
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ high-valued transformation of nonmetals in waste printed circuit boards into supercapacitor electrodes with excellent performance†
Zhen Xia,
Ruitong Gao *a,
Zhaojun Chen*a,
Hui Dua and
Zhenming Xub
*a,
Zhaojun Chen*a,
Hui Dua and
Zhenming Xub
aCollege of Chemistry and Chemical Engineering, Institute for Sustainable Energy and Resources, Qingdao University, Qingdao, 308 Ningxia Road, Qingdao 266071, P.R. China. E-mail: gaoruitong@qdu.edu.cn; chenzj_upc@126.com; Tel: +86 15806391156 Tel: +86 18953271778
bSchool of Environmental Science and Engineering, Shanghai Jiao Tong University, 800 Dongchuan Road, Shanghai 200240, P.R. China
First published on 3rd January 2024
Abstract
Nonmetals in waste printed circuit boards after metal separation containing brominated resin and fiberglass are considered hazardous and low-recoveryvalue e-waste. However, if these nonmetals are not treated or are improperly treated, they can cause serious environmental pollution. Therefore, there is an urgent and significant need to develop an efficient recycling process for these nonmetals. Based on the concept of high-valued recycling of waste, this study in situ utilized such nonmetals to prepare a porous supercapacitor electrode through a facile carbonization, activation, and carbon thermal reduction process. The results indicated that the activation was a key role in constructing a porous structure. The optimal parameters for activation were a temperature of 800 °C, mass ratio of KOH to pyrolytic residues of 2, and an activation time of 1 h. The electrode materials exhibited a surface area of 589 m2 g−1 and hierarchical porous structures. In addition, the supercapacitors exhibited a capacitance of 77.14 mF cm−2 (62.5 mF cm−2) at 0.5 mA cm−2 (100 mV s−1). Moreover, the supercapacitors had excellent temperature resistance and adaptability. The capacitance retention was 89.36% and 90% at −50 °C and 100 °C after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles, respectively. This study provides a high-valued recycling strategy to utilize the nonmetals in e-waste as energy materials.
000 cycles, respectively. This study provides a high-valued recycling strategy to utilize the nonmetals in e-waste as energy materials.
1. Introduction
With the continuous development of modern science and technology, the demand for electronic equipment is rapidly increasing. However, this has led to a concerning issue of electronic waste due to the short lifespan and frequent replacement of devices.1 According to the statistics, the global quantity of electronic waste had reached approximately 57 Mt in 2021.2 It is predicted that the annual growth rate of electronic waste will reach up to 2 Mt, which could lead to a total mass of 74.7 Mt accumulated by the year 2030.3,4 Electronic waste not only contains toxic substances5 but also contains a significant number of valuable metals. The pollution and resources of electronic waste make it a complex problem that needs to be solved urgently. Landfill and incineration disposal methods are environmentally unfriendly and a waste of resources. Therefore, it is necessary to find appropriate solutions to deal with electronic waste.Printed circuit boards represent the main parts of electronic equipment. The production of waste printed circuit boards (WPCBs) is increasing with the rising electronic waste. In China, WPCBs account for 4–7% of electronic waste and are predicted to reach 29.92 Mt by 2030.5,6 WPCBs mainly include metallic fractions (MFs) and non-metallic fractions (NMFs).7 Due to the high economic value of the MFs, various techniques, including mechanical physical recovery technologies, pyrometallurgical treatments, and hydrometallurgical processes have been researched for recycling the metallic parts. Currently, the recovery rate of expensive metals is more than 90%.8–15 In contrast, the residual NMF is difficult to recycle. However, the NMFs contain many kinds of toxic substances. The most significant of these toxic substances is brominated flame retardants crosslinked with epoxy resins. Brominated flame retardants mainly include tetrabromodiphenol A and poly-brominated diphenyl ethers (PBDEs).16 These toxic substances can not only pollute the environment but also harm human health.17 Researches have demonstrated that PBDEs can cause neurodevelopmental defects, thyroid hormone disturbances, and even cancer.18 Hence, finding suitable ways to dispose of the NMFs is necessary.
At present, various methods, such as incineration, landfill, fillers, and pyrolysis have been developed for disposing of the NMFs. Guo et al. used the sorted NMFs as reinforcement material to replace wood powder to synthesize wood plastic materials.19,20 Zheng et al. added NMFs as a reinforced filler to polypropylene (PP), which significantly improved the tensile and bending properties of the NMFs/PP composites.21 Niu et al. applied NMFs to concrete as a substitute for inorganic fillers. This application improved the compressive strength and impact of the original material.22 Gao et al. analyzed the thermal decomposition process and mechanism of WPCBs and synthesized oil-based resin, magnetic graphitic carbon, and superhydrophobic membranes using the pyrolysis hydrocarbons of NMFs.23–27 Ma et al. utilized some debromination agents to remove bromophenol, HBr, and other harmful components in NMFs and researched the mechanism of debromination, effectively reducing the harm of NMFs.28 However, incineration and landfill methods cause serious pollution to the environment and waste resources. There is also a risk of secondary contamination with the filler method. The pyrolysis technique generates a significant quantity of pyrolysis oil, gas, and residues, which are complex in composition and pose challenges in terms of recycling.29 Therefore, a reasonable way to dispose of the NMFs and realize their high-value utilization is urgently needed.
The NMFs mainly consist of resin and fiberglass, typically containing abundant silicon and carbon sources. It was noticed that the carbonaceous and siliceous materials in NMFs are evenly mixed, which is conducive to solid–state reactions. Thus, NMFs have the potential to be transformed into electrode materials rich in SiC by in situ methods. The in situ transformation of NMFs could not only solve the problem of NMFs disposal, but also provide a new strategy for the resource utilization of NMFs. However, the research on the in situ transformation of NMFs into electrode materials rich in SiC is currently insufficient. Therefore, it is very necessary to further study the in situ transformation of NMFs.
Accordingly, the in situ transformation of NMFs into electrode materials rich in porous SiC was researched in the present study. The preparation process here included acid leaching, carbonization, activation, and carbon thermal reduction. Besides, the effects of the preparation conditions, such as activation method, activation temperature, activation time, and mass ratio of KOH to the pyrolysis residues on the pore structure of the electrode materials were investigated. Moreover, the morphology, chemical composition, and structural evolution of the pyrolysis residues, activated residues, and electrode materials were observed by SEM, XRD, FT-IR, BET, and Raman. Finally, the stability of the developed supercapacitors at normal temperature, extreme temperature, and during continuous temperature changes was investigated. This research presents a recycling strategy for effectively disposing of NMFs while realizing the utilization of the high-value constituents.
2. Experimental section
2.1 Experimental materials
The WPCBs in this study were provided by Shanghai Xinjinqiao Environmental Protection and were pre-treated by mechanical treatment. Fig. S1† shows the pretreatment process. First, the electronic components of the WPCBs were removed utilizing a dismantling device. Then the placodes of WPCBs were broken by a crusher. Finally, the MFs and NMFs in the crushed powder were separated by electrostatic separation equipment. The NMFs were utilized as the experimental materials. The main compositions of the NMFs were organics (resin, 39.593%), SiO2 (43.966%), CaO (10.097%), CuO (2.794%), Al2O3 (2.806%), Fe2O3 (0.649%), and MgO (0.095%). HNO3 and KOH were bought from Sinopharm Group Co. Ltd. All the chemicals used in the experiments were analytically pure.2.2 In situ transformation of NMFs into electrode materials
The process for the in situ transformation of the NMFs into electrode materials is shown in Fig. 1. The process for transforming the NMFs into electrode materials included acid leaching, carbonization, activation, and carbon thermal reduction steps.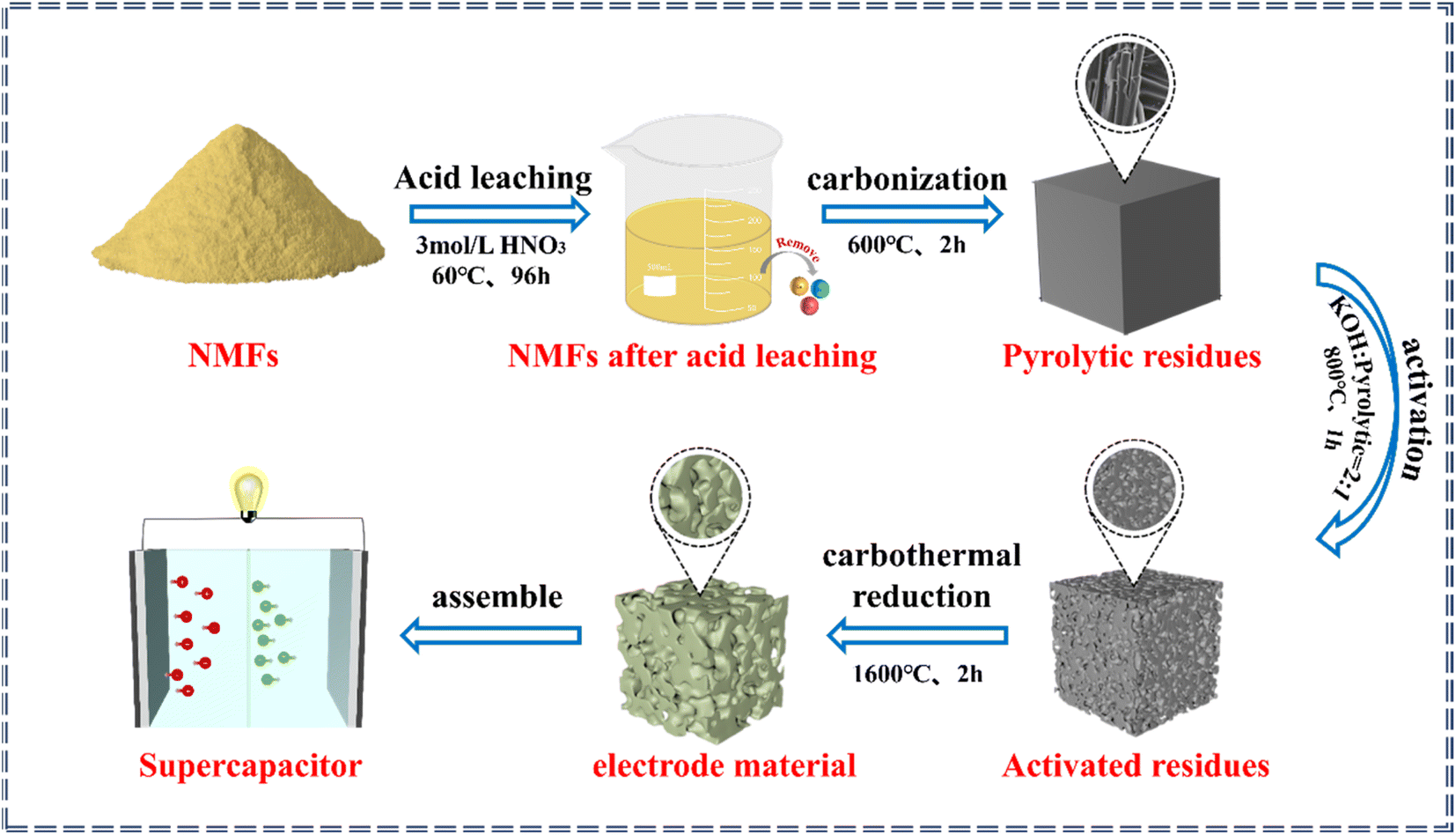 | ||
| Fig. 1 Process for the in situ transformation of NMFs into electrode materials. | ||
The first step was acid leaching. During this process, the NMFs were added into 3.0 mol L−1 HNO3 with a solid–liquid ratio of 1:5. The soaking time was 96 h at 60 °C to remove any impurities.30 Then the NMFs after acid leaching were washed with deionized water until neutral and dried for later use. The second step was carbonization. Under an Ar atmosphere, the NMFs were heated to 600 °C and kept at that temperature for 1–3 h to obtain the pyrolytic residues. The third step was activation. The mass ratio of pyrolytic residue to KOH in the mixture was in the range of 1–3 by a dry or wet activation method, and the mixture was then heated in an Ar atmosphere. The activated residues were obtained by keeping the mixture at 700–1000 °C for 0.5–2 h. The pore structures of the activated residues were regulated by adjusting the activation methods, mass ratio of KOH to pyrolytic residues, activation temperature, and activation time. The fourth step was carbothermal reduction. The activated residues with an optimal porous structure were selected for high-temperature carbothermal reduction to acquire the electrode material. Finally, the electrode material was used to assemble supercapacitors.
2.3 Electrochemical measurements
All the electrochemical properties of the WPCBs-based supercapacitor were tested on an electrochemical workstation (CHI760). The working electrodes were based on nickel foam. The electrode material, acetylene black, and adhesive (polyvinylidene chloride) were fully mixed in a mass ratio of 8:1:1, and then N,N-dimethylformamide (DFM) was added. Then the mixture was evenly smeared on the nickel foam in a smearing area of about 1 cm2, and the amount of active substance loaded on each nickel foam was about 7 mg. After that, the nickel foams coated with active substances were dried at 70 °C for 12 h, and then compressed at 10 MPa to make the working electrodes.
In the three-electrode test, the working electrode, counter electrode, and reference electrode comprised the coated foam nickel, the platinum electrode, and Hg/HgO, respectively. In the two-electrode system, two identical coated nickel foams were used as the working electrodes. In the electrochemical test, 8 M KOH aqueous solution was used for the electrolyte.31 High and low temperature test chambers were used for the variable temperature experiments.
According to the results of the GCD curves, the capacitance of the supercapacitors was obtained via formulas (1) and (2):
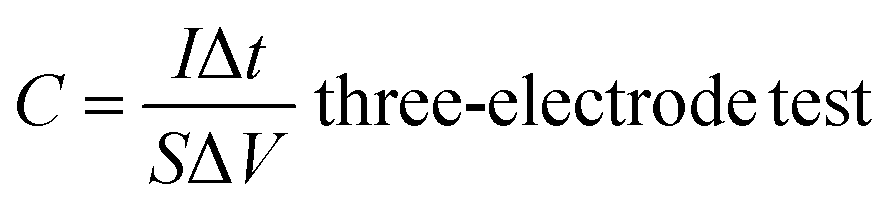 | (1) |
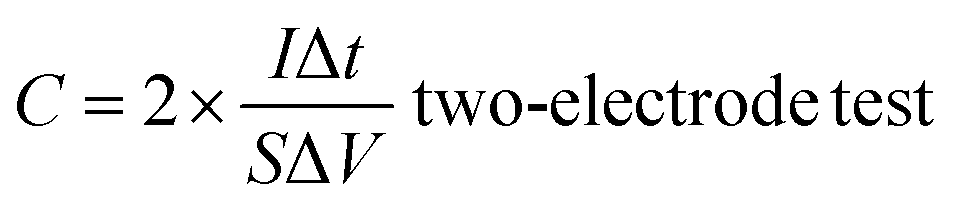 | (2) |
According to the results of the CV curves, the capacitance of the supercapacitors could be obtained with formula (3):
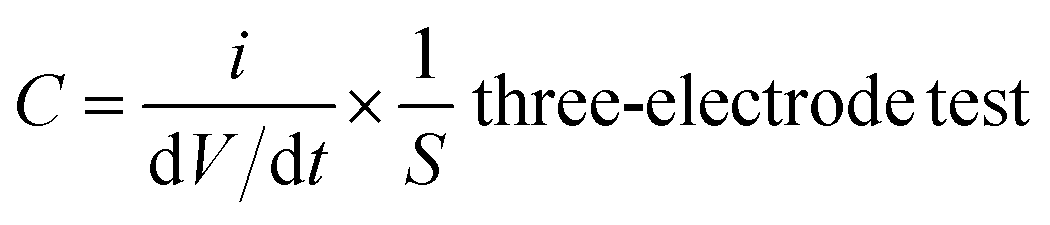 | (3) |
2.4 Characterization
X-Ray fluorescence (XRF) was used to measure the compositions of the NMFs before and after acid leaching pretreatment. Scanning electron microscopy (SEM, Sigma300) was used to observe the morphologies of the pyrolytic residues, activated residues, and electrode materials. The bonding states of the pyrolytic residues, activated residues, and electrode materials were analyzed by X-ray photoelectron spectroscopy (XPS, Nexsa). The crystal structure and phase composition of the materials at various stages were determined by X-ray diffraction (XRD, Bruker D8 Advance) and Raman spectroscopy (Raman, inVia). The organic functional groups of the materials were characterized by Fourier-transform infrared spectroscopy (FT-IR, Nicolet iS50). The specific surface area and pore-size distribution of the activated residues and electrode material were measured using the Brunauer–Emmett–Teller method (BET, Malin ASAP2460).3. Results and discussion
3.1 Acid pretreatment of the NMFs
As shown in Table 1, the compositions of the NMFs before and after acid leaching were determined by XRF (see the measurement details in the ESI, Text 1†). It could be seen that the NMFs were composed of organics (resin, 39.593%), SiO2 (43.966%), CaO (10.097%), CuO (2.794%), Al2O3 (2.806%), Fe2O3 (0.649%), and MgO (0.095%). After the acid leaching process, all the Al2O3 and MgO were completely removed, while most of the CaO, CuO, and Fe2O3 were also removed. Through this process, the NMFs after acid leaching were mainly composed of SiO2 and organics. This result suggested that most of the metal oxides in the NMFs could be removed by HNO3 leaching, which provided a purer environment for the in situ transformation of the NMFs.30| Element | SiO2 | CaO | CuO | Al2O3 | Fe2O3 | MgO | Organics |
|---|---|---|---|---|---|---|---|
| NMFs (%) | 43.966 | 10.097 | 2.794 | 2.806 | 0.649 | 0.095 | 39.593 |
| NMFs after acid leaching (%) | 63.825 | 0.719 | 0.076 | 0 | 0.077 | 0 | 35.303 |
3.2 Parameters for the in situ transformation of the NMFs
The TG and TDG curves of the NMFs are exhibited in Fig. S2(a).† When the temperature was above 400 °C, the mass of the NMFs decreased rapidly. The sample quality did not change when the temperature exceeded 600 °C. As shown in Fig. S2(b),† the pyrolytic residues were affected by the carbonization time. The specific surface area of the pyrolytic residues reached a maximum of 70 m2 g−1 when the carbonization time was 2 h. Based on the above analysis, the optimal carbonization conditions for the NMFs was 600 °C for 2 h.The size of the specific surface was affected by various activation conditions. The specific surface areas of the activated residues under different activation methods and activation ratios are presented in Fig. 2(a). The results show that the dry method had a better activation effect. In addition, when the activation proportion was 2 by the dry activation method, the maximum surface area was 1569 m2 g−1. However, the specific surface area of the activated residues was only 195 m2 g−1 when the activation proportion was 1. This was caused by the low KOH content and insufficient activation, leading to an underdeveloped porous structure. On the other hand, when the activation proportion reached 3, specific surface area decreased to 314 m2 g−1. This could be attributed to the excess amount of KOH used in the process, which caused KOH to react with carbon atoms on the skeleton. As a result, the pore walls disappeared and some micropores collapsed into mesopores and macropores, and so the specific surface area decreased.32 The activation time also greatly influenced the activation effect. The results are shown in Fig. 2(b). The maximum surface area of 1596 m2 g−1 was obtained when the activation time was 1 h. A short activation time can result in insufficient activation and an undeveloped porous structure. Therefore, when the activation temperature was reduced to 0.5 h, the activation effect was significantly reduced. At this time, the activated residues had a specific surface area of only 540 m2 g−1. The activated residues had a specific surface area of 720 m2 g−1 and 689 m2 g−1 at activation times of 1.5 h and 2 h, respectively. This was caused by the collapse of the porous structure due to the long activation time and excessive activation of KOH. Therefore, 1 h was the optimal activation time. At the same time, the effect of the activation temperature was also considered. Fig. 2(c) showed that the maximum surface area of 1596 m2 g−1 was obtained at 800 °C. This was due to the relationship between the generation of a porous structure in the activated residues and the gasification reaction of KOH.33,34 The lower temperature could not satisfy the condition of K gasification, while the higher temperature might lead to the destruction of the porous structure. In summary, the most effective activation conditions were activation by the dry method, an activation temperature of 800 °C, mass ratio of KOH to pyrolytic residues of 2, and activation time of 1 h.
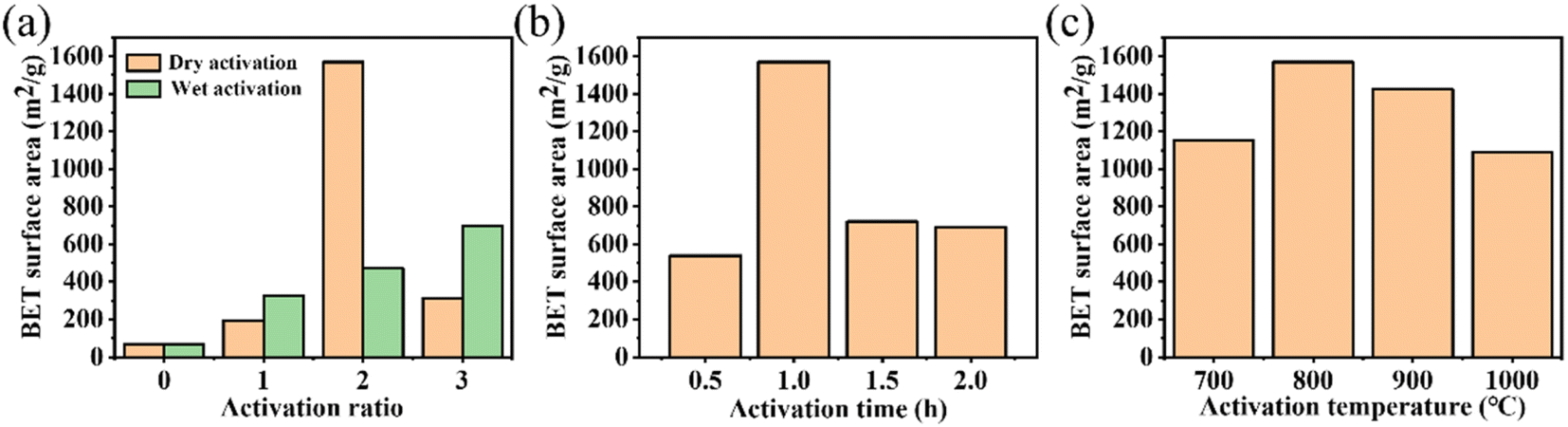 | ||
| Fig. 2 (a) The effect of the activation methods and ratios on the activation effect at 800 °C for 1 h; (b) The effect of the activation time under the condition of dry activation at 800 °C and an activation ratio of 2; (c) The effect of the activation temperature under the condition of dry activation, an activation ratio of 2, and activation time of 1 h. | ||
The formations of electrode materials by carbothermal reduction at different temperatures were analyzed by XRD, and the results are shown in Fig. S3.† When the temperature was 1300 °C, no characteristic peaks were observed. However, when the temperature rose to 1600 °C, strong diffraction peaks of SiC appeared, which proved the existence of SiC. This result was similar to the literature report.35 Therefore, 1600 °C was considered to be the optimum temperature for carbothermal reduction of the activated residues.
3.3 Characterizations of the electrode materials
The crystal structures of the pyrolytic residues, activated residues, and electrode material were analyzed by XRD, and the results are shown in Fig. 3(a). The peak at 20.9° was the characteristic peak of SiO2 (101) and that at 26.6° was the characteristic peak of SiO2 (011) (JCPDS-Card No. 65-0466). This result indicated that Si in the residues mainly existed in the form of SiO2. A weaker, wider peak was found at about 23°, indicating that C in the residues was mainly in the form of amorphous C. Compared with the pyrolytic residues, the diffraction peak of the activated residues was weaker, which proved that the structure of C was destroyed and the crystallinity was reduced after the activation by KOH. After being calcined at 1600 °C, strong diffraction peaks appeared at 35.7°, 41.3°, 60°, and 71.5°, corresponding to characteristic diffraction peaks of the face-cubic centered 3C-SiC (JCPDS-Card No. 29-1129). This proved that SiO2 and C in the residues were calcined at high temperature to successfully produce SiC through a carbothermal reduction reaction. The organic functional groups of the pyrolytic residues, activated residues, and electrode material were observed by FT-IR spectroscopy. As shown in Fig. 3(b), the samples showed infrared absorption peaks at 3440, 1620, and 1070 cm−1, corresponding to the O–H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, and C–O bonds, respectively. This indicated that the organic functional groups had not changed after KOH activation. However, after being calcined at 1600 °C, a peak was generated at 819 cm−1, belonging to the peak of Si–C.30 This confirmed that SiC was successfully prepared, corresponding to the XRD results. Fig. 3(c) shows the Raman spectra of the pyrolytic residues, activated residues, and electrode material. The characteristic band at 1340 cm−1 was the D band of carbon, while the characteristic band at 1590 cm−1 was the G-band of carbon. The D band indicated the presence of defects in graphite. The G-band was identified as belonging to the E2g phonon in the center of the Brillouin zone. This reflected the structural integrity of the sp2 hybrid bond in the graphite. Furthermore, the ID:IG ratio has been shown to be a reliable indicator of the degree of disordering of carbon materials. It could be obtained through Fig. 3(c), whereby the ID:IG value of the pyrolytic residues was 0.74. However, the ID:IG value of the activated residues was 0.86, indicating that abundant defects were produced in the residues after KOH activation. After calcination at 1600 °C, the value of ID:IG reached 1.57, and the degree of defects increased again. This might be because SiC was formed by a carbothermic reduction reaction between SiO2 and C at 1600 °C, which damaged the original structure of C. At the same time, a peak appeared at ∼798 cm−1, which belonged to the β phase of SiC.36 This proved the existence of SiC. Moreover, the content of SiC in the electrode material was about 80% (Fig. S4†).
C, and C–O bonds, respectively. This indicated that the organic functional groups had not changed after KOH activation. However, after being calcined at 1600 °C, a peak was generated at 819 cm−1, belonging to the peak of Si–C.30 This confirmed that SiC was successfully prepared, corresponding to the XRD results. Fig. 3(c) shows the Raman spectra of the pyrolytic residues, activated residues, and electrode material. The characteristic band at 1340 cm−1 was the D band of carbon, while the characteristic band at 1590 cm−1 was the G-band of carbon. The D band indicated the presence of defects in graphite. The G-band was identified as belonging to the E2g phonon in the center of the Brillouin zone. This reflected the structural integrity of the sp2 hybrid bond in the graphite. Furthermore, the ID:IG ratio has been shown to be a reliable indicator of the degree of disordering of carbon materials. It could be obtained through Fig. 3(c), whereby the ID:IG value of the pyrolytic residues was 0.74. However, the ID:IG value of the activated residues was 0.86, indicating that abundant defects were produced in the residues after KOH activation. After calcination at 1600 °C, the value of ID:IG reached 1.57, and the degree of defects increased again. This might be because SiC was formed by a carbothermic reduction reaction between SiO2 and C at 1600 °C, which damaged the original structure of C. At the same time, a peak appeared at ∼798 cm−1, which belonged to the β phase of SiC.36 This proved the existence of SiC. Moreover, the content of SiC in the electrode material was about 80% (Fig. S4†).
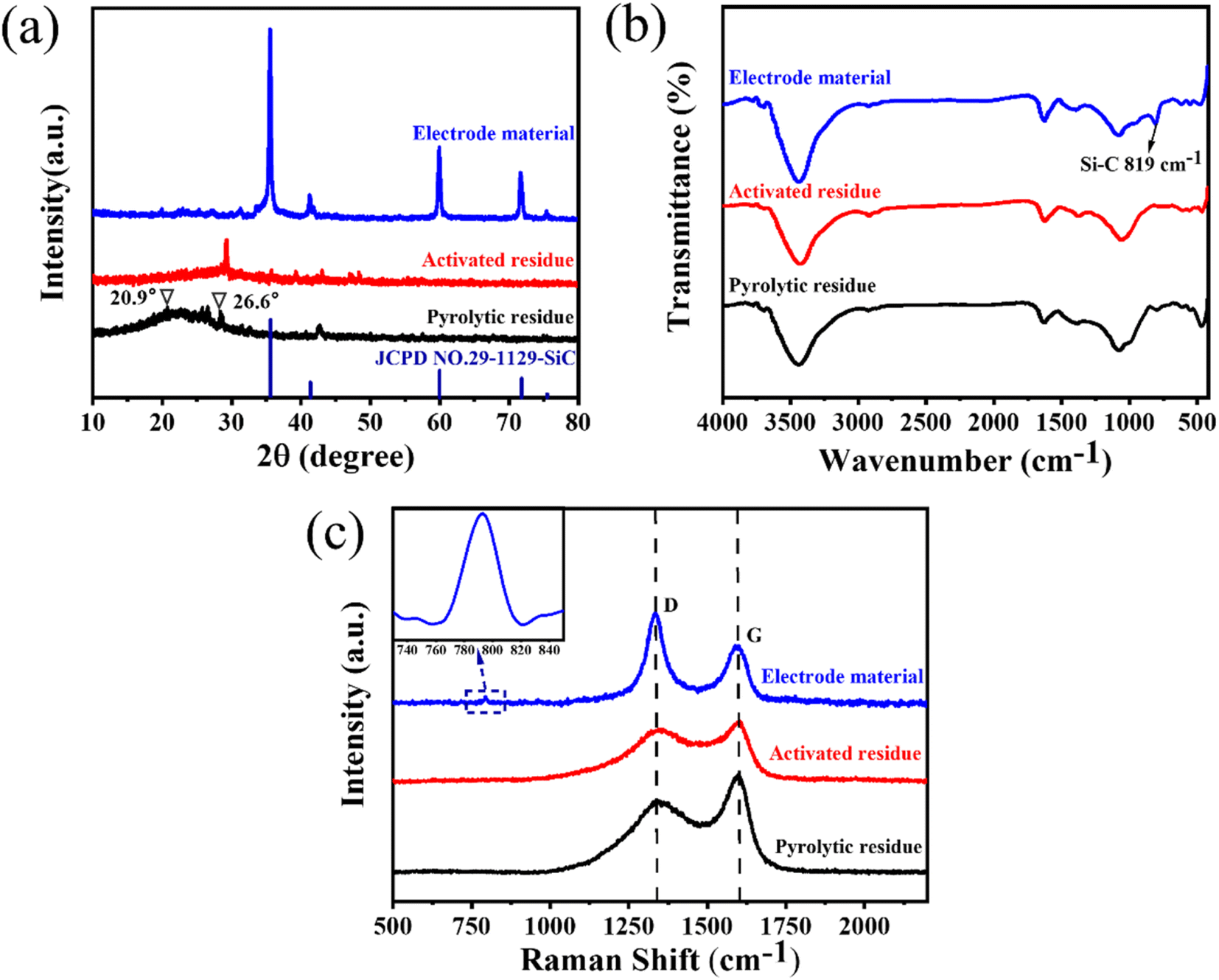 | ||
| Fig. 3 (a) XRD patterns, (b) FT-IR spectra, and (c) Raman spectra of the pyrolytic residues, activated residues, and electrode material. | ||
XPS was next used to analyze the surface chemical composition and chemical state of the pyrolytic residues, activated residues, and electrode material. The C 1s and Si 2p peaks were clearly observed in the spectrum of the pyrolytic residues, activated residues, and electrode material (Fig. S5†). In the Si 2p spectrum (Fig. 4(b)) of the pyrolytic residues and activated residues, the peaks at 104.6 and 103.2 eV were at the binding energy of Si–O.37 These indicated that Si existed in the form of SiO2 before and after activation. However, the peak for Si–O in the activated residues was shifted, which may have contributed to the structural change after activation. Besides, the peak at 283.8 eV in the C 1s spectrum (Fig. 4(a)) and peak at 100.8 eV in the Si 2p spectrum (Fig. 4(b)) of the electrode material were related to the binding energy of Si–C, indicating the successful formation of SiC.38,39
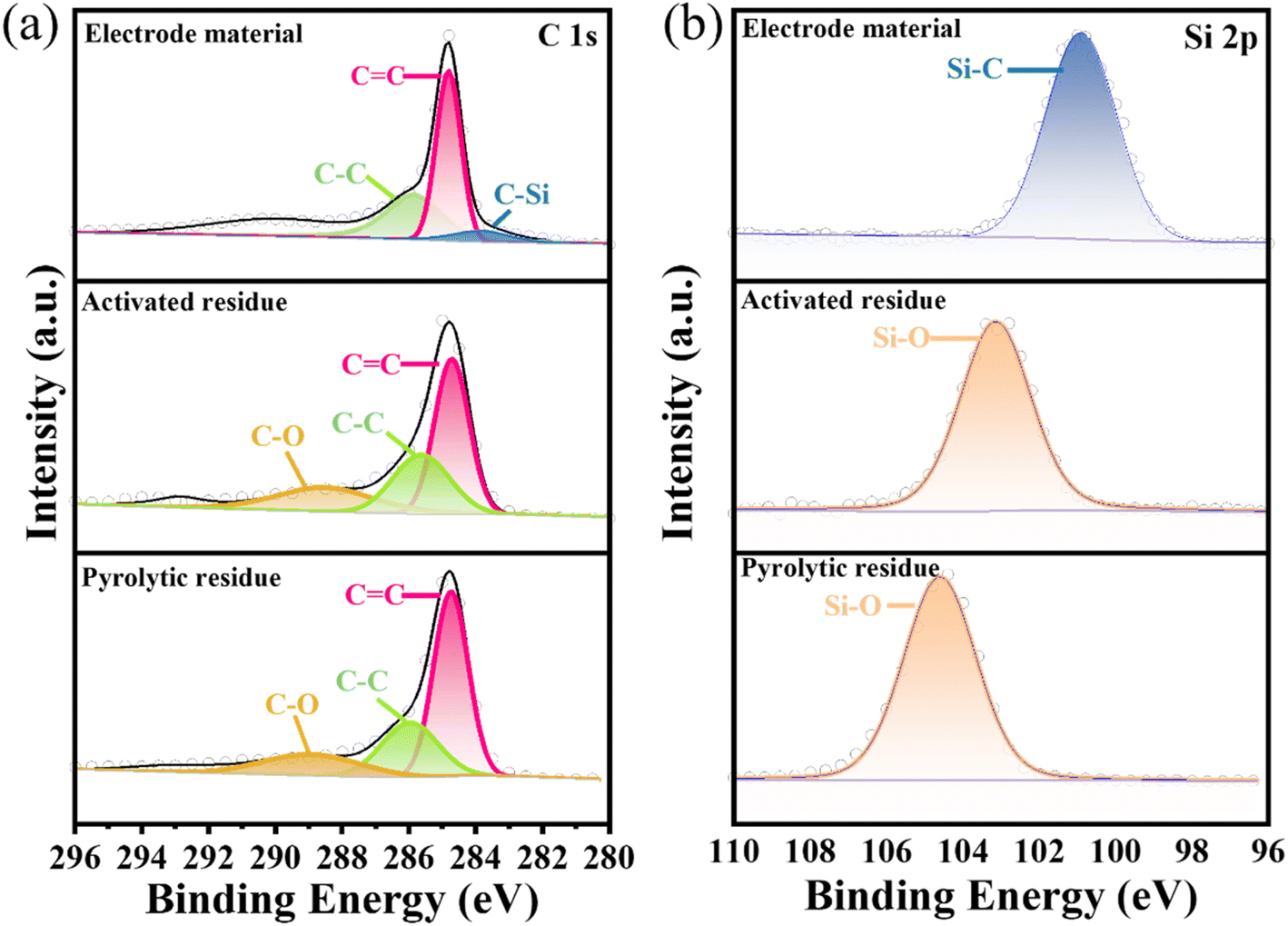 | ||
| Fig. 4 XPS spectra of (a) C 1s, and (b) Si 2p of the pyrolytic residues, activated residues, and electrode material. | ||
The morphology and size of the pyrolytic residues, activated residues, and electrode material were characterized by SEM. Fig. S6(a)† shows the SEM images of the pyrolytic residues. It could be seen that the pyrolytic residues mainly consisted of glass fiber and its coated coke. Glass fiber and bonded coke formed amorphous aggregates with particle sizes of about 300–500 μm, and the surfaces were smooth without obvious pores. Fig. S6(b)† displays the SEM results of the activated residues. Compared with the pyrolytic residues, the activated residues obtained after KOH activation had a smaller particle size and abundant pores. It could be found that the surfaces of the activated residues were rough. This was because during the activation process, KOH reacted with the carbon in the coke to form K2CO3, K2O, and H2, and the K2CO3 further reacted with carbon to form K and CO. When the activation temperature exceeded the boiling point of elemental K at 770 °C, elemental K could become a gas and enter the carbon layer, making pores, so that the original coke formed a certain porous structure.40 Fig. S6(c)† illustrates the morphology of the electrode material as obtained by SEM. Many small particles were observed and combined with the EDS mapping of C and Si distribution shown in Fig. S7(b, c)† which demonstrated the formation of a SiC-rich electrode material. Pores could also be observed on the surface of the electrode material, which indicated that the porous structure was still preserved after carbothermal reduction of the activated residues.
Next, BET analysis was used to determine the specific surface area and pore-size distribution of the activated residues and electrode material. The results are shown in Fig. 5(a). When P/P0 was less than 0.1, the adsorption capacity of the activated residues increased sharply, indicating that there were a lot of microporous structures in the activated residues. When P/P0 was greater than 0.1, the curve presented a non-horizontal adsorption platform, and the adsorption capacity increased. When P/P0 was close to 1, the curve showed an obvious upward slope, and the adsorption curve and desorption curve were not completely consistent in the range of 0.4–0.8, revealing a standard hysteresis loop.41,42 Combined with the pore-size distribution in Fig. 5(b), it could be seen that there were micropores, mesopores, and a few macropores in the activated residues. Compared with the activated residues, it could be found that the electrode material still contained a variety of pores, but the number of micropores and mesopores decreased while the number of macropores increased, which might be caused by two reasons. First, the original structure of C was destroyed by the formation of Si–C. Second, at high temperature, some micropores collapsed and were transformed into mesopores and macropores. The microporous structure of the electrode material had a high specific surface area, which was more conducive to providing more active sites for the absorption and desorption of electrolyte ions. While mesopores and macropores could better permeate the electrolyte, thus increasing the capacitive performance of the supercapacitors.43
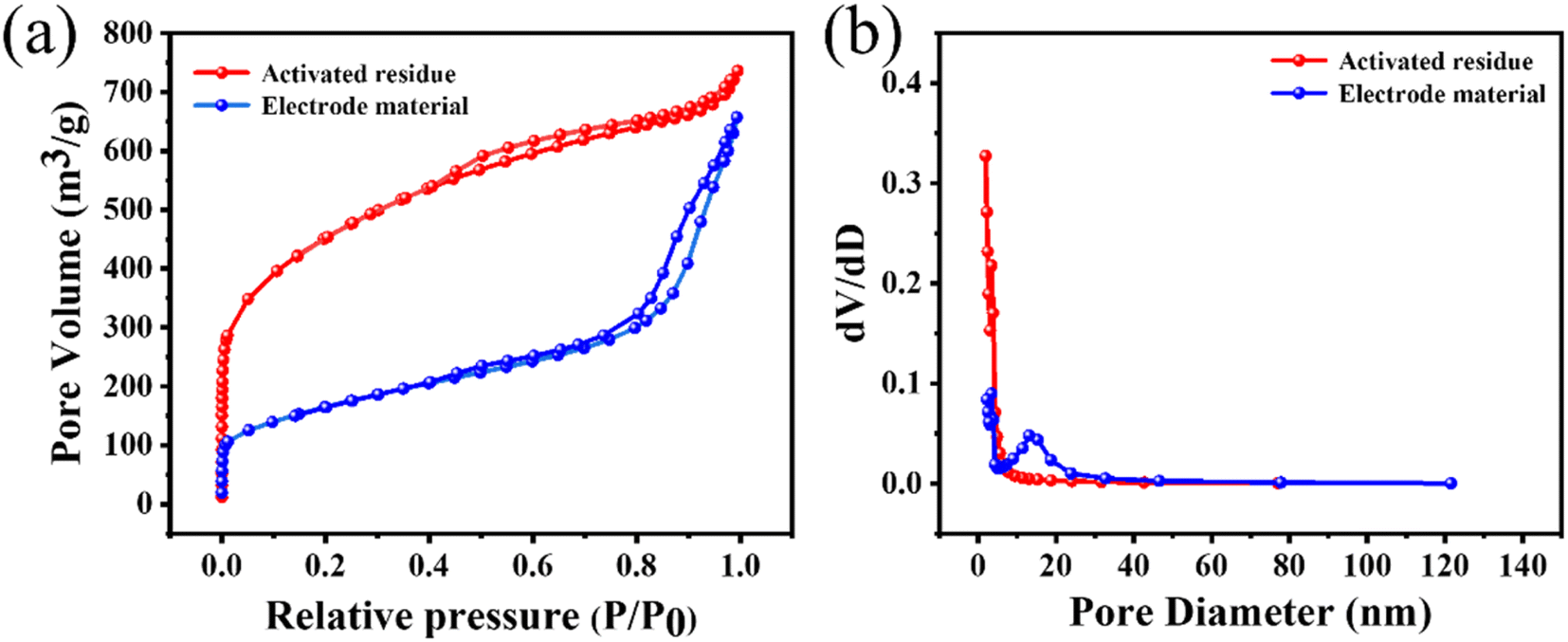 | ||
| Fig. 5 (a) N2 adsorption and desorption isotherms, and (b) pore-size distributions of the activated residues and electrode material. | ||
The above characterization results showed that the prepared electrode material contained abundant SiC and had a hierarchical porous structure. Therefore, this electrode material could be used to prepare SiC-rich supercapacitors with high performance and stability.
3.4 Electrochemical properties of the porous WPCB-based supercapacitors
The supercapacitors assembled from electrode materials that were not activated by KOH were defined as non-porous WPCB-based supercapacitors. The supercapacitors assembled by the electrode materials with hierarchical pore structures prepared by KOH activation were defined as porous WPCB-based supercapacitors. In 8 M KOH, non-porous and porous WPCB-based supercapacitors were tested with three electrodes. The CV curves of the non-porous and porous WPCB-based supercapacitors at 100 mV s−1 are shown in Fig. S8(a).† It could be found that the CV curves of both were quasi-rectangular. Fig. S8(b)† shows the GCD curves of both of them at 0.5 mA cm−2. All the curves formed a symmetrical triangular shape, which proved that the non-porous and porous WPCB-based supercapacitors had the capacitance characteristic of double layers. This also corresponded to the results of the CV tests. In addition, the capacitance of the non-porous WPCB-based supercapacitor was 3.44 mF cm−2 (1.28 mF cm−2) at 0.5 mA cm−2 (100 mV s−1), while that of the porous WPCB-based supercapacitor was 77.14 mF cm−2 (62.5 mF cm−2) at 0.5 mA cm−2 (100 mV s−1). Fig. S8(c)† shows the Nyquist comparisons of the non-porous and porous WPCB-based supercapacitors. The Warburg region of the porous WPCB-based supercapacitor was shorter than the non-porous WPCB-based supercapacitor, indicating that the porous WPCB-based supercapacitor had a lower internal resistance and faster ion migration rate. In addition, the slope of the porous WPCB-based supercapacitor was higher than that of the non-porous WPCB-based supercapacitor in the low-frequency region, which meant that the porous electrode material had a low ionic resistance in the electrolyte and faster ionic diffusion rate. The above results indicated that the porous WPCB-based supercapacitor generated after activation formed a certain pore structure, which was more conducive to ion migration and diffusion, so as to improve the capacitive performance.The electrochemical performances of the porous WPCB-based supercapacitor were evaluated by three electrodes tests at 20 °C. The CV and GCD curves (Fig. S9(a, b)†) of the supercapacitors were obtained after testing at different sweep speeds and current densities. The CV curves presented rectangular and the GCD curves triangular shapes. Therefore, the charge-storage behavior of the supercapacitors was a typical electrical double-layer capacitor. When the sweep speed reached 500 mV s−1, the CV curve could still retain a rectangle-like shape. This showed that the charge-storage process of the supercapacitors was fast and highly reversible.44 According to the GCD curves, the supercapacitors had a maximum capacitance of 77.14 mF cm−2 at 0.5 mA cm−2 (Fig. S9(c)†). The electrode area specific capacitance was still 54.29 mF cm−2 at 20 mA cm−2. The retention rate of the capacitor was 70.4%, showing that the capacitance of the supercapacitors had a satisfactory rate capability. In addition, the supercapacitors also showed excellent cyclic stability. Fig. S9(e)† shows that the capacitance retention rate was 100% at 10 mA cm−2 for 10000 cycles, and the GCD curves still showed a symmetrical triangular shape without significant changes. Nyquist plots (Fig. S9(d)†) were applied and further proved that the supercapacitors had excellent cyclic stability. The supercapacitors had almost the same slope in the low-frequency region after 10000 cycles. The equivalent resistance of the supercapacitors in series was about 0.75 Ω and remained stable before and after the cycling. The stability of impedance also indicated that the supercapacitors possessed an excellent structural stability and conductivity. Table 2 summarizes the electrochemical properties of some SiC and porous carbon supercapacitors reported in recent years. The porous WPCB-based supercapacitor in this work had a capacitance of 62.5 mF cm−2 at 100 mV s−1 and a capacitance retention rate of 100% after 10000 cycles. Compared with SiC-based supercapacitors, it thus offers the advantage of large capacitance. At the same time, it has greater stability than carbon-based supercapacitors. This result indicated that the supercapacitor prepared in this work has certain commercial prospects.
| Materials | Capacitance | Stability | Reference |
|---|---|---|---|
| Porous WPCB-based supercapacitor | 100 mV s−1 | 10000 |
This work |
| ∼62.5 mF cm−2 | ∼100% | ||
| SiC nanowires supercapacitor | 100 mV s−1 | 200000 |
45 |
| ∼240 μF cm−2 | ∼95% | ||
| SiC nanochannel supercapacitor | 10 mV s−1 | 10000 |
46 |
| ∼14.8 mF cm−2 | ∼96% | ||
| SiC nanowires supercapacitor | 50 mV s−1 | 100000 |
47 |
| ∼23 mF cm−2 | ∼90% | ||
| SiC nanoarray supercapacitor | 10 mV s−1 | 10000 |
48 |
| ∼4.8 mF cm−2 | ∼98% | ||
| SiC nanowires supercapacitor | 0.5 V s−1 | 10000 |
49 |
| ∼370 μF cm−2 | ∼100% | ||
| SiC nanowires supercapacitor | 100 mV s−1 | 10000 |
50 |
| ∼92 μF cm−2 | ∼60% | ||
| Porous carbon nanosheets supercapacitor | 2 mV s−1 | 10000 |
51 |
| 64.4 F g−1 | ∼87.6% | ||
| Porous carbon aerogels supercapacitor | 20 A g−1 | 4000 | 52 |
| 205 F g−1 | ∼92% | ||
| Porous carbon supercapacitor | 0.5 A g−1 | 3000 | 53 |
| 130 F g−1 | ∼86% | ||
| Porous carbon supercapacitor | 10 A g−1 | 4000 | 54 |
| 73.9 F g−1 | ∼84.8% |
The stability of energy-storage devices in extreme environments is a major challenge in practical applications. Therefore, the porous WPCB-based supercapacitor was tested at extreme temperatures. Fig. 6 shows the CV and GCD curves of the supercapacitors at −50 °C and 100 °C. The CV curves could still retain a rectangle-like shape at 200 mV s−1, which showed that the charge-storage process of the supercapacitors at extreme temperatures was still fast and highly reversible. Fig. 6(e) presents the cyclic stability of the supercapacitors at extreme temperatures. The results show that the capacitance retentions were 89.36% and 90% at −50 °C and 100 °C. Therefore, the porous WPCB-based supercapacitor had good temperature resistance.
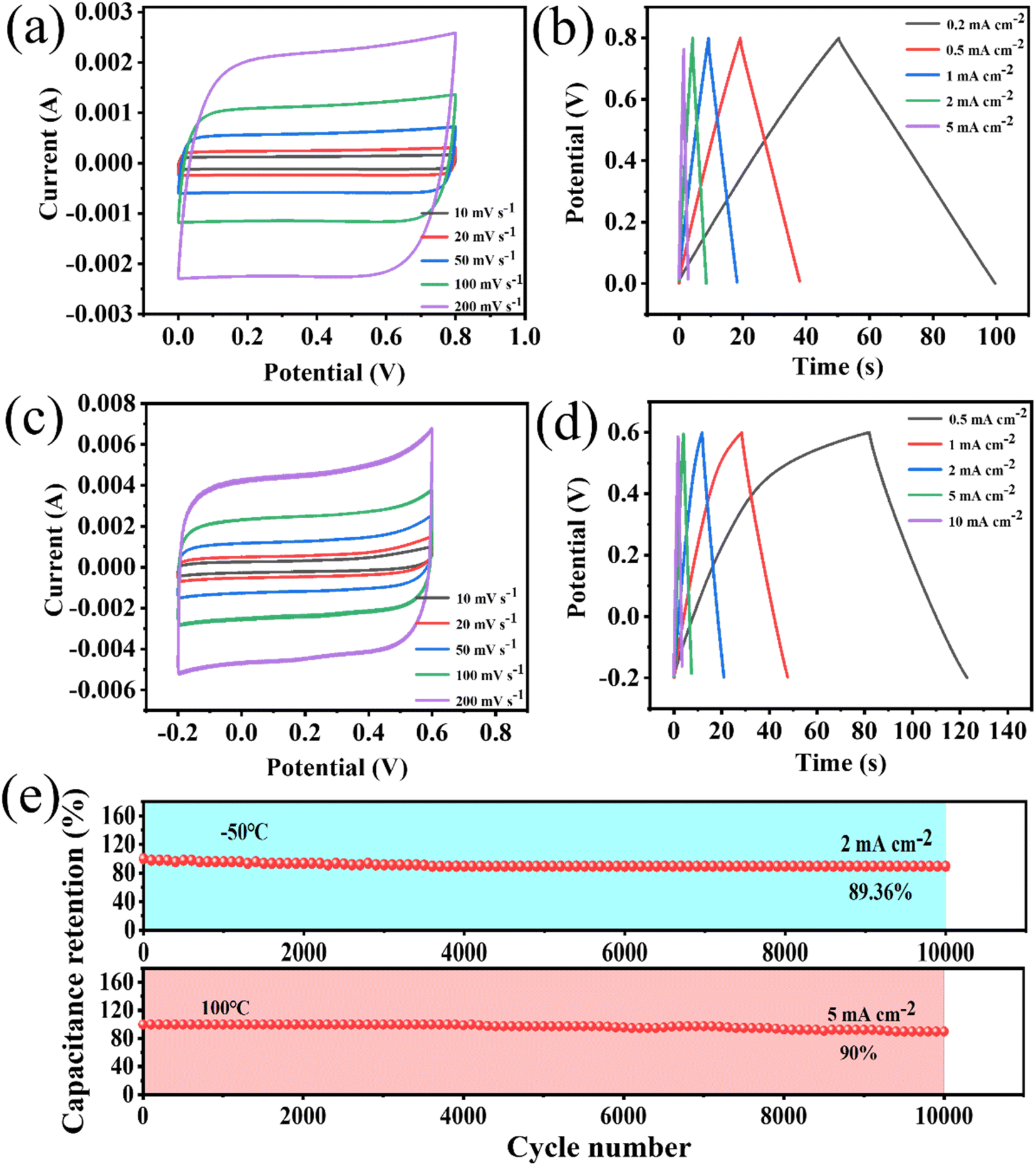 | ||
| Fig. 6 Electrochemical tests of the porous WPCB-based supercapacitor at −50 °C with two electrodes: (a) the CV curves of the porous WPCB-based supercapacitor; (b) the GCD curves of the porous WPCB-based supercapacitor. Electrochemical tests of the porous WPCB-based supercapacitor at 100 °C with two electrodes: (c) the CV curves of the porous WPCB-based supercapacitor; (d) the GCD curves of the porous WPCB-based supercapacitor; (e) the cycle stability at −50 °C and 100 °C. | ||
The adaptability of energy-storage devices to different temperatures is also an important performance to be considered. Therefore, the porous WPCB-based supercapacitor was electrochemically tested at continuously changing temperature. The supercapacitors could maintain CV curves with quasi-rectangular shapes at different temperatures in the temperature range of −50 °C to 100 °C (Fig. 7(a)). As the temperature increased, the shapes of the CV curves were slightly deformed at high voltages. This was mainly due to the surface of the porous SiC, which contained some oxygen-containing functional groups, which were decomposed at high temperatures.55 Fig. 7(b) presents the GCD curves at 2 mA cm−2 under various temperatures. The GCD curves were all symmetrical triangle shapes, which indicated that the supercapacitors had the characteristics of double-layer capacitance. The slight deformation of the GCD curve at 100 °C confirmed the existence of surface oxidation side reactions, which corresponded to the CV results. The change capacitance of the supercapacitors at various temperatures recorded from the GCD plots is shown in Fig. 7(c). With the rise in temperature from −50 °C to 50 °C, the retention rate of the capacitance increased from 69.52% to 89.4%. Nevertheless, the capacitance retention decreased from 89.4% to 76.75% going from 50 °C to 100 °C. Such degraded retention rate capabilities might be due to the side effects of the porous SiC surface at high temperature. The specific capacitances were enhanced as the temperature increased. However, the electrical conductivity of porous SiC basically remained nearly unchanged with the change in temperature.56 Therefore, the enhancement of capacitance mainly depended primarily on the increase in the electrolyte conductivity at elevated temperatures. This corresponded to a decrease in the resistance of the high-frequency regions in Fig. 7(d). A greater ion-diffusion resistance was also confirmed by the reduced slope in the low-frequency region. As shown in Fig. 7(e), the cycling stability of the supercapacitors was further examined by performing 12000 cycles at 5 mA cm−2 in the range of −50 °C to 100 °C. During the increase in temperature from −20 °C to 50 °C, all the capacitance retentions could remain higher than 97% at all temperatures for 2000 cycles. Because the electrolyte had a higher resistance at low temperature, the capacitance retention rate was low at −50 °C. The adverse reaction of the porous SiC surface at high temperature was the main reason for the low cycle retention rate at 100 °C. During the whole test, the capacitance retention could remain higher than 88% at all temperatures for 2000 cycles, indicating that the porous WPCB-based supercapacitor had excellent temperature adaptability. The results showed that the porous WPCB-based supercapacitor had strong adaptability in an environment of continuous temperature changes.
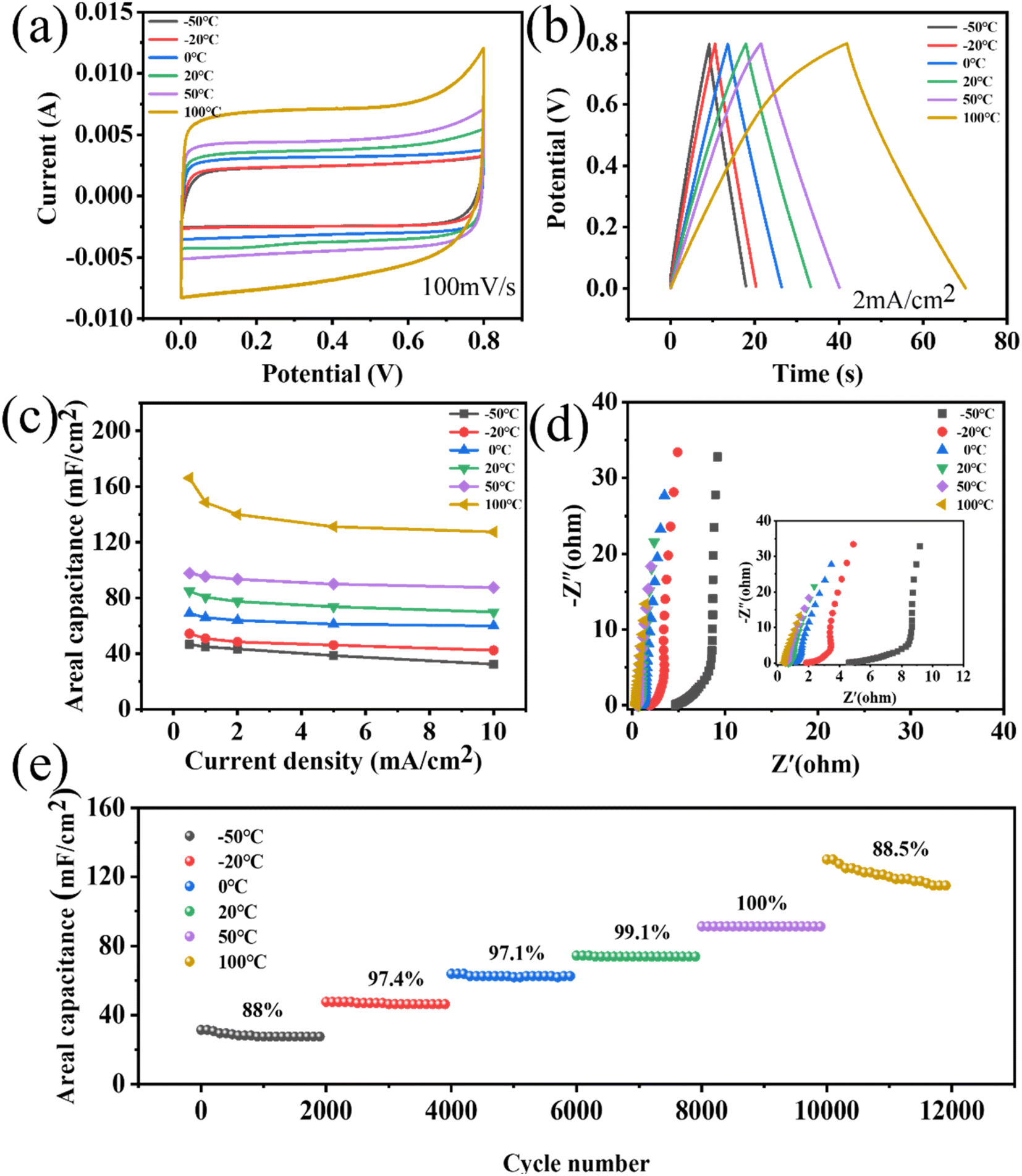 | ||
| Fig. 7 Electrochemical tests of the porous WPCB-based supercapacitor under different temperatures with two electrodes: (a) the CV curves of the porous WPCB-based supercapacitor at 100 mV s−1; (b) the GCD curves of the porous WPCB-based supercapacitor at 2 mA cm−2; (c) the capacitance of the porous WPCB-based supercapacitor at various current densities; (d) the Nyquist curves; (e) the cyclic stability varying with temperature in the range of −50 °C to 100 °C. | ||
4. Conclusion
In this study, we propose a high-value recycling strategy for the in situ transformation of NMFs into porous WPCB-based supercapacitor electrodes with excellent performance. The results indicated that the optimal parameters for activation were an activation temperature of 800 °C, mass ratio of KOH to pyrolytic residues of 2, and activation time of 1 h. The XRD, FT-IR, and Raman results showed that the electrode material was rich in SiC. The surface area of the electrode material prepared under optimal conditions was 589 m2 g−1 with hierarchical pore structures. The prepared supercapacitors exhibited a capacitance of 77.14 mF cm−2 (62.5 mF cm−2) at 0.5 mA cm−2 (100 mV s−1). In addition, at the operating temperatures of 20 °C, −50 °C, and 100 °C, the capacitance retentions of the supercapacitors for 10000 cycles were 100%, 89.36%, and 90%, respectively. Under the condition of continuous temperature change from −50 °C to 100 °C, the capacitance retention rate remained above 88% for each temperature range after 12000 cycles. The porous WPCB-based supercapacitor thus displayed good temperature resistance and temperature adaptability. Therefore, an environmentally friendly and high-value waste utilization strategy is proposed, which can provide a new way for the harmless and resource disposal of NMFs.
Author contributions
Zhen Xi: writing – original draft, conceptualization, methodology, investigation. Ruitong Gao: writing – reviewing and editing, conceptualization, methodology, funding acquisition. Zhaojun Chen: investigation supervision, resources. Hui Du: supervision, funding acquisition, resources. Zhenming Xu: visualization, supervision. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China [52172093, 21808115]; and Natural Science Foundation of Shandong Province [ZR2022QE042].References
- S. Shi, C. Nie, W. Zhou and X. Zhu, Process Saf. Environ., 2022, 162, 1025–1032 CrossRef CAS.
- X. Ji, M. Yang, A. Wan, S. Yu and Z. Yao, Int. J. Environ. Res. Public Health, 2022, 19(12), 7508 CrossRef CAS.
- C. Chaine, A. S. Hursthouse, B. McLean, I. McLellan, B. McMahon, J. McNulty, J. Miller and E. Viza, Int. J. Environ. Res. Public Health, 2022, 19(2), 766 CrossRef PubMed.
- H. Duan and X. Zhu, Energy Sources, Part A, 2022, 44, 1640–1659 CrossRef.
- H. Wang, S. Zhang, B. Li, D. A. Pan, Y. Wu and T. Zuo, Resour., Conserv. Recycl., 2017, 126, 209–218 CrossRef.
- P. Kiddee, R. Naidu and M. H. Wong, Waste Manage., 2013, 33, 1237–1250 CrossRef PubMed.
- P. Zhu, X. Liu, Y. Wang, C. Guan, Y. Yang, J. Zhu, X. Li, G. Qian and R. L. Frost, J. Environ. Chem. Eng., 2017, 5, 3439–3446 CrossRef CAS.
- J. Wu, J. Li and Z. Xu, J. Hazard. Mater., 2008, 159, 230–234 CrossRef CAS PubMed.
- E. Y. Yazici and H. Deveci, Hydrometallurgy, 2013, 139, 30–38 CrossRef CAS.
- Y. Zhou, W. Wu and K. Qiu, Waste Manage., 2011, 31, 2569–2576 CrossRef CAS.
- L. Flandinet, F. Tedjar, V. Ghetta and J. Fouletier, J. Hazard. Mater., 2012, 213–214, 485–490 CrossRef CAS.
- L. Chen, J. He, L. Zhu, Q. Yao, Y. Sun, C. Guo, H. Chen and B. Yang, Process Saf. Environ., 2023, 169, 869–878 CrossRef CAS.
- A. Priya and S. Hait, Process Saf. Environ., 2020, 143, 262–272 CrossRef CAS.
- I. Birloaga and F. Vegliò, J. Environ. Chem. Eng., 2016, 4, 20–29 CrossRef CAS.
- T. H. Bui, S. Jeon and Y. Lee, J. Environ. Chem. Eng., 2021, 9, 104661 CrossRef CAS.
- S. Mir and N. Dhawan, Resour., Conserv. Recycl., 2022, 178, 106027 CrossRef CAS.
- A. Andooz, M. Eqbalpour, E. Kowsari, S. Ramakrishna and Z. A. Cheshmeh, J. Cleaner Prod., 2022, 333, 130191 CrossRef CAS.
- J. M. L. A. Bocio, J. L. Domingo, J. Corbella, A. Teixidó and C. Casas, J. Agric. Food Chem., 2003, 51, 3191–3195 CrossRef PubMed.
- J. Guo, Q. Rao and Z. Xu, J. Hazard. Mater., 2008, 153, 728–734 CrossRef CAS PubMed.
- J. Guo, B. Cao, J. Guo and Z. Xu, Environ. Sci. Technol., 2008, 42, 5267–5271 CrossRef CAS PubMed.
- Y. Zheng, Z. Shen, C. Cai, S. Ma and Y. Xing, J. Hazard. Mater., 2009, 163, 600–606 CrossRef CAS PubMed.
- X. Niu and Y. Li, J. Hazard. Mater., 2007, 145, 410–416 CrossRef CAS.
- R. Gao, B. Liu and Z. Xu, J. Cleaner Prod., 2019, 231, 1149–1157 CrossRef CAS.
- R. Gao and Z. Xu, J. Hazard. Mater., 2019, 364, 1–10 CrossRef CAS.
- R. Gao, Y. Liu, B. Liu and Z. Xu, J. Cleaner Prod., 2019, 236, 117662 CrossRef CAS.
- R. Gao, L. Zhan, J. Guo and Z. Xu, J. Hazard. Mater., 2020, 383, 121234 CrossRef CAS.
- B. Liu, R. Gao and Z. Xu, J. Cleaner Prod., 2020, 244, 118727 CrossRef CAS.
- C. Ma, J. Yu, T. Chen, Q. Yan, Z. Song, B. Wang and L. Sun, Fuel, 2018, 230, 390–396 CrossRef CAS.
- Y. Zhu, B. Li, Y. Wei, S. Zhou and H. Wang, Process Saf. Environ., 2023, 178, 1083–1093 CrossRef CAS.
- J. Yang, J. Feng, W. Li, X. Chen, X. Liu, J. Ruan, R. Qiu, Y. Xiong and S. Tian, J. Hazard. Mater., 2019, 373, 640–648 CrossRef CAS PubMed.
- S. Chen, T. Wang, L. Ma, B. Zhou, J. Wu, D. Zhu, Y. Y. Li, J. Fan and C. Zhi, Chem, 2023, 9, 497–510 CAS.
- X. Yang, L. Sun, J. Xiang, S. Hu and S. Su, Waste Manage., 2013, 33, 462–473 CrossRef CAS PubMed.
- P. R. Choi, E. Lee, S. H. Kwon, J. C. Jung and M.-S. Kim, J. Phys. Chem. Solids, 2015, 87, 72–79 CrossRef CAS.
- Y. Kan, Q. Yue, B. Gao and Q. Li, J. Taiwan Inst. Chem. Eng., 2016, 68, 440–445 CrossRef CAS.
- O. Haibo, L. Hejun, Q. Lehua, L. Zhengjia, W. Jian and W. Jianfeng, Carbon, 2008, 46, 1339–1344 CrossRef.
- X. Li, W. Li, Q. Liu, S. Chen, L. Wang, F. Gao, G. Shao, Y. Tian, Z. Lin and W. Yang, Adv. Funct. Mater., 2020, 31, 2008901 CrossRef.
- M. R. S. Alexander, R. D., F. R. Jones, M. Stollenwerk, J. Zabold and W. Michaeli, J. Mater. Sci., 1996, 31, 1879–1885 CrossRef CAS.
- T. M. Parrill and Y. W. Chung, Surf. Sci., 1991, 243, 96–112 CrossRef CAS.
- L. Muehlhoff, M. J. Bozack, W. J. Choyke and J. T. Yates, J. Appl. Phys., 1986, 60, 2558–2563 CrossRef CAS.
- J. Bedia, C. Belver, S. Ponce, J. Rodriguez and J. J. Rodriguez, Chem. Eng. J., 2018, 333, 58–65 CrossRef CAS.
- Y. Qin, Z. Song, L. Miao, C. Hu, Y. Chen, P. Liu, Y. Lv, L. Gan and M. Liu, Chem. Eng. J., 2023, 470, 144256 CrossRef CAS.
- Y. Zhang, Z. Song, L. Miao, Y. Lv, L. Gan and M. Liu, ACS Appl. Mater. Interfaces, 2023, 15, 35380–35390 CrossRef CAS PubMed.
- L. Miao, H. Duan, D. Zhu, Y. Lv, L. Gan, L. Li and M. Liu, J. Mater. Chem. A, 2021, 9, 2714–2724 RSC.
- Z. Song, L. Miao, Y. Lv, L. Gan and M. Liu, Angew. Chem., Int. Ed., 2023, 62, e202309446 CrossRef CAS.
- J. P. Alper, M. S. Kim, M. Vincent, B. Hsia, V. Radmilovic, C. Carraro and R. Maboudian, J. Power Sources, 2013, 230, 298–302 CrossRef CAS.
- W. Li, Q. Liu, S. Chen, Z. Fang, X. Liang, G. Wei, L. Wang, W. Yang, Y. Ji and L. Mai, Mater. Horiz., 2018, 5, 883–889 RSC.
- L. Gu, Y. Wang, Y. Fang, R. Lu and J. Sha, J. Power Sources, 2013, 243, 648–653 CrossRef CAS.
- Y. Chen, X. Zhang and Z. Xie, ACS Nano, 2015, 9, 8054–8063 CrossRef CAS PubMed.
- J. P. Alper, A. Gutes, C. Carraro and R. Maboudian, Nanoscale, 2013, 5, 4114–4118 RSC.
- C. H. Chang, B. Hsia, J. P. Alper, S. Wang, L. E. Luna, C. Carraro, S. Y. Lu and R. Maboudian, ACS Appl. Mater. Interfaces, 2015, 7, 26658–26665 CrossRef CAS PubMed.
- C. Fan, Y. Tian, S. Bai, C. Zhang and X. Wu, J. Energy Storage, 2021, 44, 103492 CrossRef.
- G. Zu, J. Shen, L. Zou, F. Wang, X. Wang, Y. Zhang and X. Yao, Carbon, 2016, 99, 203–211 CrossRef CAS.
- Z. Chang, B. Yu and C. Wang, J. Solid State Electrochem., 2016, 20, 1405–1412 CrossRef CAS.
- M. Zhou, S. Yan, Q. Wang, M. Tan, D. Wang, Z. Yu, S. Luo, Y. Zhang and X. Liu, Rare Met., 2022, 41, 2280–2291 CrossRef CAS.
- M. Haque, Q. Li, A. D. Smith, V. Kuzmenko, E. Köhler, P. Lundgren and P. Enoksson, Electrochim. Acta, 2018, 263, 249–260 CrossRef CAS.
- T. Yang, S. Chen, X. Li, X. Xu, F. Gao, L. Wang, J. Chen, W. Yang, X. Hou and X. Fang, Adv. Funct. Mater., 2019, 29(11), 1806250 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra08125g |
| This journal is © The Royal Society of Chemistry 2024 |