
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirected assembly of fullerenols via electrostatic and coordination interactions to fabricate diverse and water-soluble metal cation–fullerene nanocluster complexes†
Rui He‡
a,
Chenjie Fan‡a,
Qingyuan Lianga,
Yan Wanga,
Yanyan Gaoa,
Jiakai Wua,
Qingnan Wu*a and
Fuju Tai *b
*b
aNanoAgro Center, College of Plant Protection, Henan Agricultural University, Zhengzhou 450046, China. E-mail: wuqingnan@henau.edu.cn
bNational Key Laboratory of Wheat and Maize Crop Science, College of Life Science, Henan Agricultural University, Zhengzhou 450046, China. E-mail: taifuju@henau.edu.cn
First published on 3rd January 2024
Abstract
Metal ion–nanocluster coordination complexes can produce a variety of functional engineered nanomaterials with promising characteristics to enable widespread applications. Herein, the visualization observation of the interactions of metal ions and fullerene derivatives, particularly anionic fullerenols (Fol), were carried out in aqueous solutions. The alkali metal salts only resulted in salting out of Fol to gain re-soluble sediments, whereas multivalent metal cations (Mn+, n = 2, 3) modulated further assembly of Fol to produce insoluble hybrids. These provide crucial insights into the directed assembly of Fol that two major forces involved in actuation are electrostatic and coordination effects. Through the precise modulation of feed ratios of Fol to Mn+, a variety of water-soluble Mn+@Fol coordination complexes were facilely prepared and subsequently characterized by various measurements. Among them, X-ray photoelectron spectra validated the coordination effects through the metal cation and oxygen binding feature. Transmission electron microscopy delivered valuable information about diverse morphologies and locally-ordered microstructures at the nanoscale. This study opens a new opportunity for developing a preparation strategy to fabricate water-soluble metal cation–fullerenol coordination complexes with various merits for potential application in biomedical fields.
1 Introduction
Metal ion–nanoscale cluster coordinated aggregation complexes have received great attention to produce a variety of functional engineered nanomaterials with promising characteristics of diverse architecture, great versatility, and applicability that have enabled a broad range of applications in materials science, energy, environment, and biomedicine,1–3 including pollution restorers,4,5 environmental purifiers,6 batteries and solar cells,7 catalysis,8 artificial enzymes,9 templating,10 sensing,11 anticancer drug delivery,12 and theranostics.13 As a few recent highlights, for example, the selected and precise coordination of a nanoscale polyoxometalate cluster with hexavalent actinides over trivalent lanthanides gave rise to a new ultrafiltration-based separation method for high-level waste in nuclear energy.5 The synthetic use of electron-beam lithographic techniques induced the exceptional formation of an open-hole bimetallic lead–azafullerene complex to provide a new horizon for designing advanced and well-defined carbon nanostructures.10Since the first discovery of fullerenes in 1985,14 the insertion into carbon cages or external doping of metal ions has been attempted to gain fullerene-based metal coordination complexes.15 To date, such two strategies have been successfully developed to fabricate endohedral metallofullerenes and exohedral metallofullerene complexes.15 Numerous dedicated and detailed studies have discovered that metals and nitride-, oxide-, cyano-, and sulfide-metal clusters were encapsulated inside fullerene cages to obtain conventional metallofullerenes and metal-containing cluster fullerenes.16 In most cases for investigating the electronic and molecular structure of endohedral metallofullerenes, metal-cage bonding interactions were revealed as the coordination of metal cations with the cage ligand after an appropriate number of valence electrons have transferred to the fullerene cage.16 Paramagnetic Gd@C82, Gd@C60, Gd3N@C80, ScGd2N@C80, and Sc2GdN@C80 as representative endohedral metallofullerenes can be used to develop some water-soluble derivatives via exohedral and functional modification onto the surface of metallofullerene cages as contrast agents for drastically improving the image quality of magnetic resonance imaging (MRI) diagnostic examination and potential application in the biomedical field.16 However, the yields of endohedral metallofullerenes were extremely low, equivalent to the order of 1% of the empty fullerenes.16 More researchers focused on exohedral metal–fullerene coordination complexes, including metals directly coordinating to the carbon cage, functionalized fullerene–metal ion coordination complexes, and conjugations of fullerene adducts with metal chelates.15 Among them, significant progress was achieved in the synthetic procedures and structural studies of metallic complexes with appendant groups of functionalized fullerenes as ligands. According to classification by the hydrophilic or hydrophobic polarities of appended substitutes, functionalized fullerenes are mainly divided into water-insoluble/soluble fullerene adducts. The early discoveries that triggered the interest of scientists were representative hydrophobic fullerene adducts chelating metal ions in nonaqueous solutions, among which bipyridine, terpyridine, pyrazine, and dipyrrolidine were functionalized to fullerenes.17 In 1998, a dimeric fullerene–Pt coordination complex was firstly reported, where Pt(Et3)2(OTf)2 mediated the self-assembly of dipyridylmethanofullerene to form a discrete coordination structure.18 To take a step forward, a variety of one-, two-, and three-dimensional framework structures were successfully developed, wherein various hydrophobic fullerene adduct ligands chelated with different metal ions in nonaqueous solutions to form diverse metal–fullerene frameworks for providing nanoscale porous spaces.19–22
On the other hand, hydrophilic fullerene adducts as the water-soluble derivatives of functionalized fullerenes (i.e., fullerenol is a typical representative) have been widely studied to disclose excellent bioactivities for potential biomedical applications, such as antitumor, antiviral, antibacterial, and antioxidative abilities, and efforts toward building metal-coordinated complexes in aqueous solutions and subsequent application in biomedicine are rather limited.23,24 In particular, fullerenols have a beneficial π-electron structure and numerous oxygen-containing groups for forming coordinate bonds; thus, their application to fabricate metal-coordinated complexes is eagerly anticipated.24 Some investigations mentioned the salting out of fullerenols in the cosaturated fullerenol–salt–water ternary systems in the presence of varied inorganic salts to result in precipitates of fullerenols.25–29 However, the roles of salts were largely unclear on whether metal ions-induced the further assembly of fullerenols via coordination or not. The first direct evidence about metal ions coordinating to fullerenols was revealed by Anderson et al., where fullerenols acted as a ligand analogous to a 1,2-diol or catechol to possibly form tetrahedral and octahedral coordination under the stoichiometric condition of fullerenols to metal ions excess of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10.30 Hereafter, more researchers demonstrated that the fullerenol–metal ion interactions could lead to corresponding coordination complexes. Recently, it was also clarified that fullerenols underwent self-assembly and acted as tridentate ligands to bind trivalent metal ions with 1,3,5-cyclohexanetriol coordination to be potentially utilized for co-precipitation in the removal of metal ions from contaminated water.31 As a way forward, fullerenol was grafted on the surface of graphene oxide to strengthen its adsorption capacity and selectivity for accurately and strongly chelating Pb2+ via the oxygen-containing groups of fullerenols.6 This strategy may be exploited to develop a potential method for the highly efficient removal and ultra-sensitive determination of Pb2+ in complex-matrix samples. Moreover, fullerenol/Ni2+ porous composite films were synthesized by fullerenols via Ni2+ coordination under the hard template of polystyrene microspheres and then embedding in poly(methyl methacrylate) in water. The hybrids displayed efficient nonlinear optical properties through metal–ligand charge transfer to increase electron delocalization. In a similar and earlier study, it was firstly reported that fullerenol/Fe3+ microcapsules were produced through coordination between the hydroxyl onto fullerenol cage with Fe3+ released from a rusty nail using polystyrene microspheres as templates.24 The magnetic fullerenol/Fe3+ microcapsules had good biocompatibility and colloidal stability in water and serum to exhibit effective antioxidant activities for improving the living environment of cells by scavenging the hydroxyl radical. To the best of our knowledge, fullerenols are the most compatible fullerene-based ligand species for the coordination of multivalent metal ions in aqueous media observed so far. Although various metal ions have been explored following coordination techniques to obtain fullerenol–metal ion-chelated complexes, including Ni2+,32 Pb2+,6 Fe3+,24 as well as Zn2+, Cu2+, Co2+, Cd2+, Ca2+, Mn2+, Ag+, La3+, Nd3+, and Al3+,30,31 an unsolved issue is the unavoidable formation of insoluble and random cross-linking hybrids, making it difficult to obtain water–soluble metal cation–fullerenol complexes for biomedical application. Recently, the fullerenol/Fe2+ nanocomposite was developed in aqueous media by simply mixing fullerenol (334 μM) and Fe2+ (0.556 μM) in the stoichiometric ratio of 590:1.33 Fe2+ did not disturb the further assembly process of fullerenol without a noticeable change in the morphology for the nanocomposite, but its zeta potential was indeed shifted to a more positive value. Importantly, such a nanocomposite exhibited a potentially protective effect on healthy tissues and diminished doxorubicin-induced toxic effects on liver and heart tissues. Another interesting observation was about the interaction of anionic fullerene derivatives and Hg2+ ions.34 Stoichiometric (1:1 by moles) amounts of Hg2+ was adsorbed by the water-soluble fullerene adduct, C60(S(CH2)10CO2K)5 to form hybrid metal–fullerene complexes with aggregation as stable colloidal suspensions in aqueous solution, and it was confirmed that the antidote effect to weaken the bioavailability of Hg2+ for Escherichia coli cells was comparable with that of Unithiol. In addition, various metal salts as a building block possibly fabricate the diversely structural metal ion–fullerenol complexes by tuning the coordination mode of metal ions on fullerenols, which still receives less attention.17
:10.30 Hereafter, more researchers demonstrated that the fullerenol–metal ion interactions could lead to corresponding coordination complexes. Recently, it was also clarified that fullerenols underwent self-assembly and acted as tridentate ligands to bind trivalent metal ions with 1,3,5-cyclohexanetriol coordination to be potentially utilized for co-precipitation in the removal of metal ions from contaminated water.31 As a way forward, fullerenol was grafted on the surface of graphene oxide to strengthen its adsorption capacity and selectivity for accurately and strongly chelating Pb2+ via the oxygen-containing groups of fullerenols.6 This strategy may be exploited to develop a potential method for the highly efficient removal and ultra-sensitive determination of Pb2+ in complex-matrix samples. Moreover, fullerenol/Ni2+ porous composite films were synthesized by fullerenols via Ni2+ coordination under the hard template of polystyrene microspheres and then embedding in poly(methyl methacrylate) in water. The hybrids displayed efficient nonlinear optical properties through metal–ligand charge transfer to increase electron delocalization. In a similar and earlier study, it was firstly reported that fullerenol/Fe3+ microcapsules were produced through coordination between the hydroxyl onto fullerenol cage with Fe3+ released from a rusty nail using polystyrene microspheres as templates.24 The magnetic fullerenol/Fe3+ microcapsules had good biocompatibility and colloidal stability in water and serum to exhibit effective antioxidant activities for improving the living environment of cells by scavenging the hydroxyl radical. To the best of our knowledge, fullerenols are the most compatible fullerene-based ligand species for the coordination of multivalent metal ions in aqueous media observed so far. Although various metal ions have been explored following coordination techniques to obtain fullerenol–metal ion-chelated complexes, including Ni2+,32 Pb2+,6 Fe3+,24 as well as Zn2+, Cu2+, Co2+, Cd2+, Ca2+, Mn2+, Ag+, La3+, Nd3+, and Al3+,30,31 an unsolved issue is the unavoidable formation of insoluble and random cross-linking hybrids, making it difficult to obtain water–soluble metal cation–fullerenol complexes for biomedical application. Recently, the fullerenol/Fe2+ nanocomposite was developed in aqueous media by simply mixing fullerenol (334 μM) and Fe2+ (0.556 μM) in the stoichiometric ratio of 590:1.33 Fe2+ did not disturb the further assembly process of fullerenol without a noticeable change in the morphology for the nanocomposite, but its zeta potential was indeed shifted to a more positive value. Importantly, such a nanocomposite exhibited a potentially protective effect on healthy tissues and diminished doxorubicin-induced toxic effects on liver and heart tissues. Another interesting observation was about the interaction of anionic fullerene derivatives and Hg2+ ions.34 Stoichiometric (1:1 by moles) amounts of Hg2+ was adsorbed by the water-soluble fullerene adduct, C60(S(CH2)10CO2K)5 to form hybrid metal–fullerene complexes with aggregation as stable colloidal suspensions in aqueous solution, and it was confirmed that the antidote effect to weaken the bioavailability of Hg2+ for Escherichia coli cells was comparable with that of Unithiol. In addition, various metal salts as a building block possibly fabricate the diversely structural metal ion–fullerenol complexes by tuning the coordination mode of metal ions on fullerenols, which still receives less attention.17
Based on the above studies, one proposed method for addressing the challenge to mitigate the formation of the insoluble complexes is the modulation of the random-crosslinking degree by selecting and strictly controlling the stoichiometric ratios of fullerenols to various metal ions that is tailored to the coordination requirements of water-soluble metal cation–fullerenol complexes with a variety of possibilities in the directed-assembly mode and to discriminate against insoluble hybrids. Herein, we prepared fullerenol in the anion form by improved alkaline-oxidation approach, and the visual observations were carried out for electrostatic- and coordination-directed further assembly of anionic fullerenol in various salt solutions for producing water-soluble/insoluble metal cation-fullerenol coordination complexes. Furthermore, the optimized condition of feed ratios for the preparation of diverse and water-soluble complexes were quantitative investigated by spectroscopic measurements. The resultant water-soluble metal cation–fullerenol complexes were characterized to comparatively analyze their optical, thermal, electron, and photoelectron properties and directed-assembled morphologies. Our work will not only explore the various directed assembly behavior of fullerenols but also to provide an opportunity to discover water-soluble metal cation-fullerenol coordination complexes as novel carbon structures with desirable properties for potential applications in biomedicine.
2 Experiment section
2.1 Materials
All chemicals, unless otherwise stated, were purchased from commercial sources and used without further purification. C60 (purity > 99.9%) was purchased from Henan Puyang Yongxin Reagents Company (China). Quaternary ammonium iminofullerene was prepared according to the experimental method given in recent reports, where the average formula was C60(NCH2CH2NH3+˙CF3COO−)4·10H2O (refers to IFQA hereafter) with molecular weight (MW) of 1588 g mol−1.35 The other reagents were at least analytical grade.2.2 Synthesis of negatively charged fullerenol
Fullerenol in anion form (Na+–fullerenol) was prepared using a modified literature procedure.36–38 Briefly, 300 mL 0.5 g mL NaOH solution was mixed with 10 mL 40% tetra-n-butylammonium hydroxide and 600 mL o-dichlorobenzene solution containing 25 g C60 in a 2000 mL volume of flask to be vigorously stirred by a mechanical agitator at 60–70 °C for 5 days. After stopping heating and adding 600 mL deionized water, the mixture was continued to be stirred for 1–2 days, then filtrated, washed with deionized water, and separated. The resulting aqueous solution was evaporated, washed with methanol several times, and redissolved in deionized water, followed by dialysis and drying. The purified powder was marked as Fol, and later identified as C60(OH)22(ONa)3·7H2O.2.3 Sample pretreatment of fullerene–salt–water ternary systems
Fol (4.0 mM) and IFQA (0.4 mM) stock solutions were prepared in volumetric flasks by dissolving purified powders in deionized water. To prepare the stock solutions of the salts, chlorides of alkali (LiCl, NaCl, and KCl), alkaline earth (MgCl2, CaCl2, and SrCl2), mercury (HgCl2), aluminum (AlCl3), and lanthanum (LaCl3) and sodium salts of acetate (CH3COONa), carbonate (Na2CO3), and sulfate (Na2SO4) were dissolved in deionized water in volumetric flasks. For the fullerene–salt–water ternary systems, the concentration of Fol/IFQA always remained constant while the molar concentration ratios of Fol/IFQA with respect to the salts varied between 1:1000 and 1:0. To investigate the salting-out effect of Fol in alkali metal salt, acidic, or basic solutions, Fol (4.0 mM, 0.5 mL) were diluted upon the addition a series of varying concentrations of KCl, NaCl, LiCl, HCl, Na2SO4, Na2CO3, CH3COONa, NaOH, and water (0.5 mL) in plastic centrifuge tubes, where the ratios of Fol to them were 1:1000, 1:500, 1:250, 1:100, and 1:0. To investigate the directed assembly of Fol in metal chloride solutions, Fol (0.4 mM, 1.0 mL) were diluted twice upon the addition of various concentrations of NaCl (1:1000, 1:500, 1:250, 1:100, 1:10, and 1:0), MgCl2 (1:100, 1:10, 1:7.3, 1:6, 1:1, and 1:0), AlCl3 (1:10, 1:5, 1:2, 1:1.6, 1:1, and 1:0), LaCl3 (1:10, 1:5, 1:1.8, 1:1.4, 1:1, and 1:0), KCl (1:1000, 1:500, 1:250, 1:100, 1:10, and 1:0), CaCl2 (1:50, 1:5, 1:3.8, 1:2, 1:1, and 1:0), SrCl2 (1:10, 1:5, 1:4, 1:3.5, 1:2, and 1:0), and HgCl2 (1:1000, 1:200, 1:100, 1:50, 1:10, and 1:0) in the screw bottles. To investigate the surface charge effect of the fullerene ligand on the directed assembly of metal ion–fullerene hybrids, IFQA (0.4 mM, 0.5 mL) were diluted upon the addition of a series of varying concentrations of NaCl, MgCl2, AlCl3, LaCl3, KCl, CaCl2, SrCl2, and HgCl2 (1:1000, 1:500, 1:250, 1:100, and 1:0, 0.5 mL) in plastic centrifuge tubes. After HgCl2 solution was preblended with a 10-fold equimolar amount of NaCl, 0.5 mL of the resulting solution and IFQA (0.4 mM, 0.5 mL, IFQA:HgCl2, 1:20) were successively added into a plastic centrifuge tube, followed by thorough mixing.
2.4 Visualized observation for the directed assembly/salting-out effect of Fol in salt solutions
Freshly prepared Fol with alkali metal salt, acidic, or basic solutions in plastic centrifuge tubes were immediately observed and photographed after centrifugation with 10000g at 4 °C for 5–10 min, whether there was a decoloration of the supernatant and/or some solid at the bottom or not. Fol with metal chloride solutions in screw bottles were observed and photographed after standing for 7 days. The freshly prepared IFQA with metal chloride solutions were immediately observed and photographed after centrifugation. The concentration ratios that produced precipitates/sediments were recorded.
2.5 Quantitative analysis for the directed assembly of Fol by metal ions
After the treatments in Section 2.4 by ultrasound and centrifugation, the UV-visible spectra of the 5-fold diluted supernatants were measured to record the absorption value at 350 nm. All data analysis was carried out using the three-parameter single exponential growth function as eqn (1).| y = A0 × exp(−x/R0) + y0 | (1) |
:metal chloride ratio, y is the remaining absorption value for ternary systems in comparison to metal chlorides, and R0 and y0 are the initial concentration ratio and absorption value for ternary systems in supernatants relative to corresponding metal chlorides. The critical concentration ratio was denoted as R50 with 50% reduction of the UV absorption value of ternary systems in comparison to the corresponding metal chlorides. Correspondingly, the c50 value of metal chlorides for ternary Fol–salt–water systems is defined as the critical concentration of metal chlorides at the ratio of R50. From the exponential fitting curves, the inflection points from the exponential growth interval to plateau interval were deduced as Rt, which provided the indication as the turning points of the concentration ratios to discriminate the formation of insoluble hybrids from water-soluble complexes.
2.6 Fabrication of water-soluble Mn+@Fol coordination complexes
In a general procedure, a 200 mL and specific concentration of MCln (Mg2+: 2.0 mM, Ca2+: 0.92 mM, Sr2+: 0.4 mM, La3+: 0.34 mM, or Al3+: 0.38 mM) solution was added dropwise into Fol (200 mL, 0.4 mM), and subsequently the mixture was allowed to be stirred for 30–60 min at room temperature. After filtration, the resulting filtrate was dialyzed against deionized water using a membrane with molecular weight cutoff of 3500 Da for the removal of free metal salts to harvest the purified water-soluble Mg2+@Fol, Ca2+@Fol, Sr2+@Fol, Al3+@Fol, and La3+@Fol coordination complexes.2.7 Characterization
The absorbances at 350 nm and the UV-visible spectra were recorded on an A560 spectrophotometer (AOE Instruments, China). Fourier transform infrared (FTIR) spectroscopy measurements were performed on a Thermo Scientific Nicolet iN10 MX spectrometer (Madison, USA). X-ray photoelectron spectroscopy (XPS) data were obtained by a Thermo Scientific ESCALAB 250Xi spectrometer (Thermo Scientific, USA), with Al Kα X-rays as the excitation source, working voltage of 12.5 kV, and filament current of 16 mA to perform 10 cycles of signal accumulation. Thermogravimetric analysis (TGA) was performed on a PerkinElmer STA 6000 Thermogravimetric Analyzer (TA Instruments, USA) at a heating rate of 10 °C min−1 under nitrogen (N2) atmosphere in a scanning range from room temperature to 1000 °C. The zeta potentials of the samples (20 mg L−1) in deionized water were measured using a Microtrac Nanotrac wave II particle size and zeta potential analyzer (Microtrac Instruments, USA). X-ray Diffraction (XRD) test was performed on an Ultima IV X-ray diffractometer (Rigaku, Japan) in the scanning range from 5° to 90° and at a scanning rate of 2° min−1 with Cu Kα X-rays. Transmission electron microscopy (TEM) investigation included conventional and high-resolution TEM (HRTEM) imaging and selected-area electron diffraction (SAED) to observe the size, shape, dispersity, and microstructure, which were studied on an HRTEM (JEOL JEM-2100Plus, Japan) operated at 200 kV accelerating voltage. One drop of ∼1 μM aqueous solution of the samples for TEM examination was deposited on Cu grid-supported carbon films to allow drying at room temperature for ∼2 hours.3 Results and discussion
3.1 Characterization of Na+-fullerenol
As shown in the full survey spectrum of Fol in Fig. 1a, the C1s, O1s, and Na1s peaks were observed, and the atomic ratio of C/Na was about 60:2.83. The high-resolution Na1s XPS spectrum at the binding energy of 1071 eV is presented in Fig. 1b to indicate Na+ coexistence in the tested sample, which should be introduced due to the synthesis process.36,37 The high-resolution O1s spectrum was fitted to three peaks at 531.3, 532.62, and 536.01 eV, ascribing to hydroxyl (C–OH), epoxy group (C–O–C), and alkoxide anion moiety (C–O–/C–O–Na).6,31 In the FTIR spectrum (Fig. 1d), the appendant –OH and remaining π-bonded carbons of the fullerene cages were validated as the diagnostic criteria for fullerenols with the four characteristic peaks at about 3415, 1585, 1045, and 1385 cm−1 assigned to νC–OH, νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, νO–H, and δC–OH adsorptions.38,39 The thermogravimetric (TG) and differential TG (DTG) curves are shown in Fig. 1e, where the first weight loss (∼9.5%) occurred before 136 °C, which was ascribed to bound water. The second-step decomposition (∼36.5%) was assigned to the process of dehydroxylation (C–OH, C–O–C, C–O–) of Fol between 136 °C and 619 °C. Taking into account that the molecule cannot be fractioned and the results of combination XPS and TGA, the average molecular formula for Fol was described as C60(OH)22(ONa)3·7H2O (MW = 1337 g mol−1).38 The XRD pattern was a broad diffraction peak at 2θ = 22.765° and is displayed in Fig. 1f (JCPDS 50-0926), indicating an amorphous structure of Fol at the macroscopic level.24,32
C, νO–H, and δC–OH adsorptions.38,39 The thermogravimetric (TG) and differential TG (DTG) curves are shown in Fig. 1e, where the first weight loss (∼9.5%) occurred before 136 °C, which was ascribed to bound water. The second-step decomposition (∼36.5%) was assigned to the process of dehydroxylation (C–OH, C–O–C, C–O–) of Fol between 136 °C and 619 °C. Taking into account that the molecule cannot be fractioned and the results of combination XPS and TGA, the average molecular formula for Fol was described as C60(OH)22(ONa)3·7H2O (MW = 1337 g mol−1).38 The XRD pattern was a broad diffraction peak at 2θ = 22.765° and is displayed in Fig. 1f (JCPDS 50-0926), indicating an amorphous structure of Fol at the macroscopic level.24,32
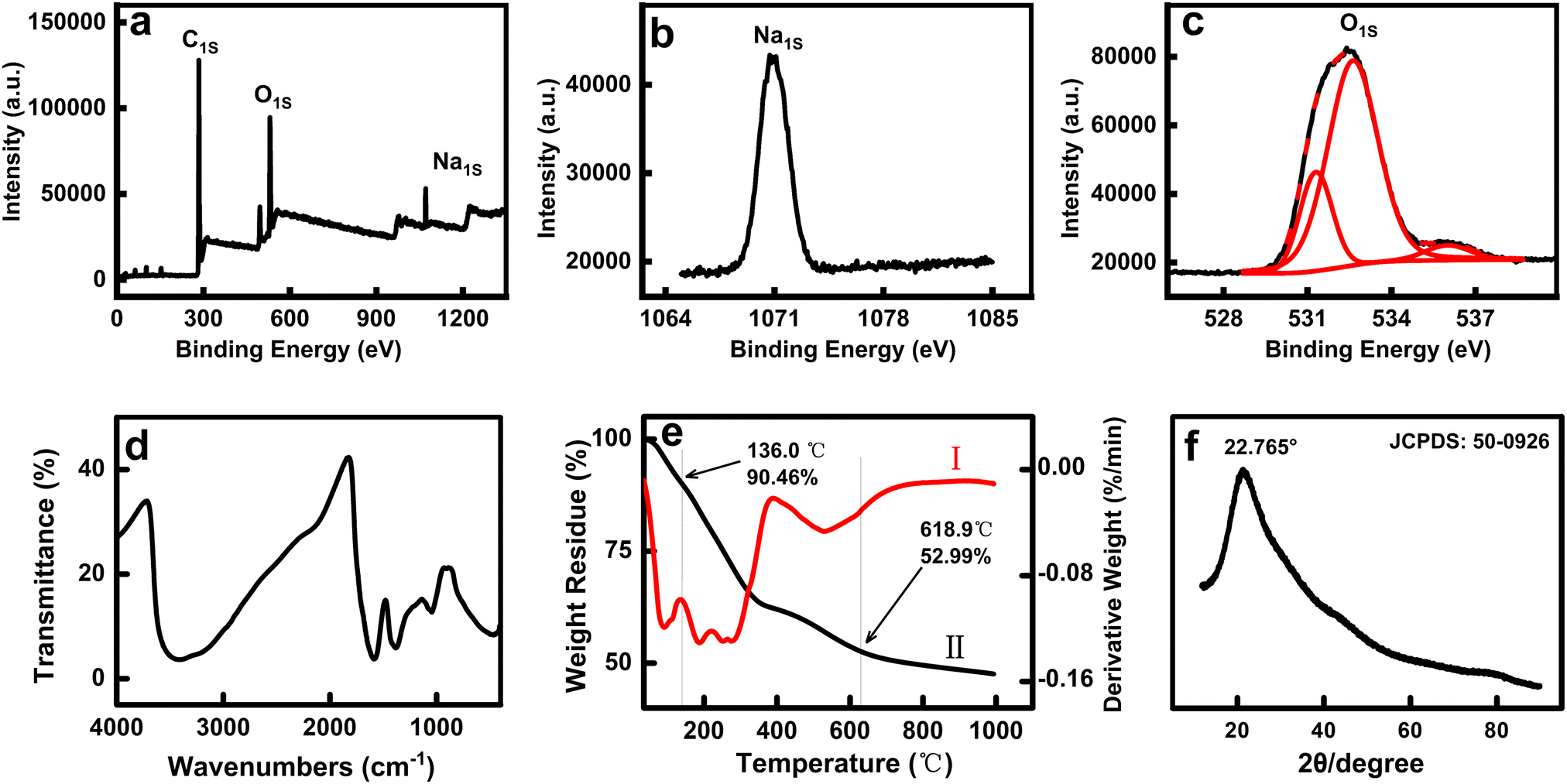 | ||
| Fig. 1 The full survey (a), high-resolution Na1s (b) and O1s (c) XPS spectra, FTIR spectrum (d), DTG (I) and TG (II) curves (e), and XRD pattern (f) of Fol. | ||
3.2 Salting-out effect of Fol in alkali salt solution
The solubility of Fol in aqueous solution of various alkali-metal salts/acid/base was studied by means of room temperature in plastic centrifuge tubes. We recorded the digital photos (Fig. 2) of all the samples with a fixed concentration of Fol but varying concentrations of acid, base, and salts. Fol (4.0 mM, 0.5 mL, the molar ratio, cFol:calkali metal ion/hydrogen ion = 1:1000, 1:500, 1:250, 1:100, and 1:0) were mixed with 0.5 mL alkali metal chlorides (KCl, NaCl, or LiCl), sodium salts (CH3COONa, Na2CO3, or Na2SO4), HCl, or NaOH. As follows from Fig. 2a–c, the dark- or light-brown precipitates of Fol were observed in the bottom of the tubes after centrifuging the aqueous solution of Fol upon the addition of KCl, NaCl, or LiCl. The increase in the precipitation (Fig. S1a–c in the ESI†) and discoloration of dark brown of the supernatants (Fig. 2a–c) indicated the reduction in Fol solubility in the presence of alkali chlorides (Fig. S2 in the ESI†), which were dependent upon the increase in the concentration of alkali chlorides, namely, the molar ratio of Fol to alkali chlorides. After the addition of other sodium salts, more or less precipitates continued to be observed in Fol–salt–water ternary systems (Fig. 2e–g). The dissolution of Fol in supernatants containing Na2CO3 and Na2SO4 had a noticeably smaller reduction than those in the presence of CH3COONa and NaCl (Fig. S2 in the ESI†). For different counter ions, the higher the anionic charge, the less was the precipitation by Na+. In addition to alkali metal salts, the similar phenomena likely occurred in the Fol–acid/base–water ternary system (Fig. 2d and h). The higher the concentration of HCl/NaOH, the more the precipitates and decoloration of Fol (Fig. S1e and f in the ESI†); correspondingly, the lower the solubility of Fol in the supernatant (Fig. S2 in the ESI†).
 | ||
| Fig. 2 The photographs of aqueous solutions of Fol (4.0 mM, 0.5 mL) upon the addition of equivalent volume of (a) KCl, (b) NaCl, (c) LiCl, (d) HCl, (e) Na2SO4, (f) Na2CO3, (g) CH3COONa, or (h) NaOH (from left to right cFol:cM+/H+ = 1:1000, 1:500, 1:250, 1:100, and 1:0) in plastic centrifuge tubes after centrifugation. c is the molarity of alkali metal ion, hydrogen ion, or Fol. The discoloration of the dark brown supernatants indicated the reductions of Fol solubility in the presence of alkali chlorides, acid, and base. | ||
Furthermore, at 0.8 mM concentration of Fol, only a small amount of precipitate was clearly observed in the Fol–salt–water ternary systems upon the addition of KCl, NaCl, or CH3COONa up to a molar ratio of 1:1000 (Fig. S3 and S4†). Under the condition of a lower concentration (0.4 mM) of Fol, precipitates were no longer produced in ternary systems with arbitrary concentrations of salts (Fig. S5†). Some evidences were demonstrated, where inorganic salts (i.e., NaCl) modulated salting out of fullerenols from saturated fullerenol–salt–water systems.25,26 On the other hand, in our present study, after decreasing Fol solubility in water–Fol ternary systems in the presence of alkali metal salts, base, or acid to oversaturate the solution, the supersaturated Fol was precipitated. It is worth noting that the above-described and solidified Fol (Fig. S1 and S4†) can be thoroughly redissolved in deionized water, and no precipitate will be produced after centrifugation again.
On the whole, we can state with confidence that the salting-out phenomena of Fol in the evaluated ternary systems was attributed to the action of alkali metal ions or acid–base regulation. When the concentration of Fol is not higher than 0.4 mM and the molar ratio of Fol to alkali metal cations is up to 1:1000, the Fol–alkali metal salt–water ternary systems still maintained dissolution as unsaturated solutions without discoloration and salting out to precipitate Fol in the solid state. Also, Fol precipitates induced by extremely high concentration of alkali metal cations will be soluble again in water, that is to say, alkali metal cations cannot cause Fol to form insoluble aggregates in water.
3.3 Directed assembly of Fol by the coordination effect
The varied metal chloride-mediated further assembly of Fol in aqueous solutions was studied by means of room temperature in screw bottles. Since metal cations are more strongly bound to smaller anionic counter ions to reduce the availability of attachment sites to derivatized fullerenes by static-electric force,40 we selected the salts of multivalent-state metal cations with chloride ion as counter ions, alkaline earth chlorides (MgCl2, CaCl2, and SrCl2), aluminum chloride (AlCl3), transition metallic chloride (HgCl2), and rare earth chloride (LaCl3) to investigate whether and how they affect the further assembly or dissolution of Fol in Fol-metallic chloride-water ternary systems relative to those of monovalent-state alkali metal chlorides. As shown in Fig. 3a and e, not surprisingly, under the condition of Fol (0.2 mM) to NaCl/KCl ratio up to 1:1000, the Fol–NaCl/KCl–water ternary systems were still unsaturated without sediments after standing for 7 days; on the other hand, the onset of sediment formation in Fol–LaCl3–water ternary system was at an extremely low Fol:LaCl3 ratio of only 1:1.4. Fol can be completely sedimented after the 10-fold-equivalent addition of LaCl3 with almost colorless supernatant (Fig. 3d and S6d†). Keskinov et al. explained the very strong salting-out effect in fullerenol–D-LaCl3–H2O ternary system at very low concentrations of fullerenols near eutonic points, where rare earth salt served as a highly-effective salting-out agent.27,28 Nevertheless, their results did not clarify whether the sediment could be further re-dissolved in deionized water. In our case, it is incomprehensible that the dissolution of the sediment from LaCl3 cannot be observed in 2 mL deionized water in comparison to treatments in Fig. 2, and it fell out of solution and precipitated in the bottom of the tubes after centrifugation again. If the so-called sediment was caused by the salting-out effect, “free” Fol can be redissolved in water. Thus, the resulting sediment from LaCl3 should not be due to the salting out effect but is attributed to the coordination effect that allows Fol–chelating La3+ as a water-insoluble hybrid, which is consistent with the findings of Heimann et al.31 As expected, it was further investigated and found that similar sedimentation events began to be formed in ternary systems in the individual presence of AlCl3, SrCl2, CaCl2, and MgCl2 with the relatively lower molar ratios (Fol:metal ion) of 1:1.6, 1:3.5, 1:3.8, and 1:7.3 in comparison to those of alkali metal chlorides (Fig. 3b, c, f, and h). When the ratios of Fol to MgCl2, CaCl2, SrCl2, and AlCl3 reached 1:100, 1:50, 1:10, and 1:10, Fol were completely sedimented (Fig. S6b, c and S6f, h†). In addition, the case under treatment of HgCl2 was somewhat unexpected, where the ratios for initial and complete sedimentation were about 1:100 and 1:1000.
 | ||
| Fig. 3 The photographs of aqueous solutions of Fol (0.4 mM, 1.0 mL) upon the addition of varied concentrations (1:1000–1:0) and equivalent volume of (a) NaCl, (b) MgCl2, (c) AlCl3, (d) LaCl3, (e) KCl, (f) CaCl2, (g) SrCl2, and (h) HgCl2 in the screw bottles after standing for 7 days. The proportion is the molar ratio of Fol to metal ion. | ||
To top it all off, all the sediments/precipitates exhibited in Fig. 3 and S6† cannot be redissolved in deionized water so that they still maintained insoluble capacities after aqueous dispersion–centrifugation process. How to reasonably clarify this phenomenon? Obviously, these sediments/precipitates were not “free” Fol but should be the salt–Fol composite. Anderson and co-workers elaborated the insoluble complexes by the irreversible reaction of hydroxyfullerene with metal salts.30,31 They were insoluble random-cross-linked polymers with the lack of an ordered structure, but there were even more metal–fullerenol complexes, where the hydroxylated sidewalls of carbon cages of fullerenols acted as the ligands. Li et al. noted a fullerenol-based porous organic–inorganic-hybrid composite film that mixed fullerenols with nickel chloride and polystyrene microsphere solution via metal coordination to embed in poly(methyl methacrylate).32 Our results are consistent with these previous researches. We give a plausible speculation as follows. Under treatments with alkali metal ions only at very high concentrations, salting out occurred to form re-soluble precipitate of Fol, which was mainly attributed to the lack of coordination activity of alkali metal cations; hence, they cannot coordinate with Fol as alkali metal cation–Fol hybrids. However, in stark contrast to the case of NaCl and KCl, treatments with LaCl3, AlCl3, SrCl2, CaCl2, and MgCl2 at an extremely low concentration did cause Fol to produce water-insoluble sediment, which was largely dependent on further directed-aggregation due to strong coordination interaction between Fol and multivalent metal cations. The monovalent alkali metal salts can only produce the solid-state Fol by means of the salting out effect at a relative high concentration ratio (>1:100), whereas multivalent metal chlorides can induce Fol to form water-insoluble hybrids by means of coordination at the lower concentration ratio (<1:10). These findings clearly underline that the most important factor for sedimenting Fol out of the solution is related to the intrinsic metal cation affinities of coordination to fullerenols rather than to their concentrations. Indeed, coordination strongly influenced the binding activities of metal ions to fullerenols.
3.4 Surface charge of fullerene ligand modulating chelation of metal ions
Besides the coordination effect, another important parameter that strongly influenced the directed assembly of fullerene ligands can be defined as the cage property factor. Thus, the surface charge of the carbon cage also played an important role for metal ions to induce the further assembly of fullerene ligands.As a new example, we recently synthesized water-soluble and cationic iminofullerene nanoparticles with the terminal amino groups in carbon chains (refers to IFQA) to provide the potential coordination binding sites.35 Under the condition of fixed concentration (0.2 mM) of IFQA, not only did the IFQA–NaCl/KCl–water ternary systems not produce precipitate (Fig. 4a and e) but also no precipitate was formed in every trial when a series of concentrations of MgCl2, CaCl2, SrCl2, AlCl3, and LaCl3 were individually added into the IFQA solution (Fig. 4b–d, f, and g). This implies that there was no coordination interaction between IFQA and these metal chlorides. An interesting phenomenon was also observed at a higher concentration (2.0 mM) of IFQA, where the ternary systems still did not produce precipitate after the addition of these chlorides up to 1000-fold equivalent amount (Fig. S7b–h†). This is reminiscent of the strikingly different behavior of Fol-induced sedimentation in the presence of the above-mentioned metal chlorides, where similar effects can be discounted for the present example on the basis that the sediment could be achieved in binding experiments of metal cations and Fol in Fig. 3. The surface charge properties of fullerene ligand strongly affect the metal cation attachment by affecting the static-electric force, which forms metal cation–fullerene coordination complex.40 Cationic IFQA can prevent the adsorption of metal cations due to the repulsive coulombic force and consequently cannot cause the coordination action.41 On the other hand, Fol has a negative zeta potential (Table 2) that readily exists as polyanion aggregates.36 The anionic Fol allowed multivalent metal cations to be easily absorbed onto the surface of carbon cages, resulting in coordination with polyhydroxyl groups on cages to subsequently form metal cation–Fol hybrids.31 Fol, as the electronegative fullerene ligand on the attachment of metallic cations, were mainly controlled by electrostatic attraction forces; on the contrary, IFQA, as an electropositive ligand, repelled the attachment of metallic cations and then prevented the formation of metal cation and IFQA complex.
 | ||
| Fig. 4 The photographs of aqueous solutions of IFQA (0.4 mM, 0.5 mL) upon the addition of equivalent volume of (a) NaCl, (b) MgCl2, (c) AlCl3, (d) LaCl3, (e) KCl, (f) CaCl2, (g) SrCl2, and (h) HgCl2 (from left to right cIFQA:cMn+ = 1:1000, 1:500, 1:250, 1:100, and 1:0) in plastic centrifuge tubes after centrifugation. c is the molarity of metal ion or IFQA. | ||
There was an apparent exception to the general correlation in the observation of electrostatic repulsion between metal cations and IFQA, where precipitation occurred after adding more than 500-fold equivalent amount of HgCl2 into IFQA (Fig. 4h and S7a†). The discoloration of brown IFQA and precipitate formation in the presence of HgCl2 indicated that the HgCl2 and IFQA complex was unexpectedly generated, i.e., HgCl2 can induce the further assembly of IFQA by coordination. Non-fading IFQA (Fig. 4a–g) indicated that IFQA did not bind metal cations to form insoluble hybrids. HgCl2 as a pseudo salt is a weakly ionized covalent compound that mainly exists in the form of HgCl2 molecules in dilute solution with no more than 3% ionization.42,43 Only a very small amount of HgCl2 was ionized as [HgCl]+, Hg2+, and Cl− ions. Since there is electrostatic repulsion between [HgCl]+ and Hg2+ with IFQA, the resulting complex should be the HgCl2–IFQA coordination complex formed by HgCl2 molecules binding directly to IFQA. On the other hand, as shown in Fig. S6h,† the HgCl2 and Fol complex was relatively easier to generate than the HgCl2–IFQA complex (the onset on precipitation, 1:100 vs. 1:500). Even though the ionization of HgCl2 was very low, under HgCl2 and more than 100 equivalents of Fol, the concentration of Hg2+ ion in the solution was comparable to that of Fol, which may result in the formation of Hg2+–Fol and/or [HgCl]+–Fol complexes by electrostatic attraction and then coordination actions. In these cases, the directed assembly behavior of fullerene derivatives in salt solutions, especially Fol, has been observed qualitatively. In the following section, the behavior will be quantificationally investigated.
3.5 Quantification of inducibility of metal chlorides for directed assembly of Fol
According to the visualization observations in Fig. 3a and e, adding the no matter how high concentration of alkali metal chlorides cannot cause further assembly of Fol that it is always impossible to produce insoluble complexes. Nevertheless, multivalent metal chlorides modulate the further assembly of Fol, which appeared to be a variation tendency heavily influenced by the type of the metal cations whose ratio for initial formation of sediment was observed approximately at 1:1–1:10 in Fig. 3.
The typical UV spectral curves of Fol–metal chloride–water ternary systems are depicted in Fig. S8,† which seem to be very similar. In all cases, a reduction of the absorbance intensity at 350 nm was associated with the gradually increased addition of metal chlorides with the Fol:metal chloride ratio between 1:0.5 and 1:900, which illustrated that Fol can be bound to precipitate and fall out of the solution. In order to precisely evaluate the inducibility of metal cations for directing the assembly of Fol, the absorbance value was used to quantify c50. In this manner, the UV-adsorption value was measured to analyze the amount of complexed precipitates and remaining Fol in the supernatant. We started with a known amount of Fol (0.4 mM, 0.5 mL) and added 0.5 mL varied multivalent metal chlorides with the initial Fol:metal chloride ratio of 1:0.5, but no precipitate was observed in all the samples. Subsequently, we kept increasing the concentrations of metal chlorides added until all of Fol was precipitated.
To validate the R50 for various metal chlorides, three-parameter single exponential growth function was proposed as eqn (1) for non-linear curve analysis. The plot shown in Fig. 5d illustrated the correlations of absorption value for the Fol–LaCl3–water ternary systems vs. LaCl3. It was exponentially recovered from −0.1683 to 0.5957 when LaCl3 was added with the Fol:LaCl3 ratio in the range from 1:2 to 1:0.7, and then reached a peak of 0.6023 at the ratio of 1:0.5. It is interesting that the absorption of the Fol–LaCl3–water ternary system is negative at the ratio of 1:2 in comparison to the LaCl3 solution. It can be seen as a further indication that the anticipated formation of insoluble LaCl3–Fol hybrids was accompanied by La3+ attachment to Fol. This caused initial LaCl3 and Fol to fall out of the solution so that the actual absorption value of the ternary system was lower than that of LaCl3 solution alone. By fitting the data to eqn (1), the R50 of LaCl3 was deduced to be 0.6462. Taking into account that the concentration of Fol was fixed at 0.2 mM, the c50 value of La3+ was calculated to be 0.3 mM (Table 1), where the average number of bound La3+ per Fol carbon cage was about 1.6, resulting in precipitation, which downregulated the absorbance by 50%. It revealed that La3+ had a high efficacy for inducing the further assembly of Fol to produce water-insoluble La3+–Fol hybrids. Turning back to Fig. 5, other metal chlorides were also tested in the same manner, and the summary of the details and key data are listed in Table 1. Very similar results were obtained in the case of Al3+, whose R50 was 0.5026, close to that of La3+. However, the R50 values of alkaline earth metal chlorides were far from the above value at 0.1356 (Sr2+), 0.04737 (Ca2+), and 0.008155 (Mg2+). In addition, the R50 of HgCl2 was 0.002030, which was substantially two orders of magnitude lower than that of La3+ and Al3+.
 | ||
| Fig. 5 The exponentially fitting curves of the absorbance recovery of 5-fold diluted supernatants for ternary Fol–metal chloride–water systems in comparison to those of the corresponding concentrations of metal chlorides accompanied by an increase in the Fol:metal chloride ratio between 1:900 and 1:0.5. The initial concentration of Fol is 0.2 mM. (a) MgCl2, (b) CaCl2, (c) SrCl2, (d) LaCl3, (e) AlCl3, and (f) HgCl2. Data are mean values ± SE for triplicate. | ||
:metal chloride ratios
| Metal chloride | Fitting formula y = −A0 × exp(−x/R0) + y0 | R50 | c50 (mM) | Average number of attachment metal ions per Fol cage | Rt |
|---|---|---|---|---|---|
| a The initial concentration of Fol is 0.2 mM; R50 is the Fol: metal chloride ratio for 50% reduction of the UV absorption value of ternary systems in comparison to corresponding metal chlorides; c50 is equal to 0.2 mM divided by R50; Rt is the Fol: metal chloride ratio for turning points from the exponential growth interval to plateau interval. | |||||
| LaCl3 | y = −24.86 × exp(−x/0.1464) + 0.6020 | 0.6462 | 0.3 | 1.6:1 |
1.1735 |
| AlCl3 | Y = −14.50 × exp(−x/0.1297) + 0.6021 | 0.5026 | 0.4 | 2.0:1 |
1.0401 |
| SrCl2 | y = −1.419 × exp(−x/0.1090) + 0.8112 | 0.1365 | 1.5 | 7.3:1 |
0.7163 |
| CaCl2 | y = −0.8085 × exp(−x/0.06946) + 0.8175 | 0.04737 | 4.2 | 21.1:1 |
0.4206 |
| MgCl2 | y = −0.3527 × exp(−x/0.04087) + 0.5578 | 0.008155 | 24.5 | 122.6:1 |
0.2007 |
| HgCl2 | y = −2.667 × exp(−x/0.0009364) + 0.6016 | 0.002030 | 98.5 | 492.7:1a |
0.0115 |
The R50 quantification can provide a relatively effective strategy to evaluate the inducibility of metal cations/HgCl2 for the directed assembly of Fol through electrostatic and/or coordination effects. For the comparison of relatively active effects of metal chlorides on binding Fol, we determined that the order of critical concentration was HgCl2 (98.5 mM) ≫ Mg2+ (24.5 mM) > Ca2+ (4.2 mM) > Sr2+ (1.5 mM) > Al3+ (0.4 mM) > La3+ (0.3 mM) (Table 1). Accordingly, except for HgCl2, the average number of attachment metal cations per Fol carbon cage on the surface was about 1.6 (La3+), 2.0 (Al3+), 7.3 (Sr2+), 21.1 (Ca2+), and 122.6 (Mg2+). Thus, the efficiency of induction for Fol to further agglomerate into water-insoluble metal cation–Fol hybrids is La3+ > Al3+ > Sr2+ > Ca2+ > Mg2+ ≫ HgCl2. The order of directed assembly of Fol follows the charge number of the metal cation as well as its ionic radius.31 For example, the inductive ability of trivalent cations is always better than those of divalent cations; for divalent cations, the inducibility is stronger for larger cations, same as that for trivalent cations.
3.6 Fabrication and characterization of water-soluble Mn+@Fol coordination complexes
Turning back to Fig. 5, for the exponential fitting curves, Rt was validated as shown in Table 1 to be 0.012 (HgCl2), 0.20 (Mg2+), 0.42 (Ca2+), 0.72 (Sr2+), 1.04 (Al3+), and 1.17 (La3+). To the right region of Rt on the curves, the absorption values remained almost constant, which meant that no insoluble hybrids precipitated under the test conditions. The sequence of the Rt value was consistent with the preliminary results of the directly visualized observation in Fig. 3 about the onset ratio of sediment formation. In other words, Rt can be regarded as the starting point for metal cations inducing Fol to produce insoluble hybrids and also indicates the inducible ability of various metal cations to drive the further assembly of Fol. Since it was validated that Fol were coordinated as water-insoluble and disordered microstructures under relatively high concentrations of various metal chlorides, we came up with a reasonable speculation: the complexes will still be formed and exist in ternary solutions, even as a “locally ordered microstructure”, at a lower concentration of various metal chlorides with the ratio exceeding Rt. This is also of potential importance where it is desirable to harvest novel water-soluble Mn+@Fol coordination complexes. In a typical approach for facile fabricating diverse and water-soluble Mn+@Fol complexes, MCln with concentration no more than that for Rt was individually added drop by drop into Fol solution at a specific concentration under room temperature. After reaction, the resultant solution was dialyzed to obtain the purified product. Our strategy had considerable merits, such as facile and mild reaction condition, versatility of reaction method, diversity of product, simplicity of after-treatment, and almost no water-insoluble byproduct.Although the four features in the FTIR spectra of Mn+@Fol remained constant in comparison to Fol (Fig. 6), the complexation between Fol and Mn+ led to clear changes in the IR absorption properties. The new and weak absorption peaks were located at 1220–1280 cm−1 and assigned to metal and oxygen (M−O) vibration bands (Fig. 6 and Table 2). It is consistent with the phenomena observed for other metal hybrids that the complexation would induce a stronger absorption band for M−O stretching.32,44,45 Another new peak at about 1710–1720 cm−1 implied the existence of oxygenous groups such as carboxylic, carbonyl, and/or hemi-acetal structure (Table 2),39 which may be caused by the oxidation effect of metal cation catalysis. In addition, C–OH stretching vibration bands were also observed to slightly shift toward high wavenumbers relative to Fol (Table 2). The blue shift of C–OH stretching in combination with the observed M−O vibration bands revealed the coordination formation of Fol with various metal ions.
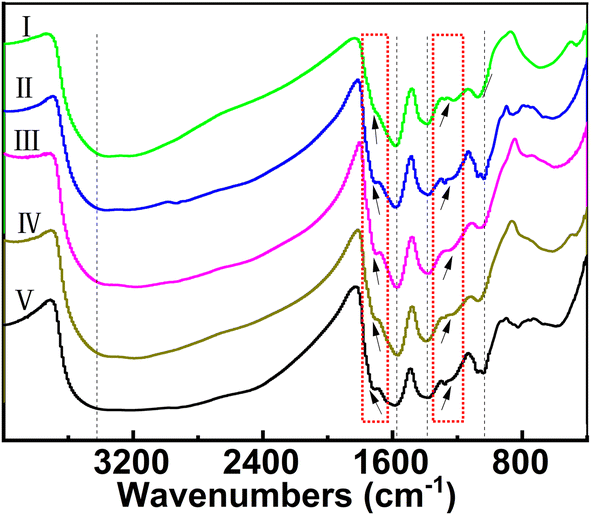 | ||
| Fig. 6 Comparative FTIR spectra of Mg2+@Fol (I), Ca2+@Fol (II), Sr2+@Fol (III), La3+@Fol (IV), and Al3+@Fol (V). | ||
| Sample | FTIR data | XPS data | Zeta potential | |||
|---|---|---|---|---|---|---|
| νCO (cm−1) |
νM–O (cm−1) | νC–OH (cm−1) | O1s binding energies of fitting C–OH/C–O–C/C–O–M (eV) | The ratio of C/Mn+ | (mV) | |
| Mg2+@Fol | 1720 | 1280, 1220 | 1070 | 531.30/532.50/533.46 | 60:1.40 |
> +200 |
| Ca2+@Fol | 1710 | 1280, 1220 | 1070, 1040 | 531.30/532.38/533.42 | 60:0.94 |
+123.2 |
| Sr2+@Fol | 1710 | 1230 | 1070 | 531.30/532.29/533.52 | 60:0.85 |
+150.5 |
| La3+@Fol | 1710 | 1280, 1220 | 1080 | 531.30/532.56/533.54 | 60:1.05 |
+166.5 |
| Al3+@Fol | 1720 | 1280, 1230 | 1080, 1040 | 531.30/532.18/533.19 | 60:2.32 |
+135.7 |
| Fol | — | — | 1045 | 531.30/532.62/536.01 | 60:2.83 |
−76.4 |
XPS investigation was carried out so as to provide valuable insights into elements and their chemical states. From the full survey spectra (Fig. 7(a1)–(e1)), the photoelectron peaks of C (285 eV) and O elements (531 eV) were always observed in Mn+@Fol complexes, and the corresponding metal elements were individually detected. For example, the high-resolution Mg1s spectrum is presented in Fig. 7(a2) with the Mg1s peak at a binding energy of 1303.85 eV. Furthermore, the peaks of Ca2p (346.98 eV), Sr3d (133.48 eV), La2p (838.08 eV), and Al2p (74.68 eV) are always present in the high-resolution spectra of the corresponding metal for other complexes (Fig. 7(b2)–(e2)). These disclosed that Mn+ with Fol can coexist in the tested samples. In addition, it should be noted that the Na1s peaks from the XPS survey spectra of Mg2+@Fol, Ca2+@Fol, Sr2+@Fol, La3+@Fol, and Al3+@Fol eventually disappeared, as shown in Fig. 7(a1)–(e1). This suggested that divalent and trivalent metal cations had stronger competitive affinity to be adsorbed and coordinate themselves to Fol after expelling and consequently replacing the remaining Na+ in Fol. The phenomena had a close similarity with the report on competitively coexisting metal ions with fullerenols to provide an indication of the relative efficacy of metal cations binding fullerenols.31 The disappearance of the Na1s peak can indirectly indicate whether the investigated metal cations and Fol were coordinated to produce the corresponding Mn+@Fol complexes. The atomic ratio of C/Mn+ was calculated by XPS to be 60:1.40 (Mg2+@Fol), 60:0.94 (Ca2+@Fol), 60:0.85 (Sr2+@Fol), 60:1.05 (La3+@Fol), and 60:2.32 (Al3+@Fol), which revealed the composition in a limited depth for Mn+@Fol complexes.32 Among them, the ratios of C/La3+ and C/Al3+ were higher than those of the corresponding feed concentration ratios, indicating that trivalent La3+ and Al3+ were more likely to bind and enrich on the surfaces of directed-assemblies after complexation.32 On the other hand, when Na+ were replaced by other Mn+ to form Mn+@Fol complexes, the high-resolution O1s spectra were fitted into three peaks for different oxidation states (Fig. 7(a3)–(e3)). The positions of C–OH and C–O–C were almost unchanged relative to those of Fol (Fig. 1c). However, the metal-oxide anion peaks (C–O–M) were shifted to lower energy region (Fig. 7(a3)–(e3)). It is consistent with the results reported in the previous literature.31 Thus, the overall XPS results implied that cationic Mn+ made bonds with polyanionic Fol to produce the novel water-soluble Mn+@Fol coordination complexes.
 | ||
| Fig. 7 XPS survey spectra of (a1) Mg2+@Fol, (b1) Ca2+@Fol, (c1) Sr2+@Fol, (d1) La3+@Fol, and (e1) Al3+@Fol, and their high-resolution XPS spectra of the core level of the corresponding metal ions (a2–e2) and O1s (a3–e3). For O1s spectra, solid black lines are the experimental data, dotted red lines are the deconvoluted spectra, and solid red lines are the fitted oxygen peaks for various oxidation states. | ||
The TGA curves of Mn+@Fol also displayed the degradation processes for weight loss of bound water and dehydroxylation similar to those of Fol; however, the contents of bound water are always significantly higher than that of Fol after complexation (Fig. 8a–e). Moreover, unlike the TG curve of Fol, Mn+@Fol always experienced drastic and continuous decompositions above 600 °C (Fig. 8a–e), which is interpreted in terms of a potential catalytic effect of multivalent metal cations on the degradation of Fol carbon cages.32
 | ||
| Fig. 8 DTG (I) and TG (II) curves of (a) Mg2+@Fol, (b) Ca2+@Fol, (c) Sr2+@Fol, (d) La3+@Fol, (e) Al3+@Fol, and (f) XRD patterns for Mg2+@Fol (I), Ca2+@Fol (II), Sr2+@Fol (III), La3+@Fol (IV), and Al3+@Fol (V). | ||
The zeta potential of Fol in deionic water was −76.4 mV, while the Mn+@Fol assemblies displayed positive charge (Table 2). The tested zeta potential changed from negative to positive before and after forming the complexes, providing evidence that cationic Mn+ were bonded to polyanionic Fol aggregates.33 The crystallinity of Mn+@Fol complexes was detected by XRD and shown in Fig. 8f, where the halo at 22.765° is the same as that for Fol (JCPDS 50-0926), indicating their non-crystallizable macroscopic structures.24
3.7 Nanoscale-ordered microstructures of water-soluble Mn+@Fol coordination complexes
Besides the optical, thermal, electron, and photoelectron properties, the directed-assembled morphologies of Mn+@Fol also changed greatly by complexation. As shown in Fig. 9(f1) and (f2), TEM observations displayed nano-sized particle-aggregated microstructure and monomodal and near-ellipsoidal morphology without a smooth surface for the as-synthesized Fol sample, which was statistically analyzed to have an average size of 25 nm with distribution in the range of 20–30 nm. After complexation, Mg2+@Fol nanoparticles maintained an ellipsoid shape similar to Fol with increased average size of 40 nm (Fig. 9(a1) and (a2)), while Ca2+@Fol appeared as irregular and uneven nanoparticles with slightly higher average size of 35 nm (Fig. 9(b1) and (b2)). With the complexation of Sr2+ to Fol, the particle size of Sr2+@Fol significantly enlarged to about 60–70 nm, the surface of the nanoparticles gradually became rough, and the shadow in the local area of the TEM image deepened, indicating the successful Sr2+-mediated modification of the Fol assembly (Fig. 9(c1) and (c2)). La3+@Fol grew as larger assemblies with approximately olive-shaped morphology (Fig. 9(d1) and (d2)), which was different from that of the Fol and M2+@Fol complexes. We described the direction of growth in accordance with the similar one-dimensional rod-like directed-assemblies to display irregular size, up to 65 nm in diameter and up to 270 nm in length. Al3+@Fol exhibited the strongest clustering trend as well-dispersed near-spherical nanoparticles with the highest average size of 370 nm (Fig. 9(e1) and (e2)). | ||
| Fig. 9 Comparative TEM (a1–f1, a2–f2) and HRTEM (a3–f3) photos of the directed-assemblies formed by (a) Mg2+@Fol, (b) Ca2+@Fol, (c) Sr2+@Fol, (d) La3+@Fol, (e) Al3+@Fol, and (f) Fol aqueous solution microfilms. Insets in a3–f3 are the microdiffraction patterns. | ||
Although the crystallinity of Mn+@Fol complexes was detected by XRD to indicate their amorphous structures, the degree of order in the nanometer length scale and microstructures of these directed-assemblies was further proved by HRTEM. The HRTEM image and SAED pattern of Fol showed a typically disordered microstructure without crystallinity (Fig. 9(f3)), which is consistent with the previously reported results.46 Nevertheless, as shown in Fig. 9(a3)–(d3), some degree of crystallinity is always observed in the HRTEM mode, which indicated the locally ordered microstructures in the nanoscale after complexation. In general, the SAED patterns of Mn+@Fol complexes displayed a weak spot or ring diffraction patterns (insets of Fig. 9(a3)–(d3)), which can be taken as a further indication of a rather “locally ordered microstructure” formation of the water-soluble Mn+@Fol complex as nanocrystalline. Mg2+@Fol nanoparticles showed a tiny weak signal for a slightly ordered microstructure with almost no morphological changes; Ca2+@Fol and Sr2+@Fol exhibited locally-ordered microstructures accompanied by slight changes in the morphology and size, while the most ordered nanoscale areas and morphological change from zero to one dimensional aggregate were confirmed in the La3+@Fol sample. The observed crystallinity should be owing to the further aggregation of Fol, preferentially with metal ions and not themselves.
The exception was the Al3+@Fol complex, which produced no diffraction spots or rings, implying that it did not have a nanoscale microstructure. As discussed elsewhere, the insoluble hybrids were observed at relatively high concentration of salts, indicating that random cross-linked complexation resulted in disordered arrangement.31 In these cases, salt treatments with higher concentrations were usually better for unordered microstructural cross-linkings. The finding achieved by the TEM experiments is in fact the “locally ordered microstructure in nanoscale” approach toward water-soluble Mn+@Fol formation under chlorides at a lower concentration (i.e., below that of Rt), where the degree of random crosslinking is insufficient. The disordered microstructure of Al3+@Fol may be due to its strongest directed assembly tendency to result in higher degree of random crosslinking relative to other Mn+@Fol.
3.8 Complexation pathway of metal ion-induced aggregation of derivatized fullerenes
The mechanism of complexation of metal-contained ions and water-soluble derivatives of fullerenes consists of two stages: electrostatic attachment and coordination, where the mode of actions are given in Fig. 10. The electrostatic action was evaluated to play a crucial role for generating metal ion–fullerene complexes, where the surface charge of water-soluble fullerene-based ligands is a critical factor for the complexation in solution through the interaction of electrostatic attraction to strongly attract the metal ions onto ligands.40 Only the negatively-charged fullerene derivatives, especially fullerenols, can attract the bare metal cations to easily form metal cation-fullerenol complexes in water. Fol as the polyanion can adsorb various Mn+ on the surface of carbon cages through electrostatic attraction. The tested zeta potential shifted from negative to positive before and after the attachment of Mn+ (Table 2), indicating the cationization after interaction Mn+ and Fol (Fig. 10a). Furthermore, cationization caused Mn+ to approach the oxygen-contained ligands onto the surface of Fol cage and consequently to be chelated through coordination to form multidentate Mn+@Fol complexes, where the formation process was very rapid. As shown in Fig. 10a, under the condition of the Fol:Mn+ ratio more than Rt, the contents of Fol in ternary Fol–MCln–H2O systems were hardly ever changed, as confirmed by the UV-visible spectra (Fig. 6 and S8†), indicating that the resultant Mn+@Fol complexes were water-soluble. Under the condition of the Fol:Mn+ ratio no more than Rt with the increase in the metal–cation concentration, the water-soluble Mn+@Fol complexes were cross-linked through further coordination to larger particles and ultimately the co-precipitation to form insoluble hybrids. Comparing the usage of tri-valent metal cations with di-valent metal cations, the morphology of the resulting water-soluble nanocluster complexes La3+@Fol and Al3+@Fol had a larger particle size and a more disordered microstructure relative to those of Mg2+@Fol, Ca2+@Fol, and Sr2+@Fol (Fig. 9); meanwhile, the results of the inducibility for the directed assembly of Fol also validated the efficiency of tri-valent cations superior to divalent cations (Fig. 5 and Table 2). These implied that higher valent ions were more likely to induce the self-assembly of Fol for the formation of complexes with a higher degree of crosslinking via electrostatic and coordination dual-actuation interactions (Fig. 10a).
 | ||
| Fig. 10 The scheme of the mode of actions to drive metal-containing ions and water-soluble derivatives of fullerenes for complexation. (a) MCln–Fol solutions; (b) HgCl2–IFQA solution; (c) HgCl2–NaCl–IFQA solution. | ||
On the other hand, cationic IFQA did not bind metal cations that lead to precipitation due to the interaction of electrostatic repulsion, which was opposite to Fol (Fig. 4, S6 and S7†). For example, the electrostatic repulsion between IFQA and Hg2+ drove them away from each other, resulting in the inability of coordination to combine into complexes (Fig. 10b). However, after adding more than 500-fold equivalent amount of HgCl2 into IFQA, the unexpected generation of the HgCl2–IFQA complex could be mainly driven by coordination (Fig. 10b), which is attributed to the weak electrolyte property of mercuric chloride that results in it being dominantly formed as HgCl2 molecules rather than Hg2+ ions (<3%) in aqueous solutions.42,43 Moreover, the insoluble hybrids were observed to fall out of solution when IFQA was added into the solution containing metal anions. As shown in Fig. 11, IFQA was treated using 20-fold equivalent amount of HgCl2 without a noticeable change in the UV-absorption value and color of the supernatant in comparison to IFQA. Interestingly, it has a distinct reduction when it was treated upon the addition of 20-fold HgCl2 and 200-fold NaCl. The sufficient difference in the color of the supernatants made it possible to observe the formation of the brown-colored precipitate after adding NaCl. This phenomenon can also continue to be explained by attractive static-electric force and coordination actions (Fig. 10c). The sufficiently stable [HgCl3]− and [HgCl4]2− can be simultaneously formed in the presence of an excess amount of Cl− in HgCl2 solution,43 which turned into electrostatic attractions between anions containing mercury and IFQA. These anions will be attached to cationic IFQA and subsequently, it can possibly lead to the generation of the water-insoluble [HgCl3]−–IFQA and/or [HgCl4]2−–IFQA coordinated complexes. In this case, electrostatic attraction continued to play a significant role in the fabrication of IFQA-coordination complexes. IFQA, as cationic fullerene ligands, can also chelate metal anions to form metal ion–fullerene complexes, but the prerequisite is that free metal cations must firstly be converted into the anionic form. Conversely, IFQA cannot directly form complexes with free metal cations in solution, or rather, metal cations cannot induce the further assembly of IFQA. It indirectly suggested that electrostatic attraction also mediated the further assembly of Fol by metal cations. It can be evaluated from the relevance and competence with the surface charge and structure of polyhydroxy functional groups that Fol can provide a good opportunity for coordinating “naked” metal cations to discover novel metal ion–fullerenol complexes.
 | ||
| Fig. 11 The photographs of aqueous solution of IFQA (0.2 mM) and IFQA in the presence of HgCl2 (4.0 mM) without and with NaCl (40 mM) in plastic centrifuge tubes after centrifuging, and the UV-visible spectra of the 5-fold dilution for their supernatants were recorded. | ||
The best condition for the fabrication of water-soluble Mn+@Fol coordination complexes appeared when the concentration ratio was close to Rt. This, in principle, led to better discrimination for the formation of insoluble hybrids from water-soluble complexes and a subsequent increase in the preparation efficiency to harvest a water-soluble product. Thus, the judicious choice of the ratio of Fol and appropriate salts, as close to, but not exceeding, Rt as possible, resulted in the invention of the procedures, allowing the suppression of the formation of insoluble cross-linking hybrids and leaving water-soluble and locally-ordered microstructural Mn+@Fol complexes as the majority of products. Furthermore, coordination effects were directly validated in XPS spectra showing the metal cation and oxygen binding feature (Fig. 7(a3)–(e3)). Thus, the most likely way for water-soluble Mn+@Fol formation is dual-actuation mode by electrostatic and coordination actions. Such Mn+@Fol complexes with variable morphologies may be stable enough to survive in normal aqueous environments.
At the present stage, the multi-valent metallic cation-induced self-assembly of Fol to obtain the water-soluble Mn+@Fol complexes was demonstrated. Some paramagnetic metal ions, such as Gd3+, Fe3+, and Mn2+, could also direct the assembly of Fol to produce paramagnetic NPs, which will be an interesting candidate as the next generation of fullerene-based MRI contrast agents with high relaxivity. The preparation and testing relaxation property of water-soluble and paramagnetic Gd3+@Fol, Mn2+@Fol, and Fe3+@Fol are ongoing projects. Furthermore, their structure–function relationship will be reported separately in the near future as well to help us understand their relaxivity mechanism for potential MRI applications in biomedical fields.
4 Conclusion
The foregoing results demonstrated that the modulation of the ratio of Fol to metal cations enabled the significant reduction of the inevitable complexation to insoluble and random crosslinking hybrids and stabilization of Mn+@Fol in aqueous solution, leading to a new class of water–soluble metal ion–nanocluster complexes. This idea opens a new opportunity for developing a preparation strategy to fabricate the water–soluble metal cation–fullerenol coordination complexes with merits of versatility, simplicity, convenience, high efficiency, and less loss of time. In future studies, finding new anionic water–soluble fullerene-based ligands will continue to be the core of fabricating water–soluble metal cation–fullerene complexes for potential applications in biomedical fields.Author contributions
He R. conceived the study, carried out some experiments, and wrote the manuscript; Fan C. carried out the experiments and prepared some parts of the manuscript; Liang Q. took part in some of the experiments; Wang Y. and Gao Y. contributed to nanomaterial preparation; Wu J. analyzed the data; Wu Q. interpreted the results, contributed to nanomaterial characterization and manuscript preparation, and supervised the project; Tai F. designed the experiments and supervised the project.Conflicts of interest
The authors declare that they have no conflict interest.Acknowledgements
The authors acknowledge the financial supports of the Natural Science Foundation of Henan Province, China (222300420461, 162300410154), the Key Scientific Research Project of Henan Higher Education Institutions of Henan Province, China (22B210003), and the Special Innovation Project and PhD Start-up Fund of Henan Agricultural University, China (KJCX2020C02, 30500458).References
- Z. Wang, Y. Zou, Y. W. Li and Y. Y. Cheng, Small, 2020, 16, 1907042 CrossRef CAS PubMed.
- J. J. Zhou, H. Y. Han and J. W. Liu, Nano Res., 2022, 15, 71–84 CrossRef CAS.
- M. X. Wu and Y. W. Yang, Adv. Mater., 2017, 29, 1606134 CrossRef PubMed.
- H. L. Zhang, A. Li, K. Li, Z. P. Wang, X. C. Xu, Y. X. Wang, M. V. Sheridan, H. S. Hu, C. Xu, E. V. Alekseev, Z. Y. Zhang, P. Yan, K. C. Cao, Z. F. Chai, T. E. Albrecht-Schonzart and S. Wang, Nature, 2023, 616, 482–487 CrossRef CAS PubMed.
- J. G. Du and G. Jiang, Dalton Trans., 2022, 51, 5118–5126 RSC.
- R. Sitko, M. Musielak, M. Serda, E. Talik, A. Gagor, B. Zawisza and M. Malecka, Sep. Purif. Technol., 2021, 277, e119450 CrossRef.
- Y. M. Lin, Z. Z. Qiu, D. Z. Li, S. Ullah, Y. Hai, H. L. Xin, W. D. Liao, B. Yang, H. S. Fan, J. Xu and C. Z. Zhu, Energy Storage Mater., 2018, 11, 67–74 CrossRef.
- L. Jiao, Y. Wang, H. L. Jiang and Q. Xu, Adv. Mater., 2018, 30, 1703663 CrossRef PubMed.
- W. Xu, D. Xiang, J. J. Xu, Y. L. Ye, D. Qiu and Z. Z. Yang, Polym. Chem., 2021, 12, 172–176 RSC.
- H. Hoelzel, S. L. Lee, K. Y. Amsharov, N. Jux, K. Harano, E. Nakamura and D. Lungerich, Nat. Chem., 2023, 15, 1444–1451 CrossRef CAS PubMed.
- Y. Shu, Q. Y. Ye, T. Dai, Q. Xu and X. Y. Hu, ACS Sens., 2021, 6, 641–658 CrossRef CAS.
- Y. Zhang, C.-Y. Sun and L. Lin, RSC Adv., 2023, 13, 23396–23401 RSC.
- X. Q. Sun, Y. Zhang, J. Q. Li, K. S. Park, K. Han, X. W. Zhou, Y. Xu, J. Nam, J. Xu, X. Y. Shi, L. Wei, Y. L. Lei and J. J. Moon, Nat. Nanotechnol., 2021, 16, 1260–1270 CrossRef CAS PubMed.
- H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162–163 CrossRef CAS.
- T. T. Wang and H. P. Zeng, Chin. J. Org. Chem., 2008, 28, 1303–1312 CAS.
- A. A. Popov, S. F. Yang and L. Dunsch, Chem. Rev., 2013, 113, 5989–6113 CrossRef CAS PubMed.
- J. J. Wang, Y. R. Yao, S. T. Yang, X. Y. Zhou, A. Yu, P. Peng and F. F. Li, Nanomaterials, 2022, 12, e1314 CrossRef.
- T. Habicher, J. F. Nierengarten, V. Gramlich and F. Diederich, Angew. Chem., Int. Ed., 1998, 37, 1916–1919 CrossRef CAS.
- J. Fan, Y. Wang, A. J. Blake, C. Wilson, E. S. Davies, A. N. Khlobystov and M. Schroder, Angew. Chem., Int. Ed., 2007, 46, 8013–8016 CrossRef CAS.
- C. H. Chen, A. Aghabali, C. Suarez, M. M. Olmstead, A. L. Balch and L. Echegoyen, Chem. Commun., 2015, 51, 6489–6492 RSC.
- P. Peng, F. F. Li, V. S. P. K. Neti, A. J. Metta-Magana and L. Echegoyen, Angew. Chem., Int. Ed., 2014, 53, 160–163 CrossRef CAS PubMed.
- A. Kraft, P. Roth, D. Schmidt, J. Stangl, K. Muller-Buschbaum and F. Beuerle, Chem.–Eur. J., 2016, 22, 5982–5987 CrossRef CAS.
- X. Y. Zhang, H. L. Cong, B. Yu and Q. Chen, Mini-Rev. Org. Chem., 2019, 16, 92–99 CrossRef CAS.
- M. J. Chen, K. Y. Yin, G. P. Zhang, H. Z. Liu, B. Ning, Y. Y. Dai, X. J. Wang, H. G. Li and J. C. Hao, ACS Appl. Bio Mater., 2020, 3, 358–368 CrossRef CAS PubMed.
- K. N. Semenov and N. A. Charykov, Russ. J. Phys. Chem., 2012, 86, 1636–1638 CrossRef CAS.
- K. N. Semenov, N. A. Charykov, V. A. Keskinov, A. S. Kritchenkov and I. V. Murin, Ind. Eng. Chem. Res., 2013, 52, 16095–16100 CrossRef CAS.
- V. A. Keskinov, K. N. Semenov, T. S. Gol'tsov, N. A. Charykov, N. E. Podol'skii, A. V. Kurilenko, Z. K. Shaimardanov, B. K. Shaimardanova and N. A. Kulenova, Russ. J. Phys. Chem., 2019, 93, 2555–2558 CrossRef CAS.
- N. A. Charykov, V. A. Keskinov, K. A. Tsvetkov, A. Kanbar, K. N. Semenov, L. V. Gerasimova, Z. K. Shaimardanov, B. K. Shaimardanova and N. A. Kulenova, Processes, 2021, 9, 349 CrossRef CAS.
- G. O. Yur'ev, V. A. Keskinov, K. N. Semenov and N. A. Charykov, Russ. J. Phys. Chem., 2017, 91, 797–799 CrossRef.
- R. Anderson and A. R. Barron, J. Am. Chem. Soc., 2005, 127, 10458–10459 CrossRef CAS PubMed.
- J. Heimann, L. Morrow, R. E. Anderson and A. R. Barron, Dalton Trans., 2015, 44, 4380–4388 RSC.
- J. R. Li, M. J. Chen, S. D. Hou, L. Y. Zhao, T. Y. Zhang, A. N. Jiang, H. G. Li and J. C. Hao, Carbon, 2022, 191, 555–562 CrossRef CAS.
- M. Seke, D. Petrovic, M. L. Borovic, I. Borisev, M. Novakovic, Z. Rakocevic and A. Djordjevic, J. Nanopart. Res., 2019, 21, 239 CrossRef CAS.
- A. V. Zhilenkov, E. A. Khakina, P. A. Troshin, I. F. Karimov and D. G. Deryabin, Pharm. Chem. J., 2019, 53, 312–317 CrossRef CAS.
- Y. J. Liu, T. T. Wang, J. H. Cao, Z. F. Zang, Q. N. Wu, H. Z. Wang, F. J. Tai and R. He, J. Agric. Food Chem., 2019, 67, 13509–13517 CrossRef CAS.
- L. O. Husebo, B. Sitharaman, K. Furukawa, T. Kato and L. J. Wilson, J. Am. Chem. Soc., 2004, 126, 12055–12064 CrossRef CAS.
- F. F. Wang, N. Li, D. Tian, G. F. Xia and N. Xiao, ACS Nano, 2010, 4, 5565–5572 CrossRef CAS PubMed.
- F. Y. Liu, F. X. Xiong, Y. K. Fan, J. Li, H. Z. Wang, G. M. Xing, F. M. Yan, F. J. Tai and R. He, J. Nanopart. Res., 2016, 18, 338 CrossRef.
- J. Wan, J. X. Ma, Y. Y. Zhang, Y. X. Xia, L. Hong and C. Yang, New J. Chem., 2021, 45, 17660–17666 RSC.
- D. Fati, V. Leeman, Y. V. Vasil'ev, T. Drewello, B. Leyh and H. Hungerbuhler, J. Am. Soc. Mass Spectrom., 2002, 13, 1448–1458 CrossRef CAS PubMed.
- F. J. Tai, S. Wang, B. S. Liang, Y. Li, J. K. Wu, C. J. Fan, X. L. Hu, H. Z. Wang, R. He and W. Wang, J. Nanobiotechnol., 2022, 20, 15 CrossRef CAS PubMed.
- J. G. Speight, Lange's Handbook of Chemistry, McGraw-Hill, New York, 16th edn, 2005, pp. 1358–1362 Search PubMed.
- S. R. Labhade and K. R. Labhade, Asian J. Chem., 2021, 33, 2069–2072 CAS.
- Z. C. Xin, Y. T. Shen, H. Hao, L. N. Zhang, X. X. Hu and J. Wang, Colloids Surf., B, 2022, 218, 112776 CrossRef CAS PubMed.
- J. Liu, R. L. Zhu, T. Y. Xu, M. W. Laipan, Y. P. Zhu, Q. Zhou, J. X. Zhu and H. P. He, J. Environ. Sci., 2018, 64, 1–9 CrossRef.
- B. Sitharaman, R. D. Bolskar, I. Rusakova and L. J. Wilson, Nano Lett., 2004, 4, 2373–2378 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra07725j |
| ‡ R. H. and C. F. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |