
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of PEG/P(U-AM-ChCl) composite hydrogels using ternary DES light polymerization and their properties
Bin Li *a,
Haiying Liua,
Mengjing Zhoua,
Aolin Wub,
Wenrui Haob,
YaJun Jianga and
Zhigang Hua
*a,
Haiying Liua,
Mengjing Zhoua,
Aolin Wub,
Wenrui Haob,
YaJun Jianga and
Zhigang Hua
aSchool of Mechanical Engineering, Wuhan Polytechnic University, Wuhan, Hubei 430023, China. E-mail: libin_027@126.com; Tel: +18827081895
bSchool of Science, Wuhan University of Technology, Wuhan, Hubei 430070, China
First published on 18th January 2024
Abstract
Deep eutectic solvents (DES) were prepared using urea (U) and acrylamide (AM) as hydrogen bond donors (HBD) and choline chloride (ChCl) as hydrogen bond acceptor (HBA), and polyethylene glycol (PEG) was selected as a filler and uniformly dispersed in DES to prepare PEG/P(U-AM-ChCl) composite hydrogels by light polymerization. The composite hydrogels were characterized by scanning electron microscopy (SEM) and Fourier transform infrared spectroscopy (FTIR). The effects of the content of PEG on the swelling properties, mechanical properties and fatigue resistance of the composite hydrogels were investigated. The results showed that the compressive strength and fatigue strength of the composite hydrogels were gradually enhanced with the increase of the PEG content in the composite hydrogels, in which the maximum compressive strength of the hydrogels with 1 wt% PEG added was increased by 1.86 times. The composite hydrogel had excellent swelling properties, and the equilibrium swelling degree of the hydrogel with 1 wt% PEG added reached 10.15. Meanwhile, the PEG/P(U-AM-ChCl) composite hydrogel had excellent self-healing properties, and the self-healing rate of the composite hydrogel with a PFG content of 1 wt% could reach 91.93% after 48 hours of healing. This study provides a convenient and efficient method to prepare composite hydrogels with superior swelling properties and self-healing properties.
1 Introduction
Hydrogels are three-dimensional polymer networks formed by cross-linking hydrophilic polymer chains.1–4 They have physical properties similar to soft tissues that do not dissolve after absorbing large amounts of water, due to the cross-linking structure within the hydrogel network.5 In addition, hydrogels are highly permeable to oxygen, nutrients and other water-soluble metabolites.6 Based on these characteristics, researchers have designed many hydrogels with different structures, compositions and properties.7 Hydrogels with different structures and properties are used in a wide range of applications in different fields, such as drug delivery,8–10 wound dressing,11 wearable sensors,12 sensors13 and so on. However, there are many problems with existing hydrogels, such as deficiencies in mechanical and self-healing properties and uncontrollable water absorption properties;14 these problems limit the promotion and application of hydrogels. Therefore, current research focuses on designing multifunctional composite hydrogels to improve hydrogel properties and meet various application requirements.15Polyethylene glycol (PEG) is a polymer of great interest to researchers. Its chemical structure consists of repeating units of the ethylene glycol molecule, and it has a number of important properties and applications.16 PEG chain has many polar hydroxyl groups, can form hydrogen bonding interactions with water molecules, has a strong hydrophilicity, can effectively absorb and retain water.17 At the same time, PEG molecules have good biocompatibility and solubility, making them soluble in a wide range of organic solvents and water. Therefore, PEG is often used to prepare drug carriers,18 biomedical material19,20 and medical lubricants, etc. More and more researchers are combining PEG molecules with hydrogels to improve the swelling and self-healing properties of hydrogel materials.1 As early as the 1920s, researchers discovered that polymers synthesized based on the reversible Diels–Alder reaction have self-healing capabilities similar to those of tissues and skin, and in the last decade, the development of self-healing hydrogels has become more and more sophisticated, especially based on autonomous interactions of the hydrogels themselves, such as dynamic chemical bonding, dynamic non-covalent bonding, and multiple repair mechanisms.
Deep eutectic solvents (DES) are a new type of ionic liquids that usually consist of a mixture of a hydrogen bonding donor (e.g., glycerol, ethylene glycol, glycine, etc.) and a hydrogen bonding acceptor (e.g., tartaric acid, oxalic acid, urea, etc.). The presence of hydrogen bonding interactions between the components of DES results in a more favourable total energy of DES relative to the lattice energy of the pure component.21 The presence of hydrogen bonding interactions gives DES a lower melting point than each of its individual source materials, which helps to maintain DES in a liquid state.22 Nowadays, DES is widely used in many research fields and has received much attention for its potential use as an environmentally friendly solvent.23 DES is also often used in the preparation of hydrogels. Compared with the traditional method of preparing hydrogels, the use of DES in the preparation of hydrogels has the advantages of low toxicity, green environmental protection and good solubility.24 DES provides both a solvent environment suitable for the reaction for the preparation of hydrogels and can also participate in the polymerization reaction as a monomer. Therefore, hydrogels with different properties can be prepared by adjusting the properties of DES.
Light polymerization (LP) is one of the methods commonly used to prepare hydrogels,25,26 it is a reaction in which UV light is used to initiate the polymerization of monomers in the presence of a photoinitiator, resulting in the formation of a gel with a three-dimensional network structure.27 In order to initiate the light polymerization reaction, it is necessary to add appropriate photoinitiators. Commonly used photoinitiators include benzoin derivatives, benzophenones, acetophenone derivatives and hydroxyalkyl phenyl ketones.5 Photoinitiators are capable of absorbing specific wavelengths of light in the ultraviolet or visible region and releasing active substances such as free radicals.28 These active substances can initiate the polymerization of monomers, resulting in the formation of cross-linked hydrogels. The use of light polymerization to prepare hydrogels has the advantages of being controllable and highly tunable, and the rate of the polymerization reaction can be adjusted by controlling conditions such as light irradiation time, light intensity, and temperature. Light polymerization is usually an instantaneous process in which liquid hydrogel precursors can be delivered and crosslinked in situ to form hydrogels in a minimally invasive manner during polymerization.29 This process provides spatial and temporal control of the liquid-to-gel transition, thus enabling the fabrication of hydrogels of various complex shapes.5
Based on previous studies, we developed a PEG/P(U-AM-ChCl) composite hydrogel with self-healing properties by adding polyethylene glycol (PEG) to DES. The polymerizable DES was prepared by mixing urea (U), acrylamide (AM) and choline chloride (ChCl) at a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1, and the PEG/P(U-AM-ChCl) composite hydrogel was prepared by adding PEG to the DES for LP. The composite hydrogels were characterized by scanning electron microscopy (SEM) and Fourier transform infrared spectroscopy (FTIR) to further investigate the effect patterns of different contents of PEG on the swelling properties, mechanical properties and anti-fatigue properties of the composite hydrogels.
:1:1, and the PEG/P(U-AM-ChCl) composite hydrogel was prepared by adding PEG to the DES for LP. The composite hydrogels were characterized by scanning electron microscopy (SEM) and Fourier transform infrared spectroscopy (FTIR) to further investigate the effect patterns of different contents of PEG on the swelling properties, mechanical properties and anti-fatigue properties of the composite hydrogels.
2 Materials and methods
2.1 Materials
Choline chloride (ChCl), acrylamide (AM), urea (U), polyethylene glycol (PEG), N, N-methylene bisacrylamide (MBA) were purchased from Shanghai Aladdin Biochemical Science and Technology Co. Ltd, and 2,4,6-trimethylbenzoyl phenyl ethyl phosphoric acid ethyl ester (TPO-L) was purchased from Sinopharm Chemical Reagent Corporation; all the raw materials were analytically pure and could be used directly. Before use, ChCl should be dried under vacuum at 70 °C for two hours to remove the absorbed water. The experimental water was distilled water. EMPITA VP-60 lamps (power 180 W, λ = 320 nm) manufactured by the famous Polish company (Lodz) were used as the UV light source.2.2 Preparation of deep eutectic solvents
Using ChCl as HBA and U and AM as HBD, the three drugs were mixed in a beaker according to the molar ratio of 1:1:1. The beaker with the mixed drugs was placed in an oil bath at 80 °C and heated (the oil surface was over the highest part of the mixed reagents), and stirred continuously with a glass rod until a transparent and clarified liquid was formed. The prepared DES was allowed to stand for a period of time and then different contents of PEG were added according to the ratio in Table 1 and stirred until homogeneous. The schematic diagram of hydrogen bonding within DES is shown in Fig. 1.
| Samples | U/AM/ChCl (molar ratio) | PEG (wt%) | MBA (wt%) | TPO-L (wt%) |
|---|---|---|---|---|
| LP1 | 1:1:1 |
0 | 1.0 | 0.5 |
| LP2 | 1:1:1 |
0.25 | 1.0 | 0.5 |
| LP3 | 1:1:1 |
0.5 | 1.0 | 0.5 |
| LP4 | 1:1:1 |
1.0 | 1.0 | 0.5 |
 | ||
| Fig. 1 Schematic diagram of hydrogen bonding within DES. | ||
2.3 Preparation of hydrogels by light polymerization
To the mixture of DES and PEG, 1 wt% MBA and 0.5 wt% photoinitiator were added and mixed well, and then the mixture was transferred to a glass test tube with the size of 100 × 12 mm, and the height of the mixture filled in the test tube was about 80 mm, and the test tube was irradiated uniformly with UV light to make the reaction of the mixture under UV irradiation, and the reaction was complete, and then the hydrogel was allowed to cool down to room temperature, and then the aqueous gel. The schematic diagram of the preparation of PEG/P(U-AM-ChCl) composite hydrogels by light polymerization is shown in Fig. 2. | ||
| Fig. 2 Schematic diagram for the preparation of SA/P (U-AM-ChCl) composite hydrogels. | ||
2.4 Properties and their characterization
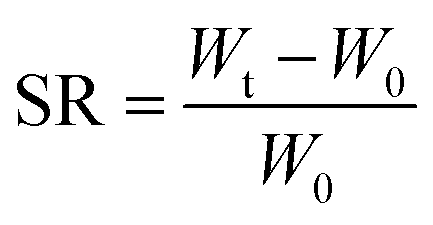 | (1) |
In eqn (1), Wt denotes the total weight of the hydrogel after time t, W0 indicates the initial weight of the dry hydrogel.
 | (2) |
In eqn (2), F is the applied force and S is the hydrogel cross-sectional area.
 | (3) |
In eqn (3), σi represents the maximum stress in the cyclic compression of the hydrogel, and σ1 represents the maximum stress in the first cyclic compression of the hydrogel.
 | (4) |
In eqn (4), P0 is the maximum tensile strength of the uncut hydrogel and Pt is the maximum tensile strength of the hydrogel after healing t time.
3 Results and discussion
3.1 Micro-morphological analysis of PEG/P(U-AM-ChCl) composite hydrogels
The LP1 hydrogel cross-section (Fig. 3a) was partially wrinkled, but no obvious pores were observed, and the LP4 hydrogel cross-section (Fig. 3b) with the addition of 1 wt% PEG showed a homogeneous mesh structure. From the cross sections of LP2 and LP3 (Fig. 3c and d), it can be seen that the pores of the hydrogel network gradually increased with the increase of PEG content. The addition of PEG decreased the crosslink density of the hydrogels, which was attributed to the fact that the PEG molecule is a soft and bendable polymer chain structure, and in the hydrogels, the addition of PEG could change the crystalline nature of the hydrogels.30 With the change of crystalline properties, the regularity of the polymer chain arrangement is weakened, making the molecular structure of the whole hydrogel looser, making it more inclined to amorphous structure.31,32 Hydrogels with amorphous structure usually have more defects and pores, and their molecules are arranged without a fixed pattern, thus forming a reticular structure. The pores of hydrogels gradually increase because PEG, as a polymer with a large molecular weight, has a large molecular size that extends the reticulation of the hydrogel,17 With the gradual filling of PEG as filler into the hydrogel network, the spatial structure of the hydrogel network was altered. | ||
| Fig. 3 SEM images of lyophilized (a) LP1, (b) LP4, (c) LP2, (d) LP3 hydrogels. | ||
3.2 Fourier infrared spectroscopy of PEG/P(U-AM-ChCl) composite hydrogels (FTIR)
Fig. 4 shows the infrared spectra of PEG, P(U-AM-ChCl) and PEG/P(U-AM-ChCl), with P(U-AM-ChCl) being the spectral line without added PEG and PEG/P(U-AM-ChCl) being the spectral line with added 1 wt% PEG content. From the figure, it can be seen that the three four spectral lines have broad absorption bands at 3200–3600 cm−1, which are formed due to the stretching vibration of the –OH group. And with the addition of PEG, the absorption bands of –OH groups gradually become broader, which is because PEG contains a large number of oxygen atoms, and the addition of DES can form more –OH groups with U and AM. The absorption peaks are observed near 1666 cm−1 in both P(U-AM-ChCl) and PEG/P(U-AM-ChCl) spectra in the figure, which is due to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups in U and AM.33 In addition, an absorption peak due to the stretching vibration of the C–H group in AM is observed near 2161 cm−1 of the P(U-AM-ChCl) absorption spectrum. Weaker absorption peaks can be observed near 1023 cm−1 of the P(U-AM-ChCl) absorption spectrum and near 1124 cm−1 of the PEG/P(U-AM-ChCl) absorption spectrum, which are due to the asymmetric stretching vibration of the –C–O–C group.34 With the addition of PEG, the absorption peak shifted from near 1023 cm−1 to near 1124 cm−1, which was attributed to the fact that the –C–O–C groups in PEG replaced the –C–O–C groups in the chain segments of the polymer network to participate in the telescopic vibration in the process of hydrogel preparation.31,32
O groups in U and AM.33 In addition, an absorption peak due to the stretching vibration of the C–H group in AM is observed near 2161 cm−1 of the P(U-AM-ChCl) absorption spectrum. Weaker absorption peaks can be observed near 1023 cm−1 of the P(U-AM-ChCl) absorption spectrum and near 1124 cm−1 of the PEG/P(U-AM-ChCl) absorption spectrum, which are due to the asymmetric stretching vibration of the –C–O–C group.34 With the addition of PEG, the absorption peak shifted from near 1023 cm−1 to near 1124 cm−1, which was attributed to the fact that the –C–O–C groups in PEG replaced the –C–O–C groups in the chain segments of the polymer network to participate in the telescopic vibration in the process of hydrogel preparation.31,32
 | ||
| Fig. 4 FTIR plots of P(U-AM-ChCl) and PEG/P(U-AM-ChCl) composite hydrogels. | ||
3.3 Analysis of swelling properties of PEG/P(U-AM-ChCl) composite hydrogels
From Fig. 5, it can be seen that all four groups of hydrogel samples reached the swelling equilibrium within 120 min, and their equilibrium swelling degrees were 6.35, 6.57, 7.59, and 10.15, respectively, and the four groups of hydrogel samples had high equilibrium swelling degrees.35 The equilibrium swelling of the PEG/P(U-AM-ChCl) composite hydrogel gradually increased with the increase of PEG content, and the equilibrium swelling of the LP4 hydrogel was maximum when the content of PEG reached 1.0 wt%. This was attributed to the fact that PEG, as a macromolecule polymer, could extend the reticular structure of the hydrogel and form pores in the hydrogel network.17 As PEG is gradually distributed into the hydrogel network, it may cause the pore structure of the hydrogel to become more enriched, providing more locations and channels for water molecules to enter the hydrogel, and therefore more capable of absorbing and retaining water molecules, thereby increasing the swelling of the hydrogel. PEG is a highly hydrophilic polymer,32 Its molecular chain contains a large number of hydroxyl groups, which can form hydrogen bonding interactions with water molecules, making PEG highly attractive to water molecules. When PEG is added to the hydrogel, its hydrophilicity helps to attract water molecules into the interior of the hydrogel, increasing the water content of the hydrogel. In addition, PEG itself as a polymer can accommodate a large number of water molecules, which increases the content of water molecules in the whole hydrogel, leading to an increase in the degree of swelling.36 PEG/P(U-AM-ChCl) composite hydrogel has good swelling properties, and the three-dimensional mesh structure of the hydrogel as well as the solubility of PEG molecules in water and lower intrinsic toxicity make our composite hydrogel have a broad application prospect in wastewater treatment.37 | ||
| Fig. 5 Dissolution kinetics curve of PEG/P(U-AM-ChCl) composite hydrogel. | ||
3.4 Mechanical and fatigue resistance analysis of PEG/P(U-AM-ChCl) composite hydrogels
The compressive stress–strain curve of PEG/P(U-AM-ChCl) composite hydrogel is shown in Fig. 6a, setting the maximum compressive deformation as 80%. The maximum compressive strength of the composite hydrogel with increasing PEG content was 0.72 MPa, 0.79 MPa, 0.99 MPa, and 1.34 MPa, in that order. This is because PEG is a polymer, and its molecular chains together form a physically crosslinked network after hydrogen-bonding interactions with AM and U molecules in the hydrogel. With the increase of PEG content, the hydrogen bonding density of the hydrogel was increased,38 this physical crosslinked network becomes more complex and stable, which helps to improve the compressive strength of the hydrogel. Secondly, the addition of PEG may affect the pore structure and molecular arrangement of the hydrogel, as shown in the SEM characterization, and the more homogeneous mesh structure of the composite hydrogel contributes to the enhancement of the cushioning properties of the hydrogel, which leads to the improvement of the compressive strength.39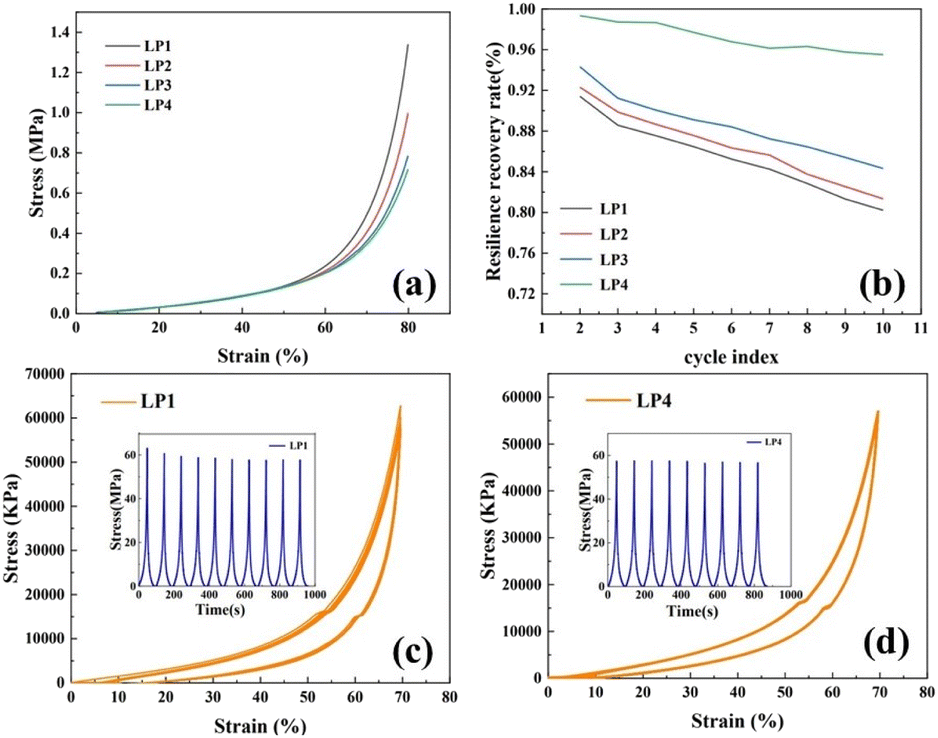 | ||
| Fig. 6 (a) Compressive stress–strain curves of PEG/P(U-AM-ChCl) composite hydrogels; (b) Maximum stress recovery of LP1–LP4 hydrogels after 10 consecutive compressive unloading; (c) and (d) Stress–strain curves for ten compression cycles at 80% strain for LP1 hydrogel and LP4 hydrogel. | ||
After ten cycles of compression, the maximum stress recovery decreased from 91.41% to 80.21% for LP1 and from 99.35% to 95.52% for LP4 (Fig. 6b). Fig. 6c and d show the compressive stress–strain curves for ten cycles of LP1 hydrogel and LP4 hydrogel at 80% strain, and the maximum stress versus time curves for LP1 and LP4 hydrogels are also shown in the figures. In the 1st to 5th cyclic compression, the maximum stress versus time curves of LP4 are more gentle compared to the maximum stress versus time curves of LP1. As shown in Fig. 6c and d, the stress–strain curves for ten cycles of compression, the curves of LP4 have a higher degree of overlap compared with those of LP1, which demonstrates that the PEG-added hydrogel has excellent fatigue resistance.40 This is because the introduction of PEG molecules into the hydrogel network increases the intermolecular interactions in the hydrogel, and the PEG molecules are able to form interactions such as hydrogen bonding or van der Waals forces with other molecules in the hydrogel, which enhances the overall stability of the hydrogel and results in enhanced fatigue resistance of the hydrogel. On the other hand, PEG molecules themselves have strong hydrophilicity,34 the swelling property of the hydrogel is greatly enhanced by the addition of PEG molecules, which helps to maintain the moisture content of the hydrogel and keep the flexibility and strength of the hydrogel, thus increasing the fatigue resistance of the composite hydrogel.38
3.5 Analysis of self-healing properties of PEG/P(U-AM-ChCl) composite hydrogels
In order to investigate the effect of different contents of PEG on the healing properties of PEG/P(U-AM-ChCl) composite hydrogels, we explored the extent of hydrogel healing at different times (6 h, 12 h, 24 h, 36 h, 48 h). Fig. 7 shows the self-healing rate of PEG/P(U-AM-ChCl) composite hydrogels at different healing times. It can be found that the self-healing rate of the composite hydrogel increased gradually with the increase of the healing time during the self-healing process from 0 to 48 h. When the healing time reached 48 h, LP1, LP2, LP3, and LP4 possessed good self-healing performance with self-healing rates of 75.13%, 86.26%, 89.88%, and 91.93%, respectively. The self-healing rate of the hydrogels was related to the intermolecular forces of PEG, AM and U. When the hydrogels were disrupted, the molecular arrangement between PEG, AM and U was disrupted, and with the increase of the healing time, the molecules were rearranged and reestablished the stable hydrogen-bonding interactions.18 The self-healing rate of the hydrogel increases with the increase of PEG content at the same healing time. This is because the PEG molecular chain contains a large number of oxygen atoms and hydroxyl groups, and with the gradual increase of the PEG content in the composite hydrogel, more dynamic hydrogen bonds can be formed inside the hydrogel,17 This leads to an increase in the self-healing rate of the hydrogels. In summary, the introduction of PEG increases the self-healing properties of PEG/P(U-AM-ChCl) composite hydrogels.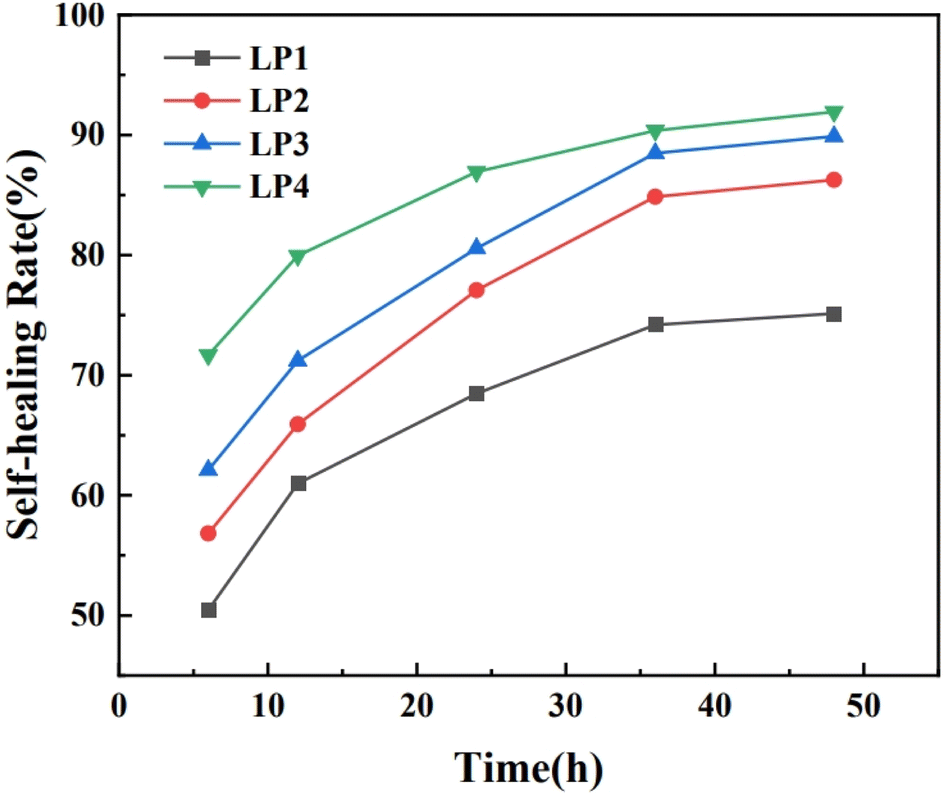 | ||
| Fig. 7 Self-healing rate of PEG/P(U-AM-ChCl) hydrogels at different healing times. | ||
Good self-healing properties are a promising approach to increase the lifetime of flexible sensor devices, and the self-healing properties of hydrogels were increased by the addition of PEG fillers while reducing the damage to the mechanical properties of hydrogels. As an electronic device attached to the skin or implanted in the human body, self-healing flexible sensors are required to be highly biocompatible, non-toxic and non-hazardous for long-term use. Therefore, the use of biocompatible and environmentally friendly raw materials is a feasible solution for the preparation of “green” self-healing hydrogels, and the good biocompatibility of PEG molecules and other characteristics of our composite hydrogels can be widely used in flexible sensors and medical devices.41
4 Conclusions
In this paper, PEG was added as a filler to DES prepared from U, AM, and ChCl, and MBA was added as a cross-linking agent and photoinitiator, and PEG/P(U-AM-ChCl) composite hydrogels with good self-healing properties were successfully prepared by photopolymerization, and the structure and properties of the composite hydrogels were investigated, and the experimental results showed that:(1) Compression experimental tests showed that the compressive strength of the composite hydrogel increased with the increase in PEG content due to the fact that the PEG molecules adsorb water molecules and maintain the moisture content of the hydrogel. This helps to maintain the flexibility and strength of the hydrogel, thus increasing the compressive properties of the composite hydrogel. After 10 cyclic compression experiments, the fatigue resistance of the hydrogel with PEG addition was stronger, with the maximum stress recovery decreasing from 91.41% to 80.21% for LP1 and from 99.35% to 95.52% for LP4.
(2) The self-healing performance of the hydrogels was significantly enhanced by the addition of PEG, and the self-healing rate of the composite hydrogel with a PEG content of 1.0 wt% reached 91.93% after 48 h. The self-healing rate of the composite hydrogel with 1.0 wt% PEG reached 91.93% after 48 h, and that of the hydrogel without PEG reached 75.13% after 48 h. It can be utilized in the fields of flexible sensors and medical devices.
(3) The PEG/P(U-AM-ChCl) composite hydrogel has good swelling properties, which makes the composite hydrogel have a good application prospect in wastewater treatment.
Author contributions
B. L., proposed the ideas, steps and details of the experiment, most of the experiments were done by H. Y. L., M. J. Z, A. L. W., where H. Y. L., was instrumental in the proper conduct of the experiments and wrote the article together with B. L., and all the authors analyzed the data, discussed the conclusions.Conflicts of interest
The authors declare that there are no competing interests regarding the publication of this article.Acknowledgements
The work is supported by the 2022 Knowledge Innovation Dawn Special Plan Project (2022010801020393). This work was finished at Wuhan Polytechnic University, Wuhan, China.Notes and references
- C. G. Garcia and K. L. Kiick, Acta Biomater., 2019, 84, 34–48 Search PubMed.
- B. Li, X. Xu, Z. Hu, Y. Li, M. Zhou, J. Liu, Y. Jiang and P. Wang, RSC Adv., 2022, 12, 19022–19028 RSC.
- E. G. Arafa, M. W. Sabaa, R. R. Mohamed, E. M. Kamel, A. M. Elzanaty, A. M. Mahmoud and O. F. Abdel-Gawad, Carbohydr. Polym., 2022, 291, 119555 CrossRef CAS PubMed.
- Z. Y. Zhang, T. R. Lin, S. X. Li, X. B. Chen, X. Y. Que, L. Sheng, Y. Hu, J. Peng, H. L. Ma, J. Q. Li, W. J. Zhang and M. L. Zhai, Macromol. Biosci., 2022, 22, e2100361 CrossRef PubMed.
- K. T. Nguyen and J. L. West, Biomaterials, 2002, 4307–4314 CrossRef CAS PubMed.
- F. Q. Luo, W. Xu, J. Y. Zhang, R. Liu, Y. C. Huang, C. S. Xiao and J. Z. Du, Acta Biomater., 2022, 147, 235–244 CrossRef CAS PubMed.
- R. Censi, W. Schuurman, J. Malda, G. di Dato, P. E. Burgisser, W. J. A. Dhert, C. F. van Nostrum, P. di Martino, T. Vermonden and W. E. Hennink, Adv. Funct. Mater., 2011, 21, 1833–1842 CrossRef CAS.
- S. Sun, Y. Cui, B. Yuan, M. Dou, G. Wang, H. Xu, J. Wang, W. Yin, D. Wu and C. Peng, Front. bioeng. biotechnol., 2023, 11, 1117647 CrossRef PubMed.
- Y. Zhang, N. Ding, T. Zhang, Q. Sun, B. Han and T. Yu, Front. Chem., 2019, 7, 682 CrossRef CAS PubMed.
- H. Pohlit, I. Bellinghausen, M. Schomer, B. Heydenreich, J. Saloga and H. Frey, Biomacromolecules, 2015, 16, 3103–3111 CrossRef CAS PubMed.
- J. X. Chen, J. Yuan, Y. L. Wu, P. Wang, P. Zhao, G. Z. Lv and J. H. Chen, J. Biomed. Mater. Res., Part A, 2018, 106, 192–200 CrossRef CAS PubMed.
- S. Zeng, J. Y. Zhang, G. Q. Zu and J. Huang, Carbohydr. Polym., 2021, 267, 118198 CrossRef CAS PubMed.
- A. Mateescu, Y. Wang, J. Dostalek and U. Jonas, Membranes, 2012, 2, 40–69 CrossRef CAS PubMed.
- T. Wang, M. Turhan and S. Gunasekaran, Polym. Int., 2004, 53, 911–918 CrossRef CAS.
- R. An, X. Zhang, L. Han, X. Wang, Y. Zhang, L. Shi and R. Ran, Mater. Sci. Eng., C, 2020, 107, 110310 CrossRef CAS PubMed.
- A. Khanal and S. Y. Fang, Chem.–Euro. J., 2017, 23, 15133–15142 CrossRef CAS PubMed.
- T. F. Ma, Y. Kong, H. B. Liu, X. Xu, Q. Y. Yue, B. Y. Gao and Y. Gao, J. Colloid Interface Sci., 2023, 633, 628–639 Search PubMed.
- L. J. Macdougall, M. M. Perez-Madrigal, J. E. Shaw, M. Inam, J. A. Hoyland, R. O'Reilly, S. M. Richardson and A. P. Dove, Biomater. Sci., 2018, 6, 2932–2937 RSC.
- S. P. Zustiak and J. B. Leach, Biomacromolecules, 2010, 11, 1348–1357 Search PubMed.
- J. Sung, D. G. Lee, S. Lee, J. Park and H. W. Jung, Materials, 2020, 13, 3277 CrossRef CAS PubMed.
- L. Liu, Y. Kong, H. Xu, J. P. Li, J. X. Dong and Z. Lin, Microporous Mesoporous Mater., 2008, 115, 624–628 Search PubMed.
- H. Vanda, Y. T. Dai, E. G. Wilson, R. Verpoorte and Y. H. Choi, C. R. Chim., 2018, 21, 628–638 Search PubMed.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- S. C. Cunha and J. O. Fernandes, TrAC, Trends Anal. Chem., 2018, 105, 225–239 CrossRef CAS.
- M. Micic, Y. J. Zheng, V. Moy, X. H. Zhang, F. M. Andreopoulos and R. M. Leblanc, Colloids Surf., B, 2003, 27, 147–158 Search PubMed.
- L. Almany and D. Seliktar, Biomaterials, 2005, 26, 2467–2477 Search PubMed.
- Y. Oh, J. Cha, S.-G. Kang and P. Kim, J. Ind. Eng. Chem., 2016, 39, 10–15 CrossRef CAS.
- N. Zivic, M. Bouzrati-Zerrelli, S. Villotte, F. Morlet-Savary, C. Dietlin, F. Dumur, D. Gigmes, J. P. Fouassier and J. Lalevée, Polym. Chem., 2016, 7, 5873–5879 RSC.
- C. Y. Gong, S. A. Shi, P. W. Dong, B. Kan, M. L. Gou, X. H. Wang, X. Y. Li, F. Luo, X. Zhao, Y. Q. Wei and Z. Y. Qian, Int. J. Pharm., 2009, 365, 89–99 CrossRef CAS PubMed.
- F. N. Tan, Y. Y. He and W. P. Sui, Adv. Mater. Res., 2014, 941–944, 1216–1220 Search PubMed.
- Q. Tang, X. Sun, Q. Li, J. Wu and J. Lin, Sci. Technol. Adv. Mater., 2009, 10, 015002 Search PubMed.
- A. Jafari, S. Hassanajili, N. Azarpira, M. Bagher Karimi and B. Geramizadeh, Eur. Polym. J., 2019, 118, 113–127 Search PubMed.
- B. V. Farahani, H. Ghasemzadeh and S. Afraz, J. Chin. Chem. Soc., 2016, 63, 438–444 CrossRef CAS.
- T. Ma, Y. Kong, H. Liu, X. Xu, Q. Yue, B. Gao and Y. Gao, J. Colloid Interface Sci., 2023, 633, 628–639 CrossRef CAS PubMed.
- T. J. Williams, A. S. Jeevarathinam, F. Jivan, V. Baldock, P. Kim, M. J. McShane and D. L. Alge, J. Mater. Chem. B, 2023, 11, 1749–1759 RSC.
- X. Yang, B. L. Dargaville and D. W. Hutmacher, Polymers, 2021, 13, 845 Search PubMed.
- D. Getya, A. Lucas and I. Gitsov, Int. J. Mol. Sci., 2023, 24, 7558 Search PubMed.
- Y. Yang, H. Sun, C. Shi, Y. Liu, Y. Zhu and Y. Song, J. Colloid Interface Sci., 2023, 629, 1021–1031 CrossRef CAS PubMed.
- T. K. Mudiyanselage, N. Weerasinghe, M. Karunaratna and N. Withanage, J. Appl. Polym. Sci., 2022, 139, e53048 CrossRef.
- Y. Chao, S. Yu, H. Zhang, D. Gong, J. Li, F. Wang, J. Chen, J. Zhu and J. Chen, ACS Appl. Bio Mater., 2023, 6, 1525–1535 CrossRef CAS PubMed.
- T. Qin, W. C. Liao, L. Yu, J. H. Zhu, M. Wu, Q. Y. Peng, L. B. Han and H. B. Zeng, J. Polym. Sci., 2022, 60, 2607–2634 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |