
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSustainable production of catechol derivatives from waste tung nutshell C/G-type lignin via heterogeneous Cu–NC catalytic oxidation†
Guozhi Zhua,
Hongmei Xiebc,
Dawei Yea,
Junjie Zhang a,
Kangping Huanga,
Bing Liaod and
Jiazhi Chen*a
a,
Kangping Huanga,
Bing Liaod and
Jiazhi Chen*a
aInstitute of Chemical Engineering, Guangdong Academy of Sciences, Guangzhou 510665, China. E-mail: chenjiazhi@gdcri.com
bDepartment of Chemical Engineering, Maoming Vocational and Technical College, Maoming, 525027, China
cSchool of Chemistry and Chemical Engineering, Guangzhou University, Guangzhou 510006, China
dGuangdong Academy of Sciences, Guangzhou 510070, China
First published on 8th February 2024
Abstract
The sustainable production of catechol derivatives is a challenging task. Catechyl (C) and guaiacyl (G) lignins coexisting in waste tung nutshells are promising feedstocks to form valuable catechol derivatives, but the depolymerization of C/G lignin typically involves a catalytic reductive process that cannot produce these oxidized aromatic chemicals. Herein, we demonstrated that the sustainable production of catechol derivative aldehydes and acids from C/G lignin could be achieved through a heterogeneous copper-catalyzed oxidative process. Under optimized conditions, the Cu–NC-800 catalyst affords a 43.5 mg g−1 yield (8.9 wt%, based on Klason lignin) of aromatic aldehydes (protocatechuic aldehyde, vanillin) and acids (protocatechuic acid, vanillic acid). XRD and XPS analyses showed that CuO and Cu2O may be the active species during the heterogeneous oxidation of the Cu–NC-800 catalyst. This study opens new opportunities for the sustainable production of catechol derivatives from C/G-type lignin.
Introduction
Catechol derivatives are important functional molecules that have wide applications in food, medicine, and polyester industries (Scheme 1A).1–3 For instance, protocatechuic aldehyde and acid have notable biological activity such as antioxidant, antibacterial, and anti-aging properties.4,5 The global market demand for protocatechuic acid was more than 250 tons in 2015 and is expected to grow by 10% annually. Vanillin is used in food, cosmetic, and bio-based polymer industries. Around 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons of vanillin is produced per year, 85% of which comes from petro-based resources.1,6 Currently, these catechol derivatives are mostly produced from the oxidation of aromatic hydrocarbons or phenols derived from nonrenewable fossil resources. In recent years, the coordinated development of the economy, energy, and environment has driven research efforts toward the utilization of renewable carbon resources.7 In this regard, the sustainable production of catechol derivatives from carbon-neutral renewable resources, such as lignocellulosic biomass, has attracted increasing interest from both industrial and academic researchers.
000 tons of vanillin is produced per year, 85% of which comes from petro-based resources.1,6 Currently, these catechol derivatives are mostly produced from the oxidation of aromatic hydrocarbons or phenols derived from nonrenewable fossil resources. In recent years, the coordinated development of the economy, energy, and environment has driven research efforts toward the utilization of renewable carbon resources.7 In this regard, the sustainable production of catechol derivatives from carbon-neutral renewable resources, such as lignocellulosic biomass, has attracted increasing interest from both industrial and academic researchers.
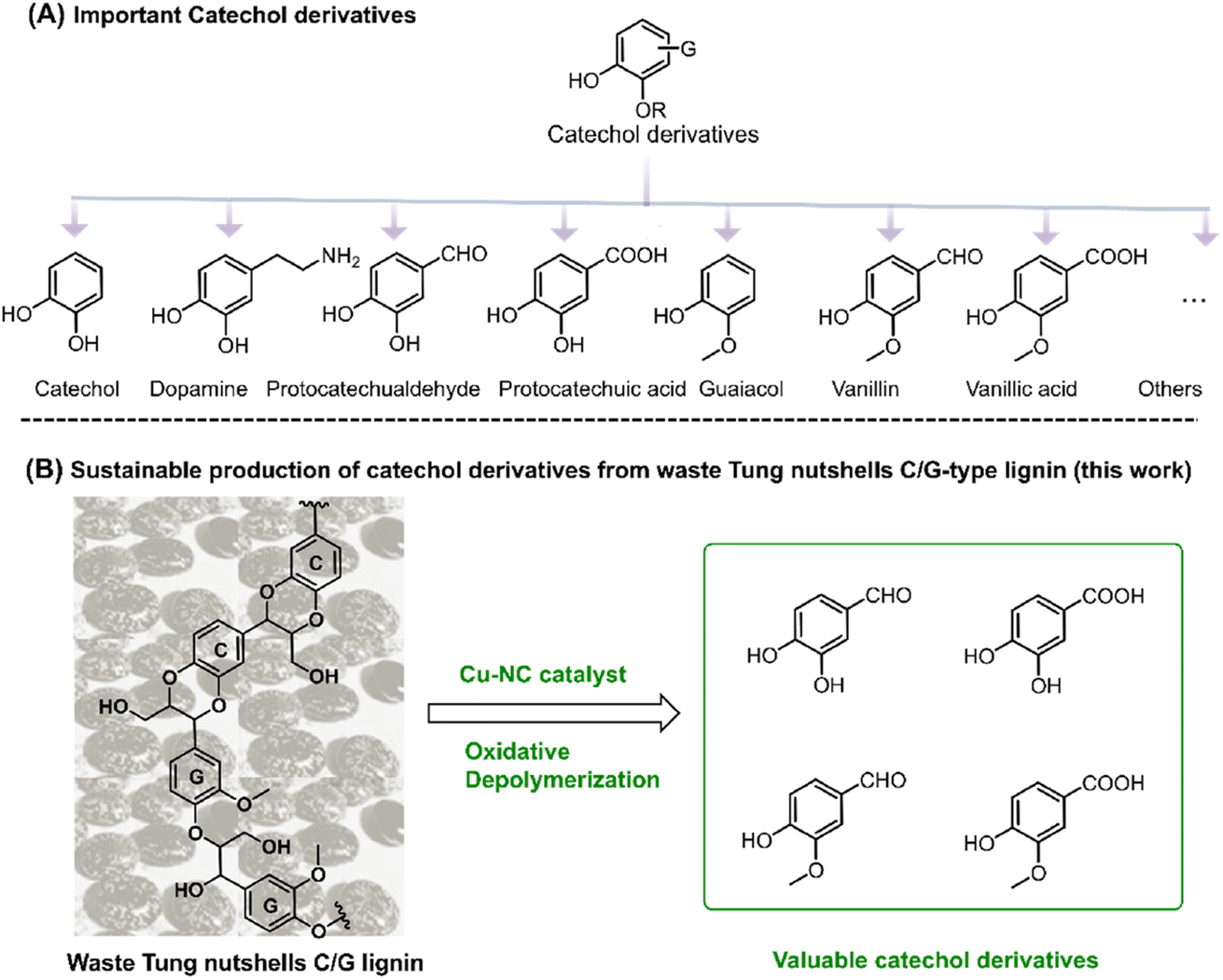 | ||
| Scheme 1 (A) Important catechol derivatives. (B) Sustainable production of catechol derivatives from tung nutshell C/G-type lignin (this work). | ||
As an important component of lignocellulose, lignin is a kind of promising feedstock to produce catechol derivatives owing to its natural aromatic skeleton.8,9 In modern industry, lignin is mainly generated as the by-product of the cellulosic bioethanol process as well as the pulp and paper industry, annually contributing approximately 60 million and 50 million tons of lignin globally, respectively.10 However, most of this lignin cannot be effectively exploited because of its structural complexity. Lignin is generally composed of three basic units, namely, guaiacyl (G), syringyl (S), and p-hydroxyphenyl (H). In 2012, catechyl (C) units were discovered as a unique structure in some plant seed coats.11,12 Representative C lignin can be found in vanilla, castor, tung, candlenut, jatropha, Chinese tallow, and other natural products.13,14 Among these, tung nutshells contain both C lignin and G lignin. Unlike the common H/G/S lignin, C lignin is formed via the polymerization of a caffeyl alcohol monomer with a linear benzodioxane structure and has gradually attracted interest in the preparation of materials and chemicals.15–17 In the last several years, Román-Leshkov, Ralph, Song, et al. have reported that catalytic reduction is an effective strategy for the depolymerization of C lignin. Ni/C, Pd/C, Ru/ZnO/C and other catalysts afforded around 71–81 mol% of propylcatechol, propanolcatechol, or propenylcatechol products in CH3OH or DES solvent at 200–250 °C, which are the typical cleavage products of C–O bonds.14,15,18–24 Although catalytic reduction enables the depolymerization of lignin to mono-phenolic compounds, it is hard to obtain the valuable catechol derivative aldehydes or acids under reduction25,26 because the formation of these aromatics corresponds to the oxidation of the Cα position.27,28 More importantly, catalytic oxidation enables the formation of highly functional aromatic compounds.29 With careful examination of the tung nutshells' lignin structure, both the catechyl (C) and guaiacyl (G) units could act as the basic structure of catechol moieties.30,31 Inspired by the lignin-to-vanillin process in industry,1,32 it is promising to employ a catalytic oxidation strategy to convert C/G lignin feedstocks to valuable catechol derivative aldehydes and acids (Scheme 1B).
Cu catalysts have demonstrated excellent performance in the catalytic aerobic oxidation of biomass conversion.33,34 Pioneer works by Wang,35 Zhang,36 Riisager,37 Westwood,38 and Tom Baker39,40 reported that Cu salt catalysts could catalyze oxidative cleavage of lignin C–C bonds to produce highly functional aromatic compounds. Very recently, we found that commercially available CuCl2 catalyst enabled the oxidative depolymerization of C/G-lignin to catechol derivative aldehydes and acids.41 However, these results are homogeneous catalytic processes, which makes it hard to effectively separate the catalyst from the aromatic products in a simple way. Metal-containing nitrogen-doped carbon (M–NC), a common catalyst, has been widely used for the aerobic oxidation of organic molecules as well as lignin.42–45 Herein, in continuation of our previous work,41 copper-containing nitrogen-doped carbon (Cu–NC) is reported in the catalytic oxidation conversion of C/G-lignin (Scheme 1B). To the best of our knowledge, this is the first report of a heterogeneous catalytic process that realizes oxidation conversion of C/G-lignin, which may provide new guidance for sustainable production of catechol derivatives from C/G lignin by an oxidation approach.
Results and discussion
Oxidation–depolymerization of tung nutshell lignin
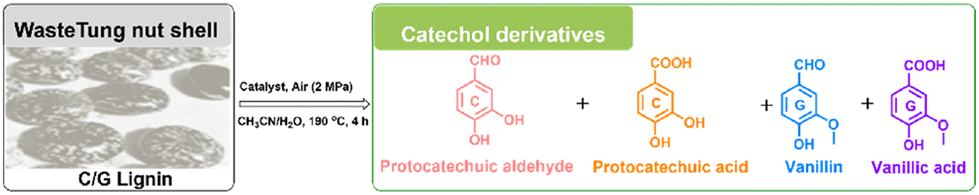
Heterogeneous catalytic oxidative depolymerization of tung nutshell lignin was conducted according to the reported CuCl2 homogeneous reaction conditions.41 These supported catalysts were used at 10 wt%, corresponding to metal loadings of 0.2–0.5 wt% based on the mass of the tung nutshell. To examine the heterogeneous catalytic oxidative performance, a variety of non-noble metal catalysts were screened. After the reaction, the mixture was filtrated to separate the solid (including the pulp and catalyst) and the liquid, where the four different catechol derivative aldehydes and acids (vanillin, vanillic acid, protocatechuic aldehyde, and protocatechuic acid) from the C/G lignin could be obtained with the furfural byproduct from the hemicellulose hydrolysis (Fig. S1†). Euphorbiaceae plant seed coats are a special lignin feedstock. The content of lignin is around 45–70 wt% based on the traditional Klason method. Notably, the lignin content is usually overestimated because many fatty acids and waxes present in seeds with high C lignin content are acid-insoluble and are always regarded as Klason lignin. Therefore, the mass ratio of monomers to tung nutshell feedstock (mg g−1) was calculated as the quantitative yield of products in this manuscript (eqn (1)). A similar expression can be found in recent literature.15,20,21 As a comparison or supplement, the yield based on the Klason lignin content was also calculated and presented in brackets (eqn (2)), and the Klason lignin content is 49 wt% in this study.
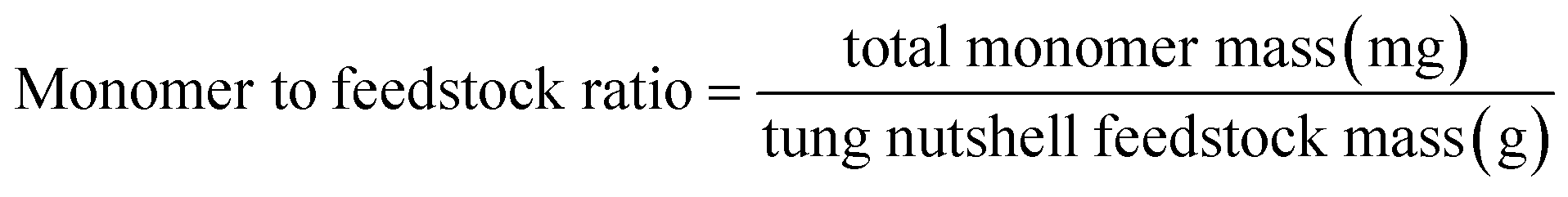 | (1) |
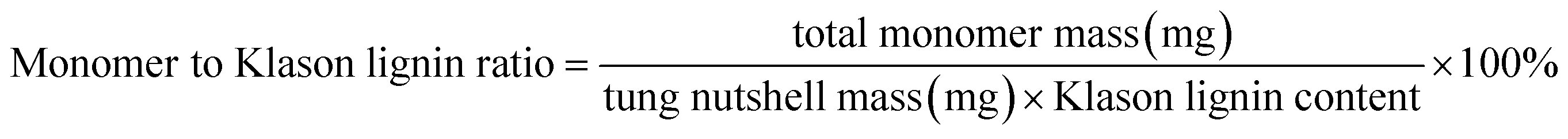 | (2) |
Around 30.0 mg g−1 (6.1 wt% based on Klason lignin) of C/G aromatic monomer was formed when using the Fe, Co, Mo, and Mn-based catalysts (Table 1, entries 3–6), which was almost no difference from a blank experiment (entry 1), implying poor catalytic activity. It should be noted that the blank yield might be related to the special solvent system and reaction atmosphere, as discussed in the ESI (Table S1).† Inspired by the homogeneous CuCl2 catalytic system, a supported Cu-based catalyst was tested (entry 7). The Cu–NC-800 catalyst showed a 43.5 mg g−1 yield (8.9 wt%) of the total C/G aromatic monomer under identical conditions. The yield was 45% higher than that of the blank experiment, indicative of good catalytic oxidative performance for the Cu-based catalyst. Compared with the previous homogeneous CuCl2 catalytic process, the Cu–NC-800 catalyst exhibited a higher molar specific activity. To exclude the effect of the carbon-based support, the NC-800 catalyst was prepared without Cu loadings. There was no obvious improvement in activity (entry 2), implying that nitrogen-doped carbon could not be the main active species. Further, commercially available noble metal Pd/C and Ru/C catalysts were also studied (entries 8 and 9). It is known that Pd/C and Ru/C are common catalysts for the reductive catalytic fractionation (RCF) of lignocellulose. In addition to RCF, supported platinum-group metals (PGMs), including Pd and Ru-based catalysts, have been also used as catalysts for aerobic oxidation.46–50 Although the Pd/C catalyst could generate more protocatechuic acid and vanillic acid compared with the blank condition, considering the economic feasibility, the Cu–NC-800 non-noble metal catalyst was selected for the current process.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Aromatic monomer yield (mg g−1) | |||||
| Vanillic acid | Vanillin | Protocatechuic acid | Protocatechuic aldehyde | Total | Furfural | ||
| a Conditions: tung nutshell (0.2 g), catalyst (20 mg), CH3CN/H2O (8/2, v/v, 20 mL), air (2 MPa), 190 °C, 4 h. | |||||||
| 1 | None | 14.6 | 14.8 | — | 0.7 | 30.1 | 3.3 |
| 2 | NC-800 | 18.8 | 15.3 | — | — | 34.1 | 1.8 |
| 3 | Fe–NC-800 | 14.0 | 13.5 | 0.9 | 0.4 | 28.8 | 3.4 |
| 4 | Co–NC-800 | 15.6 | 12.0 | — | 0.3 | 27.9 | 2.0 |
| 5 | Mo–NC-800 | 13.6 | 12.0 | 1.3 | 0.6 | 27.5 | 2.8 |
| 6 | Mn–NC-800 | 16.1 | 13.8 | — | 1.1 | 31.0 | 4.1 |
| 7 | Cu–NC-800 | 22.2 | 19.2 | 1.9 | 0.2 | 43.5 | 2.0 |
| 8 | Ru/C | 18.3 | 13.8 | 2.1 | 1.2 | 35.4 | 3.0 |
| 9 | Pd/C | 21.6 | 16.5 | 3.2 | 1.5 | 42.8 | 3.3 |
Further, to test the effects of Cu precursor, calcination temperature, and atmosphere, the Cu-based catalyst was investigated by adjusting these preparation conditions. Three common Cu(II) salt precursors (CuCl2, CuSO4, Cu(OAc)2) were selected to get Cu–NC-800-Cl, Cu–NC-800-S and Cu–NC-800, respectively. In comparison with the carbon-based support (NC-800), both the Cu–NC-800-Cl and Cu–NC-800-S catalysts showed around 38.0 mg g−1 (7.8 wt%) of aromatic monomer, with small amounts of protocatechuic aldehyde and protocatechuic acid produced (Fig. 1A, entries 2 and 3). When examining the catalytic performance of Cu–NC-800, the total yield of C/G aromatic monomer further increased to 43.5 mg g−1 (Fig. 1A, entry 4). The increasing tendency indicated that Cu(OAc)2 precursor might be more suitable for the preparation of Cu based catalyst than CuCl2 or CuSO4. Next, fixing Cu(OAc)2 as the precursor, the catalysts were prepared in a N2 inert atmosphere at 400 °C, 600 °C, and 800 °C to explore the effect of calcination temperature. When the calcination temperature is 400–800 °C, the total yield increases from 31.9 mg g−1 to 43.5 mg g−1 (Fig. 1B, entries 1–3). The Cu–NC-400N and Cu–NC-600N exhibited lower yields under identical conditions, which might be because they could not proceed effectively for the precursors' thermal decomposition to form CuO or Cu2O species at relatively low temperatures. In addition, the catalyst (Cu–NC-400H) calcination in H2 reducing atmosphere was carried out to generate the Cu0 species by the redox interaction. The Cu–NC-400H catalyst showed moderate activity, with a yield of 38.9 mg g−1 (7.9 wt%), which is higher than that of the Cu–NC-400N or Cu–NC-600N catalyst (Fig. 1B, entry 4). Based on the above control experiments, the Cu–NC-800 catalyst exhibited optimal catalytic performance. Both CuO and Cu2O might be the active species of Cu–NC-800 catalyst during the oxidative depolymerization of C/G lignin, which is demonstrated in a later part.
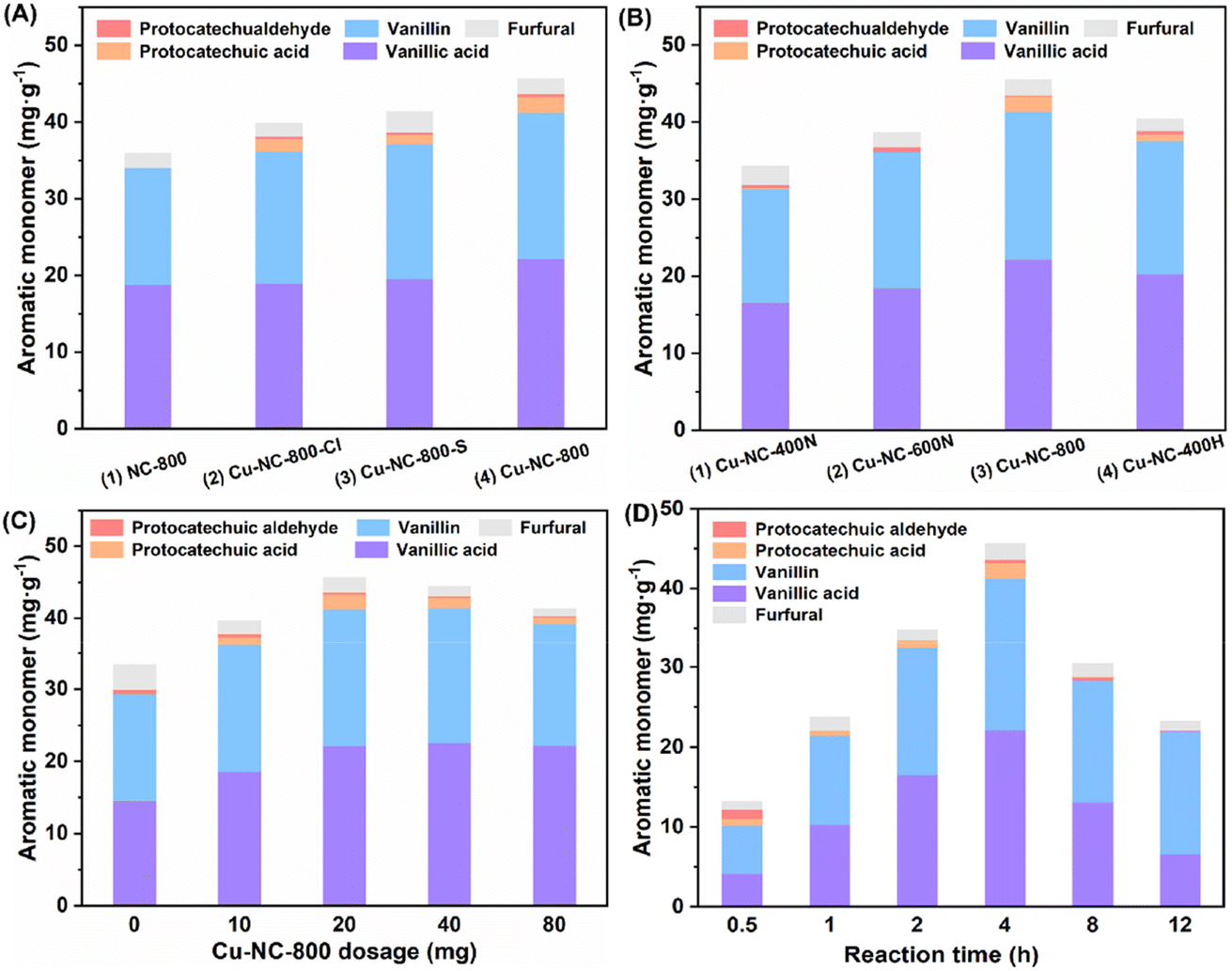 | ||
| Fig. 1 Yields of lignin-derived monomers obtained from tung nutshell oxidation–depolymerization to investigate the effects of (A) the Cu(II) salt precursor, (B) the calcination temperature and atmosphere, (C) the catalyst dosage, and (D) the reaction time. Reaction conditions unless otherwise stated: tung nutshell (0.2 g), catalyst (20 mg), CH3CN/H2O (8/2, v/v, 20 mL), air (2 MPa), 190 °C, 4 h. | ||
With the optimal Cu–NC-800 catalyst in hand, the reaction was performed using various dosages of catalyst as well as different reaction times. The effect of catalyst dosage on the activity of lignin oxidative depolymerization is shown in Fig. 1C. When the amount of catalyst was increased from 5 wt% to 40 wt%, the yield increased at first and then descended slightly. This unfavorable phenomenon might be due to the side reactions of the phenolic monomers. Meanwhile, due to the heterogeneity of the native biomass, poor material transport and interactions with the substrate cannot be excluded. Similarly, the yield of aromatic monomer first increased as the time increased from 0.5 to 4 h and then significantly decreased at a longer reaction time (Fig. 1D). The yield and reaction time had a volcano curve relationship, in which reaction at 4 h was the best, with the highest yield of 43.5 mg g−1 (8.9 wt% based on Klason lignin). It is believed that a longer reaction time would generate other undesired products, probably from repolymerization or condensation under oxidative conditions.51
2D NMR analysis
To obtain further insights into the C/G lignin structure evolution during the oxidative depolymerization reaction, 2D HSQC NMR spectra were performed. To better evaluate the catalytic oxidative depolymerization performance, a blank experiment for the HSQC analysis was performed without a catalyst under N2 atmosphere, which could approximately serve as a control experiment due to the negligible total yield of 6.0 mg g−1 (1.2 wt%) of the catechol derivatives in such conditions (Fig. S2†). The signal is mainly divided into two main regions: alkyl linkage structure (δC/δH = 50–100/2.5–6.0 ppm) and aromatic structure (δC/δH = 100–135/6.0–8.0 ppm). For the alkyl linkage structure, the signal at 56.37/3.74 ppm showed the existence of the aromatic methoxy (Ar–OCH3) group, which was from the guaiacyl (G) unit. In addition to the common β-O-4 (A), β-5 (B), and β–β (E) linkages of the G unit, there existed the cis- and trans-benzodioxane (Dc and Dt) linkages of the C unit. In the aromatic part, the signals of 110.99/6.92 ppm, 115.21/6.76 ppm, and 119.48/6.59 ppm could be assigned to the G unit. The special signals of 117.33/6.97 ppm, 116.02/6.77 ppm, and 120.79/6.95 ppm were detected and might be attributed to the C unit. The two sets of signals partially overlapped at 115–120/6.8–7.0 ppm, without the signals for the H or S unit in the aromatic regions. These results confirmed that the C/G-type lignin coexisted in tung nutshells, as supported by previous reports.41The organosolv lignin (OL) was also investigated (Fig. S3†). Based on the semi-quantitation method, the OL of tung nutshells possessed abundant C (57%) and G (42%) units. The signals are summarized in Table S2.†11,13,14,20 Next, the 2D HSQC NMR spectra of lignin oil after oxidative depolymerization reaction were studied (Fig. 2). Obviously, the signal intensities of the β-O-4 (A), β-5 (B), β–β (E), and benzodioxane (Dc and Dt) linkages became much weakened, suggesting the transformation of the alkyl linkage in C/G units. On the other hand, the signal of C/G aromatic units reduced notably, possibly due to the degradation or transformation of the catechol-type aromatics (Scheme S2†).52–54 It should be noted that many new signals at δC/δH = 110–125/7.2–7.5 ppm (blue color) could be detected. These signals were assigned to the C/G aromatic aldehydes and acids formed under such oxidative conditions and were consistent with the spectra of the four different standard compounds vanillin, vanillic acid, protocatechuic aldehyde, and protocatechuic acid in the aromatic regions (Fig. S4†). Therefore, it is believed that Cu–NC-800 catalyst exhibited aerobic oxidative activity in the depolymerization of tung nutshell C/G lignin by the cleavage of the β-O-4, β-5, β–β and benzodioxane linkages, and thus formed the oxidized catechol derivative aldehydes and acids from the C/G units.
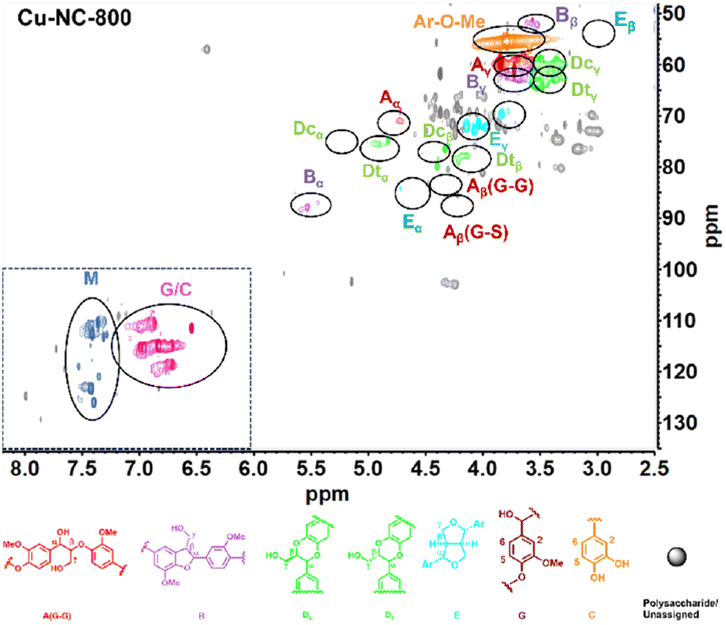 | ||
| Fig. 2 The 2D HSQC NMR spectra of tung nutshell lignin oils catalyzed by Cu–NC-800 in air; reaction conditions: tung nutshell (0.2 g), Cu–NC-800 (20 mg), CH3CN/H2O (8/2, v/v, 20 mL), air (2 MPa), 190 °C, 4 h. | ||
Structural properties of the catalysts
To understand the relationship between the catalyst properties and their oxidative depolymerization performance, various characterizations were performed for the supported Cu-based catalysts. X-ray powder diffraction (XRD) patterns of sample calcinations at different temperatures and atmospheres are depicted in Fig. 3 and S5.† The characterizations of uncalcined catalyst precursors were also provided to have a better understanding of the structural evolution. Two sets of intense characteristic diffraction peaks could be easily recognized. The 2θ of 36.4°, 42.3° and 61.3° (marked with triangles) were assigned to the (111), (200), and (220) lattice planes of Cu2O, while 35.5° and 38.7° (marked with squares) were ascribed to the (002) and (111) lattice planes of CuO, indicating that Cu(II) and Cu(I) coexisted in the Cu–NC-800N catalyst.55,56 In contrast, only metallic Cu was detected for Cu–NC-400H, proving that the complete reduction of Cu(OAc)2 precursor occurred to form the Cu0 species when calcined in H2 conditions.57,58 These results revealed that quite different Cu species would be generated for the preparation of Cu-based catalysts under N2 or H2 conditions. In addition, no obvious diffraction peaks appeared for the Cu(Phen)x(OAc)2 sample, possibly indicative of an amorphous structure. For the Cu–NC-400N and Cu–NC-600N samples, no obvious signals or only a set of weak signals of Cu2O appeared, indicating that precursor calcination at such low temperatures (400–600 °C) did not decompose or only a small amount of decomposition occurred.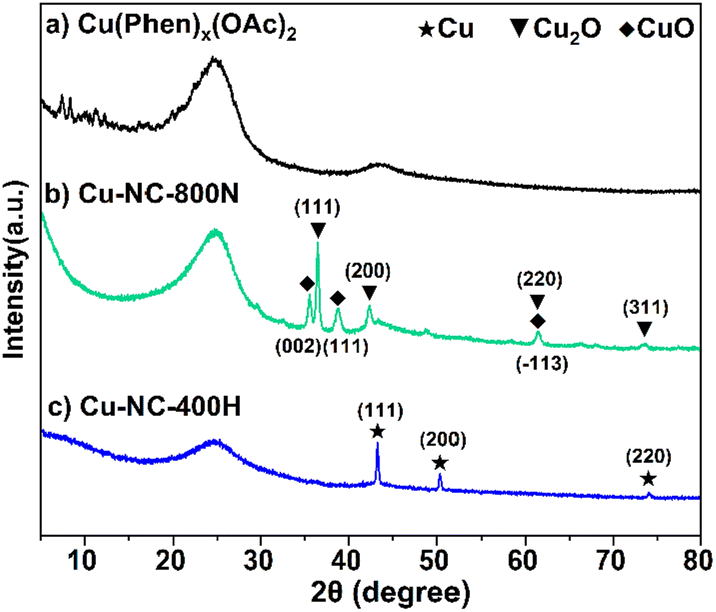 | ||
| Fig. 3 The XRD patterns of (a) Cu(Phen)x(OAc)2, (b) Cu–NC-800N, and (c) Cu–NC-400H. | ||
The TEM images are exhibited in Fig. 4 and S6.† There is no obvious difference in morphology for the above-mentioned samples, with a two-dimensional stacked layered structure (Fig. 4A–C), possibly due to the small amount of metal loading. EDX mapping of Cu–NC-800N showed that the Cu and O species are dispersed uniformly on the N-doped carbon-based material (Fig. 4D–I), without obvious agglomerates observed.
 | ||
| Fig. 4 The TEM images of (A) Cu–NC-800N, (B) Cu–NC-400H, and (C) Cu(Phen)x(OAc)2. (D) STEM-HAADF image of Cu–NC-800N, and (E–I) corresponding HRTEM-EDX mapping images of Cu, O, N, and C of Cu–NC-800N sample. | ||
To obtain further insights into the surface properties, the electronic states of Cu and N were studied by XPS measurements (Fig. 5). The full spectrum depicted in Fig. S7† further confirmed the existence of C, N, O, and Cu. It was mainly composed of C element, that is, the main structure of the catalyst is carbon, only containing a small amount of Cu and N. For the Cu 2p3/2 of the Cu(OAc)2 precursor (Fig. 5A), the two intense peaks centered at 934.4 eV and 932.2 eV could be assigned to the Cu(II) of the Cu(OAc)2 and the Cu(Phen)x(OAc)2 coordination species, respectively.59,60 The latter is possibly related to the strong electron transfer from N to Cu for the coordination of Cu2+ ions, and the trend is supported by the binding energy shift of N 1s. After calcination in N2 at 400–600 °C (Fig. S8†), both the Cu–NC-400N and Cu–NC-600N samples show similar signals as the precursor, only a weaker signal at 932.2 eV. The signals at 934.1–934.3 eV and 932.1–932.2 eV should belong to Cu(OAc)2 and Cu(Phen)x(OAc)2, respectively. In addition, these two signals may also be from the Cu2+ of CuO and Cu1+/0 of Cu2O or Cu0, respectively, because the values of these binding energies are consistent with those of CuO (934.1–934.6 eV) and Cu2O/Cu0 (932.1–932.9 eV).33,55,61,62 Therefore, partial decomposition occurred for the Cu–NC-400N and Cu–NC-600N samples. As for the Cu–NC-800N sample, the peaks at 934.4 eV and 932.9 eV are more likely to be the Cu2+ of CuO and Cu1+/0 of Cu2O/Cu0. Since the binding energy values of the latter are similar, the distinction between Cu2O/Cu0 was not achievable even by the Cu LMM Auger signal. However, according to the XRD of the Cu–NC-800N sample, CuO and Cu2O coexisted without Cu0. Therefore, combining the XRD and XPS analyses, it is probable that no decomposition or partial decomposition occurred when the precursor was calcinated in N2 at 400–600 °C. With the calcination temperature increasing to 800 °C, the precursor could effectively decompose to generate both the CuO and Cu2O species, which might be the active species for oxidative conversion. When calcinated in a H2 atmosphere, combined with the XRD and XPS data of Cu–NC-400H, the surface Cu0 species exists in the form of Cu0 and CuOx, probably due to partial surface oxidation when exposed to air.55
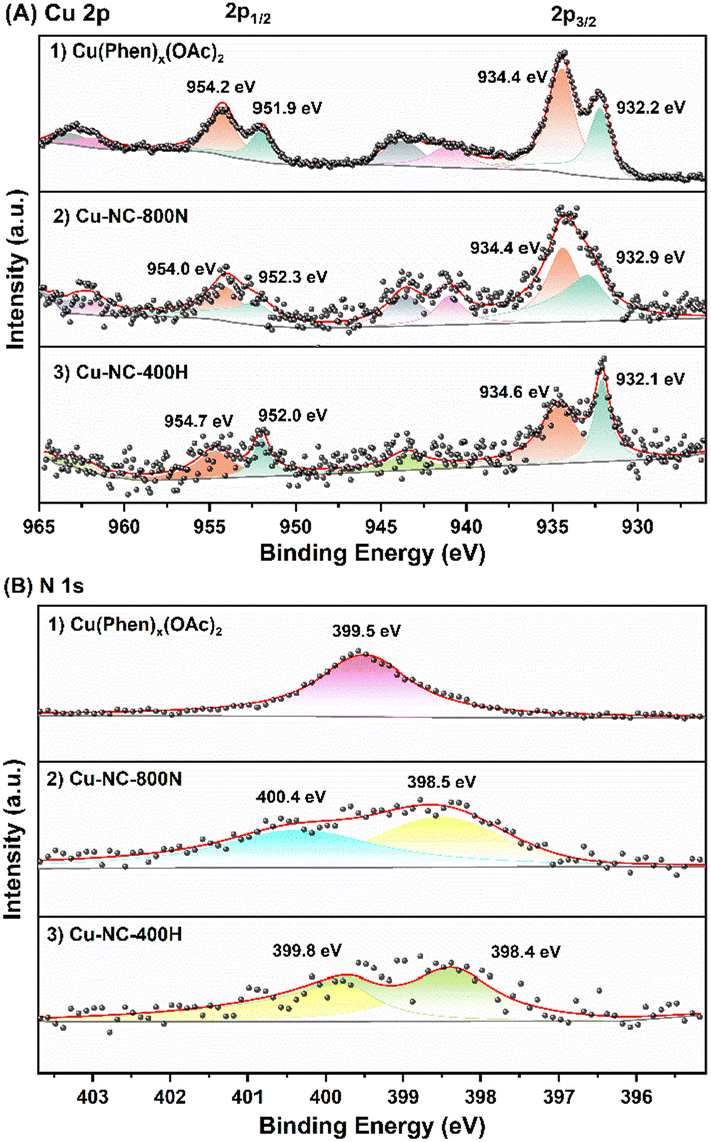 | ||
| Fig. 5 The XPS of (A) Cu 2p signals: (1) Cu(Phen)x(OAc)2, (2) Cu–NC-800N, and (3) Cu–NC-400H samples and (B) N 1s signals: (1) Cu(Phen)x(OAc)2, (2) Cu–NC-800N, and (3) Cu–NC-400H samples. | ||
The N 1s signals are shown in Fig. 5B. The peak at 399.5 eV was assigned to the Cu(Phen)x(OAc)2 coordination, supported by the 1,10-phenanthroline and metal chelate results in previous literature.63 Compared with the free 1,10-phenanthroline (binding energy of N 1s = 398.9 eV), a shift value of 0.6 eV resulted from the interaction between Cu and N.64 For Cu–NC-400N (Fig. S9†), pyridine nitrogen (N 1s = 398.3–398.5 eV) was formed,65,66 with the N of Cu(Phen)x(OAc)2 coordination (N 1s = 399.2 eV) still existing, also indicative of partial decomposition. With increasing calcination temperatures or in a H2 atmosphere, pyrrole nitrogen (N 1s = 399.8–400.6 eV) could be generated in case of the Cu–NC-600N, Cu–NC-800N and Cu–NC-400H samples. We attempted to distinguish the differences in N species and further reveal the role of N species in the aerobic oxidation reaction for different Cu–NC catalysts. However, the distinction of N species was not achievable in the current study.
Conclusions
In summary, catechol derivatives, including protocatechuic aldehyde, protocatechuic acid, vanillin, and vanillic acid, were efficiently produced from waste tung nutshells containing C/G-type lignin by heterogeneous Cu–NC catalysts. 2D HSQC NMR demonstrated that the β-O-4, β-5, β–β and benzodioxane linkages in the C/G units were almost cleaved during the catalytic aerobic oxidation. The XRD and XPS characterizations showed that CuO and Cu2O in the Cu–NC-800 catalyst might be the active species for oxidative conversion. This work introduced the heterogeneous catalytic oxidative production of catechol derivatives directly from C/G-type lignin and could inspire the oxidative valorization of C-type lignin.Experiment section
Catalyst synthesis
Pretreatment of XC-72R: 10 g of XC-72R and 350 mL of dilute nitric acid (10 wt%) were added into a 500 mL round-bottom flask. Under stirring, the mixture was refluxed at 95 °C for 4 hours to remove surface impurities. After cooling to room temperature, the mixture was vacuum filtered and washed to neutrality with distilled water. The solid was dried at 100 °C overnight and finally ground to powder (denoted as C–HNO3).Preparation of nitrogen-doped carbon-supported transition-metal catalysts: according to the method reported by Shannon S. Stahl,50 1 mmol of metal salt and 2 mmol of 1,10-phenanthroline (Phen) were dissolved in 25 mL of absolute alcohol, followed by adding 1.35 g of pretreated C–HNO3. The mixture was stirred for 24 h at room temperature, then dried at 80 °C overnight to get the precursor (denoted as M(Phen)x(OAc)2). Finally, the precursor was calcinated at 800 °C (ramp rate: 5 °C min−1) for 2 h under N2 flow to obtain the final catalyst (denoted as M–NC–800).
To screen the metal catalysts, cobalt acetate, copper acetate, iron acetate, manganese acetate and molybdenum acetylacetonate were selected, corresponding to the Co–NC-800, Cu–NC-800, Fe–NC-800, Mn–NC-800, and Mo–NC-800 catalysts, respectively (M = Co, Cu, Fe, Mn, or Mo). To investigate the anion effect of the metal salt, the copper acetate was replaced by copper chloride or copper sulfate under identical conditions, and the obtained catalyst was denoted as Cu–NC-800-Cl or Cu–NC-800-S. The calcination temperature of copper acetate was reduced to 600 °C or 400 °C (denoted as Cu–NC-600N or Cu–NC-400N) to test the temperature effect. Calcination at 400 °C with H2 flow was carried out to study the effect of redox atmosphere (denoted as Cu–NC-400H). For safety considerations, it was not calcinated at a higher temperature with H2.
Catalyst characterization
X-ray diffraction (XRD) was conducted on a PANalytical PW3040/60 powder diffractometer to test the phase analysis of the catalysts. Specific parameters: Cu Kα radiation (λ = 0.15418 nm), tube voltage of 40 kV, tube current of 40 mA, scanning speed of 5° min−1, scan range of 2θ = 5–80°. A Thermo Scientific K-Alpha spectroscope was employed to determine the X-ray photoelectron spectroscopy (XPS) signals, with a voltage of 12 kV and filament current of 6 mA. After the powder was pressed into a sheet, the sheet was attached to the sample tray. The sample was sent to the analysis chamber when the pressure in the sample chamber was less than 2.0 × 10−7 mbar. The signals were calibrated using the C 1s level (284.8 eV) as a standard reference of binding energies using Avantage software. The morphologic structure was obtained on a FEI Talos F200X transmission electron microscope with an accelerating voltage of 200 kV. The power sample was dispersed in ethanol and then dropped in an ultra-thin molybdenum net.Oxidative depolymerization of lignin
To produce tung nutshell powder feedstock (150 mesh) (Scheme S1†), the tung seeds were dried and peeled to obtain the bulk seed coats. After ultrasonic washing with EtOH/H2O, the dried seed coats were ground into powder and then sieved by the 150 mesh sieve. The heterogeneous catalytic oxidative reaction was performed in a 60 mL stainless autoclave. Typically, tung nutshell powder (200 mg, 150 mesh), Cu–NC-800 (20 mg) catalyst, and acetonitrile/water (16 mL/4 mL) co-solvent were added into the autoclave. The reactor was refilled with compressed air five times and then charged with air (2.0 MPa) after being sealed. With temperature programming, the reaction proceeded at 190 °C for 4 h under stirring. After the reaction was finished and cooled in an ice-water bath, 2,6-dimethoxyphenol (12 mg, internal standard) was directly added to the autoclave for the analysis. The qualitative and quantitative analysis procedures were based on those in our previous report.41 All the important data were carefully obtained based on repeated experiments.Author contributions
Guozhi Zhu: investigation, data curation, beneficial discussions, writing – review and editing; Hongmei Xie: investigation, data analysis, formal analysis; Dawei Ye: data analysis, methodology; Junjie Zhang: methodology; Kangping Huang: methodology, data curation; Bing Liao: methodology, resources; Jiazhi Chen: beneficial discussions, writing – review & editing, project administration, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the China Postdoctoral Science Foundation (Grant No. 2023M730737), the Natural Science Foundation of Guangdong Province of China (Grant No. 2022A1515012584), and the Guangdong Academy of Sciences (GDAS) Special Project of Science and Technology Development (Grant No. 2022GDASZH-2022010105).Notes and references
- M. Fache, B. Boutevin and S. Caillol, ACS Sustain. Chem. Eng., 2016, 4, 35–46 CrossRef CAS.
- M. Fache, E. Darroman, V. Besse, R. Auvergne, S. Caillol and B. Boutevina, Green Chem., 2014, 16, 1987–1998 RSC.
- X. Chen, J. Hou, Q. Gu, Q. Wang, J. Gao, J. Sun and Q. Fang, Polymer, 2020, 195, 122443 CrossRef CAS.
- A. L. Flourat, J. Combes, C. Bailly-Maitre-Grand, K. Magnien, A. Haudrechy, J. H. Renault and F. Allais, ChemSusChem, 2021, 14, 118–129 CrossRef CAS PubMed.
- R.-K. Zhang, Y.-S. Tan, Y.-Z. Cui, X. Xin, Z.-H. Liu, B.-Z. Li and Y.-J. Yuan, Green Chem., 2021, 23, 6515–6526 RSC.
- N. Khwanjaisakun, S. Amornraksa, L. Simasatitkul, P. Charoensuppanimit and S. Assabumrungrat, Bioresour. Technol., 2020, 299, 122559 CrossRef CAS.
- J. Yang, T. Gong, C. Li, H. Xu, S. Yu, J. Deng and Y. Fu, Carbon Neutrality, 2023, 2, 17 CrossRef.
- M. Y. Balakshin, E. A. Capanema, I. Sulaeva, P. Schlee, Z. Huang, M. Feng, M. Borghei, O. J. Rojas and T. Rosenau, ChemSusChem, 2021, 14, 1016–1036 CrossRef CAS.
- V. K. Garlapati, A. K. Chandel, S. P. J. Kumar, S. Sharma, S. Sevda, A. P. Ingle and D. Pant, Renew. Sustain. Energy Rev., 2020, 130, 109977 CrossRef CAS.
- Z. Chen and C. Wan, Renew. Sustain. Energy Rev., 2017, 73, 610–621 CrossRef CAS.
- F. Chen, Y. Tobimatsu, D. Havkin-Frenkel, R. A. Dixon and J. Ralph, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 1772–1777 CrossRef CAS.
- F. Chen, Y. Tobimatsu, L. Jackson, J. Nakashima, J. Ralph and R. A. Dixon, Plant J., 2013, 73, 201–211 CrossRef CAS.
- Y. Tobimatsu, F. Chen, J. Nakashima, L. L. Escamilla-Trevino, L. Jackson, R. A. Dixon and J. Ralph, Plant Cell, 2013, 25, 2587–2600 CrossRef CAS.
- S. Su, S. Wang and G. Song, Green Chem., 2021, 23, 7235–7242 RSC.
- S. Su, Q. Shen, S. Wang and G. Song, Int. J. Biol. Macromol., 2023, 239, 124256 CrossRef CAS PubMed.
- Q. Shen, S. Wang and G. Song, Composites, Part B, 2023, 264, 110917 CrossRef CAS.
- M. Nar, H. R. Rizvi, R. A. Dixon, F. Chen, A. Kovalcik and N. D'Souza, Carbon, 2016, 103, 372–383 CrossRef CAS.
- M. L. Stone, E. M. Anderson, K. M. Meek, M. Reed, R. Katahira, F. Chen, R. A. Dixon, G. T. Beckham and Y. Román-Leshkov, ACS Sustain. Chem. Eng., 2018, 6, 11211–11218 CrossRef CAS.
- Y. Li, L. Shuai, H. Kim, A. H. Motagamwala, J. K. Mobley, F. Yue, Y. Tobimatsu, D. Havkin-Frenkel, F. Chen, R. A. Dixon, J. S. Luterbacher, J. A. Dumesic and J. Ralph, Sci. Adv., 2018, 4, eaau2968 CrossRef PubMed.
- S. Wang, S. Su, L. Xiao, B. Wang, R. Sun and G. Song, ACS Sustain. Chem. Eng., 2020, 8, 7031–7038 CrossRef CAS.
- C. Liu, S. Wang, B. Wang and G. Song, Ind. Crops Prod., 2021, 169, 113666 CrossRef CAS.
- S. Wang, K. Zhang, H. Li, L. Xiao and G. Song, Nat. Commun., 2021, 12, 416 CrossRef CAS PubMed.
- W. Song, Q. Du, X. Li, S. Wang and G. Song, ChemSusChem, 2022, 15, e202200646 CrossRef CAS.
- S. Su, F. Cao, S. Wang, Q. Shen, G. Luo, Q. Lu and G. Song, Green Chem., 2023, 25, 8172–8180 RSC.
- Z. Shen, W. Wang, L. Pan, Z. Huang, X. Zhang, C. Shi and J. Zou, Green Chem., 2023, 25, 7782–7793 RSC.
- H. Zhang, S. Fu, X. Du and Y. Deng, ChemSusChem, 2021, 14, 2268–2294 CrossRef CAS PubMed.
- R. Behling, S. Valange and G. Chatel, Green Chem., 2016, 18, 1839–1854 RSC.
- T. Vangeel, W. Schutyser, T. Renders and B. F. Sels, Top. Curr. Chem., 2018, 376, 1–16 CrossRef CAS.
- O. Y. Abdelaziz, I. Clemmensen, S. Meier, C. A. E. Costa, A. E. Rodrigues, C. P. Hulteberg and A. Riisager, ChemSusChem, 2022, 15, e202201232 CrossRef CAS PubMed.
- S. Wang, Q. Shen, S. Su, J. Lin and G. Song, Trends Chem., 2022, 4, 948–961 CrossRef CAS.
- Y. Li, X. Meng, R. Meng, T. Cai, Y. Pu, Z. Zhao and A. J. Ragauskas, RSC Adv., 2023, 13, 12750–12759 RSC.
- W. Peng, H. Bao, H. H. Weaver, J. Gao, Y. Wang, E. Cote, W. J. Sagues, D. Xiao and Z. Tong, ChemSusChem, 2023, 16, e202300750 CrossRef CAS.
- D. A. Giannakoudakis, A. Qayyum, V. Nair, A. Khan, S. R. Pradhan, J. Prekodravac, K. Rekos, A. P. LaGrow, O. Bondarchuk, D. Łomot, K. S. Triantafyllidis and J. C. Colmenares, Mol. Catal., 2021, 514, 111664 CrossRef CAS.
- S. Li, W. Mao, L. Zhang, H. Huang, Y. Xiao, L. Mao, R. Tan, Z. Fu, N. Yu and D. Yin, Mol. Catal., 2022, 528, 112416 CrossRef CAS.
- M. Wang, L. H. Li, J. M. Lu, H. J. Li, X. C. Zhang, H. F. Liu, N. C. Luo and F. Wang, Green Chem., 2017, 19, 702–706 RSC.
- Y. Hu, L. Yan, X. Zhao, C. Wang, S. Li, X. Zhang, L. Ma and Q. Zhang, Green Chem., 2021, 23, 7030–7040 RSC.
- F. Walch, O. Y. Abdelaziz, S. Meier, S. Bjelić, C. P. Hulteberg and A. Riisager, Catal. Sci. Technol., 2021, 11, 1843–1853 RSC.
- G. Xiao, J. R. D. Montgomery, C. S. Lancefield, I. Panovic and N. J. Westwood, Chem.–Eur. J., 2020, 26, 12397–12402 CrossRef CAS PubMed.
- B. Sedaia and R. Tom Baker, Adv. Synth. Catal., 2014, 356, 3563–3574 CrossRef.
- B. Sedai, C. D. Urrutia, R. T. Baker, R. Wu, L. A. P. Silks and S. K. Hanson, ACS Catal., 2011, 1, 794–804 CrossRef CAS.
- H. Xie, G. Zhu, D. Ye, W. Cai, J. Zhang, K. Huang, Y. Mai, B. Liao and J. Chen, New J. Chem., 2023, 47, 17072–17079 RSC.
- H. Luo, L. Wang, S. Shang, G. Li, Y. Lv, S. Gao and W. Dai, Angew. Chem., Int. Ed., 2020, 59, 19268–19274 CrossRef CAS PubMed.
- T. Senthamarai, V. G. Chandrashekhar, N. Rockstroh, J. Rabeah, S. Bartling, R. V. Jagadeesh and M. Beller, Chem, 2022, 8, 508–531 CAS.
- H. Luo, L. Wang, G. Li, S. Shang, Y. Lv, J. Niu and S. Gao, ACS Sustain. Chem. Eng., 2018, 6, 14188–14196 CrossRef CAS.
- K. Sun, S. Chen, J. Zhang, G. Lu and C. Cai, ChemCatChem, 2019, 11, 1264–1271 CrossRef CAS.
- A. Villa, D. Wang, N. Dimitratos, D. Su, V. Trevisan and L. Prati, Catal. Today, 2010, 150, 8–15 CrossRef CAS.
- J. Xie, J. Nie and H. Liu, Chin. J. Catal., 2014, 35, 937–944 CrossRef CAS.
- W. Deng, H. Zhang, X. Wu, R. Li, Q. Zhang and Y. Wang, Green Chem., 2015, 17, 5009–5018 RSC.
- F. G. Sales, L. C. A. Maranhão, N. M. L. Filho and C. A. M. Abreu, Chem. Eng. Sci., 2007, 62, 5386–5391 CrossRef CAS.
- H. Luo, E. P. Weeda, M. Alherech, C. W. Anson, S. D. Karlen, Y. Cui, C. E. Foster and S. S. Stahl, J. Am. Chem. Soc., 2021, 143, 15462–15470 CrossRef CAS PubMed.
- Z. Gong and L. Shuai, Trends Chem., 2023, 5, 163–166 CrossRef CAS.
- E. A. Pillar-Little, R. C. Camm and M. I. Guzman, Environ. Sci. Technol., 2014, 48, 14352–14360 CrossRef CAS PubMed.
- E. A. Pillar-Little and M. I. Guzman, Environ. Sci. Technol., 2017, 51, 4951–4959 CrossRef CAS PubMed.
- R. Jastrzebski, B. M. Weckhuysen and P. C. A. Bruijnincx, Chem. Commun., 2013, 49, 6912–6914 RSC.
- M. Wang, X. K. Gu, H. Su, J. Lu, J. P. Ma, M. Yu, Z. Zhang and F. Wang, J. Catal., 2015, 330, 458–464 CrossRef CAS.
- P. Wang, C. H. Gong, A. Y. Tang, A. T. Gu, K. W. Chen and Y. Yi, Mater. Res. Express, 2023, 10, 025005 CrossRef.
- T. Lu, Z. Du, J. Liu, C. Chen and J. Xu, Chin. J. Catal., 2014, 35, 1911–1916 CrossRef CAS.
- X. Dai, T. Li, B. Wang, C. Kreyenschulte, S. Bartling, S. Liu, D. He, H. Yuan, A. Brückner, F. Shi, J. Rabeah and X. Cui, Angew. Chem., Int. Ed., 2023, 62, e202217380 CrossRef CAS.
- T. Yoshida, K. Yamasaki and S. Sawada, Bull. Chem. Soc. Jpn., 1979, 52, 2908–2912 CrossRef CAS.
- J. Wang, J. Ren, Q. Tang, X. Wang, Y. Wang, Y. Wang, Z. Du, W. Wang, L. Huang, L. A. Belfiore and J. Tang, Materials, 2022, 15, 1719 CrossRef CAS PubMed.
- C. Cheng, P. Li, W. Yu, D. Shen, X. Jiang and S. Gu, ACS Sustain. Chem. Eng., 2020, 8, 16217–16228 CrossRef CAS.
- D. Jeong, W. Jo, J. Jeong, T. Kim, S. Han, M.-K. Son and H. Jung, RSC Adv., 2022, 12, 2632–2640 RSC.
- T. Yoshida and S. Sawada, Bull. Chem. Soc. Jpn., 1975, 48, 3379–3380 CrossRef CAS.
- A. Nunes, L. Djakovitch, L. Khrouz, F.-X. Felpin and V. Dufaud, J. Mol. Catal. A: Chem., 2017, 437, 150–157 CrossRef CAS.
- F. A. Westerhaus, R. V. Jagadeesh, G. Wienhöfer, M.-M. Pohl, J. Radnik, A.-E. Surkus, J. Rabeah, K. Junge, H. Junge, M. Nielsen, A. Brückner and M. Beller, Nat. Chem., 2013, 5, 537–543 CrossRef CAS.
- H. Zhong, J. Wang, Y. Zhang, W. Xu, W. Xing, D. Xu, Y. Zhang and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 14235–14239 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra08052h |
| This journal is © The Royal Society of Chemistry 2024 |