
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Benzothiazole DNA gyrase inhibitors and their conjugates with siderophore mimics: design, synthesis and evaluation†
Martina Durcika,
Cristina D. Cruzb,
Mariano Andrea Scorciapino c,
Janez Ilaša,
Päivi Tammelab,
Matteo Ceccarellid,
Lucija Peterlin Mašiča and
Tihomir Tomašič*a
c,
Janez Ilaša,
Päivi Tammelab,
Matteo Ceccarellid,
Lucija Peterlin Mašiča and
Tihomir Tomašič*a
aUniversity of Ljubljana, Faculty of Pharmacy, Aškerčeva cesta 7, 1000 Ljubljana, Slovenia. E-mail: tihomir.tomasic@ffa.uni-lj.si
bDrug Research Program, Division of Pharmaceutical Biosciences, Faculty of Pharmacy, University of Helsinki, P. O. Box 56 (Viikinkaari 5 E), FI-00014 Helsinki, Finland
cDepartment of Chemical and Geological Sciences, University of Cagliari, Cittadella Universitaria di Monserrato – S. P. 8 km 0.700, 09042 – Monserrato (CA), Italy
dDepartment of Physics and IOM/CNR, Sezione di Cagliari, University of Cagliari, Cittadella Universitaria di Monserrato – S. P. 8 km 0700, 09042 – Monserrato (CA), Italy
First published on 18th January 2024
Abstract
Benzothiazole-based bacterial DNA gyrase and topoisomerase IV inhibitors are promising new antibacterial agents with potent activity against Gram-positive and Gram-negative bacterial strains. The aim of this study was to improve the uptake of these inhibitors into the cytoplasm of Gram-negative bacteria by conjugating them to the small siderophore mimics. The best conjugate 18b displayed potent Escherichia coli DNA gyrase and topoisomerase IV inhibition. The interaction analysis of molecular dynamics simulation trajectory showed the important contribution of the siderophore mimic moiety to binding affinity. By NMR spectroscopy, we demonstrated that the hydroxypyridinone moiety alone was responsible for the chelation of iron(III). Moreover, 18b showed an enhancement of antibacterial activity against E. coli JW5503 in an iron-depleted medium, clearly indicating an increased uptake of 18b in this bacterial strain.
Introduction
Antimicrobial resistance (AMR) is one of the greatest challenges of modern medicine, and the number of deaths attributable to AMR is steadily increasing.1 Due to the continued emergence of resistant bacteria, particularly in the ESKAPE (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species) group of pathogens, the development of novel therapeutic agents that act via novel mechanisms of action is of utmost importance.2 DNA gyrase and topoisomerase IV are validated targets for antibacterial drug discovery. These bacterial type IIA topoisomerases are responsible for maintaining the correct topology of the DNA molecule during its replication, transcription, decatenation, and repair.3,4 The DNA gyrase consists of two catalytic GyrA subunits and two GyrB subunits with ATPase activity, whereas the topoisomerase IV consists of two catalytic ParC subunits and two ATP-binding ParE subunits. Both enzymes exert their enzymatic activity as functional heterotetramers in the form A2B2.3,5 The clinically used fluoroquinolones6 and spiropyrimidinetriones (e.g. zoliflodacin),7 which are in clinical development, bind in complexes bordered by DNA, GyrA (ParC) and GyrB (ParE), whereas gepotidacin, a member of the NBTI class,8 binds to DNA and GyrA (ParC). The GyrB and ParE subunits, which are critical for providing energy for enzymatic reactions through ATP hydrolysis, are inhibited by the clinically used novobiocin, which has been withdrawn from therapy due to side effects and development of resistance.9 Fobrepodacin (SPR720) and DS-2969b are GyrB/ParE inhibitors currently in clinical development for the treatment of nontuberculous mycobacterial infections10 and against Clostridium difficile infections,11 respectively.Recently, we developed the benzothiazole class of ATP-competitive dual DNA gyrase and topoisomerase IV inhibitors that exhibit potent antibacterial activity against Gram-positive (G+) and Gram-negative (G−) problematic ESKAPE pathogens.12–15 These resistant G+ and G− bacteria are known to be responsible for the majority of nosocomial infections. Because they are highly resistant to clinically available antibiotics,16,17 most of these bacterial strains are also on the priority lists of WHO and CDC.18,19 The problem is challenging especially for G− species, which are protected by the additional outer membrane (OM), a real physical barrier for molecules to reach the periplasm.20 Although porins expressed in the OM constitute the main path for molecules to penetrate, their permeability might not be sufficient to have a minimal internal accumulation.21 Thus other strategies are required, such as the exploitation of active siderophore transporters.22
We have previously also designed and evaluated conjugates of the (S)-4,5,6,7-tetrahydrobenzothiazole-2,6-diamine-based GyrB/ParE inhibitors23,24 with small siderophore mimics25 as a strategy to improve low cell-wall penetration of G− bacteria.26–28 Siderophores, which are usually catechol or hydroxamic acid derivatives, are secondary microbial metabolites synthesized to sequester iron(III).29 Bacteria synthesize siderophores and release them into the host environment to take up very limited amounts of freely available iron during infection.30 Subsequently, the siderophore in complex with iron(III) is transported through the bacterial cell wall by iron uptake proteins. Conjugation of the antibacterial agent with the siderophore or siderophore mimic (e.g., catechol, hydroxypyranone, hydroxypyridone or dihydroxypyridone) can be used to hijack the bacterial iron-uptake machinery to increase the periplasmic or cytoplasmic concentration of the antibiotic and thus, enhance its antibacterial activity.22,31–33 This concept was also clinically validated, when in November 2019 the FDA approved cefiderocol, a siderophore cephalosporin antibiotic, for the treatment of complicated urinary tract infections. Cefiderocol is also being studied in phase 3 clinical trials for the treatment of nosocomial pneumonia and infections caused by carbapenem-resistant G− bacteria.34,35
In the present work, we conjugated our potent, dual-targeting benzothiazole-based GyrB/ParE inhibitors with small siderophore mimics and investigated their DNA gyrase and topoisomerase IV inhibition, antibacterial activity, and binding of iron(III) by NMR spectroscopy. In addition, the binding mode of the most potent conjugate was evaluated by molecular dynamics simulation and analysis of the interactions in the ATP-binding site of E. coli GyrB.
Results and discussion
Design and synthesis
The design of new DNA gyrase inhibitor–siderophore mimic conjugates started from our potent GyrB/ParE inhibitors A and B (Table 1) that exhibited antibacterial activity against G+ and G− ESKAPE pathogens, limited potential for resistance development and efficacy in in vivo infection models.13,14,36,37 The co-crystal structures of benzothiazole GyrB inhibitors in complex with S. aureus GyrB showed that the pyrrolamide moiety forms two hydrogen bonds with Asp81 (S. aureus GyrB numbering) and crystal water molecule as well as an extensive network of hydrophobic contacts. The benzothiazole core is engaged in the formation of cation–π interaction with Arg84 and substitution at position 4 allows for improving the physico-chemical properties and binding affinity by formation of additional interactions in the lipophilic floor of the binding site. Moreover, the carboxylate at position 6 forms a strong salt bridge with Arg144 (Fig. 1).13,14,36,37| ID | Structure | IC50 [nM] | |
|---|---|---|---|
| E. coli DNA gyrase | E. coli topoisomerase IV | ||
| A | 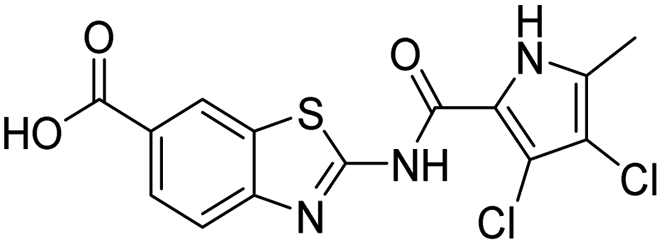 |
13 ± 0 | 500 ± 280 |
| B | 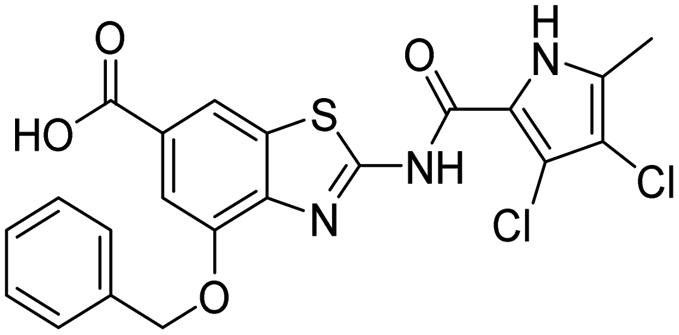 |
<10 | 350 ± 50 |
| 10 | 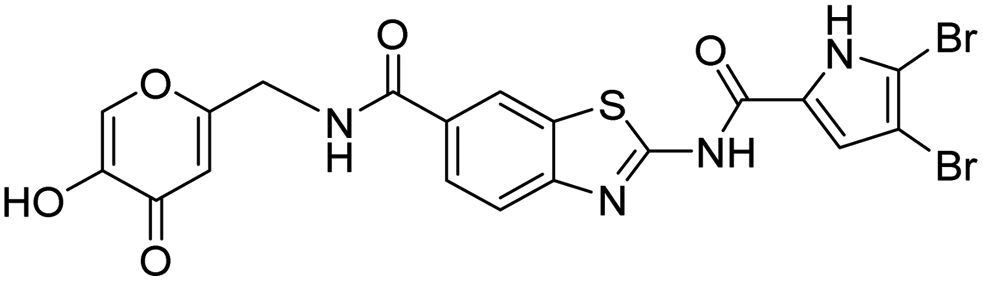 |
1420 ± 180 | >10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
| 12 | 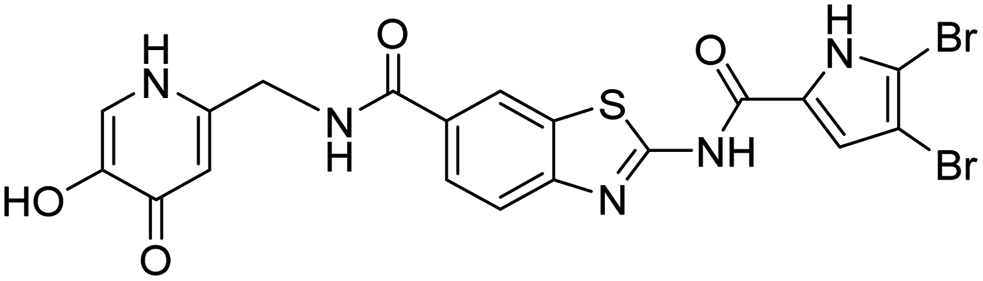 |
1700 ± 360 | >10000 |
| 18a | 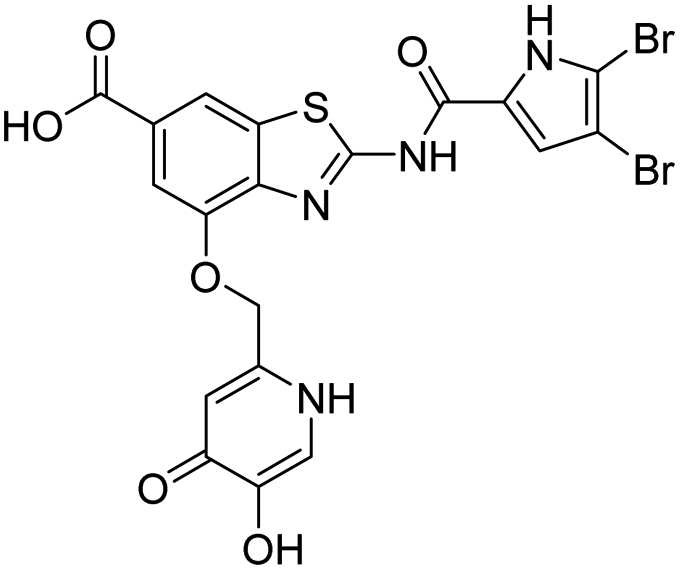 |
12 ± 5 | 1280 ± 820 |
| 18b | 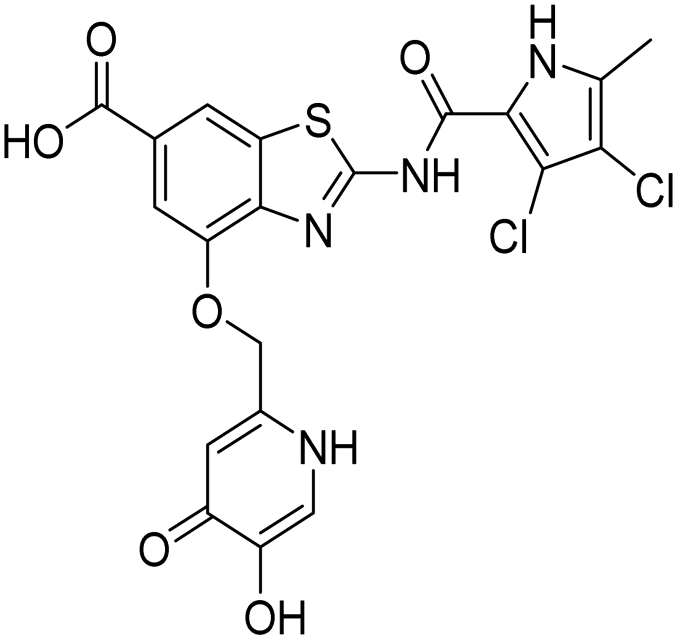 |
22 ± 6 | 430 ± 20 |
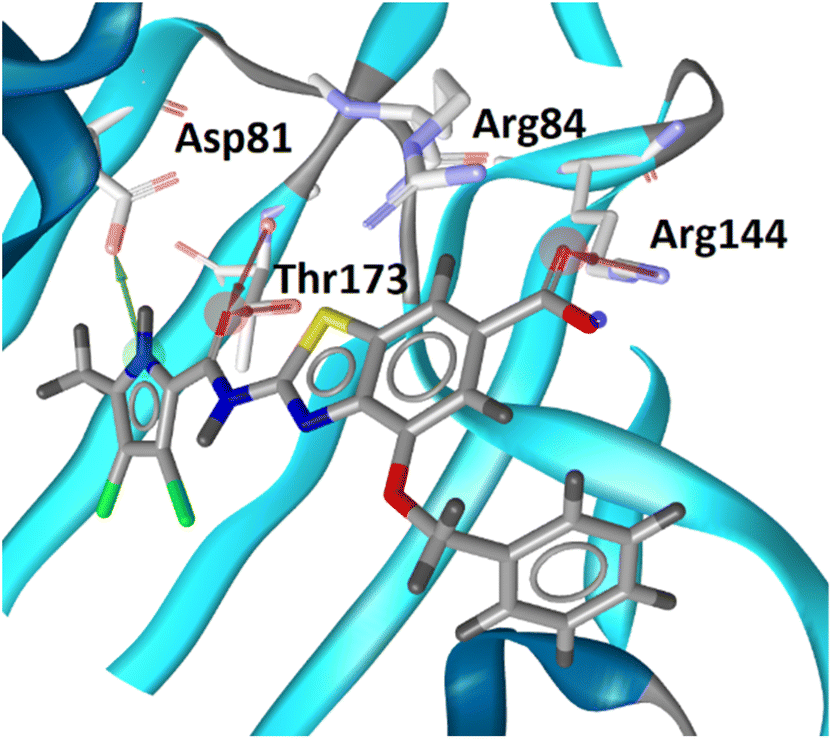 | ||
| Fig. 1 The binding mode of inhibitor B (in grey sticks) in complex with Staphylococcus aureus GyrB24 (in blue cartoon; PDB entry: 6TCK). For clarity, only amino acid residues forming hydrogen bonds or cation–π interaction are shown as grey sticks. Water molecule is presented as a red sphere. Hydrogen bonds are shown as green (hydrogen bond donor) and red (hydrogen bond acceptor) arrows. | ||
In the design of conjugates with siderophore mimics, we considered two different approaches. In the first design strategy, the 6-carboxylic acid was reacted with the 2-(aminomethyl)-5-hydroxy-4H-pyran-4-one or 2-(aminomethyl)-5-hydroxypyridin-4(1H)-one to form the amide derivatives 10 and 12. In the second strategy, a siderophore mimic was incorporated into position 4 of the benzothiazole core to obtain compounds 18a and 18b. In both approaches we anticipated that the siderophore mimic would form attractive interactions within the GyrB/ParE ATP-binding site and therefore, its cleavage from the inhibitor would not be a prerequisite for enzyme inhibition, as previously reported for some GyrA inhibitors conjugated to siderophores.38–40 We used 4,5-dibromo- or 3,4-dichloro-5-methylpyrrole moiety as both form similar interaction network in the GyrB binding site. The advantage of our approach is also that the resulting conjugates have molecular weight around 500 and therefore, better drug-like properties than previously reported ciprofloxacin–siderophore conjugates with cleavable and non-cleavable linkers.
The synthesis of the siderophore mimic building blocks 4 and 6 started from kojic acid (1) (Scheme 1).41 The hydroxyl group at position 3 of 1 was selectively protected using the 4-methoxybenzyl (PMB) chloride in the presence of potassium carbonate in N,N-dimethylformamide (DMF) to obtain compound 2. Hydroxyl group of the hydroxymethylene group at position 5 of compound 2 was then activated with methanesulfonyl chloride to mesylate 3, which was converted in nucleophilic substitution with the sodium bromide to bromide 4 or with the sodium azide to azide 5. The latter was converted to amine 6 using Staudinger reaction conditions.
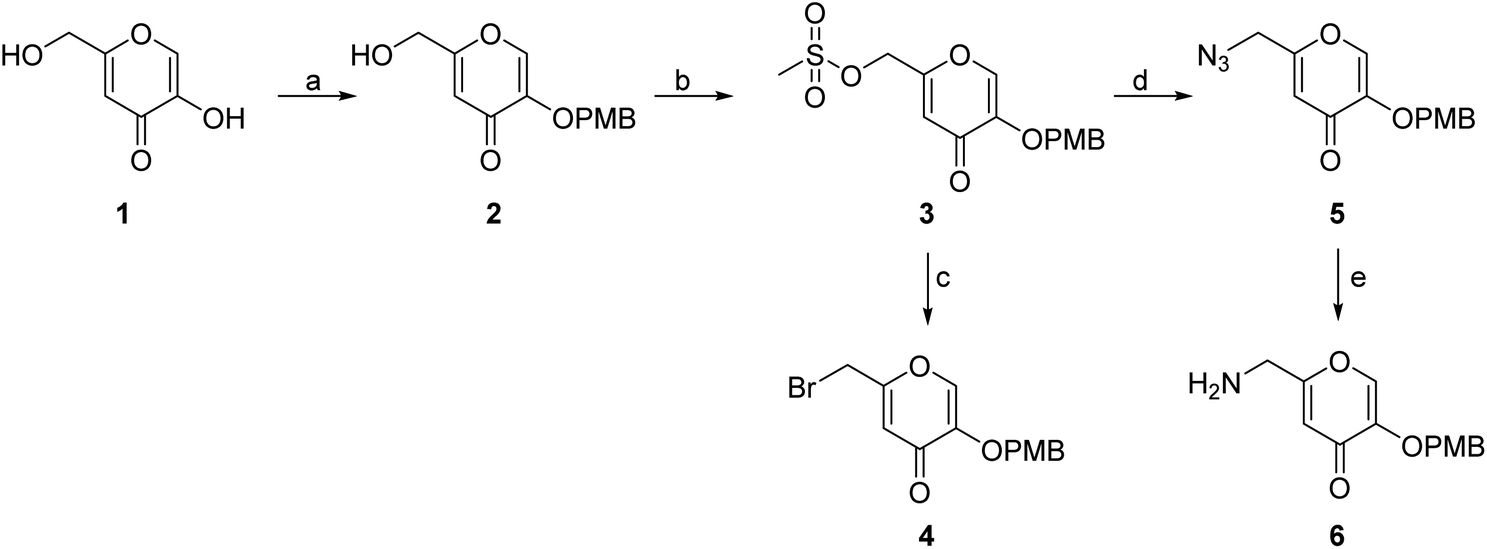 | ||
| Scheme 1 Synthesis of siderophore fragments 4 and 6. Reagents and conditions: (a) 4-methoxybenzyl chloride, K2CO3, dry DMF, 60 °C, 4 h (45.6%); (b) methanesulfonyl chloride, Et3N, DCM, 20 °C, 4 h; (c) NaBr, DMF, 20 °C, 16 h (69.0% over two steps); (d) NaN3, KI, DMF, 20 °C, 20 min (84.4%); (e) PPh3, MeOH, 20 °C, 1 h (37.1%). PMB – p-methoxybenzyl. | ||
DNA gyrase inhibitor–siderophore mimic conjugates were first prepared by attaching the siderophore mimic to the carboxylic acid group at position 6 on the benzothiazole ring (Scheme 2). Firstly, EDC/HOBt-promoted amide coupling between the 2-aminobenzothiazole-6-carboxylic acid (7) and siderophore mimic building block 6 gave compound 8, which was in the next step coupled with the 2,2,2-trichloro-1-(4,5-dibromo-1H-pyrrol-2-yl)ethan-1-one in the presence of sodium carbonate as a base in DMF to yield pyrrolamide 9. Final conjugate 10 was prepared by the removal of the 4-methoxybenzyl protecting group of 9 using HCl in acetic acid. 4H-Pyrane-4-one derivative 9 was also converted to the pyridin-4(1H)-one derivative 11 by refluxing 9 in aqueous ammonia solution. DNA gyrase–siderophore conjugate 12 was finally prepared by deprotection of the 4-methoxybenzyl group of 11 under acidolysis conditions.
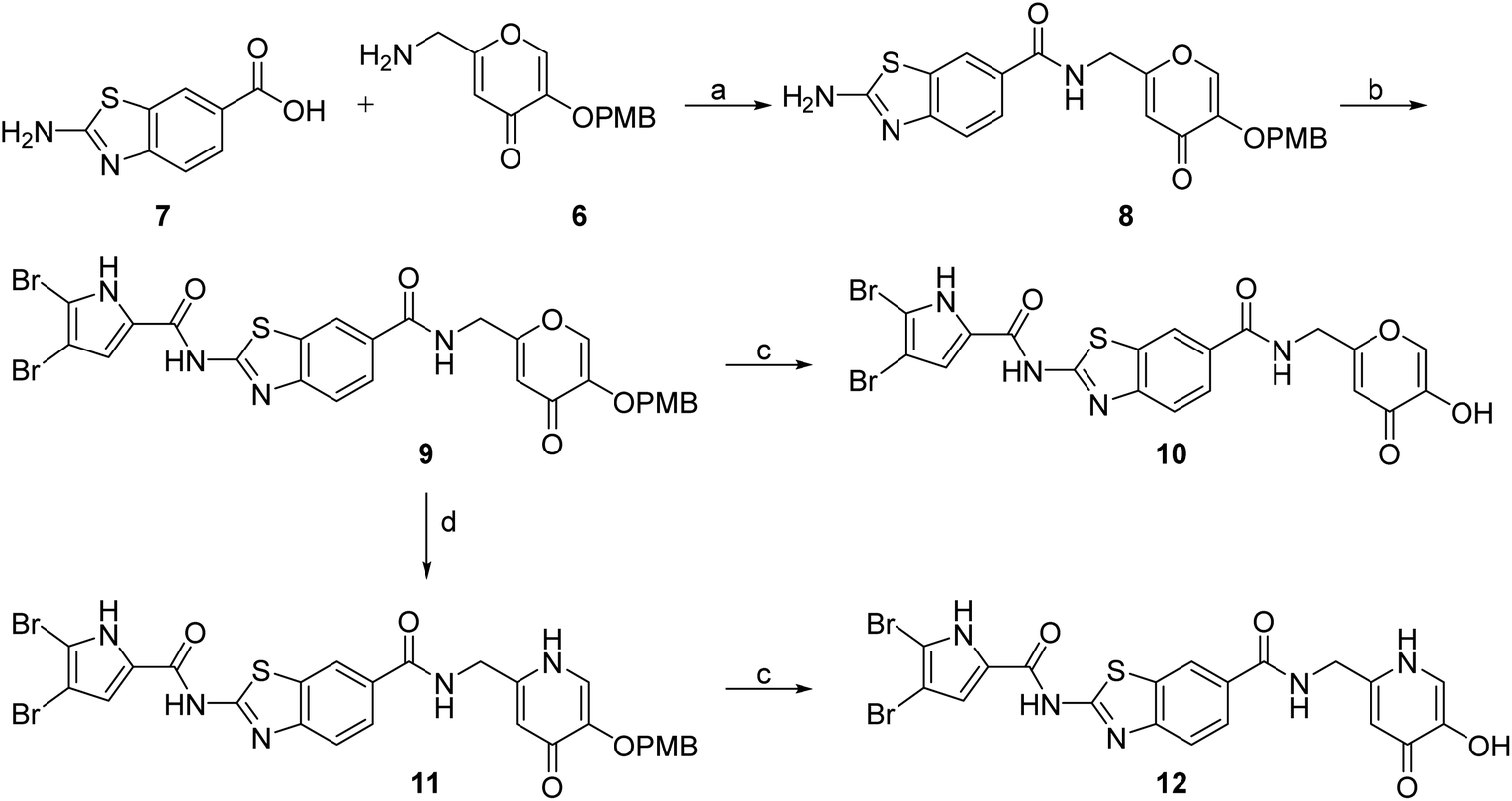 | ||
| Scheme 2 Synthesis of siderophore mimic conjugates 10 and 12. Reagents and conditions: (a) EDC, HOBt, NMM, DMF, 0 °C to 20 °C, 16 h (72.4%); (b) 2,2,2-trichloro-1-(4,5-dibromo-1H-pyrrol-2-yl)ethan-1-one, Na2CO3, DMF, 80 °C, 16 h (75.0%); (c) 1 M HCl in acetic acid, acetic acid, 20 °C, 16 h (84.8–88.9%); (d) NH3 (25% aq. solution), MeOH, 70 °C, 48 h (87.9%). | ||
In addition to the attachment of siderophore mimics to position 6 of the benzothiazole, we also considered introducing such structural elements to position 4 of this scaffold (Scheme 3). The synthesis of final compounds 18a and 18b started from methyl 2-amino-4-hydroxybenzothiazol-6-carboxylate (13)42 that was alkylated with the building block 4 under basic conditions to obtain compound 14. Pyrrolamides 15a and 15b were obtained by reacting amine 14 with the 2,2,2-trichloro-1-(4,5-dibromo-1H-pyrrol-2-yl)ethan-1-one or 2,2,2-trichloro-1-(3,4-dichloro-5-methyl-1H-pyrrol-2-yl)ethan-1-one, respectively, in the presence of sodium carbonate in DMF. 4H-Pyrane-4-one derivatives 15a and 15b were refluxed in ammonium hydroxide to obtain pyridin-4(1H)-one derivatives 16a and 16b. To prepare final conjugates 18a and 18b the order of removal of protecting groups was important. First, methyl ester of 16a and 16b was cleaved using alkaline hydrolysis to get 17a and 17b, followed by acidolysis to yield final products 18a and 18b.
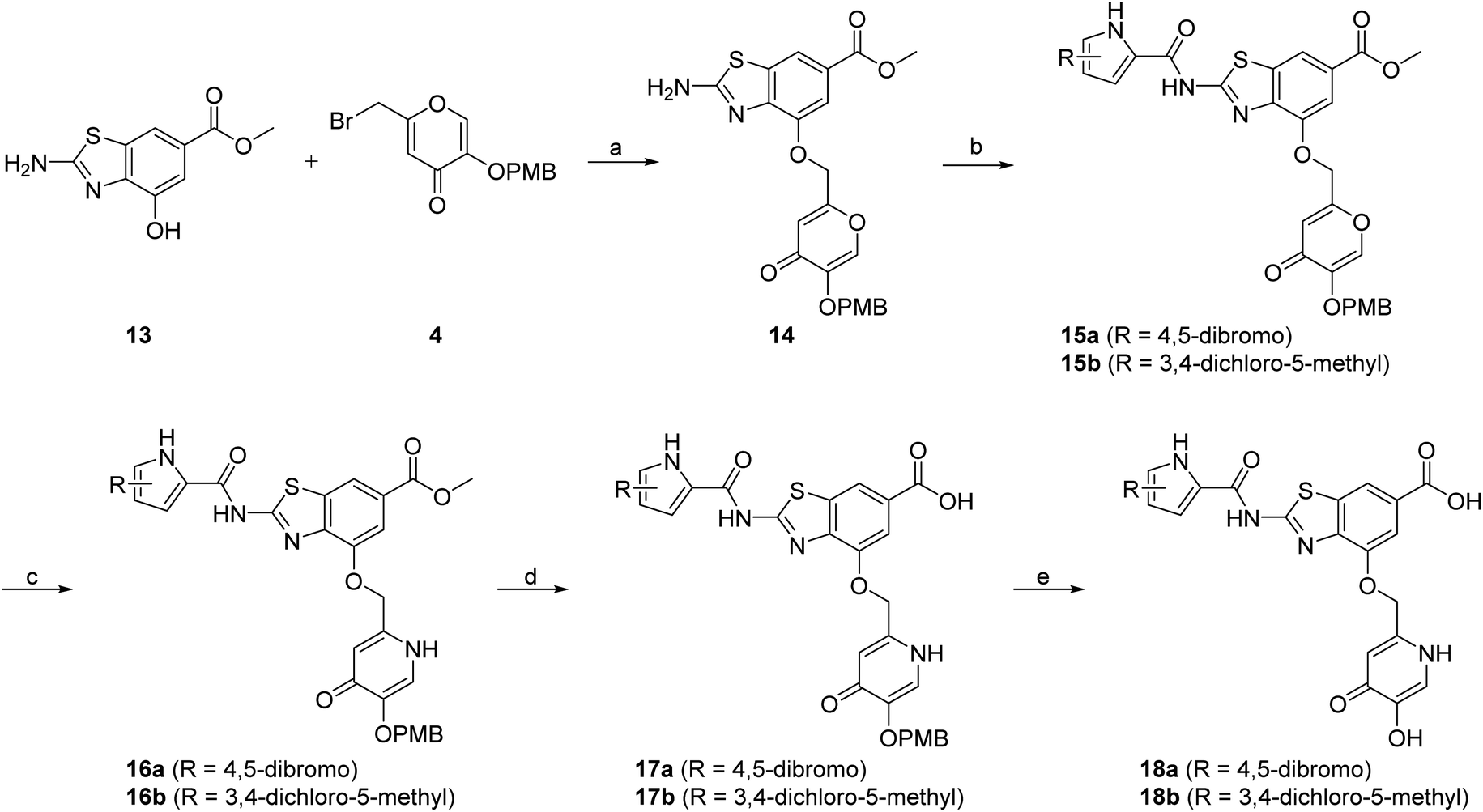 | ||
| Scheme 3 Synthesis of siderophore mimic conjugates 18a and 18b. Reagents and conditions: (a) K2CO3, CH3CN, 60 °C, 16 h (60.5%); (b) 2,2,2-trichloro-1-(4,5-dibromo-1H-pyrrol-2-yl)ethan-1-one (for 15a) or 2,2,2-trichloro-1-(3,4-dichloro-5-methyl-1H-pyrrol-2-yl)ethan-1-one (for 15b), Na2CO3, DMF, 80 °C, 16 h (24.5–77.8%); (c) NH3 (25% aq. solution), MeOH, 60 °C, 3 h, then 20 °C, 16 h (or 60 °C, 16 h – for 16b) (32.0–33.0%); (d) 1–2 M NaOH, 1,4-dioxane (for 17a) or MeOH (for 17b), 40 °C, 16 h (33.3–63.7%); (e) 1 M HCl in acetic acid, acetic acid, 20 °C, 40 h (52.4–62.3%). PMB – p-methoxybenzyl. | ||
In vitro enzyme inhibition
All final compounds were first tested for their inhibitory activity in the E. coli DNA gyrase supercoiling assay and the E. coli topoisomerase IV relaxation assay (Table 1). The enzyme inhibition data showed that the amide formation at position 6 (compounds 10 and 12) had a detrimental effect on the inhibition of E. coli topoisomerase IV (IC50 values above 10 μM) and resulted in weak inhibition of E. coli DNA gyrase with IC50 values in the low micromolar range. These results are consistent with our previous study in which bioisosteric replacements of the carboxylate group or its substitution resulted in weaker DNA gyrase and topoisomerase IV inhibition.14 In the same study, we also showed that substituents at position 4 of the benzothiazole scaffold in potent inhibitors can have different chemical properties. These observations were confirmed with compounds 18a and 18b, which carried the siderophore mimic at position 4. These compounds inhibited E. coli DNA gyrase in the low nanomolar range and displayed only slightly weaker E. coli topoisomerase IV inhibition than did the parent compound B (Table 1). With compounds 18a and 18b, we confirmed our hypothesis that it is possible to maintain potent enzyme inhibition after introduction of the siderophore mimic into the structure of the GyrB/ParE inhibitor.Molecular modeling
The binding modes of 18b in the ATP-binding sites of E. coli GyrB and ParE were studied by a combination of molecular docking calculations, molecular dynamics simulation and interaction analyzes. Inhibitor 18b was first docked to E. coli GyrB (PDB entry: 4WUB43) and ParE (PDB entry: 1S16 (ref. 44)) ATP-binding sites. These two co-crystal structures were used because they contain the flexible loop in the lipophilic floor of the enzymes that can interact with the substituent at position 4 of the benzothiazole core. The binding mode of 18b was consistent with the co-crystal structures of benzothiazole-based GyrB/ParE inhibitors in complex with S. aureus GyrB. In E. coli GyrB, the pyrrolamide moiety formed hydrogen bonds with Asp73 and Thr165 and several hydrophobic interactions in the hydrophobic pocket. A salt bridge was formed between the 6-carboxylate group and Arg136 and a cation-π interaction was formed between the benzothiazole ring and Arg76 guanidinium group. The docking binding mode of 18b in complex with E. coli GyrB was used as a starting point for the 100 ns MD simulation. The MD trajectory was then analyzed using structure-based pharmacophore modeling in LigandScout. Analysis of the MD simulation trajectory showed that the siderophore mimic bound at position 4 of the benzothiazole core is quite flexible and forms hydrogen bonds with the Ala100–Tyr109 residues of the flexible loop. It forms additional π-stacking with the Phe104 side chain (Fig. 2), which is present in 18% of the simulation time. This analysis clearly showed the important contribution of the siderophore mimic to the binding affinity.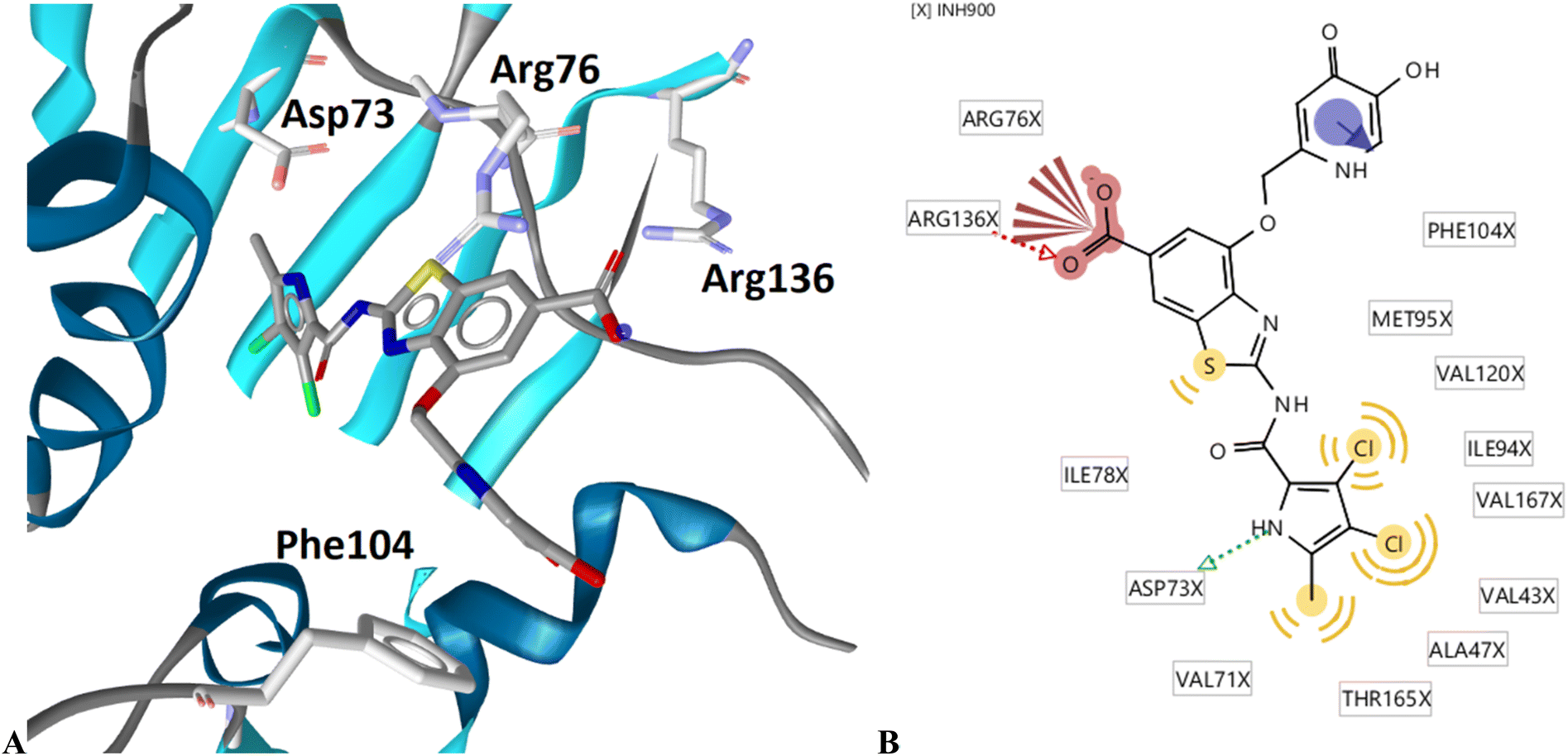 | ||
| Fig. 2 Representative snapshot from molecular dynamics trajectory. (A) Compound 18b (in grey sticks) in the binding site of E. coli GyrB (PDB entry: 4WUB). (B) Schematic representation of the pharmacophore model. Pharmacophore features are presented as follows: hydrophobic features as yellow spheres, hydrogen bond donor as a green arrow, hydrogen bond acceptor as a red arrow, negative ionizable as a red star and aromatic rings as a blue disc. | ||
Antibacterial activity
We also tested the antibacterial activity of the siderophore mimic-bearing GyrB/ParE inhibitors 10, 12 and 18b against wild type G− E. coli, P. aeruginosa and A. baumannii in iron-depleted cation-adjusted Mueller Hinton broth (ID-CAMHB) and iron-supplemented CAMHB broth (CAMHB + Fe). Cefiderocol, a siderophore cephalosporin, was used as a positive control. Compound 18a was only tested on E. coli in both media conditions. Iron-depleted conditions were used to determine the effects of the attached siderophore mimics on antibacterial activities, since bacteria under low iron conditions produce and secrete siderophores to take up iron from the extracellular environment.32 In general, compounds 10, 12, 18a and 18b displayed rather weak antibacterial activity at 50 μM in iron-supplemented and iron-depleted media, which indicated MIC values > 50 μM (Table 2). We have previously observed that our benzothiazole GyrB/ParE inhibitors are strongly effluxed from the G− bacterial cells and that this effect is especially pronounced in E. coli.14 Therefore, compounds 10, 12, and 18b were also tested against E. coli JW5503, which is a tolC deletion mutant strain with a defective efflux pump. Compound 18b showed an MIC value of 25 μM (12.7 μg mL−1) under iron-supplemented conditions. Improvement of antibacterial activity of 18b against efflux-defective E. coli JW5503 with an MIC value of 12.5 μM (6.37 μg mL−1) was observed under iron-depleted conditions. For cefiderocol, which was used as a positive control, similar twofold improvement of antibacterial activity was observed in iron-depleted medium (Table 2). These results demonstrate the contribution of the siderophore mimic to cell wall permeation rendering to a more potent antibacterial activity of 18b, however, efflux remains to be a major issue of the benzothiazole GyrB/ParE inhibitors. The antibacterial activity of 18b was weaker than that of parent compounds A and B, suggesting that the incorporation of the siderophore mimic has a limited contribution. In our recent study, we also showed that benzothiazole-based GyrB inhibitors with potent activity against G− strains have a topological surface area (TPSA) of less than 120 Å2.36,37 Replacement of the phenyl ring of inhibitor B (TPSA = 100.0 Å2) by the oxygen- and nitrogen-rich siderophore mimic in 18b results in a significantly higher TPSA of 149.4 Å2, which may be one of the reasons for the weaker antibacterial activity of 18b despite the presence of the siderophore mimic in its structure.| E. coli ATCC 25922 | P. aeruginosa ATCC 27853 | A. baumannii ATCC 19606 | E. coli JW5503 | |||||
|---|---|---|---|---|---|---|---|---|
| −Fe | +Fe | −Fe | +Fe | −Fe | +Fe | −Fe | +Fe | |
| Minimum inhibitory concentrations [μM] | ||||||||
| a −Fe: iron depleted cation adjusted Mueller Hinton broth media (ID-CAMHB), +Fe: cation adjusted Mueller Hinton broth media + Fe (CAMHB + Fe), nt.: not tested. | ||||||||
| 10 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 |
| 12 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 |
| 18a | >50 | >50 | n.t | n.t | n.t | n.t | n.t | n.t |
| 18b | >50 | >50 | >50 | >50 | >50 | >50 | 12.5 | 25 |
| Cefiderocol | 0.25 | 0.25 | 0.25 | 1 | 0.25 | 2 | 0.0625 | 0.125 |
NMR spectroscopy of 18b–iron(III) complex
Given the improved antibacterial activity of 18b against the E. coli JW5503 in iron-depleted medium, its potential for chelation of iron(III) was studied by NMR spectroscopy. To a DMSO-d6 solution of 18b, FeCl3 in steps of 0.1 equivalents of Fe3+ was added (Fig. 3). A very small shift was observed for all the resonances in the spectrum with increasing Fe3+ concentration (Fig. 3), which can be attributed to the strong acidity of the ferric chloride solution. In fact, the hydroxypyridinone resonances showed the largest shift among 18b resonances. However, while all the resonances did not change their profile along this short titration (apart from the progressive broadening due to the increasing concentration of the paramagnetic Fe3+ ion in solution), the two hydroxypyridinone resonances showed a difference. Fig. 4 shows how the single resonance (red spectrum) progressively broadened (orange) and passed through a clear convolution of two components (yellow) at about 0.2 equivalents of Fe3+. Then, the signal progressively returned to a single component (green) and started to progressively broaden (blue). This is the typical trend observed when two forms are in a relatively slow exchange, with the molar fraction of the first one progressively decreasing from 1 to 0, while the other progressively and complementarily increases from 0 to 1. One of the two forms is the free ligand, which is maximum in the absence of ferric ions. With increasing the latter, the free ligand is progressively transformed to the ligand–metal complex. Our experiment showed that the signals for a free ligand disappeared by the addition of 0.4 equivalents of FeCl3, with the half-equivalence point around 0.2. This clearly suggests the formation of complexes with stoichiometry L3M (where L is the ligand and M the metal). Moreover, in the range investigated (ligand excess), all of the other resonances appeared to be insensitive to the formation of the complex, which shows that only the hydroxypyridinone moiety is exploited in the complex formation. These two observations agree with each other. Each hydroxypyridinone is expected to use two O donors to coordinate the metal ion, thus, in the complex L3M, the three hydroxypyridinones are enough to complete the typical octahedral configuration around Fe3+.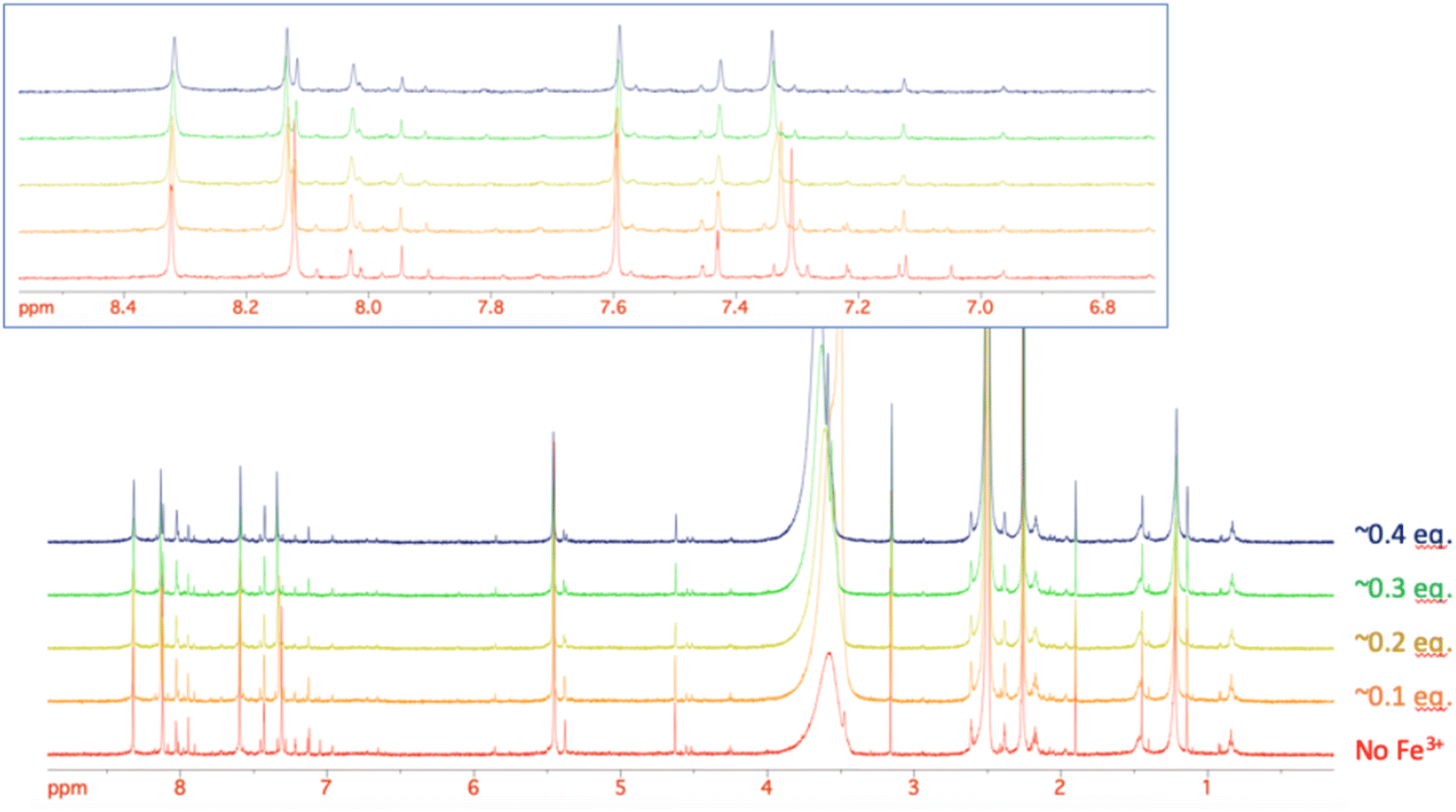 | ||
| Fig. 3 The stacking plot of NMR spectra of 18b after stepwise addition of FeCl3 to a DMSO solution of 18b. The inset shows the aromatic region of 18b. | ||
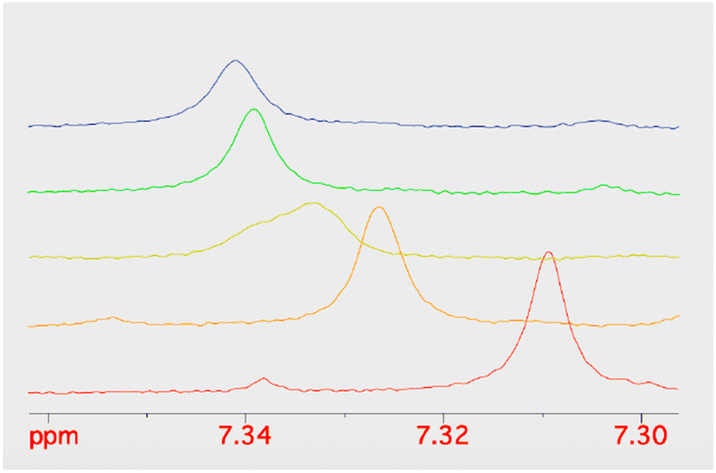 | ||
| Fig. 4 The stacking plot of NMR spectra of the pyridinone proton at 7.3 ppm of 18b after stepwise addition of FeCl3 to a DMSO solution of 18b. | ||
Conclusions
We have prepared new conjugates of our antibacterially active GyrB/ParE inhibitors with 2-(aminomethyl)-5-hydroxy-4H-pyran-4-one and 2-(aminomethyl)-5-hydroxypyridin-4(1H)-one siderophore mimics. These mimics were attached to two different positions and their incorporation to the position 4 of the benzothiazole scaffold resulted in potent DNA gyrase inhibition, whereas the inhibition of topoisomerase IV was slightly weaker compared with the parent inhibitors. The most potent inhibitor 18b showed a twofold enhancement of antibacterial activity against the efflux-defective E. coli strain JW5503, but was only weakly active against wild-type E. coli, P. aeruginosa and A. baumannii. The results with wild-type and efflux-defective E. coli strains indicated that efflux remains a problem for benzothiazole-based GyrB/ParE inhibitors and will be the focus of future optimization.Experimental section
Chemicals were obtained from Acros Organics (Geel, Belgium), Sigma-Aldrich (St Louis, MO, USA), Apollo Scientific Ltd (Stockport, UK), Fluorochem Ltd (Derbyshire, UK), and Enamine Ltd (Kyiv, Ukraine) and were used without further purification. Analytical TLC was performed on silica gel Merck 60 F254 plates (0.25 mm), using visualization with UV light and spray reagents. Column chromatography was carried out on silica gel 60 (particle size, 240–400 mesh). 1H and 13C NMR spectra were recorded at 400 MHz and 101 MHz, respectively, on a Bruker AVANCE III 400 spectrometer (Bruker Corporation, Billerica, MA, USA) in DMSO-d6 or CDCl3 solutions, with TMS as the internal standard. Mass spectra were obtained using an ADVION expression CMSL mass spectrometer (Advion Inc., Ithaca, USA) and high-resolution mass spectra were obtained using VG Analytical Autospec Q Micromass mass spectrometer (Fisons, VG Analytical, Manchester, UK) (compounds 5, 8–12) or Exactive Plus Orbitrap mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) (compounds 18a and 18b). HPLC purity analyses were performed on a 1260 Infinity II LC system (Agilent Technologies Inc., Santa Clara, CA, USA). A Waters XBridge C18 column was used (3.5 μm, 4.6 mm × 150 mm), with flow rate of 1.5 mL min−1 and sample injection volume of 10 μL. The mobile phase consisted of acetonitrile (solvent A) and 0.1% formic acid in 1% acetonitrile in ultrapure water (solvent B). The gradient (defined for solvent A) was: 0–1.0 min, 25%; 1.0–6.0 min, 25–98%; 6.0–6.5 min, 98%; 6.5–7.5 min, 98–25%; 7.5–10.5 min, 25%. All active tested compounds were more than 95% pure as established by HPLC, unless indicated otherwise.Synthesis of compounds
:1, 500 mL) and left overnight. The formed crystals were filtered off and dried. Yield: 4.21 g (45.6%); orange crystals; mp: 112 °C.:1) as eluent. Yield: 2.70 g (69.0%); white crystals; mp: 107 °C. 1H NMR (400 MHz, DMSO-d6) δ [ppm] 3.76 (s, 3H), 4.56 (s, 2H), 4.86 (s, 2H), 6.57 (s, 1H), 6.95 (d, J = 8.7 Hz, 2H), 7.35 (d, J = 8.7 Hz, 2H), 8.26 (s, 1H).−H]−): calculated 565.9128, found 565.8969. HPLC: tr 5.25 min (83.6% at 254 nm).:1) as eluent. Yield: 0.89 g (60.5%); brown solid; mp: 126 °C. 1H NMR (400 MHz, DMSO-d6) δ [ppm] 3.76 (s, 3H), 3.83 (s, 3H), 4.88 (s, 2H), 5.17 (s, 2H), 6.57 (s, 1H), 6.96 (d, J = 8.7 Hz, 2H), 7.35 (d, J = 8.7 Hz, 2H), 7.44 (d, J = 1.5 Hz, 1H), 7.99 (s, 2H), 8.02 (d, J = 1.5 Hz, 1H), 8.27 (s, 1H).:1) as eluent. Compound was obtained as a mixture of tautomers and 1H NMR signals could not be unambiguously assigned. Yield: 48 mg (32.0%); beige solid; mp > 300 °C. MS (ESI) m/z = 717.3 ([M + H]+).:1 → 9:1) as eluent to give another 32 mg of the product as white crystals. Compound was obtained as a mixture of tautomers and 1H NMR signals could not be unambiguously assigned. Yield: 92 mg (33.0%); brown solid and white crystals. MS (ESI) m/z = 643.0 ([M + H]+).−H]−).−H]−): calculated 580.8773, found 580.8772. HPLC: tr 5.72 min (95.9% at 254 nm).−H]−): calculated 506.9938, found 506.9943. HPLC: tr 5.11 min (90.0% at 254 nm).Determination of inhibitory activities against E. coli DNA gyrase and topoisomerase IV
The assays for determination of IC50 against E. coli DNA gyrase and topoisomerase IV were performed according to previously reported procedures.23 IC50 values were determined with seven concentrations of the inhibitors. GraphPad Prism program was used for calculating an IC50 value, which represents the concentration of inhibitor where the activity of the enzyme is reduced by 50%. IC50 values were determined in three independent measurements, and their average value is given as a final result.Determination of antibacterial activities
The clinical microbiology control strains of E. coli (ATCC 25922), P. aeruginosa (ATCC 27853) and A. baumannii (ATCC 19606) were obtained from Microbiologics Inc. (St. Cloud, MN, USA). The single-gene knock-out mutant strain of E. coli JW5503 (tolC knock-out) was obtained from the E. coli collection of the National BioResource Project at the National Institute of Genetics (Japan).45 To determine the antibacterial activities, broth microdilution assays were carried out in 96-well plates following the Clinical and Laboratory Standards Institute guidelines.46 Compounds were prepared as 100% DMSO stock solutions. The final concentration of DMSO was 1% and had no effect on the growth of bacteria. Preliminary screening was performed at 50 μM, and growth inhibition was measured after 24 h incubation at 37 °C. For compounds displaying >90% growth inhibition in the preliminary assay, MIC values were determined in dose–response assays (from at least two independent experiments, each with three replicates per concentration). Cation-adjusted Mueller-Hinton broth (CAMHB, BD) was used in all assays. CAMHB was prepared according to manufacturer's instructions and iron-depleted CAMHB (ID-CAMHB) according to CLSI guidelines. ID-CAMHB was used in order to mimic the conditions faced by bacteria during human infections, and induces the ferric iron transport. ID-CAMHB was prepared by adding 100 g of Chelex 100 resin (Bio-Rad) to 1 L of autoclaved CAMHB and stirred for 2 h at room temperature, in order to remove cations from the medium. Then, broth was filtered using a 0.2 μm filter to remove the resin and pH adjusted to 7.3 using 5 M hydrochloric acid. ID-CAMHB was then supplemented with calcium (CaCl2), magnesium (MgCl2) and zinc (ZnSO4) to final concentrations of 22.5 μg mL−1, 11.25 μg mL−1 and 0.56 μg mL−1, respectively, and filtered again. To evaluate the effect of iron concentration on antibacterial effect of compounds, an appropriate amount of iron(III) chloride was added to ID-CAMHB, and this supplemented media was used as control CAMHB. The iron concentration in CAMHB and ID-CAMHB were approximately 0.2 mg L−1 and ≤0.03 mg L−1, respectively. Cefiderocol, a positive control, MICs were determined against E. coli, P. aeruginosa and A. baumannii, and used as positive control in the assays. Cefiderocol was purchased from MedChemExpress Europe.000 steps) and adopted basis Newton–Raphson (10000 steps) energy minimizations. The system for MD simulation was prepared using CHARMM-GUI.49,50 Molecular mechanics parameters for compound 18b were estimated using the ParamChem tool.51–53 Structure of the 18b–GyrB complex was first embedded in a box of TIP3P water molecules. Then the system was neutralized by addition of KCl. The MD simulation was run in the NPT ensemble using the periodic boundary conditions. Temperature (300 K) and pressure (1 atm) were controlled using the Langevin dynamics and Langevin piston methods, respectively. Short-range and long-range forces were calculated every 1 and 2 time steps, respectively, with a time step of 2.0 ps. The smooth particle mesh Ewald method was used to calculate the electrostatic interactions.54 The short-range interactions were cut off at 12 Å. All of the chemical bonds between hydrogen and the heavy atoms were held fixed using the SHAKE algorithm.55 After equilibration of the complete system for 1 ns, an unconstrained 100 ns production was run.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the Slovenian Research Agency (Grant No. P1-0208, J1-3030) and by the Academy of Finland (Grant No. 321551). M. C. thanks for partial support the project INF-ACT funded within the PNRR program.References
- C. J. Murray, K. S. Ikuta, F. Sharara, L. Swetschinski, G. R. Aguilar, A. Gray, C. Han, C. Bisignano, P. Rao, E. Wool, S. C. Johnson, A. J. Browne, M. G. Chipeta, F. Fell, S. Hackett, G. Haines-Woodhouse, B. H. K. Hamadani, E. A. P. Kumaran, B. McManigal, R. Agarwal, S. Akech, S. Albertson, J. Amuasi, J. Andrews, A. Aravkin, E. Ashley, F. Bailey, S. Baker, B. Basnyat, A. Bekker, R. Bender, A. Bethou, J. Bielicki, S. Boonkasidecha, J. Bukosia, C. Carvalheiro, C. Castañeda-Orjuela, V. Chansamouth, S. Chaurasia, S. Chiurchiù, F. Chowdhury, A. J. Cook, B. Cooper, T. R. Cressey, E. Criollo-Mora, M. Cunningham, S. Darboe, N. P. J. Day, M. D. Luca, K. Dokova, A. Dramowski, S. J. Dunachie, T. Eckmanns, D. Eibach, A. Emami, N. Feasey, N. Fisher-Pearson, K. Forrest, D. Garrett, P. Gastmeier, A. Z. Giref, R. C. Greer, V. Gupta, S. Haller, A. Haselbeck, S. I. Hay, M. Holm, S. Hopkins, K. C. Iregbu, J. Jacobs, D. Jarovsky, F. Javanmardi, M. Khorana, N. Kissoon, E. Kobeissi, T. Kostyanev, F. Krapp, R. Krumkamp, A. Kumar, H. H. Kyu, C. Lim, D. Limmathurotsakul, M. J. Loftus, M. Lunn, J. Ma, N. Mturi, T. Munera-Huertas, P. Musicha, M. M. Mussi-Pinhata, T. Nakamura, R. Nanavati, S. Nangia, P. Newton, C. Ngoun, A. Novotney, D. Nwakanma, C. W. Obiero, A. Olivas-Martinez, P. Olliaro, E. Ooko, E. Ortiz-Brizuela, A. Y. Peleg, C. Perrone, N. Plakkal, A. Ponce-de-Leon, M. Raad, T. Ramdin, A. Riddell, T. Roberts, J. V. Robotham, A. Roca, K. E. Rudd, N. Russell, J. Schnall, J. A. G. Scott, M. Shivamallappa, J. Sifuentes-Osornio, N. Steenkeste, A. J. Stewardson, T. Stoeva, N. Tasak, A. Thaiprakong, G. Thwaites, C. Turner, P. Turner, H. R. van Doorn, S. Velaphi, A. Vongpradith, H. Vu, T. Walsh, S. Waner, T. Wangrangsimakul, T. Wozniak, P. Zheng, B. Sartorius, A. D. Lopez, A. Stergachis, C. Moore, C. Dolecek and M. Naghavi, Lancet, 2022, 399, 629–655 Search PubMed.
- K. Lewis, Cell, 2020, 181, 29–45 Search PubMed.
- J. J. Champoux, Annu. Rev. Biochem., 2001, 70, 369–413 Search PubMed.
- H. Hiasa, Methods Mol. Biol., 2018, 1703, 47–62 CrossRef CAS PubMed.
- T. Tomašić and L. Peterlin Masic, Curr. Top. Med. Chem., 2014, 14, 130–151 CrossRef PubMed.
- A. M. Emmerson and A. M. Jones, J. Antimicrob. Chemother., 2003, 51(suppl 1), 13–20 Search PubMed.
- P. A. Bradford, A. A. Miller, J. O'Donnell and J. P. Mueller, ACS Infect. Dis., 2020, 6, 1332–1345 CrossRef CAS PubMed.
- A. Kolarič, M. Anderluh and N. Minovski, J. Med. Chem., 2020, 63, 5664–5674 Search PubMed.
- G. S. Bisacchi and J. I. Manchester, ACS Infect. Dis., 2015, 1, 4–41 CrossRef CAS PubMed.
- A. K. Talley, A. Thurston, G. Moore, V. K. Gupta, M. Satterfield, E. Manyak, S. Stokes, A. Dane and D. Melnick, Antimicrob. Agents Chemother., 2021, 65, e0120821 CrossRef PubMed.
- A. G. Vandell, S. Inoue, J. Dennie, Y. Nagasawa, R. Gajee, J. Pav, G. Zhang, C. Zamora, N. Masuda and G. Senaldi, Antimicrob. Agents Chemother., 2018, 62, 025377 CrossRef PubMed.
- M. Gjorgjieva, T. Tomašič, M. Barančokova, S. Katsamakas, J. Ilaš, P. Tammela, L. Peterlin Mašič and D. Kikelj, J. Med. Chem., 2016, 59, 8941–8954 CrossRef CAS PubMed.
- A. Nyerges, T. Tomašič, M. Durcik, T. Revesz, P. Szili, G. Draskovits, F. Bogar, Ž. Skok, N. Zidar, J. Ilaš, A. Zega, D. Kikelj, L. Daruka, B. Kintses, B. Vasarhelyi, I. Foldesi, D. Kata, M. Welin, R. Kimbung, D. Focht, L. P. Mašič and C. Pal, PLoS Biol., 2020, 18, e3000819 CrossRef CAS PubMed.
- M. Durcik, Á. Nyerges, Ž. Skok, D. G. Skledar, J. Trontelj, N. Zidar, J. Ilaš, A. Zega, C. D. Cruz, P. Tammela, M. Welin, Y. R. Kimbung, D. Focht, O. Benek, T. Révész, G. Draskovits, P. É. Szili, L. Daruka, C. Pál, D. Kikelj, L. P. Mašič and T. Tomašič, Eur. J. Med. Chem., 2021, 213, 113200 Search PubMed.
- M. Durcik, Ž. Skok, J. Ilaš, N. Zidar, A. Zega, P. É. Szili, G. Draskovits, T. Révész, D. Kikelj, A. Nyerges, C. Pál, L. P. Mašič and T. Tomašič, Pharmaceutics, 2021, 13, 6 CrossRef CAS PubMed.
- L. B. Rice, J. Infect. Dis., 2008, 197, 1079–1081 CrossRef PubMed.
- H. W. Boucher, G. H. Talbot, J. S. Bradley, J. E. Edwards, D. Gilbert, L. B. Rice, M. Scheld, B. Spellberg and J. Bartlett, Clin. Infect. Dis., 2009, 48, 1–12 CrossRef PubMed.
- WHO Publishes List of Bacteria for Which New Antibiotics Are Urgently Needed, https://www.who.int/news/item/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed, accessed May 13, 2017 Search PubMed.
- Centers for Disease Control and Prevention (U.S.), Antibiotic Resistance Threats in the United States, 2019, Centers for Disease Control and Prevention (U.S.), 2019 Search PubMed.
- J. Vergalli, I. V. Bodrenko, M. Masi, L. Moynié, S. Acosta-Gutiérrez, J. H. Naismith, A. Davin-Regli, M. Ceccarelli, B. van den Berg, M. Winterhalter and J.-M. Pagès, Nat. Rev. Microbiol., 2020, 18, 164–176 CrossRef CAS PubMed.
- S. Acosta-Gutiérrez, I. V. Bodrenko and M. Ceccarelli, Antibiotics, 2021, 10, 635 CrossRef PubMed.
- G. L. A. Mislin and I. J. Schalk, Met. Integr. Biometal Sci., 2014, 6, 408–420 CrossRef CAS PubMed.
- T. Tomašič, S. Katsamakas, Ž. Hodnik, J. Ilaš, M. Brvar, T. Solmajer, S. Montalvão, P. Tammela, M. Banjanac, G. Ergović, M. Anderluh, L. P. Mašič and D. Kikelj, J. Med. Chem., 2015, 58, 5501–5521 CrossRef PubMed.
- A. Lamut, Ž. Skok, M. Barančoková, L. J. Gutierrez, C. D. Cruz, P. Tammela, G. Draskovits, P. É. Szili, Á. Nyerges, C. Pál, P. Molek, T. Bratkovič, J. Ilaš, N. Zidar, A. Zega, R. D. Enriz, D. Kikelj and T. Tomašič, Future Med. Chem., 2020, 12, 277–297 CrossRef CAS PubMed.
- A. Lamut, C. D. Cruz, Ž. Skok, M. Barančoková, N. Zidar, A. Zega, L. P. Mašič, J. Ilaš, P. Tammela, D. Kikelj and T. Tomašič, Bioorg. Chem., 2020, 95, 103550 CrossRef CAS PubMed.
- D. Benedetto Tiz, D. Kikelj and N. Zidar, Expert Opin. Drug Discovery, 2018, 13, 497–507 CrossRef CAS PubMed.
- H. I. Zgurskaya, C. A. Löpez and S. Gnanakaran, ACS Infect. Dis., 2015, 1, 512–522 CrossRef CAS PubMed.
- A. Nazli, D. L. He, D. Liao, M. Z. I. Khan, C. Huang and Y. He, Adv. Drug Delivery Rev., 2022, 189, 114502 CrossRef CAS PubMed.
- E. Ahmed and S. J. M. Holmström, Microb. Biotechnol., 2014, 7, 196–208 CrossRef CAS PubMed.
- R. Golonka, B. S. Yeoh and M. Vijay-Kumar, J. Innate Immun., 2019, 11, 249–262 CrossRef CAS PubMed.
- U. Bilitewski, J. A. V. Blodgett, A.-K. Duhme-Klair, S. Dallavalle, S. Laschat, A. Routledge and R. Schobert, Angew Chem. Int. Ed. Engl., 2017, 56, 14360–14382 CrossRef CAS PubMed.
- E. Gumienna-Kontecka, P. L. Carver, E. Gumienna-Kontecka and P. L. Carver, Building a Trojan Horse: Siderophore-Drug Conjugates for the Treatment of Infectious Diseases, In, Essential Metals in Medicine: Therapeutic Use and Toxicity of Metal Ions in the Clinic, De Gruyter, Berlin, Germany, 2019, vol. 19, pp. 331–358 Search PubMed.
- P. Loupias, P. Laumaillé, S. Morandat, L. Mondange, S. Guillier, K. El Kirat, S. Da Nascimento, F. Biot, N. Taudon, A. Dassonville-Klimpt and P. Sonnet, Eur. J. Med. Chem., 2023, 245, 114921 CrossRef CAS PubMed.
- J. Y. Wu, P. Srinivas and J. M. Pogue, Infect. Dis. Ther., 2020, 9, 17–40 CrossRef PubMed.
- J. Yao, J. Wang, M. Chen and Y. Cai, Front. Med., 2021, 8, 741940 CrossRef PubMed.
- A. E. Cotman, M. Durcik, D. Benedetto Tiz, F. Fulgheri, D. Secci, M. Sterle, Š. Možina, Ž. Skok, N. Zidar, A. Zega, J. Ilaš, L. Peterlin Mašič, T. Tomašič, D. Hughes, D. L. Huseby, S. Cao, L. Garoff, T. Berruga Fernández, P. Giachou, L. Crone, I. Simoff, R. Svensson, B. Birnir, S. V. Korol, Z. Jin, F. Vicente, M. C. Ramos, M. de la Cruz, B. Glinghammar, L. Lenhammar, S. R. Henderson, J. E. A. Mundy, A. Maxwell, C. E. M. Stevenson, D. M. Lawson, G. V. Janssen, G. J. Sterk and D. Kikelj, J. Med. Chem., 2023, 66, 1380–1425 Search PubMed.
- M. Durcik, A. E. Cotman, Ž. Toplak, Š. Možina, Ž. Skok, P. E. Szili, M. Czikkely, E. Maharramov, T. H. Vu, M. V. Piras, N. Zidar, J. Ilaš, A. Zega, J. Trontelj, L. A. Pardo, D. Hughes, D. Huseby, T. Berruga-Fernández, S. Cao, I. Simoff, R. Svensson, S. V. Korol, Z. Jin, F. Vicente, M. C. Ramos, J. E. A. Mundy, A. Maxwell, C. E. M. Stevenson, D. M. Lawson, B. Glinghammar, E. Sjöström, M. Bohlin, J. Oreskär, S. Alvér, G. V. Janssen, G. J. Sterk, D. Kikelj, C. Pal, T. Tomašič and L. Peterlin Mašič, J. Med. Chem., 2023, 66, 3968–3994 Search PubMed.
- C. Ji and M. J. Miller, Bioorg. Med. Chem., 2012, 20, 3828–3836 CrossRef CAS PubMed.
- W. Neumann, M. Sassone-Corsi, M. Raffatellu and E. M. Nolan, J. Am. Chem. Soc., 2018, 140, 5193–5201 CrossRef CAS PubMed.
- C. Ji and M. J. Miller, Biometals Int. J. Role Met. Ions Biol. Biochem. Med., 2015, 28, 541–551 CAS.
- M. E. Flanagan, S. J. Brickner, M. Lall, J. Casavant, L. Deschenes, S. M. Finegan, D. M. George, K. Granskog, J. R. Hardink, M. D. Huband, T. Hoang, L. Lamb, A. Marra, M. Mitton-Fry, J. P. Mueller, L. M. Mullins, M. C. Noe, J. P. O'Donnell, D. Pattavina, J. B. Penzien, B. P. Schuff, J. Sun, D. A. Whipple, J. Young and T. D. Gootz, ACS Med. Chem. Lett., 2011, 2, 385–390 CrossRef CAS PubMed.
- M. Durcik, Ž. Toplak, N. Zidar, J. Ilaš, A. Zega, D. Kikelj, L. P. Mašič and T. Tomašič, ACS Omega, 2020, 5, 8305–8311 CrossRef CAS PubMed.
- S. J. Hearnshaw, T. T.-H. Chung, C. E. M. Stevenson, A. Maxwell and D. M. Lawson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2015, 71, 996–1005 CrossRef CAS PubMed.
- S. Bellon, J. D. Parsons, Y. Wei, K. Hayakawa, L. L. Swenson, P. S. Charifson, J. A. Lippke, R. Aldape and C. H. Gross, Antimicrob. Agents Chemother., 2004, 48, 1856–1864 CrossRef CAS PubMed.
- T. Baba, T. Ara, M. Hasegawa, Y. Takai, Y. Okumura, M. Baba, K. A. Datsenko, M. Tomita, B. L. Wanner and H. Mori, Mol. Syst. Biol., 2006, 2, 2006 CrossRef PubMed.
- R. M. Humphries, J. Ambler, S. L. Mitchell, M. Castanheira, T. Dingle, J. A. Hindler, L. Koeth and K. Sei, J. Clin. Microbiol., 2018, 56, 019344 Search PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kalé and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- J. Huang, S. Rauscher, G. Nawrocki, T. Ran, M. Feig, B. L. de Groot, H. Grubmüller and A. D. MacKerell, Nat. Methods, 2017, 14, 71–73 CrossRef CAS PubMed.
- S. Jo, T. Kim, V. G. Iyer and W. Im, J. Comput. Chem., 2008, 29, 1859–1865 Search PubMed.
- J. Lee, X. Cheng, J. M. Swails, M. S. Yeom, P. K. Eastman, J. A. Lemkul, S. Wei, J. Buckner, J. C. Jeong, Y. Qi, S. Jo, V. S. Pande, D. A. Case, C. L. I. Brooks, A. D. MacKerell Jr, J. B. Klauda and W. Im, J. Chem. Theory Comput., 2016, 12, 405–413 CrossRef CAS PubMed.
- K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes, I. Vorobyov and A. D. Mackerell Jr., J. Comput. Chem., 2010, 31, 671–690 CrossRef CAS PubMed.
- K. Vanommeslaeghe and A. D. MacKerell, J. Chem. Inf. Model., 2012, 52, 3144–3154 CrossRef CAS PubMed.
- K. Vanommeslaeghe, E. P. Raman and A. D. MacKerell Jr, J. Chem. Inf. Model., 2012, 52, 3155–3168 CrossRef CAS PubMed.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- J.-P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- G. Wolber and T. Langer, J. Chem. Inf. Model., 2005, 45, 160–169 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Representative analytical data (NMR spectra, HRMS spectra, HPLC chromatograms). See DOI: https://doi.org/10.1039/d3ra08337c |
| This journal is © The Royal Society of Chemistry 2024 |