
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLithocholic acid derivatives as potent modulators of the nuclear receptor RORγt†
Somaya A. Abdel-Rahmanab,
Simone Brogi c and
Moustafa T. Gabr*a
c and
Moustafa T. Gabr*a
aDepartment of Radiology, Molecular Imaging Innovations Institute (MI3), Weill Cornell Medicine, New York, NY 10065, USA. E-mail: mog4005@med.cornell.edu
bDepartment of Medicinal Chemistry, Faculty of Pharmacy, Mansoura University, Mansoura 35516, Egypt
cDepartment of Pharmacy, University of Pisa, via Bonanno 6, 56126 Pisa, Italy
First published on 18th January 2024
Abstract
Retinoic acid receptor-related orphan receptor γt (RORγt) is a nuclear receptor found in various tissues that plays a crucial role in the differentiation and proliferation of T helper 17 (Th17) cells, as well as in their generation of the pro-inflammatory cytokine IL-17A. RORγt represents a promising therapeutic target for autoimmune diseases, metabolic disorders, and multiple tumors. Despite extensive research efforts focused on the development of small molecule RORγt modulators, no drug candidates have advanced to phase 3 clinical trials owing to a lack of efficacy or safety margin. This outcome highlights the unmet need to optimize small molecule drug candidates targeting RORγt to develop effective therapies for autoimmune and inflammatory diseases. In this study, we synthesized and evaluated 3-oxo-lithocholic acid amidates as a new class of RORγt modulators. Our evaluation entailed biophysical screening, cellular screening in different platforms, molecular docking, and in vitro pharmacokinetic profiling. The top compound from our study (3-oxo-lithocholic acid amidate, A2) binds to RORγt at an equilibrium dissociation constant (KD) of 16.5 ± 1.34 nM based on microscale thermophoresis (MST). Assessment of the efficacy of A2 in the cellular RORγt reporter luciferase assay revealed a half-maximal inhibitory concentration (IC50) value of 225 ± 10.4 nM. Unlike 3-oxo-lithocholic acid, A2 demonstrated the ability to reduce the IL-17A mRNA expression levels in EL4 cells with RORγt expression using quantitative reverse transcriptase PCR (RT-PCR). Validation of the desirable physicochemical properties and stability of A2 sets the stage for the preclinical evaluation of this new class of RORγt modulators in animal models of autoimmune diseases.
Introduction
The retinoic acid receptor-related orphan receptor γ T (RORγt) has a pivotal function in regulating the human immune system.1–3 The essential role of RORγt in promoting the differentiation of T helper 17 (Th17) cells has triggered enormous research efforts focused on the development of RORγt inhibitors.4–6 The production of interleukin-17 (IL-17) by Th17 cells is a key contributor to the development of autoimmune diseases,7 including conditions such as psoriasis,8 multiple sclerosis,9 and inflammatory bowel disease (IBD).10–12 An effective therapeutic approach involves interfering with the Th17/IL-17 pathway through the use of IL-17 monoclonal antibodies (mAbs). In this context, the clinical success of monoclonal antibodies (mAbs) targeting interleukin 17A (IL-17A) (e.g., secukinumab and ixekizumab) has validated modulation of Th17 cell differentiation for the treatment of autoimmune diseases.13–16 However, the introduction of these therapeutics has featured a lack of efficacy in a subset of patients, intolerance and/or loss of initial response, and life-threatening side effects.17–20 Multiple research groups have led extensive research efforts in the past decade to identify small molecule RORγt modulators.21–33 However, there are no small molecules that advanced to phase 3 clinical trials owing to lack of efficacy or safety margin.34 This highlights the unmet need to identify alternative small molecule RORγt inhibitors as potential therapies for autoimmune and inflammatory diseases.RORγt harbors a hydrophobic pocket for ligand binding situated within its ligand binding domain (LBD), a region that exhibits high conservation across the nuclear receptor family.35 Despite this, the receptor's transcriptional activity does not rely on ligand binding, as the apo protein maintains the C-terminal helix 12 (H12) in a conformation conducive to partial coactivator protein recruitment.36,37 Although classified as an orphan receptor without confirmed endogenous ligands, RORγt responds to the binding of naturally occurring cholesterol derivatives. Hydroxycholesterols specifically, act as effective agonists, stabilizing H12 and facilitating coactivator binding.38 In contrast, digoxin functions as an inverse agonist, stabilizing H12 in a conformation unsuitable for coactivator binding but favorable for corepressor binding, resulting in reduced gene transcription.39 In addition, various synthetic inverse agonists, including T0901317 (1, Fig. 1),40 target the same orthosteric ligand binding pocket in RORγt. The identification of a novel allosteric binding site within the RORγt LBD by potent RORγt inverse agonists MRL-871 (2, Fig. 1)41 and subsequently 3 (Fig. 1)42 is based on the ability of these ligands to engage directly with the activation function loop situated between H11 and H12 (AF-2 domain), inducing an unconventional conformation in H12. This altered conformation effectively hinders the recruitment of coactivators. Notably, reports of ligands unequivocally directed towards the allosteric pocket of RORγt are confined to compounds characterized by closely related chemotypes featuring indazole or imidazopyridine core.43
 | ||
| Fig. 1 Chemical structures of reported RORγt modulators. | ||
Numerous microbial metabolites serve as signaling molecules within the immune system and play a pivotal role in regulating immune homeostasis.44 In this context, secondary bile acids have been identified as ligands for nuclear hormone receptors that modulate the differentiation of Th17 cells.45,46 Specifically, 3-oxo-lithocholic acid (3-oxoLCA, 4, Fig. 1) and isolithocholic acid (isoLCA, 5, Fig. 1) have demonstrated an inhibitory effect on Th17 cell differentiation, which is significant in the context of autoimmune disorders.45,46 Importantly, structural analogs of lithocholic acid have proven ineffective in mimicking the impact observed with 3-oxoLCA and isoLCA on Th17 cells.45,46 While these findings have shed light on the role of bile acids in Th17 cell differentiation, questions persist, particularly regarding the underlying mechanisms responsible for the differential effects of structurally distinct bile acids on Th17 cell modulation.
Given the growing recognition of how immunomodulatory bile acid metabolites influence inflammatory conditions in humans, a more profound understanding of the structural characteristics that govern their immunomodulatory impact could pave the way for the development of therapeutic treatments targeting autoimmune and inflammatory diseases. Recent research has uncovered the ability of gut microbes in humans to conjugate bile acids with phenylalanine and tyrosine.47 Building upon this discovery, we proposed a hypothesis suggesting that these bile acids, modified by gut microbes, belong to a larger group of compounds known as bile acid amidates, which include a wide range of structurally diverse biogenic amines. Thus, we hypothesized that the conjugation of bile acids to structurally diverse scaffolds would result in compounds with improved binding profiles to the corresponding targets of the parent bile acids. Herein, we introduce the potential of the 3-oxoLCA scaffold as a RORγt ligand by evaluating a small library of compounds based on the conjugation of 3-oxoLCA to a variety of structurally diverse amines. We synthesized a series of 3-oxoLCA derivatives and their corresponding lithocholic acid (LCA) analogs as control compounds and evaluated the compounds as RORγt ligands.
Results and discussion
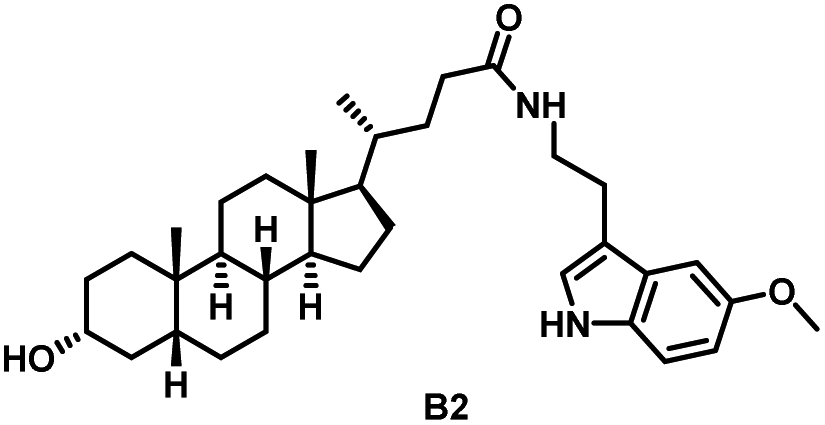
Initially, we conjugated the 3-oxoLCA and LCA cores to structurally diverse amines based on amide coupling chemistry (Scheme 1) using hexafluorophosphate benzotriazole tetramethyl uranium (HBTU) and triethylamine (TEA) in dimethylformamide (DMF). We selected these structurally diverse amines to introduce both hydrophobic and hydrogen bonding interactions into the proposed binding profiles of 3-oxoLCA and LCA to RORγt. We confirmed the identity of the synthesized bile acid amidates (A2–A6 and B2–B6, Table 1) using nuclear magnetic resonance (NMR) and high-resolution mass spectrometry (HRMS). In addition, we determined the purity by high-performance liquid chromatography (HPLC) to be ≥95%. | ||
| Scheme 1 Synthesis of bile acid amidates via amide coupling. | ||
| Compound | MST KD (nM) for RORγt |
|---|---|
 |
1083 ± 93.4 |
 |
16.5 ± 1.34 |
 |
57.2 ± 3.01 |
 |
87.4 ± 5.52 |
 |
724 ± 20.4 |
 |
618 ± 19.3 |
 |
28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 534 ± 304 534 ± 304 |
 |
9361 ± 131 |
 |
12414 ± 267 |
 |
15789 ± 334 |
 |
27103 ± 218 |
 |
24868 ± 591 |
Given that 3-oxoLCA (A1, Table 1) binds RORγt at an equilibrium dissociation constant (KD) of ∼1.1 μM,45 we analyzed the binding affinity of the synthesized bile acid amidates (A2–A6 and B2–B6) to RORγt using microscale thermophoresis (MST). We performed the assay by labeling recombinant human RORγt with the Monolith NT Protein Labeling Kit RED (NanoTemper Technologies). In our hands, 3-oxoLCA (A1) demonstrated a KD value of ∼1.08 ± 0.09 μM for RORγt binding based on MST analysis (Table 1), which is in close agreement with the reported binding affinity.45 Compounds A2–A6, based on the 3-oxoLCA scaffold, displayed submicromolar binding affinities to RORγt according to MST analysis. Specifically, compound A2 bearing a methoxy-substituted tryptamine moiety revealed the maximum enhancement in the RORγt binding affinity in comparison to 3-oxoLCA (A1) with ∼65-fold enhancement (KD value of 16.5 ± 1.34 nM, Fig. 2 and Table 1). Replacing the tryptamine scaffold in A2 with a 2-benzhydryl moiety in A3 resulted in a slight reduction in the RORγt binding affinity, indicating the loss of potential hydrogen bonding interaction in A2 with RORγt (KD = 16.5 ± 1.34 nM and 57.2 ± 3.01 nM for A2 and A3, respectively, Table 1). As illustrated in Table 1, incorporation of the biphenyl ethyl fragment into the 3-oxoLCA scaffold in A4 resulted in a remarkable enhancement in RORγt binding affinity compared with benzyl substituents in A5 and A6. This outcome indicates the significance of the elongated linker and the extended hydrophobic surface in A4 compared with those in A5 and A6 in terms of maximizing the RORγt binding affinity. Notably, we observed a similar trend regarding the outcome of the investigated moieties in modulating the RORγt binding affinity of LCA (B1), as evident from the RORγt binding affinities of B2–B6 (Table 1). However, none of the synthesized LCA derivatives (B2–B6) possessed submicromolar RORγt binding affinity according to MST analysis (Table 1). This outcome is in agreement with the reported superiority of 3-oxoLCA to LCA in RORγt binding and modulating Th17-cell proliferation.45 Thus, we conclude that further structural modifications of the 3-oxoLCA derivatives (A2–A6) rather than LCA derivatives (B2–B6) have the potential to result in the identification of novel RORγt modulators with single-digit nanomolar or subnanomolar binding affinity.
 | ||
| Fig. 2 Dose–response curve of compound A2 binding to RORγt LBD using MST. Error bars represent standard deviation (n = 3). | ||
Molecular docking studies were conducted to investigate the binding mode of the synthesized compounds and to explore the features responsible for the reduction in affinity of compounds B1–B6 with respect to compounds A1–A6 for the RORγt LBD. These two series differ from the presence of a carbonyl function in compounds A1–A6 in place of the hydroxyl function in compounds B1–B6 series. This difference is responsible for a decrease in the affinity of the B-compound series compared with that of the A-compound series. For this purpose, we coupled the molecular docking studies with the evaluation of ΔGbind, as previously described by us,48,49 because in this way, it is possible to capture the small structural differences that influenced the binding affinity. In general, the docking scores and the ΔGbind are significantly different between the two series, indicating that the presence of hydroxyl function (compounds B1–B6) is detrimental to the affinity of this series of compounds. In contrast, the presence of carbonyl function, as in compounds A1–A6, was well tolerated by the receptor, and the computational scores were favorable with respect to compounds B1–B6 (Table 2). Notably, the docking scores and the ΔGbind of compounds A1–A6 are comparable to those of the reference compound digoxin, which is a well-known antagonist of the RORγt receptor.
| Compound | GlideScore (kcal mol−1) | ΔGbind (kcal mol−1) |
|---|---|---|
| A1 | −9.165 | −88.91 |
| A2 | −10.711 | −125.38 |
| A3 | −11.683 | −121.47 |
| A4 | −10.807 | −131.75 |
| A5 | −10.898 | −114.26 |
| A6 | −10.942 | −115.89 |
| B1 | −7.288 | −79.24 |
| B2 | −9.107 | −97.73 |
| B3 | −8.371 | −93.22 |
| B4 | −9.386 | −102.15 |
| B5 | −9.477 | −91.83 |
| B6 | −10.088 | −94.71 |
| Digoxin | −12.278 | −123.69 |
In particular, as depicted in Fig. 3, compounds A1–A6 showed relevant interactions with the RORγt LBD. Starting from compound A1 (Fig. 3A), the steroid-like core is located in the hydrophobic pocket composed of the following amino acids: Leu287, Leu324, Ala327, Val361, Phe377, and Phe388, whereas the carbonyl group established an H-bond with Arg367. The introduction of pendant substituents containing aromatic functions, as in compounds A2–A6, allowed the compounds to target further residents with respect to A1. This is reflected by a dramatic increase in affinity, as observed in biophysical screening and indicated by computational scores. In fact, compounds A2–A6 were able to target, in addition to the previously mentioned residues, Trp317, Leu391, and His479 by H-bonds and/or π–π stacking and hydrophobic interactions. The carbonyl function in compounds A2–A6 preferably established an H-bond with the backbone of residue Leu287. The increase in the number of contacts of compounds A2–A6, along with the calculation of the relative binding affinity, is in good agreement with the submicromolar affinity found for these compounds for the RORγt receptor.
 | ||
| Fig. 3 Putative binding mode of compounds A1–A6 (panels A–F, respectively) into RORγt LBD (PDB ID: 3B0W). Key residues of the binding site are represented by lines and labeled. H-bonds and π–π stacking interactions are illustrated as yellow and cyan dotted lines, respectively. Pictures were generated by Maestro (Maestro, Schrödinger LLC, release 2020-3). | ||
In contrast, the docking output for compounds B1–B6 (Fig. 4) indicated that the introduction of the hydroxyl function is deleterious for their binding mode. In fact, none of the compounds could fruitfully bind to the binding site of the RORγt receptor. We observed a dramatic decrease in the number and quality of contacts with respect to compounds A1–A6. This is reflected in a significant decrease in the docking scores and the ΔGbind, indicating a decrease in affinity, as found by the MST binding analysis. In particular, the pendant substituents were no longer able to strongly target the residues Trp317 and His479, except for the biphenyl-containing compound B3, which established a π–π stacking with Trp317. The hydroxyl function could not target Leu287 or Arg367 (with exclusion of B6), whereas it targeted the side chain of Gln286 by an H-bond. This indicated that compounds B1–B6 could not penetrate deeply into the selected binding site. The significant reduction in the number of contacts along with unfavorable conformational energies, docking scores, and an increase of ΔGbind for the compounds B1–B6 culminated in a drastic reduction in the affinity for RORγt receptor in the high micromolar range, highlighting that a small structural difference could have a significant effect on binding interactions.
 | ||
| Fig. 4 Putative binding mode of compounds B1–B6 (panels A–F, respectively) into RORγt ligand binding domain (PDB ID: 3B0W). Key residues of the binding site are represented by lines and labeled. H-bonds and π–π stacking interactions are illustrated as yellow and cyan dotted lines, respectively. Pictures were generated by Maestro (Maestro, Schrödinger LLC, release 2020-3). | ||
In order to evaluate the in vitro RORγt cellular binding of the synthesized compounds, we implemented the established GAL4-RORγ reporter assay based on proprietary human cells engineered to provide high-level expression of a hybrid human RORγ. The reporter cells incorporate cDNA encoding beetle luciferase, which catalyzes the oxidation of D-luciferin, resulting in photon emission. In the reporter assay, the N-terminal DNA-binding domain (DBD) of the native RORγ receptor has been replaced with the DBD of the yeast GAL4.50,51 Ligands of RORγ (antagonists or inverse agonists) would reduce the affinity of GAL4-RORγ to the DNA site and minimize the luminescence signal. Initially, we screened A1–A6 and B1–B6 in the RORγ reporter assay at a single concentration of 1 μM using ML-209 and digoxin as positive control compounds. As shown in Fig. 5A, the outcome of the RORγ reporter assay was in agreement with that of our RORγt binding analysis using MST with compounds A1–A6, revealing a superior reduction in luminescence of reporter cells to compounds B1–B6, indicating an efficient inhibition of the binding of GAL4-RORγ to the DNA site. Among A1–A6 and B1–B6, compound A2 demonstrated the most pronounced reduction in the luminescence signal in single-dose screening (Fig. 5A). Unlike compounds A5 and A6, a potent decrease in the luminescence from the reporter cells was observed for A3 and A4 in single-dose screening (Fig. 5A). However, B1–B6 failed to induce a significant decrease in luminescence of reporter cells in single-dose screening (Fig. 5A). Dose-dependent screening of the most potent compound from our study (A2) using the RORγ reporter assay revealed a half-maximal inhibitory concentration (IC50) of 225 ± 10.4 nM (Fig. 5B). It is noteworthy to mention that the positive control (ML-209) possessed an IC50 value of 127.9 ± 18.3 nM in the RORγ reporter assay (Fig. S1, ESI†).
 | ||
| Fig. 5 (A) Single-dose screening of A1–A6 and B1–B6 at 1 μM (n = 3) in the RORγt reporter luciferase assay using ML-209 and digoxin (1 μM, n = 3) as positive controls. RLU stands for relative luminescence units from the reporter assay. *p < 0.05, **p < 0.01, ***p < 0.001, and ns denotes non-significant in comparison to vehicle control. (B) Dose–response curve of compound A2 in the RORγt reporter luciferase assay. Error bars represent standard deviation (n = 3). | ||
We further evaluated the cellular activity of the compounds by assessing the reduction of IL-17A mRNA expression levels using quantitative reverse transcriptase PCR (RT-PCR) in EL4 cells (a murine lymphoblast cell line with validated RORγt expression). Treatment of EL4 cells with A1 (3-oxoLCA), A2, and A3 at 10 μM tested concentration for 24 h revealed a potent reduction in IL-17A expression by both A2 and A3 (Fig. 6). In contrast, 3-oxoLCA (A1) induced a minimal change in IL-17A expression in EL4 cells (Fig. 6). These findings indicate the potential of the synthesized bile acid amidates as RORγt modulators compared with the parent bile acids. Notably, bile acid derivatives exhibit high cross-reactivity toward RORα and RORγt with high affinities.52 Interestingly, A2 revealed >100-fold selectivity for binding RORγt over RORα according to MST binding analysis (Fig. S2, ESI†). On the other hand, 3-oxoLCA (A1) possessed ∼2-fold selectivity for RORγt binding over RORα (Fig. S3, ESI†). The remarkable enhancement in selective RORγt binding for our new class of bile acid amidates highlights the significance of our work.
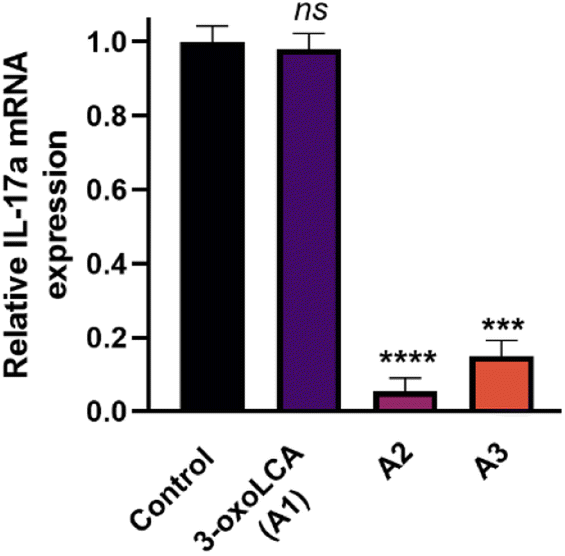 | ||
| Fig. 6 IL-17A mRNA expression in EL4 cells incubated with a single dose (10 μM, n = 3) of 3-oxoLCA (A1), A2, A3, or vehicle control (DMSO). The level of IL-17A expression was normalized to that of GAPDH expression. ***p < 0.001, ****p < 0.0001, and ns denotes non-significant in comparison to vehicle control. Error bars represent standard deviation (n = 3). | ||
In order to assess the potential of the developed compounds for preclinical evaluation, we assessed the key physicochemical properties and stability of the three best performing compounds from this study (A2, A3, and A4). As shown in Table 3, compound A2 exhibited remarkable enhancement in aqueous solubility in the protonated state compared with A3 and A4. The three compounds studied (A2, A3, and A4) possessed favorable profiles as potential drug candidates in terms of lipophilicity, human serum albumin (HSA) binding, stability in simulated intestinal fluid, and stability upon incubation with human plasma (Table 3). This likely indicates that further optimization of this class of 3-oxoLCA amidates can result in candidate molecules with desirable in vivo efficacy in models of autoimmune and inflammatory diseases.
| Compound | Solubilitya (μM) | logPb |
HSA bindingc | Chemical stabilityd (% remaining) | Plasma stabilitye (% remaining) |
|---|---|---|---|---|---|
| a Water solubility of the compounds as protonated species.b 1-Octanol–water partition coefficient.c Albumin binding.d Stability in simulated intestinal fluid (2 h incubation).e Stability in human plasma (2 h incubation). | |||||
| A2 | 72 | 2.93 | 81 | 98.1 | 95.7 |
| A3 | 12 | 3.87 | 94 | 99.3 | 97.4 |
| A4 | 9 | 3.96 | 96 | 99.1 | 96.9 |
Conclusions
In summary, we have introduced 3-oxoLCA amidates as potent modulators of RORγt. In comparison to 3-oxoLCA (A1), the top compounds from this study (A2 and A3) possessed nanomolar binding affinity to RORγt in biophysical screening, exhibited functional activity in the RORγt reporter assay at submicromolar concentrations, and inhibited the IL-17A mRNA expression in EL4 cells. We conducted a computational study to rationalize the superiority of A1–A6 compounds to B1–B6 compounds as RORγt ligands. Moreover, we verified the desirable physicochemical properties and stability of the most potent RORγt modulators. We anticipate that this study will trigger future research aiming to realize the potential of 3-oxoLCA amidates as RORγt modulators in animal models of autoimmune diseases.Experimental
General
All commercially available starting materials, reagents, and solvents were used as supplied unless otherwise stated. The reported yields are isolated yields. Proton (1H) and carbon (13C) NMR were collected on Bruker NMR spectrometers at 400 MHz for 1H and 100 MHz for 13C. Chemical shifts (δ) are reported in parts-per million (ppm) relative to the residual undeuterated solvent. High resolution mass spectra were obtained in the positive ion mode using electrospray ionization (ESI) on a double-focusing magnetic sector mass spectrometer.General method for the synthesis of A2–A6 and B2–B6
2 mmol (1 eq.) 3-oxoLCA (A1) or LCA (B1) was added in a round bottom flask in N2 atmosphere, and 5 mL of DMF. The corresponding primary amine (1.1 eq.), HBTU (1.1 eq.) and triethyl amine (TEA, 1.2 eq.) were then added. The reaction mixture was allowed to stir overnight at room temperature. Subsequently, the reaction mixture was diluted with ethyl acetate (75 mL), washed with 5% HCl, 10% Na2CO3 and brine, and dried over anhydrous Na2SO4. The organic layer was concentrated under reduced pressure to give the crude product, which was further purified by flash column chromatography using a gradient of hexane and ethyl acetate as the solvent system.13C NMR (DMSO-d6, δ ppm): 12.32, 18.76, 21.22, 22.68, 22.70, 24.24, 25.74, 26.69, 28.17, 31.17, 32.07, 32.90, 34.91, 35.35, 35.52, 36.92, 37.18, 42.33, 42.73, 43.91, 56.08, 56.16, 126.94, 127.01, 127.66, 129.73, 129.74, 138.41, 139.34, 140.52, 172.94, 212.33. HRMS (ESI): calculated for C38H51NO2 (M + H)+, 554.3993; found, 554.3980.
MST assay
For MST screening, we used Monolith NT.115 instrument from Nanotemper to assess the compounds/RORγt interaction. We procured recombinant human RORγt from WuXi and labeled it with Protein Labeling Kit RED-NHS 2nd Generation from Nanotemper (Cat #MO-L011). RORγt was produced by recombinant microbial expression from E. coli cells (BL21-CodonPlus (DE3)-RIL Competent Cells, Cat #230245 from AGILENT). We dissolved RORγt in PBS buffer (pH 7.4) with 0.1% bovine serum albumin (BSA) and 0.05% Tween 20. We kept the concentration of the fluorescently labeled RORγt constant at 25 nM. We added a volume of 5 μL of the corresponding samples in MST capillaries with a final DMSO concentration of 2%. Subsequently, we incubated the samples within the capillaries for 20 min at room temperature prior to the measurements. We detected changes in thermophoretic properties as a change in fluorescence intensity upon incubation of various concentrations of the tested compounds with fluorescently labeled RORγt. We plotted the thermophoresis signal against the compound concentration to obtain a dose–response curves, from which KD values can be deduced. MST data are represented as normalized change in fluorescence (Fnorm) upon ligand binding. Fnorm is the ratio of the fluorescence measured before and during thermophoresis.Computational details
RORγ reporter assay
We procured the RORγ reporter assay kit (Cat#IB04001) from INDIGO Biosciences (State College, PA, USA) to assess the inhibitory activity of the compounds in comparison to DMSO vehicle and control compounds. We performed the assay following the manufacturer's recommended protocol. Briefly, we dispensed 200 μL of reporter cells per well in 96-well plates and pre-incubated the cells for 6 h. We prepared samples of the tested compounds (n = 3) at 1 μM in single-dose screening or at multiple concentrations in dose-dependent screening in treatment media in the presence of 0.4% DMSO. After discarding the culture media post pre-incubation, 200 μL per well of the prepared treatment media was added. Following a 24 h incubation, treatment media were removed, and luciferase detection reagents were introduced to each well. The intensity of light emission, measured in relative light units (RLUs), from each well was quantified using a plate-reading luminometer. This assay was performed in a single run (n = 3). The statistical significance levels were denoted as follows: *p < 0.05, **p < 0.01, ***p < 0.001, and ‘ns’ indicating non-significance in comparison to the vehicle control.Quantitative IL-17A mRNA RT-PCR assay
This assay was conducted using EL4 cells (Sigma-Aldrich), cultured in DMEM (Gibco) with 10% FBS. After seeding the cells onto a 12-well plate, they were incubated in the presence of 10 μM compound (n = 3, prepared from DMSO stock solutions) or DMSO (vehicle control) for 24 h. Subsequently, the cells were activated with phorbol 12-myristate 13-acetate (PMA, 50 ng mL−1; Sigma-Aldrich) and ionomycin (1 μg mL−1; Sigma-Aldrich) for 5 h. Following activation, the cells were collected, and RNA was isolated using a RNeasy Mini Kit (Qiagen). Reverse transcription was performed using the iScript Advanced cDNA Synthesis Kit (Bio-Rad). Quantitative RT-PCR was carried out to assess mRNA levels of mouse IL-17A (n = 3) utilizing SYBR green technology (Bio-Rad) on a CFX Real-Time System (Bio-Rad). Normalization of IL-17A mRNA expression to Gapdh expression was performed, and the relative gene expression was calculated using the 2−ΔΔCt (Livak) method with the DMSO control as the calibrator. This assay was performed in a single run (n = 3). The statistical significance levels were denoted as follows: ***p < 0.001, ****p < 0.0001, and ‘ns’ indicating non-significance in comparison to the vehicle control.Water solubility
The tested compounds were suspended in 100 mL of 0.1 M HCl (pH 1.00) and the saturated solutions were transferred to a thermostat-equipped water bath (25 °C). Following the incubation time (48 h), we filtered the solutions and determined the concentration of the compounds by HPLC-ES-MS/MS.Lipophilicity (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) P)
P)
We evaluated the 1-octanol/water partition coefficient using the conventional shake-flask method. We performed the experiment at 1 mM solution of the tested compound buffered at pH 7.4. We measured the concentration of the tested compound in the water phase before and after partitioning in 1-octanol using HPLC-ES-MS/MS.
Albumin binding
Using equilibrium dialysis at a fixed compound-albumin ratio, we assessed the albumin binding profiles of the tested compounds. Initially, the compounds (100 μM) in 5% bovine serum albumin-saline solution were incubated for 24 h at 25 °C. Subsequently, the solution was dialyzed in cellulose sacs with a molecular weight cutoff of 12–14 kDa (Spectra/Por, Spectrum Medical Industries Inc., CA, USA) against 25 mL of saline solution. We equilibrated the system using mechanical shaking for 72 h at 25 °C. We assessed the concentrations of the tested compounds in the starting solution and the dialyzed solution using HPLC-ES-MS/MS.Stability in simulated fluids and human plasma
We assessed the stability of the compounds in simulated intestinal fluid (using erythromycin as a reference compound, t1/2, min = 478 min) and in human plasma (using propantheline as a reference compound, t1/2, min = 2.3 min) according to our established experimental protocols.58Conflicts of interest
There are no conflicts to declare.Notes and references
- A. M. Jetten, Y. Takeda, A. Slominski and H. S. Kang, Retinoic acid-related Orphan Receptor γ (RORγ): connecting sterol metabolism to regulation of the immune system and autoimmune disease, Curr. Opin. Toxicol., 2018, 8, 66–80 CrossRef PubMed.
- G. Eberl, RORγt, a multitask nuclear receptor at mucosal surfaces, Mucosal Immunol., 2017, 10, 27–34 CrossRef CAS PubMed.
- I. I. Ivanov, B. S. McKenzie, L. Zhou, C. E. Tadokoro, A. Lepelley, J. J. Lafaille, D. J. Cua and D. R. Littman, The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells, Cell, 2006, 126, 1121–1133 Search PubMed.
- N. Manel, D. Unutmaz and D. R. Littman, The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat, Nat. Immunol., 2008, 9, 641–649 CrossRef CAS PubMed.
- X. O. Yang, B. P. Pappu, R. Nurieva, A. Akimzhanov, H. S. Kang, Y. Chung, L. Ma, B. Shah, A. D. Panopoulos, K. S. Schluns, S. S. Watowich, Q. Tian, A. M. Jetten and C. Dong, T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma, Immunity, 2008, 28, 29–39 CrossRef CAS PubMed.
- T. Y. Park, S. D. Park, J. Y. Cho, J. S. Moon, N. Y. Kim, K. Park, R. H. Seong, S. W. Lee, T. Morio, A. L. Bothwell and S. K. Lee, RORγt-specific transcriptional interatomic inhibition suppresses autoimmunity associated with TH17 cells, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 18673–18678 CrossRef CAS PubMed.
- J. F. Zambrano-Zaragoza, E. J. Romo-Martínez, J. Durán-Avelar Mde, N. García-Magallanes and N. Vibanco-Pérez, Th17 cells in autoimmune and infectious diseases, Int. J. Inflammation, 2014, 2014, 651503 Search PubMed.
- M. Mosca, J. Hong, E. Hadeler, M. Hakimi, W. Liao and T. Bhutani, The Role of IL-17 Cytokines in Psoriasis, ImmunoTargets Ther., 2021, 10, 409–418 CrossRef CAS PubMed.
- T. Moser, K. Akgün, U. Proschmann, J. Sellner and T. Ziemssen, The role of TH17 cells in multiple sclerosis: Therapeutic implications, Autoimmun. Rev., 2020, 19, 102647 CrossRef CAS PubMed.
- L. Chen, G. Ruan, Y. Cheng, A. Yi, D. Chen and Y. Wei, The role of Th17 cells in inflammatory bowel disease and the research progress, Front. Immunol., 2023, 13, 1055914 CrossRef PubMed.
- J. Zhao, Q. Lu, Y. Liu, Z. Shi, L. Hu, Z. Zeng, Y. Tu, Z. Xiao and Q. Xu, Th17 Cells in Inflammatory Bowel Disease: Cytokines, Plasticity, and Therapies, J. Immunol. Res., 2021, 2021, 8816041 Search PubMed.
- J. Gálvez, Role of Th17 Cells in the Pathogenesis of Human IBD, ISRN Inflammation, 2014, 2014, 928461 CrossRef PubMed.
- J. G. Krueger, K. A. Wharton, T. Schlitt, M. Suprun, R. I. Torene, X. Jiang, C. Q. Wang, J. Fuentes-Duculan, N. Hartmann, T. Peters, I. Koroleva, R. Hillenbrand, M. Letzkus, X. Yu, Y. Li, A. Glueck, A. Hasselberg, B. Flannery, M. Suárez-Fariñas and W. Hueber, IL-17A inhibition by secukinumab induces early clinical, histopathologic, and molecular resolution of psoriasis, J. Allergy Clin. Immunol., 2019, 144, 750–763 CrossRef CAS PubMed.
- J. Frieder, D. Kivelevitch and A. Menter, Secukinumab: a review of the anti-IL-17A biologic for the treatment of psoriasis, Ther. Adv. Chronic Dis., 2018, 9, 5–21 Search PubMed.
- S. Darabian, M. Badii, J. P. Dutz and J. Chan, A Retrospective Study on the Effectiveness of Ixekizumab After Treatment With Secukinumab for Patients With Active Psoriatic Arthritis, J. Psoriasis Psoriatic Arthritis, 2021, 7, 13–16 CrossRef.
- C. Paul, Ixekizumab or secukinumab in psoriasis: what difference does it make?, Br. J. Dermatol., 2018, 178, 1003–1005 CrossRef CAS PubMed.
- J. F. Schwensen, A. Clemmensen, C. Sand, R. Gniadecki, L. Skov, C. Zachariae, L. Iversen, M. Rasmussen and S. F. Thomsen, Effectiveness and safety of secukinumab in 69 patients with moderate to severe plaque psoriasis: a retrospective multicenter study, Dermatol. Ther., 2017, 30, e12550 CrossRef PubMed.
- J. Berman, V. Furer, M. Berman, O. Isakov, D. Zisman, A. Haddad and O. Elkayam, Treatment with ixekizumab following secukinumab failure in patients with psoriatic arthritis: real-life experience from a resistant population, Biologics, 2021, 15, 463–470 Search PubMed.
- C. Cariti, P. Dapavo, L. Mastorino, M. Ortoncelli, N. Siliquini, M. Merli, G. Avallone, S. Giordano, R. Fabrizio, S. Susca, A. Verrone, E. Stroppiana, P. Quaglino and S. Ribero, Comparison of Secukinumab and Ixekizumab in psoriasis: a real-life cohort study on the efficacy and drug survival of 445 patients, J. Eur. Acad. Dermatol. Venereol., 2022, 36, e233–e235 CrossRef CAS PubMed.
- Y. Y. M. Huang, J. S. Ruth and S. Hsu, Loss of efficacy of secukinumab for psoriasis at 24 to 32 weeks, J. Am. Acad. Dermatol., 2016, 75, e169 CrossRef PubMed.
- A. Fouda, S. Negi, O. Zaremba, R. S. Gaidar, Y. S. Moroz, E. Rusanov, S. Paraskevas and J. Tchervenkov, Discovery, Synthesis, and In Vitro Characterization of 2,3 Derivatives of 4,5,6,7-Tetrahydro-Benzothiophene as Potent Modulators of Retinoic Acid Receptor-Related Orphan Receptor γt, J. Med. Chem., 2023, 66, 7355–7373 CrossRef CAS PubMed.
- F. A. Meijer, G. J. M. Oerlemans and L. Brunsveld, Orthosteric and Allosteric Dual Targeting of the Nuclear Receptor RORγt with a Bitopic Ligand, ACS Chem. Biol., 2021, 16, 510–519 CrossRef CAS PubMed.
- F. A. Meijer, R. G. Doveston, R. M. de Vries, G. M. Vos, A. A. Vos, S. Leysen, M. Scheepstra, C. Ottmann, L. G. Milroy and L. Brunsveld, Ligand-Based Design of Allosteric Retinoic Acid Receptor-Related Orphan Receptor γt (RORγt) Inverse Agonists, J. Med. Chem., 2020, 63, 241–259 CrossRef CAS PubMed.
- B. P. Fauber and S. Magnuson, Modulators of the nuclear receptor retinoic acid receptor-related orphan receptor-γ (RORγ or RORc), J. Med. Chem., 2014, 57, 5871–5892 CrossRef CAS PubMed.
- S. M. Bronner, J. R. Zbieg and J. J. Crawford, RORγ antagonists and inverse agonists: a patent review, Expert Opin. Ther. Pat., 2017, 27, 101–112 Search PubMed.
- P. Cyr, S. M. Bronner and J. J. Crawford, Recent progress on nuclear receptor RORγ modulators, Bioorg. Med. Chem. Lett., 2016, 26, 4387–4393 CrossRef CAS PubMed.
- J. R. Huh, E. E. Englund, H. Wang, R. Huang, P. Huang, F. Rastinejad, J. Inglese, C. P. Austin, R. L. Johnson, W. Huang and D. R. Littman, Identification of Potent and Selective Diphenylpropanamide RORγ Inhibitors, ACS Med. Chem. Lett., 2013, 4, 79–84 CrossRef CAS PubMed.
- M. Kotoku, T. Maeba, S. Fujioka, M. Yokota, N. Seki, K. Ito, Y. Suwa, T. Ikenogami, K. Hirata, Y. Hase, Y. Katsuda, N. Miyagawa, K. Arita, K. Asahina, M. Noguchi, A. Nomura, S. Doi, T. Adachi, P. Crowe, H. Tao, S. Thacher, H. Hashimoto, T. Suzuki and M. Shiozaki, Discovery of Second Generation RORγ Inhibitors Composed of an Azole Scaffold, J. Med. Chem., 2019, 62, 2837–2842 CrossRef CAS PubMed.
- J. J. Duan, Z. Lu, B. Jiang, S. Stachura, C. A. Weigelt, J. S. Sack, J. Khan, M. Ruzanov, M. A. Galella, D. R. Wu, M. Yarde, D. R. Shen, D. J. Shuster, V. Borowski, J. H. Xie, L. Zhang, S. Vanteru, A. K. Gupta, A. Mathur, Q. Zhao, W. Foster, L. M. Salter-Cid, P. H. Carter and T. G. Dhar, Structure-based Discovery of Phenyl (3-Phenylpyrrolidin-3-yl)sulfones as Selective, Orally Active RORγt Inverse Agonists, ACS Med. Chem. Lett., 2019, 10, 367–373 CrossRef CAS PubMed.
- V. B. Pandya, S. Kumar, Sachchidanand, R. Sharma and R. C. Desai, Combating Autoimmune Diseases With Retinoic Acid Receptor-Related Orphan Receptor-γ (RORγ or RORc) Inhibitors: Hits and Misses, J. Med. Chem., 2018, 61, 10976–10995 CrossRef CAS PubMed.
- J. K. Barbay, M. D. Cummings, M. Abad, G. Castro, K. D. Kreutter, D. A. Kummer, U. Maharoof, C. Milligan, R. Nishimura, J. Pierce, C. Schalk-Hihi, J. Spurlino, V. M. Tanis, M. Urbanski, H. Venkatesan, A. Wang, C. Woods, R. Wolin, X. Xue, J. P. Edwards, A. M. Fourie and K. Leonard, 6-Substituted quinolines as RORγt inverse agonists, Bioorg. Med. Chem. Lett., 2017, 27, 5277–5283 Search PubMed.
- C. Imura, A. Ueyama, Y. Sasaki, M. Shimizu, Y. Furue, N. Tai, K. Tsujii, K. Katayama, T. Okuno, M. Shichijo, K. Yasui and M. Yamamoto, A novel RORγt inhibitor is a potential therapeutic agent for the topical treatment of psoriasis with low risk of thymic aberrations, J. Dermatol. Sci., 2019, 93, 176–185 CrossRef CAS PubMed.
- J. Tian, N. Sun, M. Yu, X. Gu, Q. Xie, L. Shao, J. Liu, L. Liu and Y. Wang, Discovery of N-indanyl benzamides as potent RORγt inverse agonists, Eur. J. Med. Chem., 2019, 167, 37–48 CrossRef CAS PubMed.
- C. Gege, Retinoic acid-related orphan receptor gamma t (RORγt) inverse agonists/antagonists for the treatment of inflammatory diseases - where are we presently?, Expert Opin. Drug Discovery, 2021, 16, 1517–1535 Search PubMed.
- H. Gronemeyer, J. A. Gustafsson and V. Laudet, Principles for modulation of the nuclear receptor superfamily, Nat. Rev. Drug Discovery, 2004, 3, 950–964 CrossRef CAS PubMed.
- X. Li, M. Anderson, D. Collin, I. Muegge, J. Wan, D. Brennan, S. Kugler, D. Terenzio, C. Kennedy, S. Lin, M. E. Labadia, B. Cook, R. Hughes and N. A. Farrow, Structural studies unravel the active conformation of apo RORγt nuclear receptor and a common inverse agonism of two diverse classes of RORγt inhibitors, J. Biol. Chem., 2017, 292, 11618–11630 CrossRef CAS PubMed.
- J. M. Harris, P. Lau, S. L. Chen and G. E. Muscat, Characterization of the retinoid orphan-related receptor-alpha coactivator binding interface: a structural basis for ligand-independent transcription, Mol. Endocrinol., 2002, 16, 998–1012 CAS.
- X. Hu, Y. Wang, L. Y. Hao, X. Liu, C. A. Lesch, B. M. Sanchez, J. M. Wendling, R. W. Morgan, T. D. Aicher, L. L. Carter, P. L. Toogood and G. D. Glick, Sterol metabolism controls T(H)17 differentiation by generating endogenous RORγ agonists, Nat. Chem. Biol., 2015, 11, 141–147 CrossRef CAS PubMed.
- J. R. Huh, M. W. Leung, P. Huang, D. A. Ryan, M. R. Krout, R. R. Malapaka, J. Chow, N. Manel, M. Ciofani, S. V. Kim, A. Cuesta, F. R. Santori, J. J. Lafaille, H. E. Xu, D. Y. Gin, F. Rastinejad and D. R. Littman, Digoxin and its derivatives suppress TH17 cell differentiation by antagonizing RORγt activity, Nature, 2011, 472, 486–490 CrossRef CAS PubMed.
- N. Kumar, L. A. Solt, J. J. Conkright, Y. Wang, M. A. Istrate, S. A. Busby, R. D. Garcia-Ordonez, T. P. Burris and P. R. Griffin, The benzenesulfoamide T0901317 [N-(2,2,2-trifluoroethyl)-N-[4-[2,2,2-trifluoro-1-hydroxy-1-(trifluoromethyl)ethyl]phenyl]-benzenesulfonamide] is a novel retinoic acid receptor-related orphan receptor-alpha/gamma inverse agonist, Mol. Pharmacol., 2010, 77, 228–236 CrossRef CAS PubMed.
- M. Scheepstra, S. Leysen, G. C. van Almen, J. R. Miller, J. Piesvaux, V. Kutilek, H. van Eenennaam, H. Zhang, K. Barr, S. Nagpal, S. M. Soisson, M. Kornienko, K. Wiley, N. Elsen, S. Sharma, C. C. Correll, B. W. Trotter, M. van der Stelt, A. Oubrie, C. Ottmann, G. Parthasarathy and L. Brunsveld, Identification of an allosteric binding site for RORγt inhibition, Nat. Commun., 2015, 6, 8833 CrossRef PubMed.
- G. Ouvry, C. Bouix-Peter, F. Ciesielski, L. Chantalat, O. Christin, C. Comino, D. Duvert, C. Feret, C. S. Harris, L. Lamy, A. P. Luzy, B. Musicki, D. Orfila, J. Pascau, V. Parnet, A. Perrin, R. Pierre, G. Polge, C. Raffin, Y. Rival, N. Taquet, E. Thoreau and L. F. Hennequin, Discovery of phenoxyindazoles and phenylthioindazoles as RORγ inverse agonists, Bioorg. Med. Chem. Lett., 2016, 26, 5802–5808 CrossRef CAS PubMed.
- F. A. Meijer, I. A. Leijten-van de Gevel, R. M. de Vries and L. Brunsveld, Allosteric small molecule modulators of nuclear receptors, Mol. Cell. Endocrinol., 2019, 485, 20–34 CrossRef CAS PubMed.
- M. L. Chen, K. Takeda and M. S. Sundrud, Emerging roles of bile acids in mucosal immunity and inflammation, Mucosal Immunol., 2019, 12, 851–861 Search PubMed.
- S. Hang, D. Paik, L. Yao, E. Kim, J. Trinath, J. Lu, S. Ha, B. N. Nelson, S. P. Kelly, L. Wu, Y. Zheng, R. S. Longman, F. Rastinejad, A. S. Devlin, M. R. Krout, M. A. Fischbach, D. R. Littman and J. R. Huh, Bile acid metabolites control TH17 and Treg cell differentiation, Nature, 2019, 576, 143–148 CrossRef CAS PubMed.
- D. Paik, L. Yao, Y. Zhang, S. Bae, G. D. D'Agostino, M. Zhang, E. Kim, E. A. Franzosa, J. Avila-Pacheco, J. E. Bisanz, C. K. Rakowski, H. Vlamakis, R. J. Xavier, P. J. Turnbaugh, R. S. Longman, M. R. Krout, C. B. Clish, F. Rastinejad, C. Huttenhower, J. R. Huh and A. S. Devlin, Human gut bacteria produce TH17-modulating bile acid metabolites, Nature, 2022, 603, 907–912 Search PubMed.
- R. A. Quinn, A. V. Melnik, A. Vrbanac, T. Fu, K. A. Patras, M. P. Christy, Z. Bodai, P. Belda-Ferre, A. Tripathi, L. K. Chung, M. Downes, R. D. Welch, M. Quinn, G. Humphrey, M. Panitchpakdi, K. C. Weldon, A. Aksenov, R. da Silva, J. Avila-Pacheco, C. Clish, S. Bae, H. Mallick, E. A. Franzosa, J. Lloyd-Price, R. Bussell, T. Thron, A. T. Nelson, M. Wang, E. Leszczynski, F. Vargas, J. Gauglitz, M. Meehan, E. Gentry, T. Arthur, A. Komor, O. Poulsen, B. Boland, J. Chang, W. Sandborn, M. Lim, N. Garg, J. Lumeng, R. Xavier, B. Kazmierczak, R. Jain, M. Egan, K. Rhee, D. Ferguson, M. Raffatellu, H. Vlamakis, G. Haddad, D. Siegel, C. Huttenhower, S. Mazmanian, R. Evans, V. Nizet, R. Knight and P. C. Dorrestein, Global chemical effects of the microbiome include new bile-acid conjugations, Nature, 2020, 579, 123–129 CrossRef CAS PubMed.
- S. Brogi, M. Brindisi, S. Butini, G. U. Kshirsagar, S. Maramai, G. Chemi, S. Gemma, G. Campiani, E. Novellino, P. Fiorenzani, J. Pinassi, A. M. Aloisi, M. Gynther, R. Venskutonytė, L. Han, K. Frydenvang, J. S. Kastrup and D. S. Pickering, (S)-2-Amino-3-(5-methyl-3-hydroxyisoxazol-4-yl)propanoic Acid (AMPA) and Kainate Receptor Ligands: Further Exploration of Bioisosteric Replacements and Structural and Biological Investigation, J. Med. Chem., 2018, 61, 2124–2130 CrossRef CAS PubMed.
- K. Frydenvang, D. S. Pickering, G. U. Kshirsagar, G. Chemi, S. Gemma, D. Sprogøe, A. M. Kærn, S. Brogi, G. Campiani, S. Butini and J. S. Kastrup, Ionotropic Glutamate Receptor GluA2 in Complex with Bicyclic Pyrimidinedione-Based Compounds: When Small Compound Modifications Have Distinct Effects on Binding Interactions, ACS Chem. Neurosci., 2020, 11, 1791–1800 Search PubMed.
- S. Liu, D. Liu, R. Shen, D. Li, Q. Hu, Y. Yan, J. Sun, F. Zhang, H. Wan, P. Dong, J. Feng, R. Zhang, J. Li, L. Zhang and W. Tao, Discovery of a novel RORγ antagonist with skin-restricted exposure for topical treatment of mild to moderate psoriasis, Sci. Rep., 2021, 28, 913 Search PubMed.
- S. Rauhamäki, P. A. Postila, S. Lätti, S. Niinivehmas, E. Multamäki, K. R. Liedl and O. T. Pentikäinen, Discovery of Retinoic Acid-Related Orphan Receptor γt Inverse Agonists via Docking and Negative Image-Based Screening, ACS Omega, 2018, 3, 6259–6266 CrossRef PubMed.
- L. A. Solt and T. P. Burris, Action of RORs and their ligands in (patho)physiology, Trends Endocrinol. Metab., 2012, 23, 619–627 CrossRef CAS PubMed.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids, J. Am. Chem. Soc., 1996, 118, 11225–11236 Search PubMed.
- S. Fujita-Sato, S. Ito, T. Isobe, T. Ohyama, K. Wakabayashi, K. Morishita, O. Ando and F. Isono, Structural basis of digoxin that antagonizes RORgamma t receptor activity and suppresses Th17 cell differentiation and interleukin (IL)-17 production, J. Biol. Chem., 2011, 286, 31409–31417 CrossRef CAS PubMed.
- S. Brogi, A. Fiorillo, G. Chemi, S. Butini, M. Lalle, A. Ilari, S. Gemma and G. Campiani, Structural characterization of Giardia duodenalis thioredoxin reductase (gTrxR) and computational analysis of its interaction with NBDHEX, Eur. J. Med. Chem., 2017, 135, 479–490 CrossRef CAS PubMed.
- B. Battah, G. Chemi, S. Butini, G. Campiani, S. Brogi, G. Delogu and S. Gemma, A Repurposing Approach for Uncovering the Anti-Tubercular Activity of FDA-Approved Drugs with Potential Multi-Targeting Profiles, Molecules, 2019, 24, 4373 Search PubMed.
- H. Sirous, G. Chemi, S. Gemma, S. Butini, Z. Debyser, F. Christ, L. Saghaie, S. Brogi, A. Fassihi, G. Campiani and M. Brindisi, Identification of Novel 3-Hydroxy-pyran-4-One Derivatives as Potent HIV-1 Integrase Inhibitors Using in silico Structure-Based Combinatorial Library Design Approach, Front. Chem., 2019, 7, 574 CrossRef CAS PubMed.
- M. T. Gabr and S. Brogi, MicroRNA-Based Multitarget Approach for Alzheimer's Disease: Discovery of the First-In-Class Dual Inhibitor of Acetylcholinesterase and MicroRNA-15b Biogenesis, J. Med. Chem., 2020, 63, 9695–9704 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, and supplementary figures. See DOI: https://doi.org/10.1039/d3ra08086b |
| This journal is © The Royal Society of Chemistry 2024 |