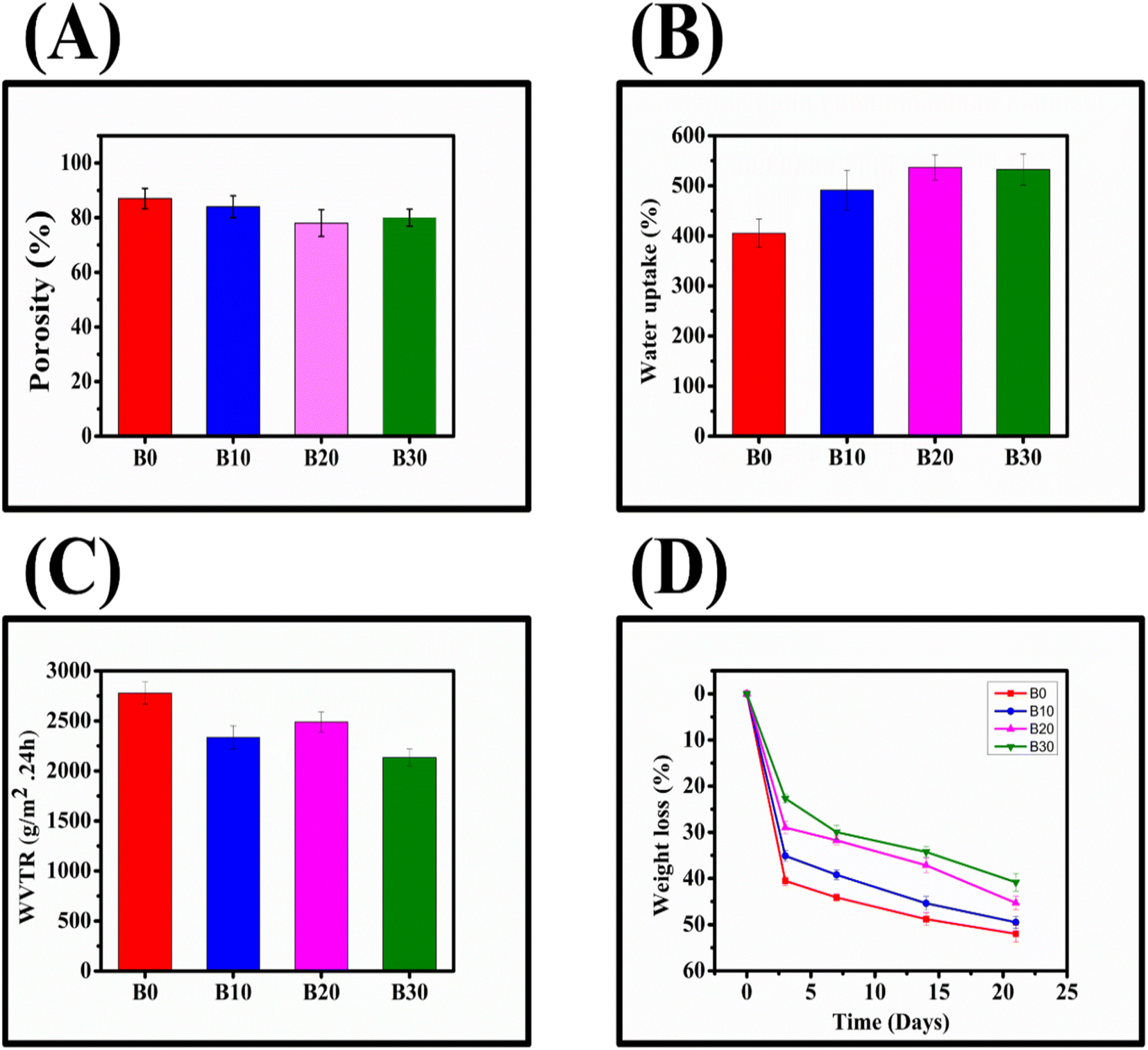
3.7. Porosity, water uptake, WVTR, and degradation of nanofibrous scaffolds
B0, B10, B20, and B30 exhibited high porosity (Fig. 9A), with respective values of 87.91 ± 3.7%, 84.12 ± 4.0%, 78.55 ± 4.9%, and 80.20 ± 3.1%, respectively. All of these percentages fall within the optimum range of the wound dressing porosity (60–90%) required for healing.96 Furthermore, after 24 hours, B0, B10, B20, and B30 demonstrated water uptake capacities of 405.26 ± 28.33%, 491.24 ± 39.67%, 536.69 ± 25.11%, and 572.47 ± 31.05%, respectively (Fig. 9B). To evaluate the ability of nanofibrous scaffolds to permeate moisture, their WVTR was measured (Fig. 9C), and B0, B10, B20, and B30 exhibited 2778.5 ± 110.7 g m−2 × 24 h, 2335.2 ± 115.6 g m−2 × 24 h, 2489.2 ± 100.1 g m−2 × 24 h, and 2134.7 ± 83.9 g m−2 × 24 h, respectively. According to Zahedi et al., wound dressings with a WVTR range of 1999.2–2500.8 g m−2 × 24 h can protect the wound from drying out or pooling by preserving the equilibrium between water evaporation and excess exudate accumulation.10 Rui et al. also reported that wound dressings with a WVTR of 2028.3 ± 237.8 g m−2 × 24 h can promote wound healing by maintaining an optimal moisture content.45 Additionally, the degradation rate of the nanofibrous scaffolds should be commensurate with the tissue regeneration rate.97 Among the nanofibrous scaffolds, B30 exhibited the lowest degradation rate (Fig. 9D), which is acceptable in terms of biological compatibility.97
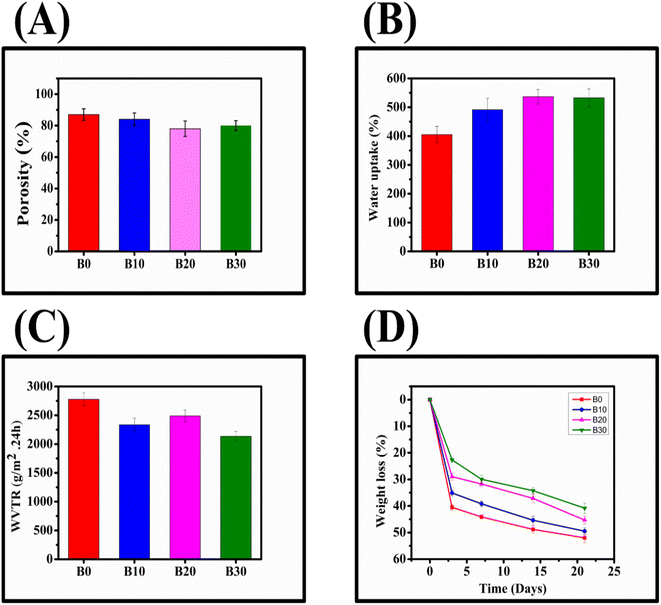 |
| | Fig. 9 Nanofibrous scaffolds porosity (A), water uptake (B), WVTR (C), degradation (D). | |
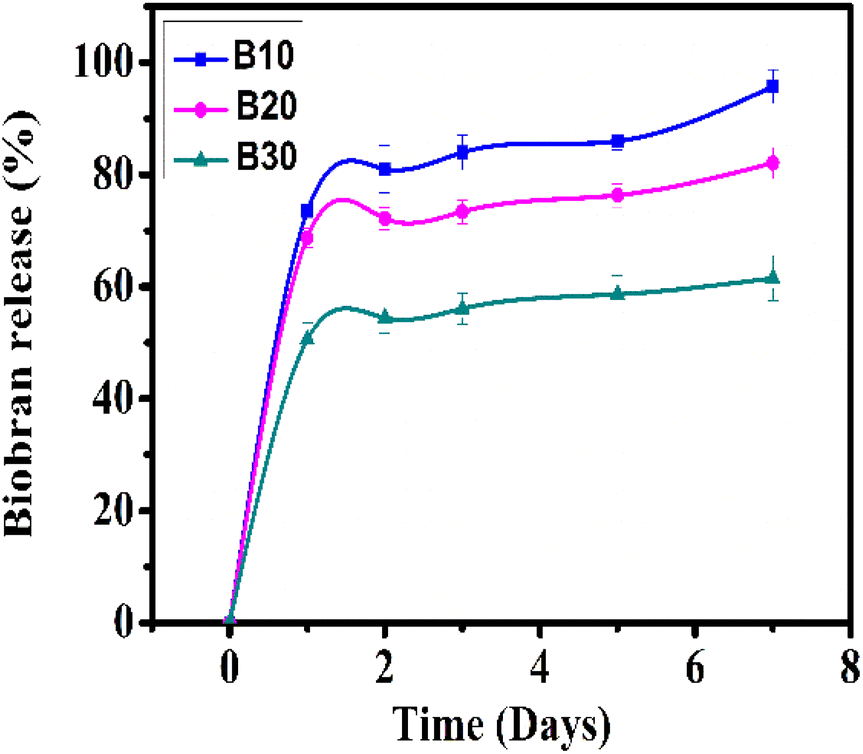
3.8. Biobran-loaded nanofibrous scaffolds release study
The cumulative Biobran release from B10, B20, and B30 is depicted in Fig. 10. All nanofibrous scaffolds showed a biphasic release behavior that consisted of an initial burst release followed by a sustained release phase. In the first stage, Biobran was rapidly released at levels of 73.45 ± 2.9%, 68.75 ± 1.7%, and 50.58 ± 3.0% from the B10, B20, and B30 samples, respectively, within the first 24 h. In the second stage (from 24 h to 7 days), it was constantly released (up to 95.74 ± 3.0%, 82.05 ± 2.8%, and 61.51 ± 4.0%) from B10, B20, and B30, respectively. This suggests that the physical interactions between the PVA and Biobran in the core effectively slowed down the Biobran release from the nanofibrous scaffolds.98–100 Notably, despite the higher hydrophilicity of B30 than B10 and B20, its Biobran release rate is comparatively slower. This can be attributed to its high crystallinity, small fiber diameter, and pore size, which also contributed to its enhanced stability during degradation.101–104 The release rate confirmed the controlled and sustained delivery of Biobran from the nanofiber scaffolds.
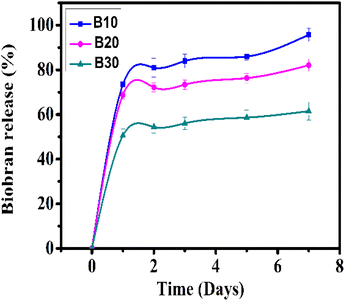 |
| | Fig. 10 Biobran cumulative release study. | |
3.9. Biocompatibility study
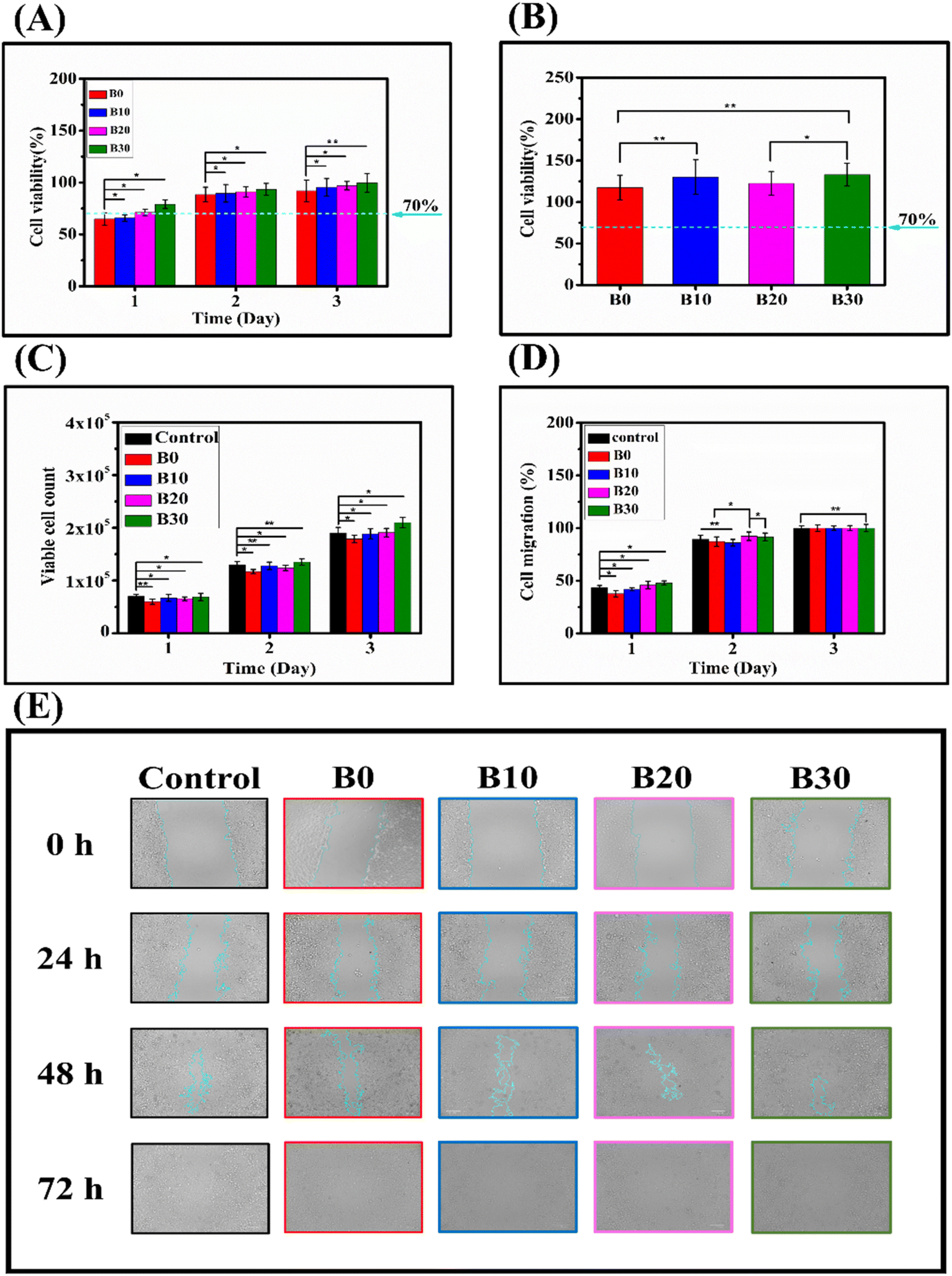
Cell viability should be below 70% to indicate the possibility of cytotoxicity.105 In this study, a direct cytotoxicity assay was conducted to evaluate the influence of nanofibrous scaffolds on the viability of WISH cells. The result of the direct culture MTT cytotoxicity assay is presented in Fig. 11A, which shows the cell viability percentages at 1, 2, and 3 days for B0, B10, B20, and B30. At 1 day, the cell viability percentages were 64.85 ± 6.0%, 65.77 ± 3.1%, 71.18 ± 3.0%, and 78.97 ± 4.2% for B0, B10, B20, and B30, respectively. At 2 days, the cell viability percentages were 88.37 ± 7.1%, 89.56 ± 8.5%, 91.0 ± 4.9%, and 93.38 ± 5.8% for B0, B10, B20, and B30, respectively. Finally, at 3 days, the cell viability percentages were 91.98 ± 10.5%, 95.37 ± 8.4%, 96.94 ± 4.1%, and 99.69 ± 9.0% for B0, B10, B20, and B30, respectively. The initially apparent cytotoxicity can be attributed to the high porosity exhibited by the nanofibrous scaffolds, which enables them to absorb a portion of the formazan crystals formed on their surface.106 Moreover, the hydrophobic surface of the scaffolds may hinder the initial cell attachment.107 These two explanations are supported by the observation that when the MTT cytotoxicity study was performed using extracts of the nanofibrous scaffolds, no cytotoxicity was noticed, as shown in Fig. 11B, which shows that the cell viability percentages were 117.4 ± 15.1%, 130.1 ± 20.8%, 122.5 ± 14.5%, and 132.9 ± 13.7% for B0, B10, B20, and B30, respectively, ruling out the possibility that there are organic solvent residues leading to toxicity or that the nanofibrous scaffolds release cytotoxic substances. Barnes et al. demonstrate that proteins and other media components can be absorbed by highly porous medical carbon absorbents used in extracorporeal therapies, leading to false-positive cytotoxicity results.108 Rafal et al. also showed false-positive MTT cytotoxicity with highly porous nanofibers, especially at 24 h.106 Consequently, the use of scaffold extract in an MTT cytotoxicity assay is a more reliable method for assessing the biocompatibility of highly porous nanofibrous scaffolds, circumventing the issue of formazan crystal absorption by restricting the physical interaction between nanofibrous materials and cytotoxicity assay reagents. Fig. 11C depicts the dye exclusion method with trypan blue that was employed to evaluate the impact of the nanofibrous scaffolds on the WISH cells in direct contact. The viable cell counts for control, B0, B10, B20, and B30 were (7.0 × 104 ± 4.0 × 103), (6.0 × 104 ± 5 × 103), (6.7 × 104 ± 6.5 × 103), (6.55 × 104 ± 3.5 × 103), and (6.9 × 104 ± 7.0 × 103) at day 1, respectively. They increased to (1.3 × 105 ± 6.0 × 103), (1.17 × 105 ± 4.0 × 103), (1.28 × 105 ± 7.0 × 103), (1.24 × 105 ± 5.0 × 103), and (1.35 × 105 ± 6.0 × 103) at day 2, respectively. Finally, they reached (1.9 × 105 ± 1.1 × 104), (1.79 × 105 ± 7.0 × 103), (1.88 × 105 ± 9.5 × 103), (1.91 × 105 ± 8.0 × 103), and (2.1 × 105 ± 1.0 × 104) at day 3, respectively. On the first day of cultivation (Fig. 11C), the nanofibrous scaffolds showed a different growth pattern of WISH cells compared to the control group. The order of cell growth was control, B30, B10, B20, and B0. This trend could be connected to the hydrophilicity of the scaffolds. The hydrophilicity of nanofibers is a decisive factor in cell–material interaction, particularly cell adhesion, as cells prefer to adhere to hydrophilic surfaces.109–111 Hydrophilic nanofibers can promote cell attachment, spreading, proliferation, and differentiation by facilitating protein adsorption and integrin binding.112,113 Conversely, hydrophobic nanofibers can lead to cell detachment, apoptosis, and inflammation by hindering protein conformation and interaction. Therefore, surface modification of nanofibers to adjust their hydrophilicity is a widely adopted strategy to enhance their biocompatibility and functionality for various biomedical applications.107,114 Furthermore, the viable cell counts grew significantly with cultivation time (Fig. 11C), showing that cell proliferation occurred.85 This result is corroborated by the findings in Fig. 11A, where cell viability on the nanofibrous scaffolds increases with the culture period. The cytotoxicity assays demonstrated a dose-dependent effect of Biobran on WISH cells, which might be attributed to the positive effect of Biobran on the physicochemical properties of the nanofibrous scaffolds, as cell adhesion and growth mechanisms depend on nanofibers' material morphology, the diameter of fibers, or even the layout of the scaffold.115,116 The B0 and B10 groups exhibited slower migration compared to other groups. However, after 3 days of culturing, all groups appeared to be the same, as depicted in Fig. 11D and E. This behavior might be attributed to the degradation rate of the scaffolds. An accelerated degradation rate can lead to an increased release of degradation byproducts. These byproducts can alter the local pH and osmolarity, potentially triggering an inflammatory response in the tissue. Such changes can have a significant impact on cell survival, proliferation, and functionality.117 Conversely, maintaining an optimal degradation rate can preserve the mechanical robustness and structural integrity of the nanofibers. This, in turn, can facilitate cell attachment, spreading, and migration, thereby promoting tissue regeneration.118 More importantly, Fig. 11D and E, indicated that the nanofibrous scaffolds did not hinder WISH cell migration, confirming their biocompatibility.
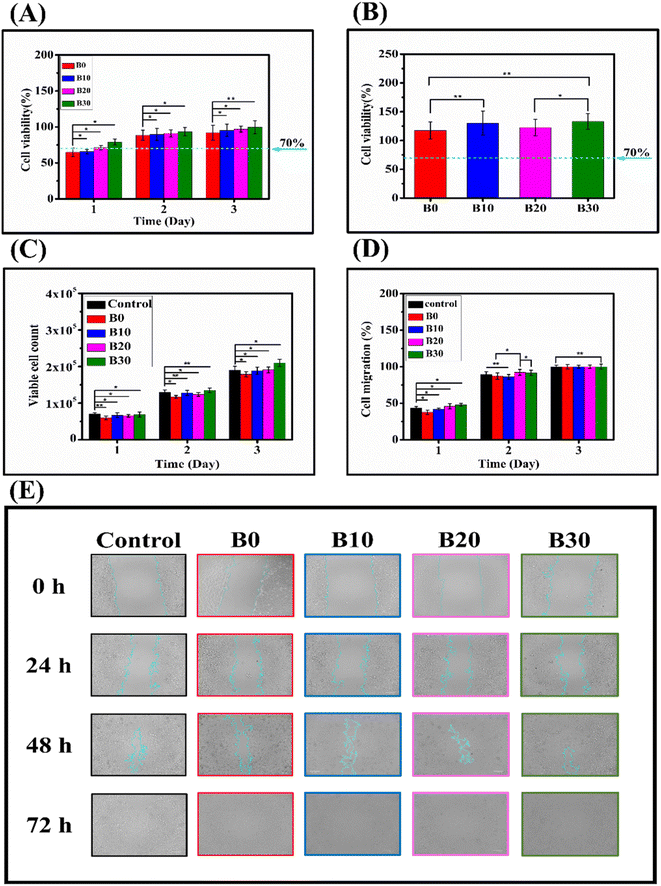 |
| | Fig. 11 MTT assay of nanofibrous scaffolds (A), MTT assay of nanofibrous scaffolds extracts (B), viable cell count using dye exclusion method assay (C), cell migration rate (D), and cell migration images (E). * indicates p < 0.05, while ** indicates p < 0.01. | |
4. Conclusions
In conclusion, this research presents a pioneering study into the feasibility of incorporating Biobran into the core/shell nanofibrous scaffold produced by coaxial electrospinning for wound healing. The scaffolds, composed of PCL in the shell and various concentrations of Biobran blended with PVA in the core, demonstrated enhanced features in terms of morphology, physical interaction, crystallinity, tensile strength, elongation at break, thermal stability, hydrophilicity, and biocompatibility, depending on the Biobran concentration. The B30 scaffold with Biobran (30 mg ml−1) had cell viability percentages of 99.69 ± 9.0% on day three, as well as strong mechanical properties comparable to those of skin and a porosity (80.20 ± 3.1%) that falls within the optimal range of the wound dressing porosity needed for healing. The B30 scaffold also had the lowest degradation rate among the nanofibrous scaffolds, which is favorable for biological compatibility. Moreover, the B30 scaffold offered outstanding moisture content (WVTR of 2134.7 ± 83.9 g m−2 × 24 h and water uptake capacity of 572.47 ± 31.05%). These findings suggest a potential application of the Biobran-loaded core/shell nanofiber scaffold in wound healing as an ideal multifunctional wound dressing.
Author contributions
E.-R. K. participated in conceptualization, supervision, resources, writing, and revision, M. S. A. E. participated in investigation, data analysis, resources, visualization, writing original draft, M. G. participated in conceptualization, supervision, resources, writing, and revision, H. M. A. S. participated in supervision, validation, visualization, revision, A. A. E.-S. participated in investigation, data analysis, resources, visualization, writing original draft, S. S. participated in investigation, data analysis, conceptualization. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
The authors declare no competing interest.
References
- J. Boateng and O. Catanzano, J. Pharm. Sci., 2015, 104, 3653–3680 CrossRef CAS PubMed.
- S. Dhivya, V. V. Padma and E. Santhini, Biomedicine, 2015, 5, 22 CrossRef PubMed.
- P. S. Murphy and G. R. D. Evans, Plastic Surgery International, 2012, 2012, 1–8 CrossRef PubMed.
- K. H. Hamza, A. A. El-Shanshory, M. M. Agwa, M. I. Abo-Alkasem, E. M. El-Fakharany, A. S. Abdelsattar, A. A. El-Bardan, T. S. Kassem, X. Mo and H. M. A. Soliman, Pharmaceutics, 2023, 15, 1518 CrossRef CAS PubMed.
- M. Abrigo, P. Kingshott and S. L. McArthur, ACS Appl. Mater. Interfaces, 2015, 7, 7644–7652 CrossRef CAS PubMed.
- C. Gao, L. Zhang, J. Wang, M. Jin, Q. Tang, Z. Chen, Y. Cheng, R. Yang and G. Zhao, J. Mater. Chem. B, 2021, 9, 3106–3130 RSC.
- A. Vasconcelos and A. Cavaco-Paulo, Appl. Microbiol. Biotechnol., 2011, 90, 445–460 CrossRef CAS PubMed.
- X. Liu, H. Xu, M. Zhang and D.-G. Yu, Membranes, 2021, 11, 770 CrossRef CAS PubMed.
- J. S. Boateng, K. H. Matthews, H. N. E. Stevens and G. M. Eccleston, J. Pharm. Sci., 2008, 97, 2892–2923 CrossRef CAS PubMed.
- P. Zahedi, I. Rezaeian, S.-O. Ranaei-Siadat, S.-H. Jafari and P. Supaphol, Polym. Adv. Technol., 2010, 21, 77–95 CrossRef CAS.
- E.-S. Ahmed, C. Wei-ming and M. Xiu-mei, J. Donghua Univ., 2014, 31, 566–571 Search PubMed.
- S. Fahimirad and F. Ajalloueian, Int. J. Pharm., 2019, 566, 307–328 CrossRef CAS PubMed.
- S. P. Miguel, R. S. Sequeira, A. F. Moreira, C. S. D. Cabral, A. G. Mendonça, P. Ferreira and I. J. Correia, Eur. J. Pharm. Biopharm., 2019, 139, 1–22 CrossRef CAS PubMed.
- R. Augustine, S. R. U. Rehman, R. Ahmed, A. A. Zahid, M. Sharifi, M. Falahati and A. Hasan, Int. J. Biol. Macromol., 2020, 156, 153–170 CrossRef CAS PubMed.
- A. A. El-Shanshory, M. M. Agwa, A. I. Abd-Elhamid, H. M. A. Soliman, X. Mo and E.-R. Kenawy, Polymers, 2022, 14, 454 CrossRef CAS PubMed.
- F. Topuz and T. Uyar, Pharmaceutics, 2018, 11, 6 CrossRef PubMed.
- J. Li, Y. Liu and H. Abdelhakim, Molecules, 2022, 27, 1803 CrossRef CAS PubMed.
- X. Chen, H. Li, W. Lu and Y. Guo, Nanomaterials, 2021, 11, 1316 CrossRef CAS PubMed.
- B. Pant, M. Park and S.-J. Park, Pharmaceutics, 2019, 11, 305 CrossRef CAS PubMed.
- S. Agarwal and S. Jiang, in Encyclopedia of Polymeric Nanomaterials, Springer Berlin Heidelberg, Berlin, Heidelberg, 2015, pp. 1323–1337 Search PubMed.
- A. Memic, T. Abdullah, H. S. Mohammed, K. Joshi Navare, T. Colombani and S. A. Bencherif, ACS Appl. Bio Mater., 2019, 2, 952–969 CrossRef CAS PubMed.
- B. Joseph, R. Augustine, N. Kalarikkal, S. Thomas, B. Seantier and Y. Grohens, Mater. Today Commun., 2019, 19, 319–335 CrossRef CAS.
- M. Ranjbar Mohammadi, S. Kargozar, S. H. Bahrami and S. Rabbani, Polym. Degrad. Stab., 2020, 174, 109105 CrossRef CAS.
- F. Chogan, T. Mirmajidi, A. H. Rezayan, A. M. Sharifi, A. Ghahary, J. Nourmohammadi, A. Kamali and M. Rahaie, Acta Biomater., 2020, 113, 144–163 CrossRef CAS PubMed.
- Y. Lin, Z. Chen, Y. Liu, J. Wang, W. Lv and R. Peng, Drug Des., Dev. Ther., 2022, 16, 2707–2728 CrossRef PubMed.
- M. Chen, J. Tian, Y. Liu, H. Cao, R. Li, J. Wang, J. Wu and Q. Zhang, Chem. Eng. J., 2019, 373, 413–424 CrossRef CAS.
- Y. P. Afsharian and M. Rahimnejad, Polym. Test., 2021, 93, 106952 CrossRef CAS.
- L. J. Bessa, P. Fazii, M. Di Giulio and L. Cellini, Int. Wound J., 2015, 12, 47–52 CrossRef PubMed.
- K. Raziyeva, Y. Kim, Z. Zharkinbekov, K. Kassymbek, S. Jimi and A. Saparov, Biomolecules, 2021, 11, 700 CrossRef CAS PubMed.
- Z. Wang, F. Qi, H. Luo, G. Xu and D. Wang, Front. Immunol., 2022, 13, 789274 CrossRef CAS PubMed.
- M. Ghoneum, Biochem. Biophys. Res. Commun., 1998, 243, 25–29 CrossRef CAS PubMed.
- M. H. Ghoneum and N. S. El Sayed, Oxid. Med. Cell. Longevity, 2021, 2021, 1–15 CrossRef PubMed.
- A. Elsaid, M. Shaheen and M. Ghoneum, Exp. Ther. Med., 2018, 15(3), 2313–2320 CAS.
- A. Pérez-Martínez, J. Valentín, L. Fernández, E. Hernández-Jiménez, E. López-Collazo, P. Zerbes, E. Schwörer, F. Nuñéz, I. G. Martín, H. Sallis, M. Á. Díaz, R. Handgretinger and M. M. Pfeiffer, Cytotherapy, 2015, 17, 601–612 CrossRef PubMed.
- M. Ghoneum, M. Matsuura and S. Gollapudp, Int. J. Immunopathol. Pharmacol., 2008, 21, 87–95 CrossRef CAS PubMed.
- S. L. Ooi, D. McMullen, T. Golombick, D. Nut and S. C. Pak, Integr. Cancer Ther., 2018, 17, 165–178 CrossRef CAS PubMed.
- D. Cholujova, J. Jakubikova and J. Sedlak, Neoplasma, 2009, 56, 89–95 CrossRef CAS PubMed.
- E. Noaman, N. K. Badr El-Din, M. A. Bibars, A. A. Abou Mossallam and M. Ghoneum, Cancer Lett., 2008, 268, 348–359 CrossRef CAS PubMed.
- H. Salama, E. Medhat, M. Shaheen, A.-R. N. Zekri, T. Darwish and M. Ghoneum, Int. J. Immunopathol. Pharmacol., 2016, 29, 647–653 CrossRef CAS PubMed.
- M. Ghoneum, S. Abdulmalek and H. H. Fadel, Nutrients, 2023, 15, 453 CrossRef CAS PubMed.
- G. Nanni, J. A. Heredia-Guerrero, U. C. Paul, S. Dante, G. Caputo, C. Canale, A. Athanassiou, D. Fragouli and I. S. Bayer, Polymers, 2019, 11, 1069 CrossRef PubMed.
- A. Nour, H. M. Refaat, A. El-Dissouky and H. M. A. Soliman, Ceram. Int., 2023, 49, 26982–26993 CrossRef CAS.
- N. Mahagani, E. Igbokwe, O. Aberefa, V. Bodiba, M. O. Daramola and S. E. Iyuke, J. Phys.: Conf. Ser., 2019, 1378, 022019 CrossRef CAS.
- Z. Yuan, L. Zhang, S. Jiang, M. Shafiq, Y. Cai, Y. Chen, J. Song, X. Yu, H. Ijima, Y. Xu and X. Mo, Smart Materials in Medicine, 2023, 4, 407–426 CrossRef.
- R. Xu, H. Xia, W. He, Z. Li, J. Zhao, B. Liu, Y. Wang, Q. Lei, Y. Kong, Y. Bai, Z. Yao, R. Yan, H. Li, R. Zhan, S. Yang, G. Luo and J. Wu, Sci. Rep., 2016, 6, 24596 CrossRef CAS PubMed.
- H. Xu, F. Zhang, M. Wang, H. Lv, D.-G. Yu, X. Liu and H. Shen, Biomater. Adv., 2022, 136, 212795 CrossRef CAS PubMed.
- B. Yu, C. He, W. Wang, Y. Ren, J. Yang, S. Guo, Y. Zheng and X. Shi, ACS Appl. Bio Mater., 2020, 3, 5383–5394 CrossRef CAS PubMed.
- M. M. Lim, T. Sun and N. Sultana, J. Nanomater., 2015, 2015, 1–10 Search PubMed.
- E. Chong, T. Phan, I. Lim, Y. Zhang, B. Bay, S. Ramakrishna and C. Lim, Acta Biomater., 2007, 3, 321–330 CrossRef CAS PubMed.
- Y. Z. Zhang, X. Wang, Y. Feng, J. Li, C. T. Lim and S. Ramakrishna, Biomacromolecules, 2006, 7, 1049–1057 CrossRef CAS PubMed.
- D. H. Reneker, A. L. Yarin, H. Fong and S. Koombhongse, J. Appl. Phys., 2000, 87, 4531–4547 CrossRef CAS.
- Y. M. Shin, M. M. Hohman, M. P. Brenner and G. C. Rutledge, Polymer, 2001, 42, 09955–09967 CrossRef CAS.
- L. Yi, Y. Wang, Y. Fang, M. Zhang, J. Yao, L. Wang and J. Marek, RSC Adv., 2019, 9, 21844–21851 RSC.
- M. Yousefzadeh and F. Ghasemkhah, in Handbook of Nanofibers, Springer International Publishing, Cham, 2019, pp. 1–58 Search PubMed.
- G. Kumar Sharma and N. Rachel James, in Recent Developments in Nanofibers Research, IntechOpen, 2023 Search PubMed.
- A.-T. Iacob, M. Drăgan, O.-M. Ionescu, L. Profire, A. Ficai, E. Andronescu, L. G. Confederat and D. Lupașcu, Pharmaceutics, 2020, 12, 983 CrossRef CAS PubMed.
- X. Cao, W. Chen, P. Zhao, Y. Yang and D.-G. Yu, Polymers, 2022, 14, 3990 CrossRef CAS PubMed.
- T. Min, L. Zhou, X. Sun, H. Du, X. Bian, Z. Zhu and Y. Wen, Food Res. Int., 2022, 157, 111256 CrossRef CAS PubMed.
- Q. Wang, Z. Zhang, L. Liu, L. Bai, R.-Y. Bao, M.-B. Yang and W. Yang, J. Mater. Chem. A, 2021, 9, 10277–10288 RSC.
- D. Kossyvaki, A. Barbetta, M. Contardi, M. Bustreo, K. Dziza, S. Lauciello, A. Athanassiou and D. Fragouli, ACS Appl. Polym.
Mater., 2022, 4, 4464–4475 CrossRef CAS.
- Y. Li, X. Zhang, Y. Si, J. Yu and B. Ding, ACS Appl. Polym. Mater., 2022, 4, 5557–5565 CrossRef.
- A. Morales-Ortega, E. Carvajal-Millan, Y. López-Franco, A. Rascón-Chu, J. Lizardi-Mendoza, P. Torres-Chavez and A. Campa-Mada, Molecules, 2013, 18, 8417–8428 CrossRef CAS PubMed.
- M. Hussain, F. Saeed, B. Niaz, A. Imran and T. Tufail, Foods, 2022, 11, 3374 CrossRef CAS PubMed.
- M. Koosha, H. Mirzadeh, M. A. Shokrgozar and M. Farokhi, RSC Adv., 2015, 5, 10479–10487 RSC.
- A. M. Abdelghany, A. A. Menazea and A. M. Ismail, J. Mol. Struct., 2019, 1197, 603–609 CrossRef CAS.
- A. Atta, A. M. Abdel Reheem and E. Abdeltwab, Surf. Rev. Lett., 2020, 27, 1950214 CrossRef CAS.
- K. Wu, Y. Li, Q. Zhou, X. Hu and S. Ouyang, J. Hazard. Mater., 2023, 443, 130298 CrossRef CAS PubMed.
- I. Zarandona, D. M. Correia, J. Moreira, C. M. Costa, S. Lanceros-Mendez, P. Guerrero and K. de la Caba, Int. J. Biol. Macromol., 2023, 227, 1070–1077 CrossRef CAS PubMed.
- S. E. Ghafouri, S. R. Mousavi, M. Khakestani, S. Mozaffari, N. Ajami and H. A. Khonakdar, Polym. Eng. Sci., 2022, 62, 4070–4081 CrossRef CAS.
- P. Jha, K. Dharmalingam, T. Nishizu, N. Katsuno and R. Anandalakshmi, Starch - Stärke, 2020, 72(1–2), 1900121 CrossRef CAS.
- A. Pandey, S. Dalal, S. Dutta and A. Dixit, J. Mater. Sci.: Mater. Electron., 2021, 32, 1341–1368 CrossRef CAS.
- P. Naderi, M. Zarei, S. Karbasi and H. Salehi, Eur. Polym. J., 2020, 124, 109502 CrossRef CAS.
- V. Švachová, V. Khunová, D. Pavliňák, Z. Fohlerová and L. Vojtová, Polym. Eng. Sci., 2017, 57, 506–512 CrossRef.
- Z. Peng and L. X. Kong, Polym. Degrad. Stab., 2007, 92, 1061–1071 CrossRef CAS.
- O. Persenaire, M. Alexandre, P. Degée and P. Dubois, Biomacromolecules, 2001, 2, 288–294 CrossRef CAS PubMed.
- K. Werner, L. Pommer and M. Broström, J. Anal. Appl. Pyrolysis, 2014, 110, 130–137 CrossRef CAS.
- J. Tomaszewska, T. Sterzyński, A. Woźniak-Braszak and M. Banaszak, Polymers, 2021, 13, 4336 CrossRef CAS PubMed.
- F. Sugiyama, A. T. Kleinschmidt, L. V. Kayser, D. Rodriquez, M. Finn, M. A. Alkhadra, J. M.-H. Wan, J. Ramírez, A. S.-C. Chiang, S. E. Root, S. Savagatrup and D. J. Lipomi, Polym. Chem., 2018, 9, 4354–4363 RSC.
- H. Abral, A. Atmajaya, M. Mahardika, F. Hafizulhaq, Kadriadi, D. Handayani, S. M. Sapuan and R. A. Ilyas, J. Mater. Res. Technol., 2020, 9, 2477–2486 CrossRef CAS.
- Z. Mahcene, A. Khelil, S. Hasni, P. K. Akman, F. Bozkurt, K. Birech, M. B. Goudjil and F. Tornuk, Int. J. Biol. Macromol., 2020, 145, 124–132 CrossRef CAS PubMed.
- C. Qiao, X. Ma, J. Zhang and J. Yao, Carbohydr. Polym., 2019, 206, 602–608 CrossRef CAS PubMed.
- G.-M. Lanno, C. Ramos, L. Preem, M. Putrinš, I. Laidmäe, T. Tenson and K. Kogermann, ACS Omega, 2020, 5, 30011–30022 CrossRef CAS PubMed.
- S. Chen, B. Liu, M. A. Carlson, A. F. Gombart, D. A. Reilly and J. Xie, Nanomedicine, 2017, 12, 1335–1352 CrossRef CAS PubMed.
- R. B. Trinca, C. B. Westin, J. A. F. da Silva and Â. M. Moraes, Eur. Polym. J., 2017, 88, 161–170 CrossRef CAS.
- Â. Semitela, A. F. Girão, C. Fernandes, G. Ramalho, I. Bdikin, A. Completo and P. A. Marques, J. Biomater. Appl., 2020, 35, 471–484 CrossRef PubMed.
- M. Doostmohammadi, H. Forootanfar, M. Shakibaie, M. Torkzadeh-Mahani, H.-R. Rahimi, E. Jafari, A. Ameri and B. Amirheidari, J. Biomater. Appl., 2021, 36, 193–209 CrossRef CAS PubMed.
- B. N. Jaffur, G. Kumar, P. Jeetah, S. Ramakrishna and S. K. Bhatia, Int. J. Biol. Macromol., 2023, 253, 126781 CrossRef CAS PubMed.
- M. C. Bertolini, N. Zamperlin, G. M. O. Barra and A. Pegoretti, SPE Polymers, 2023, 4(4), 143–155 CrossRef CAS.
- M. Pantoja, N. Boynton, K. A. Cavicchi, B. Dosa, J. L. Cashman and M. A. B. Meador, ACS Appl. Mater. Interfaces, 2019, 11, 9425–9437 CrossRef CAS PubMed.
- S. Skibiński, J. P. Czechowska, M. Guzik, V. Vivcharenko, A. Przekora, P. Szymczak and A. Zima, Sustainable Mater. Technol., 2023, e00722 CrossRef.
- Y. Wang, C. J. Hansen, C.-C. Wu, E. J. Robinette and A. M. Peterson, RSC Adv., 2021, 11, 31142–31151 RSC.
- D. Kołbuk, M. Ciechomska, O. Jeznach and P. Sajkiewicz, RSC Adv., 2022, 12, 4016–4028 RSC.
- M. Farooq, M. Khalid, Y. Yoshinori, F. Wang, M. A. Iqbal, M. N. Sarwar, G. Mayakrishnan and I. S. Kim, ACS Appl. Nano Mater., 2023, 6, 17171–17178 CrossRef CAS.
- S. Tort, D. Han and A. J. Steckl, Int. J. Pharm., 2020, 579, 119164 CrossRef CAS PubMed.
- K. Thakur, A. Rajhans and B. Kandasubramanian, Environ. Sci. Pollut. Res., 2019, 26, 32013–32028 CrossRef CAS PubMed.
- F. Shahverdi, A. Barati, E. Salehi and M. Arjomandzadegan, Int. J. Biol. Macromol., 2022, 221, 736–750 CrossRef CAS PubMed.
- A. Chanda, J. Adhikari, A. Ghosh, S. R. Chowdhury, S. Thomas, P. Datta and P. Saha, Int. J. Biol. Macromol., 2018, 116, 774–785 CrossRef CAS PubMed.
- A. Budiman, N. V. Nurani, E. Laelasari, M. Muchtaridi, S. Sriwidodo and D. L. Aulifa, Polymers, 2023, 15, 3034 CrossRef CAS PubMed.
- C. Li, Y. Liu, T. Weng, M. Yang, X. Wang and W. Chai, Pharmaceutics, 2023, 15, 1949 CrossRef CAS PubMed.
- J. Lv, Y. Fang, M. Wu, X. Ou, W. Zhang, S. Wang, H. Li, L. Shang, M. He and Y. Zhao, Mater. Today Commun., 2023, 34, 105163 CrossRef CAS.
- Y. Herdiana, N. Wathoni, S. Shamsuddin and M. Muchtaridi, Heliyon, 2022, 8, e08674 CrossRef CAS PubMed.
- S. Liu, S. Qin, M. He, D. Zhou, Q. Qin and H. Wang, Composites, Part B, 2020, 199, 108238 CrossRef CAS.
- J. Gu, S. Yagi, J. Meng, Y. Dong, C. Qian, D. Zhao, A. Kumar, T. Xu, A. Lucchetti and H. Xu, J. Membr. Sci., 2022, 654, 120571 CrossRef CAS.
- Z. J. Krysiak and U. Stachewicz, WIREs Nanomedicine and Nanobiotechnology, 2023, 15(1), e1829 CrossRef CAS PubMed.
- A. L. Urzedo, M. C. Gonçalves, M. H. M. Nascimento, C. B. Lombello, G. Nakazato and A. B. Seabra, ACS Biomater. Sci. Eng., 2020, 6, 2117–2134 CrossRef CAS PubMed.
- R. Podgórski, M. Wojasiński and T. Ciach, Sci. Rep., 2022, 12, 9047 CrossRef PubMed.
- M. Ferrari, F. Cirisano and M. C. Morán, Colloids Interfaces, 2019, 3, 48 CrossRef CAS.
- L.-M. Barnes, G. J. Phillips, J. G. Davies, A. W. Lloyd, E. Cheek, S. R. Tennison, A. P. Rawlinson, O. P. Kozynchenko and S. V. Mikhalovsky, Carbon, 2009, 47, 1887–1895 CrossRef CAS.
- P. Nitti, A. Narayanan, R. Pellegrino, S. Villani, M. Madaghiele and C. Demitri, Bioengineering, 2023, 10, 1122 CrossRef CAS PubMed.
- R. Yaseri, M. Fadaie, E. Mirzaei, H. Samadian and A. Ebrahiminezhad, Sci. Rep., 2023, 13, 9434 CrossRef CAS PubMed.
- M. Österberg, K. A. Henn, M. Farooq and J. J. Valle-Delgado, Chem. Rev., 2023, 123, 2200–2241 CrossRef PubMed.
- X. Hu, T. Wang, F. Li and X. Mao, RSC Adv., 2023, 13, 20495–20511 RSC.
- E. Biazar, M. Kamalvand and F. Avani, Int. J. Polym. Mater. Polym. Biomater., 2022, 71, 493–512 CrossRef CAS.
- S. Cai, C. Wu, W. Yang, W. Liang, H. Yu and L. Liu, Nanotechnol. Rev., 2020, 9, 971–989 CrossRef CAS.
- C. A. Bashur, L. A. Dahlgren and A. S. Goldstein, Biomaterials, 2006, 27, 5681–5688 CrossRef CAS PubMed.
- V. Chaurey, F. Block, Y.-H. Su, P.-C. Chiang, E. Botchwey, C.-F. Chou and N. S. Swami, Acta Biomater., 2012, 8, 3982–3990 CrossRef CAS PubMed.
- S. Han, K. Nie, J. Li, Q. Sun, X. Wang, X. Li and Q. Li, Stem Cells Int., 2021, 2021, 1–22 Search PubMed.
- L. Suamte, A. Tirkey and P. J. Babu, Smart Materials in Medicine, 2023, 4, 243–256 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b and
S. Shendya
*b and
S. Shendya