
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient preparation of hybrid biofuels from biomass-derived 5-(acetoxymethyl)furfural and petroleum-derived aromatic hydrocarbons†
Abhishek Kumar Yadava,
Navya Subray Bhata,
Sonal Kaushika,
Asiful H. Seikhb and
Saikat Dutta *a
*a
aDepartment of Chemistry, National Institute of Technology Karnataka (NITK), Surathkal, Mangalore-575025, India. E-mail: sdutta@nitk.edu.in
bMechanical Engineering Department, College of Engineering, King Saud University, Riyadh-11421, Saudi Arabia
First published on 18th January 2024
Abstract
Fuel candidates containing both petroleum-derived and biomass-derived molecules in their structural motifs ensure both feedstocks are used optimally and coherently. This work reports a straightforward and efficient preparation of 5-(arylmethyl)furfurals (AMFFs), 2-(arylmethyl)furans (AMFs), and 2-(arylmethyl)-5-methylfurans (AMMFs) as hybrid biofuels (or fuel oxygenates) starting from carbohydrate-derived 5-(acetoxymethyl)furfural (AcMF) and petroleum-derived aromatic hydrocarbons. The AMFFs were prepared by Friedel–Crafts reaction between AcMF and aromatic hydrocarbons (e.g., BTX, mesitylene) by employing anhydrous ZnCl2 as the catalyst. AMFs were prepared by decarbonylation of AMFFs over the Pd(OAc)2 catalyst under solvent-free conditions. In contrast, AMMFs were produced by hydrogenating AMFFs in methanol using gaseous hydrogen and the 10% Pd/C catalyst. The catalytic transformations were optimized on various parameters, and all the biofuel candidates were obtained in good to excellent isolated yields (>80%) under moderate conditions.
Introduction
The hydrocarbon-based liquid fuels will likely continue to be used in the transportation sector in the foreseeable future, especially in aviation and other difficult-to-electrify sectors.1 The past two decades have seen coordinated research for finding a sustainable carbon-based feedstock alternative to petroleum for producing liquid transportation fuels. In this regard, biomass has the commercial potential to replace petroleum, at least partially, for producing liquid transportation fuels with the desired societal, economic, and environmental incentives.2 Several catalytic strategies have been developed over the past three decades for synthesizing hydrocarbon-based drop-in liquid fuels and oxygenated fuel additives from cellulosic biomass that promise to reduce dependence on fossilized carbon.2 The commercial relevance of producing specific biofuels heavily relies on the availability, cost, and type of the biomass feedstock employed.3 Coprocessing biomass and petroleum-derived components could ensure feedstock flexibility, moderate the cost of the targeted fuel candidates, and avoid overdependence on either biomass or petroleum feedstock.2,4,5 Moreover, incorporating petroleum-derived hydrocarbons in biomass-derived oxygenated intermediates increases the energy density in the resultant hybrid fuels and improves fuel properties (e.g., stability).6,7 Acid-catalyzed hydrolysis and dehydration of abundant hexose sugars (e.g., glucose, sucrose) and polymeric carbohydrates (e.g., starch, cellulose) lead to 5-(hydroxymethyl)furfural (HMF).8 HMF has been exploited as a renewable chemical platform for producing hydrocarbon-based liquid fuels and fuel oxygenates.9–11 HMF has been reacted with petroleum-derived aromatic compounds (e.g., mesitylene) by Friedel–Crafts reaction (FCR) to form 5-(mesitylmethyl)furfural (MMFF), a potential fuel oxygenate and hybrid diesel fuel precursor.12–15 Even though aromatic hydrocarbons are typically obtained from petroleum, they can also be produced selectively from carbohydrates via furanic intermediates.16,17 Aromatic hydrocarbons can be sourced renewably from other biomass fractions, such as by depolymerizing and deoxygenating lignin or deoxygenating the bio-oil produced by pyrolyzing biomass.18,19 Even though good yields of MMFF (∼80%) have been reported in the literature using suitable catalysts, the challenge lies in the scalable, high-yielding, and economical production of HMF. Even after decades of research, HMF production is yet to reach commercialization, primarily due to its inherent hydrophilicity and poor stability (e.g., thermal, storage, hydrolytic).20In this regard, the hydrophobic, stabler analogs of HMF have shown promise to substitute HMF in its derivative chemistry.21 The esters of HMF, such as 5-(acetoxymethyl)furfural (AcMF), have received particular attention since they are hydrophobic, significantly more stable than HMF, contain only C, H, and O atoms in their moiety, and have several emerging applications.22,23 Acetic acid required for producing AcMF is biorenewable, inexpensive, thermally stable, and non-toxic. AcMF can be produced conveniently from carbohydrates, HMF, and various hydrophobic analogs of HMF.24–27
We have recently reported a solvent-free, scalable, and high-yielding preparation of AcMF from sugars and polymeric carbohydrates using a catalyst system comprising of AcOH and ZnCl2.28 Interestingly, AcMF has been used as the starting material for synthesizing hybrid biofuels (e.g., MMFF) by FCR using tin-exchanged montmorillonite as a heterogeneous catalyst.25 Removing reactive oxygen-containing functionalities in biofuels increases the energy density and improves the potential fuel candidates' stability (e.g., storage, thermal). For example, even though furfural and HMF are not used as fuel oxygenates, their partially hydrogenated furanic derivatives, such as 2-methylfuran and 2,5-dimethylfuran, have shown promise as fuel additives.29 Furanic ethers, such as 5-(ethoxymethyl)furfural has also been proposed as a fuel oxygenate.30 In this regard, MMFFs and their derivatives enjoy more flexibility in their molecular structure and physicochemical properties. Moreover, the incorporation of aromatic hydrocarbons and furaldehydes in 5-(arylmethyl)furfurals (AMFFs) help to valorize various components of biomass together for improved sustainability. As mentioned above, it is desirable to eliminate the reactive aldehyde group in AMFFs to produce fuel additives with improved stability and higher energy density. The preparation of 2-(mesitylmethyl)furan (MMF), a promising hybrid fuel, has been reported by the Pd/C catalyzed decarbonylation of MMFF.31 Therefore, the catalytic decarbonylation of AMFFs could be a convenient route to synthesizing 2-(arylmethyl)furans (AMFs) as potential biofuels (Scheme 1). However, the decarbonylation route has never been studied systematically for such hybrid biofuels. Interestingly, various 2-(arylmethyl)-5-methylfurans (AMMFs) have been hydrodeoxygenated into branched cycloalkanes for potential applications as jet fuel using a combination of Pd/C and HZSM-5 catalysts.32 Alternatively, the aldehyde group in MMFF could be catalytically reduced into a methyl group forming 2-(mesitylmethyl)-5-methylfuran (MMMF), which has never been reported in the literature. Catalytic decarbonylation and catalytic hydrogenation fall under stoichiometric reagentless organic transformations and have excellent green metrics.33 AMFs and AMMFs have significantly higher C/O and H/O ratios than AMFFs and contain no reactive aldehyde functionality. This work reports the convenient and high-yielding preparation of AMFFs by FCR between AcMF and aromatic hydrocarbons using ZnCl2 as a non-toxic, inexpensive, and readily available catalyst. AMFFs were catalytically decarbonylated into AMFs using palladium(II) acetate as the catalyst under solvent-free conditions. AMFFs were also catalytically hydrogenated in a methanolic solution over a 10% Pd/C catalyst to form AMMFs. The processes were optimized on various reaction parameters, such as temperature, catalyst loading, solvent, and duration, and all the hybrid biofuels were obtained in good to excellent isolated yields. Moreover, the recyclability of the catalysts was investigated.
 | ||
| Scheme 1 Synthesis of hybrid biofuels from 5-(acetoxymethyl)furfural and aromatic hydrocarbons. | ||
Experimental section
Materials
Petroleum ether (60–80 °C, 99%), ethyl acetate (99%), and methanol (99%) were purchased from Loba Chemie Pvt. Ltd. Benzene (99%), toluene (99%), o-xylene (99%), m-xylene (99%), p-xylene (99%), isopropylbenzene (99%), ethylbenzene (99%), mesitylene (99%), zinc chloride (anhydrous, 95%), nitromethane (99%), molecular sieve (4 Å), and silica gel (60–120 mesh) were procured from Spectrochem. Palladium(II) acetate (98%), 10% Pd/C, and Amberlyst-15 were procured from Sigma-Aldrich. 5-(Acetoxymethyl)furfural (AcMF) was prepared from fructose following a literature process.28 The solvents were dried over activated molecular sieves for 24 h before use. All the chemicals were used as received without further purification.Instrumentation
The synthesized compounds were characterized by Fourier transform infrared (FTIR) spectroscopy, nuclear magnetic resonance (NMR) spectroscopy, and high-resolution mass spectrometry (HRMS) technique. The FTIR spectrum was acquired using the ATR method on a Bruker Alpha II FTIR spectrometer fitted with silicon carbide as an IR source. The purified compounds were dissolved in dichloromethane to make a dilute solution. A drop of the solution was placed on the ATR counter and allowed to evaporate to make a thin film of the compound. Each sample was scanned 24 times with a scan rate of two scans per second and a spectral resolution of 4 cm−1. The synthesized compounds were dissolved in an appropriate deuterated solvent for NMR spectroscopy. The 1H-NMR spectra were acquired in a Bruker NanoBay® instrument operating at 400 & 300 MHz. The 13C-NMR spectra were acquired in the same instrument at the calculated frequency of 100 & 75 MHz. The Bruker Topspin software (version 4.2.0) was used to process raw NMR data. HRMS analyses of the synthesized compounds were collected using the Waters-Xevo G2-XS-QToF instrument. Gas chromatography (GC) analysis was conducted using a LECO Pegasus BT 4D spectrometer with an Rxi-5 ms column (30 m × 0.25 mm × 0.25 μm). High-purity helium gas was used as the carrier gas with a flow rate of 1.40 mL min−1. The injector temperature was fixed at 250 °C, and the split ratio was adjusted to 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The column temperature was initially set to 50 °C for 2 minutes, then it was increased up to 300 °C with a rate of 10 °C min−1.
:1. The column temperature was initially set to 50 °C for 2 minutes, then it was increased up to 300 °C with a rate of 10 °C min−1.
General synthetic procedure for synthesizing AMFFs
AcMF (0.502 g, 2.98 mmol), mesitylene (1.435 g, 4 equiv., 11.94 mmol), anhydrous ZnCl2 (0.250 g, 50 wt%), and nitromethane (5 mL) were added to a 50 mL round-bottomed flask. The reaction mixture was stirred magnetically for 3 h in a pre-heated (120 °C) oil bath. After the reaction, the reaction mixture was washed with water and saturated sodium bicarbonate solution and then extracted with ethyl acetate (3 × 15 mL). The organic layer was evaporated under reduced pressure and purified using column chromatography (petroleum ether/ethyl acetate, silica gel 60–120) to get 5-(mesitylmethyl)furfural (MMFF, 1f) as a light-yellow liquid (0.551 g, 81%). Under optimized reaction conditions, the same protocol was extended for the synthesis of 1a–1h.General synthetic procedure for synthesizing AMFs
A 50 mL round-bottomed flask was charged with MMFF 1f (0.250 g, 1.10 mmol), molecular sieve 4 Å (0.200 g), and Pd(OAc)2 (0.04 g, 16 mol%). The reaction mixture was magnetically stirred in a pre-heated oil bath at 140 °C for 3.5 h. After the reaction, the reaction mixture was diluted with ethyl acetate and filtered. The residue was washed with ethyl acetate (3 × 10 mL). The filtrate was concentrated under reduced pressure and purified through column chromatography (petroleum ether/ethyl acetate, silica gel 60–120 mesh) to get 2-(mesitylmethyl)furan (MMF, 2f) as a colorless liquid (0.176 g, 80%). The optimized reaction conditions were extended for the synthesis of 2a–2h.General synthetic procedure for synthesizing AMMFs
MMFF 1f (0.250 g, 1.10 mmol), 10% Pd/C (0.025 g, 10 wt%), Amberlyst-15 (0.125 g, 50 wt%), and methanol (10 mL) were added to 100 mL two necked round-bottomed flask. Hydrogen gas was purged into the reaction mixture five times using a hydrogen balloon, and the reaction mixture was stirred magnetically at room temperature for 2.5 h. The reaction mixture was centrifuged, decanted, and evaporated under reduced pressure. The reaction mixture was passed through a small plug of silica to get 2-(mesitylmethyl)-5-methylfuran (MMMF, 3f) as a colorless liquid (0.197 g, 84%). Similar synthetic protocols were followed for the preparation 3a–3h. The catalyst was recovered by centrifugation of the reaction mixture, followed by decanting. The catalyst was washed with methanol (2 × 10 mL) and dried in an oven for 4 h at 60 °C before subjecting it to the next catalytic cycle. Unfortunately, the catalytic activity decreased drastically on the first recycle, possibly due to the leaching of palladium nanoparticles from the carbon support. The activity remained relatively stable until the fourth recycle, affording around 40% isolated yield of 3f.Results and discussion
Initially, the authors concentrated on developing a suitable general catalytic procedure for synthesizing 1a–1h. The preparation of 1f has already been attempted from AcMF via FCR using Sn4+-exchanged montmorillonite clay as the catalyst.25 Since we have recently reported the direct preparation of AcMF from sugars and polymeric carbohydrates using a combination of acetic acid and ZnCl2,28 the efficiency of anhydrous ZnCl2 as a catalyst was explored for preparing 1a, 1b, and 1h from AcMF.34 The FCR of AcMF with various petroleum-derived arenes were performed.FCR of AcMF to AMFFs
Initially, the synthesis of 1f by the FCR between AcMF and mesitylene was optimized. The reaction was carried out in nitromethane as solvent using anhydrous ZnCl2 as the catalyst. The reaction progress was monitored using thin-layer chromatography (TLC), and products were confirmed using NMR (1H & 13C) and HRMS techniques. When the reaction was carried out at 100 °C and 110 °C, the reaction took 4 h to complete and afforded 77% and 79% yield of 1f (Fig. 1). Elevating the reaction temperature to 120 °C allowed the reaction to be completed within 3 h and increased the yield of 1f to 81%. The GC chromatogram of the crude reaction mixture did not show any significant peak except mesitylene, nitromethane, and the product 1f (ESI, Fig. S73†). Further increase of the reaction temperature to 130 °C decreased the yield of 1f to 74%. When the reaction was carried out under similar reaction conditions in excess mesitylene without nitromethane, the conversion of AcMF reached completion in 5 h. However, the isolated yield of 1f drastically decreased to 38% due to the decomposition of AcMF. | ||
| Fig. 1 The effect of reaction temperature on the yield of 1f. Reaction conditions: AcMF (0.502 g, 2.98 mmol), mesitylene (1.435 g, 4 equiv., 11.94 mmol), ZnCl2 (0.250 g, 50 wt%), and nitromethane (5 mL). | ||
After optimizing the reaction temperature, the effect of the equivalent of arene on the yield of AMFFs was investigated. Using 4 equiv. of mesitylene (compared to AcMF) gave an 81% isolated yield of 1f. Increasing the amount of mesitylene to 5 equiv. didn't improve the yield of 1f any further. However, decreasing the amount of mesitylene to 3 equiv. lowered the yield of 1f to 71%. After the reaction, the unreacted mesitylene was recovered by the column chromatography. It must be pointed out here that chromatography was chosen for product purification to minimize mass loss at the lab-scale reaction. During the process scale-up, fractional distillation can be used to separate the excess reagent and the product. Furthermore, increasing the catalyst loading to 100 wt% did not significantly change the yield (77%). The reaction was performed in a sealed glass pressure reactor to prevent moisture in the reaction mixture and the reaction afforded a 74% yield of 1f.
After process optimization of 1f, the reaction conditions were applied for the synthesis of the other derivatives. FCR of AcMF with benzene, para-xylene, and mesitylene gave exclusively one structural isomer. In contrast, toluene, ortho-xylene, meta-xylene, isopropylbenzene, and ethylbenzene gave two structural isomers, and the combined yield of the two isomers is reported (Table 1). FCR of AcMF with benzene was performed at 120 °C for 3 h in a round-bottomed ACE glass pressure vessel, which afforded a 59% isolated yield of 1a. The moderate yield of 1a was attributed to the partial charring of the reaction mixture. When the transformation was performed in an open vessel under reflux, a 50% isolated yield of 1a was obtained. Even though the conversion of AcMF was not quantitative, noticeable decomposition was observed.
|
||||
|---|---|---|---|---|
| Entry | Product code | Product(s) | Yield (%) | |
| a Reaction conditions: AcMF (0.502 g, 2.98 mmol), arene (4 equiv., 11.94 mmol), ZnCl2 (0.250 g, 50 wt%), 120 °C, 3 h.b A round-bottomed ACE glass pressure vessel was used.c Combined yield of ortho- and para-isomer. | ||||
| 1 | 1a | 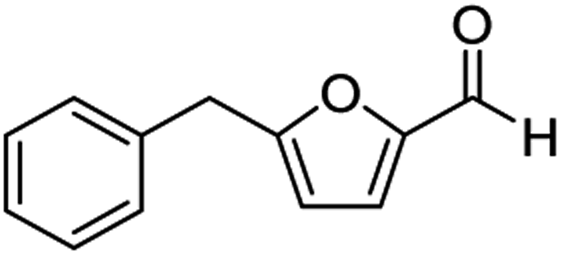 |
59b | |
| 2 | 1b | 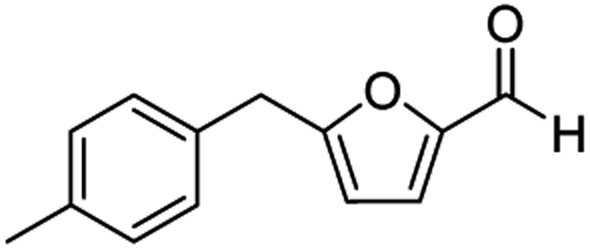 |
 |
76c |
| 3 | 1c |  |
82 | |
| 4 | 1d | 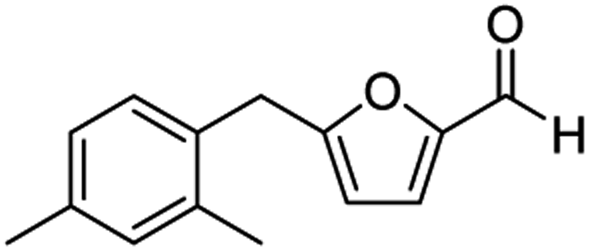 |
 |
77c |
| 5 | 1e |  |
 |
76c |
| 6 | 1f |  |
81 | |
| 7 | 1g | 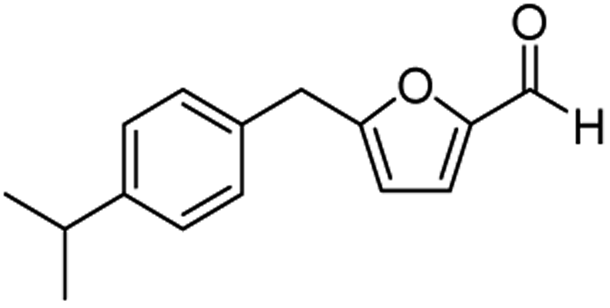 |
 |
79c |
| 8 | 1h |  |
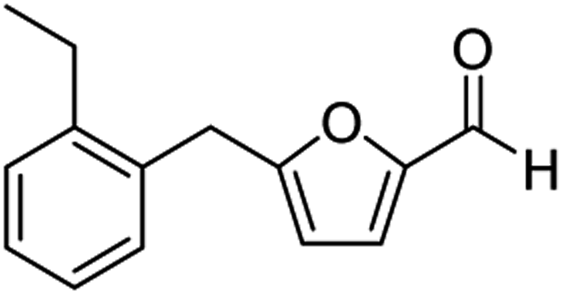 |
75c |

The structural isomers of 1b, 1g, and 1h (i.e., ortho- and para-substitution on the monoalkylated arenes) were found to form in nearly equal molar ratio based on the integration value of signals on the 1H-NMR spectra. The combined isolated yield of the two isomers were 76% for 1b, 79% for 1g, and 75% for 1h, respectively (entries 2, 7, & 8, Table 1). For 1d, the molar ratio of ortho- and para-isomers was approximately 1:3, with a combined yield of 77%. In the case of 1e, the ratio was nearly 1:2 with a 76% isolated yield (entry 5, Table 1). All the synthesized AMFFs were used as substrates for the catalytic decarbonylation and catalytic hydrogenation reactions.
Decarbonylation of AMFFs to AMFs
The decarbonylation of AMFFs was done under solvent-free conditions using conventional heating. The decarbonylation of AMFFs was optimized by using 1f. Initially, decarbonylation reaction was performed at 110 °C and 120 °C using Pd(OAc)2 (0.17 mmol, 16 mol%), afforded only 30% and 33% yield of 2f even after 24 h reaction due to incomplete conversion of 1f. When the reaction temperature was increased to 130 °C, resulted in 50% of 2f in 18 h (Fig. 2). The increase of reaction temperature to 140 °C gave a maximum 80% yield of 2f in 3.5 h under similar reaction conditions. Further increase in the reaction temperature to 150 °C diminished the yield of 2f to 72% due to partial decomposition of the product. | ||
| Fig. 2 The effect of reaction temperature on the yield of 2f. Reaction conditions: MMFF 1f (0.250 g, 1.10 mmol), Pd(OAc)2 (0.04 g, 16 mol%), and molecular sieve 4 Å (0.200 g). | ||
After optimizing the reaction temperature, the effect of catalyst loading was investigated by altering the amount of catalyst. When the catalyst amount was decreased to 8 mol%, the reaction was completed in 10 h and a 57% yield of 2f was achieved. The result may be attributed to slower kinetics of decarbonylation at lower catalyst loading and thermal decomposition of MMFF during heating for an extended period. Increasing the catalyst loading to 20 mol% did not significantly change the yield of 2f (Fig. 3). It has been observed that molecular sieves facilitate the reaction by providing more surface area for the catalyst and substrate to react. Under similar conditions, the decarbonylation of 1f was performed in the absence of molecular sieves and resulted in a 74% isolated yield of 2f. Furthermore, the molecular sieve amount was varied between 0.10 g and 0.30 g. When the reaction was carried out at 0.10 g, it afforded a 77% yield of 1f. Increasing the molecular sieve amount to 0.20 g improved the isolated yield of 1f to 80%. Further increasing the amount of the molecular sieve to 0.30 g afforded only a marginal increase in yield (82%). This shows that the yield gradually improves with increasing amounts of molecular sieves introduced in the reaction mixture. However, during process scale-up, the improvement of yield may be weighed against the cost of using and recycling molecular sieves.
 | ||
| Fig. 3 Effect of catalyst loading on the yield of 2f. Reaction conditions: MMFF 1f (0.250 g, 1.10 mmol), molecular sieve 4 Å (0.200 g), 140 °C. | ||
The decarbonylation of 1f was also performed in the ACE glass pressure reactor using solvents like cyclohexane, isooctane, and heptane, which gave 35%, 25%, and 10% yields, respectively. The substrate 1f was not fully consumed even after 24 h (monitored by TLC), which could be attributed to the in situ generation of CO being trapped in the pressure reactor and poisoning the catalyst by the chemisorption of the active sites. The high-boiling solvents, such as anisole and dodecane, were also screened for the decarbonylation of 1f. The solvents gave 20% and 32% yields of 2f, respectively, due to the incomplete conversion of 1f even after 24 h reaction time. After ensuring the best selectivity and yield for 2f, the optimized reaction conditions were applied for decarbonylating other AMFFs into AMFs. The decarbonylation of 1a, 1c, and 1f gave 73% yield of 2a, 80% yield of 2c, and 80% yield of 2f, respectively. Both ortho- and para-structural isomers of 1b, 1d, 1e, 1g, and 1h were used for the decarbonylation reaction. The combined yield of the two structural isomers was 76% for 2b, 78% for 2d, 76% for 2e, 77% for 2g, and 75% for 2h, respectively (Table 2).
|
|||||
|---|---|---|---|---|---|
| Product codea | Product(s) | Yield (%) | Product codeb | Product(s) | Yield (%) |
| a Reaction conditions: AMFF (0.250 g), Pd(OAc)2 (16 mol%), molecular sieve 4 Å (0.200 g), 140 °C, and 3.5 h.b Reaction conditions: AMFF (0.250 g), 10% Pd/C (0.025 g, 10 wt%), Amberlyst-15 (0.125 g, 50 wt%), hydrogen balloon pressure, room temperature, and 2.5 h.c Combined yield of the ortho- and para-isomers. | |||||
| 2a |  |
73 | 3a |  |
84 |
| 2b |  |
76c | 3b |  |
87c |
| 2c |  |
80 | 3c | 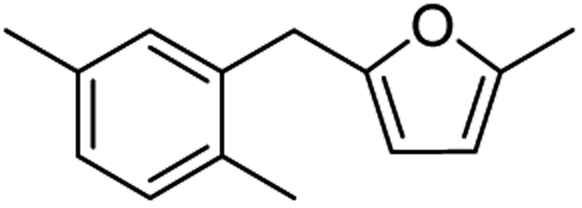 |
85 |
| 2d | 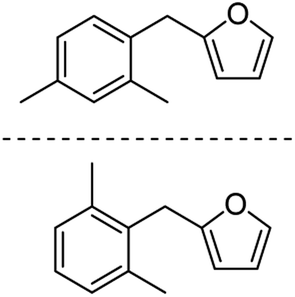 |
78c | 3d |  |
81c |
| 2e |  |
76c | 3e | 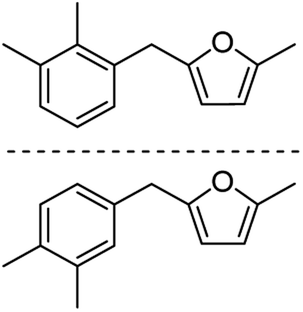 |
83c |
| 2f | 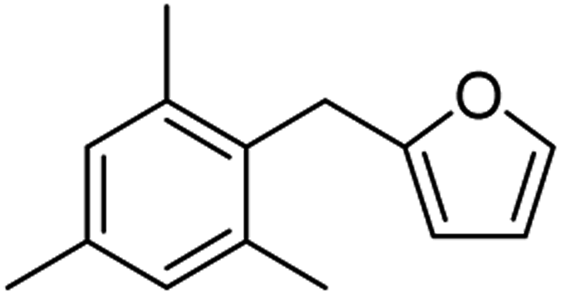 |
80 | 3f |  |
84 |
| 2g |  |
77c | 3g |  |
84c |
| 2h |  |
75c | 3h |  |
83c |

Hydrogenation of AMFFs to AMMFs
Catalytic hydrogenation of AMFFs to AMMFs was performed at room temperature in methanol under H2 balloon pressure with 10% Pd/C as the catalyst. The synthetic procedure was optimized by using 1f. The reaction progress was monitored by TLC, and products were confirmed using NMR (1H & 13C) and HRMS techniques. When the reaction was performed at room temperature, it took 2.5 h for the quantitative conversion of 1f. The reaction was optimized by varying the amount of Amberlyst-15 and 10% Pd/C catalyst loading. When the reaction was carried out using 10 wt% and 20 wt% of Amberlyst-15 (compared to the initial weight of 1f), it afforded 69% and 75% yield of 3f, respectively. Further increase of Amberlyst-15 to 30 wt% afforded a 79% yield of 3f. When the reaction was carried out at 5 wt% loading of the 10% Pd/C catalyst, it afforded a 68% yield of 3f. An increase of catalyst loading to 10 wt% afforded an 84% yield of 3f at room temperature in 2.5 h. The catalyst loading was further increased to 20 wt%, decreasing the yield to 75% of 3f. The isolated yield of 3f with fresh Amberlyst-15 and 10% Pd/C was 84%. Unfortunately, after the first cycle, the yield of 3f decreased to 48%.After optimization of 3f, the reaction conditions were extended for the synthesis of the other derivatives. The catalytic hydrogenation of 1a, 1c, and 1f afforded 84% 3a, 85% 3c, and 84% 3f, respectively. The hydrogenation reaction of both ortho- and para-isomers of 1b, 1d, 1e, 1g, and 1h were performed and afforded 87% yield of 3b, 81% yield of 3d, 83% yield of 3e, 84% yield of 3g, and 83% yield of 3h, respectively (Table 2).
Conclusions
A series of furan-based hybrid biofuels have been synthesized, starting from carbohydrate-derived AcMF and petroleum-derived aromatic hydrocarbons. The aromatic hydrocarbons were coupled with AcMF by the FCR in nitromethane solvent catalyzed by anhydrous ZnCl2. Predictably, the heavily alkylated aromatics showed more reactivity towards the FCR with AcMF. Moreover, the reactive aldehyde group in the products was removed by palladium-catalyzed decarbonylation or by palladium-catalyzed catalytic hydrogenation to produce stabler hybrid biofuels with higher C/O ratios. No apparent patterns emerged on the reactivity of the FCR products towards decarbonylation and catalytic hydrogenation reactions. All the products were obtained in satisfactory and comparable isolated yields under similar reaction conditions. The decarbonylation reaction was performed under solvent-free conditions. The catalytic hydrogenation was performed in the liquid state under mild reaction conditions. All the products were isolated in good to excellent yields. Future work will concentrate on scaling up the production of some representative fuel candidates reported in this work and studying their physicochemical and combustion characteristics. The reactivity of these novel compounds will also be studied for their downstream value addition as chemical intermediates.Author contributions
Abhishek Kumar Yadav designed the experiments, characterized the products, and prepared the ESI.† Navya Subray Bhat worked on the initial development of the project and edited the manuscript. Sonal Kaushik performed some of the experiments, optimized the reaction conditions, and analyzed the products. Asiful H. Seikh was responsible for funding acquisition and editing the manuscript. Saikat Dutta conceptualized the work, supervised, and wrote the original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the Researchers Supporting Project number (RSP2023R373), King Saud University, Riyadh, Saudi Arabia. SD thanks DST-SERB, India, for the Core Research Grant (CRG) (File No. CRG/2022/009346). The authors thank Vellore Institute of Technology (VIT), Vellore, for assisting in the HRMS data collection. The authors also thank the Central Research Facility (CRF), NITK, Surathkal, for the GC and NMR data.References
- G. Kalghatgi, H. Levinsky and M. Colket, Prog. Energy Combust. Sci., 2018, 69, 103–105 CrossRef.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- N. S. M. Aron, K. S. Khoo, K. W. Chew, P. L. Show, W.-H. Chen and T. H. P. Nguyen, Int. J. Energy Res., 2020, 44, 9266–9282 CrossRef.
- C. Lindfors, D. C. Elliott, W. Prins, A. Oasmaa and J. Lehtonen, Energy Fuels, 2023, 37, 799–804 CrossRef CAS.
- S. Dell'Orco, E. D. Christensen, K. Iisa, A. K. Starace, A. Dutta, M. S. Talmadge, K. A. Magrini and C. Mukarakate, Anal. Chem., 2021, 93, 4351–4360 CrossRef PubMed.
- H. A. Akram, M. Imran, A. Javaid, S. Latif, N. B. Rizvi, T. Jesionowski and M. Bilal, Mol. Catal., 2023, 539, 112893 CrossRef CAS.
- C. Gu, L. Chen, Y. Liu, X. Zhang, J. Liu, Q. Zhang, C. Wang and L. Ma, Mol. Catal., 2022, 524, 112317 CrossRef CAS.
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 308, 1446–1450 CrossRef CAS PubMed.
- F. Deng and A. S. Amarasekara, Ind. Crops Prod., 2021, 159, 113055 CrossRef CAS.
- Y. Xiang, S. Wen, Y. Tian, K. Zhao, D. Guo, F. Cheng, Q. Xu, X. Liu and D. Yin, RSC Adv., 2021, 11, 3585–3595 RSC.
- K. S. Arias, M. J. Climent, A. Corma and S. Iborra, Energy Environ. Sci., 2015, 8, 317–331 RSC.
- D. S. Ryabukhin, D. N. Zakusilo, M. O. Kompanets, A. A. Tarakanov, I. A. Boyarskaya, T. O. Artamonova, M. A. Khohodorkovskiy, I. O. Opeida and A. V. Vasilyev, Beilstein J. Org. Chem., 2016, 12, 2125–2135 CrossRef CAS PubMed.
- A. Kumar, B. Dahotia, J. Kumar and B. Thallada, Ind. Eng. Chem. Res., 2019, 58, 16071–16076 CrossRef CAS.
- X. Zhou and T. B. Rauchfuss, ChemSusChem, 2013, 6, 383–388 CrossRef CAS PubMed.
- A. E. Settle, L. Berstis, N. A. Rorrer, Y. Roman-Leshkóv, G. T. Beckham, R. M. Richards and D. R. Vardon, Green Chem., 2017, 19, 3468–3492 RSC.
- S. Dutta and N. S. Bhat, Biomass Convers. Biorefin., 2023, 13, 541–554 CrossRef.
- X. Zhang, J. Wu, J. Chen, L. Lu, L. Zhu and S. Wang, Chin. J. Chem. Eng., 2022, 46, 126–133 CrossRef CAS.
- Z. Luo, K. Lu, Y. Yang, S. Li and G. Li, RSC Adv., 2019, 9, 31960–31968 RSC.
- C. Rosenfeld, J. Konnerth, W. Sailer-Kronlachner, P. Solt, T. Rosenau and H. W. G. van Herwijnen, ChemSusChem, 2020, 13, 3544–3564 CrossRef PubMed.
- H. N. Anchan and S. Dutta, Biomass Convers. Biorefin., 2023, 13, 2571–2593 CrossRef CAS.
- K. Kumar, S. Pathak, A. K. Rajora, A. Halog and S. Upadhyayula, RSC Sustainability, 2023, 1, 282–293 RSC.
- K. Kumar, A. Dahiya, T. Patra and S. Upadhyayula, ChemistrySelect, 2018, 3, 6242–6248 CrossRef CAS.
- N. T. T. Huynh, K. W. Lee, J. K. Cho, Y. J. Kim, S. W. Bae, J. S. Shin and S. Shin, Molecules, 2019, 24, 4623 CrossRef CAS PubMed.
- S. Shinde, K. Deval, R. Chikate and C. Rode, ChemistrySelect, 2018, 3, 8770–8778 CrossRef CAS.
- N. R. Bocanegra, J. R. De La Rosa, C. J. Lucio-Ortiz, P. C. González, H. C. Greenwell, V. E. B. Almaráz, L. S. Rangel, B. Alcántar-Vázquez, V. Rodríguez-González and D. A. D. H. Del Río, Catal. Today, 2021, 360, 2–11 CrossRef.
- N. S. Bhat, S. L. Hegde, S. Dutta and P. Sudarsanam, ACS Sustainable Chem. Eng., 2022, 10, 5803–5809 CrossRef CAS.
- N. S. Bhat, A. K. Yadav, M. Karmakar, A. Thakur, S. S. Mal and S. Dutta, ACS Omega, 2023, 8, 8119–8124 CrossRef CAS PubMed.
- F. B. Ahmad, M. A. Kalam, Z. Zhang and H. H. Masjuki, Energy Convers. Manage.: X, 2022, 14, 100222 CAS.
- M. Zuo, L. Lin and X. Zeng, Fuel, 2023, 343, 127863 CrossRef CAS.
- J. Mitra, X. Zhou and T. Rauchfuss, Green Chem., 2015, 17, 307–313 RSC.
- P. Han, G. Nie, J. Xie, X. E, L. Pan, X. Zhang and J.-J. Zou, Fuel Process. Technol., 2017, 163, 45–50 CrossRef CAS.
- S. Dutta, Ind. Eng. Chem. Res., 2022, 61, 12884–12904 CrossRef CAS.
- S. H. Shinde and C. V. Rode, ACS Omega, 2018, 3, 5491–5501 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra08505h |
| This journal is © The Royal Society of Chemistry 2024 |