DOI:
10.1039/D3RA08257A
(Paper)
RSC Adv., 2024,
14, 8283-8292
A facile on–off fluorescence approach for fluvoxamine determination in pharmaceutical tablets; application to content uniformity testing†
Received
6th December 2023
, Accepted 3rd March 2024
First published on 11th March 2024
Abstract
A green-complied spectrofluorimetric approach for quantification of the antidepressant, fluvoxamine, has been established. The method that has been suggested relies on the development of an association complex between fluvoxamine and erythrosine B in an acetate buffer solution. After being excited at 530 nm, the quenching in erythrosine B's native fluorescence caused by complex formation with fluvoxamine was detected at a wavelength of 552 nm. The values of fluorescence quenching at the most optimal reaction conditions were rectilinear at the concentration range of 0.2–2.0 μg mL−1, with a good correlation coefficient (r = 0.9998). The detection limit for the method was 0.03 μg mL−1 while the quantitation limit was 0.09 μg mL−1. The suggested approach has been validated according to the ICH. The established approach was effectively used to determine the drug under study in its dosage form with an average percent recovery of 98.92 ± 0.87 (n = 5), with no effect caused by the existing excipients. The proposed approach was also successfully used for the content uniformity test.
1. Introduction
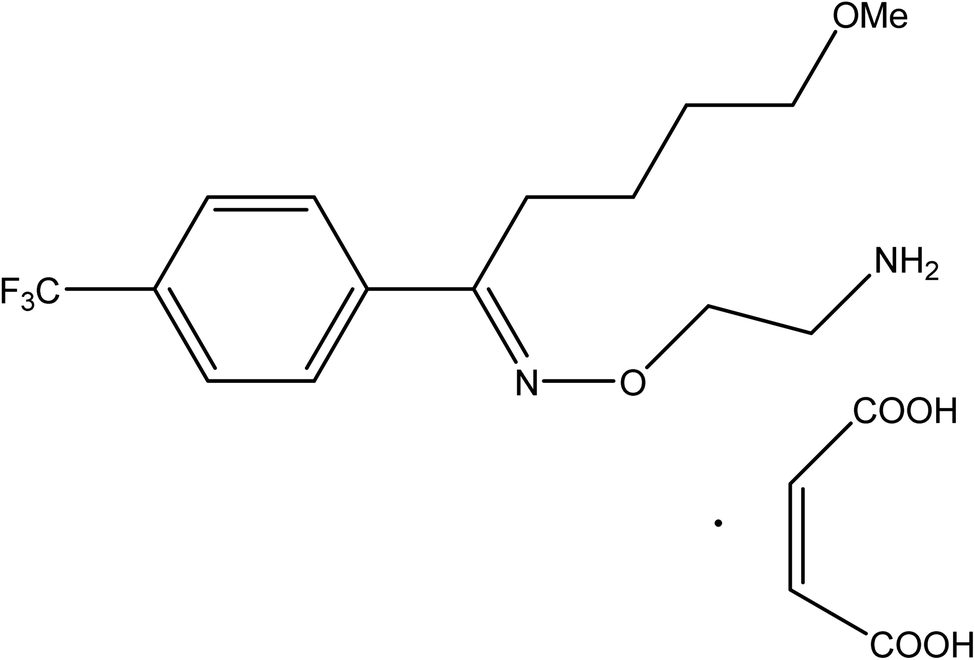
Fluvoxamine maleate (FLX, Fig. 1) is 5-methoxy-1-[4-(trifluoromethyl)phenyl]-1-pentanone-o-(2-aminoethyl) oxime maleate. FLX is a Selective Inhibitor for Serotonin Reuptake (SSRI). It has been reported to have been successfully used in the management of several types of depression.1 It helps to reduce obsessive or compulsive behavior by restoring the balance of serotonin, a natural neurotransmitter in the brain.2 Because of its selective mechanism of action, the indicated drug's adverse effects differ from those documented for tricyclic antidepressants.2
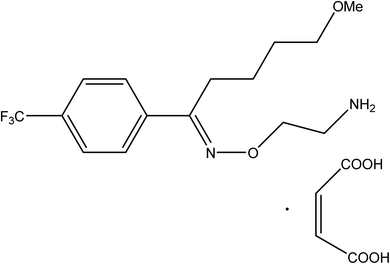 |
| | Fig. 1 Fluvoxamine maleate. | |
Reviewing the literature shows that there are numerous approaches for determining the medication under investigation. These strategies include: spectrophotometry,3–6 spectrofluorimetry,7–12 liquid chromatography (HPLC),13–18 gas chromatography (GC),19 thin layer chromatography,20–22 electrochemical methods,23–25 and capillary electrophoresis.26 HPLC necessitates the use of expensive instruments and huge quantities of highly pure liquids. Furthermore, it is tedious, time-consuming and requires lengthy sample pre-treatment procedures. Techniques like gas chromatography, electrochemistry, and electrophoresis require complicated apparatus and highly qualified personnel. Although spectrophotometric techniques are straightforward, their sensitivity is fairly low, and their dynamic concentration range is narrow.
Meanwhile, spectrofluorimetric methods are more selective due to the presence of two spectra, extremely sensitive, and need a modest instrument as well as very easy sample preparation. Nevertheless, the published spectrofluorimetric procedures for the mentioned medication suffered from certain limitations such as the use of high-priced chemical,12 damaging organic solvents,9 high degrees of temperature for extended periods of time,7–10 or possesses low sensitivity.11
There are several xanthene dyes that could be utilized as fluorescent probes through ion pairing. Merbromin is not recommended because it contain mercury atom which is hazardous compound. Eosin Y was previously used but the sensitivity of the method was lower than the current work.11 Rhodamine 6G and other basic dyes could not be used for fluvoxamine determination because of the electrostatic repulsion instead of attraction which will prevent the formation of the ion pair. On the hand, using erythrosine B as Spectrofluorimetric probe was frequently employed for the purpose of quantifying a variety of compounds with high sensitivity.27,28 Therefore, in the present study, it was utilized for the determination of FLX. The reaction relies on forming an association complex between the dye and the investigated compound. With its basic moiety (amino group), FLX can combine with erythrosine B to produce an ion pair complex, allowing its analysis using simple spectrofluorimetric approach. In an acidic medium, the amino group is protonated forming a cationic species so that being suitable for interaction with the negatively charged dye molecule. Acetate buffer was used in order to adjust the pH of the reaction medium, thus allowing the formation of the complex. The complex is formed by electrostatic interaction between lipophilic compound protonated base and the dye which possesses a negative charge. A spectrofluorimetric approach makes use of the produced complex's ability to quench erythrosine's native fluorescence.
In this work, an efficient and sensitive analytical procedure has been developed for FLX analysis. The method produced satisfactory findings when it was employed to analyze pharmaceutical formulations. The analytical performance of the method such as limits of detection and quantitation, linearity, precision and accuracy, will be carefully assessed, and confirmed in consistent with the relevant guiding rules.
The suggested method provided a major advance in simplicity and speed for FLX determination. The developed method has a remarkably simple and fast procedure, which distinguishes it from chromatographic methods. By adopting a simplified and efficient sample preparation step, the need for complex and labor-intensive processes were eliminated, saving valuable time and resources. The simplicity of the present method extends beyond sample preparation. The measurements were performed using an easy-to-use device that requires minimal effort to operate. This aspect greatly enhances the accessibility and practicality of the developed method, making it suitable for both novice and experienced researchers alike. Moreover, the remarkable simplicity of the method does not affect its performance. Despite its simplistic nature, the present approach consistently delivers accurate and reliable results.
2. Experimental
2.1. Apparatus
Measurements were performed using JASCO FP-8350 spectrofluorometer. The device has a quartz cell (1 cm), a Xe-arc lamp (150 W), and a medium-sensitivity PMT voltage. A slit width of 5 nm for the emission and excitation monochromators, and 1000 nm min−1 scanning rate were employed.
2.2. Reagents and materials
FLX was kindly supplied by MASH Premiere Pharmaceutical company (Egypt, Cairo, Badr City). Faverin® tablets (Abbott Healthcare SAS) each of which containing 50 mg FLX was purchased from the Egyptian market. Erythrosine-B was purchased from MP Biomedicals LLC (Illkirch, France) and prepared by dissolving 50 mg in 250 mL of double-distilled water to give a concentration of 2.27 × 10−4 M. By mixing the proper quantities of acetic acid (0.1 M) and sodium acetate (0.1 M), acetate buffer solutions with pH range of 3.0–4.4 was constituted. To obtain the desired pH, these solutions were combined in various ratios maintaining the overall buffer strength of 0.1 M.
2.3. Preparation of standard solutions
Ten milligrams of FLX were dissolved in 100 mL of double-distilled water to make the standard stock solution. After that, this solution had been diluted with the same solvent to prepare working solutions with concentrations between 2 and 20 μg mL−1.
2.4. General procedure for analysis
Aliquot of 1.0 mL of the medication solution (0.2–2.0 μg mL−1) was put into a series of 10 mL calibrated flasks. After that, 0.7 mL of 0.1 M acetate buffer (pH 3.5) accompanied by 0.7 mL of erythrosine B (2.27 × 10−4 M) were added. The volume was filled to the mark using double-distilled water. Following excitation at 530 nm, fluorescence intensity was measured at 552 nm. The same procedure was used to prepare a blank experiment, but without the drug solution. The fluorescence intensity difference in absence (blank) and presence of the medication (ΔF) had been plotted against the final concentration of the drug.
2.5. Procedure for FLX assay in dosage form (Faverin® tablets)
A quantity from finely powdered 10 tablets was weighed accurately to be equivalent to 50 mg FLX and carefully transferred to a volumetric glass flask (100 mL) that previously had 30 mL of distilled water in it. After around 30 minutes of sonication, the solution was completed to the mark by adding water. After filtration of the solution, the initial portion of filtrate was thrown away. Quantitative dilution of a part of the filtrate with distilled water produced a solution with a final concentration that was within the required range. Following the general methodology, the final solution's content of the medication was estimated. For testing the content uniformity.
2.6. Procedure for content uniformity test
In compliance with USP specifications (Chap. 905), the formula of FLX tablets experienced the testing for content uniformity (CU). To examine the uniformity of the contents of 10 Faverin® 50 mg tablets, a separate inspection of each tablet was carried out following the procedure outlined under “procedure for FLX assay in dosage form”.
3. Results and discussion
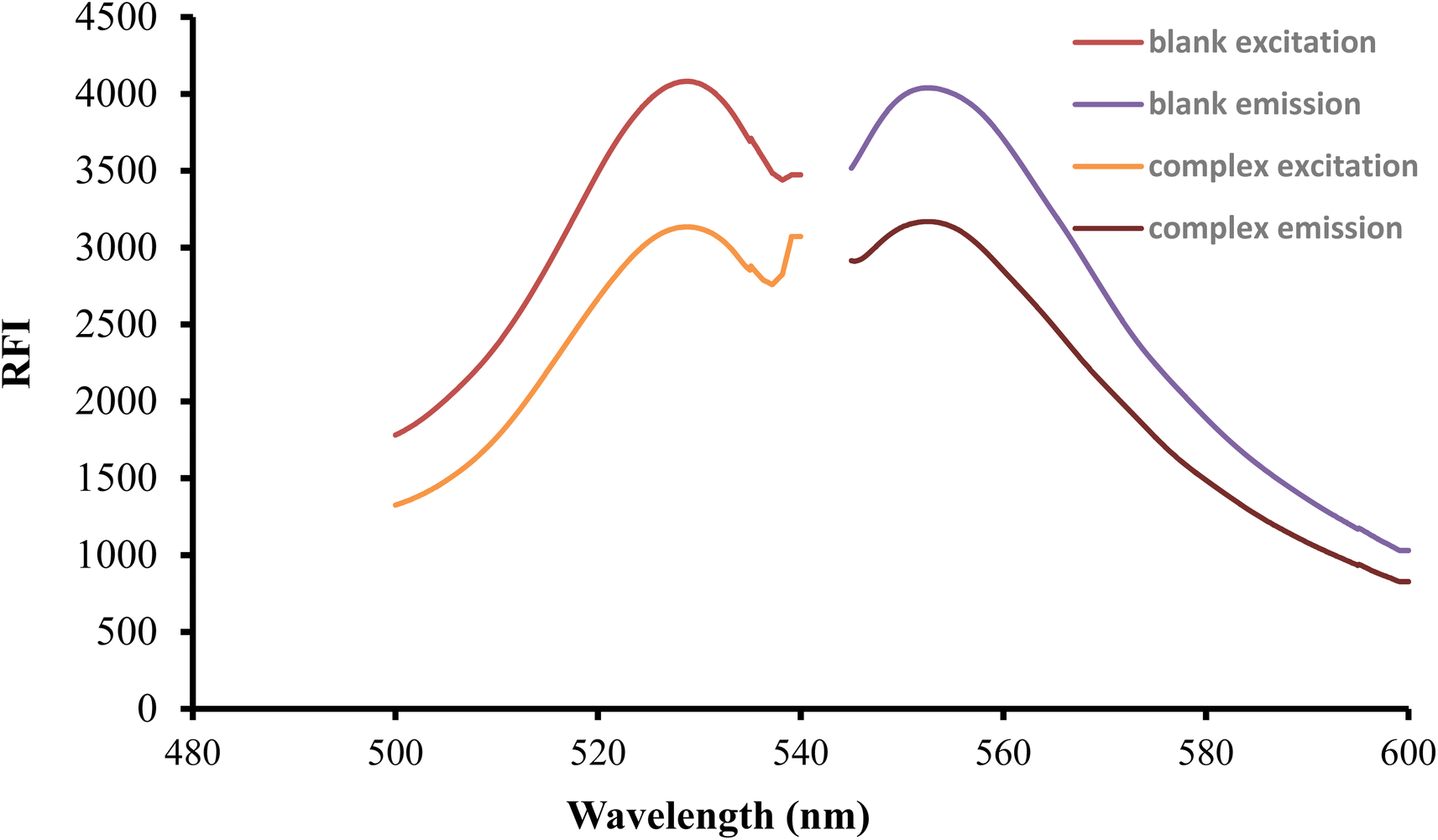
After being excited at 530 nm, the erythrosine B aqueous solution had a native fluorescence that was detected at 552 nm. When the solution of the studied medication was mixed with the reagent solution, the intensity of fluorescence was noticeably reduced as shown in Fig. 2. The studied drug and erythrosine B form an ion pair complex, which results in the fluorescence quenching action. A rectilinear relationship existed between the medication concentration and the extent of dye fluorescence intensity decline.
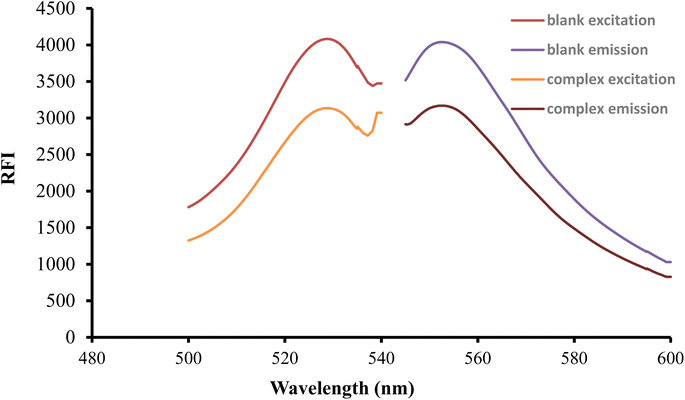 |
| | Fig. 2 Excitation and emission spectra of erythrosine-B (2.27 × 10−4 M) and its binary complex with FLX (1.5 μg mL−1). | |
3.1. Optimization of experimental conditions
The formation and stability of the reaction product were affected by numerous experimental variables. These variables were carefully examined and optimized. While each of these variables was altered individually, the others remained unchanged. Those variables included pH, erythrosine B volume, diluting solvent as well as the type and volume of the buffer solution.
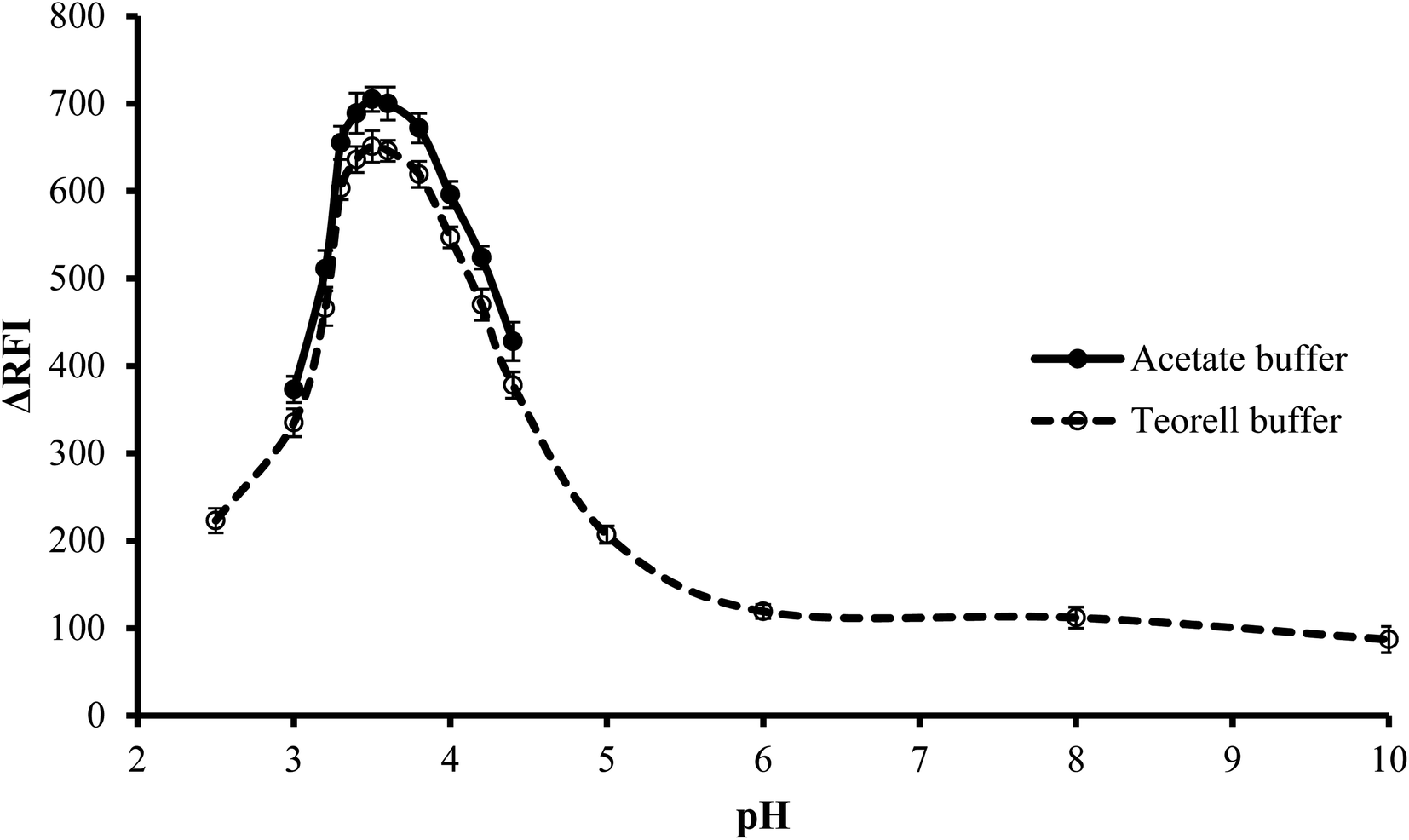
3.1.1. Effect of type of buffer. The impact of the buffer type on the fluorescence quenching of erythrosine B was investigated at the optimum pH. Two different types of buffer solutions, Teorell–Stenhagen and acetate buffers, were investigated. Fig. 3 illustrates that both buffer systems have relatively the same effect, but upon using acetate buffer, the fluorescence quenching was slightly higher and thus was chosen. The slightly lower value with Teorell–Stenhagen buffer may be due to its phosphate ions content. Compared to the acetate anion, phosphate is a stronger proton acceptor, making it a stronger competitor with erythrosine B for binding to the drug cation. Therefore, it has a higher ability to reduce the quenching effect of the drug on the fluorescence of the dye.29
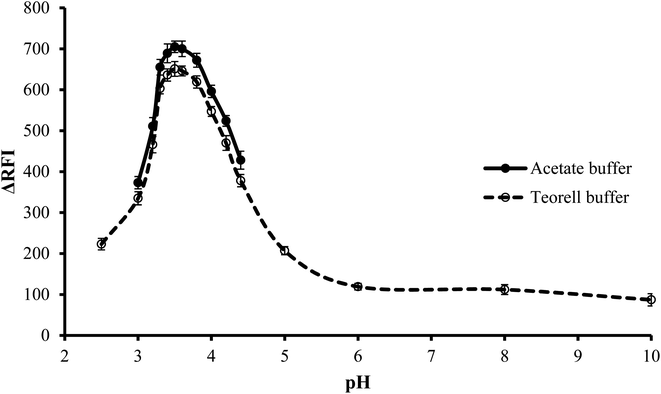 |
| | Fig. 3 Effect of pH on the RFI of the association complex formed between FLX (1.0 μg mL−1) and erythrosine-B. | |
3.1.2. Effect of pH. To conduct the investigation in a wide range of pH, Teorell–Stenhagen buffer (2.5–10) was used. As shown in Fig. 3, the highest fluorescence quenching values were in the range 3.3 to 3.8 and that there is no significant fluorescence quenching at a pH higher than 4.5. Since the region where the effect of pH is evident is the acidic region and falls within the effective pH range of acetate buffer (3.75–5.75), so the effect of pH was examined utilizing an acetate buffer with a pH range of 3.0–4.4. Although this pH range was limited, it was suitable for the present study. It was also observed that, pH 3.5 ± 0.1 produced the greatest fluorescence quenching. The results were reduced by increasing or decreasing the pH values.
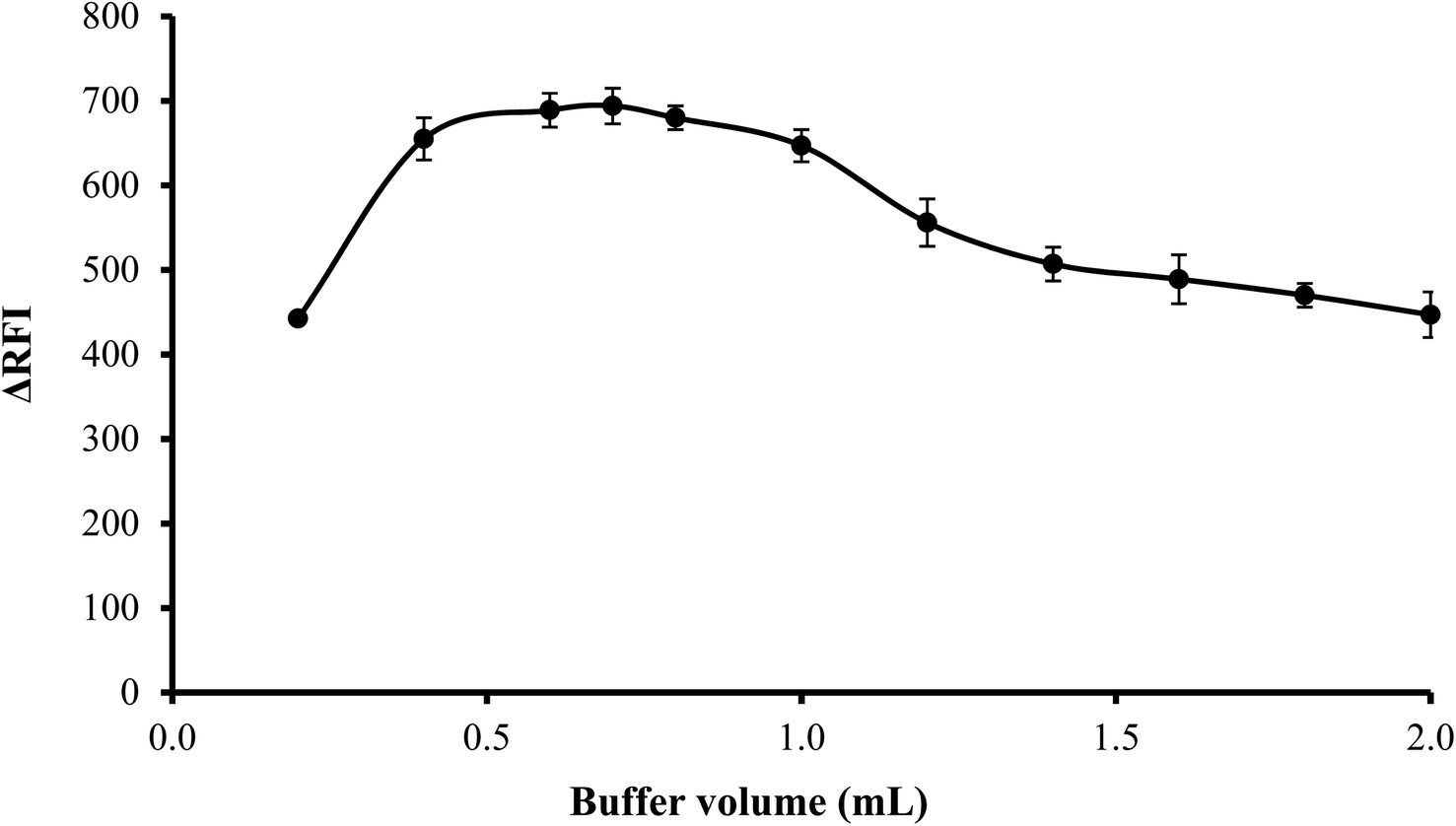
3.1.3. Effect of volume of buffer. The influence of volume of buffer on the quenching of erythrosine B's fluorescence intensity was examined over the range of 0.2–2.0 mL of acetate buffer solution (pH 3.5). Best results were observed when 0.4–1.0 mL of the buffer were used. Lower volume was not sufficient to adjust the pH of the medium and thus low values were obtained. On the other hand, higher buffer volume means higher ionic strength and in this case, the anionic counterpart (acetate ions) of the buffer will compete with erythrosine to bind with the drug cation. Thus impede the interaction of the drug with the dye. Thus, 0.7 mL of acetate buffer was adequate to obtain the highest fluorescence quenching, as indicated in Fig. 4.
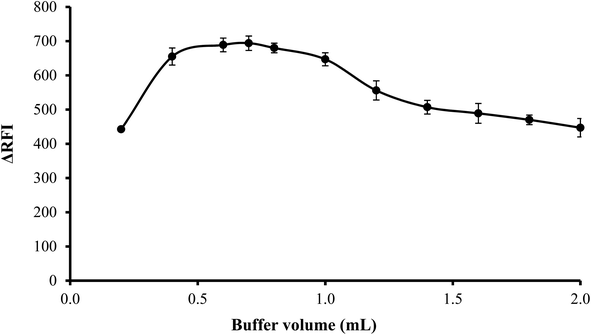 |
| | Fig. 4 Effect of buffer volume on the RFI effect of the association complex formed between FLX (1 μg mL−1) and erythrosine-B. | |
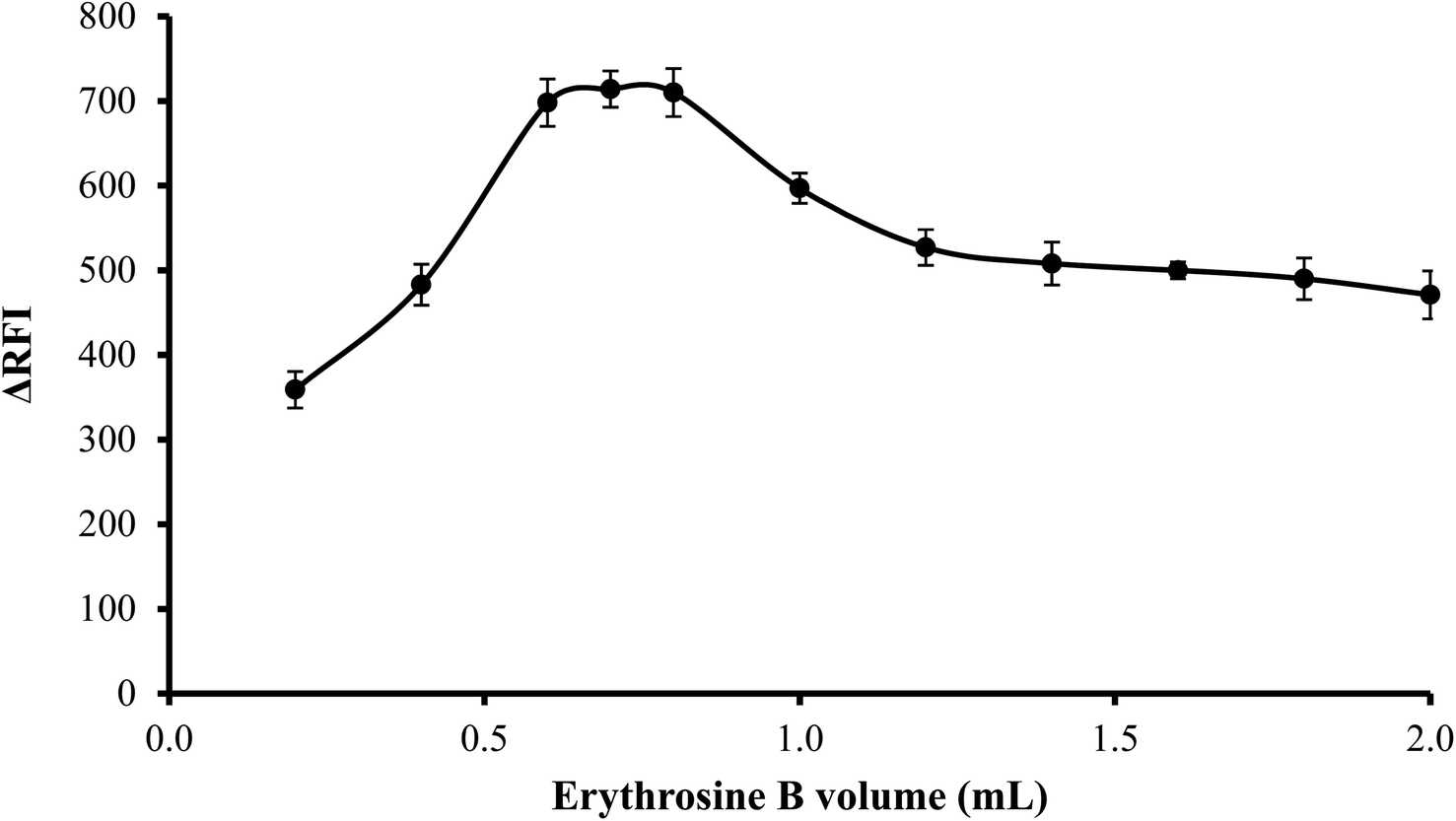
3.1.4. Effect of volume of erythrosine B. An increase in fluorescence quenching was seen as erythrosine B volume was increased up to 0.6 mL and then remained unchanged until 0.8 mL. Thus, to attain the highest readings, 0.7 mL of erythrosine B reagent was used. As shown in Fig. 5, the findings were less favorable when the reagent was used in higher volumes than 0.8 mL possibly because of self-association between the dye molecules.
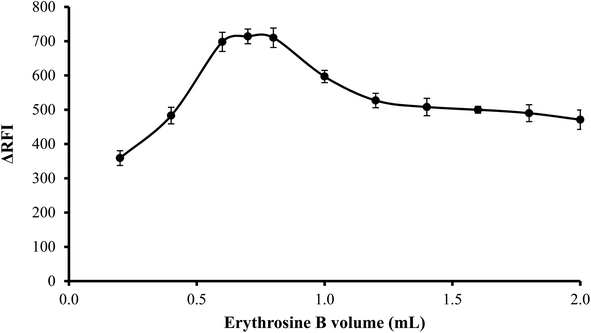 |
| | Fig. 5 Effect of erythrosine-B volume (2.27 × 10−1 M) on the RFI of the association complex formed with FLX (1 μg mL−1). | |
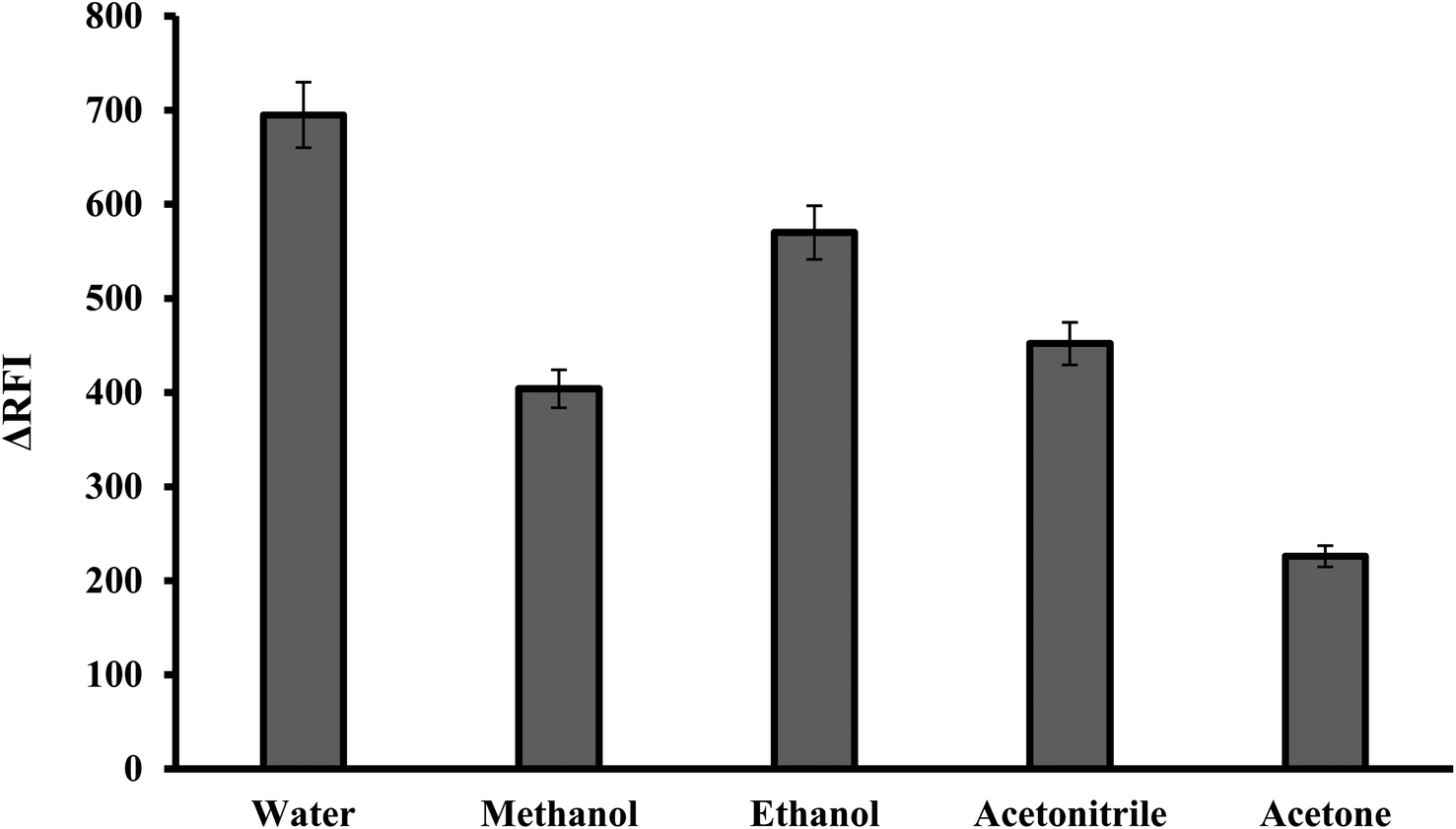
3.1.5. Effect of diluting solvents. The reaction mixture was diluted using various solvents including water, acetonitrile, ethanol, methanol and acetone. It has been found that, the best diluting solvent was water. The other solvents produced less effective outcomes, as shown in Fig. 6. It was observed that the fluorescence quenching value was linearly increased by increasing the dielectric constant of the solvent.30,31 Water and polar solvents have a high dielectric constant, meaning they have a strong ability to separate and stabilize charged species. This helps to solvate and stabilize the ion pair complex formed between the dye and the basic drug. Water was the optimal solvent because it is green, affordable and easily accessible. In comparison to many of the described approaches that use organic solvents like acetonitrile,9 the suggested method has several advantages.
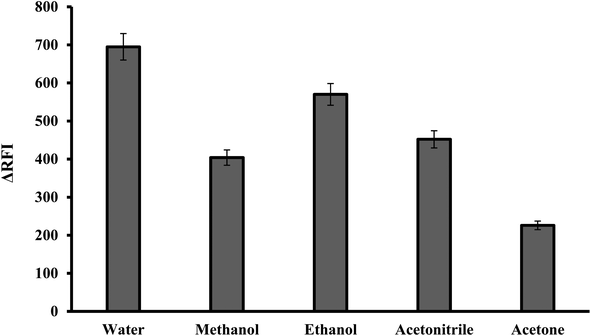 |
| | Fig. 6 Effect of diluting solvent on the RFI of the association complex formed with FLX (1.0 μg mL−1) and erythrosine-B. | |
3.2. Method validation
Following reaction conditions optimization, the suggested approach had been validated in accordance with ICH criteria.32 The results were presented in percentages, and the number of determinations was represented for each value.
3.2.1. Linearity and range. The general analytical technique was utilized to analyze several standard solutions of the investigated drug at diverse concentrations, and the fluorescence signals of the resulting solutions were measured. The calibration curve for the drug under study has been made by plotting the quenching in the relative fluorescence intensity (ΔFI) against the final concentrations of the drug. A linear regression analysis was carried out on the data, and the analytical variables were computed (Table 1). The linear regression equation was ΔFI = 0.61C − 44.36, where C is the final drug concentration in μg mL−1. With a correlation coefficient of 0.9998, drug concentrations between 0.2 − 2.0 μg mL−1 were found to have rectilinear relationships with the fluorescence quenching values.
Table 1 Regression equation and validation parameters for the proposed spectrofluorimetric methoda
| Parameters |
Fluvoxamine |
| LOD is limit of detection and LOQ is limit of quantitation. |
| Linear range (μg mL−1) |
0.2–2.0 |
| Slope (b) |
0.61 |
| Standard deviation of slope (Sb) |
0.004 |
| Intercept (a) |
−44.36 |
| Standard deviation of intercept (Sa) |
5.53 |
| Correlation coefficient (r) |
0.9998 |
| Determination coefficient (r2) |
0.9996 |
| Number of determinations |
5 |
| LOD (μg mL−1) |
0.03 |
| LOQ (μg mL−1) |
0.09 |
3.2.2. Limits of detection and quantification. Calculating the limits of detection and quantification was carried out for assessment of the proposed method's sensitivity. For calculating the limits of detection and quantitation, respectively, the following formulas were used: LOD = 3.3σ/S and LOQ = 10σ/S, where σ refers to the standard deviation of intercept and S is the calibration curve's slope. The obtained value for LOD was 0.03 μg mL−1, while LOQ was 0.09 μg mL−1.
3.2.3. Accuracy. The accuracy was investigated using the standard addition method. To previously examined tablet solutions, specific amounts of the standard drug solution were added. Three times were repeated for each added concentration. The found % recovery was very near to 100% and had a low standard deviation, demonstrating good technique accuracy and high degree of concordance between the measured and real values as represented in Table 2.
Table 2 Evaluation of accuracy of the analytical procedure by standard addition method
| Amount taken (μg mL−1) |
Amount added (μg mL−1) |
Amount found (μg mL−1) |
% recovery ± SDa |
| Mean of three determinations, SD, standard deviation. |
| 0.3 |
0.00 |
0.305 |
101.64 ± 1.14 |
| 0.3 |
0.3 |
0.602 |
100.36 ± 1.19 |
| 0.3 |
0.6 |
0.885 |
98.36 ± 0.69 |
| 0.3 |
0.9 |
1.198 |
99.86 ± 0.97 |
| 0.3 |
1.2 |
1.494 |
99.61 ± 0.36 |
3.2.4. Precision. Repeatability and intermediate precision were the two levels at which precision was evaluated. Three consecutive analyses during the same day were used to determine the repeatability (intraday precision), and three succeeding days were used to evaluate the intermediate (inter-day) precision. The low standard deviation values (less than 2.0) illustrate the great precision of the suggested strategy, as represented in Table 3.
Table 3 Evaluation of intra-day and inter-day precisions of the proposed method
| Conc. (μg mL−1) |
Intra-day precision |
Inter-day precision |
| % recovery ± SDa |
% recovery ± SDa |
| Mean of three determinations, SD, standard deviation. |
| 0.3 |
101.64 ± 1.26 |
101.52 ± 1.25 |
| 0.9 |
100.29 ± 1.96 |
100.12 ± 1.32 |
| 1.6 |
100.85 ± 1.31 |
99.69 ± 1.55 |
3.2.5. Robustness. By examining the impact of slight variations in the experimental parameters (reagent volume, buffer volume, and pH) on the method's analytical performance, the robustness of the technique was evaluated. The acquired results, which are shown in Table 4, showed that the results of the analysis were not meaningfully influenced by minor differences in any of the tested variables because the generated SD did not exceed 2%. This verifies the suggested method's reliability when doing typical tasks.
Table 4 Robustness study of the proposed method for determination of FLX (1 μg mL−1) in pure form
| Parameter |
|
% recovery ± SDa |
| Mean of three determinations, SD, standard deviation. |
| pH of solution |
3.4 |
99.56 ± 1.30 |
| 3.6 |
98.46 ± 1.37 |
| Volume of erythrosine-B (mL) |
0.6 |
98.25 ± 0.71 |
| 0.8 |
99.78 ± 1.61 |
| Volume of buffer (mL) |
0.6 |
99.72 ± 0.49 |
| 0.8 |
99.28 ± 1.27 |
3.3. Molar ratio determination
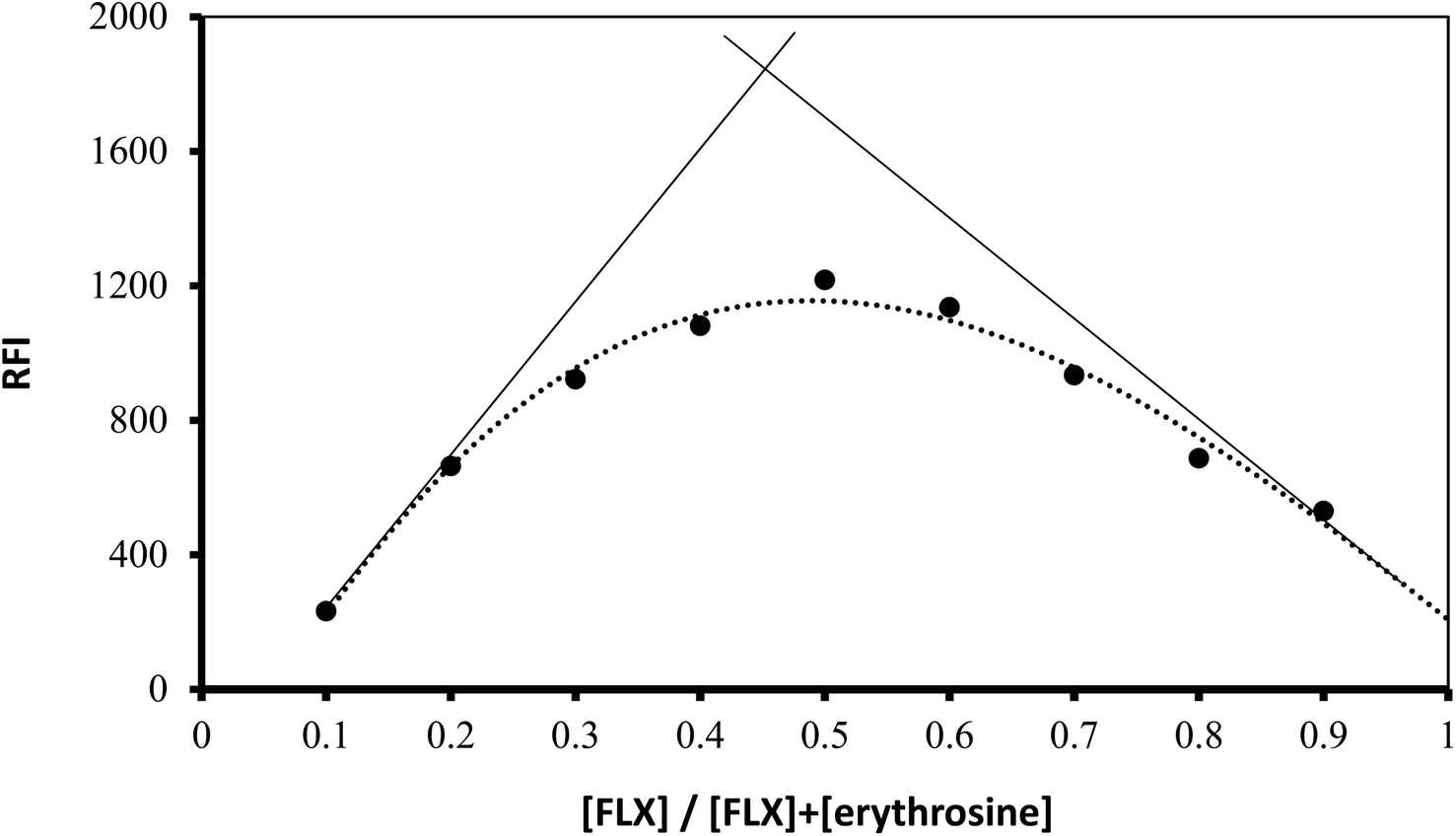
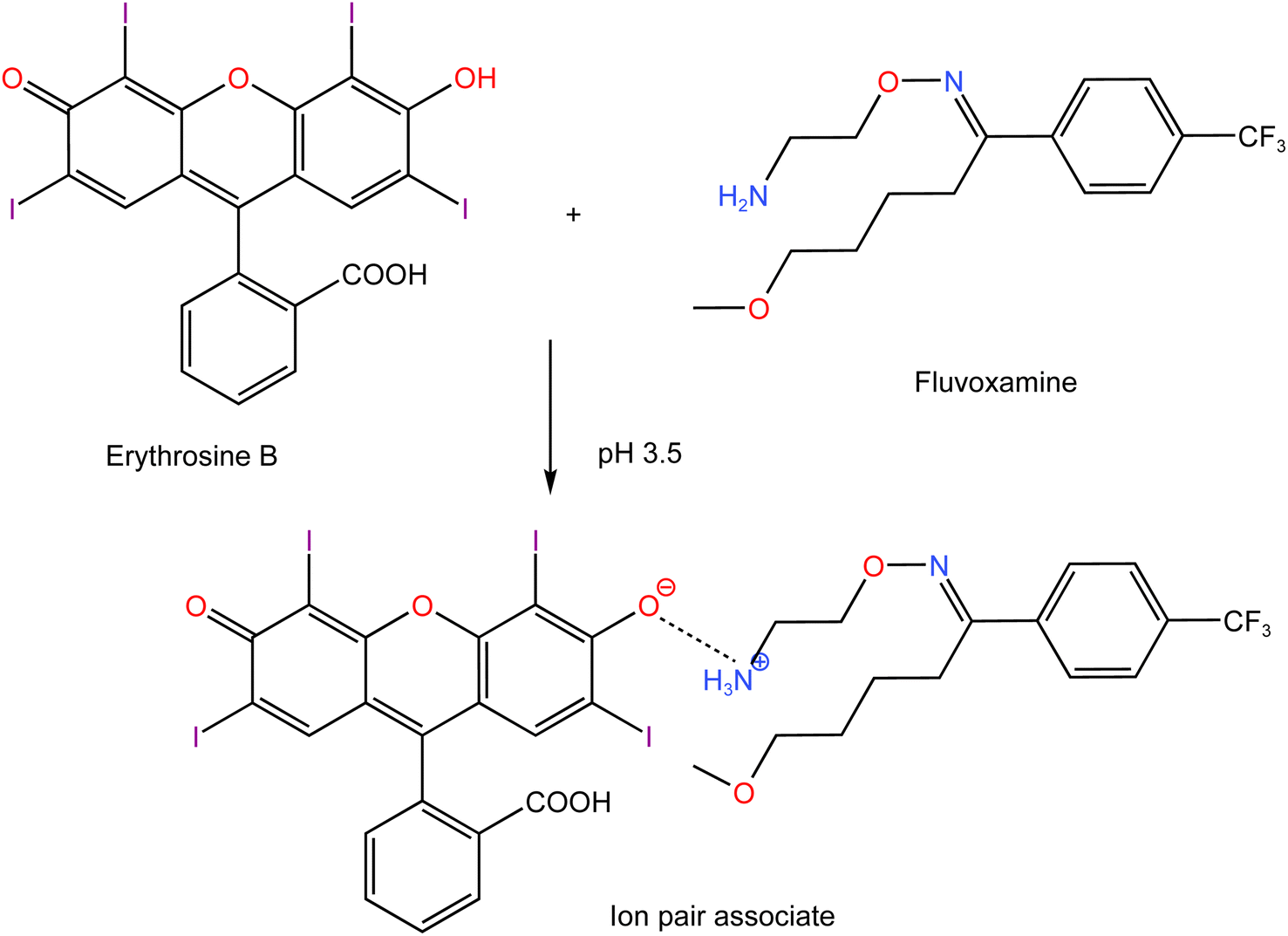
Using Job's method, the molar ratio of FLX to erythrosine-B was examined.33 FLX and dye solutions were made at an equimolar concentration of 1 × 10−4. Different mole fractions (0.1–0.9) of FLX and dye were mixed in complimentary volumes totaling 1 mL into a 10 mL flask. Following the general methodology, the whole procedure had been completed. The measured value for each solution was adjusted regarding its blank reading. As shown in Fig. 7, the results from Job's plot experiment showed that the molar ratio of FLX to dye is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. In Scheme 1, the association complex of FLX-erythrosine is explained.
:1. In Scheme 1, the association complex of FLX-erythrosine is explained.
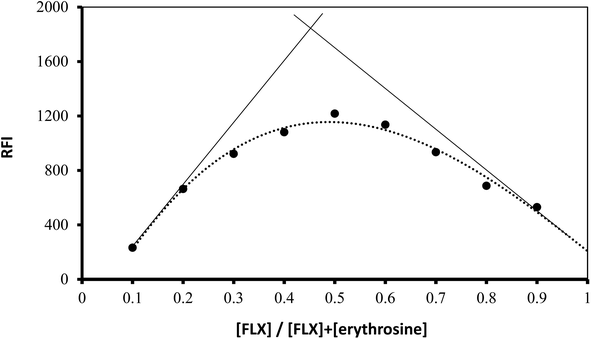 |
| | Fig. 7 Job's plot for molar ratio determination between FLX and erythrosine B using equimolar concentrations (1.0 × 10−4 M). | |
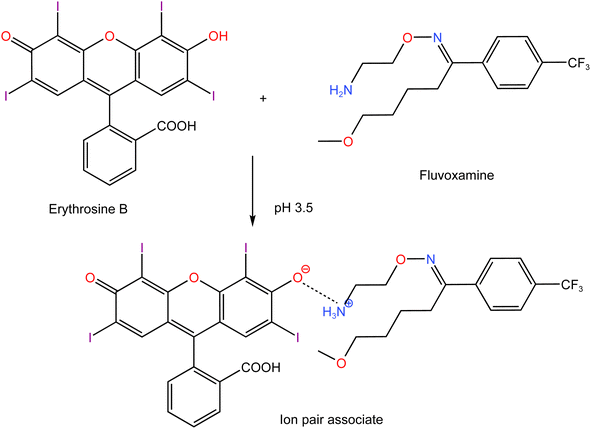 |
| | Scheme 1 Proposed pathway for ion-pair complex formation between FLX and erythrosine. | |
Erythrosine B (H2L) is an acidic dye with two pKa values (3.9 and 5.0).34 Thus, at pH 3.5, the mono valent form (HL−) was the main species of erythrosine B. Although, theoretically, there can be two kinds of enantiotropic isomers produced by the dissociation of either hydroxyl or carboxyl, the hydroxyl dissociation was predominant. Due to the presence of a strong attracting electron group near the hydroxyl group on xanthene core which decreased the electron density of oxygen on hydroxyl, it was easier to dissociate than –COOH on phenyl. FLX is a basic drug with pKa value of 8.86 and thus, at pH 3.5, the drug will be completely ionized. The nitrogen atom (–NH2) of the drug would be protonated to produce the cation (HFLX+). Therefore, at the specific pH, the drug cation (HFLX+) reacted with the dye anion (HL−) to produce a 1:1 neutral ion-association complex by the electrostatic attraction and the hydrophobic force. The interaction occurred through the positively charged nitrogen atom of protonated FLX cation and the negatively charged oxygen atom on hydroxyl group of erythrosine B. Upon this interaction, the distribution and the energy of π electron in erythrosine B conjugated system changed, which resulted in charge-transfer between (HFLX+) and (HL−). Thus, the absorption spectrum of erythrosine would be changed and the fluorescence is quenched.
3.4. Mechanism of fluorescence quenching
The quenching of the native fluorescence of erythrosine B as a result of complex formation could be arose from different pathways such as; transfer of energy, quenching by collision and excited state reactions. Stern–Volmer equation has been widely utilized to investigate the mechanism of fluorescence quenching.35–37 The intensities of fluorescence of erythrosine B in the presence and absence of FLX (F and F0, respectively) were plotted versus the molar concentration of the FLX [Q] according to the following equation:38
where, KSV is the Stern–Volmer constant. Upon plotting the ratio, F0/F versus the molar concentration of drug, the Stern–Volmer plot was obtained which show a linear line. The value of intercept of the linear line would be approximately 1.0 while its slope will equal KSV. The found value of Stern–Volmer constant for the reaction of erythrosine B and FLX was 9.26 × 103. This constant can be used to calculate quenching rate constant. The Stern–Volmer constant equals the natural radiation lifetime of erythrosine B (in absence of the quenching agent), τ0, multiplied by the quenching rate constant, kq, according to the following formula:
Erythrosine B has 89 ps as previously published fluorescence lifetime.39 Using the given equation and the abovementioned values, the calculated quenching rate constant would be 1.04 × 1015 L mol−1 s−1. This value is fairly higher than the highest Kq value (2 × 1010 L mol−1 s−1) that has been reported for quenching by collision.40 Thus, it could be clearly confirmed that static quenching is dominated upon complex formation between erythrosine B and the drug. Therefore, it can be supposed that the fluorescence quenching of the dye was due to the formation of ground state complex between erythrosine B and FLX.
3.5. Applications
3.5.1. Application to pharmaceutical formulations. The drug content of a marketed dosage form (Faverin 50 mg tablets) was assessed using the general analytical procedure. Moreover, the same product was evaluated via a published method,2 and statistics were used to compare the obtained findings of the suggested procedure to those of the reported approach in terms of precision and accuracy. Since the estimated t-test and F-test values were less than the tabulated values at a confidence limit of 95%, there was no noticeable difference between the results of the two approaches, as shown in Table 5. The obtained high % recovery with low standard deviation indicate that the coexist excipients in Faverin 50 mg tablets did not have any significant interference in the results of the method. The examined commercial tablets contain the following excipients: mannitol, maize starch, pregelatinized starch, sodium stearyl fumarate, and colloidal anhydrous silica. All these excipients are aliphatic or inorganic compounds and thus did not have native fluorescence. In addition, due to the absence of any basic center in these excipients, they could not react with erythrosine B and thus could not quench the fluorescence of the dye.
Table 5 Analysis of FLX in dosage form by reported and proposed methods
| Dosage form |
Proposed method |
Reported method |
t-testb value |
F-testb value |
| % recovery ± SDa |
% recovery ± SDa |
| Mean of five measurements. Tabulated value at 95% confidence limit, F = 6.338 and t = 2.306. |
| Faverin 50 mg tablets |
98.92 ± 0.87 |
99.20 ± 1.00 |
0.47 |
1.32 |
3.5.2. Application to content uniformity testing. It is suggested to test the uniformity of the drug content in its tablets when the drug concentration is below 25 mg or when the medication substance is less than 25% of the tablet's components. To guarantee consistency and conformity in each tablet's content, ten tablets have been investigated according to USP specifications.41 The distinct content of Faverin® 50 mg tablet was investigated via the use of content uniformity testing. According to recommendations from the United States Pharmacopoeia, the acceptability value can be calculated using the next equation:
AV = |M − ![[X with combining macron]](https://www.rsc.org/images/entities/i_char_0058_0304.gif) | + KS, | + KS, |
where M represents a reference value, represents the average percent recovery for each individual tablet's content, K represents the acceptability constant (2.4 in the instance of 10 tablets), and S represents the sample's standard deviation. The acceptance value (AV) should be below the maximum allowed AV (L1 = 15). The equation mentioned above will be adjusted according to the value of .
| If 98.5% ≤ ≤ 101.5%, then M = (AV = KS). |
| If < 98.5%, then M = 98.5% (AV = 98.5 − + KS). |
| If > 101.5%, then M = 101.5% (AV = − 101.5 + KS). |
The calculated AV in the present study was lower than the maximum allowed AV (L1 = 15), demonstrating the uniformity of Faverin tablets content, as indicated in Table 6.
Table 6 Application of the proposed method for the content uniformity test of Faverin tablets
| Tablet number |
Recovery (%) |
| L1: maximum allowed acceptance value, AV: acceptance value. |
| 1 |
100.05 |
| 2 |
102.34 |
| 3 |
102.51 |
| 4 |
98.41 |
| 5 |
98.08 |
| 6 |
97.26 |
| 7 |
100.87 |
| 8 |
101.52 |
| 9 |
101.69 |
| 10 |
96.61 |
| Mean |
99.93 |
| S |
2.18 |
| AVa |
5.81 |
| L1a |
15 |
3.6. Evaluation of method greenness
Green chemistry metrics needs to be developed and improved on a regular basis. Avoiding the use of dangerous harmful solvents, high energy consumption, chemical derivatization, and high waste production will result in a completely green analytical technique. Considerations from recent times, like the analytical eco scale score42 has been employed to assess the “ecological value” of an analytical technique. To assess how environmentally friendly the suggested system is, the eco-scale was used. Penalty points have been gathered for each of the proposed procedure's factors, such as the quantities of chemicals used, energy that were utilized and waste produced, based on the eco-scale score. A result obtained for “ultimate green analysis” is a number that represents a penalty point that was issued and subtracted from 100. These points represent the risks that were faced throughout the performing the research process. The procedure of analysis is regarded as exceptional green if it achieves a score of more than 75. Water, a non-toxic solvent, was used in the analysis, and only very little amounts of waste were produced and minimal energy were consumed. Therefore, the examined technique received a high score on the eco-scale (95), as shown in Table 7. This demonstrates that the method possesses high ecological friendliness.
Table 7 Evaluation of the greenness of the proposed method using eco score scale method
| Item |
Parameter |
Word sign |
PPT score |
| If the score is greater than 75, it represents excellent green analysis. If the score is greater than 50, it represents acceptable green analysis. If the score is less than 50, it represents inadequate green analysis, LSH is an abbreviation for less severe hazard. |
| Technique |
Fluorimetry |
|
0 |
| Reagent |
Erythrosine B |
LSH |
1 |
| Amount of reagent |
<10 mL |
|
1 |
| Solvent(s) |
Water |
Green solvent |
0 |
| Heating |
— |
|
0 |
| Temperature |
25 °C |
|
0 |
| Cooling |
— |
|
0 |
| pH |
3.7 |
|
0 |
| Energy (kW h per sample) |
<1.0 |
|
0 |
| Waste |
1–10 (mL) |
|
3 |
| Occupational hazards |
(Analytical process hermitization) |
|
0 |
| Total penalty points |
|
|
5 |
| Analytical eco-scale total scorea |
|
|
95 |
3.7. Comparison with the previous spectrofluorimetric methods
Several fluorogenic reagents were previously utilized for FLX as shown in Table 8. The suggested technique processes several benefits over the spectrofluorimetric techniques that have previously been published.7–12 The present method incorporate the use of a readily accessible and less expensive reagent (erythrosine B) rather than fluorescamine, which was previously described for FLX measurement.12 The interaction between the examined medication and erythrosine B was instantaneous at room temperature, which is fairly quick. Thus, there was no need to stand for any period of time prior to the measurements. On the other hand, in four methods of the previous techniques,7–10 the reaction mixtures were heated at high temperatures for long time, in multi-steps procedures. Moreover, the method utilized water as a green solvent so it is more ecologically friendly than other published methods, which use risky organic solvents like acetonitrile.9 In addition, it possesses high sensitivity than the method utilizes eosin dye.11
Table 8 Comparison between the suggested and reported spectrofluorimetric methods for FLX determination
| Reagent |
Temperature/time |
Solvent |
Linear range (μg mL−1) |
LOD (μg mL−1) |
Ref. |
| Ninhydrine/phenylacetaldehyde |
85 °C/5 min |
Ethanol |
0.8–14 |
0.25 |
7 |
| Acetylacetone |
100 °C/30 min |
Water |
0.2–2.0 |
0.06 |
8 |
| NBD-Cl |
50 °C/20 min |
Acetonitrile |
0.065–0.8 |
0.02 |
9 |
| NBD-Cl |
50 °C/20 min |
Methanol |
0.065–0.8 |
0.02 |
10 |
| Eosin Y |
Room temp. |
Water |
0.3–2.2 |
0.08 |
11 |
| Fluorescamine |
Room temp./5 min |
Water |
0.1–1.1 |
0.01 |
12 |
| Erythrosine B |
Room temp. |
Water |
0.2–2.0 |
0.03 |
This work |
4. Conclusion
A spectroscopic investigation was performed for quick, and sensitive quantification of FLX through spectrofluorimetry. The applied approach was relied on the formation of ion pair associate between FLX and erythrosine B dye in acetate buffer (pH 3.5). The quenching of erythrosine B's native fluorescence caused by the addition of FLX was the basis of the spectrofluorimetric measurements. The method was validated in agreement with ICH specifications. Additionally, the method was employed to determine FLX in its dose forms. The great recoveries attained demonstrate that there is no interference from the excipients that are commonly used. The developed approach has several advantages including the simplicity. The approach adopted a single sample preparation protocol that is easy and does not involve complex steps. In addition, the utilized device is simple and does not require much effort to operate. Due to the simple procedure, the proposed method has been successfully used to test the content uniformity of tablet formulations. Furthermore, the reagent used is a food additive, and water is the reaction solvent, making the method compatible with the principles of green chemistry and has high sustainability. The environmental friendliness of the suggested method was confirmed using the eco-scale score to highlight its greenness and sustainable attributes of the developed method. Thus, the suggested procedure is appropriate for determining the examined medications' dosage forms in laboratories for quality control purposes.
Conflicts of interest
The authors declare no conflict of interest.
References
- B. Blackwell and J. Simon, Drugs Today, 1986, 22, 611–633 CAS.
- R. H. Pullen and A. A. Fatmi, J. Chromatogr. B: Biomed. Sci. Appl., 1992, 574, 101–107 CrossRef CAS PubMed.
- I. A. Darwish, H. H. Abdine, S. M. Amer and L. I. Al-Rayes, Spectrochim. Acta, Part A, 2009, 72, 897–902 CrossRef PubMed.
- S. Devarajan, G. Manavalan, K. Balasubramanian, J. Annamalai and N. Madduri, J. Appl. Pharm. Sci., 2015, 5, 081–086 CrossRef.
- M. Kishore, A. Koteswarao and M. Janardhan, Res. J. Pharm. Technol., 2011, 4, 450–453 Search PubMed.
- R. A. Sayed, W. El-Alfy, O. A. Ismaiel, M. Y. El-Mammli and A. Shalaby, Ann. Pharm. Fr., 2021, 79, 375–386 CrossRef CAS PubMed.
- A. A. Abu-hassan, M. A. Omar and S. M. Derayea, Luminescence, 2020, 35, 934–940 CrossRef CAS PubMed.
- A. A. Abu-hassan, M. A. Omar and S. M. Derayea, Luminescence, 2020, 35, 1360–1365 CrossRef CAS PubMed.
- I. A. Darwish, S. M. Amer, H. H. Abdine and L. I. Al-Rayes, J. Fluoresc., 2009, 19, 463–471 CrossRef CAS PubMed.
- N. Z. Alzoman and I. A. Darwish, Molecules, 2023, 28, 5221 CrossRef CAS PubMed.
- S. M. Derayea, M. A. Omar and A. A. Abu-Hassan, R. Soc. Open Sci., 2018, 5, 170943 CrossRef PubMed.
- N. El-Enany, J. AOAC Int., 2007, 90, 376–383 CrossRef CAS PubMed.
- T. Ohkubo, R. Shimoyama, K. Otani, K. Yoshida, H. Higuchi and T. Shimizu, Anal. Sci., 2003, 19, 859–864 CrossRef CAS PubMed.
- N. Yasui-Furukori, Y. Inoue, S. Kaneko and K. Otani, J. Pharm. Biomed. Anal., 2005, 37, 121–125 CrossRef CAS PubMed.
- M. A. Saracino, L. Mercolini, G. Flotta, L. J. Albers, R. Merli and M. A. Raggi, J. Chromatogr. B, 2006, 843, 227–233 CrossRef CAS PubMed.
- S. H. Wong, H. R. Kranzler, S. Dellafera and R. Fernandes, Biomed. Chromatogr., 1994, 8, 278–282 CrossRef CAS PubMed.
- M. Bagli, M. L. Rao, T. Sobanski and G. Laux, J. Liq. Chromatogr. Relat. Technol., 1997, 20, 283–295 CrossRef CAS.
- S. T. Ulu, Chromatographia, 2006, 64, 169–173 CrossRef CAS.
- J. Berzas Nevado, M. Villasenor Llerena, A. Contento Salcedo and E. A. Nuevo, J. Chromatogr. Sci., 2000, 38, 200–206 CAS.
- S. Pawar and S. Dhaneshwar, J. Planar Chromatogr.--Mod. TLC, 2012, 25, 338–343 CrossRef CAS.
- C. Schweitzer, H. Spahn and E. Mutschler, J. Chromatogr. B, 1986, 382, 405–411 CrossRef CAS PubMed.
- T. Gondová, D. Halamová and K. Špacayová, J. Liq. Chromatogr. Relat. Technol., 2008, 31, 2429–2441 CrossRef.
- T. Madrakian, M. Soleimani and A. Afkhami, Sens. Actuators, B, 2015, 210, 259–266 CrossRef CAS.
- R. F. Ajayi, E. Nxusani, S. F. Douman, A. Jonnas, P. G. L. Baker and E. I. Iwuoha, J. Nano Res., 2016, 44, 208–228 CAS.
- A. A. Diva, S. Fathi and F. Chekin, J. Anal. Chem., 2019, 74, 809–815 CrossRef.
- J. B. Nevado, A. C. Salcedo, M. V. Llerena and E. A. Nuevo, Anal. Chim. Acta, 2000, 417, 169–176 CrossRef.
- S. M. Derayea, A. A. Hamad, R. Ali and H. R. H. Ali, Microchem. J., 2019, 149, 104024 CrossRef CAS.
- K. M. Badr El-Din, H. Salem, S. Derayea, A. Abdelaziz and D. M. Nagy, Luminescence, 2023, 38, 1583–1590 CrossRef CAS PubMed.
- T. Alev-Behmoaras, J. J. Toulmé and C. J. B. Hélène, Biochimie, 1979, 61, 957–960 CrossRef CAS PubMed.
- M. Pannipara, A. M. Asiri, K. A. Alamry, M. N. Arshad and S. A. El-Daly, J. Fluoresc., 2014, 24, 1629–1638 CrossRef CAS PubMed.
- R. M. Melavanki, R. A. Kusanur, J. S. Kadadevaramath and M. V. Kulkarni, J. Fluoresc., 2010, 20, 1175–1180 CrossRef CAS PubMed.
- ICH Harmonised Tripartite Guideline, Validation of analytical procedures: text and methodology, 2005, 05 Search PubMed.
- C. Y. Huang, in Methods in Enzymology, 1982, vol. 87, pp. 509–525 Search PubMed.
- D. Snigur, M. Fizer, A. Chebotarev, O. Lukianova and O. Zhukovetska, Dyes Pigm., 2022, 198, 110028 CrossRef CAS.
- M. Bielska, A. Sobczynska and K. Prochaska, Dyes Pigm., 2009, 80, 201–205 CrossRef CAS.
- P. Garg, B. Kaur, G. Kaur, S. Saini and G. R. Chaudhary, Colloids Surf., A, 2021, 610, 125697 CrossRef CAS.
- W. Zou, M. Song, J. He, P. Qiu, Z. Sun, Z. Su and Y. Bai, Anal. Bioanal. Chem., 2021, 413, 1429–1440 CrossRef CAS PubMed.
- J. G. Xu and Z. B. Wang, Fluorimetric Analysis Method, Science Press, Beijing, 3rd edn, 2006 Search PubMed.
- N. Boens, W. Qin, N. Basarić, J. Hofkens, M. Ameloot, J. Pouget, J.-P. Lefèvre, B. Valeur, E. Gratton and M. VandeVen, Anal. Chem., 2007, 79, 2137–2149 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer-Verlag, Berlin, Heidelberg, 3rd edn, 2006 Search PubMed.
- M. Rockville, The United States Pharmacopoeia 30, the National Formulary 25 US Pharmacopeial Convention, Electronic version, 2007 Search PubMed.
- K. Aken, L. L. Strekowski and L. Patiny, Beilstein J. Org. Chem., 2006, 2, 3 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b,
Ahmed Abdulhafez Hamadc and
Dalia M. Nagya
*b,
Ahmed Abdulhafez Hamadc and
Dalia M. Nagya