
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFerrocenylselenoether and its cuprous cluster modified TiO2 as visible-light photocatalyst for the synergistic transformation of N-cyclic organics and Cr(VI)†
Zhuo Yanga,
Jinshan Wanga,
Aimin Lib,
Chao Wangac,
Wei Ji*a,
Elísabet Pires c,
Wenzhong Yanga and
Su Jing*a
c,
Wenzhong Yanga and
Su Jing*a
aSchool of Chemistry and Molecular Engineering, Nanjing Tech University, Nanjing 211816, China. E-mail: buffycomji@njtech.edu.cn; sjing@njtech.edu.cn
bState Key Laboratory of Pollution Control and Resource Reuse, School of the Environment, Nanjing University, Nanjing 210023, China
cInstituto de Síntesis Química y Catálisis Homogénea, CSIC-Universidad de Zaragoza, Pedro Cerbuna 12, E-50009 Zaragoza, Spain
First published on 3rd January 2024
Abstract
In this study, fcSe@TiO2 and [Cu2I2(fcSe)2]n@TiO2 nanosystems based on ferrocenylselenoether and its cuprous cluster were developed and characterized by X-ray photoelectron spectroscopy (XPS), high-resolution transmission electron microscopy (HR-TEM), energy dispersive X-ray spectroscopy (EDX), and electron paramagnetic resonance (EPR). Under optimized conditions, 0.2 g L−1 catalyst, 20 mM H2O2, and initial pH 7, good synergistic visible light photocatalytic tetracycline degradation and Cr(VI) reduction were achieved, with 92.1% of tetracycline and 64.5% of Cr(VI) removal efficiency within 30 minutes. Mechanistic studies revealed that the reactive species ˙OH, ˙O2−, and h+ were produced in both systems through the mutual promotion of Fenton reactions and photogenerated charge separation. The [Cu2I2(fcSe)2]n@TiO2 system additionally produced 1O2 from Cu+ and ˙O2−. The advantages of the developed nanosystems include an acidic surface microenvironment provided by Se⋯H+, resourceful product formation, tolerance of complex environments, and excellent adaptability in refractory N-cyclic organics.
Introduction
Nitrogen-containing cyclic organics, N-cyclic organics in brief, are among the most common functional compounds in biological, pharmaceutical, and material fields.1,2 With the rapid development of industry and global consumption, the N-cyclic organics are continuously discharged into the environment.3,4 Due to their high stability and high chemical oxygen demand, it is difficult to rely solely on degradation by microorganisms under natural conditions.5 These N-cyclic organics have become major harmful pollutants in soil and aquatic ecosystems, which have toxic effects on living organisms, including teratogenesis, carcinogenesis, and mutagenesis.6–9 Simultaneously, the highly toxic and non-biodegradable Cr(VI) is one of the most coexisting toxic metals and can cause severe threats to the natural environment and public health.10–12 It is very significant to develop environmentally friendly technologies to simultaneously transform N-cyclic organics and the coexisting toxic Cr(VI) from complex ecosystems.The common methods available to treat N-cyclic organics in the environment include chemical oxidation,13–16 biodegradation17 and physical adsorption.18 As an approach of advanced oxidation processes (AOPs), Fenton chemistry has gradually shown the prominent prospect of industrial application in wastewater treatment.5,19,20 In Fenton chemistry methodology, organic pollutants can be degraded by highly oxidative hydroxyl radicals (˙OH) produced by Fe(II) and H2O2 reactions. To solve the slow Fe2+ recovery that inhibits the efficiency of Fenton chemistry, the efficient redistribution of local electrons in the catalyst system and/or the excess electrons generated under an external field play an important role.20 Recently, the design of heterogeneous photo-Fenton system based on semiconductors has witnessed rapid development.21 Ding and Wang demonstrated a nanocomposite catalyst, Fe3O4@β-CD/g-C3N4, that enriched the photoinduced electrons to promote Fe(II) regeneration on the catalyst surface.22 Zhu et al. designed the electron self-sufficient core–shell BiOCl@Fe(III)–BiOCl nanosheet photocatalyst to promote the Fe(III)/Fe(II) recycling in the photo-Fenton reaction.23 In this regard, the most reported photo-Fenton systems used inorganic iron salts as Fenton reagents, which suffer from low stability, iron sludge formation, and strict acidic pH limitation. Also, the charge-separation efficiency is another key point for enhancing the photocatalytic performance. It is imminent to develop novel photocatalytic nanoplatforms with features, such as suitable catalysts with stable active metal sites, environmental benignity, high adaptability, and resource regeneration.
Ferrocene is a sandwich-shaped organometallic compound containing ferrous ion, owning the advantages of kinetic stability, easy modification, biocompatibility, and redox capability. Ferrocenyl compounds have been confirmed as efficient Fenton reactors,24–26 and the visible absorption bands of ferrocene allow the photo-promoted Fe(II)–Fe(III) redox pair recycle through the Fenton reaction to improve ˙OH release.27,28 Alternatively, stable copper(I) clusters based on selenoethers can effectively enhance photocatalytic performance.29–31 These systematic studies have prompted us to further investigate the applications of ferrocenyl derivatives in waste water treatment. In the current study, we sensitized TiO2 with ferrocenylselenoether fcSe and its cuprous iodide cluster [Cu2I2(fcSe)2]n as heterogeneous photocatalysts (Scheme 1). Choosing Tetracycline (TC) and Cr(VI) as template reactions, the obtained fcSe@TiO2 and [Cu2I2(fcSe)2]n@TiO2 were applied in the visible light photo-Fenton simultaneous efficient transformation at neutral aqueous solution. The two nanosystems were further proved to possess excellent adaptability in the degradation of seven representative N-cyclic organic contaminants, that are, antibiotics Ciprofloxacin and imidazole, textile dyes Toluidine Blue, Methyl Orange, Methylene Blue, Malachite Green, and Basic Violet. The innovations of two photocatalytic nanosystems, fcSe@TiO2 and [Cu2I2(fcSe)2]n@TiO2 in this study are: (1) Fenton reaction inhibited the recombination of the photogenerated charges; (2) the effective recycling between ferrocene ([Fe2+(η5-C5H5)2]) and ferrocenium ([Fe3+(η5-C5H5)2]) pumped by photoexcited TiO2; (3) the incorporation of selenium atoms in nanosystems lead to an acidic microenvironment for efficient Fenton reaction in neutral aqueous solution.
 | ||
| Scheme 1 (a) In situ preparation and (b) photocatalytic transformation study of N-cyclic organics and Cr(VI) of fcSe@TiO2 and [Cu2I2(fcSe)2]n @TiO2. | ||
Experimental section
Materials
All starting materials were analytical-grade reagents and purchased from Aladdin or Source Leaf and used without further purification unless otherwise specified. TiO2 was commercial P25 (75% anatase, 25% rutile). 1,2,3-Triselena[3]ferrocenophane fcSe3 (fc = [Fe(η5-C5H4)(η5-C5H4)]) was prepared according to the literature methods.32Characterization
1H NMR spectra were recorded on a Bruker DRX spectrometer (400 MHz) at room temperature. The ESI-mass spectra were determined by a Micromass Quattro II triple-quadrupole mass spectrometer and analyzed using the MassLynx software. The single-crystal X-ray diffraction measurement was performed using a Bruker SMART APEX II CCD diffractometer. Transmission electron microscopy (TEM, Hitachi HT7700), high-resolution transmission electron microscopy (HRTEM, JEOL 2100F), and energy-dispersive X-ray (EDX) spectroscopic analysis were performed to gain the structure information. The elemental analysis and oxidation state study of the samples were carried out by X-ray photoelectron spectroscopy (XPS, PHI 5000 VersaProbe), and the binding energies were corrected for specimen charging effects using the C 1s level at 284.8 eV as the reference. UV-vis diffuse reflectance spectroscopy (UV-VIS DRS) was performed on a PerkinElmer Lambda 950 UV/vis/nir spectrophotometer with BaSO4 as a reference. FT-IR spectroscopy was performed using a Fourier transform infrared spectrometer with KBr as a reference. Inductively coupled plasma emission spectrometer (ICP) experiments were performed on an Agilent 7900 ICP-MS. Brunauer–Emmett–Teller (BET) results were obtained on an ASAP 2460. Electron paramagnetic resonance (EPR) analysis was performed using Bruker A300. Malvern Zetasizer Nano ZS90 was used to determine the zeta potentials and the hydrodynamic diameters of liposomes.The visible light source is a Xenon Lamp MC-PF300C coupled with a power meter (MC-PM100C) for photo-Fenton reaction, purchased from Beijing Merry Change Technology Co., Ltd, and equipped with a 420 nm filter to filter out UV light. Gaseous products in photocatalytic degradation were measured by gas chromatography (GC-9790plus, FULI) with a flame ionization detector (FID) and a thermal conductivity detector (TCD). The liquid products were measured by ion chromatography (Dionex Aquion Rfic), LC-MS (MSQ PLUS/U3000), and HPLC (Agilent 1260).
Preparation of L0, fcSe, fcSe@TiO2, and [Cu2I2(fcSe)2]n@TiO2
As shown in Scheme 2a, 1,1′-bis((4-carboxybenzyl)seleno)ferrocene fcSe was synthesized via a similar route described by us previously.27 The detailed synthetic procedures, 1H NMR and MS spectra of the compounds, crystal structure and crystal data and structure refinement details of L0 and fcSe are provided in ESI (Fig. S1–S5 and Tables S1, S2†). The data for L0 (CCDC 2216502) is available from the Cambridge Crystallographic Data Center via https://www.ccdc.cam.ac.uk/. | ||
| Scheme 2 Synthetic routes of (a) fcSe, (b) fcSe@TiO2 and [Cu2I2(fcSe)2]n@TiO2. | ||
The stepwise in situ self-assembly of fcSe and [Cu2I2(fcSe)2]n on the TiO2 P25 surface as shown in Scheme 2b.
Photocatalytic activity evaluation
In the photocatalytic degradation experiment, TC (20 mg L−1), Ciprofloxacin (20 mg L−1), Toluidine Blue (20 mg L−1), Methyl Orange (20 mg L−1), Methylene Blue (20 mg L−1), Malachite Green (20 mg L−1), Basic Violet (20 mg L−1), and imidazole (1 g L−1) were selected as model N-cyclic organic pollutants to represent the antibiotics and textile dyes in the industrial wastewater, respectively. The initial pH of the reaction system was adjusted by 1 mol L−1 NaOH or HCl solution.In the photocatalytic reaction, 0.010 g catalyst was added into 50 mL aqueous solution containing the target N-cyclic organic. After stirring for 40 min in the dark at room temperature, 100 μL of 30 wt% H2O2 was added, and at the same time, the visible light photo-Fenton reaction was started. During the photo-Fenton reaction, 3.0 mL sample was collected from the suspension at an interval of 5 min and immediately filtered using 0.45 μm membrane filters.
The concentration of most of the target N-cyclic organics was detected by the UV-vis absorption intensity, and the detection wavelengths for TC, CIP, TB, MO, MB, MG and BV were 357 nm, 279 nm, 640 nm, 463 nm, 664 nm, 617 nm, and 585 nm, respectively. The concentration of imidazole was detected by HPLC with ZORBAX Eclipse Plus C18 column and acetonitrile as eluent (0.8 ml min−1 flow rate).
The degradation efficiency was calculated using eqn (1):
 | (1) |
The apparent rate constant k of catalysts was estimated by a pseudo-first-order kinetics equation (eqn (2)):
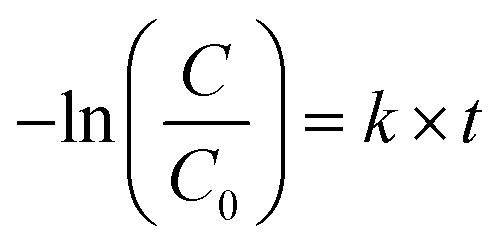 | (2) |
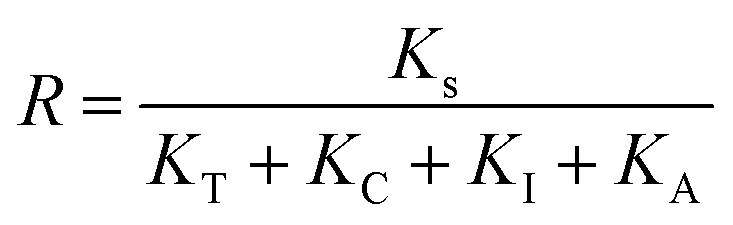 | (3) |
The photoreduction of Cr(VI) to Cr(III) was analyzed using a pretreatment method of solvent extraction separation and enrichment combined with ICP analyses.
The conversion rate of CO and HCOOH was calculated by equation (eqn (4)):
 | (4) |
Results and discussion
Characterizations of fcSe@TiO2 and [Cu2I2(fcSe)2]n@TiO2
The X-ray photoelectron spectra (XPS) confirmed the successful assembly of fcSe and [Cu2I2(fcSe)2]n on the surface of TiO2 P25 (Fig. 1a). In the high-resolution XPS Spectra of fcSe@TiO2, stable Fe 2p state (Fig. 1b) was obtained with the binding energy values at 708.2 and 721.2 eV, revealing Fe2+ species of the ferrocene unit; the Se 3d was fitted to the peak with a binding energy value of 55.9 eV (Fig. 1c); the energy separation 5.7 eV of the Ti 2p doublet peaks, Ti 2p3/2 (458.5 eV) and Ti 2p1/2 (464.2 eV), perfectly agrees with the characteristic signals for TiO2 Ti4+ state described in literature works (Fig. S6†).29,34 In the high-resolution XPS spectra of [Cu2I2(fcSe)2]n@TiO2, as well as the similar Fe 2p, Se 3d, and Ti 2p spectra, the confirmed Cu 2p spectrum (Cu 2p3/2 933.1 eV and Cu 2p1/2 953.8 eV) (Fig. 1d) and I 3d spectrum (I 3d5/2 620.1 eV and I 3d3/2 631.0 eV) (Fig. 1e) indicated the successful incorporation of CuI.29,34,35 Transmission electron microscopy (TEM) images suggest that the construction of fcSe@TiO2 and [Cu2I2(fcSe)2]n@TiO2 maintained the overall dimensions or morphology of TiO2 nanoparticles with a diameter of 20 nm (Fig. S7, S8† and 2a–c). The high-resolution transmission electron microscopy (HR-TEM) of [Cu2I2(fcSe)2]n@TiO2 in Fig. 2a–c shows a coated layer with approximately 5.00 nm thickness on the TiO2 surface; well-defined lattice fringes in substrate TiO2 with the lattice spacing was estimated to be ca. 0.325 nm, these are equal to the lattice parameter in the (110) facet of anatase TiO2.36 [Cu2I2(fcSe)2]n@TiO2 was also analyzed using Scanning TEM (STEM) coupled with energy dispersive X-ray spectroscopy (EDX) to confirm the presence of Ti, Cu, Fe, I, and Se, which suggests the successful preparation of core–shell nanoparticles [Cu2I2(fcSe)2]n@TiO2 (Fig. 2d and 2e–i). The quantitative mapping analysis manifested that the molar ratio of Cu/Fe = 2.0 far exceeded the bulk molar ratio of Cu/Fe = 1.0 measured by ICP, suggesting that the content of Cu decreased from the exterior to the interior. In the FTIR spectra of fcSe@TiO2 and [Cu2I2(fcSe)2]n@TiO2 (Fig. S9†), the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration is at 1677 cm−1, as well as asymmetric and symmetric stretching bands of the carboxylate anion (COO−) due to the splitting of carboxylate groups when complexed with surface Ti centers, υas at 1600 cm−1 and υs at 1424 cm−1.37 The value of Δυa–s = 176 cm−1 in the current two nanosystems suggests fcSe is chemically adsorbed on the TiO2 surface through the bidentate bridging form,38 which further facilitates electron transfer between ferrocene and TiO2 surface.39–41
O stretching vibration is at 1677 cm−1, as well as asymmetric and symmetric stretching bands of the carboxylate anion (COO−) due to the splitting of carboxylate groups when complexed with surface Ti centers, υas at 1600 cm−1 and υs at 1424 cm−1.37 The value of Δυa–s = 176 cm−1 in the current two nanosystems suggests fcSe is chemically adsorbed on the TiO2 surface through the bidentate bridging form,38 which further facilitates electron transfer between ferrocene and TiO2 surface.39–41
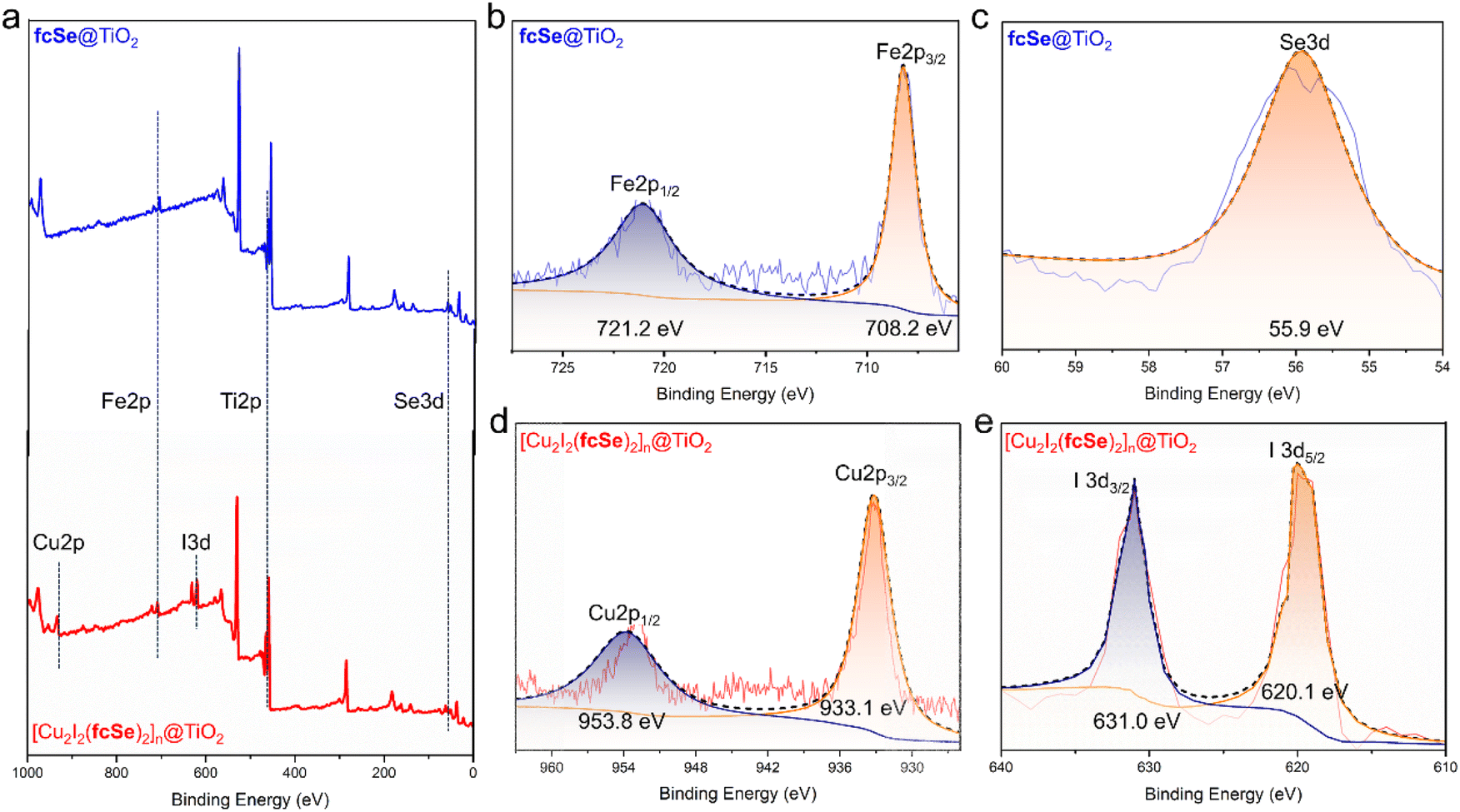 | ||
| Fig. 1 (a) XPS survey spectrum of fcSe@TiO2 and [Cu2I2(fcSe)2]n@TiO2, high-resolution XPS spectra of (b) Fe 2p and (c) Se 3d in fcSe@TiO2, (d) Cu 2p and (e) I 3d in [Cu2I2(fcSe)2]n@TiO2. | ||
 | ||
| Fig. 2 (a–c) TEM and HRTEM images, (d) EDX-mapping spectrum and (e–i) elemental mapping image of [Cu2I2(fcSe)2]n@TiO2. | ||
The relative BET specific surface areas of fcSe@TiO2 and [Cu2I2(fcSe)2]n@TiO2 were found to be 36.1 m2 g−1 and 48.3 m2 g−1 (Fig. S10†), slightly decreased from that of pure TiO2 (54.2 m2 g−1), indicating that the surface modification did not produce holes and cavities.
The UV-vis diffuse reflectance spectra (DRS) of fcSe@TiO2 and [Cu2I2(fcSe)2]n@TiO2 in Fig. 3a revealed the broad and strong visible absorption, primarily attributed to the presence of the ferrocene moiety.29,30 Tauc plot was applied to calculate the band gap energy Eg via the Kubelka–Munk method (Fig. 3b and S11†), giving 2.3 eV for fcSe@TiO2 and 2.2 eV for [Cu2I2(fcSe)2]n@TiO2. The introduction of fcSe and [Cu2I2(fcSe)2]n significantly decreases the Eg of free TiO2 (3.2 eV) and increases the visible light absorption, owing to the potential to accelerate the photocatalytic reaction.
 | ||
| Fig. 3 (a) UV-vis-DRS spectra and (b) the band gap estimation of fcSe@TiO2 and [Cu2I2(fcSe)2]n@TiO2. | ||
Visible light photocatalytic degradation of TC
The photocatalytic activities of fcSe@TiO2 and [Cu2I2(fcSe)2]n@TiO2 on TC degradation were initially evaluated by the degradation of TC (20 mg L−1) under the role of each constituent, including visible light (λ > 420 nm), H2O2 (20 mM) and catalysts (0.2 g L−1). As shown in Fig. 4a and calculated by eqn (1), according to the decreased adsorption of TC at 357 nm, the TC degradation efficiency (93.1%) was obtained by fcSe@TiO2 + H2O2 + hν within 30 min, exhibiting a significant enhancement as compared with TiO2 + H2O2 + hν (67.8%) and fcSe + H2O2 + hν (23.8%). Calculated by eqn (2), the apparent rate constant k value of fcSe@TiO2 + H2O2 + hν was 0.064 min−1, 2 times of TiO2 + H2O2 + hν (0.033 min−1) and 8 times of fcSe + H2O2 + hν (0.009 min−1) (Fig. 4b). | ||
| Fig. 4 (a) Degradation rate and (b) the corresponding pseudo-first-order kinetic curves of TC under various experimental conditions in the fcSe@TiO2 system (blue line). (c) Photo-Fenton degradation efficiency and (d) the corresponding pseudo-first-order kinetic curves of TC on various experimental conditions in the [Cu2I2(fcSe)2]n@TiO2 system (red line). | ||
Without visible light irradiation, fcSe@TiO2 +H2O2, TC removal efficiency decreased to 46.8% with k = 0.011 min−1, suggesting that visible light could significantly promote Fenton chemistry by providing photogenerated electrons in the recovery of Fe(II) from Fe(III). As for the photocatalytic process without H2O2 addition (fcSe@TiO2 + hν), 45.7% TC degradation was observed, indicating that H2O2 is not the only source of reactive species. The system of hν + H2O2 could hardly degrade TC, illustrating the vital role of fcSe@TiO2. Therefore, three constituents (visible light, H2O2, and fcSe@TiO2) are indispensable for this photo-catalytic process and their interaction is responsible for the efficient degradation of TC. Similar results were obtained for [Cu2I2(fcSe)2]n@TiO2 (Fig. 4c and d). The TC degradation efficiency (91.3%) and k = 0.062 min−1 were obtained by [Cu2I2(fcSe)2]n@TiO2 + H2O2 + hν within 30 min.
We next moved on to screen different factors influencing the TC degradation efficiency by the two studied nanosystems, including catalyst dosage, H2O2 concentration, and initial pH on TC degradation (Fig. 5). In the three studied catalyst dosages (0.1 g L−1, 0.2 g L−1, and 0.3 g L−1), the best TC degradation efficiency was found to be 93.1% for fcSe@TiO2 and 91.3% for [Cu2I2(fcSe)2]n@TiO2 at 0.2 g L−1 dosage (Fig. 5a and d). As shown in Fig. 5b and e, 10 mM/20 mM/30 mM H2O2 concentrations were studied, the TC degradation efficiency showed no significant enhancement for fcSe@TiO2 (from 92.2% to 93.1%) and medium enhancement for [Cu2I2(fcSe)2]n@TiO2 (from 83.4% to 91.3%). These agree with literature works, excessive H2O2 can cause oversaturation of the reaction sites and result in the scavenging of ˙OH.42–44
 | ||
| Fig. 5 TC Degradation rate under different initial conditions. (a) Catalyst dosage, (b) H2O2 concentration and (C) initial pH in fcSe@TiO2 system (blue line). (d) Catalyst dosage, (e) H2O2 concentration, and (f) initial pH in [Cu2I2(fcSe)2]n@TiO2 system (red line). | ||
It is known that most reported Fenton systems are limited by strict acidic pH requirements.20 However, our two catalysts have much wider pH tolerance. The Z-average hydrodynamic diameters and zeta potentials of fcSe@TiO2 and [Cu2I2(fcSe)2]n@TiO2 at pH = 3, 5, 7, and 9 were obtained from dynamic light scattering (DLS) measurements (Table S3†). The larger Z-average hydrodynamic diameters and more positive zeta potentials were obtained in an acidic solution. The results of the study indicated that fcSe has the ability to form Se⋯ H+ bond, which may effectively create an acidic microenvironment that could potentially enhance the Fenton reaction. As shown in Fig. 5c and f, the degradation efficacy of TC was maintained around 90% under acidic or neutral conditions, pH = 3, 5, 7. Under alkaline conditions, pH = 9, TC rates decreased to 70.5% for fcSe@TiO2 and 73.1% for [Cu2I2(fcSe)2]n@TiO2, which is due to electrostatic repulsion and the additional decomposition of H2O2.45 So, the following studies are all based on the optimal conditions, which are 0.2 g L−1 catalyst and 20 mM H2O2 and initial pH = 7.
In actual wastewater systems, it is inevitable that various ionic species exist and pose a negative effect on the photocatalytic performance of the catalyst.46,47 Herein, five different anions Cl−, Cr2O72−, HCO3−, H2PO4−, SO42− and three different cations K+, Na+, NH4+ at a concentration of 10 mM, were selected to study their influence on TC degradation. It was found that in the fcSe@TiO2 system, Cl−, Cr2O72−, SO42− and K+, Na+, NH4+ showed negligible effect on TC degradation (Fig. 6a); the same phenomena was observed for Cr2O72−, HCO3−, Na+, NH4+ in the [Cu2I2(fcSe)2]n@TiO2 system (Fig. 6b). A significant inhibiting effect was observed in the presence of H2PO4−, the TC degradation efficiency decreasing to 71.9% for fcSe@TiO2 and 75.1% for [Cu2I2(fcSe)2]n@TiO2, which was attributed to the H2PO4− scavenger of ˙OH and/or the attachment to the surface of TiO2.48,49 The introduction of Cl− slightly inhibited the degradation of TC (79.8%) in the [Cu2I2(fcSe)2]n@TiO2 system, which can be due to the less oxidizable ˙Cl and ˙ClOH− formed from Cl− and ˙OH.50 Therefore, it is delightful to find that fcSe@TiO2 and [Cu2I2(fcSe)2]n@TiO2 have good tolerance of most coexisting inorganic anions and cations, so no pre-treatment is needed to remove them before the photodegradation of TC wastewater.
 | ||
| Fig. 6 Effect of coexisting ions on the degradation of TC in (a) fcSe@TiO2 (blue) and (b) [Cu2I2(fcSe)2]n@TiO2 system (red). Synergetic photodegrade of TC and photoreduction of Cr(VI) in (c) fcSe@TiO2 (blue) and (d) [Cu2I2(fcSe)2]n@TiO2 system (red). Experimental conditions: TC concentration = 20 mg L−1, H2O2 concentration = 20 mM, catalyst dosages = 0.2 g L−1, initial pH = 7. | ||
We then further explored simultaneous photodegradation of TC and reduction of Cr(VI). Under neutral conditions, 92.1% of TC and 64.5% of Cr(VI) removal efficiency were achieved with fcSe@TiO2, and 89.2% of TC removal and 41.2% of Cr(VI) reduction efficiency with [Cu2I2(fcSe)2]n@TiO2, along with unchangeable efficient degradation efficiency of TC (Fig. 6c and d). Moreover, the presence of TC degradation increased the Cr(VI) reduction rate 3.76-fold in fcSe@TiO2/H2O2/hν system and 3.72-fold in [Cu2I2(fcSe)2]n@TiO2/H2O2/hν system. According to the reported studies,10,51 three aspects could interpret the good synergetic activity: (1) the reduction of Cr(VI) benefits from the acidic microenvironment obtained from Se in fcSe; (2) the photogenerated electrons effectively reduce Cr(VI); (3) TC degradation consumes reactive species and suppresses the re-oxidation of Cr(III).
The reusability and stability of fcSe@TiO2 and [Cu2I2(fcSe)2]n@TiO2 catalysts were investigated by successive TC degradation and Cr(VI) reduction processes (Fig. S12†). The catalytic performance for TC degradation remained invariant during the initial four successive cycles, then was slightly reduced to around 88% in the fifth cyclic run. The same phenomena were observed for Cr(VI) reduction. From the relevant characterizations of fcSe@TiO2 and [Cu2I2(fcSe)2]n@TiO2 catalysts after the fifth degradation process, no obvious change in the size or shape of nanoparticles was observed in TEM images (Fig. S13 and S14†), while FTIR spectra indicated physical adsorption on the surface of the photocatalysts which may slightly decrease the catalytic performance (Fig. S15†). The leaching of Cu and Fe concentration was a key issue related to the stability of nanocatalysts. Our ICP analyses revealed low concentrations of leachable iron in the solutions in comparison to literature reports;52,53 after five cycles, only 0.048 mg L−1 of fcSe@TiO2 (1.46% of 3.28 mg L−1) and 0.009 mg L−1 of [Cu2I2(fcSe)2]n@TiO2 (0.30% of 3.10 mg L−1). Leachable copper in the solution after five cycles is 0.141 mg L−1 of [Cu2I2(fcSe)2]n@TiO2 (3.98% of 3.55 mg L−1). Based on the ICP analysis, it is not surprising to find the increased stability of [Cu2I2(fcSe)2]n@TiO2 compared to fcSe@TiO2.
The photocatalytic removal efficiency of TC of fcSe@TiO2 and [Cu2I2(fcSe)2]n@TiO2 were compared with the reported catalysts in the literature works (Table S4†). Considering the catalyst dosage, the irradiation time and interfering ions, our two catalysts exhibited excellent degradation ability of TC (93.1% and 91.3%) under the conditions of low catalyst dosage (0.2 g L−1), short reaction time (30 min), and good tolerance of most coexisting inorganic ions. Moreover, it was worth noting that good synergetic redox photocatalytic activity of the photodegrade of TC and photoreduction of Cr(VI) to Cr(III). In summary, all these results demonstrated that fcSe@TiO2 and [Cu2I2(fcSe)2]n@TiO2 are efficient and stable photocatalysts, with potential industrial applications. Although various copper compounds have been studied as efficient H2O2 activators in Fenton-like reactions,54–59 [Cu2I2(fcSe)2]n@TiO2 catalyst system shows no significant improvement as compared with fcSe@TiO2.
Mechanism study
To study the reactive species generated in the two heterogeneous photo-catalytic systems, radical trapping experiments were firstly performed by employing different scavengers, isopropanol (IPA) for ˙OH, ammonium oxalate (AO) for h+, TEMPOL for ˙O2− and carotene for 1O2 respectively.60 In the fcSe@TiO2 system (Fig. 7a), the TC degradation efficiency decreased from 93.1% to 84.6% by IPA, 81.8% by AO, 78.7% by TEMPOL, and 89.0% by carotene. In the [Cu2I2(fcSe)2]n@TiO2 system (Fig. 7b), the TC degradation efficiency decreased from 91.3% to 73.4% by IPA, 69.4% by AO, 76.2% by TEMPOL, and 76.9% by carotene. From the collective results of the above degradation rates under various scavengers (Fig. 7c), combining the fractional contribution R of each active specie calculated from first-order rate constant (ks), eqn (3) (Fig. 7d–f), it can be indicated that ˙OH, ˙O2− and h+ were responsible for TC degradation in the fcSe@TiO2 system, while additional 1O2 existed in the [Cu2I2(fcSe)2]n@TiO2 system.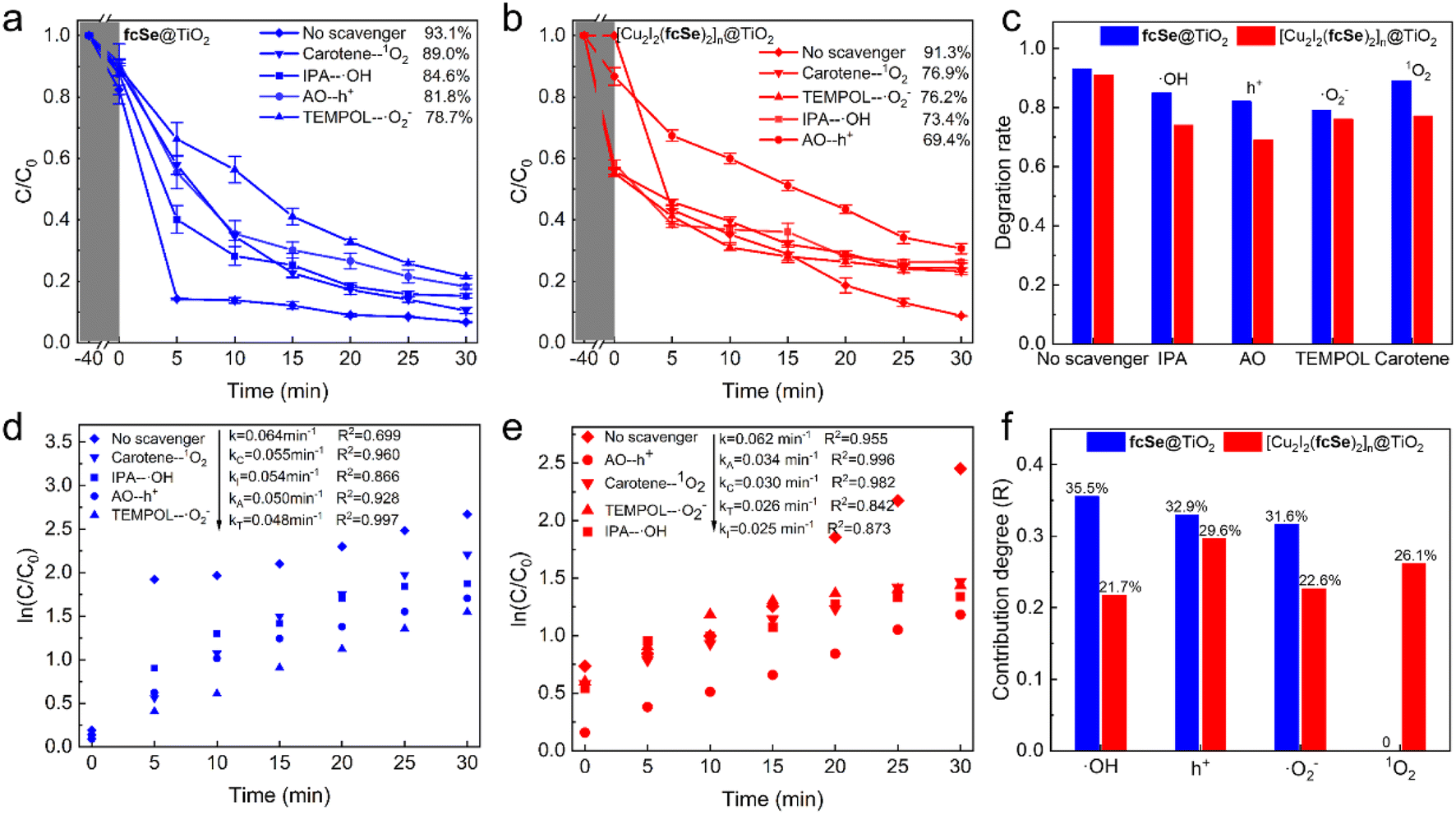 | ||
| Fig. 7 Effects of different scavengers on the photo-catalytic degradation rate of TC in (a) fcSe@TiO2 (blue), (b) [Cu2I2(fcSe)2]n@TiO2 system (red) and (c) collective results in bar graph. The according pseudo-first-order kinetic curves (d) in fcSe@TiO2 system (blue), (e) [Cu2I2(fcSe)2]n@TiO2 system (red) and (f) the fractional contribution of different reactive species R in the bar graph. Experimental conditions are 25–30 °C, pH = 7, catalyst dosage = 0.2 g L−1, H2O2 concentration = 20 mM. | ||
The spin-trapping EPR experiment with 5,5-dimethyl-1-pyrroline N-oxide (DMPO) was performed to attest the generation of ˙OH and ˙O2− radicals in the two systems, singlet oxygen sensor green (SOSG) was selected as 1O2 probe through the change of the intense fluorescence emission centered at 525 nm due to the cycloaddition reaction with 1O2.61 As depicted in Fig. 8a–f, no obvious signals were detected in the dark. After 5 min of visible light irradiation, the characteristic peaks of DMPO-˙OH and DMPO-˙O2− adducts were observed, respectively. fcSe@TiO2 system displayed 2.0 times and 1.2 times higher DMPO-˙OH and DMPO-˙O2− intensities signals than those in the [Cu2I2(fcSe)2]n@TiO2 system. Thus, it could be concluded that fcSe@TiO2 is able to generate a higher concentration of ˙OH and ˙O2− radicals than [Cu2I2(fcSe)2]n@TiO2, which is in congruence with their different removal efficiencies in the first five minutes as shown in Fig. 4a. In fcSe@TiO2 system there is no significant change of SOSG fluorescence emission near 525 nm before and after illumination, implying the absence of 1O2; while in [Cu2I2(fcSe)2]n@TiO2 system the significantly enhanced emission indicated 1O2 produced. When the ˙O2− scavenger TEMPOL39,61 was added into the [Cu2I2(fcSe)2]n@TiO2 system, the fluorescence intensity of SOSG in response to 1O2 decreased sharply to about 8.0%, indicating the generation of 1O2 from ˙O2− (Fig. S16†).
 | ||
| Fig. 8 EPR spectra of (a) DMPO-˙OH, (b) DMPO-˙O2− and (c) fluorescence change of SOSG in response to 1O2 generated using fcSe@TiO2 (blue line). EPR spectra of (d) DMPO-˙OH, (e) DMPO-˙O2− and (f) fluorescence change of SOSG in response to 1O2 generated using [Cu2I2(fcSe)2]n@TiO2 (red line). Experimental conditions are 25–30 °C, pH = 7, catalyst dosage = 0.2 g L−1, H2O2 concentration = 20 mM. | ||
According to the results of reactive species capture, EPR, and 1O2 detection, we infer that fcSe and [Cu2I2(fcSe)2]n on TiO2 tune the generation of reactive species through photogenerated charge and the photo-Fenton process further affect the pathways in TC photocatalytic degradation, and finally determine the degradation rate.
To further reveal the photocatalytic degradation pathways of TC, the intermediates and final products formed during the 5 min, 10 min, and 30 min process were analyzed through GC and LC-MS, and chemical structures of the corresponding degradation products are summarized in Table S5.† Based on the observed m/z peaks and related literature, probable degradation pathways are presented in Scheme 3.62–64
 | ||
| Scheme 3 Proposed photo-Fenton degradation pathways of TC in fcSe@TiO2 and [Cu2I2(fcSe)2]n@TiO2 system. | ||
In the fcSe@TiO2 system, three possible initial degradation pathways were generated by the independent attack of ˙O2−, ˙OH, and h+. For pathway I, TC molecule underwent 1,3-dipolar addition of double bond to give I1 (m/z = 461) (blue part in Scheme 3), due to the attacks of ˙O2−. After that, compound I1 was further demethylated to form compound I2 (m/z = 433) with loss of both methyl groups in the amino moiety. Through the dehydration-oxidation induced by h+, pathway II was dealcoholization on methylcyclohexan-1-one to give I3 (m/z = 427) (yellow part in Scheme 3); I3 was oxidatively degraded, and removed the methyl groups on the dimethylamine. Then, a series of ring opening and the side chain degradation to obtain I4 (m/z = 353); I3 could also be converted into I5 (m/z = 337) by deamidation along with the ring opening. In pathway III, I6 (m/z = 417) intermediate was generated through demethylation of the dimethylamino group when TC reacted with ˙OH (red part in Scheme 3). Subsequently, I7 (m/z = 447) was formed by ring opening and deamination of the compound I6.
The significant difference in the [Cu2I2(fcSe)2]n@TiO2 system was that there only existed pathway II and III. As electron-rich functional groups, double bond, amide group and dimethylamino group in TC molecule were more vulnerable to the attack of electrophilic 1O2. Therefore, compound I7 was much easier to generate when I6 was attacked by 1O2 in pathway III.
The TC stepwise degradation goes through ring opening, decarboxylation, dealcoholization and dealkylation processes, and finally, to form HCOOH, CO, CO2, H2O, NO3−. Conversion rate ηCO+HCOOH was then calculated by eqn (4), to give 7.2% in fcSe@TiO2 and 6.1% in [Cu2I2(fcSe)2]n@TiO2 (Table 1). The yield of CO and HCOOH was low under visible light irradiation, which meant the selectivity of TC to these products was poor. When white light was applied, much-enhanced transformation of TC to CO and HCOOH (22.8%) was found in fcSe@TiO2.
| Visible light (λ > 420 nm) | White light | |||
|---|---|---|---|---|
| fcSe@TiO2 | [Cu2I2(fcSe)2]n@TiO2 | fcSe@TiO2 | [Cu2I2(fcSe)2]n@TiO2 | |
| a Experimental conditions are 25–30 °C, pH = 7, catalyst dosage = 0.2 g L−1, H2O2 concentration = 20 mM.b Conversion rate of CO and HCOOH ηCO+HCOOH was calculated by eqn (4). | ||||
| Removal efficiency (%) | 93.1 | 91.3 | 97.4 | 96.2 |
| ηCO+HCOOH (%)b | 7.2 | 6.1 | 22.8 | 7.4 |
Based on the above analyses, the proposed degradation mechanism of TC over fcSe@TiO2 or [Cu2I2(fcSe)2]n@TiO2 catalysts is presented in Fig. 9. Firstly, visible-light irradiated fcSe@TiO2 or [Cu2I2(fcSe)2]n@TiO2 to produce photoexcited e− and h+. The h+ in the valence band (VB) is directly involved in the degradation process and H+ evolution from H2O. At the same time, a Fenton-like reaction between the ferrocene group and H2O2 released ˙OH and ferrocenium group; in this step, Se could form hydrogen bonds with H+ to form an acid microenvironment, which further improves the efficiency of the photo-Fenton reaction. Then, the O–Ti bonds between fcSe and TiO2 surface allow part e− migration to promote the reduction conversion of ferrocenium ([Fe3+(η5-C5H5)2])/ferrocene ([Fe2+(η5-C5H5)2]). Meanwhile, the remaining electrons accumulated in the CB could effectively reduce Cr(VI) and react with O2 to form reactive ˙O2− radicals.64,65 The redox cycle between different valence states CuI/CuII could also contribute to the generation of 1O2 from ˙O2−.66 These processes benefit the inhibition of the recombination of e−/h+, and produce reactive species (h+, ˙OH, ˙O2− and 1O2) for synergistical transformation of TC and Cr(VI).
 | ||
| Fig. 9 Proposed TC degradation mechanism in fcSe@TiO2 and [Cu2I2(fcSe)2]n@TiO2 system. | ||
Screen the application scope of N-cyclic organics
A distinguished system in water treatment is expected to have widespread applicability in the degradation of different pollutants. We then evaluated fcSe@TiO2 and [Cu2I2(fcSe)2]n@TiO2 in the degradation of seven representative N-cyclic organic contaminants. As shown in Table S6,† the degradation efficiency in the fcSe@TiO2 system within 30 min was 86.2% for Ciprofloxacin, 93.9% for Methylene Blue, 94.2% for Toluidine Blue, 92.0% for Pigment Green, 92.3% for Basic Violet, 17.1% for Methyl Orange, and 16.6% for imidazole; while in the [Cu2I2(fcSe)2]n@TiO2 system, 64.9% for Ciprofloxacin, 41.4% for Methylene Blue, 88.9% for Toluidine Blue, 93.7% for Pigment Green, 86.2% for Basic Violet, 16.0% for Methyl Orange and 25.9% for imidazole, respectively. Within these studied organics, only Methyl Orange and imidazole showed very low degradation efficiency, possibly due to the stable bonds in small molecular structures. Resourceful HCOOH and CO were found in final products with the conversion rate ηCO+HCOOH from 3.0% to 20.8%. These demonstrated that as-prepared fcSe@TiO2 and [Cu2I2(fcSe)2]n@TiO2 possessed excellent adaptability to various N-cyclic organic pollutants.Conclusions
Two novel visible light photocatalysts, fcSe@TiO2 and [Cu2I2(fcSe)2]n@TiO2, were successfully constructed to achieve efficient simultaneous transformation of refractory N-cyclic organic contaminants and Cr(VI) reduction under neutral conditions. Within just 30 minutes, 92.1% of TC and 64.5% of Cr(VI) removal efficiency were achieved with fcSe@TiO2, 89.2% of TC removal and 41.2% of Cr(VI) reduction efficiency with [Cu2I2(fcSe)2]n@TiO2. Mechanistic studies inferred that fcSe and [Cu2I2(fcSe)2]n on TiO2 tune the generation of reactive species through photogenerated charge and photo-Fenton process. The Fenton reaction inhibited the recombination of the photogenerated charges; meanwhile, the efficient photogenerated electrons could accelerate ferrocenium/ferrocene reduction conversion in the Fenton cycle. Se atom can also form interactions with H+, thereby providing an acidic microenvironment, which would be the key factor for the acidic Fenton reaction, acidic Cr(VI) reduction, and H+ evolution from H2O over a hole in the valence band. The two nanosystems exhibited good tolerance towards most coexisting inorganic anions and cations, as well as stability after five cycling runs. This study provides an efficient and practical approach for visible light photocatalytic transformation of N-cyclic organics and Cr(VI) under mild reaction conditions, with resourceful products, complex environment tolerance, and wide adaptability in wastewater treatment.Author contributions
Zhuo Yang, Jinshan Wang, Chao Wang: photocatalysts preparation and photocatalytic study. Wei Ji: experiments design and original draft writing. Su Jing: conceptualization, methodology, supervision, writing-reviewing and editing. Aimin Li, Elísabet Pires, Wenzhong Yang: results and discussion and manuscript comment.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work is funded by the Primary Research & Development Plan of Jiangsu Province (BE2019708 and BE2021712), and the National Natural Science Foundation of China (Grant No. 22175090).References
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- D. Sun, D. N. Confair and J. A. Ellman, Acc. Chem. Res., 2021, 54, 1766–1778 CrossRef PubMed.
- F.-K. Zhao, L. Yang, H. Yen, Q.-Y. Feng, M. Li and L.-D. Chen, Nat. Commun., 2023, 14, 6094 CrossRef CAS PubMed.
- S. M. Zainab, M. Junaid, N. Xu and R. N. Malik, Water Res., 2020, 187, 116455 CrossRef CAS PubMed.
- P. Chaturvedi, B. S. Giri, P. Shukla and P. Gupta, Bioresour. Technol., 2021, 319, 124161 CrossRef CAS PubMed.
- P. Kovalakova, L. Cizmas, T. J. McDonald, B. Marsalek, M.-B. Feng and V. K. Sharma, Chemosphere, 2020, 251, 126351 CrossRef CAS PubMed.
- Q.-L. Yang, Y. Gao, J. Ke, P. L. Show, Y.-H. Ge, Y.-H. Liu, R.-X. Guo and J.-Q. Chen, Bioengineered, 2021, 12, 7376–7416 CrossRef PubMed.
- J. Sharma, S. Sharma and V. Soni, Reg. Stud. Mar. Sci., 2019, 7, 24 Search PubMed.
- T. Yu, H. Chen, T. Hu, J. Feng, W.-L. Xing, L. Tang and W.-W. Tang, Appl. Catal., B, 2024, 342, 123401 CrossRef CAS.
- S. Guo, W. Yang, L. You, J. Li, J. Y. Chen and K. Zhou, Chem. Eng. J., 2020, 393, 124758 CrossRef CAS.
- X. Yin, W. Liu and J. Ni, Chem. Eng. J., 2014, 248, 89–97 CrossRef CAS.
- Y. Wang, L. Rao, P. Wang, Z. Shi and L. Zhang, Appl. Catal., B, 2020, 262, 118308 CrossRef CAS.
- B. Zhu, S. Chen, C. Li, G. Jiang, F. Liu, R. Zhao and C. Liu, Appl. Catal., B, 2022, 318, 121881 CrossRef CAS.
- C. Shi, S. Yu and C. Li, Chem. Eng. J., 2022, 441, 136052 CrossRef CAS.
- S. Wu, H. Hu, Y. Lin, J. Zhang and Y. H. Hu, Chem. Eng. J., 2020, 382, 122842 CrossRef CAS.
- W. Shi, C. Hao, Y. Fu, F. Guo, Y. Tang and X. Yan, Chem. Eng. J., 2022, 433, 133741 CrossRef CAS.
- H. Tan, D. Kong, Q. Li, Y. Zhou, X. Jiang, Z. Wang, R. E. Parales and Z. Ruan, Environ. Pollut., 2022, 305, 119299 CrossRef CAS PubMed.
- C. Xia, H. Huang, D. Liang, Y. Xie, F. Kong, Q. Yang, J. Fu, Z. Dou, Q. Zhang and Z. Meng, Chem. Eng. J., 2022, 443, 136398 CrossRef CAS.
- D. Meyerstein, Nat. Rev. Chem, 2021, 5, 595–597 CrossRef CAS PubMed.
- Z. Tang, P. Zhao, H. Wang, Y. Liu and W. Bu, Chem. Rev., 2021, 121, 1981–2019 CrossRef CAS PubMed.
- Y. Zhu, R. Zhu, Y. Xi, J. Zhu, G. Zhu and H. He, Appl. Catal., B, 2019, 255, 117739 CrossRef CAS.
- H. Wang, C.-Y. Zhang, X.-L. Zhang, S.-J. Wang, Z.-X. Xia, G.-F. Zeng, J. Ding and N.-Q. Ren, Chem. Eng. J., 2022, 429, 132445 CrossRef CAS.
- Z.-H. Wu, J. Shen, W.-L. Li, J.-S. Li, D.-H. Xia, D.-F. Xu, S.-Y. Zhang and Y.-F. Zhu, Appl. Catal., B, 2023, 330, 122642 CrossRef CAS.
- C.-A. Li, W. Ji, J. Qu, S. Jing, F. Gao and D.-R. Zhu, Dalton Trans., 2018, 47, 7463–7470 RSC.
- H.-Y. Zhou, M. Li, J. Qu, S. Jing, H. Xu, J.-Z. Zhao, J. Zhang and M.-F. He, Organometallics, 2016, 35, 1866–1875 CrossRef CAS.
- J. Qu, Q. Xia, W. Ji, S. Jing, D. Zhu, L. Li, L. Huang, Z. An, C. Xin, Y. Ni, M. Li, J. Jia, Y. Song and W. Huang, Dalton Trans., 2018, 47, 1479–1487 RSC.
- J. Zhou, X. Zhu, Q. Cheng, Y. Wang, R. Wang, X. Cheng, J. Xu, K. Liu, L. Li, X. Li, M. He, J. Wang, H. Xu, S. Jing and L. Huang, Inorg. Chem., 2020, 59, 9177–9187 CrossRef CAS PubMed.
- T. Zhou, Q. Cheng, L. Zhang, D. Zhang, L. Li, T. Jiang, L. Huang, H. Xu, M. Hu and S. Jing, Chem. Eng. J., 2022, 438, 135637 CrossRef CAS.
- W. Ji, J. Qu, C. A. Li, J. W. Wu, S. Jing, F. Gao, Y. L. Lv, C. Liu, D. R. Zhu, X. M. Ren and W. Huang, Appl. Catal., B, 2017, 205, 368–375 CrossRef CAS.
- G.-J. Zha, W. Ji, Z.-H. Qi, W.-J. Qiu, A.-M. Li, D.-R. Zhu and S. Jing, J. Catal., 2022, 405, 313–321 CrossRef CAS.
- W. Ji, J. Qu, S. Jing, D. R. Zhu and W. Huang, Dalton Trans., 2016, 45, 1016–1024 RSC.
- M. R. Burgess, S. Jing and C. P. Morley, J. Organomet. Chem., 2006, 691, 3484–3489 CrossRef CAS.
- F. Guo, H. Zhang, H. Li and Z. Shen, Appl. Catal., B, 2022, 306, 121092 CrossRef CAS.
- V. Kumaravel, S. Rhatigan, S. Mathew, J. Bartlett, M. Nolan, S. J. Hinder, P. K. Sharma, A. Singh, J. A. Byrne, J. Harrison and S. C. Pillai, J. Phys. Chem. C, 2019, 123, 21083–21096 CrossRef CAS.
- Y. Xu, J. Wan, L. Huang, J. Xu, M. Ou, Y. Liu, X. Sun, S. Li, C. Fang, Q. Li, J. Han, Y. Huang and Y. Zhao, Energy Storage Mater., 2020, 33, 432–441 CrossRef.
- D. Dambournet, Acc. Chem. Res., 2020, 55, 696–706 CrossRef PubMed.
- A. B. Grafton and C. M. Cheatum, Chem. Phys., 2021, 512, 3–12 CrossRef.
- Y. Ma, Y. Lu, G. Hai, W. Dong, R. Li, J. Liu and G. Wang, Sci. Bull., 2020, 65, 658–669 CrossRef CAS PubMed.
- A. Vittadini, A. Selloni, F. P. Rotzinger and M. Grätzel, J. Phys. Chem. B, 2000, 104, 1300–1306 CrossRef CAS.
- M. Nilsing, P. Persson and L. Ojamäe, Chem. Phys. Lett., 2005, 415, 375–380 CrossRef CAS.
- M. W. Mara, D. N. Bowman, O. Buyukcakir, M. L. Shelby, K. Haldrup, J. Huang, M. R. Harpham, A. B. Stickrath, X. Zhang, J. F. Stoddart, A. Coskun, E. Jakubikova and L. X. Chen, J. Am. Chem. Soc., 2015, 137, 9670–9684 CrossRef CAS PubMed.
- L. Yang, L. Li, Z. Liu, C. Lai, X. Yang, X. Shi, S. Liu, M. Zhang, Y. Fu, X. Zhou, H. Yan, F. Xu, D. Ma and C. Tang, Chemosphere, 2022, 294, 133736 CrossRef CAS PubMed.
- S. Huang, Q. Zhang, P. Liu, S. Ma, B. Xie, K. Yang and Y. Zhao, Appl. Catal., B, 2020, 263, 118336 CrossRef CAS.
- Y. Yao, G. Wu, F. Lu, S. Wang, Y. Hu, J. Zhang, W. Huang and F. Wei, Environ. Sci. Pollut. Res., 2016, 23, 21833–21845 CrossRef CAS PubMed.
- H. Qi, X. Sun and Z. Sun, Chem. Eng. J., 2021, 403, 126270 CrossRef CAS.
- F. Chen, Q. Yang, X. Li, G. Zeng, D. Wang, C. Niu, J. Zhao, H. An, T. Xie and Y. Deng, Appl. Catal., B, 2017, 200, 330–342 CrossRef CAS.
- Q. Li, G. Wei, Y. Yang, L. Gao, L. Zhang, Z. Li, X. Huang and J. Gan, Chem. Eng. J., 2021, 424, 130537 CrossRef CAS.
- N. Li, Y. Wang, X. Cheng, H. Dai, B. Yan, G. Chen, L. Hou and S. Wang, Water Res., 2022, 222, 118896 CrossRef CAS PubMed.
- Y. Ren, L. Lin, J. Ma, J. Yang, J. Feng and Z. Fan, Appl. Catal., B, 2015, 165, 572–578 CrossRef CAS.
- Y. Liu, X. He, Y. Fu and D. D. Dionysiou, Chem. Eng. J., 2016, 284, 1317–1327 CrossRef CAS.
- J. Gong, W. Zhang, T. Sen, Y. Yu, Y. Liu, J. Zhang and L. Wang, ACS Appl. Nano Mater., 2021, 4, 4513–4521 CrossRef CAS.
- R. Liu, Y. Xu and B. Chen, Environ. Sci. Technol., 2018, 52, 7043–7053 CrossRef CAS PubMed.
- J. Sun, J. Wan, Y. Wang, Z. Yan, Y. Ma, S. Ding, M. Tang and Y. Xie, J. Hazard. Mater., 2022, 429, 128299 CrossRef CAS PubMed.
- E. Ma, K. Wang, Z. Hu and H. Wang, J. Colloid Interface Sci., 2021, 603, 685–694 CrossRef CAS PubMed.
- G. Pan and Z. Sun, Chemosphere, 2021, 283, 131257 CrossRef CAS PubMed.
- H. Hu, K. Miao, X. Luo, S. Guo, X. Yuan, F. Pei, H. Qian and G. Feng, J. Cleaner Prod., 2021, 318, 128632 CrossRef CAS.
- J. Xu, X. Zheng, Z. Feng, Z. Lu, Z. Zhang, W. Huang, Y. Li, D. Vuckovic, Y. Li, S. Dai, G. Chen, K. Wang, H. Wang, J. K. Chen, W. Mitch and Y. Cui, Nat. Sustain., 2021, 4, 233–241 CrossRef PubMed.
- N. Schmachtenberg, S. Silvestri, J. da S. Salla, G. L. Dotto, D. Hotza, S. L. Jahn and E. L. Foletto, J. Environ. Chem. Eng., 2019, 7, 102954 CrossRef CAS.
- Y. Tian, N. Jia, H. Ma, G. Liu, Z. Xiao, Y. Wu, L. Zhou, J. Lei, L. Wang, Y. Liu and J. Zhang, J. Hazard. Mater., 2021, 419, 126359 CrossRef CAS PubMed.
- Y. Yang, G. Zeng, D. Huang, C. Zhang, D. He, C. Zhou, W. Wang, W. Xiong, B. Song, H. Yi, S. Ye and X. Ren, Small, 2020, 16, 2001634 CrossRef CAS PubMed.
- B. Chen, Y. Yang, Y. Wang, Y. Yan, Z. Wang, Q. Yin, Q. Zhang and Y. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 18533–18544 CrossRef CAS PubMed.
- C. Liu, H. Dai, C. Tan, Q. Pan, F. Hu and X. Peng, Appl. Catal., B, 2022, 310, 121326 CrossRef CAS.
- D. Xing, Z. Cui, Y. Liu, Z. Wang, P. Wang, Z. Zheng, H. Cheng, Y. Dai and B. Huang, Appl. Catal., B, 2021, 290, 120029 CrossRef CAS.
- K. Dou, C. Y. Peng, R. C. Wang, H. P. Cao, C. Yao, J. F. Qiu, J. L. Liu, N. Tsidaeva and W. Wang, Chem. Eng. J., 2023, 455, 140813 CrossRef CAS.
- C. Y. Yang, J. Wang, R. Wang, W. X. Zhu, L. Zhang, T. Du, J. Sun, M. Q. Zhu, Y. Z. Shen and J. L. Wang, Appl. Catal., B, 2023, 322, 122084 CrossRef CAS.
- Q. Wang, D. Zhou, K. Lin and X. Chen, Chem. Eng. J., 2021, 419, 129667 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2216502. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra07390d |
| This journal is © The Royal Society of Chemistry 2024 |