
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Review on anti-alzheimer drug development: approaches, challenges and perspectives
Abdallah E. Abdallah
 *
*
Pharmaceutical Medicinal Chemistry & Drug Design Department, Faculty of Pharmacy (Boys), Al-Azhar University, 11884, Cairo, Egypt. E-mail: abdulla_emara@azhar.edu.eg
First published on 5th April 2024
Abstract
Alzheimer is an irreversible progressive neurodegenerative disease that causes failure of cerebral neurons and disability of the affected person to practice normal daily life activities. There is no concrete evidence to identify the exact reason behind the disease, so several relevant hypotheses emerged, highlighting many possible therapeutic targets, such as acetylcholinesterase, cholinergic receptors, N-methyl D-aspartate receptors, phosphodiesterase, amyloid β protein, protein phosphatase 2A, glycogen synthase kinase-3 beta, β-secretase, γ-secretase, α-secretase, serotonergic receptors, glutaminyl cyclase, tumor necrosis factor-α, γ-aminobutyric acid receptors, and mitochondria. All of these targets have been involved in the design of new potential drugs. An extensive number of these drugs have been studied in clinical trials. However, only galantamine, donepezil, and rivastigmine (ChEIs), memantine (NMDA antagonist), and aducanumab and lecanemab (selective anti-Aβ monoclonal antibodies) have been approved for AD treatment. Many drugs failed in the clinical trials to such an extent that questions have been posed about the significance of some of the aforementioned targets. On the contrary, the data of other drugs were promising and shed light on the significance of their targets for the development of new potent anti-alzheimer drugs.
 Abdallah E. Abdallah | Abdallah E. Abdallah received his Bachelor's degree in pharmaceutical sciences in 2004, Master's degree in pharmaceutical chemistry in 2015, and PhD degree in pharmaceutical chemistry in 2018. Now, he is an associate professor at Faculty of Pharmacy, Al-Azhar university, Egypt. He is interested in the discovery of new therapeutic candidates, in particular, anticancer, immunomodulatory, and anti-SARS-CoV agents, especially in fields that are of high significance to society. Furthermore, he is interested in complex diseases that are difficult to treat e.g., Alzheimer disease. |
1. Introduction
1.1. Background
Alzheimer disease (AD) is an irreversible and progressive neurodegenerative disorder leading to ultimate damage and death of brain neurons.1,2 It causes cognitive impairment, behavioral defects, psychological disorders, memory loss, and disturbances in daily life activities such as eating, drinking, reading, writing, walking, communication, etc.3 It significantly affects not only the life of patients, but also that of their family.4 It mainly affects old people at or above 65 years old, but nowadays it seems to touch younger adults as a consequence of modern lifestyles.3 AD is the primary cause of dementia and affects approximately 10% of people over the age of 65 and 50% over the age of 85.5 It was reported that 6.7 million American people lived with clinical AD in 2023, and this figure is expected to reach 8.5 million by the year 2030.6 In a separate report, the estimate of worldwide people with AD reaches 74.7 million, with a care cost of about US$2 trillion by 2030.51.2. Etiology
Unfortunately, the etiology of AD is still incompletely understood. There is no definite reason evident to be behind the disease. Alternatively, AD is considered to be related to different reasons, among which are genetic factors and environmental effects such as mental stress, food habits, and lifestyle.7 However, histopathological studies revealed relevant multifactorial disorders from which different hypotheses originated to shed light on the likely mechanisms and effective targets of the disease. The emerged hypotheses include the following: (a) cholinergic hypothesis, (b) amyloid cascade hypothesis, (c) tau hypothesis, (d) mitochondrial cascade hypothesis, (e) oxidative stress hypothesis (f) excitotoxicity hypothesis, (g) neuroinflammatory hypothesis, and (h) others (like genetic factors, environmental factors, etc.).These facts draw attention to amyloid cascade hypothesis as a considerable theory of AD pathogenesis.24 Over decades, researchers considered Aβ aggregation as the main cause of all AD pathological changes such as neurotoxicity, neuronal inflammation, and neuron loss.16,18 Accordingly, several attempts at developing anti-Alzheimer drugs targeting Aβ have been made.25,26 But repeated failures of Aβ-targeted clinical trials pose a question concerning the significance of the amyloid hypothesis.16 On the basis of such deficiencies in the amyloid hypothesis as well as some recent findings, researchers suggested that the major factor underlying the development and progression of AD is tau rather than Aβ.27
In contrast to amyloid plaques that aggregate extracellularly, tau-based neurofibrillary tangles are accumulated intracellularly. It was proposed that tau pathology is the crucial factor and occurs earlier than Aβ aggregation in sporadic AD.29 So, the tau hypothesis is likely to reveal more predominant effects on neurons than the amyloid hypothesis.27
1.2.3.1. Mitochondrial hypothesis. Mitochondrial function changes over time under the effects of genetic and/or environmental factors. Oxidative stress is a fundamental element that disrupts mitochondrial function and induces mitochondrial fragmentation during the pathogenesis of AD.34 The mitochondrial hypothesis states that in sporadic, late-onset AD, changes in mitochondrial function affect APP expression and amyloid accumulation in a manner that triggers the pathogenesis of amyloid cascade.35 Correlations have been increasingly recognized between mitochondrial function, Aβ amyloidosis, and tau phosphorylation.36
1.3. Challenges
There are some challenges in the treatment of AD, such as a lack of evidence of the exact mechanism and the primary target on which researchers can work.56 Furthermore, no neuronal protective or regenerative drug is available nowadays.57 There is no cure for damaged neurons, but there are attempts to stop or delay the worsening of AD. The development of AD drugs was based on different hypotheses that shed light on the histopathological disorders in AD. However, the results of clinical trials questioned almost all of these hypotheses and revealed that none of them can be considered the sole approach to treatment. The data from clinical trials also reflect the complexity of the disease, which has different essential factors contributing to its pathogenesis. Furthermore, AD takes a relatively longer time to complete the clinical study in comparison to most other therapeutic fields.58 One more challenge is that AD is a progressive disease. Each stage may require its own specific study because early-stage drugs are likely to be ineffective in later stages. AD is also chronic, which reflects the requirement of drug safety for long-term use.However, there are advances in anti-alzheimer drugs and their mechanisms of action. The following section concerns the drugs approved for the treatment of AD, and the drugs passed to clinical trials. The results of these clinical studies and their significance are shown below, considering the mechanisms of action extended to a wide range of targets identified in this review.
2. Discussion
In terms of their mechanisms of action, anti-alzheimer drugs can be classified as the following.2.1. Cholinergic drugs
Three drugs are currently FDA-approved for the treatment of AD as ChEIs: galantamine, donepezil, and rivastigmine. Donepezil and galantamine are selective AChEIs,62 while rivastigmine has a dual inhibitory effect on AChE and BChE.63
2.1.1.1. AChEIs. Galantamine is a natural alkaloid based on benzofuro[4,3-cd]azepine (Fig. 1). It is a reversible AChE inhibitor with no effect on BChE. Furthermore, its cholinergic activity is enhanced by binding to allosteric nicotinic sites. It is exposed to minor biotransformation that includes demethylation of about 5–6% of the dose. It is excreted mainly in the urine.64
 | ||
| Fig. 1 The chemical structures of approved cholinesterase inhibitors. | ||
Donepezil is another reversible AChE inhibitor that is based on an inden-1-one nucleus (Fig. 1). It is indicated for symptomatic treatment of patients suffering from mild-to-moderate AD. Donepezil is extensively bound to plasma proteins (about 96%), so its elimination half-life is prolonged to 70 h.64
2.1.1.2. Dual AChE and BChE inhibitors. Rivastigmine is a pseudoirreversible noncompetitive carbamate inhibitor of AChE and BChE (Fig. 1). Although its half-life is limited to only 2 h, its inhibition of ChEs is extended to approximately 10 h. This prolonged effect is attributed to the slow dissociation of the drug enzyme complex. In April 2000, rivastigmine was approved by the Food and Drug Administration (FDA) for the treatment of mild-to-moderate AD.64
Tacrine is another dual AChE and BChE inhibitor, based on an acridine nucleus (Fig. 1). Due to its hepatotoxicity, it has been withdrawn from the market.65 This class of drugs represents just a symptomatic treatment without preventing the progression of the disease.66–69
Metrifonate, dimethyl (2,2,2-trichloro-1-hydroxyethyl)phosphonate, is an irreversible inhibitor for both ChEs, with higher selectivity for BChE than AChE. It is a prodrug converted nonenzymatically to the active metabolite dichlorvos, which is responsible for the sustained cholinesterase inhibition (see Fig. 2). Its clinical evaluation in mild-to-moderate AD revealed its toxicity, so its use was suspended for AD patients.64
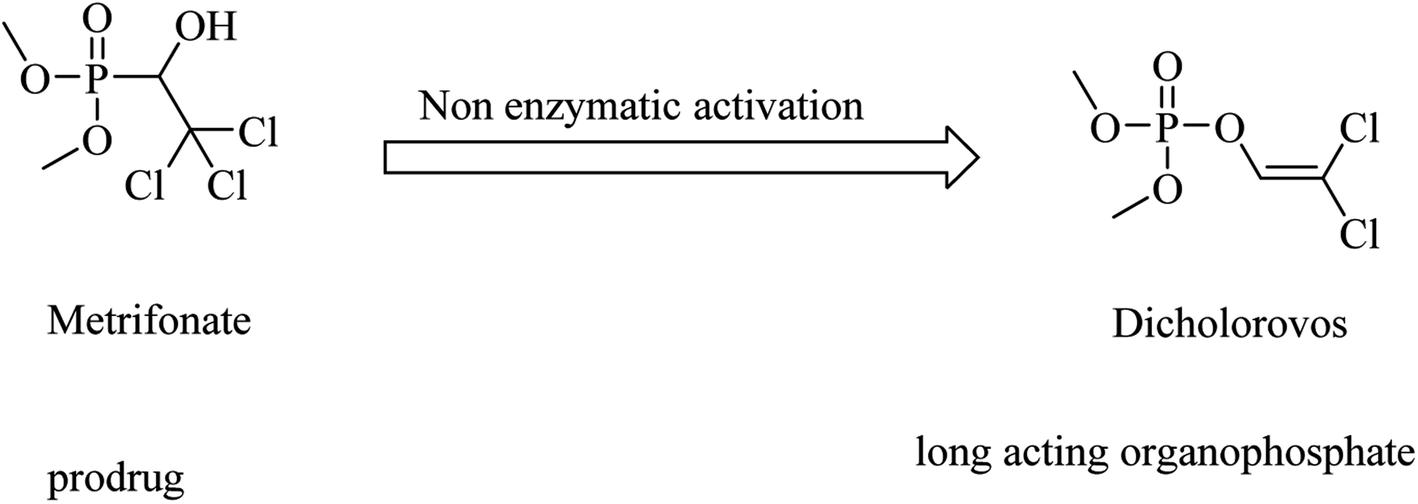 | ||
| Fig. 2 Chemical structures of organophosphate molecules as cholinesterase inhibitors. | ||
ABT-126 (Fig. 3) was identified as a selective α7 nicotinic receptor agonist and suggested as a monotherapy in mild to moderate AD. In phase 2a clinical trials, ABT-126 improved cognition to some extent in patients with mild to moderate AD, while it failed in phase 2b study to reveal any significant improvement72 and showed no therapeutic effect.73 With respect to safety profile, ABT-126 was generally well tolerated.73
 | ||
| Fig. 3 The chemical structures of some nicotinic receptor agonists. | ||
Similarly, α4β2 neuronal nicotinic receptors are associated with cognitive functions such as learning, memory, and attention.74 ABT-089 (Fig. 3) was developed as a selective α4β2 partial agonist,75 but it showed no therapeutic effects against alzheimer in clinical trials.76
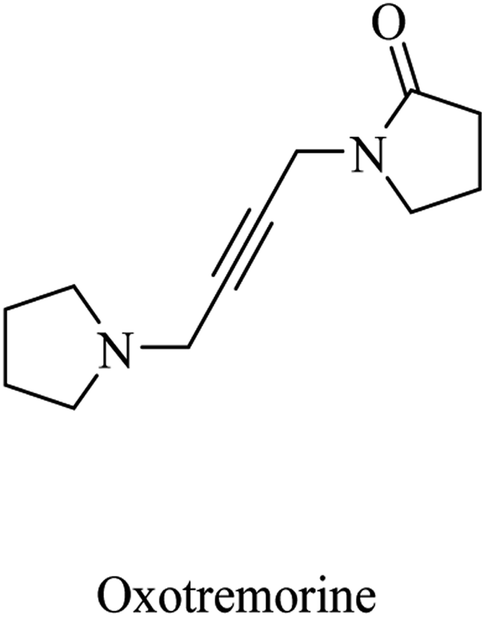 | ||
| Fig. 4 Oxotremorine chemical structure. | ||
Xanomeline is a selective muscarinic agonist based on the pyridinylthiadiazole scaffold (Fig. 5). It showed a significant improvement in AD patients in clinical trials.78 On the other side, it triggered a lot of unwanted side effects. However, it was considered a lead for the development of anti-alzheimer drugs.79
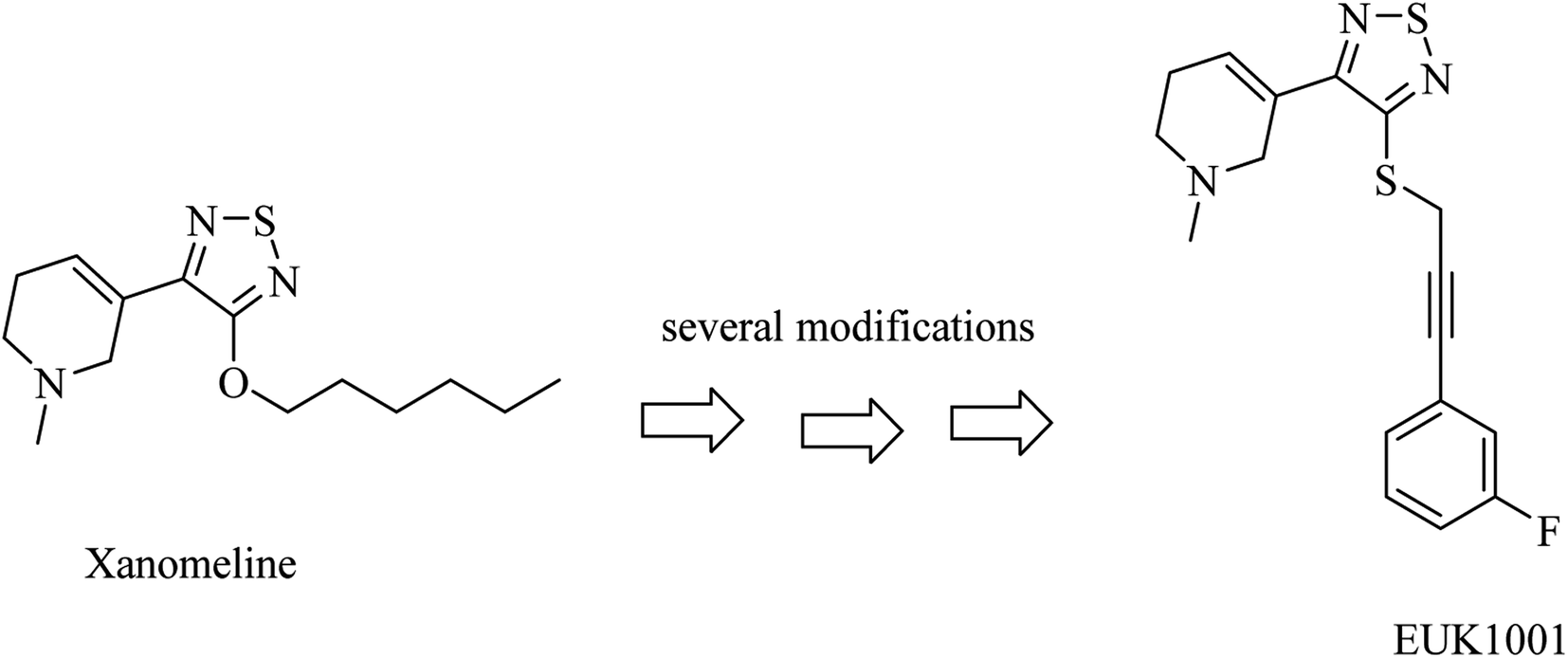 | ||
| Fig. 5 Development of EUK1001 as a xanomeline analog. | ||
EUK1001 is a fluorinated derivative of xanomeline, as can be noticed from Fig. 5. EUK1001 was found to decrease AD-like neurodegenerative disorder in presenilin-deficient mice, which has no Aβ pathology. Furthermore, EUK1001 revealed a significant improvement in cognitive functions in AD mice as well as a reduction in Aβ42.79 Meanwhile, another study indicated that EUK1001 improved memory function in aged mice.80 According to these data, EUK1001 was suggested as a promising compound for the treatment of AD.81
2.2. Glutamatergic drugs
Memantine (Fig. 6), an adamantine derivative, was approved by the FDA, and hence it is currently used for the treatment of AD as an NMDA antagonist.82 It clinically enhances cognitive ability and improves behavioral disturbance with an excellent safety profile, whether it is used alone or in combination with donepezil.83–85 Memantine blocks the NMDA receptor, reducing calcium ion influx into the neurons, so that it prevents the activation of toxic intracellular events.40 It was found to have a low-to-moderate affinity for NMDA.86 On the other side, antagonists with high affinity, such as phencyclidine (Fig. 6), revealed severe adverse effects that make their use for alzheimer not practical.86 | ||
| Fig. 6 The chemical structure of some glutamatergic drugs. | ||
2.3. Phosphodiesterase inhibitors (PDEIs)
Inhibition of PDE was suggested to prevent and improve AD by increasing the levels of cyclic guanosine monophosphate (cGMP) and cyclic adenosine monophosphate (cAMP).87 | ||
| Fig. 7 The chemical structures of vinpocetine and cilostazol. | ||
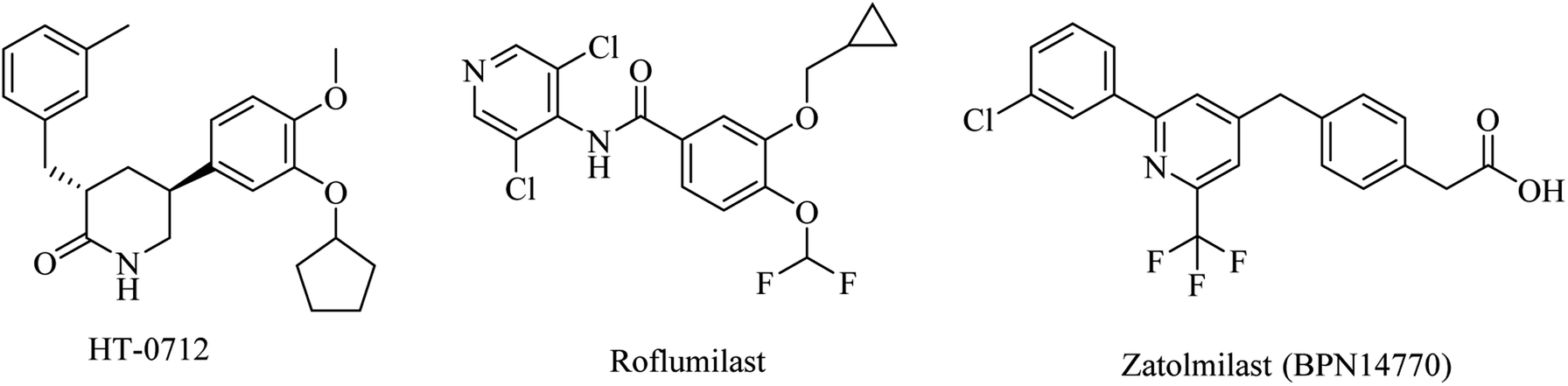 | ||
| Fig. 8 The chemical structures of some PDE4 inhibitors. | ||
Meanwhile, a phase I study showed that a combination of roflumilast, a N-pyridinylbenzamide-based molecule (Fig. 8), and donepezil improved verbal learning performance.96 In a recent study, roflumilast reduced neuroinflammation, amyloidogenesis, oxidative stress, cholinergic impairments, and phosphorylated tau levels in the rat hippocampus exposed to streptozotocin-induced sporadic AD.98
Zatolmilast (BPN14770), a phenylacetic acid derivative (Fig. 8), was reported as a PDE4D-negative allosteric inhibitor. It was found to show an improvement in memory and cognitive functions. Two clinical studies revealed that BPN14770 is safe and well tolerated. It was designed not to completely inhibit the enzyme in order to reduce the emetic effect.96
Clinical trials of denbufylline, a xanthine PDE4 inhibitor (Fig. 9), were inconclusive and preliminary.99
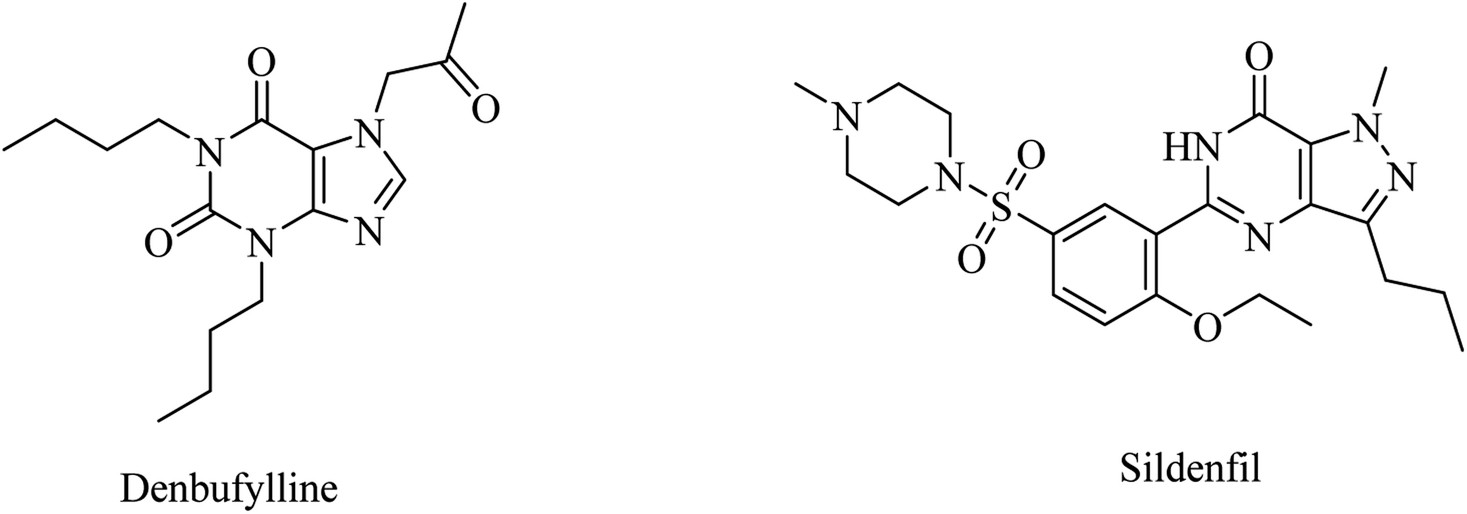 | ||
| Fig. 9 The chemical structure of denbufylline and sildenafil. | ||
 | ||
| Fig. 10 The chemical structure of some PDE9 inhibitors. | ||
 | ||
| Fig. 11 Xanthine based broad spectrum PDEI. | ||
2.4. Anti-Aβ drugs
Anti-Aβ drugs have been designed to degrade the amyloid plaques either chemically or immunologically by activating phagocytosis or microglia. This is likely to prevent the neuronal damage triggered by the amyloid plaques.57,102–1042.4.1.1. Passive immunotherapy. For years, a lot of molecules have been designed and evaluated for their effects on senile amyloid plaques as a significant hallmark of AD pathogenesis. Many therapeutic targets directly or indirectly linked to Aβ protein have been involved. As a result of such a huge effort, aducanumab and lecanemab were approved by the FDA.57,106,107
Aducanumab is a selective anti-Aβ monoclonal antibody with the ability to clear Aβ plaques. On the basis of these proven data, the FDA approved it in June 2021 as the first drug underlying the pathophysiology of Alzheimer's disease (AD). On the other side, the correlation between clearance of Aβ plaques and improvement in cognitive functions lacks evidence. Moreover, the data obtained from two phase III studies were controversial and insufficient to prove aducanumab efficacy. So, the accelerated approval of aducanumab by the FDA generated a debate among scientists. Some researchers consider this approval an obstacle to progress and pose a question concerning the cost and safety profile of aducanumab. A further argument in support of the insignificance of aducanumab is the rejection of it by the European Medicines Agency in December 2021. Now, Biogen is designing a confirmatory study, named ENVISION, required by the FDA, and it is expected to be complete in 2026.108–112
Lecanemab is a monoclonal antibody directed against both soluble and insoluble forms of Aβ polypeptides.113 Lecanemab received FDA-accelerated approval in January 2023.113–116 After further investigation, it received traditional approval from FDA in July 2023 for the treatment of early-stage AD.106 Lecanemab showed both amyloid clearance and a slowing of clinical decline in early AD.117
On the contrary, solanezumab,118,119 bapineuzumab,120,121 ponezumab,122 and gantenerumab119,123,124 failed in clinical trials as monoclonal anti-Aβ antibodies.57 Similarly, clinical trials on the natural anti-Aβ antibodies obtained from the blood of healthy adults or Alzheimer patients revealed no clinical effects on AD.57
2.4.1.2. Active immunotherapy. One important aspect of immunotherapy is vaccination. It takes successive stages of clinical trials. Firstly, clinical trials of vaccines composed of purified Aβ-42 polypeptide (AN1792)125 were disappointing due to the toxicity detected in about 6% of AN1792-treated patients, such as cytotoxic T-cell-induced meningoencephalitis.126,127 Furthermore, AN1792 failed to clear amyloid plaques; however, it activated the production of Aβ antibodies in AD patients' blood.128 Due to the detected toxicity linked to the full-length Aβ1-42 vaccine, the concept has been modified to develop a new generation such as vanutide cridificar (ACC-001) that was designed to stimulate antibodies against N-terminal Aβ1-7. The results obtained were acceptable with regard to safety but showed no therapeutic effects.57 Therefore, the next clinical trials involved a combination of ACC-001 and QS-21 adjuvant. The results revealed a good safety profile and high levels of anti-amyloid beta IgG in mild to moderate AD patients after long-term exposure to the combination.129,130 This high level of anti-amyloid-beta IgG declined after some time.131 In another trial, ABvac40 vaccine was designed to initiate the production of antibodies against the C-terminal end of Aβ40. It showed good tolerability and developed Aβ40 specific antibodies.57 ABvac40 phase II clinical studies are still being processed.132
 | ||
| Fig. 12 Tramiprosate and its optimized prodrug valiltramiprosate. | ||
Valiltramiprosate (ALZ-801) was designed as an optimized prodrug (Fig. 12) to address the extensive gastrointestinal metabolism and gastrointestinal irritation detected for the active drug tramiprosate.133
Phase I clinical trials of ALZ-801 revealed good tolerability and no serious side effects.133 ALZ-801 was also found to pass across the blood–brain barrier (BBB) efficiently, showing an intracranial concentration of about 40% of its plasma levels.138 ALZ-801 phase II and phase III studies on APOE4 carries are ongoing.139
2.5. Tau protein targeting drugs
Tau protein represents a crucial factor in AD pathogenesis. Nevertheless, the benefits of clinical trials on molecules targeting tau hyperphosphorylation were insignificant in terms of efficacy and/or safety.57In addition to its action as an NMDA antagonist, memantine (Fig. 6) was found to activate PP2A, leading to inhibition of tau hyperphosphorylation.140
Sodium selenate is another phosphate modifier that increases the activity of PP2A.140 The data of a phase IIa clinical trial, in patients with mild to moderate AD, revealed some benefits for sodium selenate on diffusion MRI. But it failed to show any considerable improvement in other measures such as cognition and CSF levels of Aβ and tau proteins.141
Methylthioninium chloride (MTC) (Fig. 13), known as methylene blue, was the first molecule considered to inhibit tau aggregation without affecting the physiological function of tau as a stabilizer for neuronal microtubules.142 On the other side, the poor pharmacokinetics and intolerability of MTC were likely to be the reasons behind inefficacy in phase 2 clinical trials.57
 | ||
| Fig. 13 The chemical structure of MTC. | ||
Tideglusib, a thiadiazolinedione derivative (Fig. 14), is developed to irreversibly inhibit GSK3β without interaction with the ATP binding domain.145,146 In animal models of AD, it caused a reduction in neurofibrillary tangles and amyloid plaques with an improvement in memory.147,148 These data were confirmed by the preliminary data of a larger-scale phase II clinical trial in which tideglusib caused an improvement in cognitive functions along with a reduction in CSF β-secretase levels in a subgroup of mild AD patients. However, the precise analysis of the entire study revealed the insignificance of the improvement caused by tideglusib.149
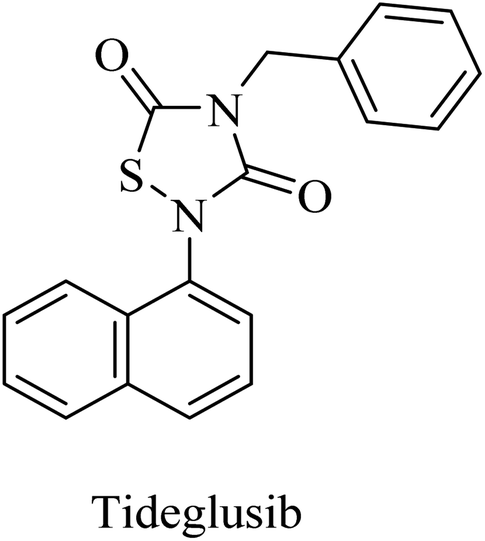 | ||
| Fig. 14 Structure of tideglusib. | ||
Lithium chloride (or ‘lithium’) was identified as a GSK3β inhibitor.150 Preliminary results of lithium clinical trials showed some improvement with few side effects in elderly patients with AD. Further studies are required to evaluate the efficacy and safety of lithium in AD patients.140
2.6. β-secretase acting drugs
 | ||
| Fig. 15 The chemical structures of some BACE1 inhibitors. | ||
LY2886721 is a picolinamide-based (Fig. 15) BACE1 inhibitor. Its oral administration in a transgenic mouse model-based study showed a significant reduction in the levels of Aβ and soluble APPβ (sAPPβ). Similar promising results were obtained in another study that revealed a great and continuous reduction in the levels of Aβ in CSF after oral use of LY2886721 in a cannulated beagle dog model. In addition to this, LY2886721 was found to have good BBB penetration, showing high CSF concentrations.16,153 The drug LY2886721 has been exposed to clinical trials because of its good pharmacology and safety profile. In a phase II clinical study, LY2886721 induced an abnormal elevation in liver enzymes, so the study was terminated.154
Umibecestat (CNP520) is another picolinamide-based (Fig. 16) oral BACE1 inhibitor.155 CNP520 was designed to meet the requirements of prevention treatment. In the preclinical studies, CNP520 revealed a considerable reduction in acute and chronic Aβ along with an acceptable safety profile.156 In 2019, CNP520 passed the phase II clinical trial, showing promising efficacy and tolerability. But it failed in phase III as it caused cognitive worsening in the treatment groups.157 The Alzheimer's Prevention Initiative Generation Program (Generation Study 1) announced that CNP520 worsens cognitive functions.155
 | ||
| Fig. 16 Some picolinamide based BACE1 inhibitors. | ||
Another picolinamide-based (Fig. 16) oral BACE-1 inhibitor is verubecestat (MK8931), which has been reported to reduce Aβ levels in AD patients.158,159 Nevertheless, in clinical trials, it caused no improvement in cognitive function in mild to moderate AD patients.158 Furthermore, some studies reported a greater decline in cognitive functions among patients receiving verubecestat than those receiving placebo.160
Atabecestat (JNJ-54861911), a picolinamide derivative (Fig. 17), was designed by Pharmaceutical Janssen as an oral BACE-1 inhibitor. In 2013, some subsequent clinical phase I studies of atabecestat have been conducted. It was launched in Belgium with a single increasing dose model, followed by a second study in addition to a similar trial performed in Japan. These studies were conducted on a limited number of healthy elderly volunteers and concluded promising results for atabecestat, which reduced Aβ aggregation after single or multiple doses.161–163 A subsequent phase IIb/III clinical trial (NCT02569398) was conducted to investigate the efficiency and safety of the drug, but it was terminated in 2018 because of the observed adverse effects, which include hepatotoxicity.164,165 In addition, other adverse effects on cognition, sleep, depression, and anxiety were reported for atabecestat in some other studies.164,166
 | ||
| Fig. 17 The chemical structure of some BACE1 inhibitors. | ||
Lanabecestat (Fig. 17) was developed as an anti-alzheimer drug that has the ability to pass the BBB and improve the clinical features, along with preventing the progression of the disease through inhibition of BACE1.167 The data obtained from some studies reflected adverse effects that include psychiatric disorders, weight loss, and change in hair color, while there was no considerable improvement in primary or secondary efficacy measures.167,168
Elenbecestat (E2609) is a pyrazinecarboxamide-based (Fig. 18) BACE1 inhibitor. It showed a dose-dependent reduction in the levels of Aβ in CSF. However, it showed no difference from placebo with regard to some other AD-related measures, such as the Alzheimer's Disease Composite Score (ADCOMS, p = 0.38) and CDR-SB (p = 0.55). Two 24 months studies of elenbecestat in a large number of mild AD patients with positive amyloid pathology biomarkers were discontinued in the sense that the toxicity induced outweighed the benefits obtained.169
 | ||
| Fig. 18 The chemical structure of elenbecestat. | ||
In general, and on the basis of the published data relating to the effects of the oral BACE1 inhibitors in transgenic mouse models of AD, it can be concluded that BACE1 inhibitors are able to reduce the levels of Aβ in brain and CSF in a dose-dependent manner, but there is no solid evidence that they can improve cognitive functions effectively. In addition to the unfavorable toxicities observed in such studies and linked to BACE1 inhibitors, seventeen BACE1 inhibitors have failed in clinical trials to show considerable improvements in AD patients. Many of these trials were discontinued due to toxicity and/or cognitive worsening.170
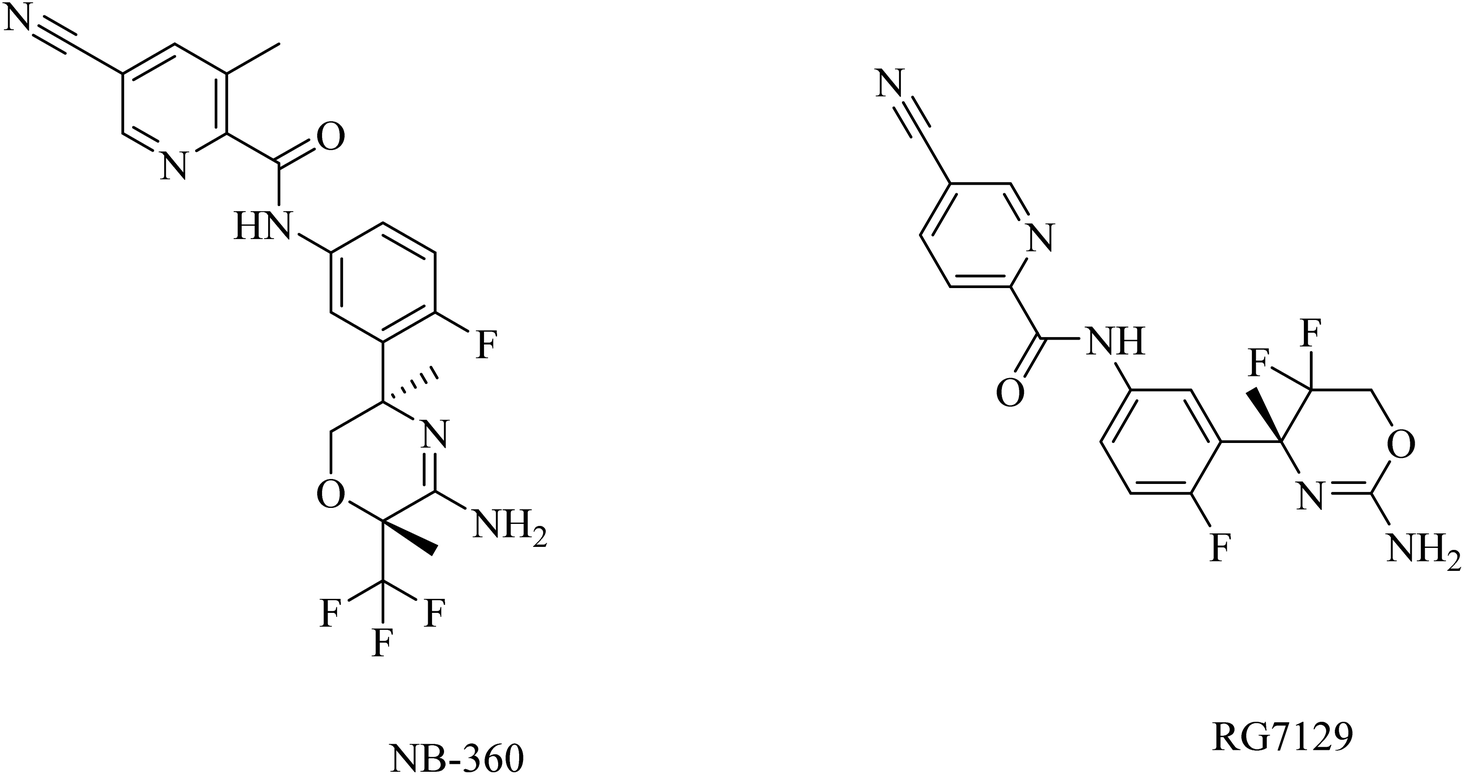 | ||
| Fig. 19 Picolinamide based inhibitors for both BACE1 and BACE2. | ||
RG712 (Fig. 19) was generated by Roche as a substituted phenylpicolinamide derivative carrying an oxazoline moiety. It was developed as an oral BACE inhibitor, and it showed an IC50 of 30 nM in the preclinical studies with minor selectivity over BACE2.174 It was evaluated in combination with an anti-Aβ antibody (gantenerumab) in some studies that involved AD transgenic mouse models, and it was found to reduce the amyloid plaques with slowing down the disease progression.174 However, the hepatotoxicity induced by RG712 prevented the completion of any further related clinical trials conducted by Roch.175
2.7. γ-Secretase acting drugs
In addition to the role of γ-secretase in the generation of Aβ from APP, it also proteolyzes many other type I integral membrane proteins, in particular the Notch receptor, which plays a role in many essential steps during cell differentiation.176 So targeting γ-secretase as a significant approach to the treatment of AD should avoid affecting Notch signaling in order to show a good safety profile.177 | ||
| Fig. 20 The chemical structure of some γ-secretase inhibitors. | ||
MK-0752 (Fig. 20) is another GSI that does not differentiate between γ-secretase and Notch receptor.154 In a phase I clinical trial, evaluation of its safety, pharmacokinetics, and pharmacodynamics on the basis of a single oral dose has been conducted in 27 healthy volunteers.181 The data obtained revealed that MK-0752 is safe and reaches its maximum plasma concentration in approximately 3–4 h with a t1/2 of 20 h as well as greatly reducing Aβ1-40 levels in CSF for 12 h.154 However, the safety profile was disappointing in a phase II study because it did not differentiate between γ-secretase and Notch.154
Avagacestat (BMS-708163) is a benzenesulfonamide-based (Fig. 21) potent and selective GSI developed by Bristol-Myers Squibb.182 It revealed a 193-fold selectivity for APP over Notch blockade. It inhibited Aβ40 production with an IC50 of 0.30 nM, resulting in lowering the levels of Aβ40 in CSF, brain, and plasma as studied in dogs and rats.183 Phase I study demonstrated that the safety, tolerability, pharmacokinetics, and pharmacodynamics properties of BMS-708163 were promising after oral administration in healthy, young volunteers (NCT01454115). Accordingly, it was decided to be evaluated by further clinical trials. In phase II trials, BMS-708163 was evaluated in 209 outpatients with a median age of 75 years, diagnosed with mild-to-moderate AD. Different doses were examined in this study, and the results revealed that it cannot be tolerated at a dose of 100 mg or above. Additionally, a worsening of cognition was observed at such doses. Meanwhile, the dose up to 50 mg day−1 showed results similar to those of placebo.184 The obtained results have been confirmed by a subsequent study conducted on CSF biomarker-negative volunteers, which demonstrated deterioration in the health conditions of patients with the occurrence of nausea, vomiting, diarrhea, rash, and nonmelanoma skin cancers. Meanwhile, there were no therapeutic benefits to the lower doses.185
 | ||
| Fig. 21 Benzenesulfonamide based GSI. | ||
Begacestat (GSI-953), 2,5-disubstituted thiophene sulfonamide (Fig. 22), is considered a second generation GSI. It was generated by Wyeth (now Pfizer).186 Researchers at Wyeth applied the high throughput screening technique to get the molecule LI (Fig. 22).187 L1 was then modified to afford the more optimized L2, which in turn led to the discovery of L3, which showed much potent inhibition for Aβ production,183 as can be seen in Fig. 22. The most important character of L2 and L3 is that they revealed much better selectivity for APP over Notch. But unfortunately, they were metabolically unstable, and this was attributed to the easy oxidation of the methylene group. So, the alkyl side chain was contracted to afford a series of new molecules, from which GSI-953 was discovered (Fig. 22). GSI-953 was found to be relatively more resistant to rapid metabolism, showing a half-life of more than 90 min. Meanwhile, GSI-953 displayed comparatively better properties with respect to inhibition of Aβ production and selectivity for APP over Notch, as presented in Fig. 22.183,186 GSI-953 revealed promising data in phase I clinical trials as it inhibited Aβ production at nanomolar concentrations, and showed 16-fold selectivity for APP over Notch. In a study conducted on a human APP-overexpressing Tg2576 transgenic mouse model, oral use of GSI-953 led to a significant reduction in Aβ levels in CNS, brain, and plasma. Similar results have been obtained when the effects of oral use of GSI-953 were evaluated in healthy human volunteers.188
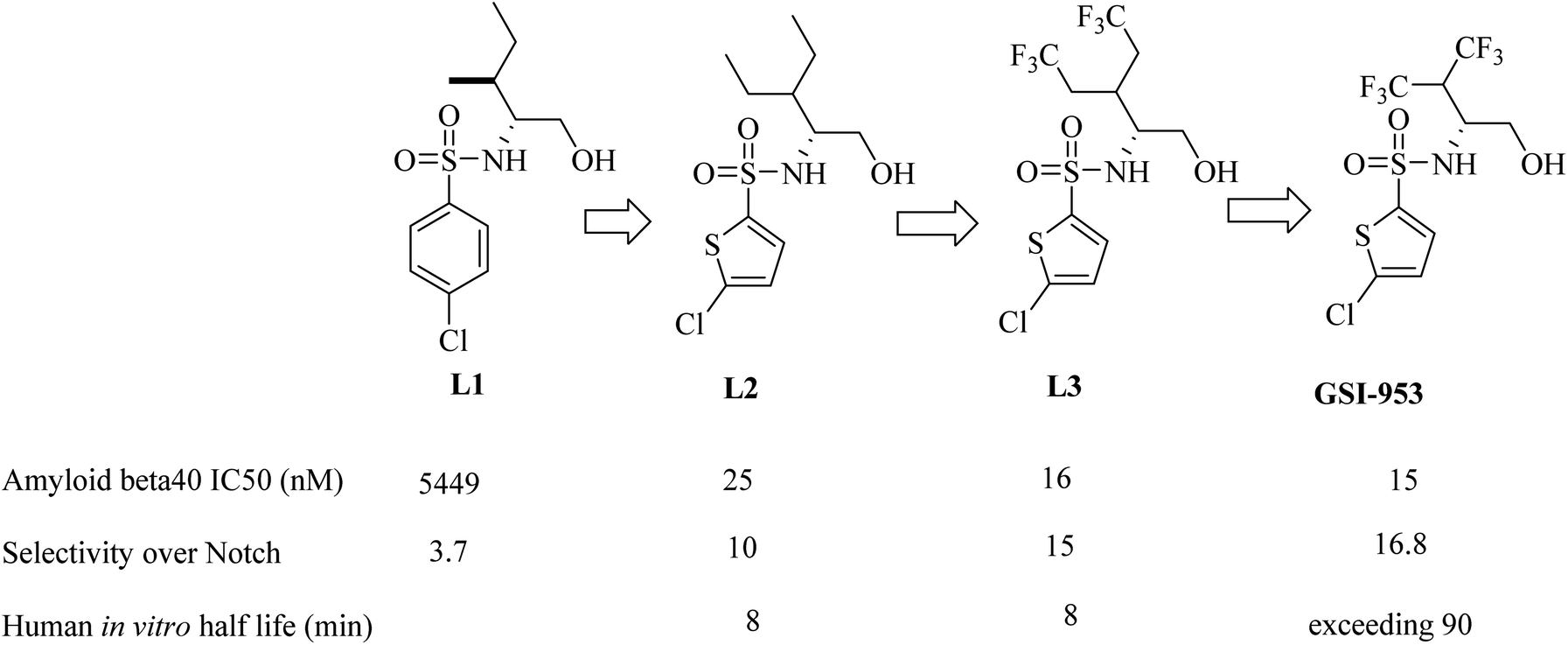 | ||
| Fig. 22 Lead optimization in the development of GSI-953. | ||
PF-3084014 is an imidazolylpentamide-based Notch-sparing GSI (Fig. 23). It was reported as a non-competitive reversible human GSI with an IC50 of 1.3 nM.189,190 With respect to selectivity, PF-3084014 displayed minor inhibition of Notch signaling (IC50 = 19.15 nM). The selectivity index of PF-3084014 was found to be 1473 for APP over Notch. So it was considered a Notch sparing GSI. It showed good penetration to the BBB with a reduction in Aβ levels in animals. However, there is a lack of data about its effect on amyloid plaque deposition in transgenic mice, as well as no data available about its behavioral effects in AD animal models.154
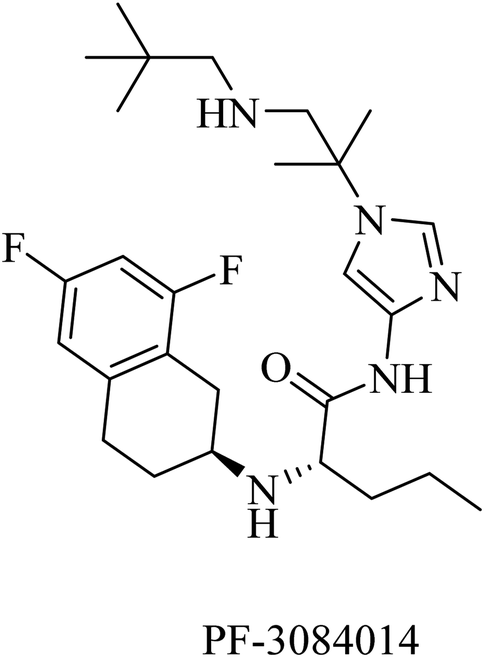 | ||
| Fig. 23 Notch sparing GSI. | ||
In clinical trials, GSIs such as semagacestat and avagacestat reduced Aβ production in AD patients.184,191 However, the multiple effects of such inhibitors prevented further progress in the clinical trials due to inhibition of Notch signaling,192 which induces adverse effects such as gastrointestinal disturbance, infection, worsening cognition, and the risk of skin cancer.193 To address this problem, a new class of γ-secretase acting molecules that specifically regulate or modulate this enzyme are required.193
2.7.2.1. First-generation GSMs. Nonsteroidal anti-inflammatory drugs (NSAIDs) represented the first generation of GSI as they were found to modulate the activity of γ-secretase in a manner that reduces Aβ42 and increases the soluble Aβ38 without inhibiting Notch. Ibuprofen, indomethacin, and sulindac sulfide (Fig. 24) are examples of this class of NSAIDs.194,195
 | ||
| Fig. 24 First generation of NSAIDs as GSMs. | ||
Unfortunately, ibuprofen, indomethacin, and sulindac sulfide lack good brain penetration, so they showed insignificant data in clinical trials associated with AD models.196
2.7.2.2. Second-generation GSMs. The poor brain penetration of NSAIDs was required to be resolved in the second-generation GSMs, which can be classified into different categories, including NSAID-derived carboxylic acid GSMs, non-NSAID-derived imidazole GSMs, and natural product-derived GSMs.194
2.7.2.3. NSAID-derived carboxylic acid GSMs. Tarenflurbil ((R)-flurbiprofen), an aryl propionic acid derivative (Fig. 25), was strongly suggested as a clinical candidate for AD treatment.197 It showed no Notch inhibition-related adverse effects;198 however, it still showed insufficient brain penetration and minor efficacy, leading to its failure in phase III clinical study.199–201
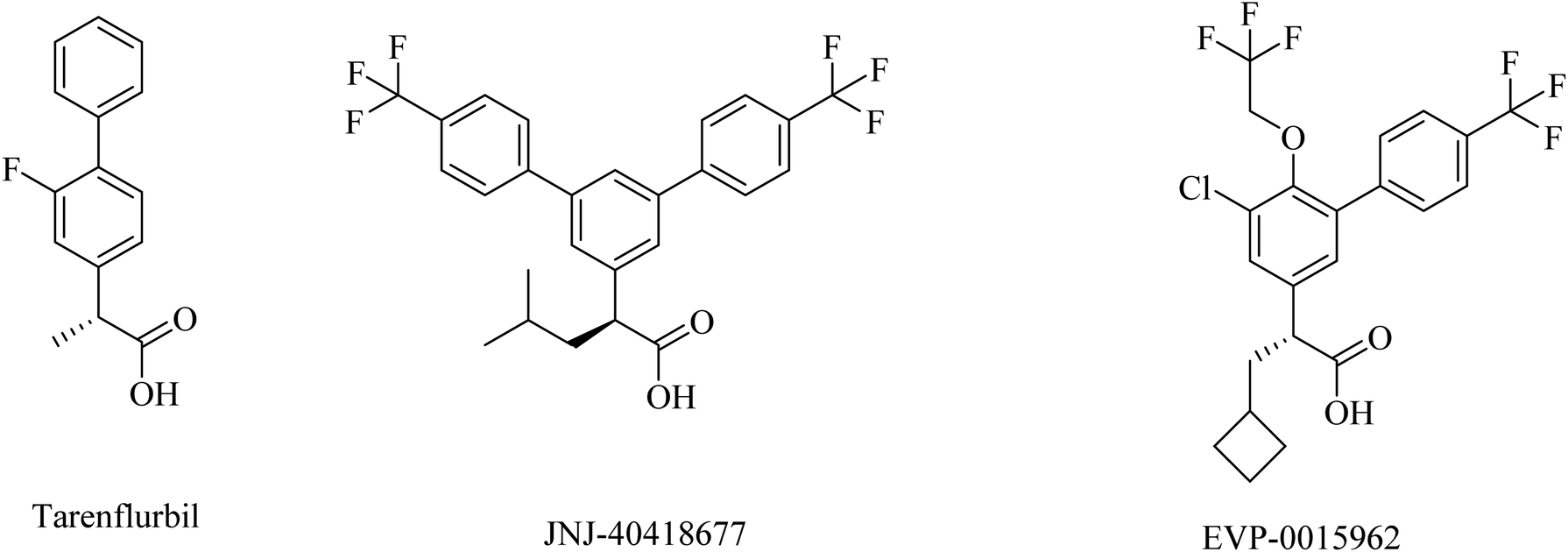 | ||
| Fig. 25 NSAID derived carboxylic acid GSMs. | ||
JNJ-40418677 was generated by Janssen as an analog of flurbiprofen by addition of substituents on the core aryl ring (Fig. 25). JNJ-40418677 has sufficient lipophilicity to penetrate the brain and reveal its therapeutic activities. It was found to reduce Aβ42 levels and elevate the levels of Aβ38 in the brain without affecting the levels of total Aβ in the brain. The safety profile of JNJ-40418677 still needs to be evaluated.202
EVP-0015962 is a cyclobutyl group containing analog of (R)-flurbiprofen (Fig. 25), it was generated by Forum Pharmaceuticals. It displayed good data, such as a considerable reduction in Aβ42 levels when administered orally with good efficacy in animals.203 Nevertheless, the high lipophilicity was considered an unfavorable property of this drug.196 So its clinical trials were discontinued after phase II.204
Itanapraced (CHF5074) is another carboxylic acid derivative (Fig. 26) designed by Chiesi Pharmaceuticals as a GSM. It was found to enhance memory and reduce microglial activation in Tg2576 mice.205 It did not show a reduction in Aβ42 levels either in plasma or CSF; instead, it decreased the levels of CD40, which is considered a marker for microglia activation. So CHF5074 is now classified as a microglia modulator.196,206
 | ||
| Fig. 26 The chemical structure of itanapraced. | ||
Biogen's research was focused on the discovery of NSAID derived carboxylic acid GSMs. In 2011, Biogen reported203 the discovery of a potential preclinical GSM candidate (BIIB042). Its development was based on the optimization of the previously examined L4, as shown in Fig. 27. This lead compound showed considerable potency, but it had poor brain penetration. So, the researchers worked on improving this criterion. More than one modification has been done on the basis of deduced SAR, which revealed that the activity was improved by the creation of a chiral center by adding a methyl group at the α position of the carboxyl group in addition to a rigid piperidinyl moiety, as can be noticed in Fig. 27. This optimization afforded the preclinical candidate BIIB042, which showed a reduction in Aβ42 levels with a concomitant elevation in Aβ38 levels and no effect on Aβ40 levels. Several clinical studies have been conducted by Biogen, which has published about 66 in vivo efficacy studies of BIIB042. The published data indicated that BIIB042 decreased Aβ42 levels and increased Aβ38 levels, but showed no effect on Aβ40 levels in the brains and plasma of mice and rats. These pharmacodynamic properties have been confirmed by similar results concluded from a study conducted on monkeys administered orally a single dose of BIIB042.203
 | ||
| Fig. 27 Biogen's NSAID-derived carboxylic acid GSM 1 and an optimized modulator BIIB042. | ||
In addition to these promising data, BIIB042 did not inhibit Notch signaling in an in vitro model.207 In a more advanced study conducted on human APP-overexpressing Tg2576 mice, BIIB042 reduced the levels of Aβ42 and decreased the amyloid plaque burden.208 BIIB042 is a candidate drug for AD.209
2.7.2.4. Non NSAID imidazole GSMs. This class of compounds attracted the attention of researchers of Eisai group as they focused on the development of non-carboxylic acid GSMs. At first, nearly all non-carboxylic acid GSMs had a general structural feature of an arylimidazole moiety linked via an olefin to a lactam, as in E2012, or a heterocycle, as in E2212 (Fig. 28). They developed E2012 as a substantial GSM with an IC50 of 83 nM and it was the first non-carboxylic acid molecule to be studied clinically as a GSM.154 The clinical trials revealed a question concerning the safety of E2012, which induced cataracts in rats.210 As a consequence, its clinical trials have been terminated and exposed to a series of modifications by Eisai, which finally developed E2212 (Fig. 28).211
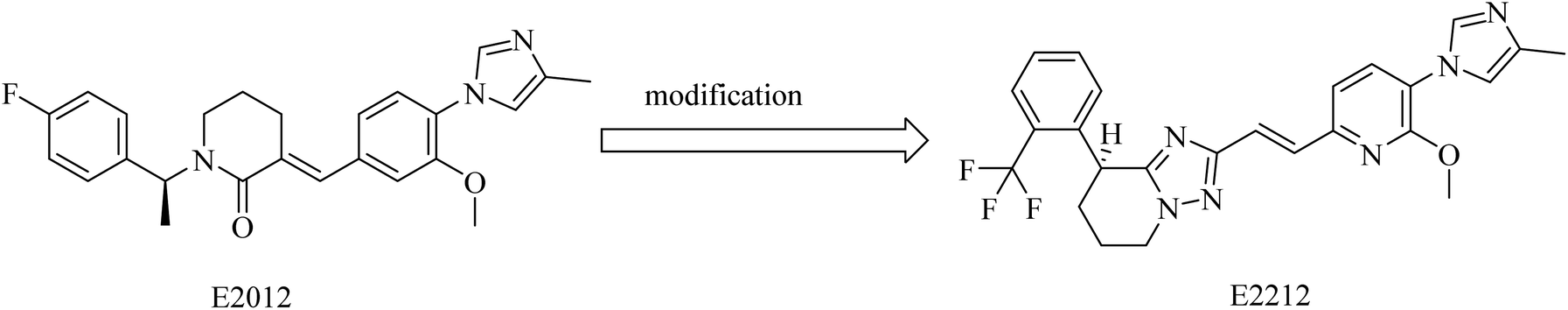 | ||
| Fig. 28 Non NSAID imidazole GSMs: E2012 and E2212. | ||
It was reported that E2012 was modified to E2212 for improving the drug-likeness.154 E2212, in its phase I clinical trials, showed comparatively better safety in comparison to E2012.198 Diarrhea was the most observed adverse effect for E2212.198 It also revealed promising efficacy by reducing the levels of Aβ42 by 54%,212 showing an IC50 of 17 nM.213 Although the structure of E2212 has never been revealed, it is predicted by several Eisai process chemistry patents. The further development of this compound has not been reported.154
Compound BMS-869780 (Fig. 29) was developed by Bristol-Myers Squibb as a non-NSAID imidazole GSM. It showed promising activity in mouse and rat brains by a significant reduction of Aβ42 levels after oral administration.214 Although there was no evidence that BMS-869780 triggers side effects related to Notch inhibition, further clinical studies have been terminated because of a potential toxicity issue, as the daily dose required to provide therapeutic effects in human AD was very high (700 mg). BMS-869780 was then modified to BMS-932481 (IC50 of 7 nM) in order to improve the pharmacodynamics, as shown in Fig. 29. It can be noticed that the chloroimidazole nucleus was replaced by the methyltriazole moiety.
 | ||
| Fig. 29 Non NSAID derived GSMs. | ||
BMS-932481 was evaluated for pharmacokinetics, which appeared to be promising. However, further studies have been terminated due to safety issues.154
PF-06648671 (Fig. 30) was developed by Pfizer, and it was found to be of good tolerability at single doses given to healthy persons. It decreased the plasma levels of Aβ40 and Aβ42. Meanwhile, it increased the levels of Aβ37 and Aβ38.215 However, it was discontinued due to Pfizer's discontinuation of R&D in neurology in 2018.
 | ||
| Fig. 30 The chemical structure of the non NSAID imidazole GSM developed by Pfizer. | ||
In 2017, researchers at Merck reported the discovery of a new series of 17 tetrahydroisoxazole molecules as GSMs developed on the basis of Eisai's E2012 (ref. 216) and their own preceding GSM molecules, among which compounds is L5 (ref. 196) (Fig. 31). The double bond and the lactam carbonyl in Eisai's E2012 are sites of potential metabolism, so they were replaced by a tetrahydrobenzisoxazole moiety, as shown in Fig. 31. Another benefit of the tetrahydrobenzisoxazole group is avoiding the stereochemistry of the double bond. The synthetic compounds showed variable activities, from which SAR has been deduced to develop some compounds with good selectivity and potency. The most promising candidates were compounds 1 and 2, showing an IC50 of 39 nM and 29 nM against Aβ42, respectively (Fig. 31). It can be noticed that 1 and 2 were far more potent than E2012 and L5. The promising criteria of 1 and 2 encouraged their evaluation in a preclinical rat model. Compound 1 reduced CSF Aβ42 levels by 58%, compared to only a 20% reduction in Aβ levels reported for compound 2 after 3 h of a single oral dose in rats. Interestingly, compound 2 was more potent than 1 in vitro. To find an explanation for this controversy, the levels of both drugs in the plasma and brain of rats have been assessed. The concentration of compound 1 in the brain was found to be sixfold that of compound 2, which gave an account of the better results of compound 1 in animal model study. Meanwhile, the efflux ratio of 2 was four-fold higher than that of 1. Despite these promising data, we did not find any further data concerning the safety and efficacy of compound 1 or its future evaluation in clinical trials.196
 | ||
| Fig. 31 Merck Research Laboratory's oxadiazine L5 and tetrahydrobenzisoxazole analogs 1 and 2. | ||
2.7.2.5. Natural products as gamma secretase modulators. Compound SPI-014 (Fig. 32), isolated from the extract of Actaea racemosa (black cohosh), was found to exhibit activities like GSMs. Several modifications have been done on SPI-014, and a lot of semisynthetic analogs have been developed. The modifications included the substitution of both sugar and acetate moieties with more stable groups to enhance drug-like properties, as can be seen in Fig. 32. These modifications led to the discovery of a new drug molecule, SPI-1865 (Fig. 32). Despite the improvement in the pharmacodynamics of the modified molecule, it did not pass the clinical trials due to the unanticipated off-target adrenal toxicity reported.217
 | ||
| Fig. 32 Natural and semisynthetic analogs as GSMs. | ||
2,3-Bis((Z)-4-methoxybenzylidene)succinonitrile (N1) (Fig. 33) was extracted from the marine sponge-derived fungus Dichotomomyces cejpii. It was found to decline the excessive production of Aβ42 in an AD cellular assay.218
 | ||
| Fig. 33 Natural GSM. | ||
Dihydroergocristine (DHEC) (Fig. 34) is an FDA-approved natural drug that revealed significant data with respect to selective inhibition of APP. It has a Leu-Phe-Pro motif, which plays a crucial role in binding to the allosteric site of the enzyme. This allosteric site represents the region to which APP-C99 interacts to be cleaved. So DHEC competes with APP for binding to γ-secretase, reducing the production of Aβ42 in the brain.219 DHEC was found to reduce the levels of Aβ at micromolar concentrations when examined in vivo.219
 | ||
| Fig. 34 The chemical structure of DHEC. | ||
2.8. α-Secretase activators
Activation of α-secretase was suggested as a potential therapeutic strategy in order to inhibit the aggregation of amyloid plaques.220 This is because of the role of α-secretase in enhancing the proteolysis of APP in the non-amyloidogenic pathway and, hence, decreasing the formation of amyloid plaques. A series of membrane-bound proteases (a disintegrin and metalloprotease family) regulate the α-secretase.221 ADAM10, ADAM17, and ADAM9 have been suggested as α-secretases.222Synthetic retinoids were proven to improve the nonamyloidogenic proteolysis of APP. One of the important synthetic retinoids in this regard is acitretin, a vitamin A analog (Fig. 35). Acitretin was evaluated in a phase II clinical study and was found to increase ADAM10 expression as well as reduce the levels of Aβ in APP/PS-1 transgenic mice.223 Furthermore, it enhances the stimulation of the mature ADAM10, resulting in higher activity of α-secretase in neuroblastoma cells.223 One of the encouraging features of acitretin is its ability to cross the BBB easily, and its level is not affected by glycoprotein (P-gp).224 On the other side, it was linked to some severe toxicity, such as alopecia, peeling, cheilitis, and hepatotoxicity.154
 | ||
| Fig. 35 The chemical structure of acitretin. | ||
Etazolate (EHT-0202), a pyrazolopyridine derivative (Fig. 36), is a gamma-aminobutyric acid-A (GABAA) receptor modulator. It was found to stimulate α-secretase and enhance sAPPα production.225 EHT-0202 has been assessed in a phase II clinical study in mild-to-moderate AD subjects. It showed cognitive improvement along with a good safety profile and tolerability.225,226
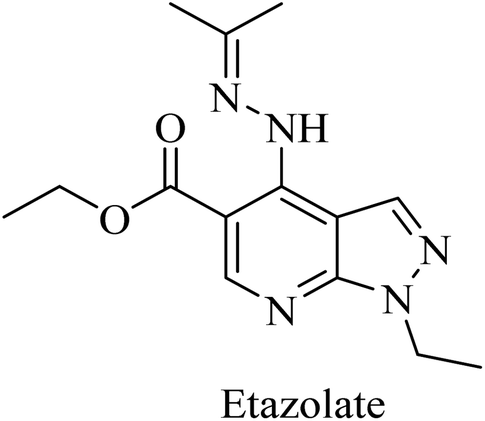 | ||
| Fig. 36 The chemical structure of etazolate. | ||
2.9. 5HT-receptors
One of the receptors that emerged lately as potential targets for cognitive disorders and AD is serotonin.227,228 In this regard, 5-HT6R and 5-HT7R are the most extensively studied serotonin receptors because of their high distribution in the brain and association with cognitive properties in vivo.229 Furthermore, 5-HT6R signaling was found to be associated with changes in cholinergic and glutamatergic functions in the brain, with little peripheral effect.230 However, the clinical trials against AD revealed no evidence for the therapeutic activity of any of the selective 5-HT6R or 5-HT7R drugs.57PF-05212377 (SAM-760), a benzimidazole derivative, and idalopirdine, an indole-based molecule, are selective 5-HT6R antagonists (Fig. 37).57 Clinical studies demonstrated that SAM-760 had no therapeutic benefits with regard to cognitive disorders; however, it showed good safety and tolerability.57,231
 | ||
| Fig. 37 The chemical structures of selective 5-HT6R antagonists. | ||
Similarly, idalopirdine was ineffective for AD patients, with a risk of elevated liver enzymes and vomiting.232 These data reveal the insignificance of idalopirdine for the treatment of AD.233
2.10. Glutaminyl cyclase (QC) inhibitors
The importance of QC in the development of anti-alzheimer drugs originated from the information that QC catalyzes the formation of cerebral pyroglutamate-Aβ 3 (AβpE3), which is considered one of the most neurotoxic forms of Aβ.234,235 So, inhibition of QC is suggested as a potential therapeutic approach to AD treatment.234 The work on the development of QC inhibitors has drawn attention in the last decade, where the design developed a zinc-binding group in order to coordinate the Zn2+ ion incorporated in the active site in addition to other common features.235 One of the outstanding works in this regard was done by the Probiodrug company, which rationally developed some promising QC inhibitors and identified the imidazole nucleus as a zinc chelating weak QC inhibitor.235PQ-912, a benzimidazole-based molecule (Fig. 38), showed competitive inhibition for QC with a Ki value of 25 nM. It was found to interact through coordination of the zinc ion in the QC's active site.235 PQ912 was evaluated for pharmacokinetic properties, which were found to be acceptable with good safety and tolerability in doses up to 200 mg.236 The studies have identified the maximum tolerated dose to support further studies at lower doses.237 Despite the promising cognitive improvement reported, many studies on PQ912 have been discontinued due to high dose toxicity.238
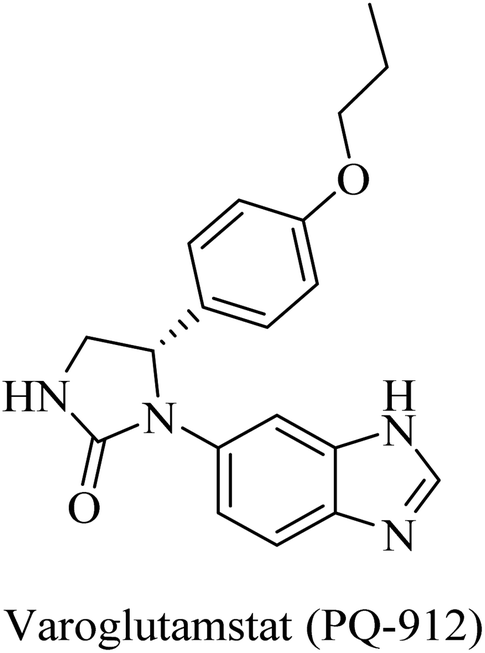 | ||
| Fig. 38 Benzimidazole based QC inhibitor. | ||
The results obtained from clinical evaluation of PQ912 have proven that QC is a significant AD druggable target. Furthermore, QC inhibition was found to improve synaptic functions by decreasing the toxic effects of AβpE3. In addition to this, long-term use is likely to modify the disease and reduce the neuroinflammations associated with AD.234,235
2.11. Anti-inflammatory agents
Chronic inflammation of the cerebral neurons is one of the pathological hallmarks linked to AD.239 It was suggested that the origin of neuroinflammation is the activation of glial cells by triggering factors such as neural environment or neuronal injury. One of the crucial factors in this scenario is TNF-α, which plays a pivotal role in neuronal excitotoxicity, neuroinflammation, and synapse loss. Another role for TNF-α associated directly with AD pathogenesis is enhancing amyloidogenesis and upregulating BACE-1 expression.240–242Etanercept is a competitive TNF-α inhibitor. It is a fully human dimeric fusion protein consisting of the extracellular ligand-binding domain of the human 75 kilodalton TNF-α receptor linked to the Fc portion of human immunoglobulin G1 (IgG1).243 Clinical studies revealed that it had no therapeutic benefits in clinical studies against alzheimer.243 Etanercept limitation is related to its pharmacokinetics; it is unable to cross the brain–blood barrier.243
Thalidomide, another TNF-α inhibitor, is a small molecule composed of a phthalimide nucleus attached to a glutarimide moiety (Fig. 39). It also showed no therapeutic benefits in the clinical trial against alzheimer due to poor tolerability.244
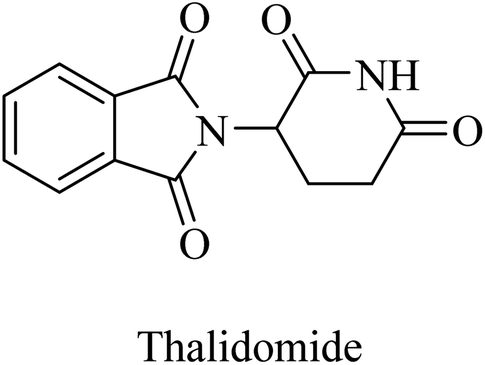 | ||
| Fig. 39 The chemical structure of thalidomide. | ||
2.12. GABA
Etazolate, a pyrazolopyridine derivative (Fig. 36), is a GABAA receptor modulator that was found to show neuroprotective properties against the toxic effects of Aβ. Moreover, it revealed cognitive improvement and anti-inflammatory activity in traumatic brain injury.247,248 Further investigation into the mechanisms of its neuroprotective effect revealed GABAA receptor activation as well as stimulating α-secretase cleavage of APP. The importance of its GABAA role is highlighted by the full block of its neuroprotective effect by GABAA antagonists.226
Other examples of GABAA agonists that showed promising results are muscimol (5-(aminomethyl)isoxazol-3(2H)-one) and propofol (2,6-diisopropylphenol) (Fig. 40).249,250 Muscimol pretreatment was found to effectively inhibit the Aβ25–35-induced neuronal death in cultured rat cortical neurons.250
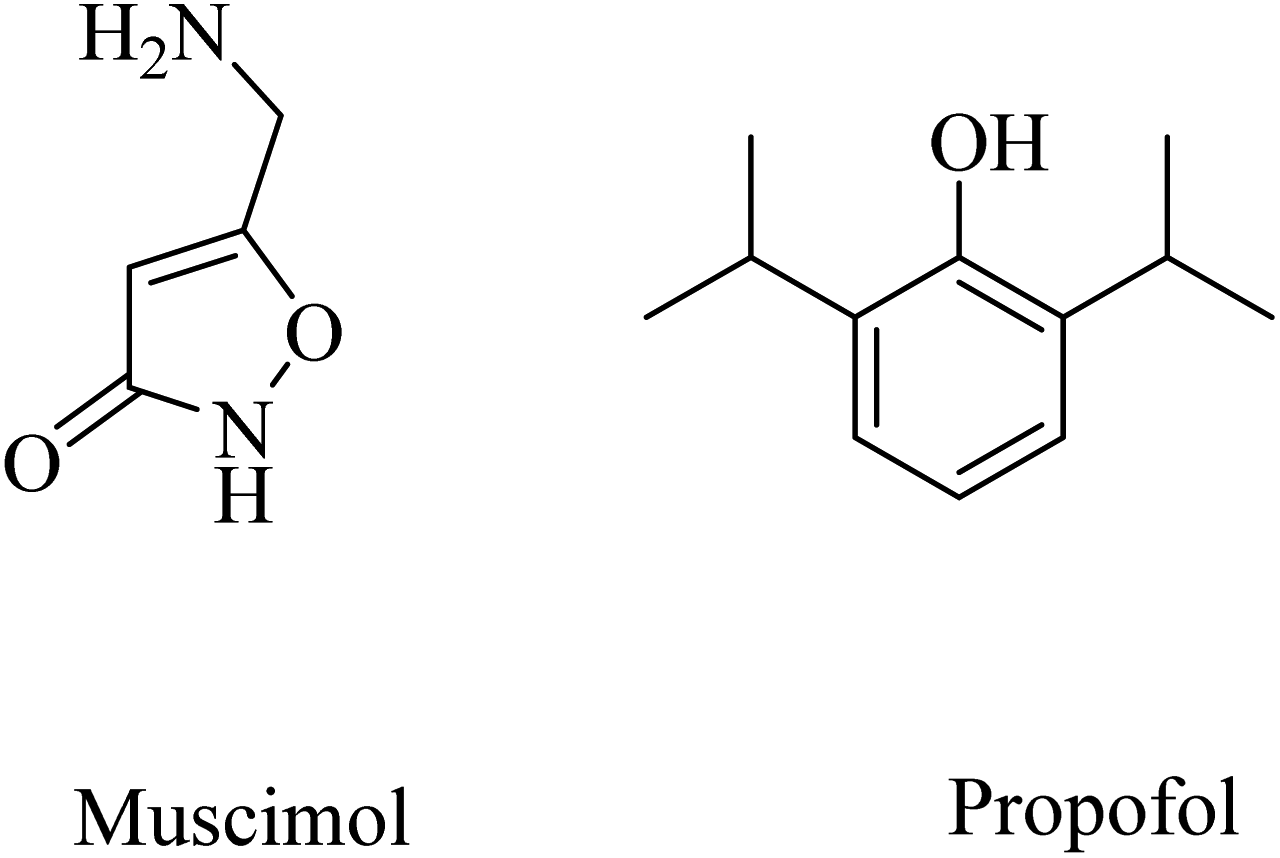 | ||
| Fig. 40 The chemical structures of some GABAA agonists. | ||
Long-term use of propofol for the treatment of aged mice was found to reduce Aβ40 and Aβ42 levels in the brain and decrease the expression level of BACE1, thereby decreasing the aggregation of amyloid plaques.250 These data reflected the importance of chronic GABAA receptor activation by propofol in neuroprotection and decreasing Aβ levels in brain. Furthermore, propofol was proven to improve cognitive function in both WT and APP/PS1 mice.249
SGS742 (CGP36742), a phosphinic acid derivative (Fig. 41), was the first GABAB antagonist evaluated for AD in clinical trials. Its effects on rodents and monkeys were outstanding, as evidenced by significant improvements in cognition and learning tasks.251 In addition, SGS742 was found to be well tolerated not only in experimental animals but also in human volunteers. In a phase II clinical trial, 8 weeks of oral administration of SGS742 revealed considerable attention improvement and memory enhancement in patients suffering mild cognitive impairment.251–253
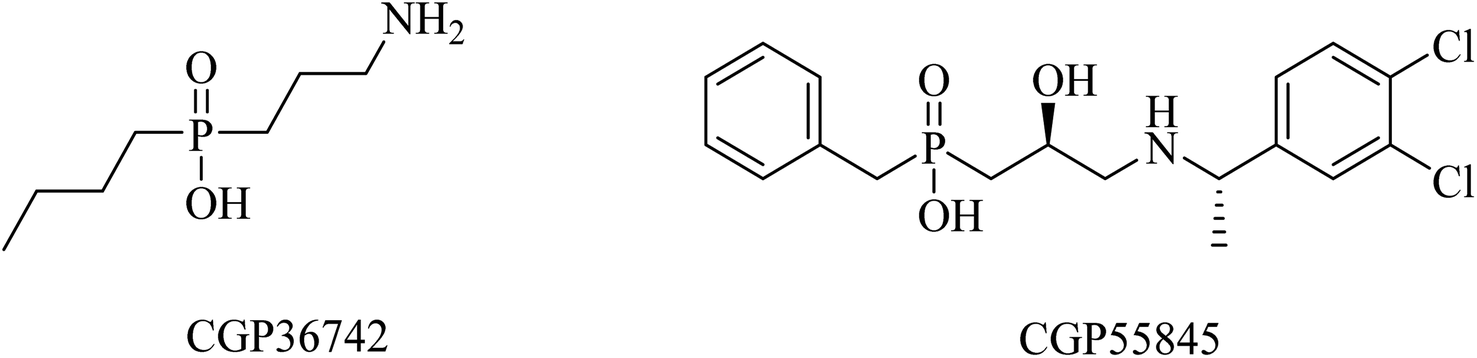 | ||
| Fig. 41 GABAB antagonists. | ||
CGP55845 is another phosphinic acid-based (Fig. 41) GABAB receptor antagonist. In an aged rat model with cognitive impairment, CGP55845 was found to completely improve the olfactory discrimination learning deficits and retrieve performance.254 These data indicated the significance of GABAB receptors as a potential target for improving cognitive disorders.255
2.13. Antioxidants
 | ||
| Fig. 42 The chemical structure of vitamin E. | ||
 | ||
| Fig. 43 Natural antioxidants. | ||
Another herbal antioxidant that showed promising data with regard to improvement of AD is silibinin (silybin) (Fig. 43), a flavonoid derived from the herb milk thistle (Silybum marianum). Silibinin was reported to prevent memory impairment and to eliminate the oxidative stress induced by Aβ in mice, so it is likely to be a potential candidate for AD treatment.262
Curcumin is a di-phenolic molecule (Fig. 44) extracted from turmeric and is known for diversity in its biological effects, such as antioxidant, anticarcinogenic, and anti-inflammatory. In the last decade, its pharmacological effects on AD have been discovered. It exhibited neuroprotection, and inhibition of Aβ aggregation, and Aβ-induced inflammation, so it is likely to be helpful in treating AD as one of the neurodegenerative diseases.263 Curcumin has also been found to inhibit AChE in in vitro studies.263
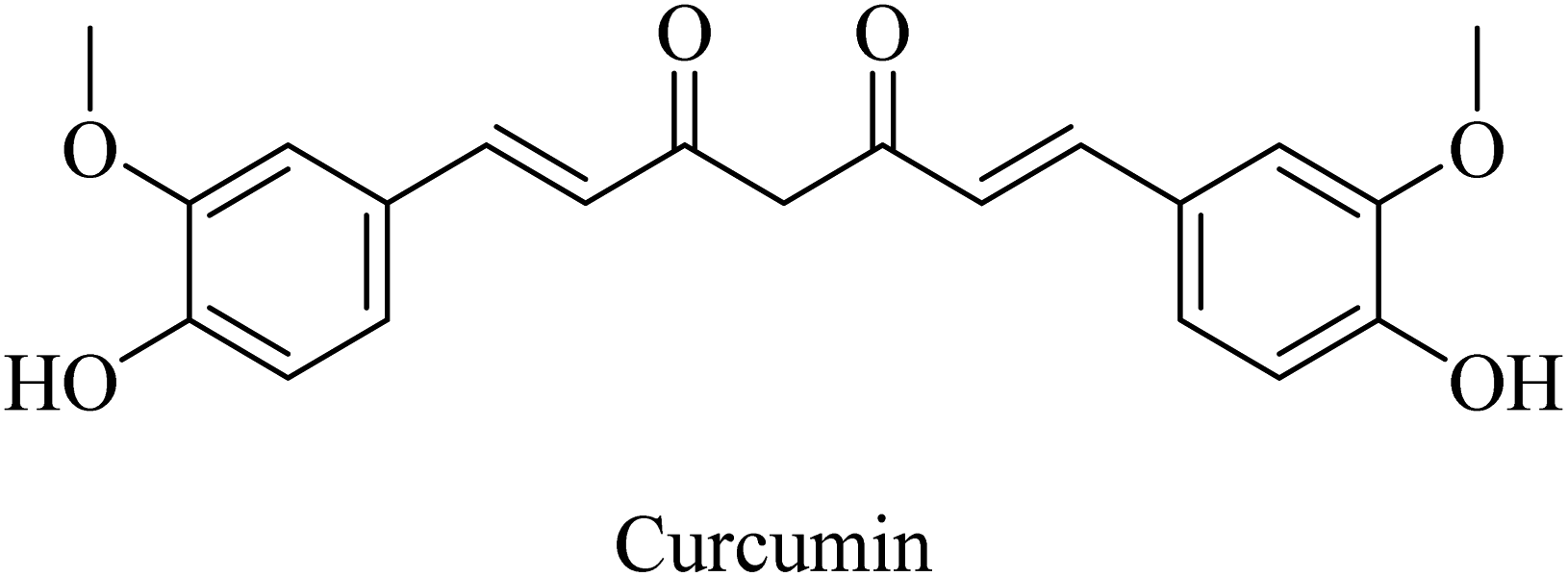 | ||
| Fig. 44 The chemical structure of curcumin. | ||
Similarly, the polyphenolic quercetin (Fig. 45) exhibited neuroprotective properties that encourage researchers to utilize it as a lead compound for developing drugs against neurodegenerative disorders such as AD. However, the oral bioavailability of quercetin is low, so the clinical trials were impeded. It was reported to reduce β-amyloidosis, astrogliosis, tauopathy, and microgliosis in the hippocampus and amygdala.264
 | ||
| Fig. 45 The poly phenolic quercetin. | ||
Luteolin (Fig. 46) is a flavonoid compound that was found to act as a neuroprotective agent in a streptozotocin-induced alzheimer's rat model. This reported effect was suggested to be due to luteolin's antioxidant properties, which are mediated by blocking free radicals and dispersing amyloid plaques. Accordingly, luteolin is proposed as a potential therapeutic candidate for neuronal disorders, e.g., AD; however, further investigation is still required.265
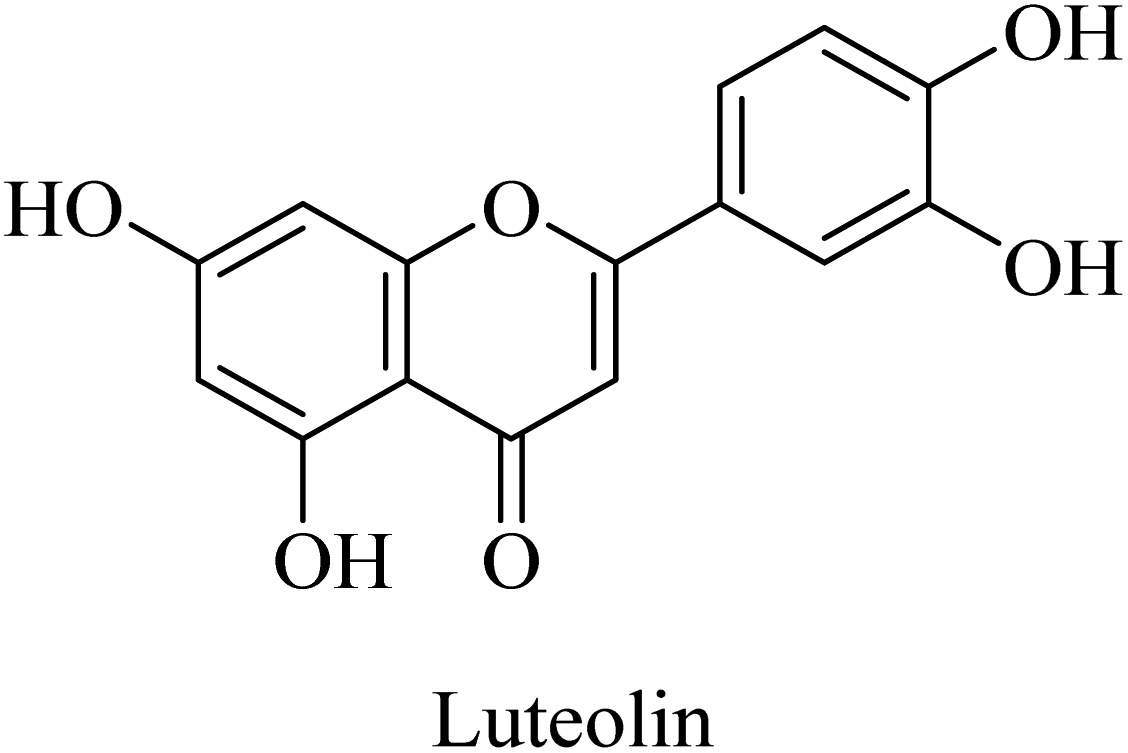 | ||
| Fig. 46 The chemical structure of luteolin. | ||
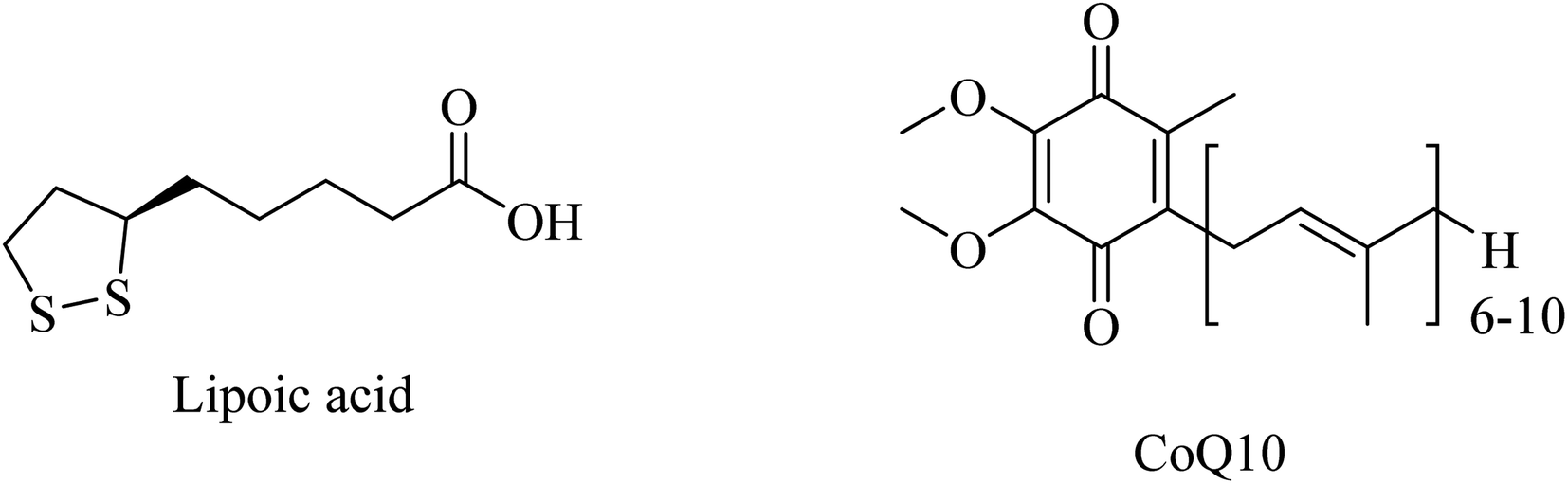 | ||
| Fig. 47 The chemical structure of lipoic acid and CoQ10. | ||
Administration of LA (Fig. 47) for long periods has been reported to decrease the expression of lipid peroxidation markers without reducing the Aβ levels in the brains of AD mice. Additionally, LA was found ineffective in improving cognition.266
CoQ10 (Fig. 47) plays a protective role, preventing mitochondrial damage by oxidative stress as well as protecting the whole neuronal cell through reducing Aβ overproduction and intracellular neurofibrillary tangles. Furthermore, CoQ10 is an essential factor for the production of ATP by mitochondria, so it is recommended as a significant antioxidant for AD prevention.261
 | ||
| Fig. 48 The chemical structure of melatonin and selegiline. | ||
Selegiline (Fig. 48), a selective monoamine oxidase-B inhibitor, was suggested for neurodegenerative disease treatment due to its possible antioxidant properties in addition to rapid generation of the potent vasodilator nitric oxide in cerebral blood vessels.273 In 1997, it was reported to protect neurons and decrease the progression rate of AD in patients with moderately severe impairment from AD.258 In 2000, the analysis of 15 clinical trials revealed that there was no solid evidence to identify selegiline as a potential treatment for AD.261
The discussed drugs are outlined in the following table (Table 1) along with their mechanisms of action and references.
| Serial | Drug | Mechanism of action | Reference |
|---|---|---|---|
| 1 | Galantamine | AChEI | 62 |
| 2 | Donepezil | AChEI | 62 |
| 3 | Rivastigmine | AChEI and BChEI | 63 |
| 4 | Tacrine | AChEI and BChEI | 65 |
| 5 | Metrifonate | Irreversible AChEI and BChEI | 64 |
| 6 | ABT-126 | Selective α7 nicotinic receptor agonist | 72 |
| 7 | ABT-089 | Selective α4β2 nicotinic partial agonist | 75 |
| 8 | Oxotremorine | CNS muscarinic agonist | 77 |
| 9 | Xanomeline | CNS muscarinic agonist | 79 |
| 10 | EUK1001 | CNS muscarinic agonist | 79 |
| 11 | Memantine | NMDA antagonist | 82 |
| 12 | Phencyclidine | NMDA antagonist | 86 |
| 13 | Vinpocetine | PDE1 inhibitor | 89 |
| 14 | Cilostazol | PDE3 inhibitor | 87 |
| 15 | HT-0712 | PDE4 inhibitor | 96 |
| 16 | Roflumilast | PDE4 inhibitor | 96 |
| 17 | Zatolmilast (BPN14770) | PDE4 inhibitor | 96 |
| 18 | Denbufylline | PDE4 inhibitor | 99 |
| 19 | Sildenafil | PDE5 inhibitor | 87 and 96 |
| 20 | BI-409306 | PDE9 inhibitor | 100 and 101 |
| 21 | PF-04447943 | PDE9 inhibitor | 100 and 101 |
| 22 | Propentofylline | Broad-spectrum PDE inhibitor | 87 |
| 23 | Aducanumab | Selective anti-Aβ monoclonal antibody | 108–112 |
| 24 | Lecanemab | Selective anti-Aβ monoclonal antibody | 113–116 |
| 25 | Solanezumab | Monoclonal anti-Aβ antibody | 118 and 119 |
| 26 | Bapineuzumab | Monoclonal anti-Aβ antibody | 120 and 121 |
| 27 | Ponezumab | Monoclonal anti-Aβ antibody | 122 |
| 28 | Gantenerumab | Monoclonal anti-Aβ antibody | 119 |
| 29 | AN1792 | Purified Aβ-42 polypeptide vaccine | 125 |
| 30 | ACC-001 | N-terminal Aβ1-7 vaccine | 57 |
| 31 | QS-21 | Adjuvant combined with ACC-001 | 129 and 130 |
| 32 | ABvac40 | C-terminal end of Aβ40 vaccine | 57 |
| 33 | Tramiprosate | Inhibitor for Aβ42 oligomer formation | 133–136 |
| 34 | Valitramiprosate (ALZ-801) | Inhibitor for Aβ42 oligomer formation (prodrug) | 133 |
| 35 | Sodium selenate | PP2A activator | 140 |
| 36 | Methylthioninium chloride | PP2A activator | 142 |
| 37 | Tideglusib | GSK3β inhibitor | 145 and 146 |
| 38 | Lithium chloride | GSK3β inhibitor | 150 |
| 39 | LY2811376 | BACE-1 inhibitor | 151 |
| 40 | LY2886721 | BACE-1 inhibitor | 16 and 153 |
| 41 | Umibecestat (CNP520) | BACE-1 inhibitor | 155 |
| 42 | Verubecestat (MK8931) | BACE-1 inhibitor | 158 and 159 |
| 43 | Atabecestat (JNJ-54861911) | BACE-1 inhibitor | 161–163 |
| 44 | Lanabecestat | BACE-1 inhibitor | 167 |
| 45 | Elenbecestat (E2609) | BACE-1 inhibitor | 169 |
| 46 | NB-360 | Dual BACE-1 and BACE-2 inhibitor | 171 |
| 47 | RG712 | Dual BACE-1 and BACE-2 inhibitor | 174 |
| 48 | Semagacestat (LY450139) | γ-Secretase inhibitor | 178 |
| 49 | MK-0752 | γ-Secretase inhibitor | 154 |
| 50 | Avagacestat (BMS-708163) | Selective γ-secretase inhibitor | 182 |
| 51 | Begacestat (GSI-953) | Selective γ-secretase inhibitor | 186 |
| 52 | PF-3084014 | Notch sparing γ-secretase inhibitor | 189 and 190 |
| 53 | Ibuprofen | First generation GSM | 194 and 195 |
| 54 | Indomethacin | First generation GSM | 194 and 195 |
| 55 | Sulindac | First generation GSM | 194 and 195 |
| 56 | Tarenflurbil | Second generation GSM | 197 |
| 57 | JNJ-40418677 | Second generation GSM | 202 |
| 58 | EVP-0015962 | Second generation GSM | 203 |
| 59 | Itanapraced (CHF5074) | Second generation GSM | 205 |
| 60 | BIIB042 | Second generation GSM | 203 and 208 |
| 61 | E2012 | Second generation GSM | 154 and 210 |
| 62 | E2212 | Second generation GSM | 211 |
| 63 | BMS-869780 | Second generation GSM | 214 |
| 64 | BMS-932481 | Second generation GSM | 154 |
| 65 | PF-06648671 | Second generation GSM | 215 |
| 66 | SPI-014 | Second generation GSM | 217 |
| 67 | SPI-1865 | Second generation GSM | 217 |
| 68 | Dihydroergocristine | Second generation GSM | 219 |
| 69 | Acitretin | α-Secretase activator | 223 |
| 70 | Etazolate (EHT-0202) | GABAA modulator & α-secretase activator | 225 |
| 71 | PF-05212377 (SAM-760) | Selective 5-HT6R antagonist | 57 and 231 |
| 72 | Iadalopirdine | Selective 5-HT6R antagonist | 233 |
| 73 | Varoglutamstat (PQ-912) | QC inhibitor | 235 |
| 74 | Etanercept | Competitive TNF-α inhibitor | 243 |
| 75 | Thalidomide | Immunomodulator & TNF-α inhibitor | 244 |
| 76 | Muscimol | GABAA agonist | 250 |
| 77 | Propofol | GABAA agonist | 250 |
| 78 | SGS742 (CGP36742) | GABAB antagonist | 251 |
| 79 | CGP55845 | GABAB antagonist | 254 |
| 80 | Vitamin E | Antioxidant | 257 and 258 |
| 81 | Caffeine | Antioxidant | 261 |
| 82 | Silibinin | Antioxidant | 262 |
| 83 | Curcumin | Antioxidant | 263 |
| 84 | Quercetin | Antioxidant | 264 |
| 85 | Luteolin | Antioxidant | 265 |
| 86 | α-Lipoic acid | Mitochondrial-targeted antioxidant | 261 and 266 |
| 87 | Coenzyme Q10 | Mitochondrial-targeted antioxidant | 261 |
| 88 | Melatonin | Antioxidant | 267–271 |
| 89 | Selegiline | Antioxidant | 258 and 261 |
3. Conclusion and perspectives
Herein, AD potential therapeutic targets have been discussed, along with the clinically studied relevant drugs. As can be seen above, only a few drugs have been approved for the treatment of AD. Galantamine, donepezil, and rivastigmine (ChEIs), memantine (NMDA antagonist), and aducanumab and lecanemab (selective anti-Aβ monoclonal antibodies) are the currently approved drugs. This limited number of clinically used drugs against AD does not reflect the large number of proteins and enzymes identified as significant therapeutic targets of AD or the extensive number of drugs studied in the clinical trials of AD. But this image reflects the complexity of the disease and the lack of concrete evidence for the exact, definite cause of the disease.Accordingly, AD can be considered a multifactorial disease that requires a deeper understanding of its etiology to give priority to the most crucial targets. At the same time, early diagnosis of the disease is of great importance to maximize the benefits of the treatment; especially, it was found that amyloid plaques and neurofibrillary tangles can be detected decades before the appearance of symptoms.274 As we saw earlier, lecanemab has been approved for the early stages of AD. So, the identification of early detectable biomarkers of AD is highly significant. Furthermore, there is some evidence that the approach to prevent further formation of amyloid plaques is not enough for the treatment of AD in the sense that the neurodegeneration is triggered by the neurotoxic effects of the already aggregated Aβ. This may provide an explanation for the failure of almost all clinical trials based on the prevention of Aβ aggregation. So, all aspects should be taken into account in any further clinical study. The clinical data given in this work reveals GSM and QC inhibitors as the most promising classes involving inhibition of the formation of toxic Aβ. Additionally, α-secretase activators enhance proteolysis of APP in the non-amyloidogenic pathway. At the same time, the current study presented many therapeutic classes that have been proven to protect neurons from the toxic effects of Aβ. They include the neuroprotective GABAA agonists, antioxidants, anti-inflammatory, and immunomodulatory. Furthermore, the significant effects of PP2A activators and GSK-3β inhibitors in preventing neurofibrillary tangles should also be considered. For future work, it may be beneficial in such a case to apply molecular hybridization in order to develop potential drugs that work on more than one target linked firmly to AD. Otherwise, a combination of AD drugs is highly recommended. Clinical trials of drugs acting on Aβ and tau protein in combination with neuroprotective agents may change the current situation and reveal a significant protocol for AD treatment.
For the design of new anti-alzheimer small molecules, two isosteric nuclei showed very significant therapeutic properties, including reduction of the production of further toxic Aβ, activation of the cleavage of APP to soluble Aβ rather than the insoluble one, neuroprotection against the toxic effects of accumulated Aβ, and symptomatic improvement in cognitive functions. These two promising isosteric nuclei are benzimidazole and pyrazolopyridine. We saw above that their derivatives revealed QC inhibition, α-secretase activation, GABAA modulation, and 5HT antagonist activity, showing very promising clinical results. The importance of these nuclei was highlighted by the therapeutic effects of their isostere, xanthine nucleus. We found that xanthine derivatives exhibited PDE inhibition as well as antioxidant activity. The most significant of them is benzimidazole in the sense that imidazole is considered a zinc binding group in QC inhibition, and on it a class of anti-alzheimer drugs were built. This class is called non-NSAID-derived imidazole GSMs, as explained earlier. Accordingly, these nuclei can be used as a scaffold for building new candidates for potential multi-target anti-alzheimer activity, taking together the clinical results and molecular structure of their derivatives discussed in the current study.
Conflicts of interest
There is no any conflict of interest.Acknowledgements
The author thanks the permanent scientific committee for the promotion of professors and associate professors specializing in pharmaceutical chemistry, organic chemistry, and analytical chemistry at Al-Azhar University for suggesting writing this article.References
- J. C. de la Torre, Alzheimer's disease is incurable but preventable, J. Alzheimer's Dis., 2010, 20(3), 861–870 Search PubMed.
- F. Di Domenico, et al., The triangle of death in Alzheimer's disease brain: the aberrant cross-talk among energy metabolism, mammalian target of rapamycin signaling, and protein homeostasis revealed by redox proteomics, Antioxid. Redox Signaling, 2017, 26(8), 364–387 CrossRef CAS PubMed.
- J. Folch, et al., Current research therapeutic strategies for Alzheimer’s disease treatment, Neural Plast., 2016, 2016, 8501693 Search PubMed.
- M. Agrawal, et al., Nose-to-brain drug delivery: An update on clinical challenges and progress towards approval of anti-Alzheimer drugs, J. Controlled Release, 2018, 281, 139–177 CrossRef CAS PubMed.
- I. Bhushan, et al., Alzheimer's disease: Causes & treatment–A review, Ann. Biotechnol., 2018, 1(1), 1002 Search PubMed.
- K. B. Rajan, et al., Population estimate of people with clinical Alzheimer's disease and mild cognitive impairment in the United States (2020–2060), Alzheimer's Dementia, 2021, 17(12), 1966–1975 CrossRef PubMed.
- A. Di Stefano, et al., Drug delivery strategies for Alzheimer's disease treatment, Expert Opin. Drug Delivery, 2011, 8(5), 581–603 CrossRef CAS PubMed.
- G. Benzi and A. Moretti, Is there a rationale for the use of acetylcholinesterase inhibitors in the therapy of Alzheimer's disease?, Eur. J. Pharmacol., 1998, 346(1), 1–13 CrossRef CAS PubMed.
- J. J. Buccafusco and A. V. Terry, Multiple central nervous system targets for eliciting beneficial effects on memory and cognition, J. Pharmacol. Exp. Ther., 2000, 295(2), 438–446 CAS.
- H. C. Dringenberg, Alzheimer's disease: more than a ‘cholinergic disorder'—evidence that cholinergic–monoaminergic interactions contribute to EEG slowing and dementia, Behav. Brain Res., 2000, 115(2), 235–249 CrossRef CAS PubMed.
- E. K. Perry, et al., Correlation of cholinergic abnormalities with senile plaques and mental test scores in senile dementia, Br. Med. J., 1978, 2(6150), 1457–1459 CrossRef CAS PubMed.
- P. T. Francis, et al., Neurochemical studies of early-onset Alzheimer's disease: possible influence on treatment, N. Engl. J. Med., 1985, 313(1), 7–11 CrossRef CAS PubMed.
- M. Carreiras and J. Marco, Recent approaches to novel anti-Alzheimer therapy, Curr. Pharm. Des., 2004, 10(25), 3167 CrossRef CAS PubMed.
- A. Martorana, Z. Esposito and G. Koch, Beyond the cholinergic hypothesis: do current drugs work in Alzheimer's disease?, CNS Neurosci. Ther., 2010, 16(4), 235–245 CrossRef CAS PubMed.
- F. Hosseini, et al., Design, synthesis, and biological evaluation of novel 4-oxobenzo [d] 1, 2, 3-triazin-benzylpyridinum derivatives as potent anti-Alzheimer agents, Bioorg. Med. Chem., 2019, 27(13), 2914–2922 CrossRef CAS PubMed.
- Y. Zhang, et al., Amyloid β-based therapy for Alzheimer's disease: Challenges, successes and future, Signal Transduction Targeted Ther., 2023, 8(1), 248 CrossRef CAS PubMed.
- G. G. Glenner and C. W. Wong, Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein, Biochem. Biophys. Res. Commun., 1984, 120(3), 885–890 CrossRef CAS PubMed.
- L. Chen, Hypotheses of Alzheimer's Disease Pathogenesis, 2022 Search PubMed.
- K. L. Chagas Monteiro, et al., BACE-1 Inhibitors Targeting Alzheimer's Disease, Curr. Alzheimer Res., 2023, 20(3), 131–148 CrossRef CAS PubMed.
- A. S. Gurjar, et al., Design, synthesis, in silico and in vitro screening of 1, 2, 4-thiadiazole analogues as non-peptide inhibitors of beta-secretase, Bioorg. Chem., 2014, 57, 90–98 CrossRef CAS PubMed.
- S. Salloway, et al., Disease-modifying therapies in Alzheimer’s disease, Alzheimer’s Dementia, 2008, 4(2), 65–79 CrossRef CAS PubMed.
- S. Azimi, et al., Discovery of imidazopyridines containing isoindoline-1, 3-dione framework as a new class of BACE1 inhibitors: design, synthesis and SAR analysis, Eur. J. Med. Chem., 2017, 138, 729–737 CrossRef CAS PubMed.
- K. R. Sadleir, et al., Presynaptic dystrophic neurites surrounding amyloid plaques are sites of microtubule disruption, BACE1 elevation, and increased Aβ generation in Alzheimer's disease, Acta Neuropathol., 2016, 132, 235–256 CrossRef CAS PubMed.
- J. A. Hardy and G. A. Higgins, Alzheimer's disease: the amyloid cascade hypothesis, Science, 1992, 256(5054), 184–185 CrossRef CAS PubMed.
- X. Liu, et al., Clusterin transduces Alzheimer-risk signals to amyloidogenesis, Signal Transduction Targeted Ther., 2022, 7(1), 325 CrossRef PubMed.
- B.-L. Sun, et al., Critical thinking on amyloid-beta-targeted therapy: challenges and perspectives, Sci. China: Life Sci., 2021, 64, 926–937 CrossRef CAS PubMed.
- F. Kametani and M. Hasegawa, Reconsideration of amyloid hypothesis and tau hypothesis in Alzheimer's disease, Front. Neurosci., 2018, 12, 25 CrossRef PubMed.
- R. B. Maccioni, et al., The revitalized tau hypothesis on Alzheimer's disease, Arch. Med. Res., 2010, 41(3), 226–231 CrossRef CAS PubMed.
- A. F. Arnsten, et al., Hypothesis: Tau pathology is an initiating factor in sporadic Alzheimer's disease, Alzheimer's Dementia, 2021, 17(1), 115–124 CrossRef CAS PubMed.
- K. Iqbal, Tau and Alzheimer's disease: Past, Present and Future, Cytoskeleton, 2023 Search PubMed.
- E. Lauretti, O. Dincer and D. Praticò, Glycogen synthase kinase-3 signaling in Alzheimer's disease, Biochim. Biophys. Acta, Mol. Cell Res., 2020, 1867(5), 118664 CrossRef CAS PubMed.
- J.-M. Sontag and E. Sontag, Protein phosphatase 2A dysfunction in Alzheimer's disease, Front. Mol. Neurosci., 2014, 7, 16 CAS.
- W. Qian, et al., PP2A regulates tau phosphorylation directly and also indirectly via activating GSK-3β, J. Alzheimer’s Dis., 2010, 19(4), 1221–1229 CAS.
- X. Wang, et al., Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer's disease patients, Am. J. Pathol., 2008, 173(2), 470–482 CrossRef CAS PubMed.
- R. H. Swerdlow, J. M. Burns and S. M. Khan, The Alzheimer's disease mitochondrial cascade hypothesis: progress and perspectives, Biochim. Biophys. Acta, Mol. Basis Dis., 2014, 1842(8), 1219–1231 CrossRef CAS PubMed.
- R. H. Swerdlow, J. M. Burns and S. M. Khan, The Alzheimer's disease mitochondrial cascade hypothesis, J. Alzheimer’s Dis., 2010, 20(s2), S265–S279 Search PubMed.
- W. R. Markesbery, Oxidative stress hypothesis in Alzheimer's disease, Free Radicals Biol. Med., 1997, 23(1), 134–147 CrossRef CAS PubMed.
- D. Praticò, Oxidative stress hypothesis in Alzheimer's disease: a reappraisal, Trends Pharmacol. Sci., 2008, 29(12), 609–615 CrossRef PubMed.
- J. W. Olney, D. F. Wozniak and N. B. Farber, Excitotoxic neurodegeneration in Alzheimer disease: new hypothesis and new therapeutic strategies, Arch. Neurol., 1997, 54(10), 1234–1240 CrossRef CAS PubMed.
- M. T. Kabir, et al., NMDA receptor antagonists: repositioning of memantine as a multitargeting agent for Alzheimer's therapy, Curr. Pharm. Des., 2019, 25(33), 3506–3518 CrossRef CAS PubMed.
- W. Danysz, et al., Neuroprotective and symptomatological action of memantine relevant for Alzheimer's disease—a unified glutamatergic hypothesis on the mechanism of action, Neurotoxic. Res., 2000, 2(2–3), 85–97 CrossRef CAS PubMed.
- A. C. Paula-Lima, J. Brito-Moreira and S. T. Ferreira, Deregulation of excitatory neurotransmission underlying synapse failure in Alzheimer's disease, J. Neurochem., 2013, 126(2), 191–202 CrossRef CAS PubMed.
- S. Meftah and J. Gan, Alzheimer's disease as a synaptopathy: Evidence for dysfunction of synapses during disease progression, Front. Synaptic Neurosci., 2023, 15, 1129036 CrossRef CAS PubMed.
- F. J. Rodriguez-Jimenez, et al., Alzheimer's disease and synapse Loss: What can we learn from induced pluripotent stem Cells?, J. Adv. Res., 2023, 54, 105–118 CrossRef CAS PubMed.
- P. Dourlen, et al., The new genetic landscape of Alzheimer's disease: from amyloid cascade to genetically driven synaptic failure hypothesis?, Acta Neuropathol., 2019, 138, 221–236 CrossRef CAS PubMed.
- S. W. Pimplikar, Neuroinflammation in Alzheimer's disease: from pathogenesis to a therapeutic target, J. Clin. Immunol., 2014, 34, 64–69 CrossRef CAS PubMed.
- V. Calsolaro and P. Edison, Neuroinflammation in Alzheimer’s disease: current evidence and future directions, Alzheimer’s Dementia, 2016, 12(6), 719–732 CrossRef PubMed.
- B. Tamburini, et al., Emerging Roles of Cells and Molecules of Innate Immunity in Alzheimer's Disease, Int. J. Mol. Sci., 2023, 24(15), 11922 CrossRef CAS PubMed.
- C. K. Jung, et al., Fibrillar amyloid plaque formation precedes microglial activation, PLoS One, 2015, 10(3), e0119768 CrossRef PubMed.
- C. A. Colton, Heterogeneity of microglial activation in the innate immune response in the brain, J. Neuroimmune Pharmacol., 2009, 4, 399–418 CrossRef PubMed.
- P. Eikelenboom, et al., Neuroinflammation–an early event in both the history and pathogenesis of Alzheimer's disease, Neurodegener. Dis., 2010, 7(1–3), 38–41 CrossRef CAS PubMed.
- L. Bertram, C. M. Lill and R. E. Tanzi, The genetics of Alzheimer disease: back to the future, Neuron, 2010, 68(2), 270–281 CrossRef CAS PubMed.
- J. Andrade-Guerrero, et al., Alzheimer's Disease: An Updated Overview of Its Genetics, Int. J. Mol. Sci., 2023, 24(4), 3754 CrossRef CAS PubMed.
- R. E. Tanzi, The genetics of Alzheimer disease, Cold Spring Harbor Perspect. Med., 2012, 2(10), a006296 Search PubMed.
- M. Lozupone, et al., The Impact of Apolipoprotein E (APOE) Epigenetics on Aging and Sporadic Alzheimer's Disease, Biology, 2023, 12(12), 1529 CrossRef CAS PubMed.
- M. Wong-Guerra, et al., Revisiting the neuroinflammation hypothesis in Alzheimer's disease: a focus on the druggability of current targets, Front. Pharmacol., 2023, 14, 1161850 CrossRef CAS PubMed.
- C. E. Conti Filho, et al., Advances in Alzheimer's disease's pharmacological treatment, Front. Pharmacol., 2023, 14, 1101452 CrossRef PubMed.
- J. B. Langbaum, et al., Recommendations to address key recruitment challenges of Alzheimer's disease clinical trials, Alzheimer's Dementia, 2023, 19(2), 696–707 CrossRef PubMed.
- A. Basiri, et al., Design and synthesis of new piperidone grafted acetylcholinesterase inhibitors, Bioorg. Med. Chem. Lett., 2017, 27(2), 228–231 CrossRef CAS PubMed.
- L. Wang, et al., Design, synthesis, biological evaluation, and molecular modeling studies of chalcone-rivastigmine hybrids as cholinesterase inhibitors, Bioorg. Med. Chem., 2017, 25(1), 360–371 CrossRef CAS PubMed.
- Y. Furukawa-Hibi, et al., Butyrylcholinesterase inhibitors ameliorate cognitive dysfunction induced by amyloid-β peptide in mice, Behav. Brain Res., 2011, 225(1), 222–229 CrossRef CAS PubMed.
- D. Dimitrova, D. Getova and K. Saracheva, Effects of 3R, 16S-2-hydroxyethyl apovincaminate (HEAPO), donepezil and galantamine on learning and memory retention in naïve Wistar rats, Acta Pharm., 2023, 73(1), 91–105 CrossRef CAS PubMed.
- N. A.-E. El-Sayed, et al., Design, synthesis, in vitro and in vivo evaluation of novel pyrrolizine-based compounds with potential activity as cholinesterase inhibitors and anti-Alzheimer's agents, Bioorg. Chem., 2019, 93, 103312 CrossRef PubMed.
- C. O. Wilson, et al., Wilson and Gisvold's Textbook of Organic Medicinal and Pharmaceutical Chemistry, 2004 Search PubMed.
- A. Kumar and A. Singh, A review on Alzheimer's disease pathophysiology and its management: an update, Pharmacol. Rep., 2015, 67(2), 195–203 CrossRef CAS PubMed.
- L. Huang, et al., Discovery of indanone derivatives as multi-target-directed ligands against Alzheimer's disease, Eur. J. Med. Chem., 2014, 87, 429–439 CrossRef CAS PubMed.
- J. Wang, et al., Synthesis and evaluation of multi-target-directed ligands for the treatment of Alzheimer's disease based on the fusion of donepezil and melatonin, Bioorg. Med. Chem., 2016, 24(18), 4324–4338 CrossRef CAS PubMed.
- Z. Gazova, et al., Multi-target-directed therapeutic potential of 7-methoxytacrine-adamantylamine heterodimers in the Alzheimer's disease treatment, Biochim. Biophys. Acta, Mol. Basis Dis., 2017, 1863(2), 607–619 CrossRef CAS PubMed.
- A. S. M. Arshad, et al., Synthesis, characterization and crystal structure of new tetrahydro-β-carboline as acetylcholinesterase inhibitor, J. Mol. Struct., 2020, 1200, 127070 CrossRef.
- M. B. Colovic, et al., Acetylcholinesterase inhibitors: pharmacology and toxicology, Curr. Neuropharmacol., 2013, 11(3), 315–335 CrossRef CAS PubMed.
- J. L. Yakel, Cholinergic receptors: functional role of nicotinic ACh receptors in brain circuits and disease, Pflug. Arch. Eur. J. Physiol., 2013, 465(4), 441–450 CrossRef CAS PubMed.
- L. M. Gault, et al., ABT-126 monotherapy in mild-to-moderate Alzheimer's dementia: randomized double-blind, placebo and active controlled adaptive trial and open-label extension, Alzheimer’s Res. Ther., 2016, 8(1), 1–13 CrossRef PubMed.
- H. Florian, et al., Efficacy and safety of ABT-126 in subjects with mild-to-moderate Alzheimer's disease on stable doses of acetylcholinesterase inhibitors: a randomized, double-blind, placebo-controlled study, J. Alzheimer’s Dis., 2016, 51(4), 1237–1247 CAS.
- O. Sabri, et al., Cognitive correlates of α4β2 nicotinic acetylcholine receptors in mild Alzheimer's dementia, Brain, 2018, 141(6), 1840–1854 CrossRef PubMed.
- L. E. Rueter, et al., ABT-089: pharmacological properties of a neuronal nicotinic acetylcholine receptor agonist for the potential treatment of cognitive disorders, CNS Drug Rev., 2004, 10(2), 167–182 CrossRef CAS PubMed.
- R. A. Lenz, et al., Adaptive, dose-finding phase 2 trial evaluating the safety and efficacy of ABT-089 in mild to moderate Alzheimer disease, Alzheimer Dis. Assoc. Disord., 2015, 29(3), 192–199 CrossRef CAS PubMed.
- M. Hernandez, et al., Different muscarinic receptor subtypes mediating the phasic activity and basal tone of pig isolated intravesical ureter, Br. J. Pharmacol., 1993, 110(4), 1413–1420 CrossRef CAS PubMed.
- N. C. Bodick, et al., Effects of xanomeline, a selective muscarinic receptor agonist, on cognitive function and behavioral symptoms in Alzheimer disease, Arch. Neurol., 1997, 54(4), 465–473 CrossRef CAS PubMed.
- Z. Li, et al., Xanomeline derivative EUK1001 attenuates Alzheimer's disease pathology in a triple transgenic mouse model, Mol. Med. Rep., 2017, 16(5), 7835–7840 CrossRef CAS PubMed.
- Y. Cui, et al., Enhancement of memory function in aged mice by a novel derivative of xanomeline, Cell Res., 2008, 18(11), 1151–1153 CrossRef CAS PubMed.
- W. Si, et al., A novel derivative of xanomeline improves fear cognition in aged mice, Neurosci. Lett., 2010, 473(2), 115–119 CrossRef CAS PubMed.
- B. C. Tang, Y. T. Wang and J. Ren, Basic information about memantine and its treatment of Alzheimer’s disease and other clinical applications, Ibrain, 2023, 9(3), 340–348 CrossRef PubMed.
- S. Matsunaga, et al., The efficacy and safety of memantine for the treatment of Alzheimer's disease, Expert Opin. Drug Saf., 2018, 17(10), 1053–1061 CrossRef CAS PubMed.
- T. Kishi, et al., Memantine for Alzheimer's disease: an updated systematic review and meta-analysis, J. Alzheimer’s Dis., 2017, 60(2), 401–425 CAS.
- P. Xia, et al., Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses, J. Neurosci., 2010, 30(33), 11246–11250 CrossRef CAS PubMed.
- M. R. Farlow and N. M. D. A. receptor antagonists, A new therapeutic approach for Alzheimer's disease, Geriatrics, 2004, 59(6), 22–27 Search PubMed.
- O. Sanders and L. Rajagopal, Phosphodiesterase inhibitors for Alzheimer's disease: a systematic review of clinical trials and epidemiology with a mechanistic rationale, J. Alzheimer’s Dis. Reports, 2020, 4(1), 185–215 Search PubMed.
- M. Shekarian, et al., Neuroprotective effects of vinpocetine, as a phosphodiesterase 1 inhibitor, on long-term potentiation in a rat model of Alzheimer's disease, BMC Neurosci., 2023, 24(1), 1–10 CrossRef PubMed.
- A. E. Medina, Therapeutic utility of phosphodiesterase type I inhibitors in neurological conditions, Front. Neurosci., 2011, 5, 21 CAS.
- A. A. Ali, et al., Vinpocetine mitigates aluminum-induced cognitive impairment in socially isolated rats, Physiol. Behav., 2019, 208, 112571 CrossRef CAS PubMed.
- H. I. Ahmed, S. A. Abdel-Sattar and H. S. Zaky, Vinpocetine halts ketamine-induced schizophrenia-like deficits in rats: impact on BDNF and GSK-3β/β-catenin pathway, Naunyn-Schmiedeberg’s Arch. Pharmacol., 2018, 391, 1327–1338 CrossRef CAS PubMed.
- V. Fattori, et al., Vinpocetine reduces diclofenac-induced acute kidney injury through inhibition of oxidative stress, apoptosis, cytokine production, and NF-κB activation in mice, Pharmacol. Res., 2017, 120, 10–22 CrossRef CAS PubMed.
- G. Svab, et al., The mitochondrial targets of neuroprotective drug vinpocetine on primary neuron cultures, brain capillary endothelial cells, synaptosomes, and brain mitochondria, Neurochem. Res., 2019, 44, 2435–2447 CrossRef CAS PubMed.
- L. J. Thal, et al., The safety and lack of efficacy of vinpocetine in Alzheimer's disease, J. Am. Geriatr. Soc., 1989, 37(6), 515–520 CrossRef CAS PubMed.
- X. Wei, et al., Targeting phosphodiesterase 4 as a therapeutic strategy for cognitive improvement, Bioorg. Chem., 2023, 130, 106278 CrossRef CAS PubMed.
- J. Prickaerts, P. R. Heckman and A. Blokland, Investigational phosphodiesterase inhibitors in phase I and phase II clinical trials for Alzheimer's disease, Expert Opin. Invest. Drugs, 2017, 26(9), 1033–1048 CrossRef CAS PubMed.
- A. Zagórska, et al., Drug Discovery and Development Targeting Dementia, Pharm., 2023, 16(2), 151 Search PubMed.
- N. Hasan, et al., Roflumilast reduces pathological symptoms of sporadic Alzheimer's disease in rats produced by Intracerebroventricular Streptozotocin by inhibiting NF-κB/BACE-1 mediated Aβ production in the Hippocampus and activating the cAMP/BDNF signalling pathway, Neurotoxic. Res., 2022, 40(2), 432–448 CrossRef CAS PubMed.
- T. Treves and A. Korczyn, Denbufylline in dementia: a double-blind controlled study, Dementia Geriatr. Cognit. Disord., 1999, 10(6), 505–510 CrossRef CAS PubMed.
- L. Frölich, et al., Evaluation of the efficacy, safety and tolerability of orally administered BI 409306, a novel phosphodiesterase type 9 inhibitor, in two randomised controlled phase II studies in patients with prodromal and mild Alzheimer's disease, Alzheimer’s Res. Ther., 2019, 11, 1–11 CrossRef PubMed.
- E. M. Schwam, et al., A multicenter, double-blind, placebo-controlled trial of the PDE9A inhibitor, PF-04447943, in Alzheimer's disease, Curr. Alzheimer Res., 2014, 11(5), 413–421 CrossRef CAS PubMed.
- S. Da Mesquita, et al., Meningeal lymphatics affect microglia responses and anti-Aβ immunotherapy, Nature, 2021, 593(7858), 255–260 CrossRef CAS PubMed.
- L. Vaillant-Beuchot, et al., Accumulation of amyloid precursor protein C-terminal fragments triggers mitochondrial structure, function, and mitophagy defects in Alzheimer's disease models and human brains, Acta Neuropathol., 2021, 141, 39–65 CrossRef CAS PubMed.
- B. R. Troutwine, et al., Mitochondrial function and Aβ in Alzheimer's disease postmortem brain, Neurobiol. Dis., 2022, 171, 105781 CrossRef CAS PubMed.
- G. E. Vitek, B. Decourt and M. N. Sabbagh, Lecanemab (BAN2401): an anti–beta-amyloid monoclonal antibody for the treatment of Alzheimer disease, Expert Opin. Invest. Drugs, 2023, 32(2), 89–94 CrossRef CAS PubMed.
- H. D. Larkin, Lecanemab gains FDA approval for early Alzheimer disease, JAMA, J. Am. Med. Assoc., 2023, 329(5), 363 Search PubMed.
- J. Park, C. Simpson and K. Patel, Lecanemab: A Humanized Monoclonal Antibody for the Treatment of Early Alzheimer Disease, Ann. Pharmacother., 2023, 10600280231218253 CrossRef PubMed.
- M. Vaz, et al., Role of aducanumab in the treatment of Alzheimer's disease: Challenges and opportunities, Clin. Interventions Aging, 2022, 797–810 CrossRef PubMed.
- S. Walsh, et al., Aducanumab for Alzheimer’s Disease?, Br. Med. J., 2021, 374, n1682 CrossRef PubMed.
- J. Sevigny, et al., The antibody aducanumab reduces Aβ plaques in Alzheimer's disease, Nature, 2016, 537(7618), 50–56 CrossRef CAS PubMed.
- S. A. Beshir, et al., Aducanumab therapy to treat Alzheimer’s disease: A narrative review, Int. J. Alzheimer’s Dis., 2022, 2022, 9343514 Search PubMed.
- M. Tagliapietra, Aducanumab for the treatment of Alzheimer's disease, Drugs Today, 2022, 58(10), 465–477 CrossRef PubMed.
- S. M. Hoy, Lecanemab: first approval, Drugs, 2023, 83(4), 359–365 CrossRef CAS PubMed.
- J. Cummings, et al., Lecanemab: appropriate use recommendations, J. Prev. Alzheimers Dis., 2023, 1–16 Search PubMed.
- C. H. Van Dyck, et al., Lecanemab in early Alzheimer's disease, N. Engl. J. Med., 2023, 388(1), 9–21 CrossRef CAS PubMed.
- N. H. Parikh, et al., Current trends and updates in the treatment of Alzheimer's disease, in Alzheimer's Disease and Advanced Drug Delivery Strategies, Elsevier, 2024, pp. 373–390 Search PubMed.
- E. McDade, et al., Lecanemab in patients with early Alzheimer's disease: Detailed results on biomarker, cognitive, and clinical effects from the randomized and open-label extension of the phase 2 proof-of-concept study, Alzheimer’s Res. Ther., 2022, 14(1), 1–17 CrossRef PubMed.
- L. S. Honig, et al., Trial of solanezumab for mild dementia due to Alzheimer's disease, N. Engl. J. Med., 2018, 378(4), 321–330 CrossRef CAS PubMed.
- S. Salloway, et al., A trial of gantenerumab or solanezumab in dominantly inherited Alzheimer's disease, Nat. Med., 2021, 27(7), 1187–1196 CrossRef CAS PubMed.
- A. I. Abushouk, et al., Bapineuzumab for mild to moderate Alzheimer's disease: a meta-analysis of randomized controlled trials, BMC Neurol., 2017, 17(1), 1–13 CrossRef PubMed.
- R. Vandenberghe, et al., Bapineuzumab for mild to moderate Alzheimer's disease in two global, randomized, phase 3 trials, Alzheimer’s Res. Ther., 2016, 8(1), 1–13 CrossRef PubMed.
- L. Lannfelt, et al., Perspectives on future Alzheimer therapies: amyloid-β protofibrils-a new target for immunotherapy with BAN2401 in Alzheimer's disease, Alzheimer’s Res. Ther., 2014, 6(2), 1–8 Search PubMed.
- S. Ostrowitzki, et al., A phase III randomized trial of gantenerumab in prodromal Alzheimer's disease, Alzheimer’s Res. Ther., 2017, 9(1), 1–15 CrossRef PubMed.
- R. J. Bateman, et al., Gantenerumab: an anti-amyloid monoclonal antibody with potential disease-modifying effects in early Alzheimer's disease, Alzheimer’s Res. Ther., 2022, 14(1), 1–17 CrossRef PubMed.
- R. L. Patton, et al., Amyloid-β peptide remnants in AN-1792-immunized Alzheimer's disease patients: a biochemical analysis, Am. J. Pathol., 2006, 169(3), 1048–1063 CrossRef CAS PubMed.
- E. Masliah, et al., Aβ vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease, Neurology, 2005, 64(1), 129–131 CrossRef CAS PubMed.
- I. Ferrer, et al., Neuropathology and pathogenesis of encephalitis following amyloid β immunization in Alzheimer's disease, Brain Pathol., 2004, 14(1), 11–20 CrossRef CAS PubMed.
- S. R. Robinson, et al., Lessons from the AN 1792 Alzheimer vaccine: lest we forget, Neurobiol. Aging, 2004, 25(5), 609–615 CrossRef CAS PubMed.
- M. Hull, et al., Long-Term extensions of randomized vaccination trials of ACC-001 and QS-21 in mild to moderate alzheimer's disease, Curr. Alzheimer Res., 2017, 14(7), 696–708 CrossRef CAS PubMed.
- F. Pasquier, et al., Two phase 2 multiple ascending–dose studies of vanutide cridificar (ACC-001) and QS-21 adjuvant in mild-to-moderate Alzheimer's disease, J. Alzheimer’s Dis., 2016, 51(4), 1131–1143 CAS.
- C. Van Dyck, et al., Vanutide Cridificar (ACC-001) and QS-21 Adjuvant in Individuals with Early Alzheimer's Disease: Amyloid Imaging Positron Emission Tomography and Safety Results from a Phase 2 Study, J. Prev. Alzheimers Dis., 2016, 3(2), 75–84 CAS.
- E. Molina, et al., AB1601 topline results–Phase 2 study of ABvac40 in patients with amnestic mild cognitive impairment (a-MCI) or very mild Alzheimer's Disease (Vm-AD), Alzheimer's Dementia, 2022, 18, e065633 CrossRef.
- J. A. Hey, et al., Clinical pharmacokinetics and safety of ALZ-801, a novel prodrug of tramiprosate in development for the treatment of Alzheimer's disease, Clin. Pharmacokinet., 2018, 57, 315–333 CrossRef CAS PubMed.
- M. Tolar, et al., Aducanumab, gantenerumab, BAN2401, and ALZ-801—the first wave of amyloid-targeting drugs for Alzheimer's disease with potential for near term approval, Alzheimer’s Res. Ther., 2020, 12, 1–10 CrossRef PubMed.
- S. Manzano, et al., A review on tramiprosate (Homotaurine) in alzheimer’s disease and other neurocognitive disorders, Front. Neurol., 2020, 11, 614 CrossRef PubMed.
- P. Kocis, et al., Elucidating the Aβ42 anti-aggregation mechanism of action of tramiprosate in Alzheimer’s disease: integrating molecular analytical methods, pharmacokinetic and clinical data, CNS Drugs, 2017, 31, 495–509 CrossRef CAS PubMed.
- S. Abushakra, et al., Clinical benefits of tramiprosate in alzheimer's disease are associated with higher number of APOE4 alleles: the “APOE4 gene-dose effect, J. Prev. Alzheimer's Dis., 2016, 3(4), 219–228 CAS.
- M. Tolar, S. Abushakra and M. Sabbagh, The path forward in Alzheimer's disease therapeutics: Reevaluating the amyloid cascade hypothesis, Alzheimer's Dementia, 2019 Search PubMed.
- S. Abushakra, et al., Prevalence of Amyloid-Related Imaging Abnormalities in APOE4/4 Homozygotes with Early Alzheimer’s Disease: Baseline Findings from Ongoing Clinical Trials of Oral Anti-amyloid Agent ALZ-801 (Valiltramiprosate)(P5-6.003), Neurology, 2023, 100(17_supplement_2) DOI:10.1212/WNL.0000000000203550.
- E. E. Congdon and E. M. Sigurdsson, Tau-targeting therapies for Alzheimer disease, Nat. Rev. Neurol., 2018, 14(7), 399–415 CrossRef CAS PubMed.
- C. B. Malpas, et al., A phase IIa randomized control trial of VEL015 (Sodium Selenate) in mild-moderate Alzheimer's disease, J. Alzheimer’s Dis., 2016, 54(1), 223–232 CAS.
- V. Melis, et al., Effects of oxidized and reduced forms of methylthioninium in two transgenic mouse tauopathy models, Behav. Pharmacol., 2015, 26(4), 353 CrossRef CAS PubMed.
- L. Martin, et al., Tau protein kinases: involvement in Alzheimer's disease, Ageing Res. Rev., 2013, 12(1), 289–309 CrossRef CAS PubMed.
- J. J. Lucas, et al., Decreased nuclear β-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3β conditional transgenic mice, EMBO J., 2001, 20(1–2), 27–39 CrossRef CAS PubMed.
- J. M. Domínguez, et al., Evidence for irreversible inhibition of glycogen synthase kinase-3β by tideglusib, J. Biol. Chem., 2012, 287(2), 893–904 CrossRef PubMed.
- A. Martinez, et al., First non-ATP competitive glycogen synthase kinase 3 β (GSK-3β) inhibitors: thiadiazolidinones (TDZD) as potential drugs for the treatment of Alzheimer's disease, J. Med. Chem., 2002, 45(6), 1292–1299 CrossRef CAS PubMed.
- B. DaRocha-Souto, et al., Activation of glycogen synthase kinase-3 beta mediates β-amyloid induced neuritic damage in Alzheimer's disease, Neurobiol. Dis., 2012, 45(1), 425–437 CrossRef CAS PubMed.
- L. Serenó, et al., A novel GSK-3β inhibitor reduces Alzheimer's pathology and rescues neuronal loss in vivo, Neurobiol. Dis., 2009, 35(3), 359–367 CrossRef PubMed.
- S. Lovestone, et al., A phase II trial of tideglusib in Alzheimer's disease, J. Alzheimer’s Dis., 2015, 45(1), 75–88 CAS.
- V. Stambolic, L. Ruel and J. R. Woodgett, Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells, Curr. Biol., 1996, 6(12), 1664–1669 CrossRef CAS PubMed.
- E. Portelius, et al., β-site amyloid precursor protein-cleaving enzyme 1 (BACE1) inhibitor treatment induces Aβ5-X peptides through alternative amyloid precursor protein cleavage, Alzheimer’s Res. Ther., 2014, 6, 1–8 CrossRef PubMed.
- P. C. May, et al., Robust central reduction of amyloid-β in humans with an orally available, non-peptidic β-secretase inhibitor, J. Neurosci., 2011, 31(46), 16507–16516 CrossRef CAS PubMed.
- P. C. May, et al., The potent BACE1 inhibitor LY2886721 elicits robust central Aβ pharmacodynamic responses in mice, dogs, and humans, J. Neurosci., 2015, 35(3), 1199–1210 CrossRef PubMed.
- D. Kumar, et al., Secretase inhibitors for the treatment of Alzheimer's disease: Long road ahead, Eur. J. Med. Chem., 2018, 148, 436–452 CrossRef CAS PubMed.
- L.-K. Huang, S.-P. Chao and C.-J. Hu, Clinical trials of new drugs for Alzheimer disease, J. Biomed. Sci., 2020, 27(1), 1–13 CrossRef PubMed.
- U. Neumann, et al., The BACE-1 inhibitor CNP 520 for prevention trials in Alzheimer's disease, EMBO Mol. Med., 2018, 10(11), e9316 CrossRef PubMed.
- A. Graf, et al., Fts3-01-01: Umibecestat (Cnp520) Is Not Associated With Changes In Hippocampal Morphology In Rats Or Changes In Csf Ad Biomarkers In Humans Treated For 3 Months, Alzheimer's Dementia, 2019, 15, P872 Search PubMed.
- M. F. Egan, et al., Randomized trial of verubecestat for mild-to-moderate Alzheimer's disease, N. Engl. J. Med., 2018, 378(18), 1691–1703 CrossRef CAS PubMed.
- T. Voss, et al., Progression from Prodromal Alzheimer’s Disease to Mild Alzheimer’s Disease Dementia in the Verubecestat APECS Study: Adjudicating Diagnostic Transitions, J. Alzheimer’s Dis., 2023, 92(1), 341–348 CAS.
- M. F. Egan, et al., Randomized trial of verubecestat for prodromal Alzheimer's disease, N. Engl. J. Med., 2019, 380(15), 1408–1420 CrossRef CAS PubMed.
- M. Timmers, et al., BACE1 dynamics upon inhibition with a BACE inhibitor and correlation to downstream Alzheimer's disease markers in elderly healthy participants, J. Alzheimer’s Dis., 2017, 56(4), 1437–1449 CAS.
- M. Timmers, et al., Profiling the dynamics of CSF and plasma Aβ reduction after treatment with JNJ-54861911, a potent oral BACE inhibitor, Alzheimer’s Dement.: Transl. Res. Clin. Interv., 2016, 2(3), 202–212 CrossRef PubMed.
- M. Timmers, et al., Pharmacodynamics of atabecestat (JNJ-54861911), an oral BACE1 inhibitor in patients with early Alzheimer's disease: randomized, double-blind, placebo-controlled study, Alzheimer’s Res. Ther., 2018, 10, 1–18 CrossRef PubMed.
- D. Henley, et al., Preliminary Results of a Trial of Atabecestat in Preclinical Alzheimer's Disease, The N. Engl. J. Med., 2019, 380(15), 1483–1485 CrossRef PubMed.
- S. De Jonghe, et al., Biopsy pathology and immunohistochemistry of a case of immune-mediated drug-induced liver injury with Atabecestat, Hepatology, 2021, 73(1), 452–455 CrossRef CAS PubMed.
- R. Sperling, et al., Findings of efficacy, safety, and biomarker outcomes of atabecestat in preclinical Alzheimer disease: a truncated randomized phase 2b/3 clinical trial, JAMA Neurol., 2021, 78(3), 293–301 CrossRef PubMed.
- A. M. Wessels, et al., Efficacy and safety of lanabecestat for treatment of early and mild Alzheimer disease: the AMARANTH and DAYBREAK-ALZ randomized clinical trials, JAMA Neurol., 2020, 77(2), 199–209 CrossRef PubMed.
- J. A. Zimmer, et al., Lanabecestat: Neuroimaging results in early symptomatic Alzheimer's disease, Alzheimer’s Dement.: Transl. Res. Clin. Interv., 2021, 7(1), e12123 CrossRef PubMed.
- D. Caouette, Eisai and Biogen to Discontinue Phase III Clinical Studies of BACE Inhibitor Elenbecestat in Early Alzheimer's Disease, Eisai’s press release, 2019 Search PubMed.
- B. P. Imbimbo and M. Watling, Investigational BACE inhibitors for the treatment of Alzheimer's disease, Expert Opin. Invest. Drugs, 2019, 28(11), 967–975 CrossRef CAS PubMed.
- U. Neumann, R. Machauer and D. R. Shimshek, The β-secretase (BACE) inhibitor NB-360 in preclinical models: From amyloid-β reduction to downstream disease-relevant effects, Br. J. Pharmacol., 2019, 176(18), 3435–3446 CrossRef CAS PubMed.
- U. Neumann, et al., A novel BACE inhibitor NB-360 shows a superior pharmacological profile and robust reduction of amyloid-β and neuroinflammation in APP transgenic mice, Mol. Neurodegener., 2015, 10(1), 1–15 CrossRef PubMed.
- D. R. Shimshek, et al., Pharmacological BACE1 and BACE2 inhibition induces hair depigmentation by inhibiting PMEL17 processing in mice, Sci. Rep., 2016, 6(1), 21917 CrossRef CAS PubMed.
- H. Jacobsen, et al., Combined treatment with a BACE inhibitor and anti-Aβ antibody gantenerumab enhances amyloid reduction in APPLondon mice, J. Neurosci., 2014, 34(35), 11621–11630 CrossRef CAS PubMed.
- C. G. Parsons and G. Rammes, Preclinical to phase II amyloid beta (Aβ) peptide modulators under investigation for Alzheimer's disease, Expert Opin. Invest. Drugs, 2017, 26(5), 579–592 CrossRef CAS PubMed.
- F. Tadros Hakem, Y. Farid Fouad and R. K. Arafa, Gamma Secretase as an Important Drug Target for Management of Alzheimer's Disease: A Comprehensive Review, Curr. Top. Med. Chem., 2024, 24(2), 109–127 CrossRef PubMed.
- B. De Strooper, T. Iwatsubo and M. S. Wolfe, Presenilins and γ-secretase: structure, function, and role in Alzheimer disease, Cold Spring Harbor Perspect. Med., 2012, 2(1) CAS.
- P. C. May, et al., O3-06-07 Multi-compartmental pharmacodynamic assessment of the functional gamma-secretase inhibitor LY450139 in PDAPP transgenic mice and non-transgenic mice, Neurobiol. Aging, 2004,(25), S65 CrossRef.
- D. B. Henley, et al., Development of semagacestat (LY450139), a functional γ-secretase inhibitor, for the treatment of Alzheimer's disease, Expert Opin. Pharmacother., 2009, 10(10), 1657–1664 CrossRef CAS PubMed.
- R. Doody, Alzheimer’s Disease Cooperative Study Steering Committee, E. Siemers, G. Sethuraman, R. Mohs and Semagacestat Study Group, et al.) A phase 3 trial of semagacestat for treatment of Alzheimer's disease, N. Engl. J. Med., 2013, 369, 341–350 CrossRef CAS PubMed.
- R. Rosen, et al., Efficacy and tolerability of vardenafil in men with mild depression and erectile dysfunction: the depression-related improvement with vardenafil for erectile response study, Am. J. Psychiatry, 2006, 163(1), 79–87 CrossRef PubMed.
- G. Tong, et al., Multicenter, randomized, double-blind, placebo-controlled, single-ascending dose study of the oral γ-secretase inhibitor BMS-708163 (Avagacestat): tolerability profile, pharmacokinetic parameters, and pharmacodynamic markers, Clin. Ther., 2012, 34(3), 654–667 CrossRef CAS PubMed.
- S. C. Mayer, et al., Discovery of begacestat, a Notch-1-sparing γ-secretase inhibitor for the treatment of Alzheimer's disease, J. Med. Chem., 2008, 51(23), 7348–7351 CrossRef CAS PubMed.
- V. Coric, et al., Safety and tolerability of the γ-secretase inhibitor avagacestat in a phase 2 study of mild to moderate Alzheimer disease, Arch. Neurol., 2012, 69(11), 1430–1440 CrossRef PubMed.
- V. Coric, et al., Targeting prodromal Alzheimer disease with avagacestat: a randomized clinical trial, JAMA Neurol., 2015, 72(11), 1324–1333 CrossRef PubMed.
- C. R. Hopkins, ACS Chemical Neuroscience Molecule Spotlight on Begacestat (GSI-953), ACS, 2012, 3–4 CAS.
- D. C. Cole, et al., (S)-N-(5-Chlorothiophene-2-sulfonyl)-β, β-diethylalaninol a Notch-1-sparing γ-secretase inhibitor, Bioorg. Med. Chem. letters, 2009, 19(3), 926–929 CrossRef CAS PubMed.
- R. L. Martone, et al., Begacestat (GSI-953): a novel, selective thiophene sulfonamide inhibitor of amyloid precursor protein γ-secretase for the treatment of Alzheimer's disease, J. Pharmacol. Exp. Ther., 2009, 331(2), 598–608 CrossRef CAS PubMed.
- B. P. Imbimbo, Alzheimer's disease: γ-secretase inhibitors, Drug Discovery Today: Ther. Strategies, 2008, 5(3), 169–175 Search PubMed.
- D. M. Barten and C. F. Albright, Therapeutic strategies for Alzheimer's disease, Mol. Neurobiol., 2008, 37, 171–186 CrossRef CAS PubMed.
- R. S. Doody, et al., A phase 3 trial of semagacestat for treatment of Alzheimer's disease, N. Engl. J. Med., 2013, 369(4), 341–350 CrossRef CAS PubMed.
- Y. Mitani, et al., Differential effects between γ-secretase inhibitors and modulators on cognitive function in amyloid precursor protein-transgenic and nontransgenic mice, J. Neurosci., 2012, 32(6), 2037–2050 CrossRef CAS PubMed.
- J.-Y. Hur, γ-Secretase in Alzheimer's disease, Exp. Mol. Med., 2022, 54(4), 433–446 CrossRef CAS PubMed.
- C. J. Crump, D. S. Johnson and Y.-M. Li, Development and mechanism of γ-secretase modulators for Alzheimer's disease, Biochemistry, 2013, 52(19), 3197–3216 CrossRef CAS PubMed.
- S. Weggen, et al., A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity, Nature, 2001, 414(6860), 212–216 CrossRef CAS PubMed.
- S. Mekala, G. Nelson and Y.-M. Li, Recent developments of small molecule γ-secretase modulators for Alzheimer's disease, RSC Med. Chem., 2020, 11(9), 1003–1022 RSC.
- T. Morihara, et al., Selective inhibition of Aβ42 production by NSAID R-enantiomers, J. Neurochem., 2002, 83(4), 1009–1012 CrossRef CAS PubMed.
- Y. Yu, et al., Safety, tolerability, pharmacokinetics, and pharmacodynamics of the novel γ-secretase modulator, E2212, in healthy human subjects, J. Clin. Pharmacol., 2014, 54(5), 528–536 CrossRef CAS PubMed.
- R. C. Green, et al., Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial, JAMA, J. Am. Med. Assoc., 2009, 302(23), 2557–2564 CrossRef CAS PubMed.
- J. L. Eriksen, et al., NSAIDs and enantiomers of flurbiprofen target γ-secretase and lower Aβ42 in vivo, J. Clin. Invest., 2003, 112(3), 440–449 CrossRef CAS PubMed.
- G. K. Wilcock, et al., Efficacy and safety of tarenflurbil in mild to moderate Alzheimer's disease: a randomised phase II trial, Lancet Neurol., 2008, 7(6), 483–493 CrossRef CAS PubMed.
- B. Van Broeck, et al., Chronic treatment with a novel γ-secretase modulator, JNJ-40418677, inhibits amyloid plaque formation in a mouse model of Alzheimer's disease, Br. J. Pharmacol., 2011, 163(2), 375–389 CrossRef CAS PubMed.
- K. Rogers, et al., Modulation of γ-secretase by EVP-0015962 reduces amyloid deposition and behavioral deficits in Tg2576 mice, Mol. Neurodegener., 2012, 7, 1–18 Search PubMed.
- J. Zhao, et al., Targeting amyloidogenic processing of APP in Alzheimer's disease, Front. Mol. Neurosci., 2020, 13, 137 CrossRef CAS PubMed.
- S. Sivilia, et al., Multi-target action of the novel anti-Alzheimer compound CHF5074: in vivo study of long term treatment in Tg2576 mice, BMC Neurosci., 2013, 14(1), 1–14 CrossRef PubMed.
- B. P. Imbimbo, et al., Pharmacokinetics and pharmacodynamics of CHF5074 after short-term administration in healthy subjects, Alzheimer Dis. Assoc. Disord., 2013, 27(3), 278–286 CrossRef CAS PubMed.
- H. Peng, et al., Discovery of BIIB042, a potent, selective, and orally bioavailable γ-secretase modulator, ACS Med. Chem. Lett., 2011, 2(10), 786–791 CrossRef CAS PubMed.
- R. H. Scannevin, et al., BIIB042, a novel γ-secretase modulator, reduces amyloidogenic Aβ isoforms in primates and rodents and plaque pathology in a mouse model of Alzheimer's disease, Neuropharmacology, 2016, 103, 57–68 CrossRef CAS PubMed.
- W. Wuli, et al., Human-induced pluripotent stem cells and herbal small-molecule drugs for treatment of Alzheimer's disease, Int. J. Mol. Sci., 2020, 21(4), 1327 CrossRef CAS PubMed.
- K. Nakano-Ito, et al., E2012-induced cataract and its predictive biomarkers, Toxicol. Sci., 2014, 137(1), 249–258 CrossRef CAS PubMed.
- P. Nie, A. Vartak, and Y.-M. Li, γ-Secretase inhibitors and modulators: Mechanistic insights into the function and regulation of γ-Secretase, in Seminars in cell & developmental biology, Elsevier, 2020 Search PubMed.
- A. Hall and T. R. Patel, γ-Secretase modulators: current status and future directions, Prog. Med. Chem., 2014, 53, 101–145 CAS.
- T. T. Wager, et al., Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties, ACS Chem. Neurosci., 2010, 1(6), 435–449 CrossRef CAS PubMed.
- J. H. Toyn, et al., Identification and Preclinical Pharmacology of the-Secretase Modulator BMS-869780, Int. J. Alzheimer’s Dis., 2014, 2014, 431858 Search PubMed.
- J. E. Ahn, et al., Pharmacokinetic and Pharmacodynamic Effects of a γ-Secretase Modulator, PF-06648671, on CSF Amyloid-β Peptides in Randomized Phase I Studies, Clin. Pharmacol. Ther., 2020, 107(1), 211–220 CrossRef CAS PubMed.
- Z. Zhao, et al., Discovery of a tetrahydrobenzisoxazole series of γ-secretase modulators, ACS Med. Chem. Lett., 2017, 8(10), 1002–1006 CrossRef CAS PubMed.
- M. A. Findeis, et al., Natural product and natural product-derived gamma secretase modulators from Actaea Racemosa extracts, Medicines, 2015, 2(3), 127–140 CrossRef CAS PubMed.
- H. Harms, et al., Aβ-42 lowering agents from the marine-derived fungus Dichotomomyces cejpii, Steroids, 2015, 104, 182–188 CrossRef CAS PubMed.
- X. Lei, et al., The FDA-approved natural product dihydroergocristine reduces the production of the Alzheimer's disease amyloid-β peptides, Sci. Rep., 2015, 5(1), 16541 CrossRef CAS PubMed.
- S. F. Lichtenthaler and C. Haass, Amyloid at the cutting edge: activation of α-secretase prevents amyloidogenesis in an Alzheimer disease mouse model, J. Clin. Invest., 2004, 113(10), 1384–1387 CrossRef CAS PubMed.
- B. De Strooper, R. Vassar and T. Golde, The secretases: enzymes with therapeutic potential in Alzheimer disease, Nat. Rev. Neurol., 2010, 6(2), 99–107 CrossRef CAS PubMed.
- R. E. Tanzi and L. Bertram, Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective, Cell, 2005, 120(4), 545–555 CrossRef CAS PubMed.
- F. Tippmann, et al., Up-regulation of the α-secretase ADAM10 by retinoic acid receptors and acitretin, FASEB J., 2009, 23(6), 1643–1654 CrossRef CAS PubMed.
- D. Holthoewer, et al., Acitretin, an enhancer of alpha-secretase expression, crosses the blood–brain barrier and is not eliminated by P-glycoprotein, Neurodegener. Dis., 2012, 10(1–4), 224–228 CrossRef CAS PubMed.
- M. Marcade, et al., Etazolate, a neuroprotective drug linking GABAA receptor pharmacology to amyloid precursor protein processing, J. Neurochem., 2008, 106(1), 392–404 CrossRef CAS PubMed.
- B. Vellas, et al., EHT0202 in Alzheimer's disease: a 3-month, randomized, placebo-controlled, double-blind study, Curr. Alzheimer Res., 2011, 8(2), 203–212 CrossRef CAS PubMed.
- G. S. V. Higa, et al., 5-HT-dependent synaptic plasticity of the prefrontal cortex in postnatal development, Sci. Rep., 2022, 12(1), 21015 CrossRef CAS PubMed.
- D. V. Eremin, et al., Serotonin Receptors as a Potential Target in the Treatment of Alzheimer's Disease, Biochem., 2023, 88(12), 2023–2042 CAS.
- K. Kucwaj-Brysz, et al., Chemical update on the potential for serotonin 5-HT6 and 5-HT7 receptor agents in the treatment of Alzheimer's disease, Bioorg. Med. Chem. Letters, 2021, 49, 128275 CrossRef CAS PubMed.
- M. Andrews, B. Tousi and M. N. Sabbagh, 5HT6 antagonists in the treatment of Alzheimer’s dementia: current progress, Neurol. Ther., 2018, 7, 51–58 CrossRef PubMed.
- T. Fullerton, et al., A Phase 2 clinical trial of PF-05212377 (SAM-760) in subjects with mild to moderate Alzheimer's disease with existing neuropsychiatric symptoms on a stable daily dose of donepezil, Alzheimer’s Res. Ther., 2018, 10, 1–10 CrossRef PubMed.
- S. Matsunaga, H. Fujishiro and H. Takechi, Efficacy and safety of idalopirdine for Alzheimer's disease: a systematic review and meta-analysis, Int. Psychogeriatr., 2019, 31(11), 1627–1633 CrossRef PubMed.
- A. Atri, et al., Effect of idalopirdine as adjunct to cholinesterase inhibitors on change in cognition in patients with Alzheimer disease: three randomized clinical trials, JAMA, J. Am. Med. Assoc., 2018, 319(2), 130–142 CrossRef CAS PubMed.
- D. Chen, et al., Development and evolution of human glutaminyl cyclase inhibitors (QCIs): an alternative promising approach for disease-modifying treatment of Alzheimer’s disease, Front. Aging Neurosci., 2023, 15, 1209863 CrossRef CAS PubMed.
- J. R. Coimbra, et al., An overview of glutaminyl cyclase inhibitors for Alzheimer's disease, Future Med. Chem., 2019, 11(24), 3179–3194 CrossRef CAS PubMed.
- I. Lues, et al., A phase 1 study to evaluate the safety and pharmacokinetics of PQ912, a glutaminyl cyclase inhibitor, in healthy subjects, Alzheimer’s Dement.: Transl. Res. Clin. Interv., 2015, 1(3), 182–195 CrossRef PubMed.
- P. Scheltens, et al., Safety, tolerability and efficacy of the glutaminyl cyclase inhibitor PQ912 in Alzheimer's disease: results of a randomized, double-blind, placebo-controlled phase 2a study, Alzheimer’s Res. Ther., 2018, 10(1), 1–14 CrossRef PubMed.
- D. K. Vijayan and K. Y. Zhang, Human glutaminyl cyclase: Structure, function, inhibitors and involvement in Alzheimer's disease, Pharmacol. Res., 2019, 147, 104342 CrossRef CAS PubMed.
- Z.-Z. Si, et al., Targeting neuroinflammation in Alzheimer's disease: From mechanisms to clinical applications, Neural Regener. Res., 2023, 18(4), 708 CrossRef CAS PubMed.
- T. Song, et al., Mitochondrial dysfunction, oxidative stress, neuroinflammation, and metabolic alterations in the progression of Alzheimer's disease: A meta-analysis of in vivo magnetic resonance spectroscopy studies, Ageing Res. Rev., 2021, 72, 101503 CrossRef CAS PubMed.
- C. L. Chen, et al., Alzheimer's disease THErapy with NEuroaid (ATHENE): A randomized double-blind delayed-start trial, J. Am. Med. Dir. Assoc., 2022, 23(3), 379–386 CrossRef PubMed.
- D. Plantone, et al., The Role of TNF-α in Alzheimer's Disease: A Narrative Review, Cells, 2023, 13(1), 54 CrossRef PubMed.
- B. Goffe and J. C. Cather, Etanercept: an overview, J. Am. Acad. Dermatol., 2003, 49(2), 105–111 CrossRef PubMed.
- B. Decourt, et al., Poor safety and tolerability hamper reaching a potentially therapeutic dose in the use of thalidomide for Alzheimer's disease: results from a double-blind, placebo-controlled trial, Curr. Alzheimer Res., 2017, 14(4), 403–411 CrossRef CAS PubMed.
- S. Jo, et al., GABA from reactive astrocytes impairs memory in mouse models of Alzheimer's disease, Nat. Med., 2014, 20(8), 886–896 CrossRef CAS PubMed.
- Z. Wu, et al., Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzheimer's disease model, Nature communications, 2014, 5(1), 4159 CrossRef CAS PubMed.
- M. Bauer, et al., Delta-like 1 participates in the specification of ventral midbrain progenitor derived dopaminergic neurons, J. Neurochem., 2008, 104(4), 1101–1115 CrossRef CAS PubMed.
- J. Drott, et al., Etazolate improves performance in a foraging and homing task in aged rats, Eur. J. Pharmacol., 2010, 634(1–3), 95–100 CrossRef CAS PubMed.
- H. Shao, et al., Chronic treatment with anesthetic propofol improves cognitive function and attenuates caspase activation in both aged and Alzheimer's disease transgenic mice, J. Alzheimer’s Dis., 2014, 41(2), 499–513 CAS.
- Y. Zhang, et al., Chronic treatment with anesthetic propofol attenuates β-amyloid protein levels in brain tissues of aged mice, Transl. Neurodegener., 2014, 3(1), 1–7 CAS.
- W. Froestl, et al., SGS742: the first GABAB receptor antagonist in clinical trials, Biochem. Pharmacol., 2004, 68(8), 1479–1487 CrossRef CAS PubMed.
- D. Getova and N. Bowery, Effects of high-affinity GABA B receptor antagonists on active and passive avoidance responding in rodents with gamma-hydroxybutyrolactone-induced absence syndrome, Psychopharmacology, 2001, 157, 89–95 CrossRef CAS PubMed.
- K. Helm, et al., GABAB receptor antagonist SGS742 improves spatial memory and reduces protein binding to the cAMP response element (CRE) in the hippocampus, Neuropharmacology, 2005, 48(7), 956–964 CrossRef CAS PubMed.
- C. L. LaSarge, et al., Blockade of GABA (B) receptors completely reverses age-related learning impairment, Neuroscience, 2009, 164(3), 941–947 CrossRef CAS PubMed.
- Y. Li, et al., Implications of GABAergic neurotransmission in Alzheimer's disease, Front. Aging Neurosci., 2016, 8, 31 Search PubMed.
- C. Foy, et al., Plasma chain-breaking antioxidants in Alzheimer’s disease, vascular dementia and Parkinson’s disease, Q. J. Med., 1999, 92(1), 39–45 CrossRef CAS PubMed.
- T. Montiel, et al., Role of oxidative stress on β-amyloid neurotoxicity elicited during impairment of energy metabolism in the hippocampus: protection by antioxidants, Exp. Neurol., 2006, 200(2), 496–508 CrossRef CAS PubMed.
- E. E. Devore, et al., Dietary antioxidants and long-term risk of dementia, Arch. Neurol., 2010, 67(7), 819–825 CrossRef PubMed.
- M. Sano, et al., A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer's disease, N. Engl. J. Med., 1997, 336(17), 1216–1222 CrossRef CAS PubMed.
- D. Dias-Santagata, et al., Oxidative stress mediates tau-induced neurodegeneration in Drosophila, J. Clin. Invest., 2007, 117(1), 236–245 CrossRef CAS PubMed.
- Y. Feng and X. Wang, Antioxidant therapies for Alzheimer’s disease, Oxid. Med. Cell. Longevity, 2012, 2012, 472932 Search PubMed.
- P. Lu, et al., Silibinin prevents amyloid β peptide-induced memory impairment and oxidative stress in mice, Br. J. Pharmacol., 2009, 157(7), 1270–1277 CrossRef CAS PubMed.
- K. Goozee, et al., Examining the potential clinical value of curcumin in the prevention and diagnosis of Alzheimer's disease, Br. J. Nutr., 2016, 115(3), 449–465 CrossRef CAS PubMed.
- L. G. Costa, et al., Mechanisms of neuroprotection by quercetin: counteracting oxidative stress and more, Oxid. Med. Cell. Longevity, 2016, 2016, 2986796 Search PubMed.
- H. Wang, et al., Ameliorating effect of luteolin on memory impairment in an Alzheimer's disease model, Mol. Med. Rep., 2016, 13(5), 4215–4220 CrossRef CAS PubMed.
- J. F. Quinn, et al., Chronic dietary α-lipoic acid reduces deficits in hippocampal memory of aged Tg2576 mice, Neurobiol. Aging, 2007, 28(2), 213–225 CrossRef CAS PubMed.
- J. Zhou, et al., Melatonin impairs NADPH oxidase assembly and decreases superoxide anion production in microglia exposed to amyloid-β1–42, J. Pineal Res., 2008, 45(2), 157–165 CrossRef CAS PubMed.
- Y.-q. Deng, et al., Effects of melatonin on wortmannin-induced tau hyperphosphorylation, Acta Pharmacol. Sin., 2005, 26(5), 519–526 CrossRef CAS PubMed.
- S. J. Liu and J. Z. Wang, Alzheimer-like tau phosphorylation induced by wortmannin in vivo and its attenuation by melatonin, Acta Pharmacol. Sin., 2002, 23(2), 183–187 CAS.
- D. L. Wang, et al., Melatonin attenuates isoproterenol-induced protein kinase A overactivation and tau hyperphosphorylation in rat brain, J. Pineal Res., 2004, 37(1), 11–16 CrossRef CAS PubMed.
- X.-C. Li, et al., Effect of melatonin on calyculin A-induced tau hyperphosphorylation, Eur. J. Pharmacol., 2005, 510(1–2), 25–30 CrossRef CAS PubMed.
- Z. Feng and J.-t. Zhang, Protective effect of melatonin on β-amyloid-induced apoptosis in rat astroglioma c6 cells and its mechanism, Free Radicals Biol. Med., 2004, 37(11), 1790–1801 CrossRef CAS PubMed.
- T. Thomas, Monoamine oxidase-B inhibitors in the treatment of Alzheimers disease, Neurobiol. Aging, 2000, 21(2), 343–348 CrossRef CAS PubMed.
- A. Wuestefeld, et al., Age-related and amyloid-beta-independent tau deposition and its downstream effects, Brain, 2023, awad135 Search PubMed.
| This journal is © The Royal Society of Chemistry 2024 |