
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA concise update on the synthetic transformation of aurones via asymmetric cycloaddition, annulation, and Michael/Mannich reactions
Deepa
John
,
Kevin
George
 and
Ethiraj Kannatt
Radhakrishnan
*
and
Ethiraj Kannatt
Radhakrishnan
*
Department of Chemistry, Vellore Institute of Technology, Vellore, India. E-mail: johndeepa265@gmail.com; kvngeorge03@gmail.com; ethukr@gmail.com
First published on 20th February 2024
Abstract
This review provides a comprehensive overview of the significance of aurone cores in organic chemistry, highlighting their crucial role as synthetic intermediates. With their innate electrophilic reactivity and convenient accessibility, aurone cores play a vital role in catalysing the development of novel methodologies and facilitating the creation of intricate compounds. The objective of this review is to present a current and insightful compilation that summarizes the progress in aurone synthetic transformations, focusing on diverse cycloaddition ([3 + 2], [4 + 2], [4 + 3], [10 + 2]) and annulation reactions.
Introduction
Aurones 1 (Fig. 1) belong to a subgroup of secondary metabolites categorized within the extensive realm of flavonoids. They are particularly abundant in fruits and flowers, playing a crucial role in botanical pigmentation.1–4 Structurally, aurones feature a benzofuranone heterocyclic pattern and are characterized by incorporating a phenyl unit through a carbon–carbon exocyclic double bond. Notably, the aurone framework holds a special position in medicinal chemistry due to its architectural significance, often linked with various pharmacologically active compounds.5–8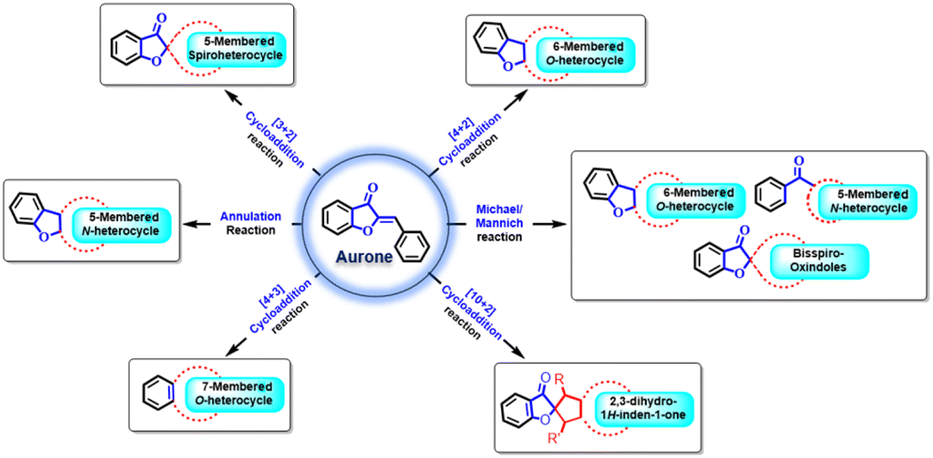 | ||
| Fig. 1 Overview of synthetic transformation of aurones. | ||
A significant breakthrough occurred in 1943 when Geissman and Heaton isolated the first aurone derivative from the flowers of Coreopsis grandiflora.5 Subsequent investigations have unveiled an array of remarkable biological activities demonstrated by aurone derivatives. These activities include anticancer,9–11 antioxidant,12,13 metal ion-chelating,14,15 antitumor,16 anti-tyrosinase,17 antiparasitic,18,19 antibacterial,20,21 antifungal,22 antiviral,23 anti-hormonal,24 anti-inflammatory,25,26 anti-diabetic,27 and anti-obesity.28 These diverse attributes have spurred significant interest in the scientific community, leading to the integration of aurone-based structures into various medicinal products (Fig. 2).24,29
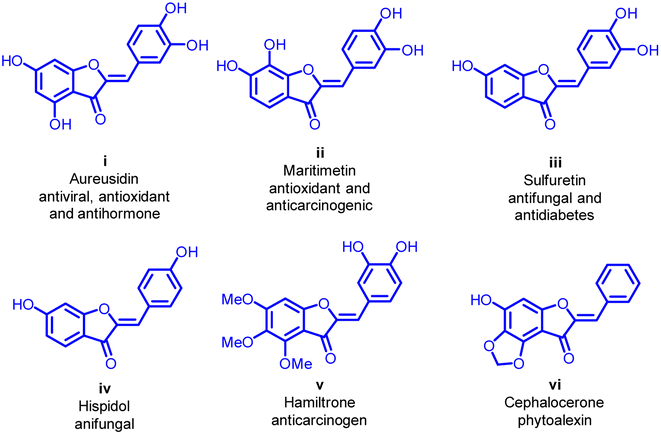 | ||
| Fig. 2 Some aurone molecules being considered as potential drug candidates exhibit significant physiological activity. | ||
In modern synthetic organic research, there is a strong focus on developing new strategies and chemical reactions. Among the numerous types of reactions discovered in the last 90 years, cyclization and cycloaddition reactions have emerged as highly valuable and extensively studied transformations. These reactions have seen remarkable advancements, leading to the creation of complex and diversely functionalized carbocycles and heterocycles. These compounds are particularly important in natural products and pharmaceuticals. For instance, methods involving [2 + 1], [2 + 2], [3 + 2], [4 + 2], [4 + 3], [5 + 2], etc. cycloaddition reactions have been established for constructing small- to medium-sized rings.30 Within this context, the cycloaddition and annulation reactions of aurones have garnered significant interest among synthetic chemists. Their significant electrophilic reactivity and easy availability make them incredibly valuable for developing new methodologies and synthesizing complex natural products. They're particularly useful for creating spiro benzofuranone-carbocyclic or -heterocyclic structures through various cycloaddition and annulation reactions, often assisted by different organo and transition metal catalysts.31–39
Past literature has extensively explored aurones from different angles. In 2003, Boumendjel conducted a meticulous study on aurone synthesis and their biological properties.1 Notably, Boumendjel and Kirsch independently echoed these efforts in 2012.24,40 Subsequently, in 2021, Wenming Zhou and co-workers presented a comprehensive review encompassing aurone synthesis and their biological implications.41 To the best of our knowledge, there has not been any review explaining the synthetic transformation of aurones. Hence, this review aims to offer a timely and insightful compilation that encapsulates the latest advancements in aurone synthetic transformations from 2012 to 2023.
[3 + 2] Cycloaddition reactions (32CA)
The discovery of 32CA reactions dates back to the late 19th century, but it was in 1961 that Huisgen recognized their broad applicability, significance, and underlying mechanism. Huisgen and his team dedicated considerable effort and research to these reactions, leading to their rapid development. Consequently, Huisgen earned the title “father” of “1,3-dipolar cycloadditions”. These reactions involve the interaction between a zwitterionic molecule known as a “1,3-dipole” and a multiple-bond system called a “dipolarophile”. By adding the multiple bond system to a three-atom component (TAC), which is a neutral species consisting of three closely connected heavy nuclei with a minimum electron density of 8 electrons, these reactions have proven highly valuable in synthesizing five-membered heterocyclic compounds.42–45In 2013, Moncef Msadek and co-workers developed a successful strategy for creating a unique category of extensively substituted pyrazole structures 5. They achieved this through a 1,3-cycloaddition reaction involving aurone 1 and aryl diazomethane 3 (Scheme 1). Notably, these reactions exhibited exceptional specificity in terms of the regiochemical outcomes.46
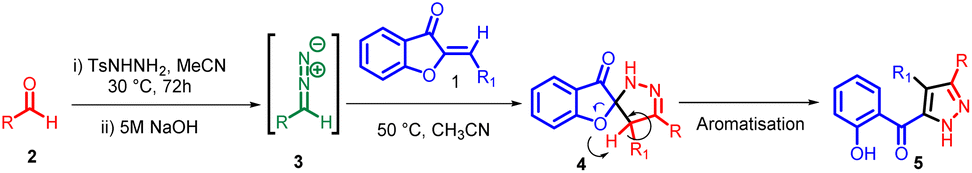 | ||
| Scheme 1 One-pot synthesis of substituted pyrazole derivatives. | ||
Chang Guo et al., in 2014 reported a remarkably selective N-heterocyclic carbene (NHC) 8 catalyzed formal [3 + 2] annulation involving α,β-unsaturated aldehydes 7 and aurones 1, resulting in the formation of spiro-heterocycles (Scheme 2). The spiro heterocycles produced via this method adopt a novel NHC protocol and feature a quaternary stereogenic centre with notably high optical purity. The enantioselectivity observed in the formation of spiroheterocycles 12 is attributed to the NHC precatalyst. Likewise, the reverse configuration of spiroheterocycles can be achieved with equivalent yield and enantioselectivity by employing the enantiomer of the NHC precatalyst.
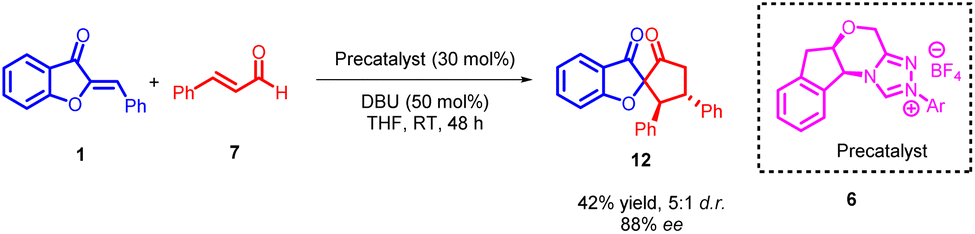 | ||
| Scheme 2 NHC catalyzed [3 + 2] annulations reaction of enals to form spiroheterocycles. | ||
The catalytic cycle, as depicted in Scheme 3, commences with the generation of NHC organocatalyst 8 through the deprotonation of precatalyst salt 6. Introduction of NHC organocatalyst 8 to enal 7 yields NHC-homoenolate 9, prompting a Michael addition from the rear face and subsequent coordination through a hydrogen-bonding interaction, as seen in intermediate 10. Importantly, the stereochemistry observed aligns best with the proposed reaction model illustrated by intermediate 10. Following the creation of a carbon–carbon bond, a tautomerization process leads to the formation of acyl azolium 11. This species is believed to undergo C-acylation, resulting in the generation of the final product 12 while simultaneously regenerating NHC organocatalyst 8.47
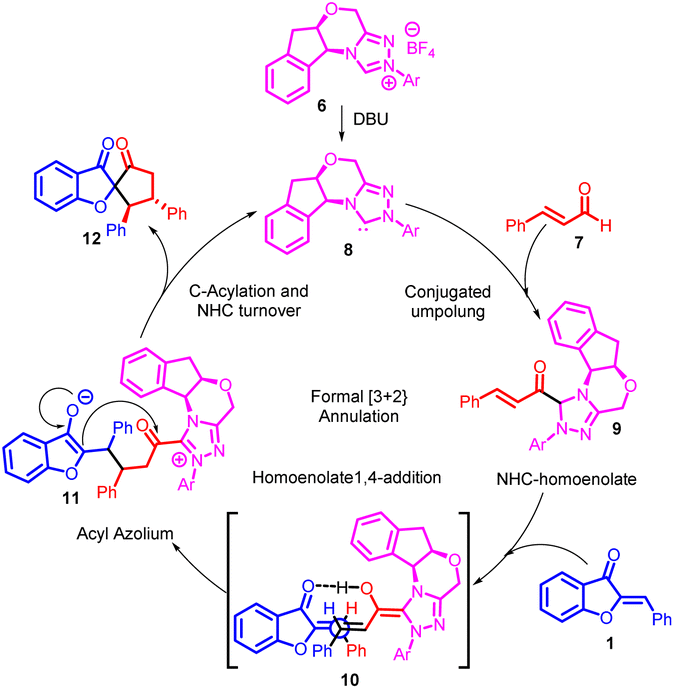 | ||
| Scheme 3 Proposed mechanism for the NHC catalyzed annulation of enals. | ||
In 2013, Wei-Qiang Hu et al., introduced a 1,3-dipolar cycloaddition approach involving aurone 1 and azomethine ylides 14. This innovative reaction was effectively facilitated by basic imidazolium salts 13, resulting in the efficient construction of spirocycles containing highly substituted pyrrolidines. The products were obtained with outstanding yields (Scheme 4).
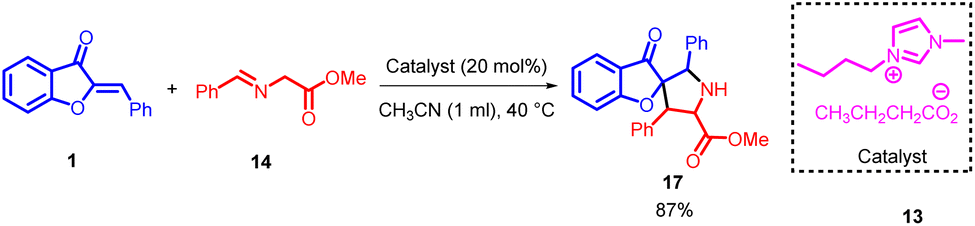 | ||
| Scheme 4 1,3-Dipolar cycloaddition of aurone and azomethine ylides to form pyrrolidine and 1-indanone derivatives. | ||
A plausible bifunctional catalytic mechanism has been proposed for this process. In this mechanism, a nucleophilic butyric anion 13 acts as a base, capturing a proton from the azomethine ylides 14. Simultaneously, an electrophilic imidazolium cation functions as an electron acceptor, completing the reaction cycle. This mechanistic insight is illustrated in Scheme 5. Activation of azomethine ylides is attributed to the involvement of the butyric anion, while the imidazolium cation is believed to activate the aurone 1.48
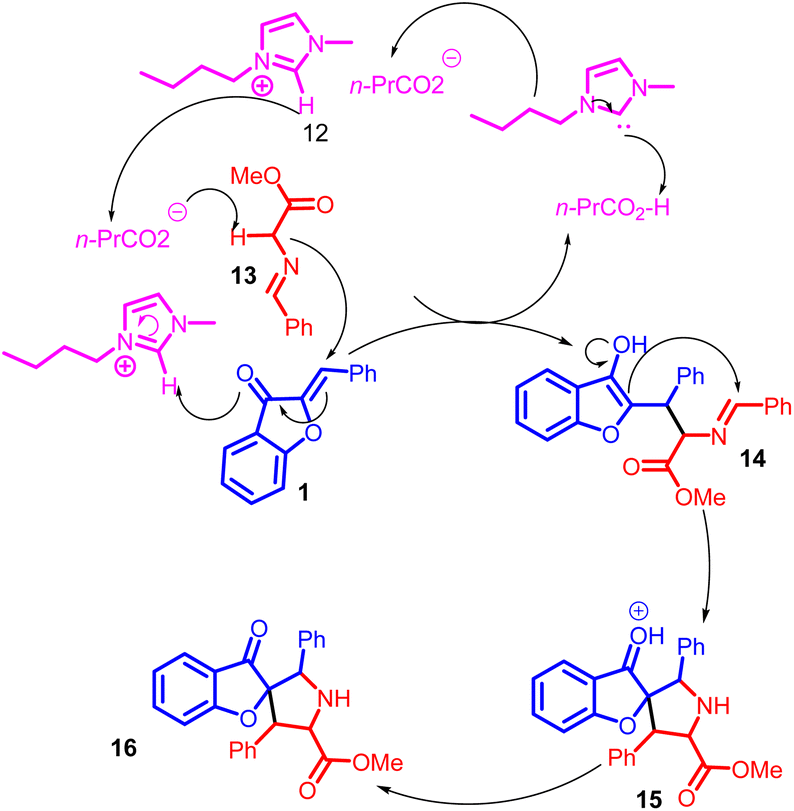 | ||
| Scheme 5 Proposed mechanism for the synthesis of pyrrolidine and 1-indanone derivatives. | ||
In 2016, Tao Ding and co-workers achieved a significant breakthrough by establishing a remarkably efficient catalytic system capable of constructing both spiro[pyrrolidine benzofuran-3-one] 20 and spiro[pyrrolidine-benzofuran-2-one] 22 compounds. They pioneered the development of the initial 1,3-dipolar cycloaddition reaction involving aurone 1 and azomethine ylides 19, employing simple functional ionic liquids as catalysts (Scheme 6). This innovative approach led to the synthesis of a diverse range of spiro[pyrrolidine-benzofuran-3-one] 20 compounds, characterized by highly substituted pyrrolidine structures featuring a spiro quaternary stereogenic centre. Remarkably high yields (ranging from 73% to 99%) were achieved using this method. Moreover, the same catalytic system demonstrated its versatility by facilitating the 1,3-dipolar cycloaddition between aurone 1 and pyrazolidinone-based dipoles 21, yielding spiro[pyrrolidine-benzofuran-2-one] derivatives 22 with yields ranging from 25% to 85%.
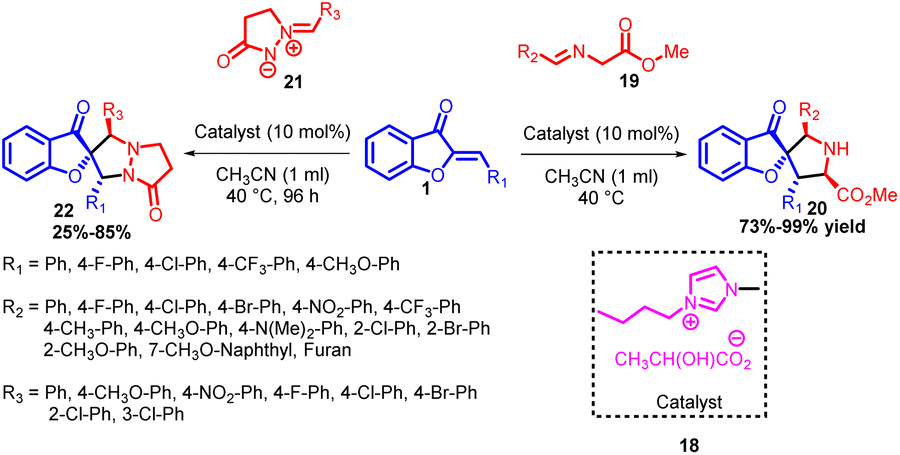 | ||
| Scheme 6 1,3-Dipolar cycloaddition of aurones derivatives and azomethine ylides. | ||
This catalytic system's efficiency was evident in its wide applicability to diverse substrate ranges; all achieved under mild reaction conditions. Importantly, these studies not only offer a vital approach for producing two distinct classes of highly substituted pyrrolidines featuring spiro[pyrrolidinebenzofuranone] motifs but also present an avenue for their in situ generation from benzofuranone derivatives.49
Łukasz Albrecht and co-workers in 2016 introduced a novel and highly selective strategy for synthesizing pyrrolidine derivatives featuring a benzofuran-3(2H)-one framework. This pioneering method relies on a [3 + 2] cycloaddition process that involves aurone 1 and imines 24, derived from salicylaldehyde and diethyl aminomalonates (Scheme 7). Initial experimentation with complex catalysts yielded unsatisfactory results. However, a significant improvement in the reaction outcomes was achieved by substituting these advanced catalysts with a simpler cinchona alkaloid, known as quinine.
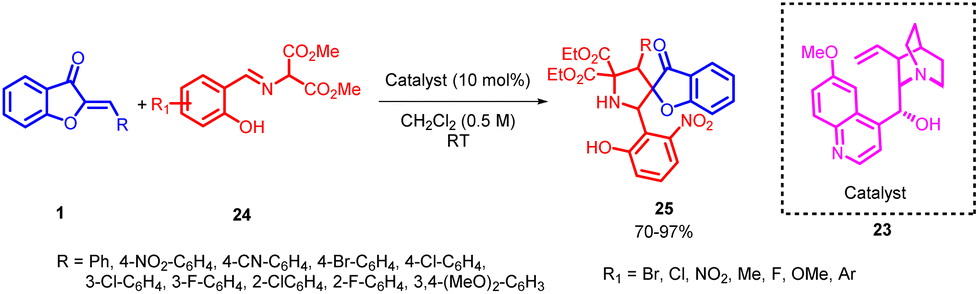 | ||
| Scheme 7 Asymmetric organocatalyzed synthesis of pyrrolidine derivatives bearing a benzofuran-3(2H)-one. | ||
Further exploration, which involved examining different cinchona alkaloids and their derivatives under basic conditions, highlighted the critical role of the C-9 hydroxyl group in recognizing substrates and, consequently, creating a well-defined stereochemical environment for the cycloaddition reaction. This method demonstrates impressive versatility with a wide range of applicable substrates, emphasizing efficiency in both chemical synthesis and stereochemical precision while maintaining a straightforward operational process. Furthermore, it is evident that lowering the reaction temperature had an adverse impact on the stereoselectivity of both diastereoselection and enantioselection. A similar effect was observed when altering the concentration of the reaction, with both increasing and diluting the reaction solution leading to a decrease in the stereoselectivity of the cycloaddition process. These findings provide valuable insights into the optimization of conditions to achieve desired stereochemical outcomes in cycloaddition reactions. These compounds are synthesized with excellent yields and meticulous attention to stereochemical accuracy. The resulting compounds feature two biologically significant heterocyclic components and three neighbouring stereogenic centers, one of which is quaternary.50
Azaoxyallyl cation intermediates, derived from α-halo hydroxamates in the presence of organic or inorganic bases, have emerged as versatile substitutes for “1,3-dipole” entities. These intermediates have been effectively utilized in diverse cycloaddition reactions, including [3 + 1], [3 + 2], [3 + 3], and [4 + 3] processes, involving sulphur ylides, aldehydes, indoles, 2-alkenylindoles, nitrones, and electron-rich dienes. Building upon prior research in this synthetic methodology of azaoxyallyl cations, Pan Lin Shao and co-workers in 2017 introduced the pioneering [3 + 2] cycloaddition reaction between in situ generated azaoxyallyl cations 27 and aurone 1 under mild reaction conditions (Scheme 8). The proposed mechanism for this reaction is provided in Scheme 9.51
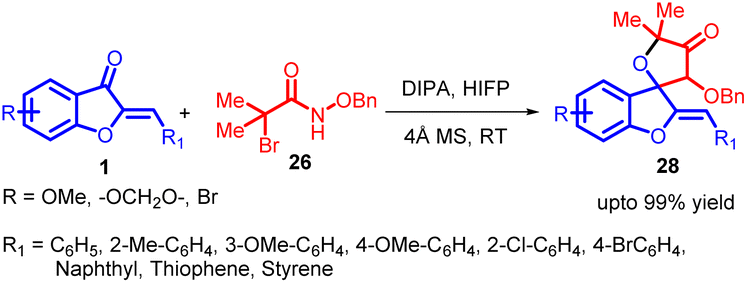 | ||
| Scheme 8 Cycloaddition of azoxyallyl cations with aurones to form spiro-4-oxazolidinones. | ||
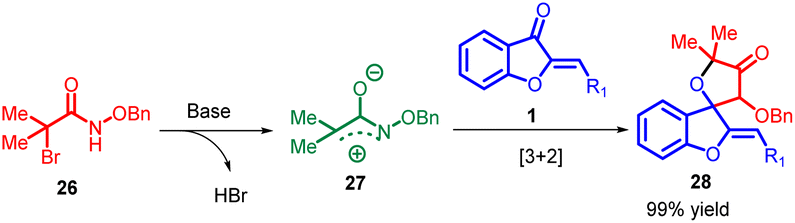 | ||
| Scheme 9 Proposed mechanism for the synthesis of spiro-4-oxazolidinones. | ||
This approach led to the successful synthesis of a range of spiro-4-oxazolidinones 28 with exceptional yields, some reaching up to 99%. The impressive efficiency of this method, combined with its straightforward implementation, positions it as an appealing strategy for spiro-4-oxazolidinone 28 synthesis.
In 2018, Zhi-Peng Wang and co-workers introduced a transition metal-free cyclization strategy employing NaOH for the reaction between methyl isocyanates 29 and aurone analogs 1 (Scheme 10). This method led to the efficient synthesis of diverse 2,3 and 4-polysubstituted pyrroles 30 with high efficacy.
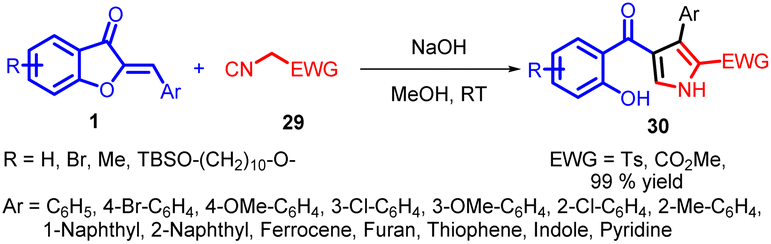 | ||
| Scheme 10 Transition metal-free synthesis of polysubstituted pyrrole via cyclization of methyl isocyanoacetate with aurone. | ||
In an endeavour to enhance comprehension of the reaction profile, deuterium labelling studies were carried out. d1-1a (88% deuterium) and d2-29a (99% deuterium) were prepared for the cyclization. As shown in Scheme 11, under the optimized reaction conditions, 30 was obtained in 99% yield, replacement of the solvent with anhydrous MeCN, 30a could also be formed in high yield, no deuterium labelling was observed on both of the products (Reaction A and B, Scheme 11). However, the use of CD3OD as reaction media resulted in significant deuterium labelling (96% deuterium) at the 5-position on the pyrrole ring, and further investigation revealed that the proton at 5-position can also be labelled by deuterium (67% deuterium) when 3.0 equivalents of D2O was added with anhydrous MeCN as solvent (Reaction C and D, Scheme 11). This intriguing finding implies that an active proton facilitates proton transfer, aligning with the pronounced solvent effect observed.
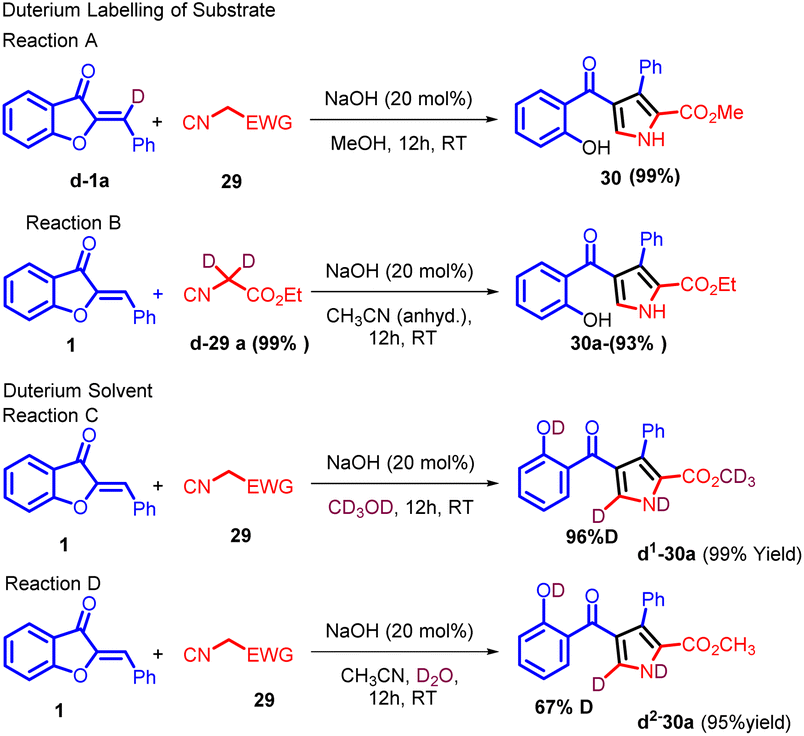 | ||
| Scheme 11 Deuterium labelling studies. | ||
This transformation employs readily available, versatile starting materials, an economical catalyst, and proceeds under mild conditions, all without reliance on transition metals. The method holds practical promise for synthesizing pharmaceutically relevant polysubstituted pyrroles 30, particularly regarding substitution patterns and compatibility with functional groups.52
In 2018, Zhi-Peng Wang et al., devised an enantioselective formal [3 + 2] cycloaddition process involving isocyanoacetates 32 and aurone analogs 1 (Scheme 12). This reaction employed a chiral Ag-complex catalyst. Despite the potential for the 1,3-dipole enolate derived from isocyanoacetate 32 to engage in a [4 + 3] reaction sequence, the study exclusively unveiled the [3 + 2] cyclization pathway, resulting in the formation of spiropyrroline 35. Intriguingly, no evidence of a [4 + 3] annulation product 36 emerged.
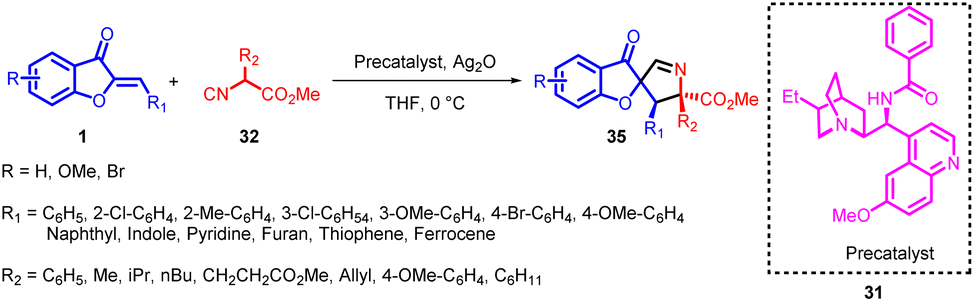 | ||
| Scheme 12 Catalytic asymmetric [3 + 2] cycloaddition reaction between aurones and isocyanoacetates to form spiropyrrolines. | ||
While the precise mechanism of the reactions described in this study is still not fully understood, a plausible transition-state model, 34, has been proposed. This model is based on experimental findings and the absolute configuration of the major isomer 35. In this proposed model, the α-proton of isocyanoacetate 32 is readily deprotonated by the tertiary amine on the precatalyst, facilitated by the activation of Ag(I), which is chelating to the terminal carbon of the isocyano group. Additionally, the central metal Ag(I) could form chelation bonds with the amide nitrogen and the Lewis base phosphorus on the precatalyst. Due to steric hindrance, the ester motif is positioned beneath the bulky phenyl groups. Consequently, the isocyanoacetate would approach the aurone olefin from the Re-face. Simultaneously, the oxygen atom of the aurone is involved in hydrogen bonding with the precatalyst and coordinated to Ag(I) to stabilize the transition state 34. As a result of these interactions, a 5-endo-dig cyclization occurs, leading to the formation of the spiropyrroline product, complete with three newly formed stereocenters (Scheme 13).
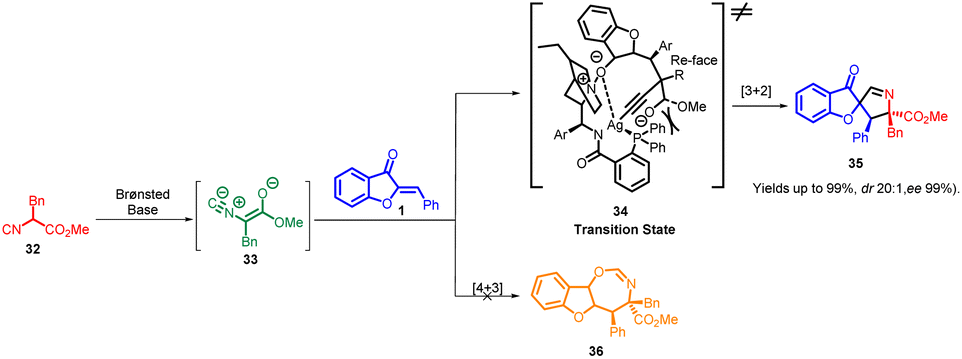 | ||
| Scheme 13 Proposed mechanism for the synthesis of spiropyrrolines. | ||
When employing precatalysts 31 along with Ag2O, excellent yield and stereoselectivity were achieved. Furthermore, reducing the reaction temperature to 0 °C improved both yield and the levels of diastereoselectivity and enantioselectivity. However, changing the solvent did not have a positive impact on enantioselectivity. The protocol demonstrated remarkable tolerance for a diverse array of isocyanoacetates 32 and aurones 1, encompassing varying electronic and steric attributes. This led to the formation of optically active spiropyrrolines featuring three adjacent stereogenic centres, with exceptional outcomes in terms of yields and diastereo- and enantioselectivities (achieving yields up to 99%, dr exceeding 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and ee surpassing 99%).53
:1, and ee surpassing 99%).53
In 2018, Ting Du et al., developed an asymmetric [3 + 2] cycloaddition process involving aurones 1 and imino esters 38 (Scheme 14). Various quaternary ammonium salts, including amide, urea, and thiourea derivatives, were tested as potential catalysts, each serving as an H-donor. Interestingly, the use of a L-isoleucine-derived quaternary ammonium salt as the catalyst resulted in a 92% yield of product 39, accompanied by a 73% enantiomeric excess (ee) and a 6:1 diastereomeric ratio (dr). In contrast, other quaternary ammonium salts employing urea as the double H-donor did not significantly improve enantioselectivity, although they did enhance diastereoselectivity to 14:1. It is well-established that thiourea exhibits both higher H-bond strength and acidity compared to urea. Utilizing catalyst 37, derived from L-isoleucine, led to a remarkable improvement in enantioselectivity, resulting in a 98% ee and an 11:1 dr. These findings underscore the influence of catalyst structure and composition on the stereochemical outcomes of the reaction. The reactions exhibited high efficiency, enabling the straightforward synthesis of diverse chiral pyrrolidine derivatives 39 that featured a benzofuran-3(2H)-one framework. Notably, these reactions occurred with exceptional enantioselectivity and satisfactory diastereoselectivity, all conducted under mild reaction conditions.54
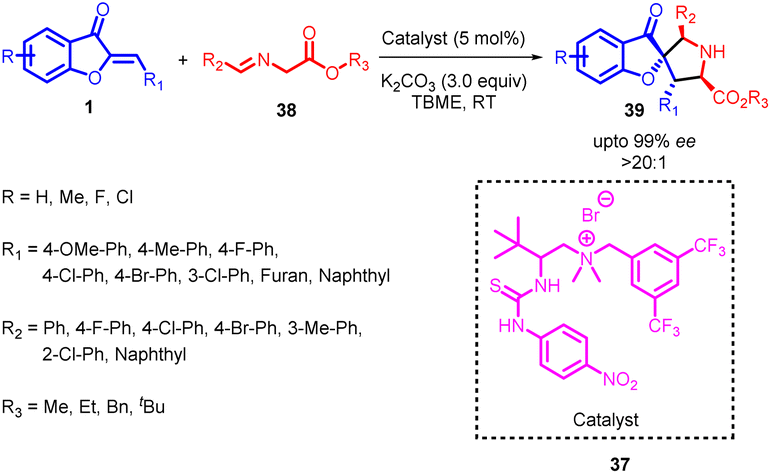 | ||
| Scheme 14 Enantioselective 1,3-dipolar cycloaddition of imino esters with benzofuranone derivatives catalyzed by thiourea quaternary ammonium salt. | ||
In 2020, Yulai Hu and co-workers reported an effective and regioselective [3 + 2] dipolar cycloaddition reaction involving benzoaurone 1b and in situ generated nitrile imine 40 (Scheme 15). This protocol provides facile access to various interesting spiro naphthofuranone-pyrazoline 41 compounds in good to excellent yields. The proposed mechanism (Scheme 16) involves the reaction of hydrazonoyl chloride 40a with K2CO3, leading to the formation of nitrile imines with distinct resonance structures A–D. These nitrile imines then react with benzoaurone 1b. Notably, the resonance allenic C of the nitrile imine plays a significant role in the [3 + 2] cycloaddition, participating via an asynchronous transition state (42). This transition state involves a two-centre interaction between the carbenoid carbon of C and the electrophilic centre of benzoaurone 1b. Consequently, the reaction yields the spiro naphthofuranone-pyrazoline compound 41a, exhibiting high regioselectivity.55
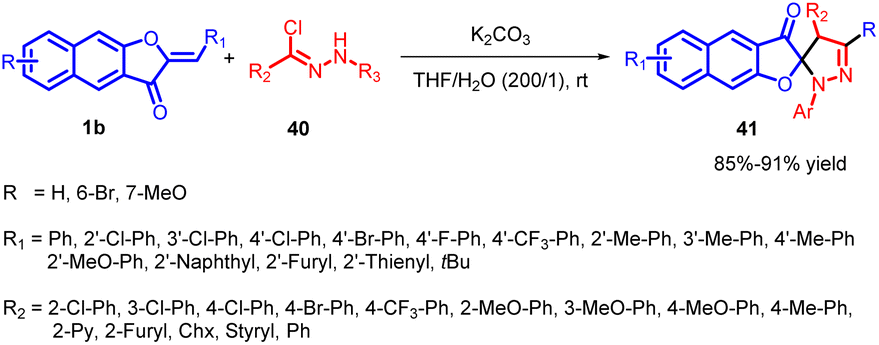 | ||
| Scheme 15 Synthesis of the spiro-pyrazolines with nitrile imines via [3 + 2] cycloaddition. | ||
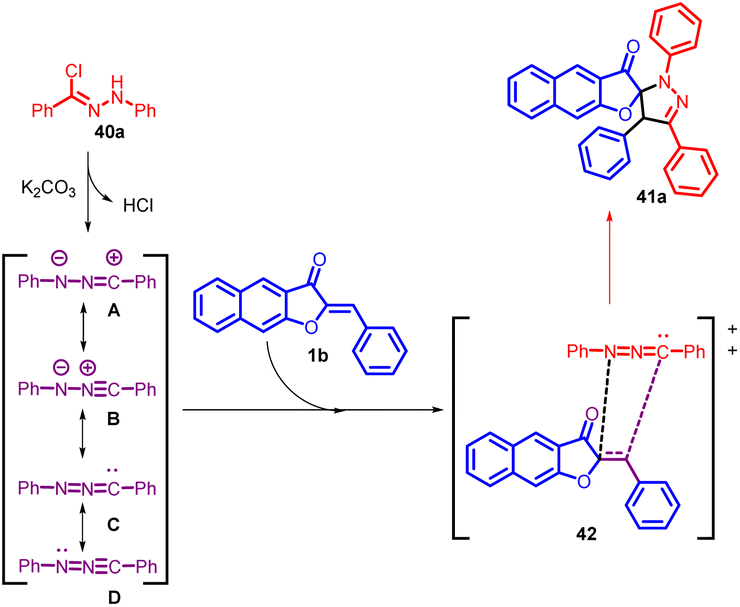 | ||
| Scheme 16 Proposed mechanism for the [3 + 2] cycloaddition. | ||
In 2021, Prakash K. Warghude and co-workers developed a remarkably selective [3 + 2] annulation method involving Morita–Baylis–Hillman (MBH) carbonates of isatin 43 with aurone 1 (Scheme 17). This approach was successful in producing diverse spiro-heterocycles, including spiro-oxindole cyclopentadiene 50 derivatives. The reactivity of Lewis bases, particularly DMAP, played a crucial role in driving this synthetic transformation.
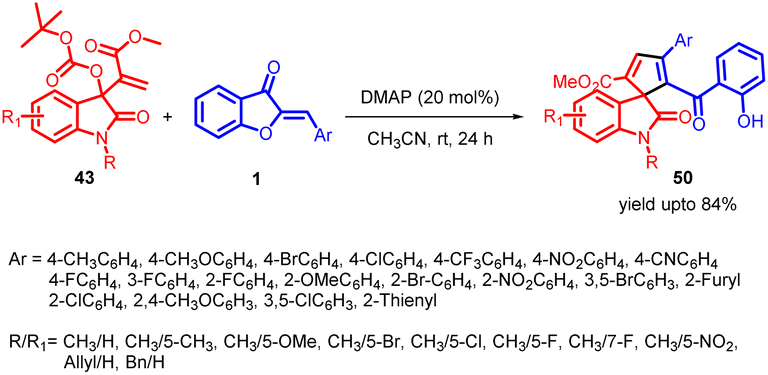 | ||
| Scheme 17 Synthesis of multifunctional cyclopentadiene- and cyclopentene-spirooxindoles via [3 + 2] annulation. | ||
According to their observations, the process begins with nucleophilic DMAP 44 attacking MBH carbonate 43, forming quaternary ammonium salt 45 while liberating carbon dioxide and tert-butoxide. However, this initial step is found to be thermodynamically unstable (ΔG = 12.7 kcal mol−1). The tert-butoxide generated in situ subsequently removes a proton from quaternary ammonium salt 45, leading to the formation of an allylic nitrogen-ylide 46. The resulting ylide 46 then reacts with aurone 1, generating intermediate 47 (reaction free energy ΔG = −20.0 kcal mol−1). This intermediate readily undergoes an intramolecular Michael addition, facilitated by its highly favourable energy profile (ΔG = −31.0 kcal mol−1), leading to the formation of intermediate 48 and the elimination of DMAP. Continuing the reaction, DMAP abstracts a proton from the intermediate 48, causing ring opening and the formation of a phenoxide intermediate 49. Finally, this intermediate undergoes protonation, leading to the desired compound 50, while also regenerating DMAP to complete the catalytic cycle (Scheme 18).56
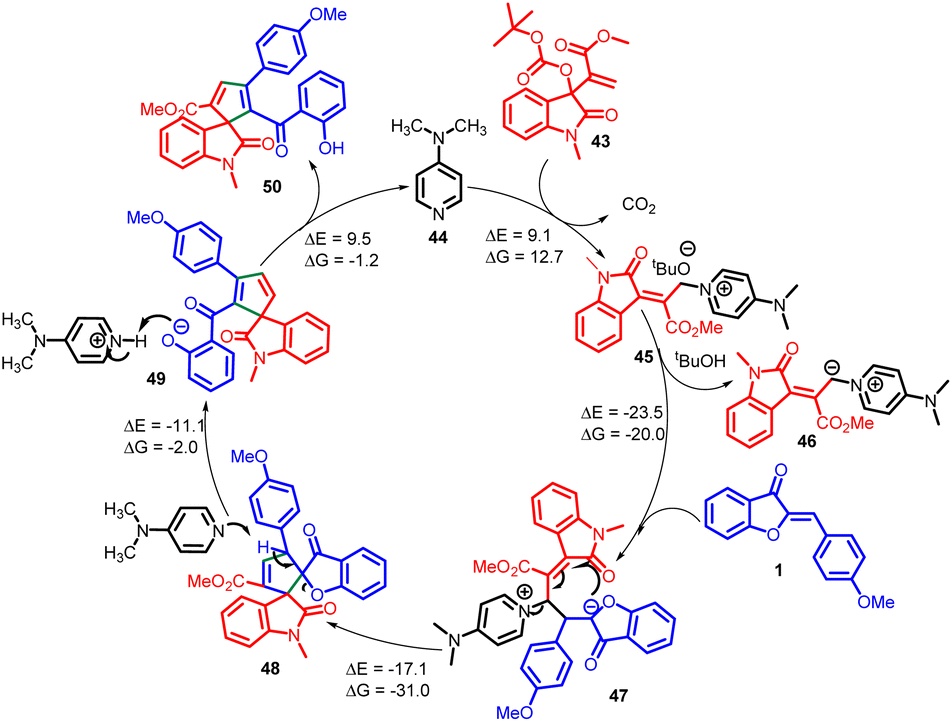 | ||
| Scheme 18 Proposed reaction mechanism for the formation of spirooxindole cyclopentadiene. | ||
In 2021, Rui-Li Wang et al., developed a highly enantioselective method for synthesizing chiral spiro[benzofuran-pyrrolidine] 57 derivatives (Scheme 19). They achieved this by utilizing chiral dinuclear zinc catalysts and starting from aurones 1 and N-2,2,2-trifluoroethylisatin ketimines 52. The reaction involved a [3 + 2] cycloaddition mechanism, progressing through a domino Michael/Mannich reaction. The researchers obtained the desired products with excellent diastereoselectivities and enantioselectivities, all achieved under mild reaction conditions.
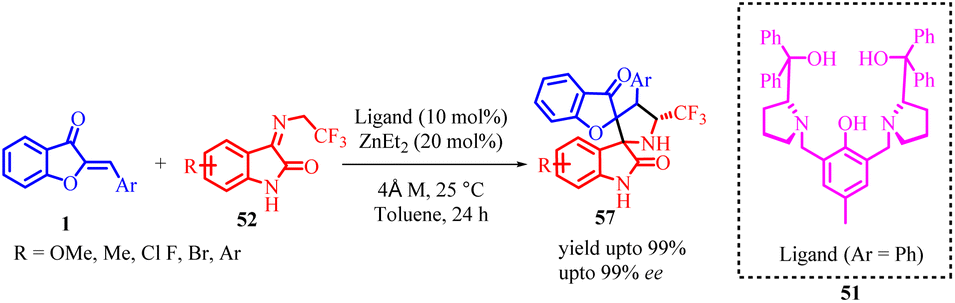 | ||
| Scheme 19 Dinuclear zinc-catalyzed enantioselective formal [3 + 2] cycloaddition of N-2,2,2-trifluoroethylisatin ketimines with low reactivity aurone derivatives. | ||
In the proposed reaction mechanism (Scheme 20), the synthesis begins with the formation of a dinuclear zinc complex 51in situ through the reaction of the ligand with two equivalents of Et2Zn. The N-2,2,2-trifluoroethylisatin ketamine 52 is then deprotonated by the dinuclear zinc complex, resulting in the formation of the activated azomethine ylide 53 and the release of ethane. Subsequently, the aurone coordinates to both zinc atoms, generating intermediate 54. This coordination enables a Michael addition reaction to occur, leading to the formation of complex 55. Following that, an intramolecular Mannich reaction takes place within the same catalytic system, facilitating the cycloaddition process. The catalytic cycle restarts as intermediate 56 undergoes a proton exchange with another imine. This step is succeeded by the release of the activated azomethine ylide 52 and the desired spiro[benzofuran-pyrrolidine] product 57.
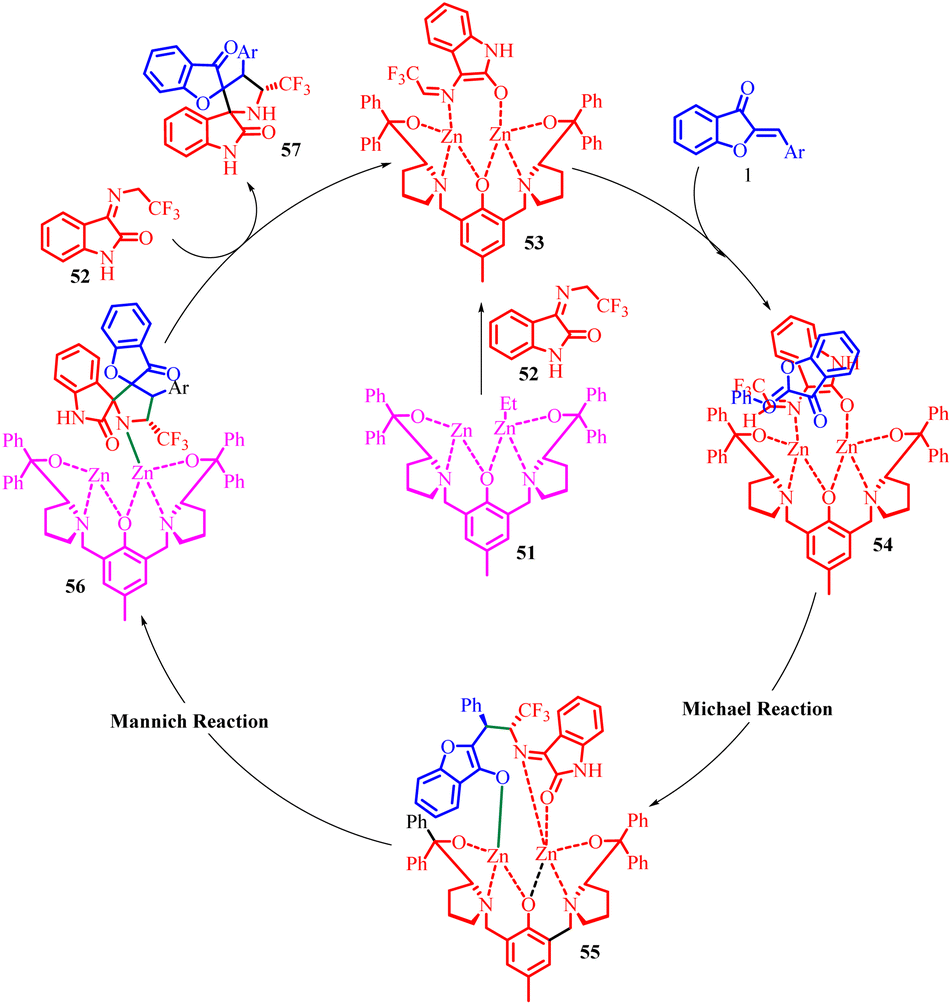 | ||
| Scheme 20 Proposed mechanism for the synthesis of spiro[benzofuran-pyrrolidine]. | ||
The catalytic asymmetric control of this chemical transformation was assessed using a variety of chiral ligands with different substituents and backbones. These included various ProPhenol ligands, AzePhenol ligands, and non-C2 symmetric ligands. The outcomes of these experiments revealed that the choice of chiral ligands significantly influenced the enantioselectivity. Notably, the alternative ligands examined did not lead to an improvement in enantioselectivity. To enhance both the yield and enantioselectivity of compound 57, several additives were introduced, such as 4 Å molecular sieves (MS), Ph3PO, Ph3PS, and NEt3. The inclusion of 40 mg of 4 Å MS as an additive resulted in the completion of the 1,3-dipolar cycloaddition reaction within 12 hours, yielding the product in an impressive 85% yield with a 94% enantiomeric excess (ee). Further optimization involved a solvent screening, revealing that toluene was the most suitable solvent, leading to a slight improvement in the ee value. Subsequent experimentation at an elevated temperature (25 °C) yielded the desired product in a 90% yield with a 95% ee, ultimately establishing the optimal reaction conditions.
These reactions not only present a new methodology for building the chiral spiro[benzofuran-pyrrolidine] structure 57 but also enhance the application of the chiral dinuclear zinc cooperative strategy in synthesizing complex compounds. This research widens the scope of constructing intricate molecules with specific chirality, offering valuable insights for the development of novel catalytic systems.57
In 2021, Ying-Chun Chen and co-workers58 introduced a novel method for the asymmetric α-regioselective [3 + 2] annulation reactions of aurone derivatives 1 with isatin-derived Morita–Baylis–Hillman (MBH) carbonates 59 (Scheme 21). This reaction was carried out under the catalysis of a chiral DMAP-type catalyst 58, resulting in the efficient synthesis of a diverse range of spiro-benzofuran 64 frameworks with remarkable levels of diastereo- and enantioselectivity. Upon comparing various chiral DMAP-type catalysts, it was evident that those bearing 3,5-bis(trifluoromethyl)phenyl groups in catalyst 58 exhibited higher enantioselectivity and superior stereocontrol. Conversely, alternative catalysts displayed moderate catalytic activity and enantioselectivity. To elucidate the catalytic process further, a proposed mechanism was developed based on the product's absolute configuration.
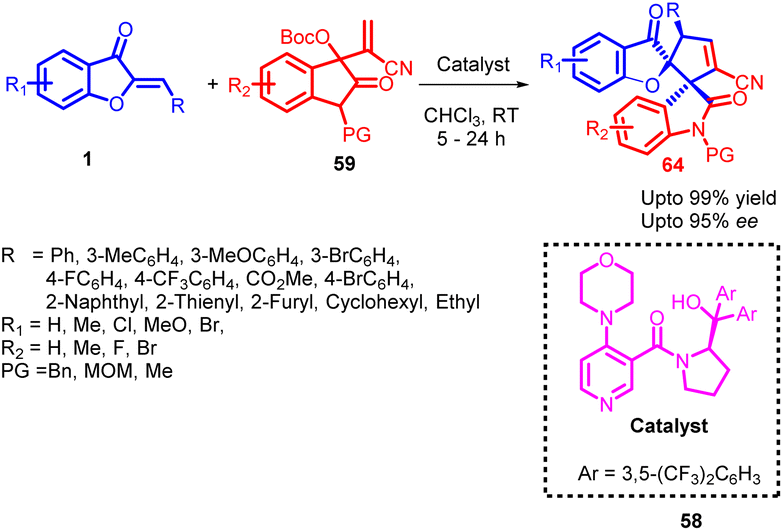 | ||
| Scheme 21 Asymmetric [3 + 2] annulations of aurone derivatives for the construction of spiroheterocycles. | ||
The absolute configuration of the product played a crucial role in proposing a plausible catalytic mechanism for this asymmetric annulation reaction (Scheme 22). According to their proposed mechanism, catalyst 58 acts as an activator for the MBH carbonate 59, leading to the formation of a zwitterionic allylic ylide 60. Subsequently, this intermediate 60 attacks the aurone substrate 1 from the Si-face, with a possible hydrogen-bonding interaction between the hydroxy group of 58 and the carbonyl group of 1 facilitating the process. This step affords an intermediate 62. Finally, through cyclization and the subsequent release of catalyst 58; the desired product 64 was generated.
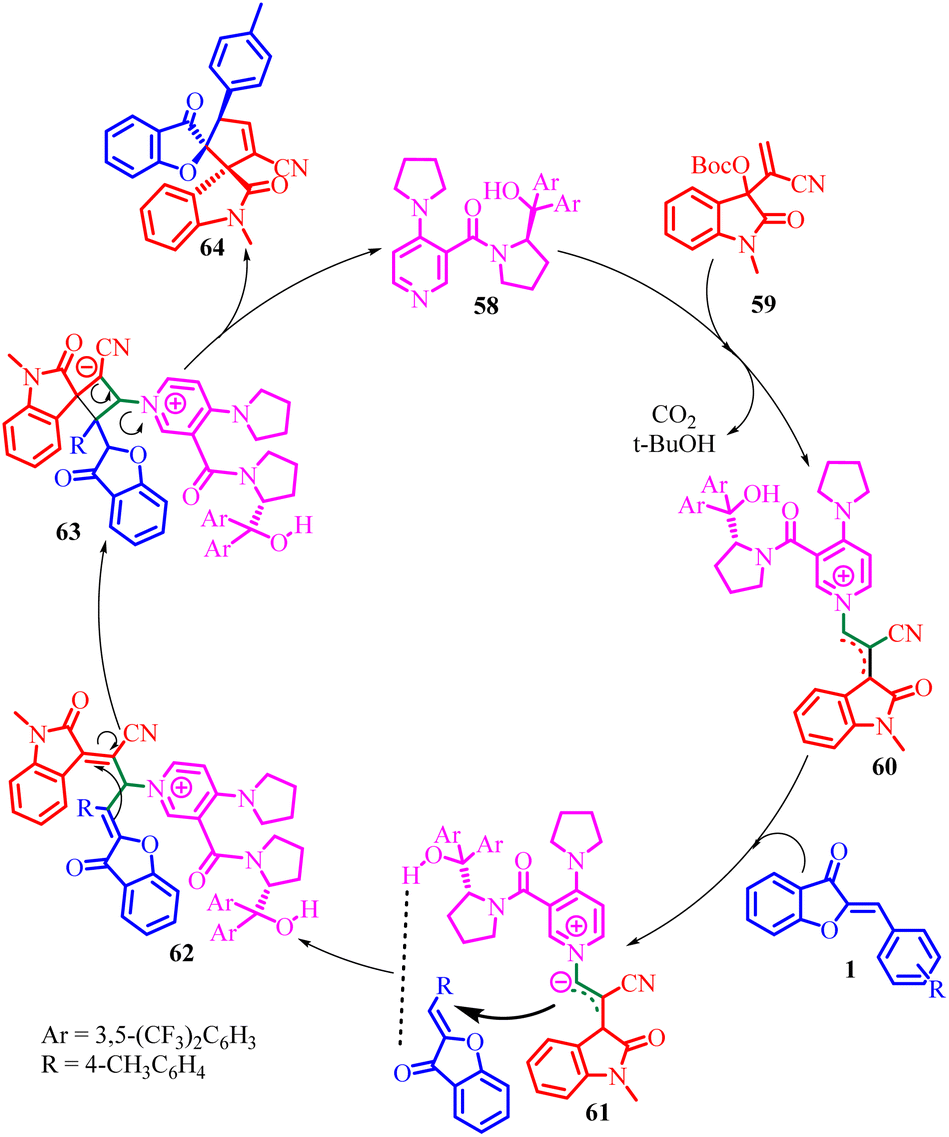 | ||
| Scheme 22 Proposed mechanism for the asymmetric [3 + 2] annulations of aurone. | ||
The synthetic strategy developed in this study enables the rapid construction of intricate spiroheterocyclic compounds with excellent enantioselectivity, utilizing readily available starting materials. This approach holds great potential for further applications in the fields of synthetic chemistry and medicinal chemistry.58
In 2022, Jie Huang et al., developed an efficient method for synthesizing poly-substituted pyrazoles 68 using tosyldiazomethane (TsDAM) 65 and (Z)-aurones 1 as starting materials (Scheme 23). This protocol involved the use of Cs2CO3 as a catalyst in water/protic solvents. The reaction proceeded through a series of steps, as depicted in Scheme 24 of their study. Initially, 2-diazo-1-aryl-2-tosylethanone 65 reacted with water in the presence of a base, leading to the formation of an intermediate 66. This intermediate underwent 1,3-dipolar cycloaddition with (Z)-aurones 1, resulting in the formation of intermediate 67. Subsequently, intermediate 67 underwent aromatization, causing the aurone ring to open and yielding fully aromatized poly-substituted pyrazole derivatives 68.
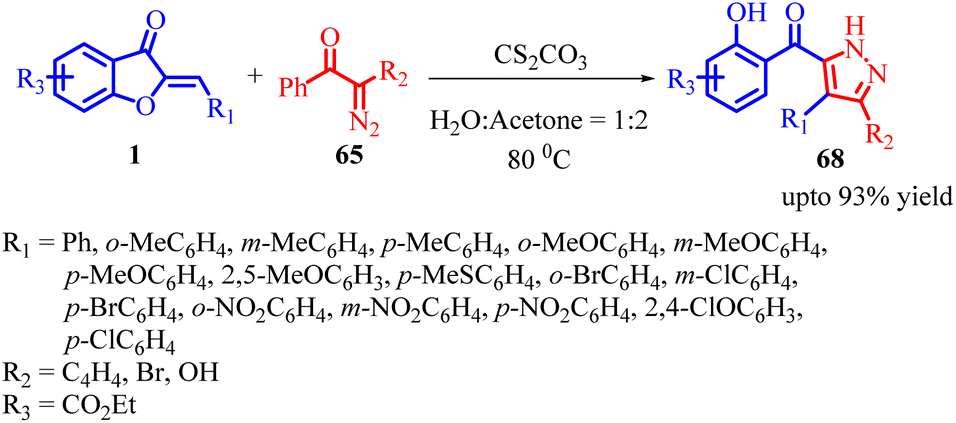 | ||
| Scheme 23 Base-catalysed cascade [3 + 2] spiroannulation/aromatization of tosyldiazomethane (TsDAM) with (Z)-aurones. | ||
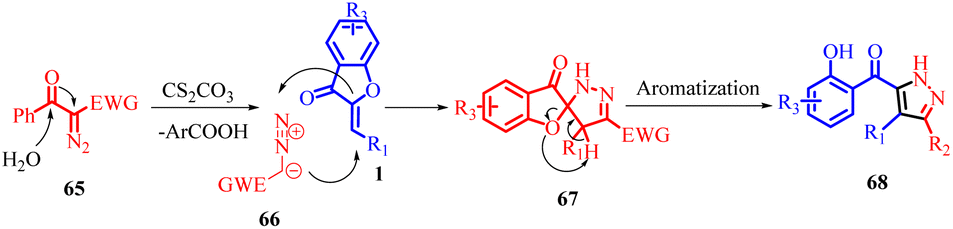 | ||
| Scheme 24 Proposed mechanism for base catalyzed [3 + 2] spiroannulation/aromatization. | ||
This method was successful in synthesizing novel and interesting pyrazoles 68. The domino reaction between (Z)-aurones 1 and in situ generated TsDAM proceeded smoothly, providing the desired tosyl-substituted pyrazoles 68 in good to excellent yields. The reaction yield varied depending on the substitution pattern of the (Z)-aurones 1. Generally, para-substituted (Z)-aurones yielded higher yields compared to ortho- and meta-substituted ones, primarily due to steric effects. It is noteworthy that electron-donating groups yielded slightly better results than electron-withdrawing groups. However, when nitro (a strong electron-withdrawing group) substrates were used, the yields were low. Among the three nitro substrates, the para-substituted nitro substrate showed the lowest yield. Additionally, various diazo compounds are used as reactants alongside 2-diazo-1-phenyl-2-tosylethan-1-one as dipole component. It was found that 2-diazo-1-phenyl-2-tosylethan-1-one performed better than 2-diazo-3-oxobenzoate in terms of reaction yield.59
In 2022, Wang et al., introduced an innovative method involving DBU catalysis for a [3 + 2] cycloaddition of aurones 1 and 3-homoacyl coumarins 69. This strategy enabled the creation of spiro-fused pentacyclic spiro-benzofuranones 72 that possess a combination of spiro[benzofuranone-cyclopentane] and coumarin components. This technique proved effective in producing a variety of functionalized spiro-benzofuranone derivatives 72 with high yields and impressive diastereoselectivity, demonstrating its versatility for synthesis (Scheme 25).60
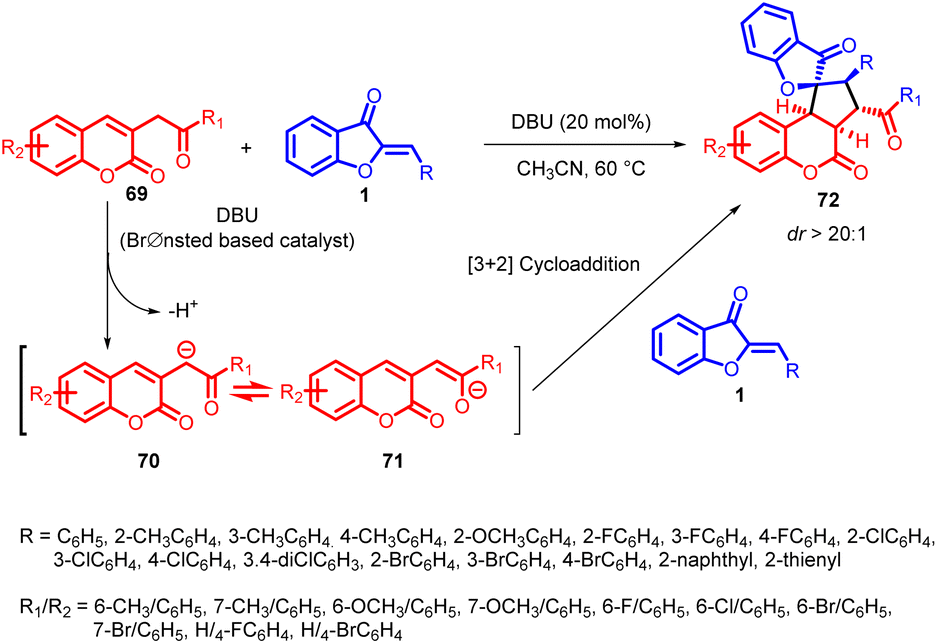 | ||
| Scheme 25 Synthesis of spiro[benzofuranone-cyclopentane] derivatives via annulations of aurones derivatives with various 3-homoacyl coumarins derivatives. | ||
Indanone-fused polycyclic frameworks are commonly found in bioactive natural compounds and are significant in medicinal chemistry due to their unique structural properties. Ji-Ya Fu and co-workers in 2023 have successfully developed a regioselective reaction, known as [3 + 2] cycloaddition, between 2-benzylidene-1-indenones 73 and aurone 1 (Scheme 26). By employing 1,4-diazabicyclo [2.2.2]octane (DABCO) as a base under mild conditions, they have been able to create a novel class of indanone-fused cyclopentane backbones. This reaction method utilizes the electrophilic C3-position of 2-benzylidene-1-indenones 73 and the nucleophilic C4-position of aurone 1 to construct cyclopentane rings with five consecutive chiral centres. The functional olefins act as both nucleophiles and electrophiles, participating in the [3 + 2] cycloaddition reaction. This metal-free approach enables the efficient synthesis of indanone-fused cyclopentane frameworks with five continuous chiral centres, allowing for the production of diverse indanone-fused cyclopentane polycycles with multiple substituents in high yields.
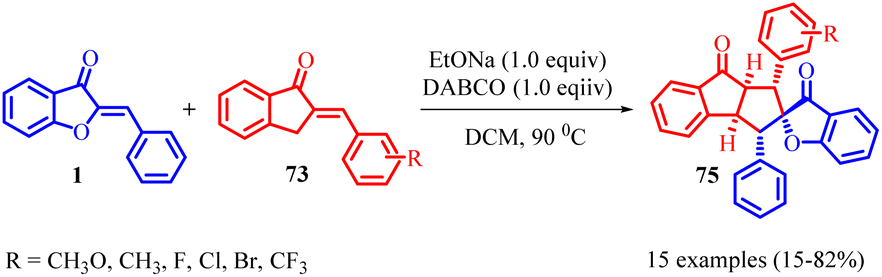 | ||
| Scheme 26 [3 + 2] Cycloaddition reaction of 2-benzylideneinden-1-ones with 3-benzylidenebenzofuran-2(3H)-one. | ||
The proposed mechanism involves the conversion of 2-benzylidene-1-indenone 73 into an intermediate compound in the presence of the base, followed by a [3 + 2] cycloaddition reaction between the intermediate and aurone 1 to yield the desired product (Scheme 27). The successful synthesis of the desired fused tricyclic and spirocyclic scaffolds was achieved by subjecting less reactive aurone 1 to standard reaction conditions in the presence of 2-benzylideneinden-1-one. Interestingly, the reaction predominantly yielded a crossover product. Expanding the scope of this transformation, a range of 2-benzylidene-1-indenones 73 containing para-, meta-, and ortho-substituted aryl rings were employed. This systematic exploration resulted in the synthesis of a diverse collection of fused tricyclic and spirocyclic compounds, demonstrating the versatility and applicability of this reaction methodology.61
 | ||
| Scheme 27 Proposed mechanism for [3 + 2] cycloaddition of aurones with 2-benzylideneinden-1-ones. | ||
[4 + 2] Cycloaddition reaction
The inception of the [4 + 2] cycloaddition reaction dates back to 1928, when Otto Diels and his student, Kurt Alder, documented their findings on the cycloaddition of cyclopentadiene with quinone. Subsequently, there has been a continuous growth in the body of literature focusing on various facets of the [4 + 2] cycloaddition. This reaction, known as the Diels–Alder reaction in honor of its pioneers, has evolved into the most versatile means of producing six-membered carbon and heterocyclic structures. In the contemporary landscape, this method remains unsurpassed for the synthesis of such structures. Presently, the application of this reaction extends beyond the use of conjugated 1,3-dienes; conjugated nitroalkenes have emerged as frequent candidates as hetero-analogues of dienes. Hetero-analogs of ethene play a pivotal role in this cycloaddition process, encompassing molecular units that incorporate nitrogen, oxygen, sulphur, selenium, and other elements.62–64Jin-Pei Cheng and co-workers developed a highly enantioselective [4 + 2] cycloaddition method employing allenoates 77 and aurone 1 as substrates (Scheme 28). This transformation was facilitated by the application of the (DHQD)2AQN catalyst 76, the structure of which is provided. This innovative approach enabled the controlled synthesis of optically active dihydropyran-fused benzofurans, showcasing significant enantioselectivity. The research delved into the investigation of cycloaddition using various catalysts. Through a comprehensive comparative analysis, it was established that (DHQD)2AQN 76 proved to be the optimal catalyst in terms of both catalytic reactivity and enantioselectivity. Subsequently, the reaction was subjected to optimization by examining factors such as solvent, concentration, and additives, all in the presence of (DHQD)2AQN 76. Screening various solvents revealed that THF delivered the most favourable results among the solvents tested. A slight increase in reaction concentration led to a reduction in reaction time. The introduction of 4 Å molecular sieves to the reaction mixture significantly improved regioselectivity but had a notable adverse effect on enantioselectivity.
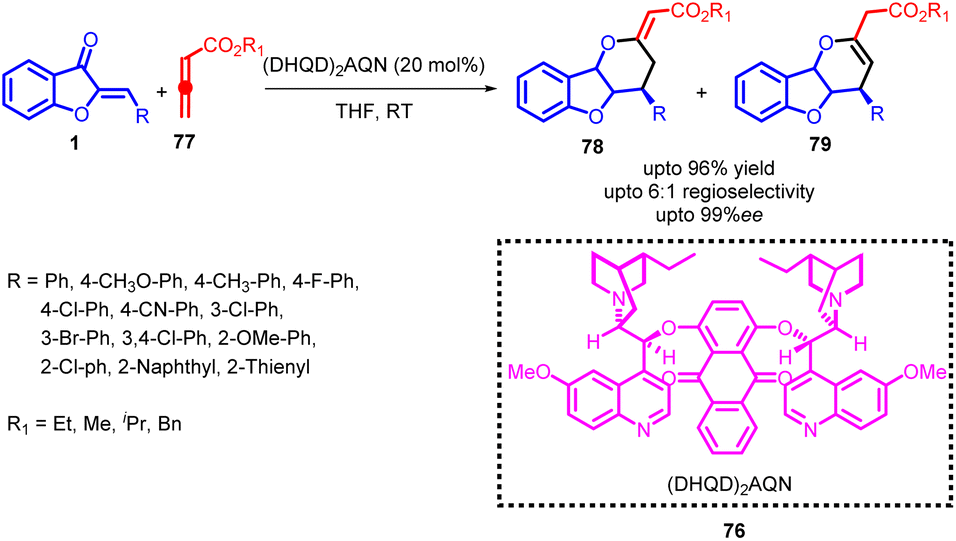 | ||
| Scheme 28 Enantioselective [4 + 2] cycloaddition of allenoates and 2-olefinic benzofuran-3-ones. | ||
The absolute configuration of the products was assigned based on the crystallographic analysis. Considering the molecular structure of the resulting product, a plausible transition state 80 was conceptualized (Fig. 3). The (DHQD)2AQN catalyst 76, adopting an open conformation, interacted with the aurone 1. This substrate was stabilized by π–π stacking interactions between the phenyl ring of benzofuranone and the quinoline moiety. The attack occurred at the Re-face of the substrate, with the zwitterion intermediate formed by the combination of the allenoate and the quinuclidine nitrogen atom. A notable feature of this methodology was its successful cyclization strategy, which enabled the creation of an all-carbon tertiary chiral centre within the dihydropyran-fused benzofuran framework. Consequently, a series of chiral dihydropyran-fused benzofuran derivatives were obtained with satisfactory regioselectivity.65
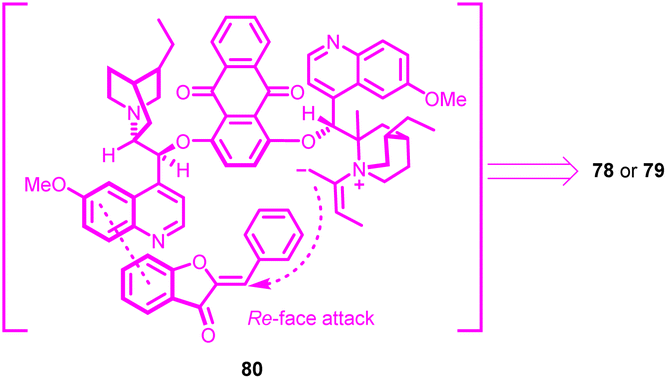 | ||
| Fig. 3 The transition state. | ||
In 2015, Rasmas Mose and co-workers showcased an organocatalytic approach for cross-dienamine activation, leading to the synthesis of chiral multifunctionalized norcamphor compounds 83 with a wide range of substitution patterns (Scheme 29). The mechanistic insights into the organocatalytic intramolecular aldol reaction catalysed by primary amines derived from cinchona alkaloids suggest a closely related transition state 84 for this intermolecular aminocatalyzed Diels–Alder reaction. Initially, the cinchona alkaloid aminocatalyst undergoes condensation with cyclopentenone to form a cross-dienamine intermediate. The dienophile is then activated and guided to the appropriate position via hydrogen bonding upon protonation of the quinuclidine part of the catalyst. Following the cycloaddition step, the resulting enamine is hydrolyzed to produce the corresponding ketone while releasing the aminocatalyst. Considering the diastereoselectivity achieved, it appears that the reaction likely proceeds through an asynchronous concerted cycloaddition, given the high diastereomeric ratio of more than 20:1.
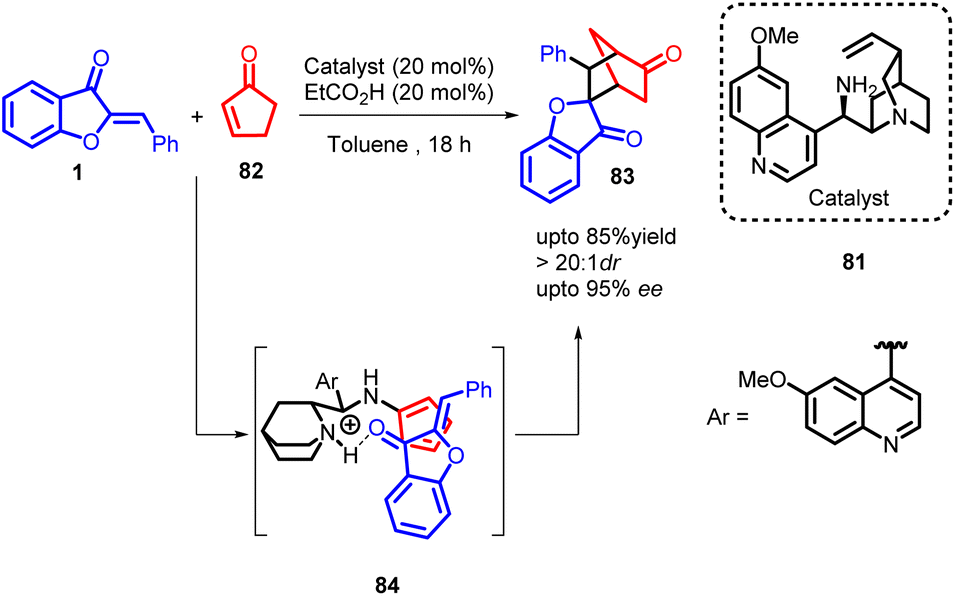 | ||
| Scheme 29 Synthesis of multifunctionalized norcamphor scaffolds by asymmetric organocatalytic Diels–Alder reactions. | ||
The investigation demonstrated that the catalyst derived from quinine outperformed all other catalysts employed, leading to a favourable yield of compound 83 with high enantioselectivity. This method presents a direct and versatile approach for generating 5,6-substituted norcamphor derivatives 83 through an asymmetric organocatalytic process. Importantly, the reaction conditions exhibited a remarkable level of versatility, enabling the use of common electron-deficient olefins in the cycloaddition reaction without requiring extensive modifications. The resulting norcamphor scaffolds 83 were consistently obtained with good to high yields and displayed a high degree of stereoselectivity. This approach provides an efficient and streamlined route for synthesizing a significant class of essential privileged structures.66
In 2018 L. C. Yang et al., reported a different Lewis-acid-assisted palladium catalysed method for the syntheses of [5,5] and [6,5] spiro heterocycles 91 (Scheme 30). There was an extraordinary switch from alkoxide-p-allyl to dienolate reactivity which was achieved by using Pd–Ti relay catalysis and also Umpolung reactivity of the vinylethylene carbonates 85 was represented.
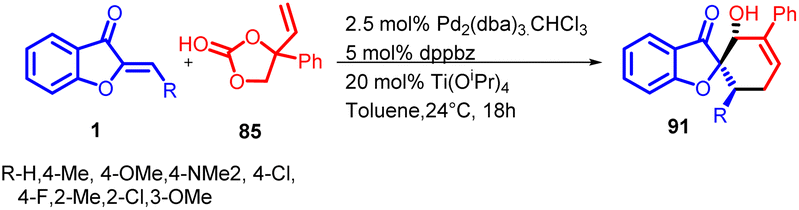 | ||
| Scheme 30 Palladium–titanium relay catalysis enables synthesis of spiro-heterocycle. | ||
The mechanism for the [4 + 2] cycloaddition is shown in Scheme 31, which describe palladium–titanium relay catalysis. The Pd-π-allyl intermediate 86 is formed from 85 and Pd (0). On one hand, 86 could undergo standard [3 + 2] cycloaddition with the electrophilic aurone 1a (using Mg as a Lewis acid) to produce 89. On the other hand, 86 may react with Ti(OiPr)4 to form an alternative intermediate 87, in which ligand exchange between Ti and Pd is proposed to free up the alkoxide–Pd coordination and enable β-hydride elimination to produce the titanium-dienolate 88. The palladium–hydride isopropoxide complex released from this step then undergoes reductive elimination to complete the palladium catalytic cycle. In the titanium catalytic cycle, the formation of 88 converts the electrophilic Pd-π-allyl into a nucleophilic titanium-dienolate. The aurone 1a is also likely activated by the titanium-based Lewis acid, and undergoes a reaction with 87 in a formal vinylogous Michael addition. The resultant intermediate 90 undergoes intramolecular aldol followed by protonation to yield the desired product 91. In the presence of a chiral titanium complex, the formation of 90 may proceed in an enantioselective fashion.67
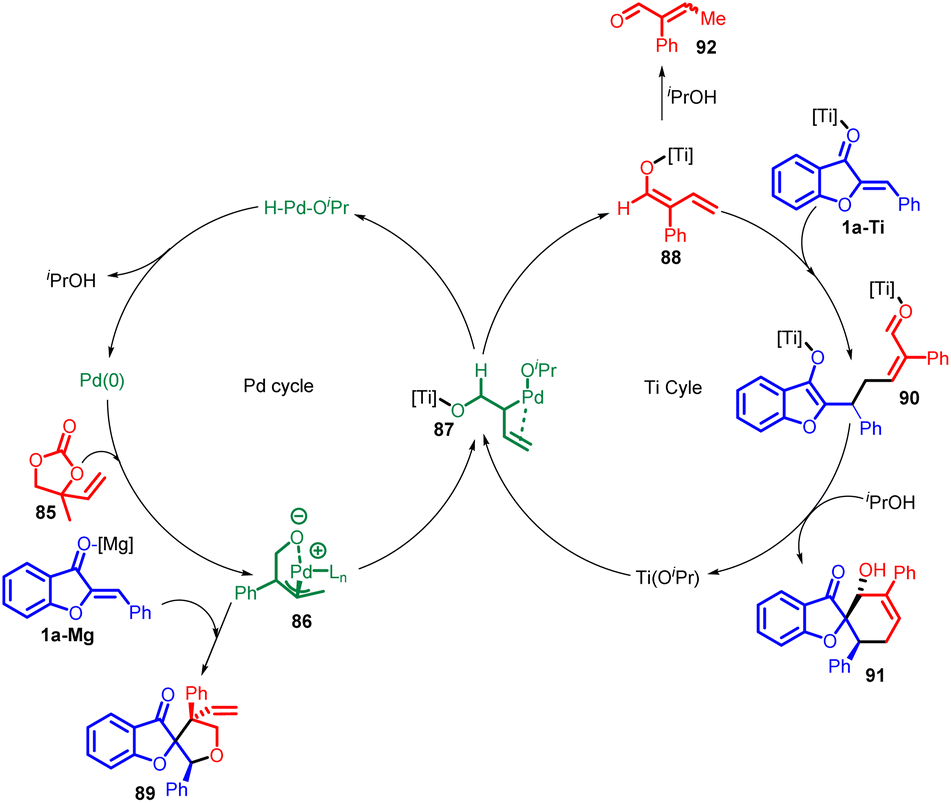 | ||
| Scheme 31 Proposed mechanism for the synthesis of spiro-heterocycle. | ||
[4 + 3] Cycloaddition reaction
The [4 + 3] cycloaddition reaction involves a formal cycloaddition between an allylic cation and a diene, resulting in the creation of a seven-membered ring. Apart from its intrinsic significance as a potentially pericyclic reaction involving a reactive entity, this reaction pattern is notable for forming a seven-membered ring structure. The notation [4 + 3] indicates the total count of atoms participating in the ring-forming process. Electron-wise, the reaction corresponds to a [4 + 2] cycloaddition since the allylic cation dienophile possesses only two π electrons. As such, this reaction can be seen as a counterpart of the Diels–Alder reaction in terms of electron count and mechanism.68–70Zuo and Trost in 2019 reported a significant advancement in the field of organic synthesis involving a palladium-catalysed enantioenriched asymmetric [4 + 3] cycloaddition to create seven-membered scaffolds. This innovative process led to the production of numerous benzo[b]oxepines 95 using a wide range of donors and acceptors, showcasing exceptional levels of chemo-, regio-, diastereo-, and enantioselectivity (Scheme 32).
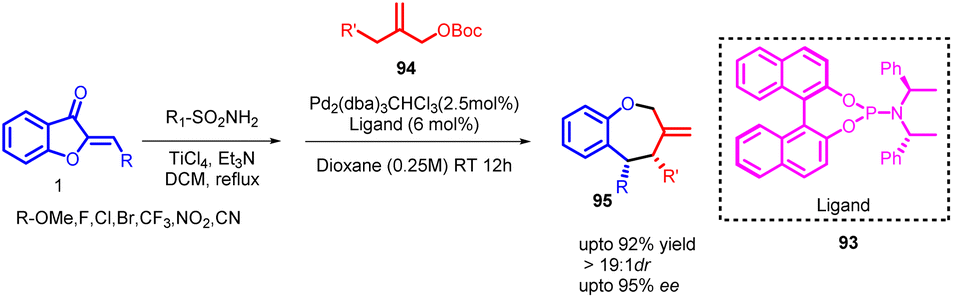 | ||
| Scheme 32 Synthesis of tetrahydroazepines and benzo[b]oxepines through palladium-catalyzed [4 + 3] cycloaddition reactions. | ||
The reaction follows a sequence involving the zwitterionic Pd-trimethylenemethane (Pd-TMM) intermediate 96 (Fig. 4). This intermediate is formed in situ through the ionization of the donor. Subsequently, base-assisted deprotonation occurs, followed by nucleophilic addition to the acceptors. This series of steps leads to the creation of the zwitterionic Pd-TMM intermediate. The nucleophilic attack onto the Pd-π-allyl intermediate can occur from either end of the allyl anion, resulting in the formation of different cyclic products. The regioselectivity of this process heavily relies on the substantial contrast in nucleophilicity and steric hindrance between these positions. Notably, the nucleophilic attack at the terminal position of the π-allyl intermediate is favoured over the internal nucleophilic attack, leading to the formation of a solitary [4 + 3] cycloadduct. The enantioselective and diastereoselective [4 + 3] product by induction of chiral palladium species.
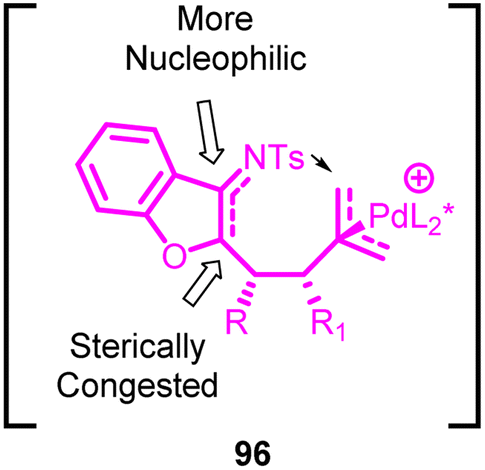 | ||
| Fig. 4 Transition state. | ||
Unlike the conventional TMM cycloaddition process, this approach exhibits distinctive regioselectivity. The practical advantage of employing two easily accessible reactants significantly influences the feasibility of this synthetic strategy. Furthermore, the resulting seven-membered products constitute foundational structures in numerous vital bioactive compounds.71
[10 + 2] Cycloaddition reaction
Exploring higher-order cycloaddition processes offers an intriguing strategy for constructing intricate polycyclic frameworks through a convenient one-step reaction. Despite the well-established nature of the Diels–Alder and 1,3-dipolar cycloaddition reactions involving 6π-electrons, the advancement of higher-order cycloadditions encompassing more than 6π-electrons has been relatively limited. This restraint is likely attributed to challenges posed by intricate regio- and chemoselectivity, along with potential competing pathways. Through a comprehensive examination of existing literature, it has been identified that cyclic polyene species known as isobenzofulvenes, initially investigated by Hafner and Bauer in 1968, exhibit notable potential as highly reactive 8π- or 10π-synthons in diverse cycloaddition reactions.62 Classified as a pericyclic reaction, the [10 + 2] cycloaddition follows a cyclic transition state and involves the reconfiguration of pi-electrons. This reaction has gained notable attention for its capacity to efficiently generate intricate and complex structures within a single step. It finds application in the synthesis of a wide array of compounds, particularly in the construction of innovative and distinctive cyclic architectures, holding significant promise across diverse realms of chemistry.72,73In 2020, Yang Yang et al., introduced a groundbreaking advancement in the realm of organic synthesis. They presented a chemoselective and asymmetric cross [10 + 2] cycloaddition reaction. This novel process involved a diverse range of activated electron-deficient alkenes, such as aurones 1. Even when utilizing catalyst loadings as low as 1 mol%, the reaction displayed impressive results, yielding a variety of fused molecular frameworks that exhibited multifunctional characteristics. These outcomes were achieved with remarkable levels of diastereo- and enantioselectivity (Scheme 33).
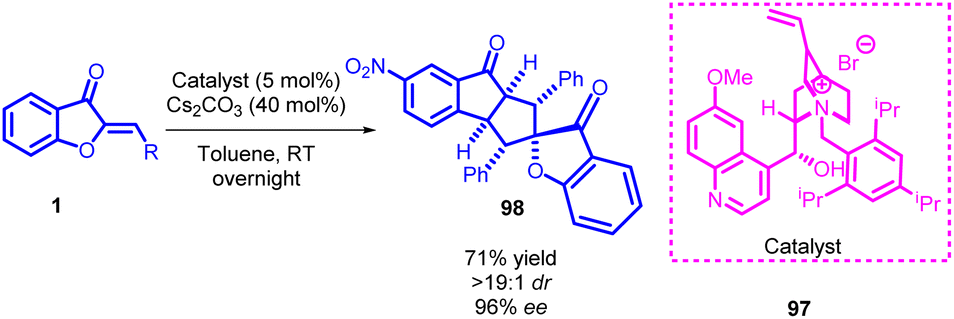 | ||
| Scheme 33 Asymmetric cross [10 + 2] cycloadditions of aurones and activated alkenes. | ||
The research unveiled that activated aurones 1 can engage in highly selective cross-formal [10 + 2] cycloaddition reactions with various electron-deficient alkene substrates. Under chiral phase transfer catalytic conditions, aurones could be effectively converted into dearomative 1-hydroxyl isobenzofulvene-type species. Notably, despite the spatial separation between the reactive site and the chiral ion pair complex, exceptional diastereo- and enantioselectivity were consistently achieved. This was accomplished through the utilization of a newly devised cinchona-based ammonium salt 97 featuring a sizable substituent. Impressively, this catalyst design operated at notably low loadings (1 mol%), leading to a diverse array of fused frameworks characterized by substantial structural intricacy and adaptability.73
Annulation reaction
The term “annulation” originates from the Latin word “annulatus,” meaning “ringed,” and it pertains to the creation of rings in organic chemistry. This concept involves the process of introducing a ring structure onto an existing system, regardless of its cyclic or non-cyclic nature. The newly added ring can vary in size, although 5- and 6-membered rings are the most common. This wide-ranging definition encompasses reactions that might not traditionally be seen as annulation reactions, such as Diels–Alder reactions, acid-catalyzed polyolefinic cyclizations, photochemical, radical, and thermal cyclizations. Annulation reactions can be categorized based on how a 3-ketoalkyl chain is attached, which includes Michael reactions, nucleophilic additions (using Grignards, ylides, etc.), and alkylations. Additionally, annulation reagents can be further classified into those designed to add single rings sequentially (known as mon-annulation reagents) and those that introduce multiple ring segments simultaneously, referred to as bis- or tris-annulation reagents.74In 2018, Xueji Ma et al., introduced an efficient method for the annulation of α-imino rhodium carbenes with aurones 1 (α,β-unsaturated ketones) (Scheme 34). This innovative approach resulted in the synthesis of versatile 2,3-dihydropyrrole derivatives 103 with multiple substituents. Importantly, this method was also effective with typical linear α,β-unsaturated ketones.
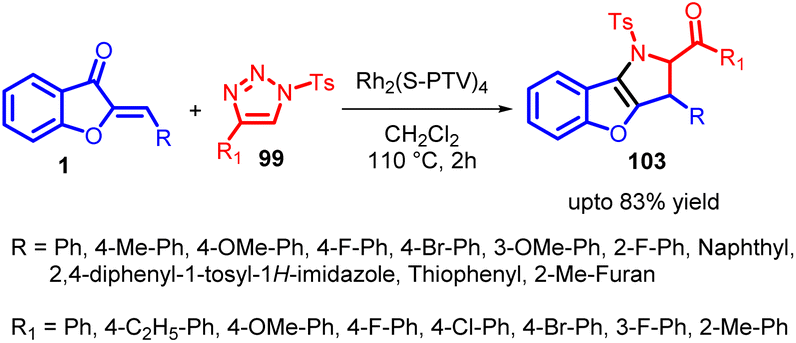 | ||
| Scheme 34 Rhodium-catalyzed annulation of α-imino carbenes with α,β-unsaturated ketones. | ||
The proposed catalytic cycle for generating the desired products (compounds 103 and 104) is delineated in Scheme 35. This depiction draws upon both experimental observations and established scientific literature. Initially, a triazole compound 99 triggers the generation of α-imino rhodium carbene species 100, facilitated by the presence of a Rh(II) catalyst. Subsequently, the oxygen atom of the α,β-unsaturated ketone participates in a nucleophilic attack on the carbene species, leading to the formation of oxonium ylide species 101. The imino group of species 101 then engages in a nucleophilic attack on the carbon atom of the carbonyl group, resulting in a 1,2-addition process that yields oxazole compound 102. However, due to its inherent instability, compound 102 undergoes cleavage of the C–O bond, leading to rearrangement and ultimately generating the multi-substituted 2,3-dihydropyrrole 103. Alternatively, species 101 can undergo intramolecular ring closure reactions, forming seven-membered heterocyclic products. Notably, the entire experimental study did not detect any products arising from 1,4-addition reactions. This high degree of selectivity can be attributed to the strong electron-attracting nature of carbonyl groups toward amino groups, promoting the formation of five-membered rings as the preferred outcome over seven-membered rings.
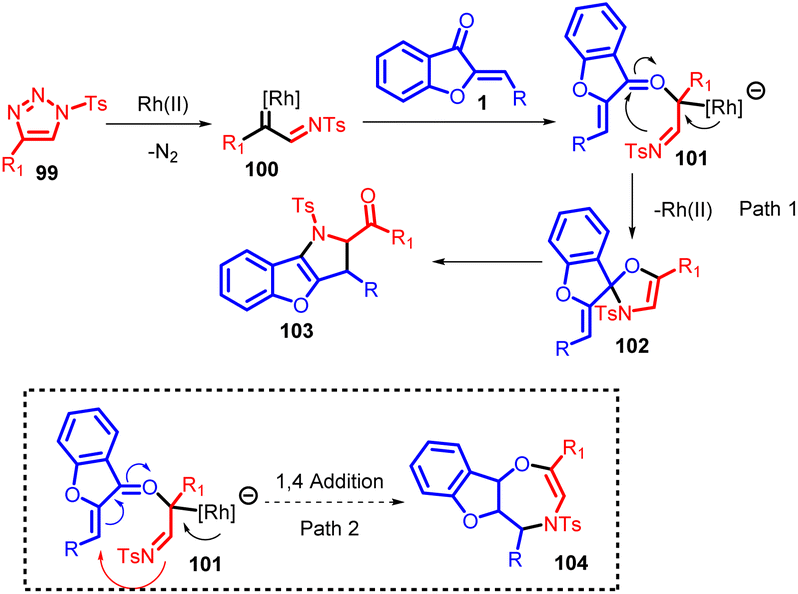 | ||
| Scheme 35 Proposed mechanism for the synthesis of 2,3-dihydropyrrole. | ||
The methodological procedure outlined in this study underscores the evident nucleophilic behaviour of α,β-unsaturated ketones towards α-imino rhodium carbenes. This observation holds significant potential for rapidly generating molecular complexity, a valuable trait for synthesizing diverse bioactive pharmaceutical compounds.75
Michael/Mannich reaction of aurones
The Mannich–Michael reaction stands as a potent organic transformation, combining the fundamental Mannich reaction and Michael addition. This sequential process enables the synthesis of intricate molecules bearing diverse functionalities and stereocenters. In the Mannich reaction, a primary or secondary amine reacts with a carbonyl compound (aldehyde or ketone) and a proton-source compound (like enolizable ketones or imines). This leads to the nucleophilic addition of the amine to the carbonyl, followed by the elimination of a leaving group, resulting in β-amino carbonyl compounds.76 Conversely, Michael addition involves a nucleophile (often enolates or enols) reacting with an α,β-unsaturated carbonyl compound (α,β-unsaturated ketone or ester), forming a new carbon–carbon bond.77,78 In the Mannich–Michael reaction, an amine reacts with an α,β-unsaturated carbonyl compound in the presence of a proton-source substance. The amine's initial interaction with the carbonyl generates an intermediate through Mannich reaction, followed by Michael addition with another molecule of the carbonyl compound. This yields complex molecules with new carbon–carbon bonds and often multiple stereocenters. The Mannich–Michael reaction's versatility facilitates intricate structure creation and multiple bond and stereocenter formation in one step, finding application in synthesizing natural products, pharmaceuticals, and intricate molecules.79In 2014, Song Ye and co-workers conducted a study wherein they utilized bifunctional N-heterocyclic carbenes (NHCs) 105 possessing a free hydroxy group as highly effective catalysts (Scheme 36). The aim was to facilitate the [3 + 4] annulation reaction between enals and aurones, yielding benzofuran-fused γ-lactones 110 with favourable outcomes in terms of yield, diastereoselectivity, and enantioselectivity. Importantly, this bifunctional NHC catalysis prevented the competitive formation of corresponding [3 + 2] cycloadducts. Their experiments demonstrated that the [3 + 4] cycloadducts were kinetically favoured over the thermodynamically preferred [3 + 2] cycloadducts. This behaviour changed when a non-bifunctional NHC catalyst was introduced, indicating that the [3 + 4] cycloadducts could transform into the [3 + 2] cycloadducts under these conditions.
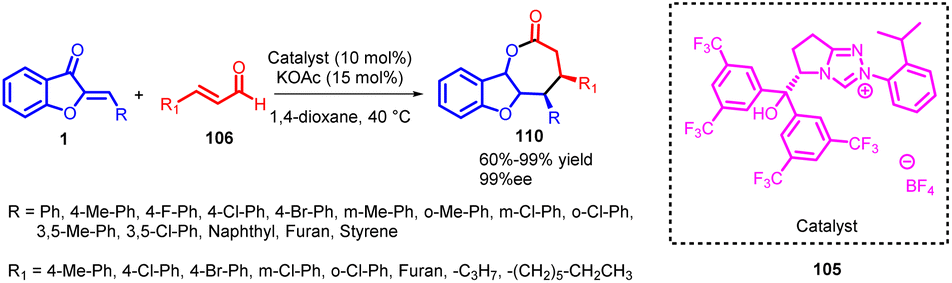 | ||
| Scheme 36 Bifunctional N-heterocyclic carbene catalyzed [3 + 4] annulation of enals and aurones. | ||
The suggested mechanism for this catalytic process is illustrated in Scheme 37. Initially, the NHC 105 added to enals, generating the vinyl Breslow intermediate 107. This intermediate then underwent a Michael addition reaction with aurones, leading to the formation of adduct 108. The presence of a possible hydrogen bonding interaction between the hydroxy group of the NHC 105 and aurones 1 was proposed to enhance reactivity and regulate both diastereoselectivity and enantioselectivity. Subsequently, intramolecular acylation of adduct 109 occurred, resulting in the formation of the ultimate cycloadduct 110 and concurrently regenerating the NHC catalyst. Another potential intramolecular hydrogen bonding interaction between the hydroxy and acyl azonium groups was suggested to transform the carbonyl into a stronger Lewis acid. This transformation facilitated the harder Lewis basic O-acylation of the enolate, favouring the production of the kinetically favoured [3 + 4] cycloadduct 110 while discouraging the formation of the [3 + 2] cycloadduct, which would result from a softer Lewis basic C-acylation pathway.
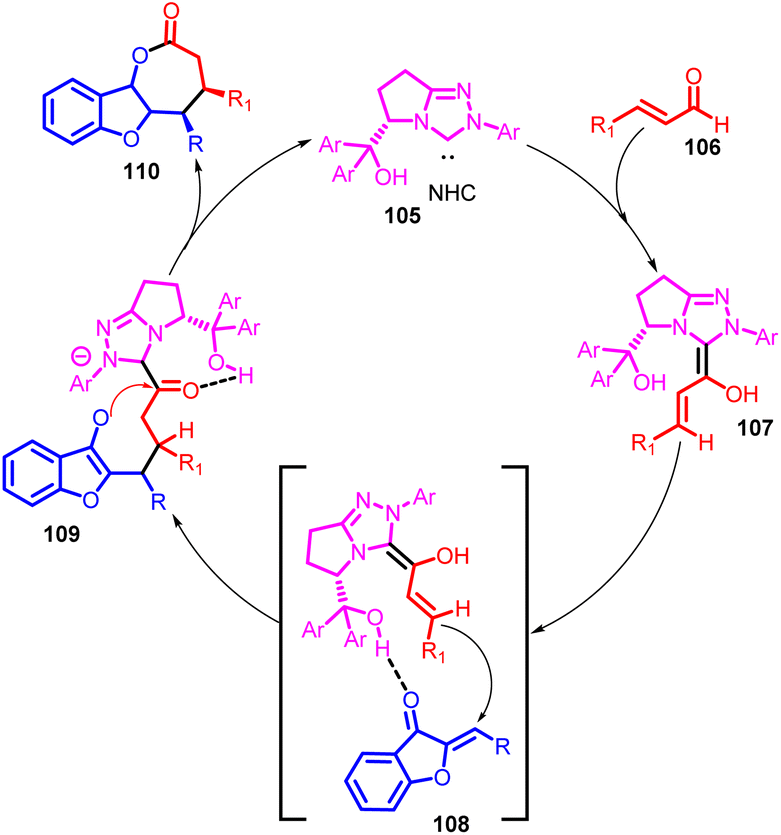 | ||
| Scheme 37 Proposed catalytic cycle for the NHC catalyzed [3 + 4] annulation of enals and aurone. | ||
Pre-NHCs with hydroxy groups exhibiting higher acidity showed improved yields and similar levels of diastereoselectivity and enantioselectivity. During solvent evaluation, the most favorable results were obtained in 1,4-dioxane, leading to enhanced diastereomeric ratios (dr) and enantiomeric excess (ee). Additionally, a minor improvement in diastereoselectivity and enantioselectivity was observed when potassium acetate was used as the base, and a slight excess of the base had a positive impact on reaction yield. Ultimately, the reaction could be conducted at 40 °C, resulting in a high yield without compromising diastereoselectivity or enantioselectivity. These findings provide valuable insights for optimizing the reaction conditions for this process.80
In 2016, Satavisha Kayal et al., introduced a novel cascade reaction that combines Michael addition and cyclization processes to achieve the synthesis of 3,2′-pyrrolidinyl bispirooxindole 113 derivatives with high enantioselectivity. This innovative approach involves the interaction between 3-isothiocyanato oxindoles 112 and exocyclic α,β-unsaturated ketones (aurone) 1, all catalyzed by a quinine-derived bifunctional tertiary amino-squaramide catalyst 111 (Scheme 38). The resulting products, which possess three contiguous stereogenic centres, are exclusively obtained as a single diastereomer, typically with excellent yields and remarkable levels of enantioselectivity.81
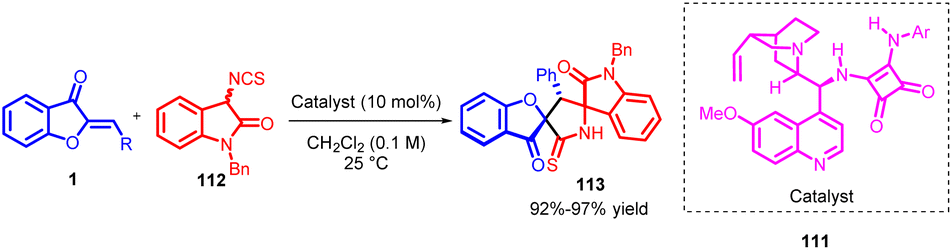 | ||
| Scheme 38 Synthesis of 3,2-pyrrolidinyl bispirooxindoles from 3-isothiocyanato oxindoles and α,β-unsaturated ketones. | ||
In 2019, Wenjin Yan and co-workers synthesized a series of compounds showcasing inventive bispiro[benzofuranoxindole-pyrrolidine] 116 cores. This achievement was realized through the utilization of a thiourea-catalysed 114 1,3-dipolar cycloaddition procedure (Scheme 39). A comprehensive assessment of catalysts helped in the identification of a more selective thiourea catalyst. This catalyst achieved an exceptional enantiomeric excess of 99%. Subsequent investigation into the impact of solvents revealed that alkyl halides were superior in terms of stereoselective performance compared to other solvents. Notably, 1,2-dichloroethane emerged as the optimal solvent, resulting in a remarkable 99% yield and an enantiomeric excess exceeding 99%. Given the remarkable efficiency of catalyst 114, the study explored the use of lower catalyst loadings. Surprisingly, even with only 1 mol% of catalyst, the reaction maintained excellent enantioselectivity, diastereoselectivity, and yield, albeit with a slightly extended reaction time. These findings highlight the remarkable potential of this catalyst and its versatility in achieving high-quality results with reduced catalyst quantities. These products were characterized by their impressive yields and selectivity in terms of stereoisomer formation.
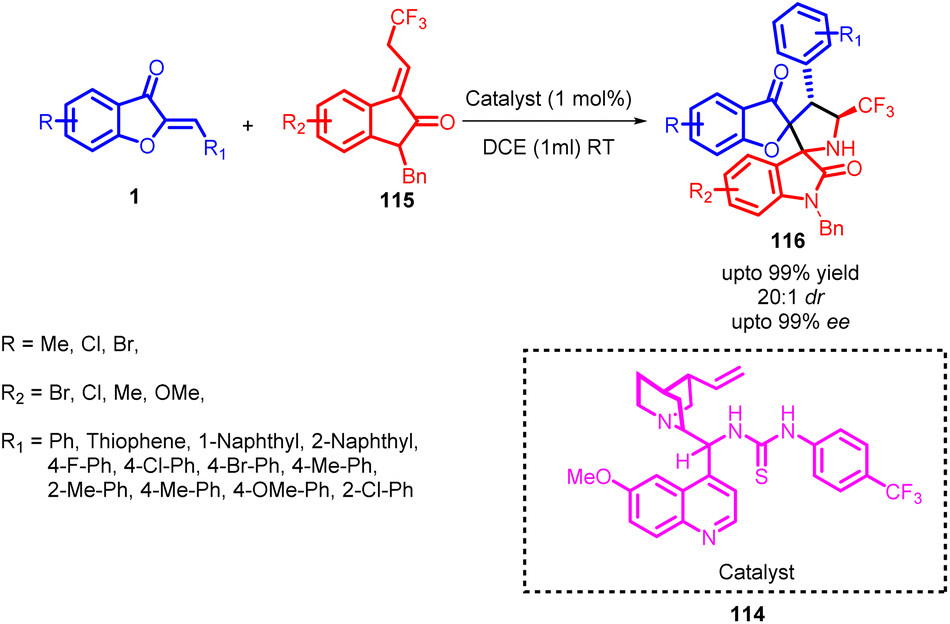 | ||
| Scheme 39 Synthesis of bispiro[benzofuran-oxindole-pyrrolidine]s through organocatalytic cycloaddition. | ||
The reaction pathway involved the Michael/Mannich transformation of N-2,2,2-trifluoroethylisatin ketimines 115 with aurones 1, ultimately giving rise to the production of cycloadducts. By analysing the X-ray crystallography of the resulting product's structure, a potential transitional state 117 (Fig. 5) was suggested for the gradual cycloaddition process. Initially, the activated azomethine ylide engages in a Michael addition to the aurone substrate, yielding an intermediate. This intermediate subsequently carries out an intramolecular Mannich reaction, directing its attack towards the Re-face of the imine. This sequence ultimately leads to the formation of the cycloadduct.82
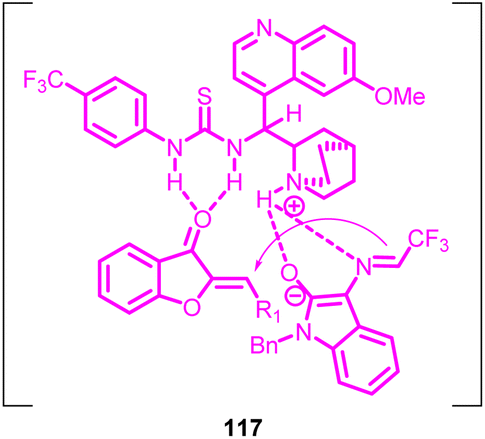 | ||
| Fig. 5 Intermediate. | ||
In 2022, Ze Zhang and co-workers introduced a successful iodine-promoted domino reaction involving aurones 1 and amidines 118. This reaction unfolds in a sequential fashion, involving a series of steps including Michael addition, iodination, intramolecular nucleophilic substitution-driven cyclization, and spiro ring opening to achieve dehydrogenative aromatization. By adopting this innovative approach, a range of 1,2,4-trisubstituted 5-(O-hydroxybenzoyl)imidazoles were effectively synthesized with moderate to good yields, utilizing readily available starting materials (Scheme 40).
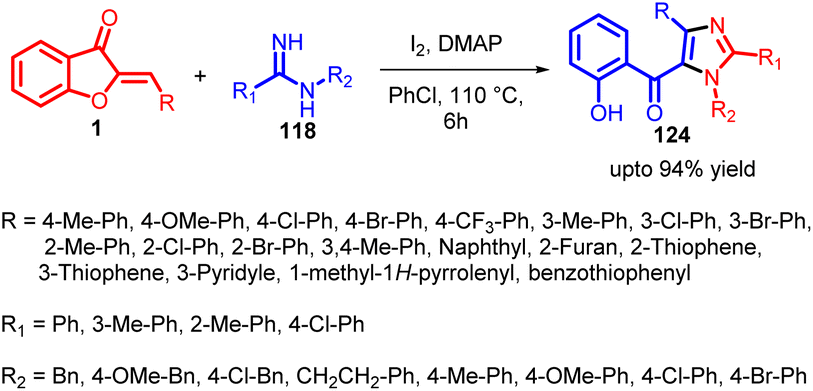 | ||
| Scheme 40 Synthesis of fully substituted 5-(o-hydroxybenzoyl)imidazoles. | ||
A plausible mechanism (Scheme 41) for this iodine-promoted domino reaction to generate fully substituted 5-(o-hydroxybenzoyl)imidazole 124 is postulated. Initially, an aza-Michael addition transpires between the reactants 1 and 118 in the presence of I2, resulting in the formation of an adduct that exists in an equilibrium between ketone/imine 119 and enol/enamine 120. Subsequent iodination of this adduct by I2 leads to the creation of intermediate iodide 121, which undergoes an intermolecular nucleophilic substitution event to yield the intermediate spiroimidazoline 123. LC-MS analysis detects intermediates 119/120 and 121 during these stages. Concomitantly, 2 equivalents of HI are generated and captured by DMAP. Further, a base-mediated spiro ring opening of 123 facilitates dehydrogenative oxidation and aromatization, eventually yielding the desired product, imidazole 124. Additionally, the interaction between I2 and the oxygen atom within the spiro ring can potentially facilitate cleavage of the C–O bond.
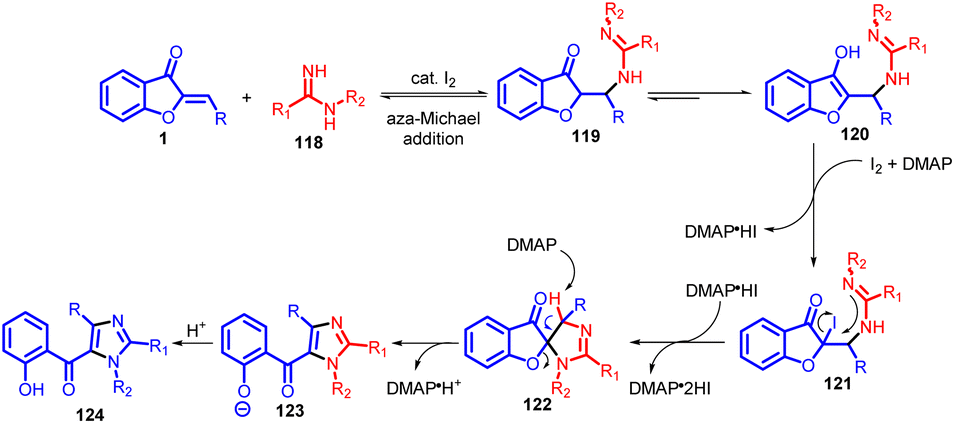 | ||
| Scheme 41 Proposed mechanism. | ||
This approach boasts several advantages, including being metal-free, operating under mild reaction conditions, promoting high atom economy, accommodating a broad substrate range, not necessitating the use of specialized or costly starting materials, and not requiring an excess of oxidants. The strategy of spiro ring opening for the dehydrogenative aromatization of spiro compounds may have the potential for extension to the construction of diverse conjugated heterocycles.83
In 2022, Hui Xu et al., introduced an innovative and effective I2/FeCl3-catalyzed domino reaction, combining aurones with enamino esters through a sequence of Michael addition, iodination, intramolecular nucleophilic substitution, and spiro ring opening processes (Scheme 42). This method initiates with a Michael addition involving 1 and 125 in the presence of either I2 or FeCl3, forming adduct 126. This compound tautomerizes into the enol-form adduct 127. Subsequently, 127 engages with I2 through an electrophilic substitution to generate intermediate 128, which then undergoes intramolecular nucleophilic substitution to yield spiro intermediate 129. In the course of iodination from 127 to 128 and the subsequent cyclization of 128 into 129, the generated HI is both further oxidized by Fe(III) to regenerate I2 and simultaneously produce Fe(II). Oxygen then oxidizes Fe(II) back into Fe(III) to complete the catalytic cycle. Following this, aromatization occurs, leading to the final product 131via an E1-elimination of 130, formed through spiro ring opening of intermediate 129 with the assistance of Lewis acid I2 or FeCl3 to facilitate C–O bond cleavage (Scheme 43).
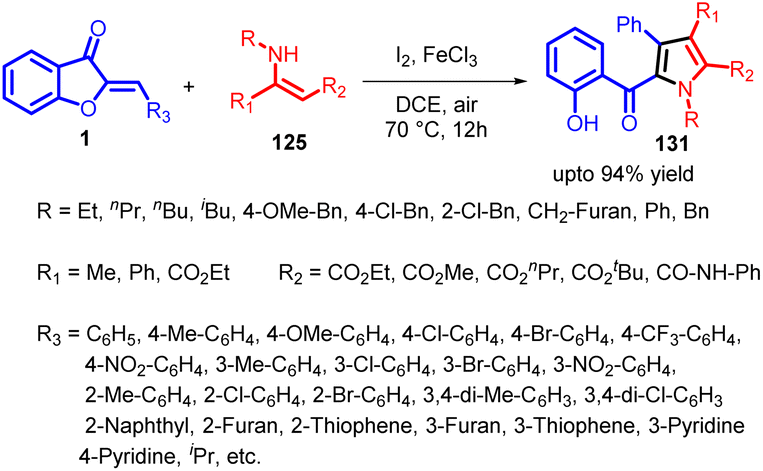 | ||
| Scheme 42 I2/FeCl3 catalysed domino reactions of aurones with enamino esters. | ||
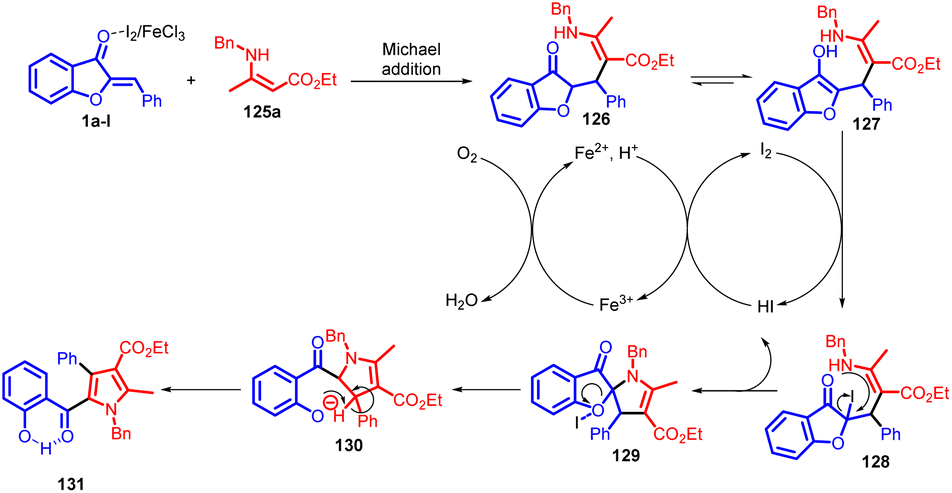 | ||
| Scheme 43 Proposed mechanism for the domino reaction to form pyrroles. | ||
This method facilitates the synthesis of a diverse range of highly functionalized pyrroles (131) with moderate to high yields and good functional group compatibility. The presence of a phenolic hydroxy group in the products offers the potential for subsequent functionalization. Notably, this novel reaction offers the benefits of operating under mild conditions, promoting high atom economy, and being suitable for large-scale synthesis. As a result, it is expected to emerge as a significant route for constructing conjugated nitrogen-containing heterocycles.84
General reactions
Anil K. Patel et al., in 2010 accomplished the synthesis of 3-(4-aryl-benzofuro[3,2-b]pyridin-2-yl)coumarins 133 using Kröhnke's reaction (Scheme 44). This transformation was achieved through the utilization of 3-coumarinoyl methyl pyridinium salts 132 and 2-arylidene aurones 1 as reactants. The reaction was facilitated by the presence of ammonium acetate and acetic acid as additives.85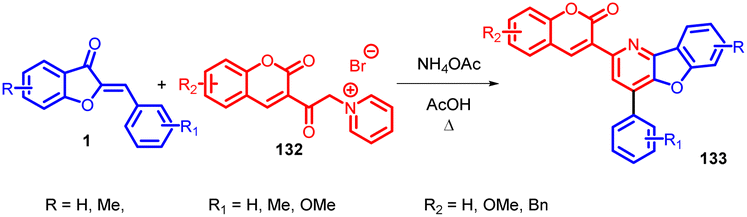 | ||
| Scheme 44 Synthesis of 3-(4-arylbenzofuro[3,2-b]pyridine-2-yl)coumarins from aurones. | ||
In 2014, Min Wang et al.,86 devised a method utilizing NHC catalysis for the divergent annulation of enals 137 with aurones 1. This innovative approach yields either benzofuran-containing ε-lactones 141a & 141b or spiro-heterocycles 142a & 142b with remarkable diastereo- and enantioselectivity (Scheme 45). Notably, the selectivity is directed by the chiral catalyst's structural framework.
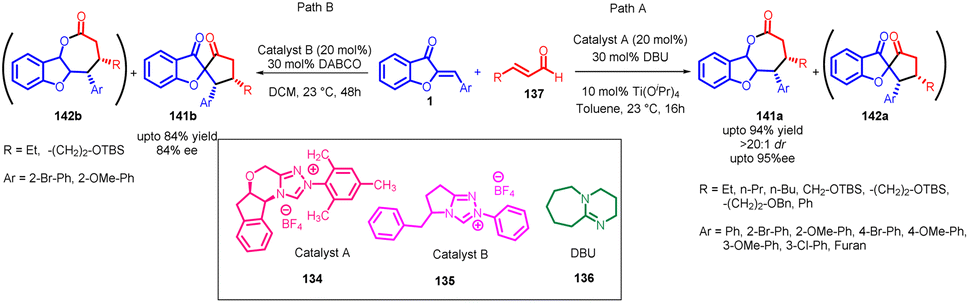 | ||
| Scheme 45 Stereoselective synthesis of ε-lactones or spiro-heterocycles through NHC-catalysed annulation. | ||
Building upon the distinctive stereochemical outcomes and drawing insights from earlier research by the Scheidt and Ye groups, a plausible mechanism was postulated. The process involves the generation of a homoenolate intermediate 138 through interaction between the enal and carbene catalyst. This intermediate then attacks the heterocyclic enone, yielding enolate 140. The configuration of the newly formed stereocenters is orchestrated by the influence of the chiral catalyst, depicted in the transitional state model 139 (Scheme 46). It is worth noting that the enantioselectivity displayed similar levels for both the ε-lactones and spirocycle series, suggesting a shared intermediate. In cases where the enals bear linear aliphatic substituents (R), an impressive diastereomeric ratio of 20:1 was achieved. Conversely, when R is replaced with a bulkier aromatic group, steric interactions between –R, –Ar, and the heterocyclic backbone might trigger alternative conformations, leading to reduced diastereoselectivity.86
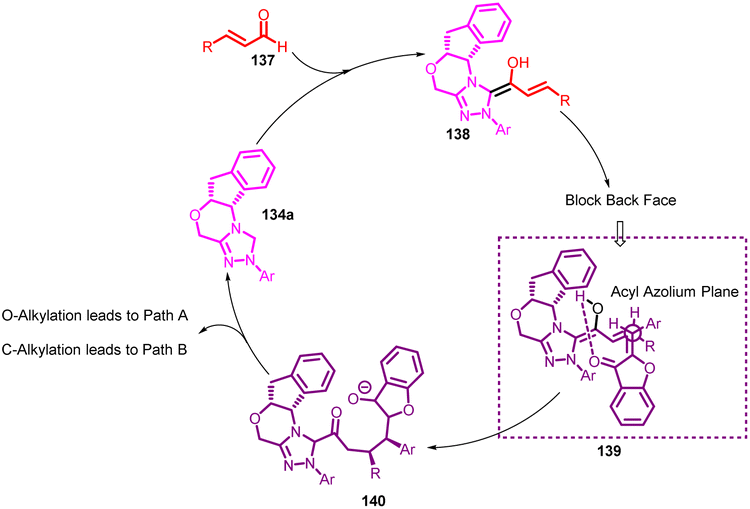 | ||
| Scheme 46 Proposed mechanism for the stereoselective synthesis of ε-lactones or spiro-heterocycles. | ||
In 2015, Rakesh R. Giri et al., synthesised 4-methyl-3-phenyl-6-(4-phenyl-benzofuro[3,2-b] pyridin-2-yl) coumarins 148via Michael addition approach, involving the nucleophilic attack of the active methylene group from 4-methyl-3-phenyl-6-coumarinoyl methyl pyridinium salt 143 onto the α,β-unsaturated carbonyl moiety presents in aurones 1 (Schemes 47 and 48).
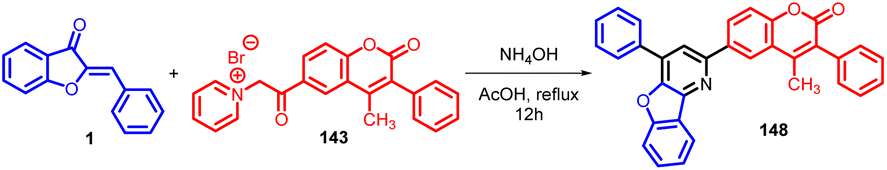 | ||
| Scheme 47 Synthesis of modified pyridine-substituted coumarins from aurones. | ||
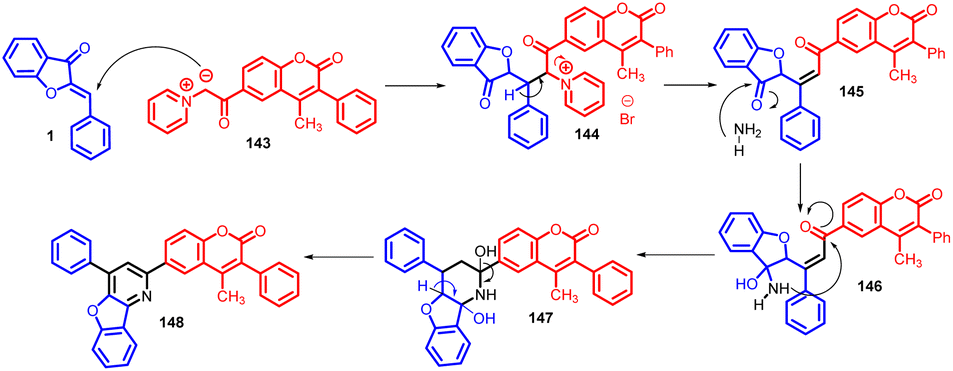 | ||
| Scheme 48 Proposed mechanism for the synthesis of modified pyridine substituted coumarines. | ||
Subsequently, the synthesized compounds were subjected to antimicrobial testing against a range of microorganisms including Staphylococcus aureus, Bacillus subtilis, Escherichia coli, Salmonella typhi, Candida albicans, and Aspergillus niger. The outcome of these assays revealed noteworthy antimicrobial efficacy of the synthesized compounds against the tested strains.87
During investigations into the synthesis and chemical properties of spiroheterocycles, Moncef Msaddek and co-workers in 2014 revealed that when they subjected aurone 1 derivatives to a reaction with 2-diazopropane 149, it led to the formation of spirocyclopropane 150 compounds. While the anticipated outcome primarily comprised the formation of cycloadducts, there was an unexpected result in certain instances: the generation of oxadiazole 151 compounds. This unforeseen occurrence added an interesting dimension to their research findings (Scheme 49).88
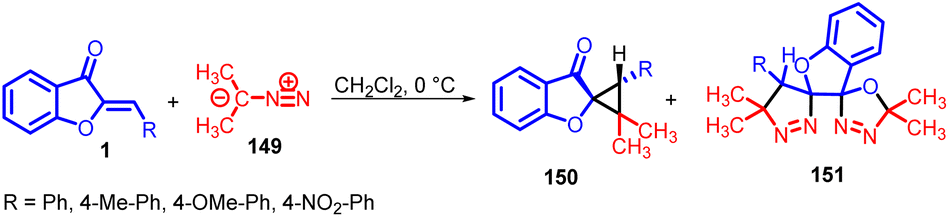 | ||
| Scheme 49 Synthesis of spirocyclopropane and oxadiazole from aurones. | ||
Łukasz Albrecht and coworkers in 2016 reported an enantioselective synthesis of spirocyclic tetrahydrothiophene 154 compounds featuring a benzofuran-3(2H)-one structural motif. This innovative approach makes use of aurone 1 and 2-thioacetaldehyde, which is formed in situ from 1,4-dithiane-2,5-diol 153, as the initial building blocks (Scheme 50). The process follows a sequential cascade mechanism involving a thio-Michael-aldol reaction sequence.
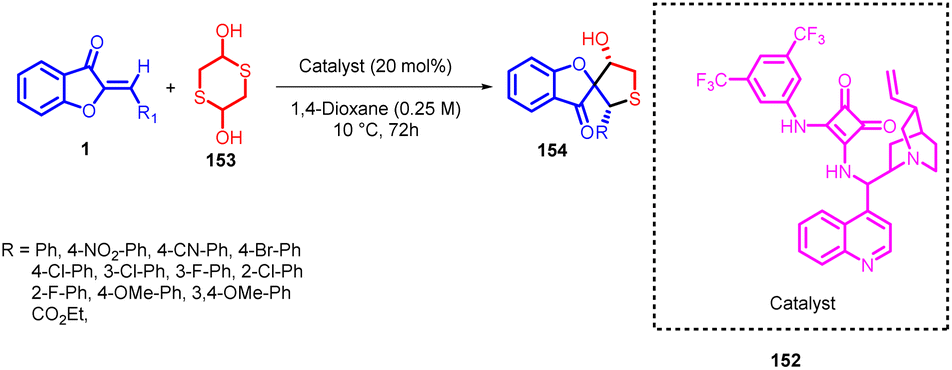 | ||
| Scheme 50 Enantioselective synthesis of spirocyclic tetrahydrothiophene derivatives bearing a benzofuran-3(2H)-one scaffold. | ||
The stereochemical outcome of this reaction was finely controlled through the utilization of a bifunctional catalyst derived from cinchona alkaloids, with a squaramide functional group. This strategy allowed for precise manipulation of the desired stereochemistry in the resulting compounds.89
Shanshan Ma et al., in 2020 developed a novel phosphine promotion to facilitate a domino reaction between aurone derivatives 1 and MBH carbonate 155 (Scheme 51). This strategy enables the efficient synthesis of [6-5-5-6-6] pentacyclic compounds that incorporate benzothiophene and tetrahydroquinoline frameworks 163 and 164. These intricate structures are typically challenging to construct using conventional synthetic methods. The researchers successfully prepared a diverse array of [6-5-5-6-6] pentacyclic compounds 163 and 164 in a highly efficient manner.
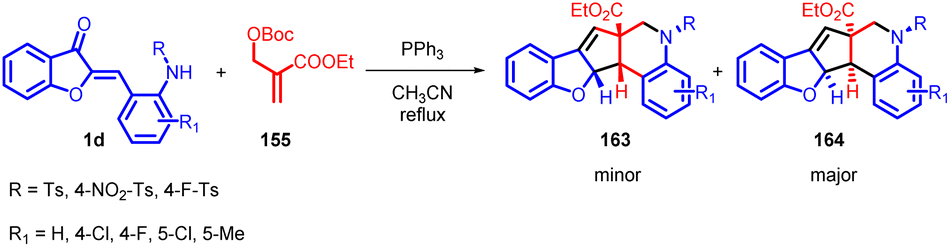 | ||
| Scheme 51 Synthesis of [6-5-5-6-6] pentacyclic via phosphine catalysed domino reaction. | ||
The proposed mechanism involves the interaction between MBH carbonate 155 and PR3 to generate a phosphorus ylide 156, along with tBuOH and CO2 (Scheme 52). The ensuing deprotonation of 1d by phosphorus ylide 157 leads to the formation of intermediate 158. Subsequent steps encompass the nucleophilic addition of 158 to 157, accompanied by the removal of PPh3, resulting in intermediate 159. This intermediate is subject to nucleophilic attack by PPh3 at the terminal alkene carbon, initiating a cycloaddition reaction that yields intermediate 161. A proton transfer process leads to the formation of intermediate 162. Ultimately, an intramolecular Wittig reaction involving intermediate 162 brings forth the desired products 163 and 164, completing the synthetic pathway.90
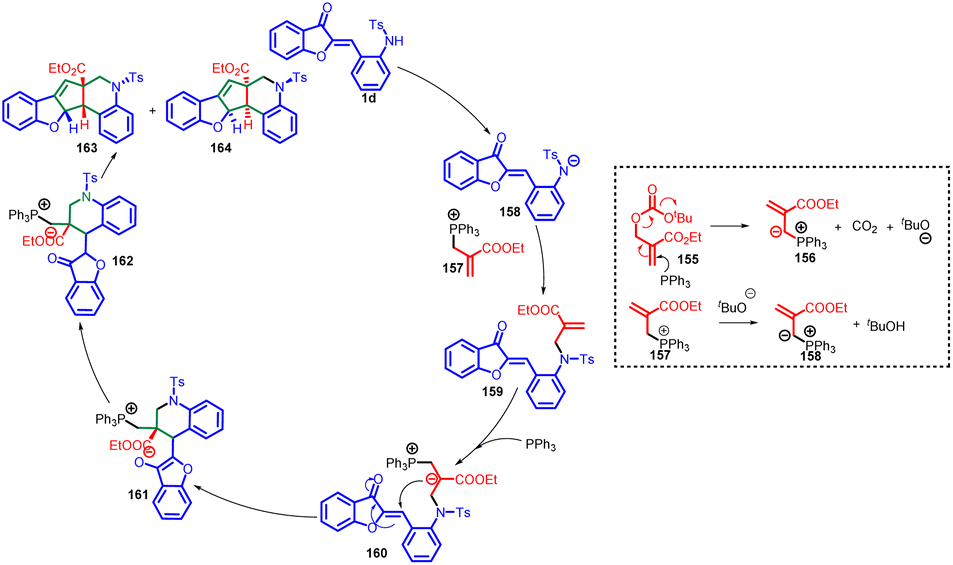 | ||
| Scheme 52 Proposed mechanism for the synthesis of [6-5-5-6-6] pentacyclic skeleton. | ||
Arjun Kafle et al., attempted to prepare azido-substituted aurones via a copper catalysed azidation in 2020 (Scheme 53). But the intended product could not be produced instead an uncommon triazole product 165 was formed. During the further studies it was noted that copper catalyst is not required for this synthesis instead simple thermal treatment with sodium azide in a polar aprotic solvent was enough to achieve this uncommon product.
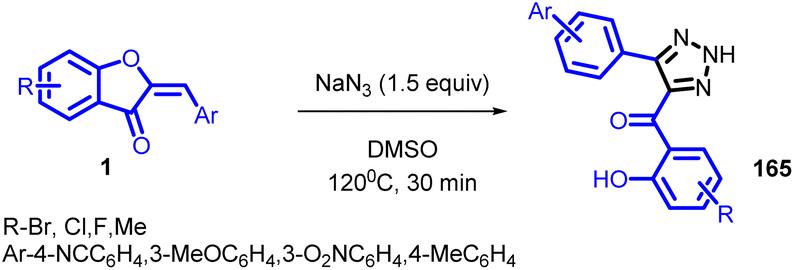 | ||
| Scheme 53 Triazole synthesis from aurones. | ||
The mechanism of the reaction is unclear. It uncertain that some form of Michael addition/elimination/cyclization sequence, a cycloaddition/elimination sequence, or elimination to an intermediate ynone followed by cycloaddition is occurring. Elimination/cycloaddition would seem to be the simplest mechanistic explanation for this reaction in view that ynones are reacting with sodium azide to produce triazoles. Although attempts for aynone intermediate was unsuccessful. The product triazoles have two distinct sites for alkylation chemistry and offer several opportunities for modification. It is believed that, given this potential and their accessibility, they may prove to be novel, untested scaffolds for more development.91
Conclusion
In summary, this concise review offers an insightful update on the versatile synthetic capabilities of aurones, primarily attributed to their distinctive electronic properties. These properties render aurones valuable candidates for a wide array of chemical reactions, including cycloadditions, annulations, and Michael/Mannich reactions. The pivotal role of the α,β-unsaturated ketone within aurones is evident, facilitating favoured nucleophilic addition at the carbonyl carbon and β-carbons, while also promoting electrophilic addition at the carbonyl oxygen. Notably, cycloaddition reactions with aurones have yielded intricate and intriguing molecular structures, encompassing benzofuranone-appended spiro-heterocyclic scaffolds through [3 + 2] cycloadditions, benzofuranone-fused pyrans via [4 + 2] cycloadditions, and even oxepane ring formation via [4 + 3] cycloadditions. Furthermore, aurones demonstrate exceptional selectivity in cross-formal [10 + 2] cycloaddition reactions with electron-deficient alkene substrates, resulting in the formation of dearomative 1-hydroxyl isobenzofulvene-type species. Additionally, under various conditions, Michael/Mannich reactions of aurones yield diverse products, including benzofuranone-appended spiro-heterocyclic compounds, benzofuran-fused oxepanones, and ring-opened derivatives. An in-depth understanding of these synthetic transformations, including their proposed mechanisms, not only enhances our comprehension of these processes but also encourages the scientific community to explore the myriad possibilities offered by aurone cores, thus propelling advancements in the field of organic synthesis.Abbreviations
| (DHQD)2AQN | Hydroquinidine(anthraquinone-1,4-diyl)diether |
| DABCO | 1,4-Diazabicyclo[2.2.2]octane |
| DBU | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| DCE | 1,2-Dichloroethane |
| DCM | Dichloromethane |
| DMAP | Dimethylaminopyridine |
| dr | Diastereoselectivity |
| ee | Enantiomeric excess |
| LC-MS | Liquid chromatography-mass spectrometry |
| MBH | Morita–Baylis–Hillman |
| NHC | N-Heterocyclic carbene |
| Pd-TMM | Pd-trimethylenemethane |
| TAC | Three atom components |
| TBME | tert-Butyl methyl ether |
| THF | Tetrahydrofuran |
| TsDAM | Tosyldiazomethane |
| 32CA | [3 + 2] Cycloaddition |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
DJ and KG thanks VIT, Vellore for the research fellowships. KRE thanks VIT Management for providing infrastructure facilities and VIT Seed Grant No. SG20230044 for carrying out this work. The authors thank Dr S. Kannadasan Department of Chemistry, VIT Vellore and Dr P. Shanmugam, Chief Scientist, Organic and Bioorganic Chemistry, CSIR-CLRI Chennai, for giving valuable suggestions.References
- A. Boumendjel, Aurones: A Subclass of Flavones with Promising Biological Potential, Curr. Med. Chem., 2003, 10, 2621–2630 CrossRef CAS.
- C. Taylor and Y. Bolshan, Metal-free Methodology for the Preparation of Sterically Hindered Alkynoylphenols and its Application to the Synthesis of Flavones and Aurones, Tetrahedron Lett., 2015, 56, 4392–4396 CrossRef CAS.
- I. Hawkins and S. T. Handy, Synthesis of Aurones under Neutral Conditions Using a Deep Eutectic Solvent, Tetrahedron, 2013, 69, 9200–9204 CrossRef CAS.
- H. Harkat, A. Blanc, J.-M. Weibel and P. Pale, Versatile and Expeditious Synthesis of Aurones via AuI-Catalyzed Cyclization, J. Org. Chem., 2008, 73, 1620–1623 CrossRef CAS PubMed.
- T. A. Geissman and C. D. Heaton, Anthochlor Pigments. IV. The Pigments of Coreopsis grandiflora, Nutt. I, J. Am. Chem. Soc., 1943, 65, 677–683 CrossRef CAS.
- A. Alsayari, A. M. Bin Muhsinah, Z. Hassan, M. J. Ahsan, J. A. Alshehri and N. Begum, Aurone: A Biologically Attractive Scaffold as Anticancer Agent, Eur. J. Med. Chem., 2019, 166, 417–431 CrossRef CAS.
- L. M. Lazinski, G. Royal, M. Robin, M. Maresca and R. Haudecoeur, Bioactive Aurones, Indanones, and Other Hemiindigoid Scaffolds: Medicinal Chemistry and Photopharmacology Perspectives, J. Med. Chem., 2022, 65, 12594–12625 CrossRef CAS PubMed.
- H.-M. Sim, C.-Y. Lee, P. L. R. Ee and M.-L. Go, Dimethoxyaurones: Potent inhibitors of ABCG2 (breast cancer resistance protein), Eur. J. Pharm. Sci., 2008, 35, 293–306 CrossRef CAS.
- X. Zheng, H. Wang, Y.-M. Liu, X. Yao, M. Tong, Y.-H. Wang and D.-F. Liao, Synthesis, Characterization, and Anticancer Effect of Trifluoromethylated Aurone Derivatives, J. Heterocycl. Chem., 2015, 52, 296–301 CrossRef CAS.
- N. J. Lawrence, D. Rennison, A. T. McGown and J. A. Hadfield, The Total Synthesis of an Aurone Isolated from Uvaria hamiltonii: Aurones and Flavones as Anticancer Agents, Bioorg. Med. Chem. Lett., 2003, 13, 3759–3763 CrossRef CAS.
- S. Demirayak, L. Yurttas, N. Gundogdu-Karaburun, A. C. Karaburun and I. Kayagil, Synthesis and Anti-cancer Activity Evaluation of New Aurone Derivatives, J. Enzyme Inhib. Med. Chem., 2015, 30, 816–825 CrossRef CAS PubMed.
- A. Detsi, M. Majdalani, C. A. Kontogiorgis, D. Hadjipavlou-Litina and P. Kefalas, Natural and Synthetic 2′-hydroxy-chalcones and Aurones: Synthesis, Characterization and Evaluation of the Antioxidant and Soybean lipoxygenase Inhibitory Activity, Bioorg. Med. Chem., 2009, 17, 8073–8085 CrossRef CAS.
- T. Narsinghani, M. C. Sharma and S. Bhargav, Synthesis, Docking Studies and Antioxidant Activity of Some Chalcone and Aurone Derivatives, Med. Chem. Res., 2013, 22, 4059–4068 CrossRef CAS.
- S. Das and J. P. N. Rosazza, Microbial and Enzymatic Transformations of Flavonoids, J. Nat. Prod., 2006, 69, 499–508 CrossRef CAS PubMed.
- N. A. Alshaye, E. U. Mughal, E. B. Elkaeed, Z. Ashraf, S. Kehili, Y. Nazir, N. Naeem, N. Abdul Majeed and A. Sadiq, In silico, in vitro VEGFR-2 Inhibition, and Anticancer Activity of a 3-(hydrazonomethyl)naphthalene-2-ol Derivative, J. Biomol. Struct. Dyn., 2022, 41, 1–16 Search PubMed.
- W. Huang, M.-Z. Liu, Y. Li, Y. Tan and G.-F. Yang, Design, Syntheses, and Antitumor Activity of Novel Chromone and Aurone derivatives, Bioorg. Med. Chem., 2007, 15, 5191–5197 CrossRef CAS PubMed.
- D. Muhammad, J. Hubert, N. Lalun, J.-H. Renault, H. Bobichon, M. Nour and L. Voutquenne-Nazabadioko, DFT Studies on the Antioxidant Activity of Naringenin and Its Derivatives: Effects of the Substituents at C3, Phytochem. Anal., 2015, 26, 137–144 CrossRef CAS PubMed.
- O. Kayser, A. F. Kiderlen and S. L. Croft, Natural products as antiparasitic drugs, Parasitol. Res., 2003, 90, S55–S62 CrossRef PubMed.
- O. Kayser, A. Kiderlen and R. Brun, In Vitro Activity of Aurones against Plasmodium falciparum Strains K1 and NF54, Planta Med., 2001, 67, 718–721 CrossRef CAS.
- H. Olleik, S. Yahiaoui, B. Roulier, E. Courvoisier-Dezord, J. Perrier, B. Pérès, A. Hijazi, E. Baydoun, J. Raymond, A. Boumendjel, M. Maresca and R. Haudecoeur, Aurone Derivatives as Promising Antibacterial Agents Against Resistant Gram-positive Pathogens, Eur. J. Med. Chem., 2019, 165, 133–141 CrossRef CAS.
- S. Venkateswarlu, G. K. Panchagnula, A. L. Gottumukkala and G. V. Subbaraju, Synthesis, Structural Revision, and Biological Activities of 4′-chloroaurone, a Metabolite of Marine Brown Alga Spatoglossum variabile, Tetrahedron, 2007, 63, 6909–6914 CrossRef CAS.
- C. L. Sutton, Z. E. Taylor, M. B. Farone and S. T. Handy, Antifungal Activity of Substituted Aurones, Bioorg. Med. Chem. Lett., 2017, 27, 901–903 CrossRef CAS PubMed.
- A.-L. Liu, H.-D. Wang, S. M. Lee, Y.-T. Wang and G.-H. Du, Structure–Activity Relationship of Flavonoids as Influenza Virus Neuraminidase Inhibitors and Their In vitro Anti-viral Activities, Bioorg. Med. Chem., 2008, 16, 7141–7147 CrossRef CAS PubMed.
- R. Haudecoeur and A. Boumendjel, Recent Advances in the Medicinal Chemistry of Aurones, Curr. Med. Chem., 2012, 19, 2861–2875 CrossRef CAS PubMed.
- Y. T. Han, Z. Wang and E. J. Bae, Synthesis of the Proposed Structure of Damaurone D and Evaluation of Its Anti-inflammatory Activity, Chem. Pharm. Bull., 2015, 63, 907–912 CrossRef CAS PubMed.
- B. P. Bandgar, S. A. Patil, B. L. Korbad, S. C. Biradar, S. N. Nile and C. N. Khobragade, Synthesis and Biological Evaluation of a Novel Series of 2,2-bisaminomethylated Aurone Analogues as Anti-inflammatory and Antimicrobial Agents, Eur. J. Med. Chem., 2010, 45, 3223–3227 CrossRef CAS PubMed.
- G. Zhu, Y. Luo, X. Xu, H. Zhang and M. Zhu, Anti-Diabetic Compounds from the Seeds of Psoralea Corylifolia, Fitoterapia, 2019, 139, 104373 CrossRef CAS PubMed.
- P. T. V. Nguyen, H. A. Huynh, D. V. Truong, T.-D. Tran and C.-V. T. Vo, Exploring Aurone Derivatives as Potential Human Pancreatic Lipase Inhibitors through Molecular Docking and Molecular Dynamics Simulations, Molecules, 2020, 25(20), 4657–4671 CrossRef CAS.
- B. Boucherle, M. Peuchmaur, A. Boumendjel and R. Haudecoeur, Occurrences, Biosynthesis and Properties of Aurones as High-End Evolutionary Products, Phytochemistry, 2017, 142, 92–111 CrossRef CAS PubMed.
- J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Formal [4+1] Annulation Reactions in the Synthesis of Carbocyclic and Heterocyclic Systems, Chem. Rev., 2015, 115, 5301–5365 CrossRef CAS.
- A. J. C. Padmashrija, S. Kannadasan and P. Shanmugam, Synthesis of 3-Spiro Cycloalkenes Fused 7-Aza-2-indalones from 3,3’-di-or N3,3’-tri-allyl/homoallyl/pentenyl 7-Aza-2-indalones via Ring Closing Metathesis Using Grubbs-II Catalyst, ChemistrySelect, 2023, 8, e202302522 CrossRef.
- Z. Gu, J.-J. Xie, G.-F. Jiang and Y.-G. Zhou, Catalytic Asymmetric Conjugate Addition of Tritylthiol to Azadienes with a Bifunctional Organocatalyst, Asian J. Org. Chem., 2018, 7, 1561–1564 CrossRef CAS.
- Z. Gu, J. Zhou, G.-F. Jiang and Y.-G. Zhou, Synthesis of Chiral γ-Aminophosphonates Through the Organocatalytic Hydrophosphonylation of Azadienes with phosphites, Org. Chem. Front., 2018, 5, 1148–1151 RSC.
- J. Lin, T.-Y. Zheng, N.-Q. Fan, P. Zhang, K. Jiang and Y. Wei, Pyrrole Synthesis Through Cu-Catalyzed Cascade [3 + 2] Spiroannulation/aromatization of Oximes with Azadienes, Org. Chem. Front., 2021, 8, 3776–3782 RSC.
- K. George and S. Kannadasan, Synthesis of Quinoline/Oxindole Appended Phenylvinyl-2,5-dihydrofuran Derivatives via Ring closingEnynr Metathesis, Tetrahedron, 2023, 142, 133544–133558 CrossRef CAS.
- P. P. Kaishap, G. Duarah, B. Sarma, D. Chetia and S. Gogoi, Ruthenium(II)-Catalyzed Synthesis of Spirobenzofuranones by a Decarbonylative Annulation Reaction, Angew. Chem., Int. Ed., 2018, 57, 456–460 CrossRef CAS PubMed.
- Q. Luo, X.-Y. Wei, J. Yang, J.-F. Luo, R. Liang, Z.-C. Tu and Y.-X. Cheng, Spiro Meroterpenoids from Ganoderma applanatum, J. Nat. Prod., 2017, 80, 61–70 CrossRef CAS.
- Q. Luo, L. Di, W.-F. Dai, Q. Lu, Y.-M. Yan, Z.-L. Yang, R.-T. Li and Y.-X. Cheng, Applanatumin A, a New Dimeric Meroterpenoid from Ganoderma applanatum That Displays Potent Antifibrotic Activity, Org. Lett., 2015, 17, 1110–1113 CrossRef CAS.
- Y. Li, X. Li and J.-P. Cheng, Catalytic Asymmetric Synthesis of Chiral Benzofuranones, Adv. Synth. Catal., 2014, 356, 1172–1198 CrossRef CAS.
- C. Zwergel, F. Gaascht, S. Valente, M. Diederich, D. Bagrel and G. Kirsch, Aurones: Interesting Natural and Synthetic Compounds with Emerging Biological Potential, Nat. Prod. Commun., 2012, 7, 389–394 CrossRef CAS PubMed.
- G. Sui, T. Li, B. Zhang, R. Wang, H. Hao and W. Zhou, Recent Advances on Synthesis and Biological Activities of Aurones, Bioorg. Med. Chem., 2021, 29, 115895 CrossRef CAS PubMed.
- M. Ríos-Gutiérrez and L. R. Domingo, Unravelling the Mysteries of the [3+2] Cycloaddition Reactions, Eur. J. Org. Chem., 2019, 2019, 267–282 CrossRef.
- R. Kavitha Preethi, S. Kannadasan and P. Shanmugam, Azomethine ylide (3+2) cycloaddition of 3-alkylidene-7-aza-2-indolone: Synthesis of 3,3’-dispiropyrrolidine- and 3,3’dispiropyrrolizidine bis 7-aza-2-oxindoes, Tetrahedron Lett., 2023, 120, 154448–154451 CrossRef CAS.
- H.-W. Frühauf, Organotransition metal [3+2] cycloaddition reactions, Coord. Chem. Rev., 2002, 230, 79–96 CrossRef.
- R. Jasiński and E. Dresler, On the Question of Zwitterionic Intermediates in the [3+2] Cycloaddition Reactions: A Critical Review, Organics, 2020, 1, 49–69 CrossRef.
- A. Haouas, N. Ben Hamadi, A. Nsira and M. Msadek, A Facile One Pot Synthesis of Substituted Pyrazole Derivatives, J. Chem. Res., 2013, 37, 435–437 CrossRef CAS.
- C. Guo, M. Schedler, C. G. Daniliuc and F. Glorius, N-Heterocyclic Carbene Catalyzed Formal [3+2] Annulation Reaction of Enals: An Efficient Enantioselective Access to Spiro-Heterocycles, Angew. Chem., Int. Ed., 2014, 53, 10232–10236 CrossRef CAS PubMed.
- W.-Q. Hu, Y.-S. Cui, Z.-J. Wu, C.-B. Zhang, P.-H. Dou, S.-Y. Niu, J.-Y. Fu and Y. Liu, Construction of Spirocycles Containing Highly Substituted Pyrroli-dine and 1-indanone Motifs with Spiro Quaternary Stereogenic Centers via 1,3-dipolar Cycloaddition of 2-alkylidene-1-indanone and Azomethine Ylides Promoted by Simple Imidazolium Salts, RSC Adv., 2015, 5, 70910–70914 RSC.
- C.-B. Zhang, P.-H. Dou, J. Zhang, Q.-Q. Wei, Y.-B. Wang, J.-Y. Zhu, J.-Y. Fu and T. Ding, 1,3-Dipolar Cycloaddition of Benzofuranone Derivativesand Azomethine Ylides Promoted by Simple Functional Ionic Liquids: Direct Access to Highly Substituted Pyrrolidine and Spirocyclic Benzofuranone, ChemistrySelect, 2016, 1, 4403–4407 CrossRef CAS.
- D. Kowalczyk, J. Wojciechowski and Ł. Albrecht, Asymmetric Organocatalysis in the Synthesis of Pyrrolidine Derivatives Bearing a Benzofuran-3(2H)-one Scaffold, Synthesis, 2016, 49, 880–890 CrossRef.
- P.-L. Shao, Z.-R. Li, Z.-P. Wang, M.-H. Zhou, Q. Wu, P. Hu and Y. He, [3 + 2] Cycloaddition of Azaoxyallyl Cations with Cyclic Ketones: Access to Spiro-4-oxazolidinones, J. Org. Chem., 2017, 82, 10680–10686 CrossRef CAS PubMed.
- Z.-P. Wang, Y. He and P.-L. Shao, Transition-Metal-Free Synthesis of Polysubstituted Pyrrole Derivatives via Cyclization of Methyl isocyanoacetate with Aurone Analogues, Org. Biomol. Chem., 2018, 16, 5422–5426 RSC.
- Z.-P. Wang, S. Xiang, P.-L. Shao and Y. He, Catalytic Asymmetric [3 + 2] Cycloaddition Reaction Between Aurones and Isocyanoacetates: Access to Spiropyrrolines via Silver Catalysis, J. Org. Chem., 2018, 83, 10995–11007 CrossRef CAS PubMed.
- T. Du, Z. Li, C. Zheng, G. Fang, L. Yu, J. Liu and G. Zhao, Highly Enantioselective 1,3-dipolar Cycloaddition of Imino Esters with Benzofuranone Derivatives Catalyzed by Thiourea-Quaternary Ammonium Salt, Tetrahedron, 2018, 74, 7485–7494 CrossRef CAS.
- Y. Su, C. Ma, Y. Zhao, C. Yang, Y. Feng, K.-H. Wang, D. Huang, C. Huo and Y. Hu, Regioselective synthesis of spiro naphthofuranone-pyrazoline via a [3+2] cycloaddition of benzoaurones with nitrile imines, Tetrahedron, 2020, 76, 131355 CrossRef CAS.
- P. K. Warghude, A. S. Sabale, R. Dixit, K. Vanka and R. G. Bhat, An Easy and Practical Approach to Access Multifunctional Cylcopentadiene- and Cyclopentene-spirooxindoles via [3 + 2] Annulation, Org. Biomol. Chem., 2021, 19, 4338–4345 RSC.
- R.-L. Wang, S.-K. Jia, Y.-J. Guo, Y. Yi, Y.-Z. Hua, H.-J. Lu and M.-C. Wang, Dinuclear Zinc-Catalyzed Enantioselective Formal [3 + 2] Cycloaddition of N-2,2,2-trifluoroethylisatin ketimines with Low Reactivity Aurone Derivatives, Org. Biomol. Chem., 2021, 19, 8492–8496 RSC.
- B. Liu, R. Yan, X. Li, W. Du and Y. Chen, Asymmetric [3+2] Annulations of Thioaurone and Aurone Derivatives for the Construction of Spiroheterocycles, Asian J. Org. Chem., 2021, 10, 784–787 CrossRef CAS.
- J. Huang, S. Sun, P. Ma, C. Yom, Y. Xing, Y. Wu and G. Wang, Base-Catalyzed Cascade [3 + 2] Spiroannulation/Aromatization of In situ Generated Tosyldiazomethane (TsDAM) with (Z)-Aurones: An efficient Access to Polysubstituted Pyrazole Derivatives, Tetrahedron Lett., 2022, 99, 153840 CrossRef CAS.
- H. Lin, J. Gu, S. Luo, Q. Gu, X. Cao, Y. Ge, C. Wang, C. Yuan and H. Wang, DBU-Catalyzed [3+2] Cycloaddition of Benzoaurones with 3-Homoacyl Coumarins: Synthesis of Spiro[Benzofuranone-Cyclopentane] Compounds, ChemistrySelect, 2022, 7(24), e202201599–e202201604 CrossRef CAS.
- Y.-H. Deng, C.-B. Zhang, J.-J. Sun, W.-L. Xu and J.-Y. Fu, A Regioselective [3 + 2] Cycloaddition Reaction of 2-benzylidene-1-indenones with Functional Olefins to Access Indanone-Fused 2D/3D Skeletons, Org. Biomol. Chem., 2023, 21, 4388–4392 RSC.
- R. Jasiński, On the Question of Stepwise [4+2] Cycloaddition Reactions and Their Stereochemical Aspects, Symmetry, 2021, 13(10), 1911–1927 CrossRef.
- S. E. Denmark and A. Thorarensen, Tandem [4+2]/[3+2] Cycloadditions of Nitroalkenes, Chem. Rev., 1996, 96, 137–166 CrossRef CAS.
- W. Oppolzer, Intramolecular [4+2] and [3+2] Cycloadditions in Organic Synthesis, Angew. Chem., Int. Ed., 1977, 16, 10–23 CrossRef.
- F. Wang, C. Luo, Y.-Y. Shen, Z.-D. Wang, Xi. Li and J.-P. Cheng, Highly Enantioselective [4 + 2] Cycloaddition of Allenoates and 2-Olefinic Benzofuran-3-ones, Org. Lett., 2015, 17, 338–341 CrossRef CAS PubMed.
- R. Mose, M. E. Jensen, G. Preegel and K. A. Jørgensen, Direct Access to Multifunctionalized Norcamphor Scaffolds by Asymmetric Organocatalytic Diels–Alder Reactions, Angew. Chem., 2015, 127, 13834–13838 CrossRef.
- L.-C. Yang, Z. Y. Tan, Z.-Q. Rong, R. Liu, Y.-N. Wang and Y. Zhao, Pd-Titanium Relay Catalysis Enables Switch of Alkoxide-π-Allyl to Dienolate Reactivity for Spiro-Heterocycle Synthesis, Angew. Chem., Int. Ed., 2018, 57, 7860–7864 CrossRef CAS PubMed.
- M. Harmata, The (4+3)-cycloaddition reaction: simple allylic cations as dienophiles, Chem. Commun., 2010, 46, 8886 RSC.
- J. H. Rigby and F. C. Pigge, [4 + 3] Cycloaddition Reactions, in Organic Reactions, John Wiley & Sons, Inc., Hoboken, NJ, USA, 1997, vol. 51, pp. 351–478 Search PubMed.
- K. Selvaraj, S. Chauhan, K. Sandeep and K. C. K. Swamy, Advances in [4+3]-Annulation/Cycloaddition Reactions Leading to Homo- and Heterocycles with Seven-Membered Rings, Chem.–Asian J., 2020, 15, 2380–2402 CrossRef CAS PubMed.
- B. M. Trost and Z. Zuo, Highly Regio-, Diastereo-, and Enantioselective Synthesis of Tetrahydroazepines and Benzo[b]oxepines via Palladium-Catalyzed [4+3] Cycloaddition Reactions, Angew. Chem., Int. Ed., 2020, 59, 1243–1247 CrossRef CAS.
- J.-Q. Zhao, S. Zhou, H.-L. Qian, Z.-H. Wang, Y.-P. Zhang, Y. You and W.-C. Yuan, Higher-Order [10 + 2] Cycloaddition of 2-alkylidene-1-indanones Enables the Dearomatization of 3-nitroindoles: Access to Polycyclic Cyclopenta[b]indoline Derivatives, Org. Chem. Front., 2022, 9, 3322–3327 RSC.
- Y. Yang, Y. Jiang, W. Du and Y. Chen, Asymmetric Cross [10+2] Cycloaddtions of 2-Alkylidene-1-indanones and Activated Alkenes under Phase-Transfer Catalysis, Chem.–Eur. J., 2020, 26, 1754–1758 CrossRef CAS PubMed.
- M. E. Jung, A Review of Annulation, Tetrahedron, 1976, 32, 3–31 CrossRef CAS.
- X. Ma, L. Liu, J. Wang, X. Xi, X. Xie and J. Wang, Rhodium-Catalyzed Annulation of α-Imino Carbenes with α,β-Unsaturated Ketones: Construction of Multisubstituted 2,3-Dihydropyrrole/pyrrole Rings, J. Org. Chem., 2018, 83, 14518–14526 CrossRef CAS PubMed.
- Y. Shi, Q. Wang and S. Gao, Recent Advances in the Intramolecular Mannich Reaction in Natural Products Total Synthesis, Org. Chem. Front., 2018, 5, 1049–1066 RSC.
- T. Poon, B. P. Mundy and T. W. Shattuck, The Michael Reaction, J. Chem. Educ., 2002, 79, 264–267 CrossRef CAS.
- G. Huang and X. Li, Applications of Michael Addition Reaction in Organic Synthesis, Curr. Org. Synth., 2017, 14, 568–571 CrossRef CAS.
- E. Venturini Filho, M. K. Antoniazi, R. Q. Ferreira, G. F. S. dos Santos, C. Pessoa, C. J. Guimarães, J. B. Vieira Neto, A. M. S. Silva and S. J. Greco, A Green Multicomponent Domino Mannich-Michael Reaction to Synthesize Novel Naphthoquinone-Polyphenols with Antiproliferative and Antioxidant Activities, Eur. J. Org Chem., 2022, 39, e202200442 CrossRef.
- Z.-Q. Liang, Z.-H. Gao, W.-Q. Jia and S. Ye, Bifunctional N-Heterocyclic Carbene Catalyzed [3+4] Annulation of Enals and Aurones, Chem.–Eur. J., 2015, 21, 1868–1872 CrossRef CAS PubMed.
- S. Kayal and S. Mukherjee, Catalytic Enantioselective Cascade Michael/Cyclization Reaction of 3-isothiocyanato oxindoles with Exocyclic α,β-Unsaturated Ketones En Route to 3,2’-pyrrolidinyl bispirooxindoles, Org. Biomol. Chem., 2016, 14, 10175–10179 RSC.
- B. Li, F. Gao, X. Feng, M. Sun, Y. Guo, D. Wen, Y. Deng, J. Huang, K. Wang and W. Yan, Highly Efficient Enantioselective Synthesis of Bispiro[benzofuranoxindole-pyrrolidine]s through Organocatalytic Cycloaddition, Org. Chem. Front., 2019, 6, 1567–1571 RSC.
- H. Xu, H. Chen, X. Hu, G. Xuan, P. Li and Z. Zhang, Synthesis of Fully Substituted 5-(o-Hydroxybenzoyl)imidazoles via Iodine-Promoted Domino Reaction of Aurones with Amidines, J. Org. Chem., 2022, 87, 16204–16212 CrossRef CAS PubMed.
- H. Xu, M.-J. Li, H. Chen, F.-H. Huang, Q.-Y. Zhu, G.-W. Wang and Z. Zhang, I2/FeCl3-Catalyzed Domino Reaction of Aurones with Enamino Esters for the Synthesis of Highly Functionalized Pyrroles, Org. Lett., 2022, 24, 8406–8411 CrossRef CAS PubMed.
- A. K. Patel, N. H. Patel, M. A. Patel and D. I. Brahmbhatt, Synthesis of Some 3-(4-Aryl-benzofuro[3,2-b]pyridine-2-yl)coumarins and Their Antimicrobial screening, J. Heterocycl. Chem., 2012, 49, 504–510 CrossRef CAS.
- M. Wang, Z.-Q. Rong and Y. Zhao, Stereoselective Synthesis of e-lactones or Spiro-Heterocycles through NHC-Catalyzed Annulation: Divergent Reactivity by Catalyst Control, Chem. Commun., 2014, 50, 15309–15312 RSC.
- R. R. Giri, H. B. Lad, V. G. Bhila, C. V. Patel and D. I. Brahmbhatt, Modified Pyridine-Substituted Coumarins: A New Class of Antimicrobial and Antitubercular Agents, Synth. Commun., 2015, 45, 363–375 CrossRef CAS.
- A. Haouas, N. Ben Hamadi and M. Msaddek, Synthesis of New Spirocyclopropane and Oxadiazole Products, J. Heterocycl. Chem., 2015, 52, 1765–1768 CrossRef CAS.
- D. Kowalczyk, J. Wojciechowski and Ł. Albrecht, Enantioselective Synthesis of Spirocyclic Tetrahydrothiophene Derivatives Bearing a Benzofuran-3(2H)-one Scaffold. Unusual Supramolecular Crystal Structure with High Z’, Tetrahedron Lett., 2016, 57, 2533–2538 CrossRef CAS.
- S. Ma, A. Yu, S. Zhang, L. Zhang and X. Meng, Construction of [6-5-5-6-6] Pentacyclic Skeleton via Phosphine Catalyzed Domino Reaction and Mechanism Study, J. Org. Chem., 2020, 85, 7884–7895 CrossRef CAS PubMed.
- A. Kafle, S. Bhattarai and S. T. Handy, An Unusual Triazole Synthesis from Aurone, Synthesis, 2020, 52, 2337–2346 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |