
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in the application of alkynes in multicomponent reactions
Seyedmohammad Hosseininezhad
a and
Ali Ramazani
 *ab
*ab
aThe Organic Chemistry Research Laboratory (OCRL), Department of Chemistry, Faculty of Science, University of Zanjan, Zanjan 45371-38791, Iran. E-mail: hossinichemistry1397@gmail.com; M.hosseininezhad@stu.nit.ac.ir; aliramazani@gmail.com; aliramazani@znu.ac.ir
bThe Convergent Sciences & Technologies Laboratory (CSTL), Research Institute of Modern Biological Techniques (RIMBT), University of Zanjan, Zanjan 45371-38791, Iran
First published on 2nd January 2024
Abstract
Alkynes have two active positions to carry out chemical reactions: C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C and C–H. These two positions are involved and activated in different reactions using different reagents. In this study, we investigated the reactions of alkynes that are involved in multi-component reactions through the C–C and C–H positions and examined the progress and gaps of each reaction by carefully studying the mechanism of the reactions. Firstly, we investigated and analyzed the reactions involving the CC position of alkynes, including the reactions between derivatives of alkynes with RN3, sulfur compounds (RSO2R′, DMSO, S8, DABCO(SO2)2 and DABSO), barbituric acids, aldehydes and amines, COOH, α-diazoesters or ketones, and isocyanides. Then, we examined and analyzed the important reactions involving the C–H position of alkynes and the progress and gaps in these reactions, including the reaction between alkyne derivatives with amines and aldehydes for the synthesis of propargylamines, the reaction between alkynes with CO2 and the reaction between alkynes with CO.
C and C–H. These two positions are involved and activated in different reactions using different reagents. In this study, we investigated the reactions of alkynes that are involved in multi-component reactions through the C–C and C–H positions and examined the progress and gaps of each reaction by carefully studying the mechanism of the reactions. Firstly, we investigated and analyzed the reactions involving the CC position of alkynes, including the reactions between derivatives of alkynes with RN3, sulfur compounds (RSO2R′, DMSO, S8, DABCO(SO2)2 and DABSO), barbituric acids, aldehydes and amines, COOH, α-diazoesters or ketones, and isocyanides. Then, we examined and analyzed the important reactions involving the C–H position of alkynes and the progress and gaps in these reactions, including the reaction between alkyne derivatives with amines and aldehydes for the synthesis of propargylamines, the reaction between alkynes with CO2 and the reaction between alkynes with CO.
 Seyedmohammad Hosseininezhad | Seyedmohammad Hosseininezhad was born in 1997 in Zanjan, Iran. He received his B.Sc in Pure Chemistry from Payam Noor University (PNU) in 2020 as a first-ranked student. Then, he received his M.Sc in Organic Chemistry with a GPA of 4 out of 4 from Babol Noshirvani University of Technology (BNUT) in 2023. During this period, he worked on intramolecular hydrogen bonding, multicomponent reactions, and synthesis of shift bases. After graduating with the M.Sc. Degree, he immediately joined Prof. Ramazani's group at the University of Zanjan (ZNU) as a research assistant to increase his knowledge in the fields of organic chemistry, medicinal chemistry, and synthesis of heterocyclic and organic compounds with modern methods. |
 Ali Ramazani | Professor Dr Ali Ramazani completed his PhD under the supervision of Professor Issa Yavari in the Department of Chemistry at the Tarbiat Modares University (TMU) in the Tehran-Iran. He currently works as a Full Professor in Chemistry at the University of Zanjan in the Zanjan-Iran. His studies are focused on organic synthesis, medicinal chemistry and nanochemistry and he has published more than 600 papers and patents. He is an Editorial Board Member of the international Journal Nanochemistry Research. He has received several national and international awards, including the 2013 Khwarizmi International Award, several top-cited author awards and best-paper awards from leading ISI Journals, Best Researcher Awards, and the Best Lecturer Awards at the University of Zanjan. Google Scholar: https://scholar.google.co.in/citations?user=XDWRu9gAAAAJ%26hl=en. |
1 Introduction
Alkynes are versatile molecules that can be widely applied for transformations in organic synthesis.1 Also, alkynes and their derivatives are among the most valuable chemical motifs owing to their versatile reactivities and abundance.2 These basic chemicals can serve as molecular building blocks for the development of new organic reactions and the construction of functional materials.3 Alkynes have at least one C–C triple bond in which the carbon atoms are sp hybridized. The C atoms are connected by one σ bond and two π bonds, in which the two p orbitals in the C atoms overlap.4 In recent years, several novel organic transformations involving alkynes have been explored for the construction of valuable molecular scaffolds.5–13 However, because sp-hybridized carbon atoms are less reactive, activating the CC bonds in alkynes is key to their synthetic transformation. In addition, CC bond substituents in alkynes can be selectively activated by either both π- and σ-activation (terminal alkynes) or π-activation (internal alkynes) alone.14 Alternatively, alkynes play a significant role as a coupling agent in transition metal-catalysed directed C–H activation reactions (Fig. 1). Also, because of the linear geometry and low steric requirements of alkynes, they can be effectively coordinated with metal d-orbitals. All these applications make alkynes useful coupling partners in terms of their numerous beneficial transformations.15
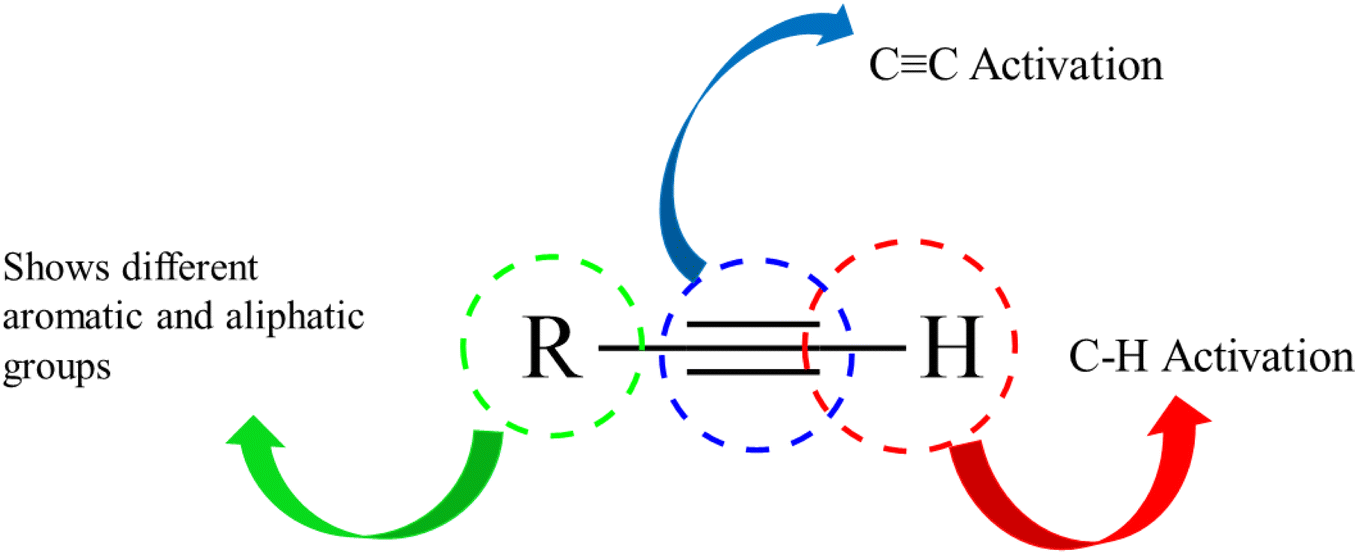 | ||
| Fig. 1 Structure of an alkyne showing its two active positions: C–H and CC. | ||
Creating molecular complexity and molecular diversity from available and simple compounds is considered one of the basic challenges in organic synthesis. Therefore, the use of methods that use simple compounds to form complex molecules while creating several links in chemical processes in a single process is progressing. In this regard, multicomponent reactions provide these conditions.16 The applications of multicomponent reactions include the synthesis of natural products,17 polymer chemistry,18 agrochemistry,19 medicinal chemistry, and drug discovery.20 In addition, through multicomponent reactions, it is possible to synthesize different and important molecular skeletons, such as γ-lactams, quinoxalinones, benzodiazepinedione, benzimidazoles, and thiazoles.21,22 These molecular skeletons have been demonstrated to possess medicinal properties, for example, thiazole scaffolds have anti-cancer, anti-inflammatory, and antibacterial properties.23 Among the advantages of multi-component reactions, the most notable are the purification and separation of products at low cost, the formation of low tracer waste, high atom economy, and high efficiency of products.24 However, many practical features of these methods and the role of multicomponent reactions in solving delicate chemical problems still have to be demonstrated.25 Alternatively, the mechanism of multi-component reactions is often complex, and thus most of the processes in these reactions have reversible equilibrium, whereas the last stage is irreversible.26
In this review, we focus on some C–H and CC activation reactions of alkynes. In the case of the activation of C–C alkynes, we review the gaps, developments, and mechanisms of reactions of alkynes with carboxylic acids, RN3, sulfur-containing compounds, barbituric acids, aldehydes, amines, alpha-diazo esters or ketones, and isocyanides. Then, for the activation of C–CH alkynes, we investigate the gaps, advances and reaction mechanisms of alkynes with amines and aldehydes for the synthesis of propargylamines, reaction between alkynes with CO2, and reaction between alkynes with CO.
2 C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) C activation
C activation
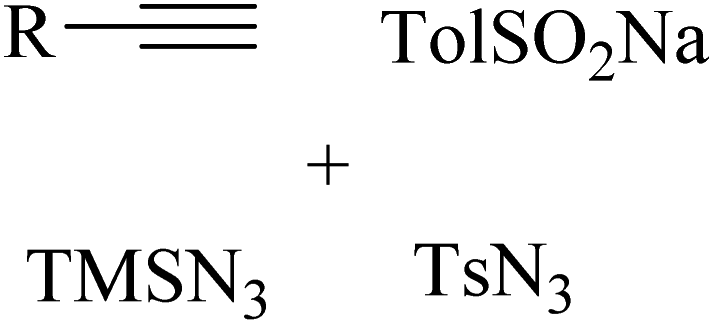
2.1 Reaction of RN3
Click chemistry was discovered by Sharpless et al.27 These reactions refer to reactions that are easy to perform, generate products with high efficiency and little or no side products, are not affected by the nature of different groups, and perform well under different conditions.28 The features of click reactions have are presented in Fig. 2.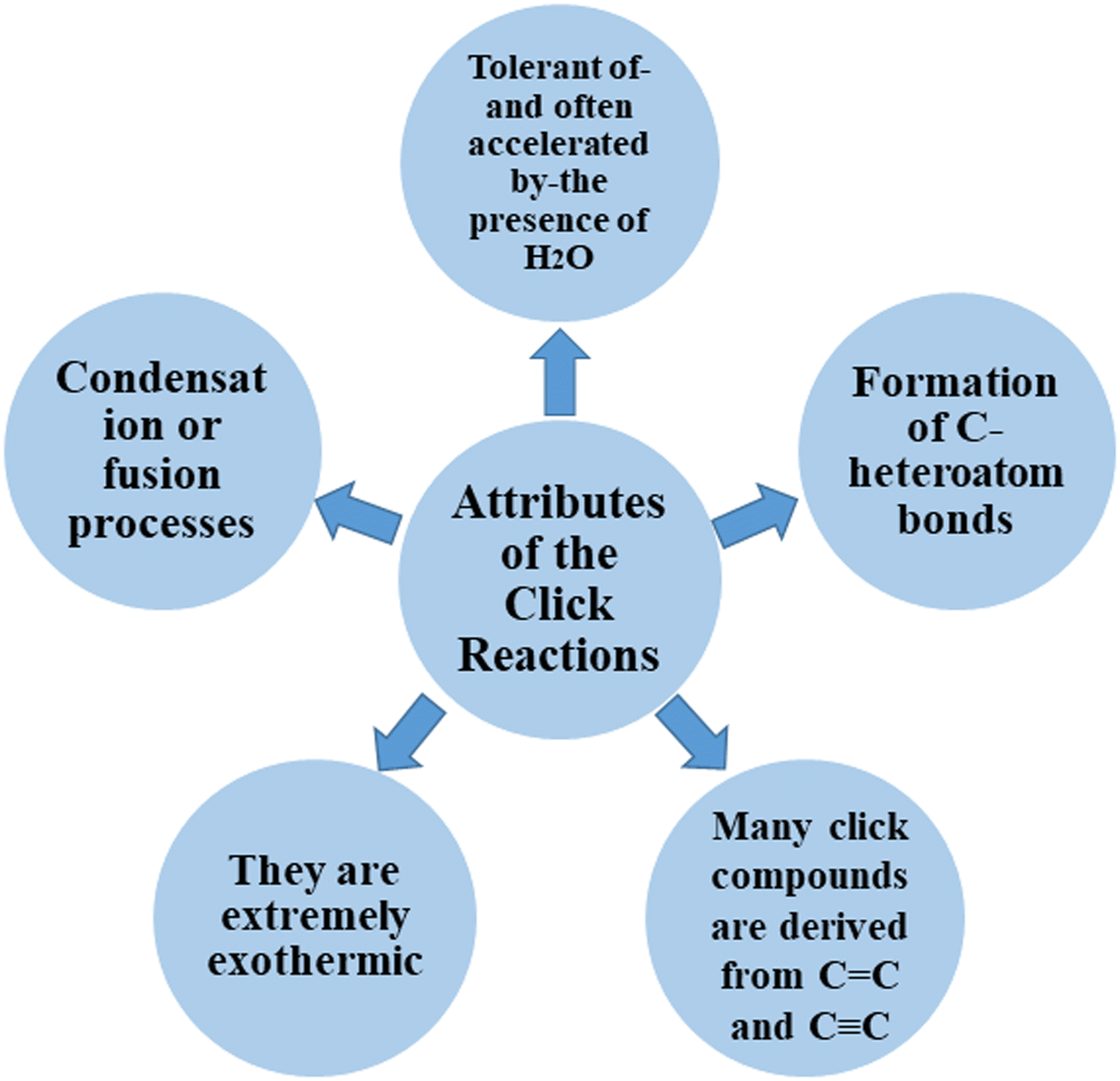 | ||
| Fig. 2 General attributes of click reactions. | ||
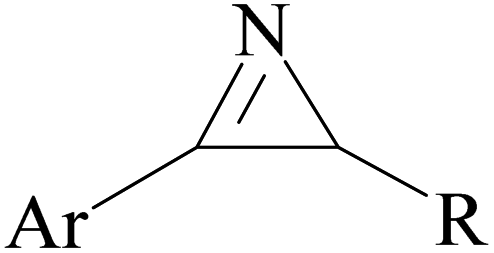
Among the known reactions, a small number has the characteristics of click reactions, including the azide–alkyne cycloaddition (AAC) reaction, which produces 1,2,3-triazoles (T) (Fig. 3). This is one of the most important click reactions.28 It should be noted that 1,2,3-triazoles have wide biological activities such as anti-fungal and anti-bacterial, anti-virus, anti-tuberculosis, anti-cancer.29 The AAC reaction has the following characteristics: (1) high chemical potential energy of azides and alkynes, (2) slow reaction rate (long-term heating for inactive alkynes), (3) in the presence of solvent, electrophiles and nucleophiles are stable (azide and alkyne groups) and (4) they are accelerated by catalysts such as Cu(I) (CuAAC).28 One of the most widely used click reactions is the CuAAC reaction; however, despite its widespread use, it has disadvantages because copper is cytotoxic and the excessive consumption of copper can cause Alzheimer's, neurological disorders, and hepatitis.30
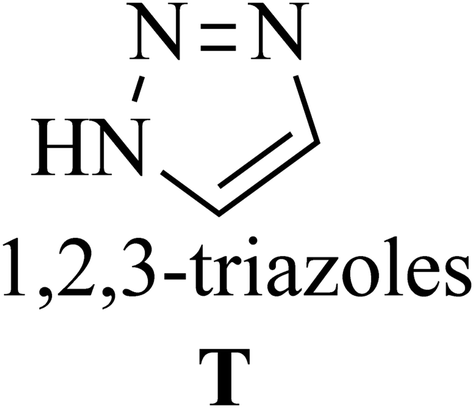 | ||
| Fig. 3 Structure of 1,2,3-triazoles. | ||
Click chemistry is used in drug discovery, organic synthesis, pharmaceutical industry, and polymer chemistry, materials science, and bioconjugation.29 Meanwhile, drug discovery based on click chemistry classified as shown in Fig. 4.
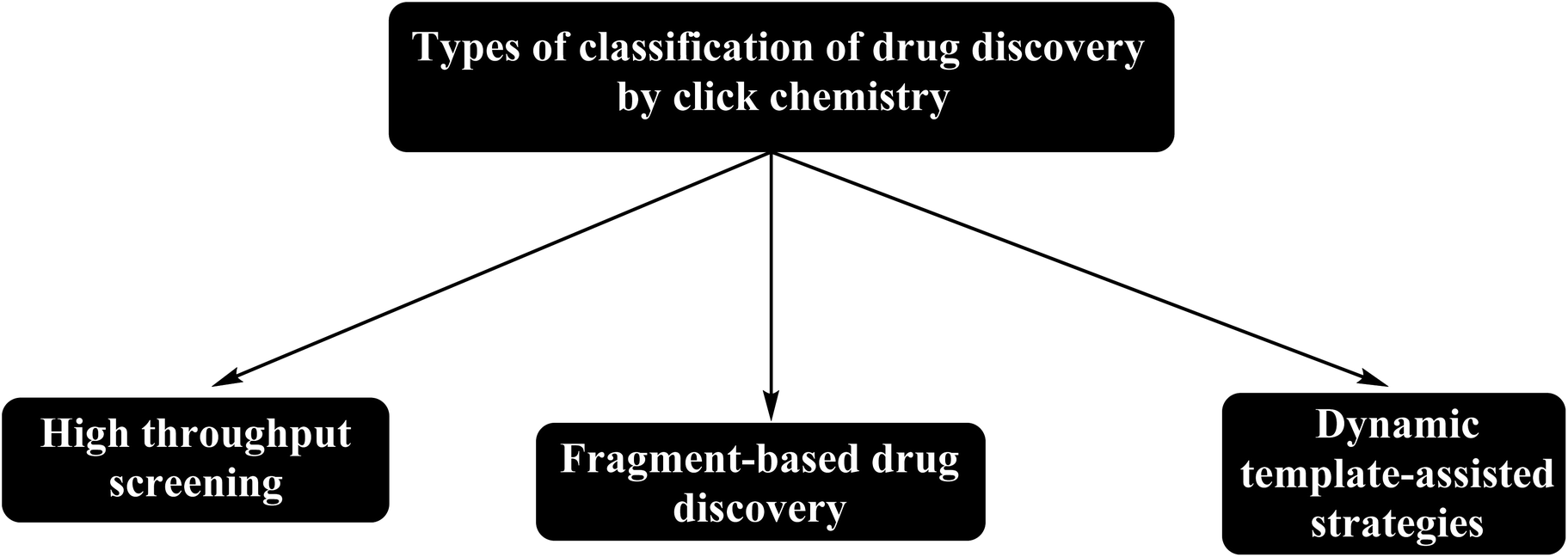 | ||
| Fig. 4 Classification of drug discovery based on click chemistry. | ||
A series of N1- and N2-oxyalkylated 1,2,3-triazole derivatives 4 and 5 was synthesized via the reaction of ethers 3, alkynes 1, and TMSN3 2 in the presence of CuI and TBHP (as an oxidant) at 80 °C for 16 h by Bao and co-workers (Scheme 1).31
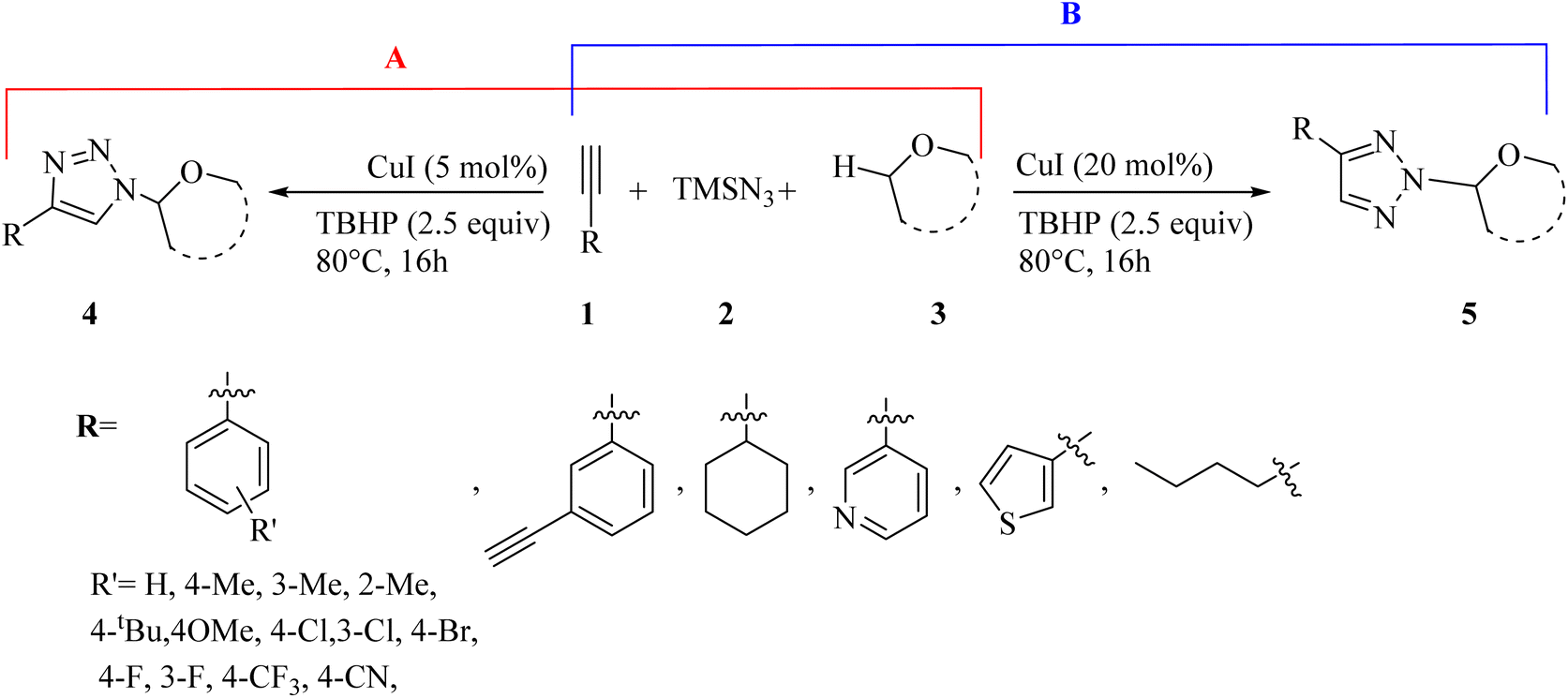 | ||
| Scheme 1 (A) Synthesis of N1-oxyalkylated triazoles 4 and (B) synthesis of N2-oxyalkylated triazoles 5.31 | ||
In the first stage for the formation of 4, the radical species t-BuO˙ and t-BuOO˙ are produced through the Cu-catalyzed homolytic decomposition of TBHP. This process causes A to convert to B. Next, B is oxidized, producing α-alkoxyalkyl cation C. Then, C undergoes nucleophilic attack by TMSN3 to produce oxyalkylated azide D, which in the presence of phenylacetylene, undergoes the click reaction and produces 4 (Scheme 2A). E produces 5 via the copper-promoted 1,2-shift of the ether group. This process leads to a sequential electronic transfer process (Scheme 2B).
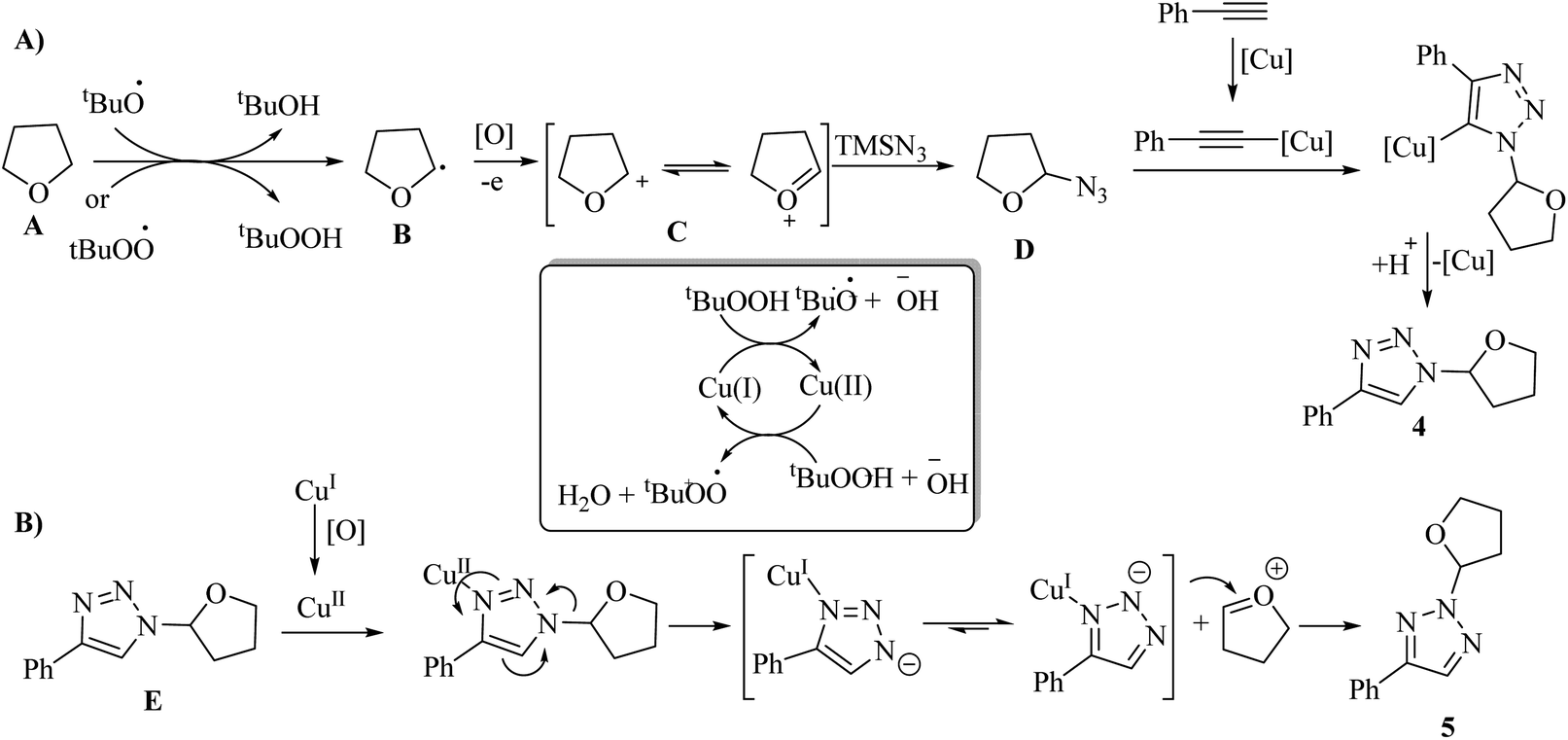 | ||
| Scheme 2 Possible reaction mechanism for the synthesis of (A) N1-oxyalkylated triazoles and (B) N2-oxyalkylated triazoles.31 | ||
In the case of 4 (Scheme 1A), the Me group at different positions of the benzene ring (ortho, meta, and para) in aromatic alkynes produced the desired triazole derivatives in 70%, 75%, and 66% yield, respectively. tBu and OMe groups at the para position of phenylacetylene produced the desired products in 76% and 66% yield, respectively. Also, phenylacetylene with CF3, Cl, Br, and F substituents gave the corresponding triazole derivatives in 57–71% yield. Notably, aliphatic alkynes (hex-1-yne and ethynylcyclohexane) and heterocyclic aromatic alkynes (3-ethynylpyridine and 3-ethynylthiophene) produced the corresponding triazole derivatives in 65% and 60% yield and 67% and 72%, respectively. In the case of 5 (Scheme 1B), aromatic alkyne derivatives with various substituents produced the corresponding triazole derivatives in 64–80% yield. In addition, aliphatic and heterocyclic aromatic alkynes (3-ethynylthiophene) produced the corresponding triazole derivatives in 66–78% yield.
In another study, Bao and co-workers32 synthesized a selective assembly of N1- and N2-alkylated 1,2,3-triazole derivatives 9 and 10 via the reaction between ether 7, alkynyl carboxylic acid derivatives 6, and azidotrimethylsilane (TMSN3) 8 in the presence of CuCl2 in TBHP at 80 °C for 12 h (Scheme 3).
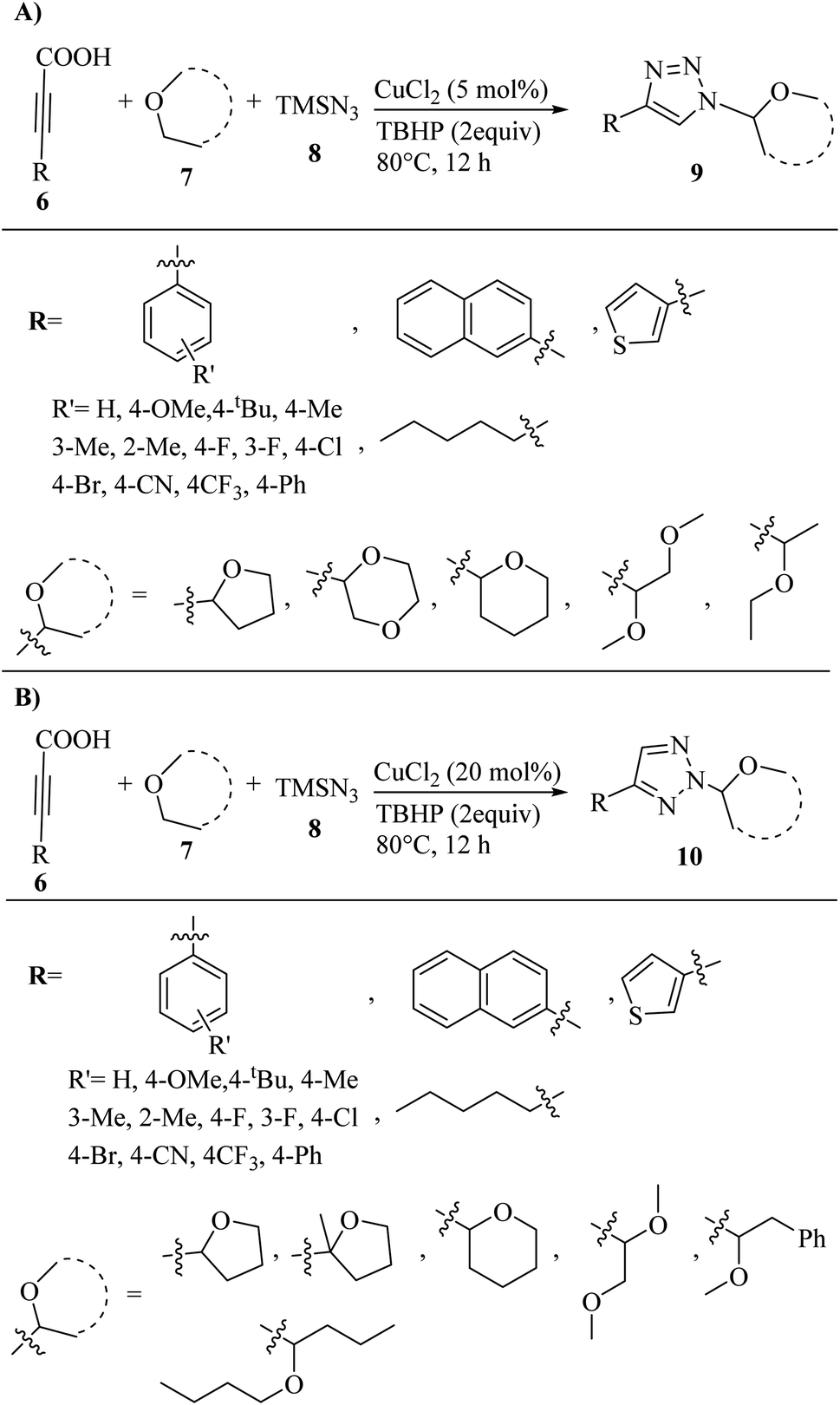 | ||
| Scheme 3 (A) Synthesis of N1-alkylated 1,2,3-triazoles 9 via CuCl2 and (B) synthesis of N2-alkylated 1,2,3-triazoles 10 via CuCl2.32 | ||
Two pathways were proposed for the formation of 9. In the first pathway, t-BuO˙ and t-BuOO˙ abstract the α-H atom from A, producing alkoxyalkyl radical B. Then, B is oxidized to produce C. Next, B undergoes nucleophilic attack by TMSN3, generating D. Subsequently, an alkynyl copper intermediate is produced via the reaction between the alkynyl carboxylic acid and Cu salt and via the reaction with one molecule of HCl, releasing CO2. The alkynyl copper intermediate reacts with D and generates E via a click reaction. Finally, D is quenched by a proton to give 9 (Scheme 4A). In the second pathway, propiolic acids 6 react with 8 via the copper-catalyzed decarboxylative cycloaddition, which generates F. Next, F undergoes alkylation to generate 9 (Scheme 4A). Then, the copper-mediated 1,2-shift of the ether group in 9 generates 10 (Scheme 4B).
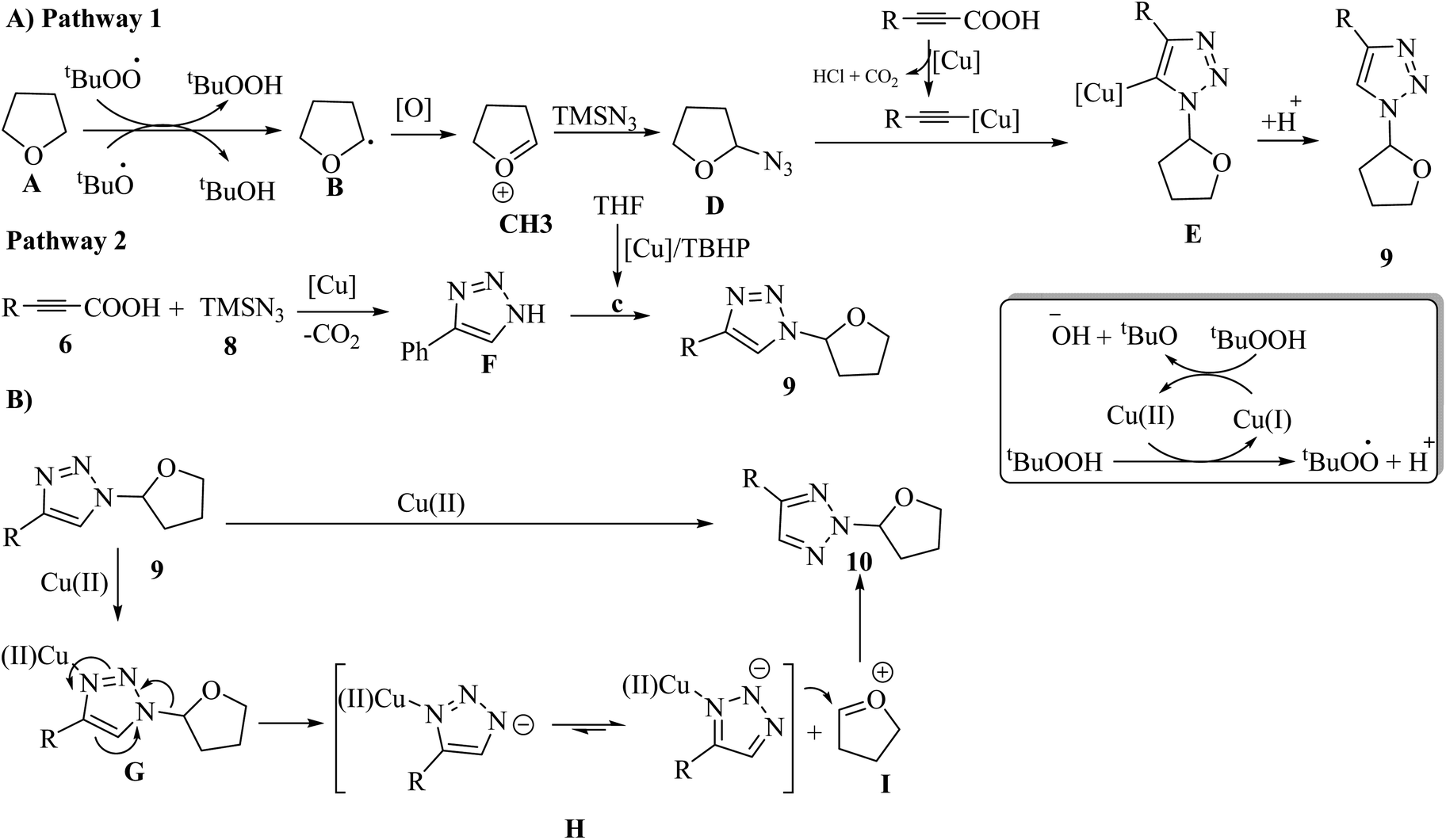 | ||
| Scheme 4 Possible reaction mechanism for the synthesis of (A) N1-alkylated 1,2,3-triazoles and (B) N2-alkylated 1,2,3-triazoles.32 | ||
The above-mentioned method is a selective and efficient approach for the synthesis of the desired triazole derivatives in 40–92% yield. In the case of 9 (Scheme 3A), arylpropiolic acids with electron-poor or -rich aryl rings react well and the desired triazole derivatives were obtained in 57–92% yield. Also, 2-naphthalenyl propiolic acid, 3-(thiophen-3-yl)propiolic acid and biphenyl propiolic acid produced the corresponding triazole derivatives in 61%, 75%, and 57% yield, respectively. In addition, alkylpropiolic acid (oct-2-ynoic acid) also reacted and produced the desired 1,2,3-triazole derivative in 60% yield. Furthermore, the tolerance of ether derivatives was examined in the above-mentioned method, and besides THF, other cyclic ether derivatives such as tetrahydro-2H-pyran and 1,4-dioxane produced the corresponding triazole derivatives in 60% and 40% yield, respectively. Also, acyclic ethers (1,2-dimethoxyethane and methylphenylethyl ether) produced the desired triazole derivatives in 40% and 71% yield, respectively. In the case of 10 (Scheme 3B), similar to the preparation of N1-alkylated 1,2,3- triazoles, substituted arylpropiolic acid derivatives containing electron-withdrawing and -donating groups produced the corresponding triazole derivatives in 70–85% yield. Specifically, 3-(thiophen-3-yl)propiolic acid produced the desired product in 73% yield, while oct-2-ynoic acid also produced the desired 1,2,3-triazole derivatives in 70% yield. Furthermore, cyclic and acyclic ether derivatives produced the corresponding triazole derivatives in 40–68% yield.
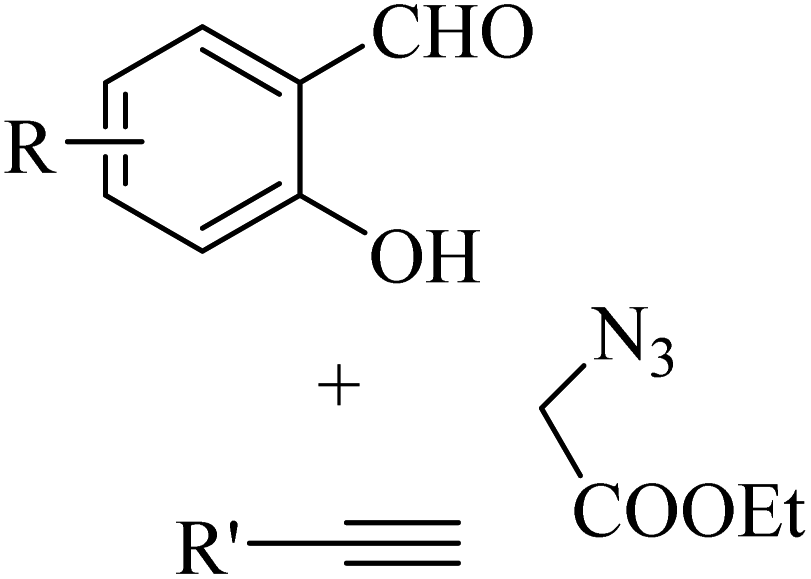
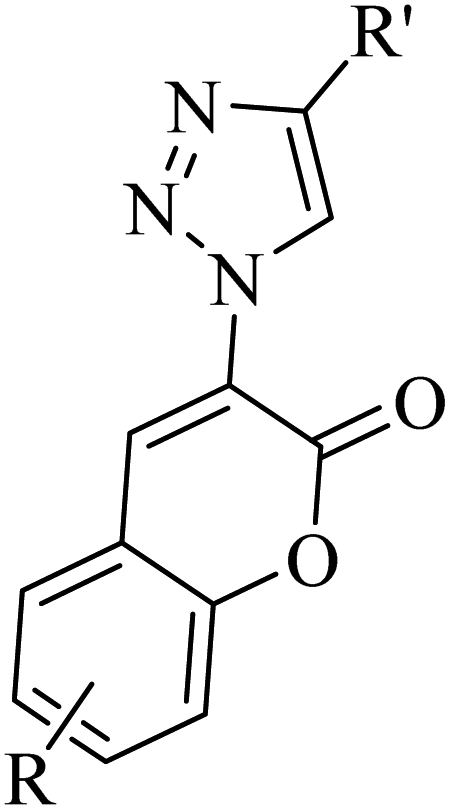
He and co-workers33 synthesized 3-triazolylcoumarin derivatives 14 via a one-pot, copper-catalyzed multicomponent reaction. The desired product was produced from the reaction of arylacetylenes 13, salicylaldehydes 11, ethyl 2-azidoacetate 12, and CuI in EtOH and K3PO4 at 50 °C for 5 h (Scheme 5).
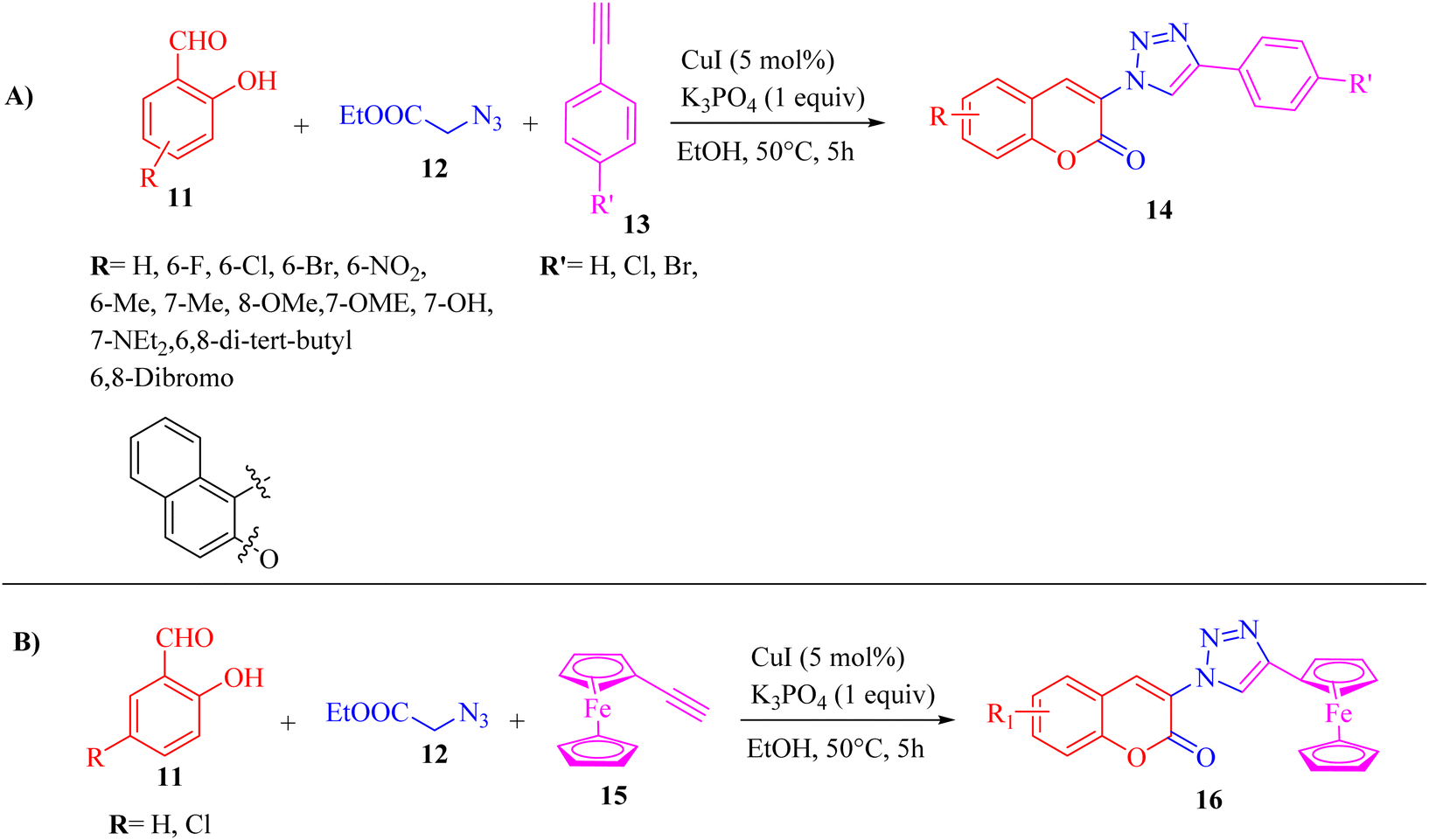 | ||
| Scheme 5 (A) Synthesis of 3-triazolylcoumarin derivatives 14 and (B) synthesis of 3-(4-ferrocenyl)-trizaolycoumarins 16.33 | ||
Initially, 12 and 13 generates A via the copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) reaction. Then, A reacts with an aldehyde group of 11 in the presence of K3PO4 via Knoevenagel condensation, generating intermediate B. Next, in the presence of K3PO4 the intermediate C is generated. Subsequently, 14 is generated via the intramolecular nucleophilic addition of C, followed by transesterification (Scheme 6).
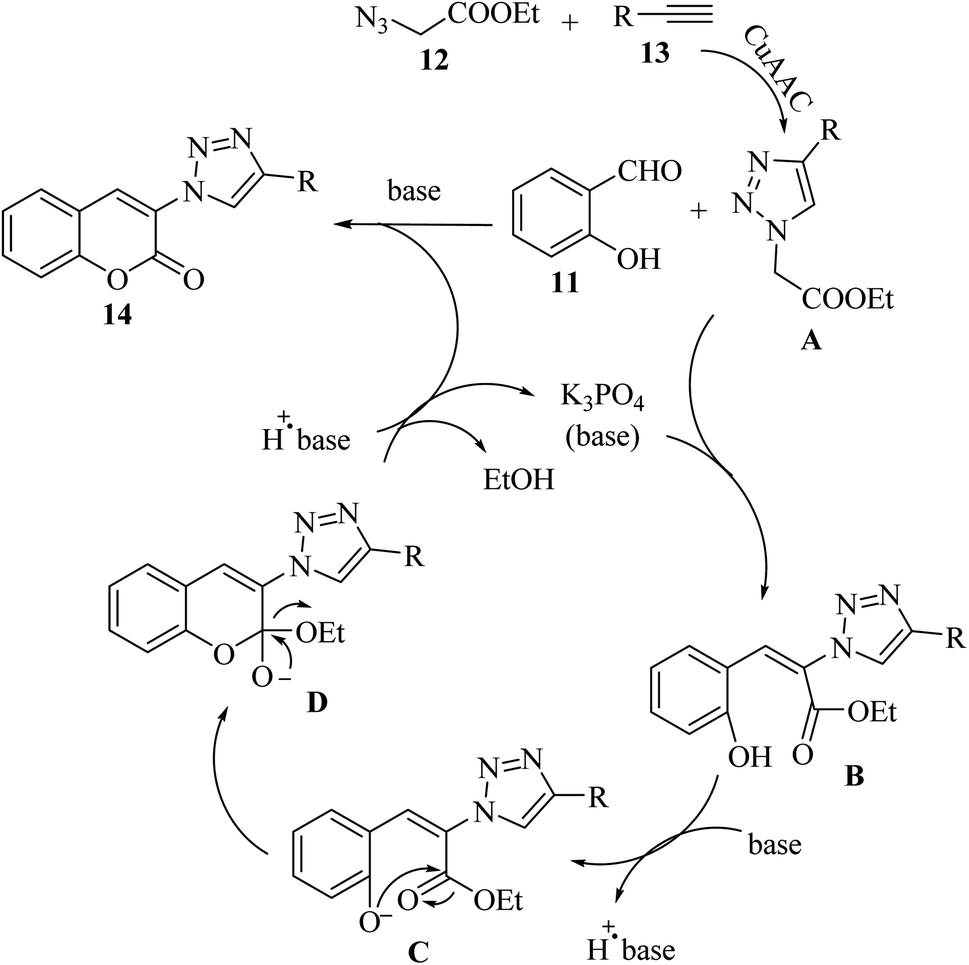 | ||
| Scheme 6 Possible reaction mechanism for the synthesis of 3-triazolylcoumarin derivatives.33 | ||
The above-mentioned method has advantages such as eco-friendly nature, convenience and production of substituted 3-trizolylcoumarin derivatives in moderate to excellent yields. According to this study, halide substituents in arylacetylenes were compatible to produce the desired triazolylcoumarin derivatives in good yield in the range of 67% to 82%. Also, ferrocenylacetylene 15 was investigated via the one-pot optimized condition, which produced the desired products 16 in a good yield (R = H, 85% and R = Cl, 87%).
Saikia and co-workers34 synthesized 1,4-disubstituted 1,2,3-triazole derivatives 20 via the reaction of benzyl bromide derivatives 19, NaN3 18, and alkyne derivatives 17 in the presence of Cu(II) precatalyst in MeOH at 65 °C for 8–10 min under microwave irradiation (Scheme 7).
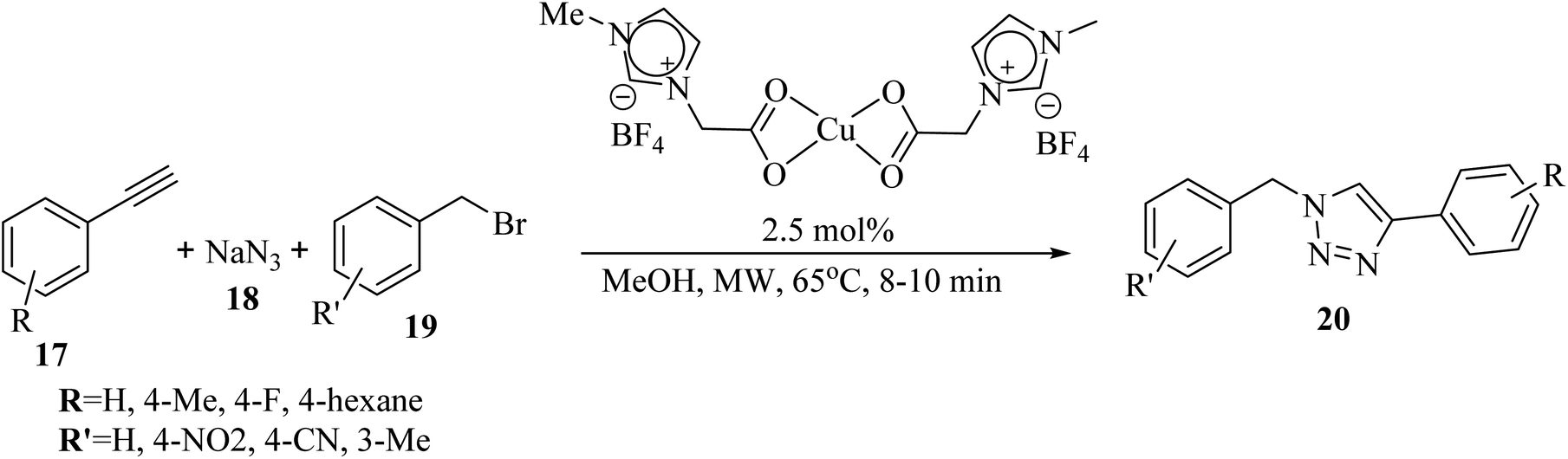 | ||
| Scheme 7 Synthesis of 1,4-disubstituted 1,2,3-triazole derivatives 20 using a ionic liquid-supported Cu(II) catalyst.34 | ||
In the first stage, the reaction between 18 and organic halides in MeOH forms azides. Then, the Cu(II) catalyst is added and reduced by MeOH to Cu(I) species. Subsequently, 17 is added and forms T. Finally, 20 is generated via the protonolysis of T (Scheme 8).
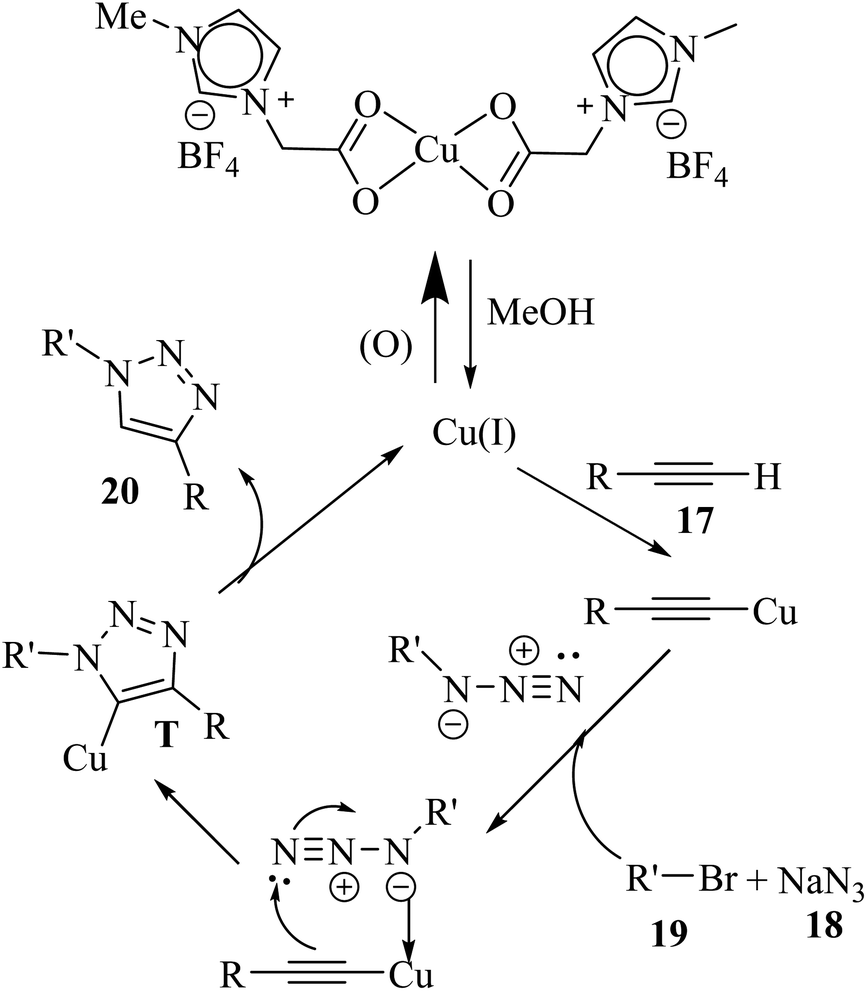 | ||
| Scheme 8 Possible reaction mechanism for the synthesis of 1,4-disubstituted 1,2,3-triazole derivatives.34 | ||
Different types of benzyl bromide and alkyne derivatives were investigated, and results showed that the corresponding products were produced in 85–95% yield. Also, aromatic alkyne derivatives bearing an electron-withdrawing substitute such as F did not have an effect on the reaction result.
Zhang and co-workers35 synthesized 5-trifluoromethylthio-1,2,3-triazole derivatives 23 using S8, alkynes 21, (trifluoromethyl)trimethylsilane (TMSCF3), and 4-methoxybenzyl azide (PMBN3) 22 (Scheme 9).
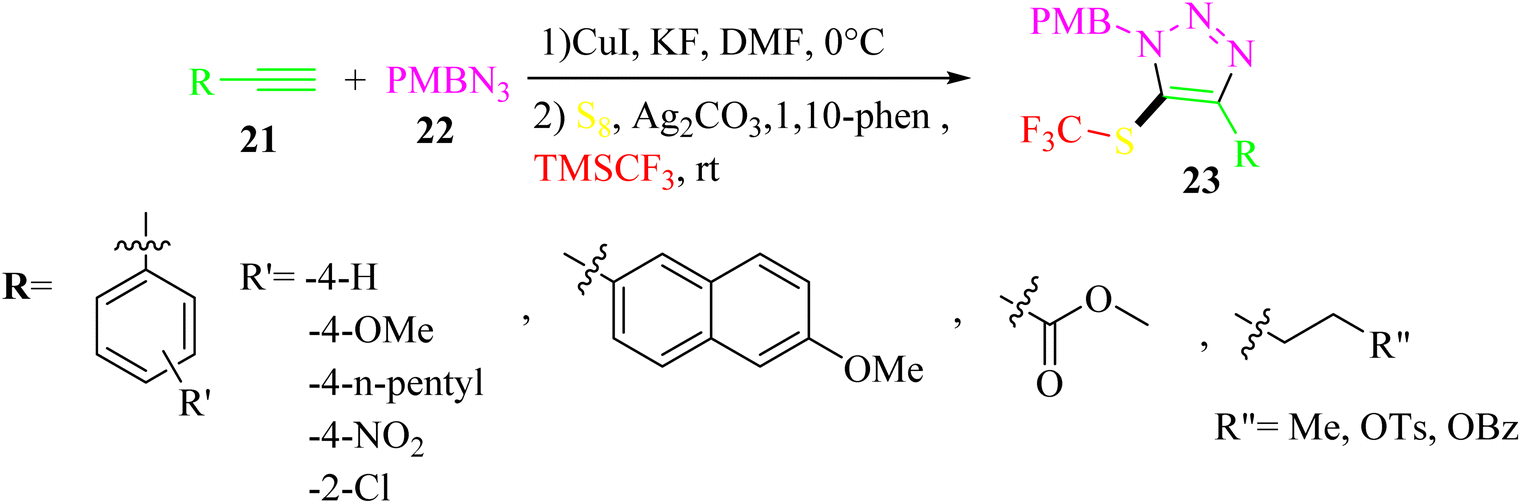 | ||
| Scheme 9 Synthesis of 5-trifluoromethylthio-1,2,3-triazole derivatives 23.35 | ||
The desired products were obtained in two steps. In the first step, PMBN3, alkynes, CuI, and KF in DMF were mixed at 0 °C for 3 h. In step 2, S8, Ag2CO3 as a silver salt, 1,10-phen as a ligand, and TMSCF3 were mixed at room temperature for 12 h.
S8 and TMSCF3 convert to [(phen)Cu(SCF3)]2 in the presence of CuI, 1,10-phen, and KF, which reacts with intermediate S and generates key intermediate K. Finally, 23 is prepared via the reductive elimination of K (Scheme 10).
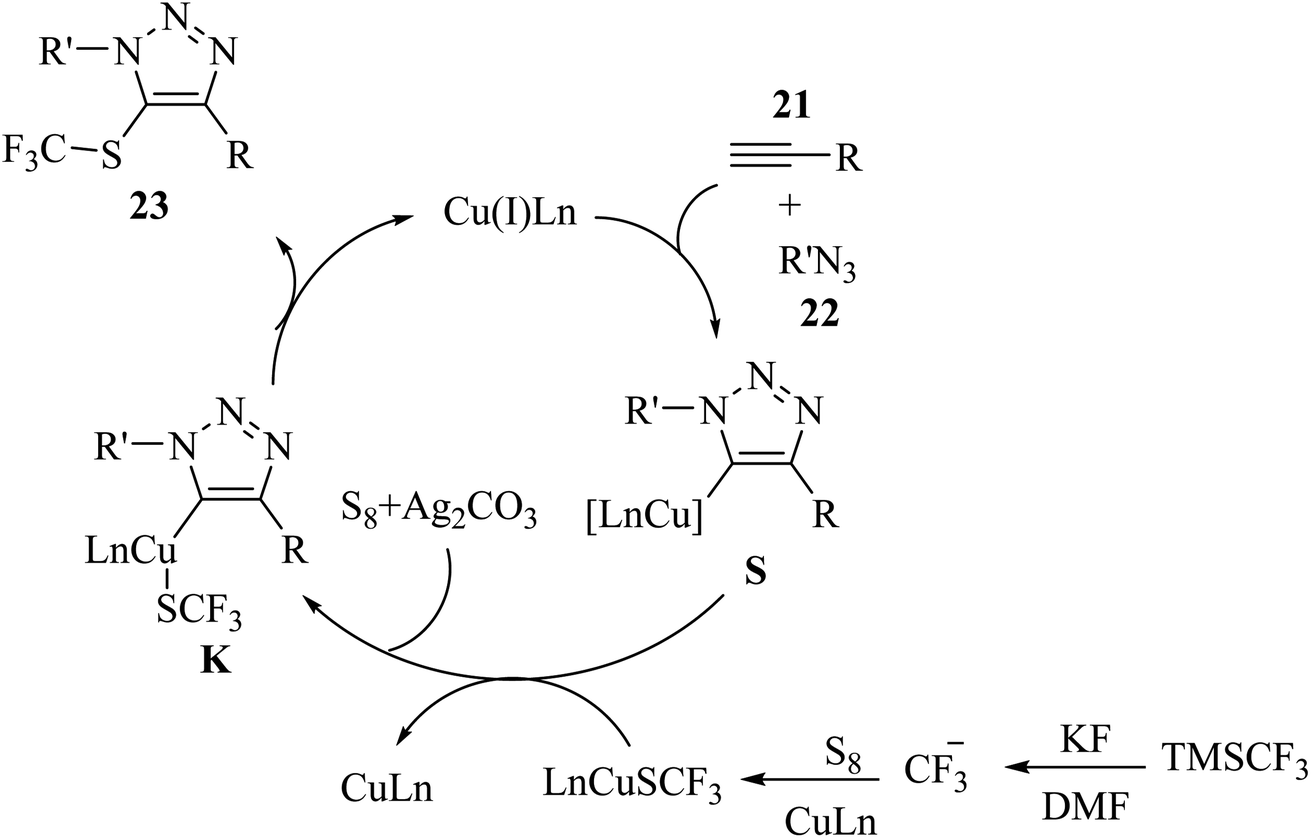 | ||
| Scheme 10 Possible reaction mechanism for the synthesis of 5-trifluoromethylthio-1,2,3-triazole derivatives 23.35 | ||
Alkyne derivatives with electron-donating substituents on the aryl gave the desired products in 74–75% yield, whereas electron-withdrawing groups led to lower yields with an isolated yield of 30–48%. Also, alkyl alkynes produced the corresponding triazole derivatives in moderate yields in the range of 36–56%.
Khandar et al.36 synthesized 1,4-disubstituted 1,2,3-triazoles 28 via the reaction of R′N3 26, alkyne derivatives 27 and benzyl halide derivatives (24 and 25) using a novel binuclear copper(II)–oxalate complex as the catalyst in H2O at 70 °C (Scheme 11). Various benzyl chloride derivatives 24 with both electron-donating and -withdrawing groups (H, 2-Me, and 4-NO2) were reacted with phenylacetylene and produced the corresponding triazole derivatives in 99%, 77%, and 87% yields, respectively. It seems that 2-Me substituted benzyl chloride resulted in a much lower yield due to the steric hindrance effect. Also, substituting phenylacetylene with dimethyl propargyl alcohol and propargyl alcohol decreased the reaction yield (34% and 64%, respectively). In addition, the reaction of benzyl bromide 25 with dimethyl propargyl alcohol and propargyl alcohol led to the production of the corresponding products in 73% and 74% yield, respectively.
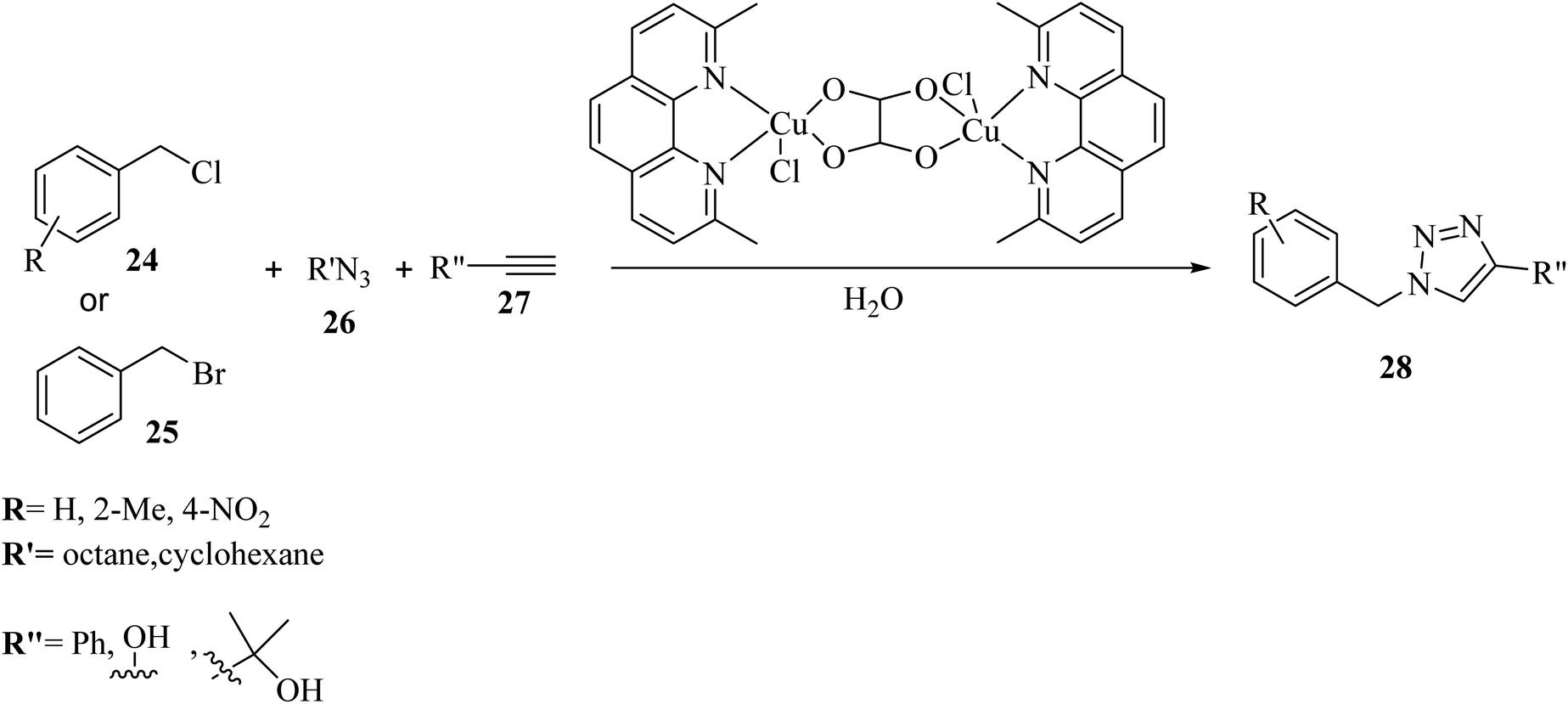 | ||
| Scheme 11 Synthesis of 1,4-disubstituted 1,2,3-triazoles 28 via the binuclear copper(II)–oxalate complex containing 2,9-dimethyl-1,10-phenanthroline.36 | ||
Liu and co-workers37 synthesized sulfuryl-substituted N-vinyl-1,2,3-triazole derivatives 32 via the reaction of vinyl azide surrogate 30, S- and O-nucleophiles 31 and alkynes 29 in the presence of CuI/Pd(PPh3)4, and Et3N in DMSO at 60 °C for 2 h (Scheme 12).
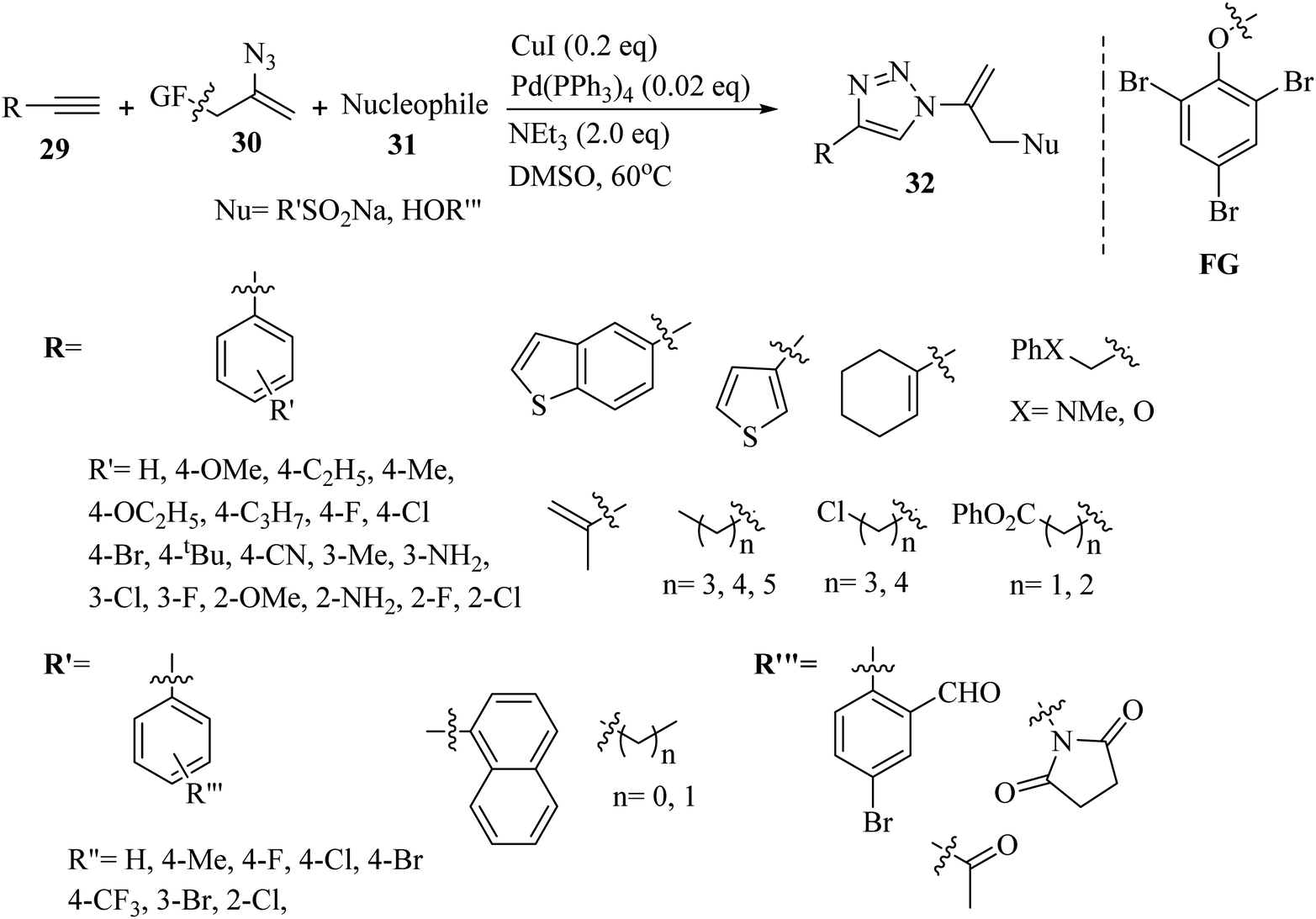 | ||
| Scheme 12 Synthesis of N-vinyl-1,2,3-triazoles 32 via CuI.37 | ||
Active palladium species Q was prepared via the dissociation of Pd(PPh3)4 catalyst, which underwent oxidative addition with 30 to generate complex R. Then, intermediate R reacted with 31 and generated functionalized vinyl azide S. Next, activated acetylide T and intermediate S reacted via a click reaction and produced Cu(I) triazoles U. Finally, 32 was generated via the protolysis of U by trace H2O in solvent or air (Scheme 13).
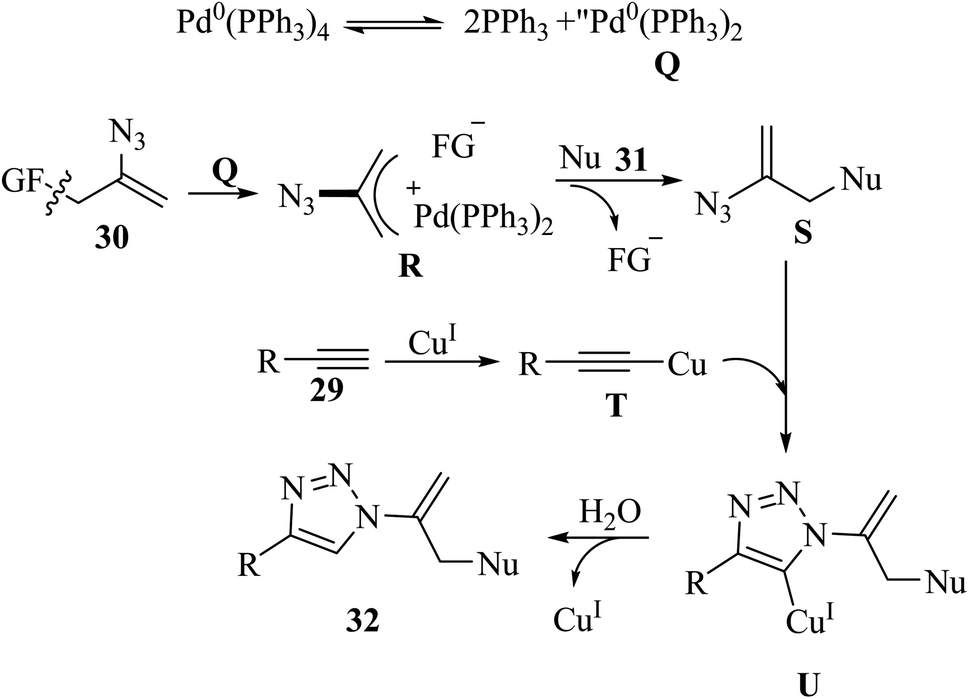 | ||
| Scheme 13 Possible reaction mechanism for the synthesis of sulfuryl-substituted N-vinyl-1,2,3-triazole derivatives 32.37 | ||
In the case of 29, para-substituted aromatic alkyne derivatives bearing various groups gave the desired triazole derivatives in 82% to 88% yield. Also, substrates with meta-substituents such as halogen, Me, and amino gave the desired triazole derivatives in 78% to 88% yield. In addition, ortho-substituents on the benzene ring of the alkyne derivatives were reacted and produced the desired triazole derivatives in 80% to 83% yield. Also, S-containing aromatic alkynes in the reaction were tolerated and the corresponding triazole derivatives obtained in 75–77% yield. Furthermore, alkenyl and aliphatic alkynes were tested and the corresponding triazole derivatives obtained in 72% to 82% yield. A variety of aliphatic alkynes with heteroatoms was reacted and produced the corresponding triazole derivatives in 74% to 79% yield. Various sulfinates were reacted with phenylacetylene and the results showed that aryl sulfinates with para-substituents produced the corresponding triazole derivatives in 78% to 89% yield. Ortho- and meta-substituted aryl sulfinates were also reacted and produced the desired triazole derivatives in 83% to 82% yield, respectively. In addition, 2-naphthyl sulfinate produced the corresponding product in 85% yield. Alkyl-substituted sulfonate derivatives were also evaluated in the above-mentioned method and produced the corresponding triazole derivatives in 74% to 77% yield. Finally, a variety of O-nucleophiles 31 was reacted and the corresponding triazole derivatives obtained in 69% to 83% yield.
Wang and co-workers38 synthesized 5-aryl-1,4-disubstituted 1,2,3-triazole derivatives 36 via one-pot in situ oxidative-coupling. The compounds were prepared using arylboronic acid derivatives 35, organic azide derivatives 33 and alkyne derivatives 34 in the presence of O2/chloramine-T as an oxidant, CuCl as a catalyst, and TEA in toluene at 50 °C (Scheme 14).
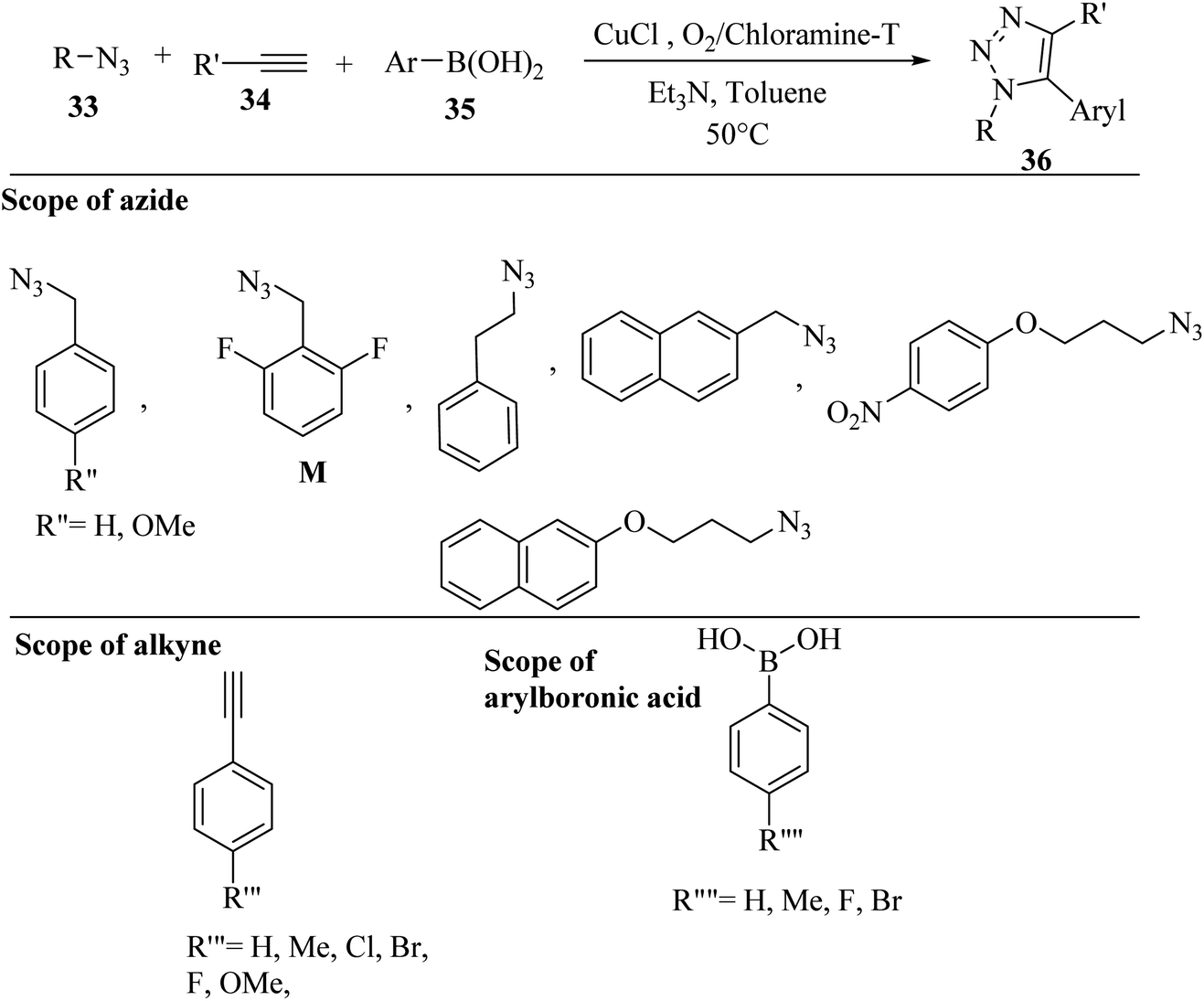 | ||
| Scheme 14 Synthesis of 1,2,3-triazole derivatives 36 using CuCl.38 | ||
Intermediate L was produced via the reaction between 34 and CuCl in the presence of TEA. Then, intermediate L reacted with 33 via the CuAAC reaction and generated triazolide copper Z. Next, Z reacted with 35 under oxidative conditions and generated X. It should be noted that transmetalation must take place, and then the protonation of Z (Scheme 15).
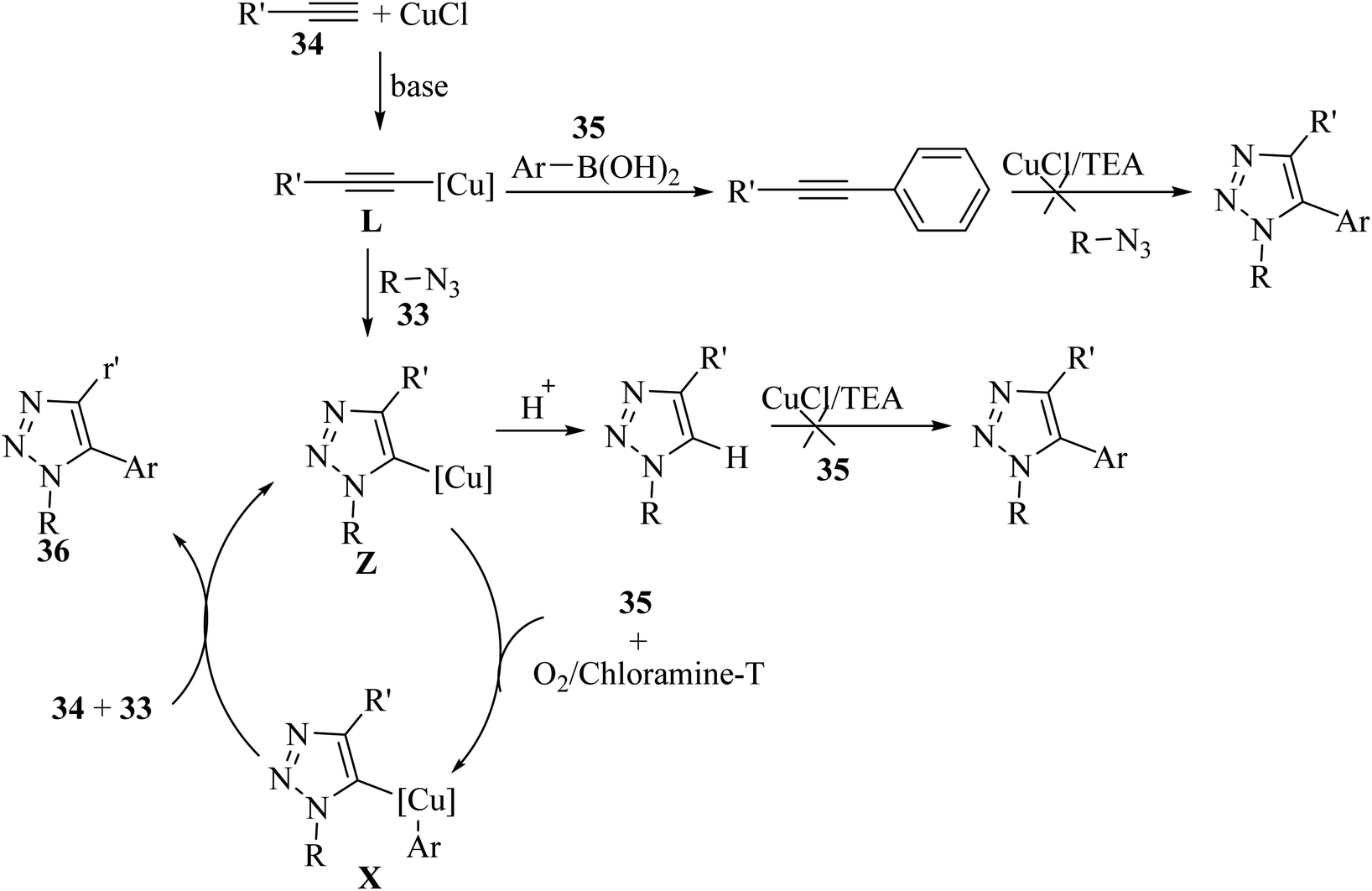 | ||
| Scheme 15 Possible reaction mechanism for the synthesis of 5-aryl-1,4-disubstituted 1,2,3-triazole derivatives 36.38 | ||
The benefits of this method include the use of a low reaction temperature, low-cost catalyst, and less-toxic additive. The reaction among aryl alkynes with various substituents, arylboronic acids, and benzyl azides produced 5-aryl-1,2,3-triazoles in 80–90% yield. The reaction among various alkyl azides with different substituted groups, benzyl azides, and arylboronic acids produced triazole derivatives in moderate to good yield (68–80%). Azide M with 2,5-difluorobenzyl motifs that exist in the clinic drug rufinamide reacted with arylboronic acids and terminal alkynes and produced triazole derivatives, showing that this method has potential applications for drug screening.
Camberlein and co-workers39 synthesized 1,5-disubstituted 1,2,3-triazole derivatives 40 via the reaction between NaN3 39, alkyne derivatives 38, and bromides 37 in the presence of Cp2Ni (precatalyst), Cs2CO3, and xantphos ligand in DMF under microwave irradiation at 50 °C for 4 h (Scheme 16). Various alkynes (aromatic and aliphatic) 38 were evaluated, and the desired triazole derivatives were obtained in 27% to 84% yield. According to the regioselectivity of para-NO2 phenylacetylene (ratio 66![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :34) with respect to phenylacetylene (ratio 89:11), and then para-F phenylacetylene (ratio 91:9) and para-OMe phenylacetylene, it had a positive mesomeric effect on the phenyl ring, but did not affect the yield. Interestingly, heteroaryl and alkyl chains can be employed in the above-mentioned reaction. In the case of the scope of bromide derivatives 37, aromatic benzyl bromides bearing electron-donating or electron-withdrawing groups after flash chromatography purification (64–79%) and semi-preparative SFC (31–84%) purification produced the corresponding 1,5-regioisomers in high yields. The ortho isomer in this series was obtained in the lowest yield due to the steric effect. The regioisomerism of the phenyl substitution did not influence the regioselectivity (ratio = 85:15 to 97:3) and only had a minor effect on the yield. Also, functional groups (ester and carbamate-protected amine) were tolerated in the reaction and produced the corresponding triazole derivatives in 34% and 90% yield, respectively.
:34) with respect to phenylacetylene (ratio 89:11), and then para-F phenylacetylene (ratio 91:9) and para-OMe phenylacetylene, it had a positive mesomeric effect on the phenyl ring, but did not affect the yield. Interestingly, heteroaryl and alkyl chains can be employed in the above-mentioned reaction. In the case of the scope of bromide derivatives 37, aromatic benzyl bromides bearing electron-donating or electron-withdrawing groups after flash chromatography purification (64–79%) and semi-preparative SFC (31–84%) purification produced the corresponding 1,5-regioisomers in high yields. The ortho isomer in this series was obtained in the lowest yield due to the steric effect. The regioisomerism of the phenyl substitution did not influence the regioselectivity (ratio = 85:15 to 97:3) and only had a minor effect on the yield. Also, functional groups (ester and carbamate-protected amine) were tolerated in the reaction and produced the corresponding triazole derivatives in 34% and 90% yield, respectively.
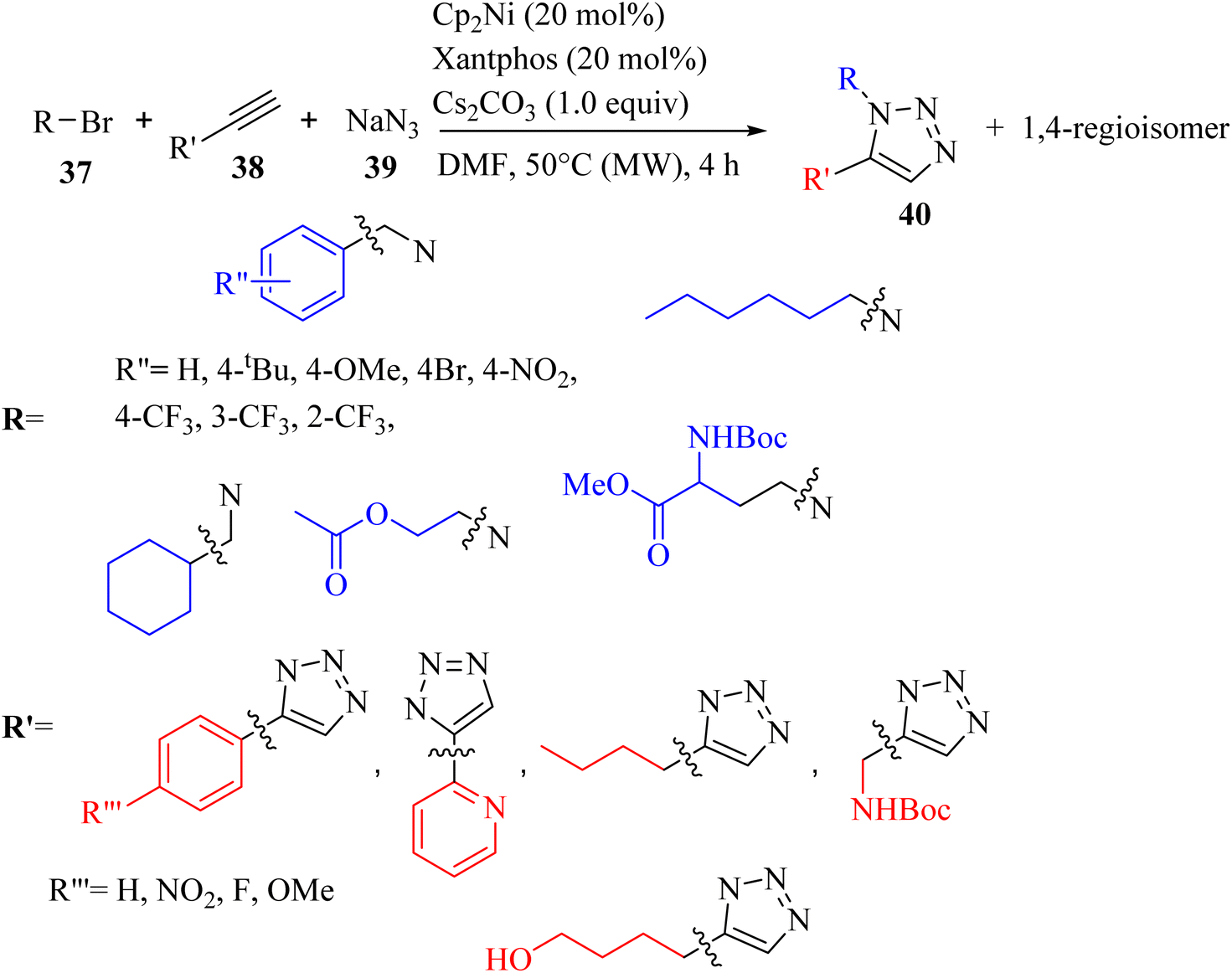 | ||
| Scheme 16 Synthesis of 1,2,3-triazole derivatives using the nickelocene (Cp2Ni) precatalyst.39 | ||
Yuan et al.40 synthesized 5-iodo-1,4-substituted 1,2,3-triazoles 44 via the reaction among terminal alkyne derivatives 41, organic azide derivatives 42, and NaI 43 in the presence of CuI, DIPEA as a base, chloramine-T as an oxidant, and L as a ligand in THF under an N2 atmosphere at 50 °C (Scheme 17).
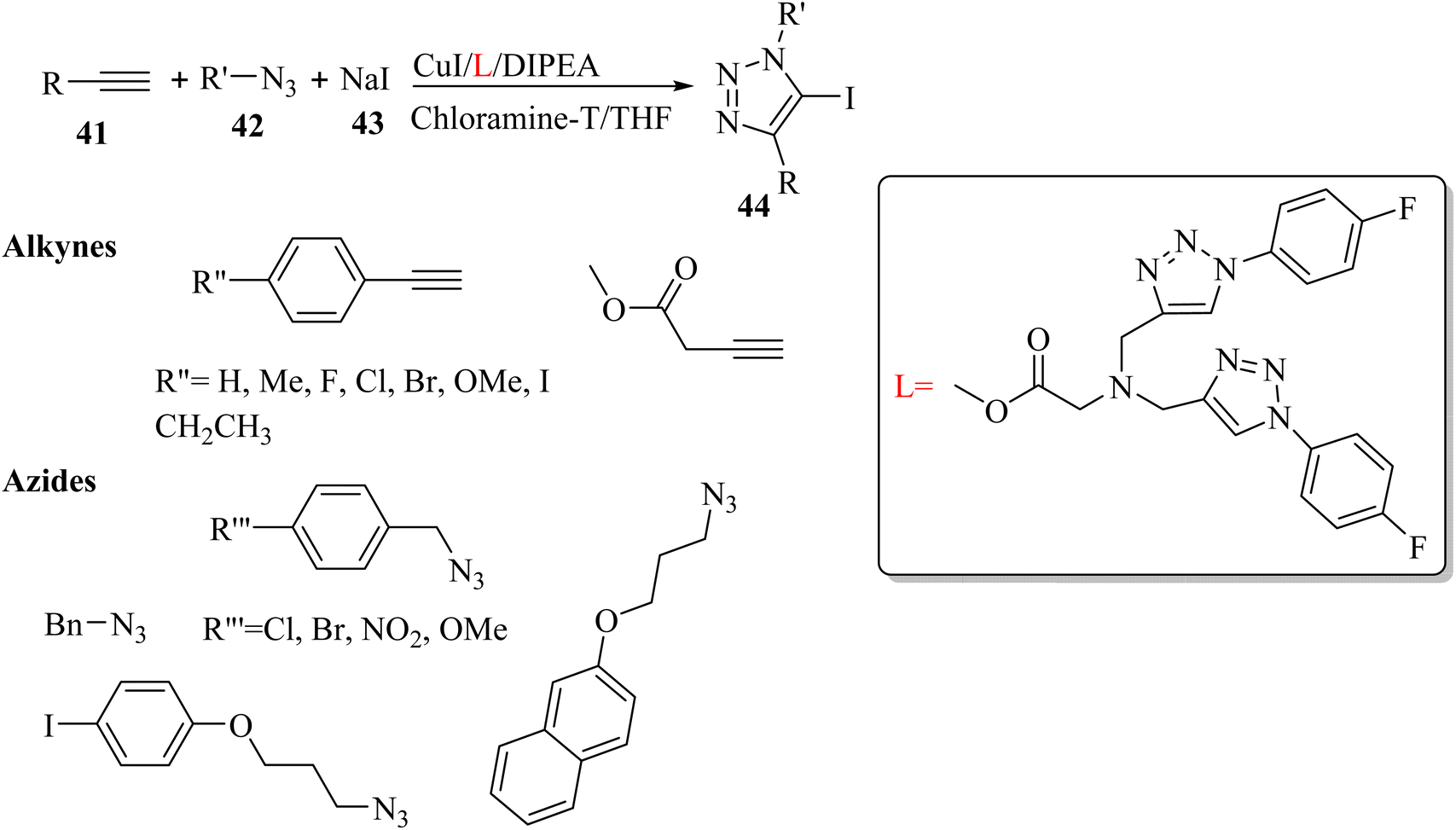 | ||
| Scheme 17 Synthesis of 5-iodo-1,4-substituted 1,2,3-triazoles 44 by CuI.40 | ||
Two reaction pathways were proposed by the authors. It should be noted that 43 was oxidized to electrophilic “I+” by chloramine-T. In pathway B, iodoalkyne(III) O may be generated via iodination directly on 41, whereas in pathway A, iodination may occur on intermediate copper triazolide(II) P (Scheme 18).
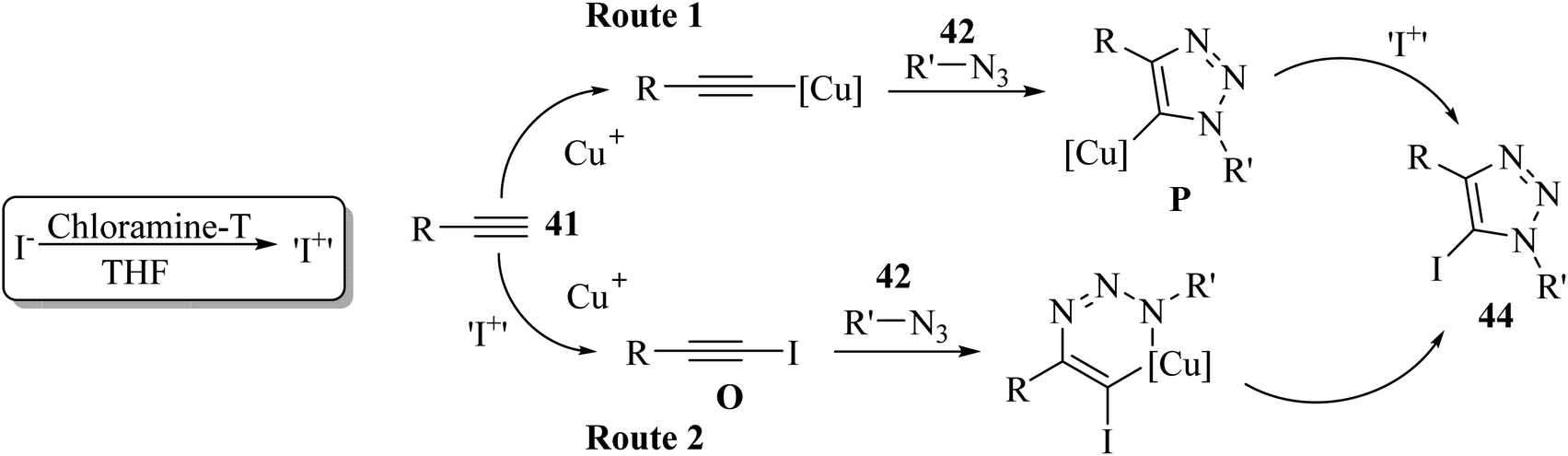 | ||
| Scheme 18 Possible reaction mechanism for the synthesis of 5-iodo-1,4-substituted 1,2,3-triazoles 44.40 | ||
Also, 41 and 42 were investigated. Phenylacetylenes bearing various groups were reacted and produced the desired triazole derivatives in 71% to 78% yield. Also, benzyl azides with electron-withdrawing and -donating substituents were tested and the desired triazole derivatives obtained in 67% to 84% yield. In addition, alkyl azides were tolerated in the reaction and the desired triazole derivatives obtained in 73% to 74% yield.
In 2021, Wang and co-workers41 synthesized triazoles bearing β-hydroxy chalcogenides (49 and 49′) via the reaction among terminal alkyne derivatives 45, azide derivatives 46, epoxide derivatives (47 and 47′), and Se/K2S 48 in the presence of CuCl, Me4Phen as a ligand, KOH as a base, and TBAI as an additive in acetone/H2O [1/1(v/v)] at 25 °C for 6 h under an air atmosphere (Scheme 19).
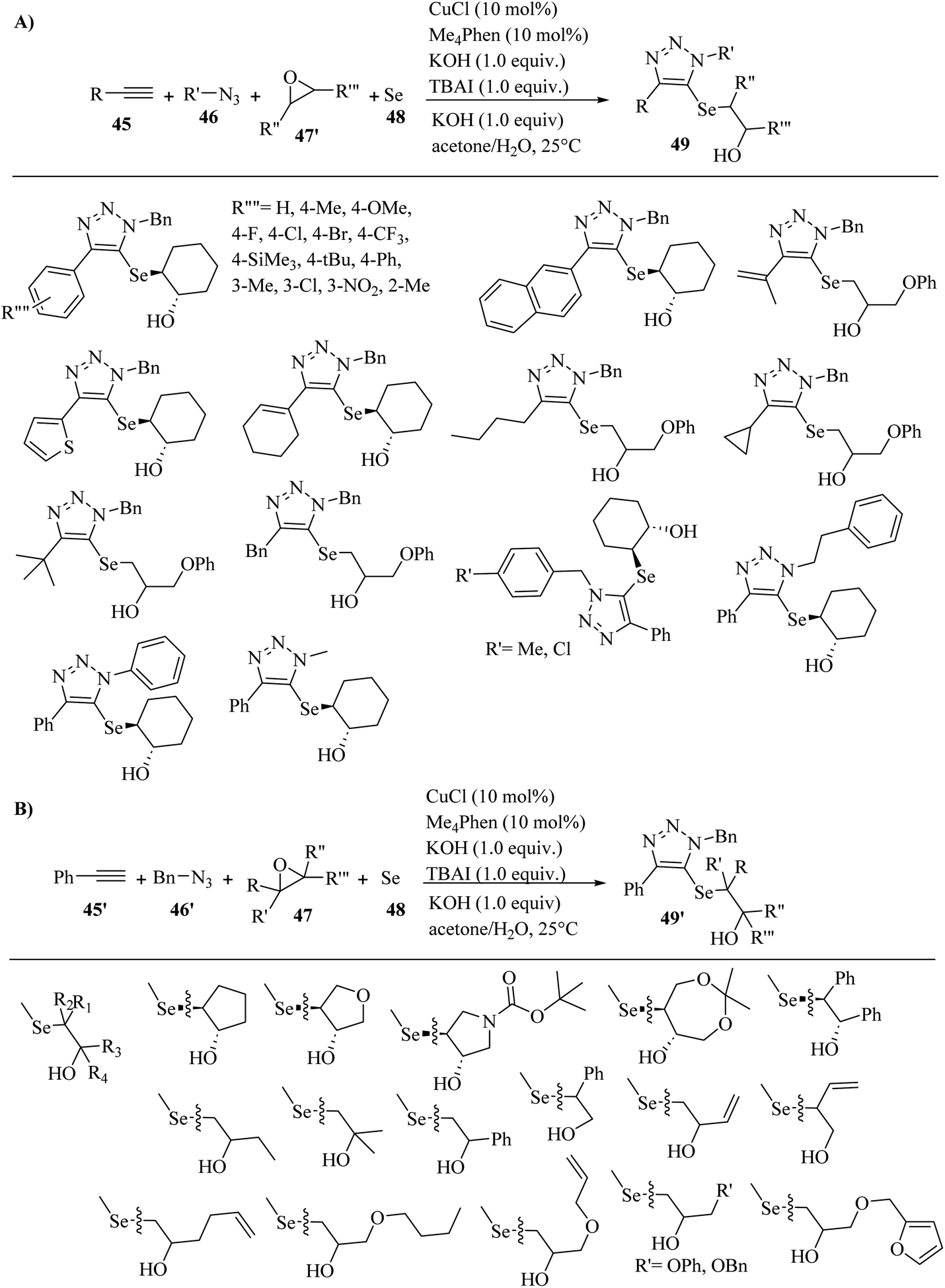 | ||
| Scheme 19 Synthesis of triazole derivatives using CuCl catalyst. (A) Substrate scope of alkyne derivatives and azide derivatives. (B) Substrate scope of epoxide derivatives.41 | ||
Initially, Se2− and SeO32− were produced via the disproportionation reaction of Se in the presence of KOH. Then, 45 reacted with copper salt A and generated copper(I) acetylide B. Subsequently, B reacted with 46 and produced triazolyl-copper D by [3 + 2] cycloaddition. In this stage, cycloaddition included the coordination of 46 with B to afford a six-membered ring intermediate, and then generated 5-cuprated triazole via reductive elimination. Next, copper(III) species E was generated via the ligand exchange of D with Se2−/S2− and oxidation by SeO32−/O2. In the next step, E generated triazolylchalcogenide anion F via reductive elimination together with the release of the copper(I) catalyst. Finally, 49 or 49′ was produced by nucleophilic attack of F to 47 (Scheme 20).
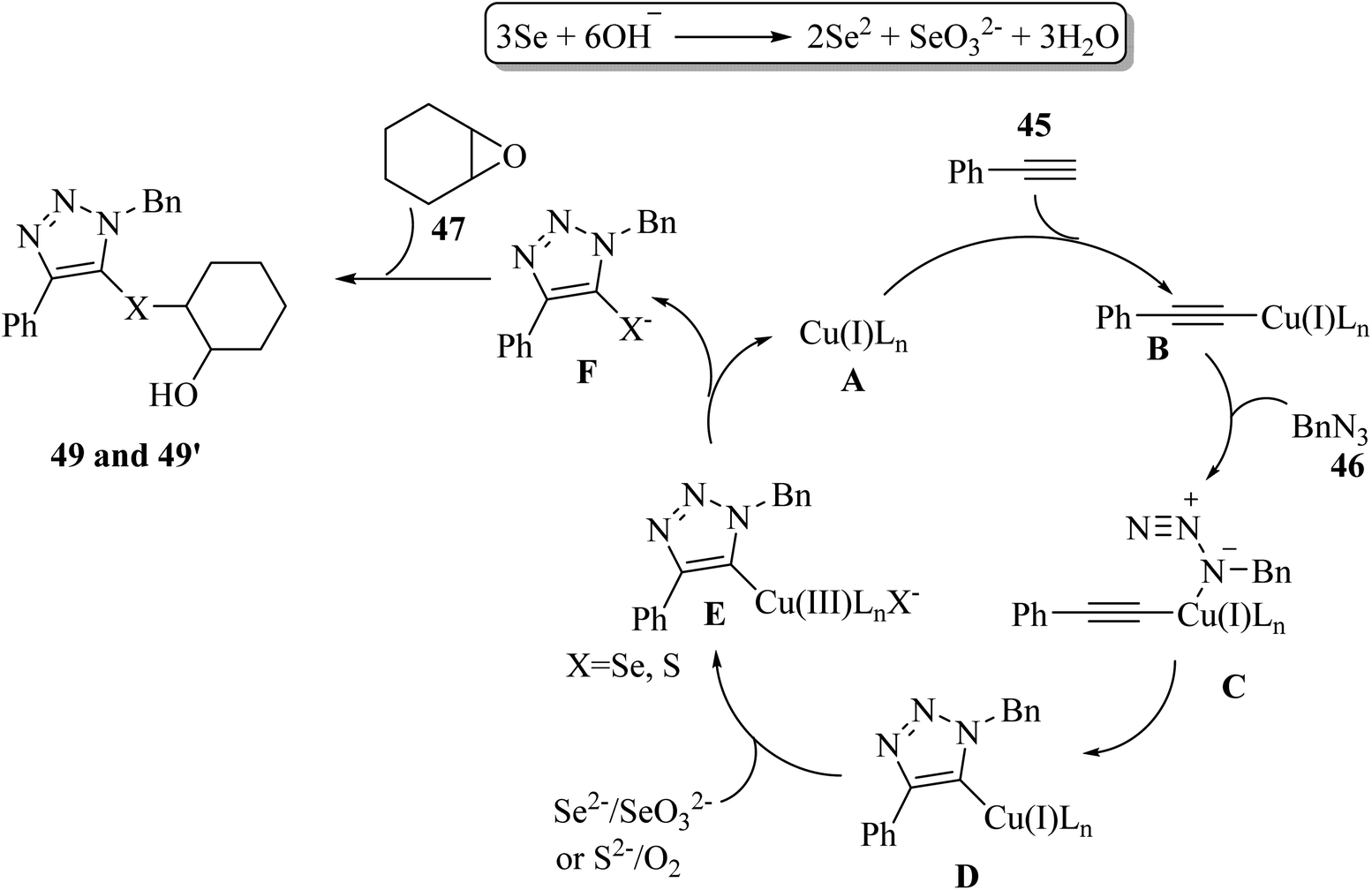 | ||
| Scheme 20 Possible reaction mechanism for the synthesis of triazoles bearing β-hydroxy chalcogenides.41 | ||
In the case of 45 (Scheme 11a), phenylacetylenes with both electron-withdrawing and -donating groups (Me, OMe, F, Cl, Br, CF3, Me3Si, tBu, Ph, and NO2) on the benzene ring were reacted and produced the desired triazole derivatives in 39% to 81% yield and >20:1 dr. Importantly, the electronic effect of phenylacetylenes affected the yield. Phenylacetylene derivatives containing CF3 and NO2 gave the products in low yields (45% and 39%, respectively). In addition, thienylacetylene and naphthylacetylene reacted well and produced the corresponding products in 82% and 65% yield, respectively. Also, alkenyl-substituted alkyne derivatives showed high reactivity under the above-mentioned conditions and produced the corresponding products in 64–65% yield. Moreover, alkyl-substituted alkyne derivatives containing 3,3-dimethylbut-1-yne, ethynylcyclopropane, prop-2-yn-1-ylbenzene, and hex-1-yne were reacted with elemental selenium, epoxide, and azide and produced the corresponding products in 70%, 66%, 72%, and 70% yield, respectively. In the case of 46 (Scheme 19A), Cl- and Me-substituted benzyl azides were tested and produced the corresponding triazole derivatives in 68% and 76% yield, respectively. Also, phenethyl azide was tested and produced the corresponding triazole in 68% yield. In addition, phenyl azide was tested and produced the desired triazole in 40% yield. Especially, (trimethylsilyl)methyl azide also reacted well and produced the 1-Me substituted 1,2,3-triazole in 54% yield and >20:1 dr. In the case of the scope of 47 (Scheme 19B), epoxide derivatives containing linear, aromatic, aliphatic, and cyclic epoxides reacted with elemental selenium 48, phenylacetylene 45′, and benzyl azide 46′ and produced the corresponding 5-selenotriazoles 49′ in 47% to 79% yield.
In 2023, Sun et al.42 synthesized ditriazolyl diselenides (53 and 53′) via the reaction among azides (51 and 51′), terminal alkyne derivatives 50, and elemental selenium 52 in the presence of Cu(CH3CN)4PF6, Me4phen, KOH, and tetrabutylammonium bromide in dioxane/H2O at 25 °C for 12 h (Scheme 21).
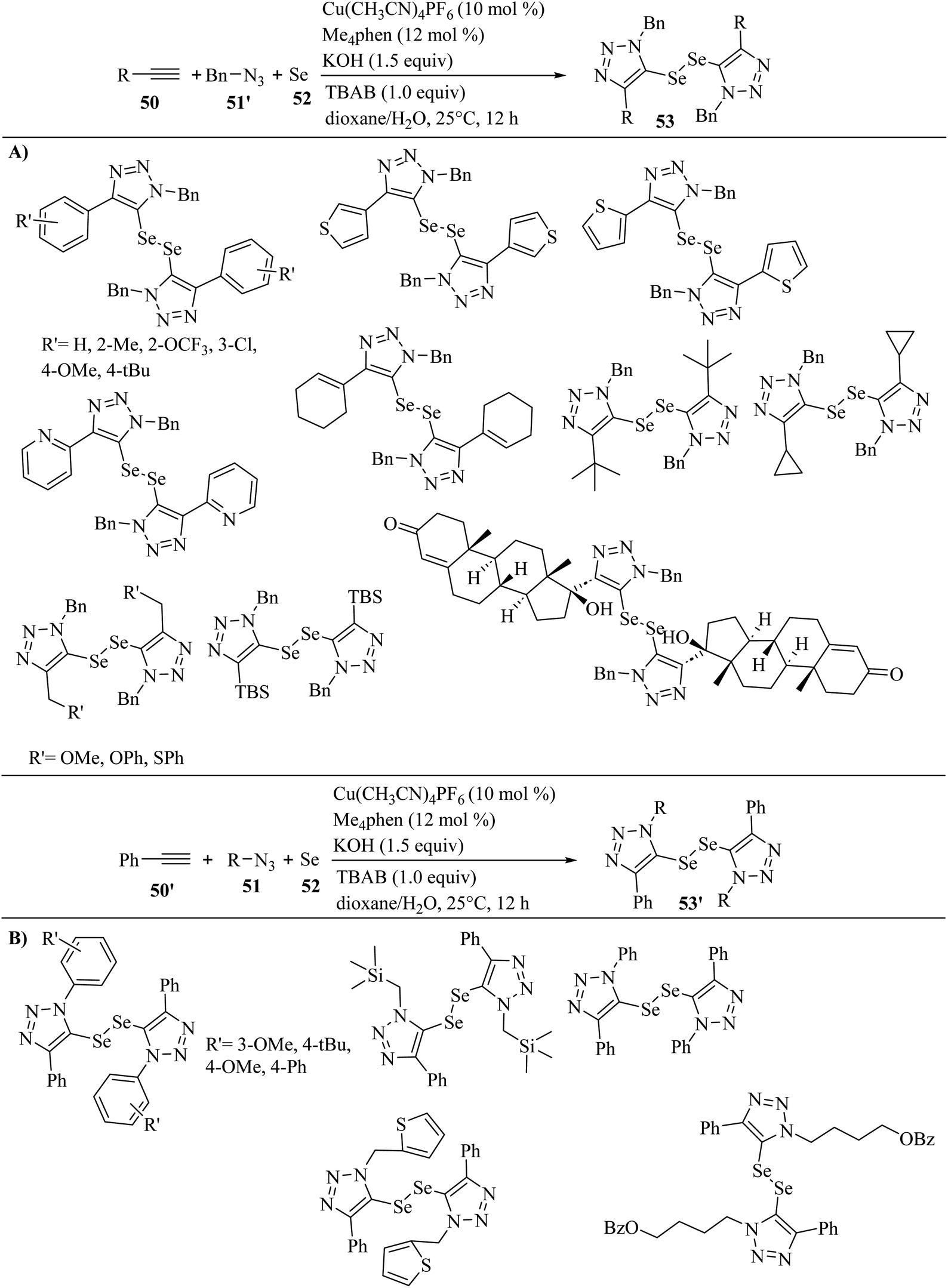 | ||
| Scheme 21 Synthesis of ditriazolyl diselenides in the presence of Cu(CH3CN)4PF6. (A) Substrate scope of alkynes and (B) substrate scope of azides.42 | ||
SeO32− and Se2− were produced via the disproportionation of 52 in the presence of KOH. Then, Cu(I) reacted with 50 and generated alkynyl copper S. Next, S in the presence of 51 underwent cycloaddition and generated 5-cuprated triazole U. After that, copper(III) species V was produced via the exchange of U with Se22− and oxidation by SeO32−. Finally, intermediate V generated 53 or 53′ and Cu(I) via reductive elimination (Scheme 22).
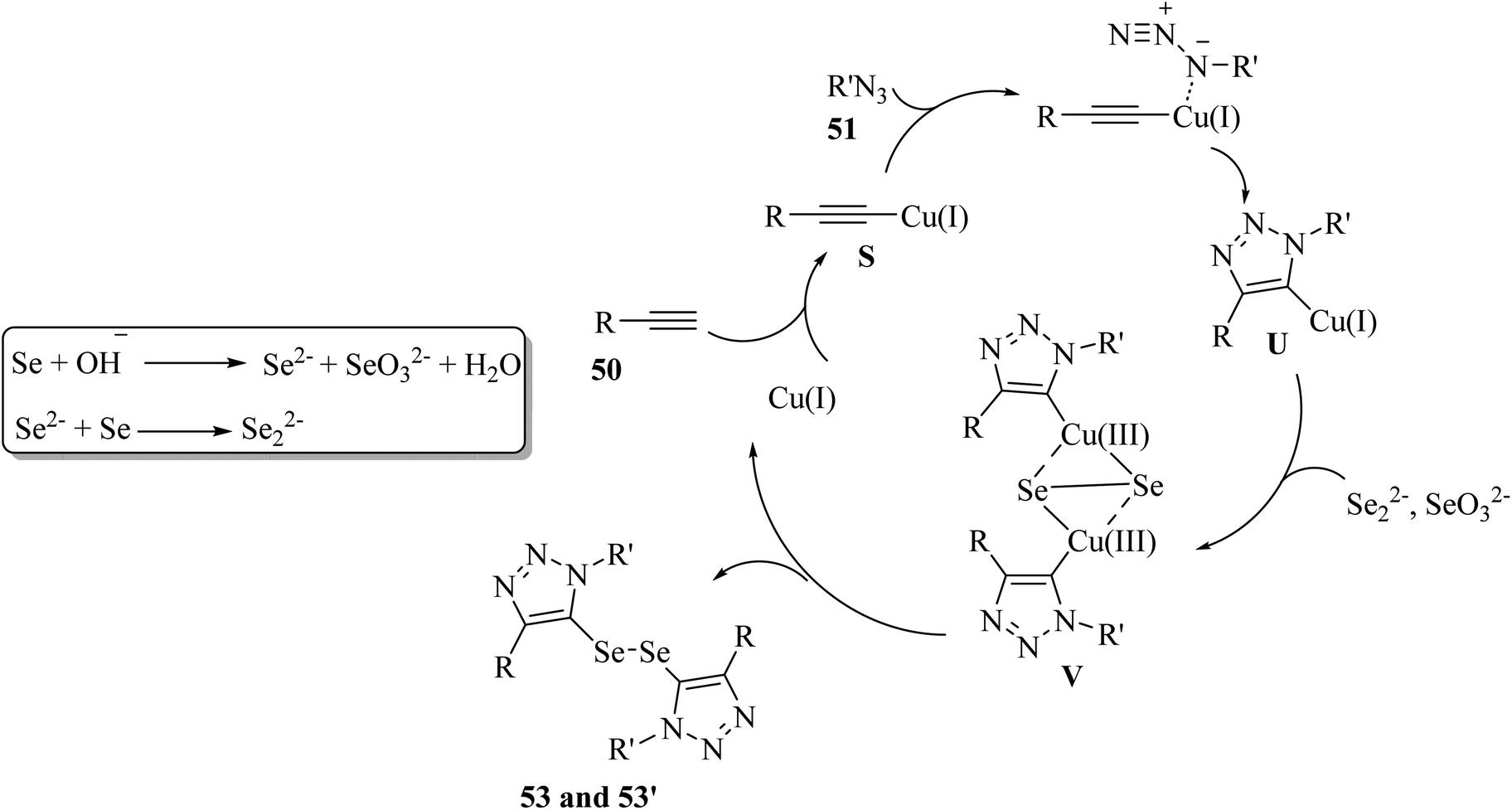 | ||
| Scheme 22 Possible reaction mechanism for the synthesis of ditriazolyl diselenides.42 | ||
The above-mentioned reaction produced the corresponding products with high atom-economy. In the case of 50 (Scheme 21A), phenylacetylenes containing Me, tBu, OMe, OCF3, and Cl on the phenyl ring were tested and produced the corresponding products in 40–75% yield. Also, ethynylthiophenes showed high reactivity and produced the corresponding ditriazolyl diselenides in 53–57% yield. Unfortunately, 2-ethynylpyridine did not produce the corresponding product. Alkenyl-substituted alkyne in the above-mentioned reaction was tested and produced the corresponding ditriazolyl diselenide in 62% yield. Also, alkyl-substituted alkynes in the above-mentioned method were tolerated in the present transformation. Moreover, phenoxy-, phenylthio-, and methoxy-incorporated alkyl alkynes reacted well and the corresponding ditriazolyl diselenides obtained in 51–73% yield. Also, butyldimethylsilyl (TBS)-substituted alkyne was compatible and gave the desired product in 55% yield. It should be noted that ethisterone-derived alkyne demonstrated high reactivity and afforded the desired diselenide in 51% yield. In the case of 51 (Scheme 21B), tBu-, OMe-, and phenyl-substituted benzyl azide derivatives 51 reacted with 50′ and 52 and produced the corresponding ditriazolyl diselenides 53′ in 47–59% yield. Also, 2-(azidomethyl)-thiophene reacted smoothly in the above-mentioned reaction and afforded the desired ditriazolyl diselenide in 73% yield. Subsequently, (trimethylsilyl)-methyl azide reacted well with elemental selenium and phenylacetylene to obtain the desired ditriazolyl diselenide in 48% yield. 4-Azidobutyl benzoate was suitable in the above-mentioned reaction and produced the desired ditriazolyl diselenide in 57% yield. Unfortunately, phenyl azide failed to give the corresponding product in this method.
A summary of the reaction of alkyne derivatives with various RN3 derivatives, and also the limitation of some reactions are presented in Table 1.
| Reactants | Product | Catalyst | Solvent | Method employed | Reaction time | Yield | Limitations | Ref. |
|---|---|---|---|---|---|---|---|---|
 |
 |
CuBr | (1) MeOH | (1) RT, N2 | (1) 8 h | 23–70% | Alkyl-substituted alkynes had poor reactivity and produced trace amount of corresponding product | Wang et al.43 |
| (2) Toluene | (2) hν | (2) 2–6 h | ||||||
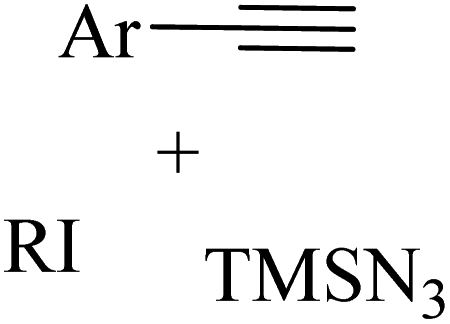 |
 |
Fe(OTf)3 | (1) DME | (1) TBPB, RT | (1) 5–20 min | 30–78% | Reaction of ethynylbenzene and 1-iododecane did not produce the desired product | Xiong et al.44 |
| (2) Toluene | (2) 120 °C | (2) 10 min | ||||||
 |
 |
Ag2SO4 | DMF | K2CO3, H2O, 70 °C | 8 h | 17–86% | Alkyne containing electron-donating group produced the products in lower yields than those with an electron-withdrawing group | Chen et al.45 |
 |
 |
Ag3PO4 | DMSO | H2O, 70 °C | 4–10 h | 52–85% | (1) CC triple bond cleavage |
Liu et al.46 |
| (2) High product yield | ||||||||
| (3) High efficiency | ||||||||
| (4) An unprecedented 4-component reaction | ||||||||
| (5) Good functional group tolerance | ||||||||
 |
 |
CuI | EtOH | K3PO4, 50 °C | 5 h | Trace-90% | (1) Four new bonds formation | He et al.47 |
| (2) Large-scale synthesis | ||||||||
| (3) Eco-friendly | ||||||||
| (4) Mild conditions | ||||||||
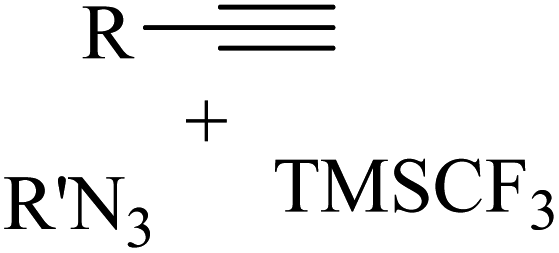 |
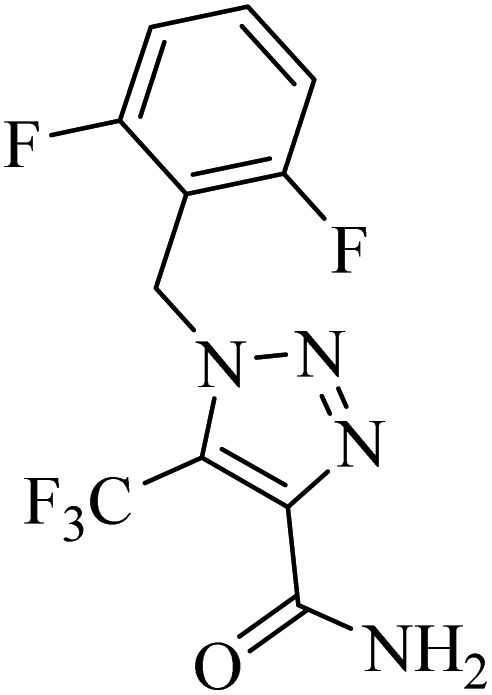 |
CuI | DMF | Phen, Ag2CO3, Et3N, Ar, RT | 15 h | Up to 87% | – 2 equiv. AgNO3 as the oxidant | Cheung et al.48 |
| – Only CF3- substitution | ||||||||
| – Only alkyl azides as valid reactants |
2.2 Reaction of sulfur-containing compounds
Ansari et al.49 synthesized (Z)-β-phenoxy vinylsulfones 57 via the reaction between phenols 56, 1-phenyl-1-propyne 54, and sodium sulfonate derivatives 55 in the presence of I2 and K2CO3 in EtOH at 60 °C (Scheme 23).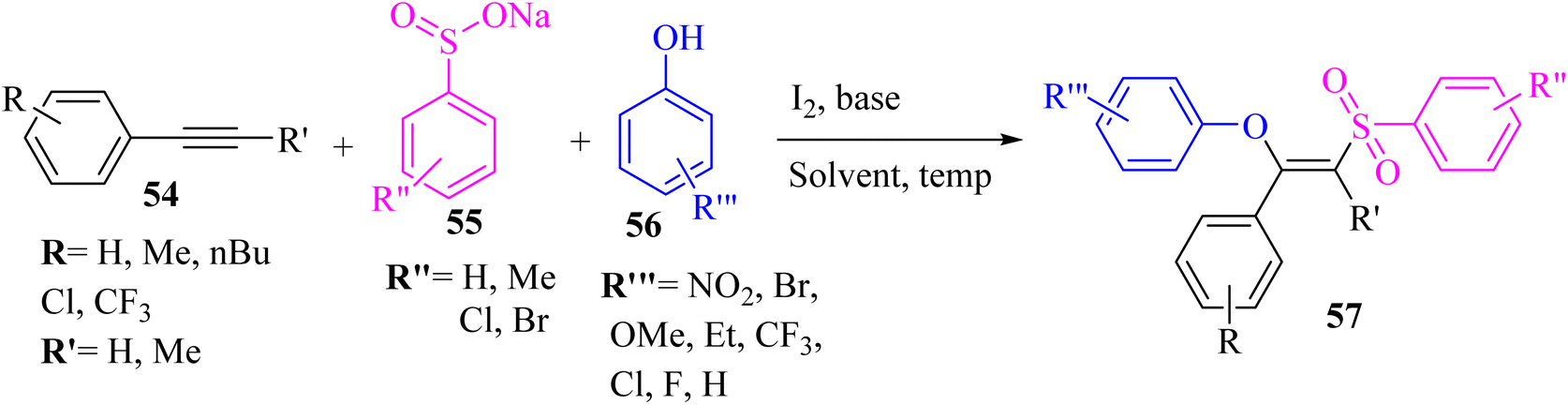 | ||
| Scheme 23 Synthesis of (Z) β-phenoxy vinylsulfone derivatives 57.49 | ||
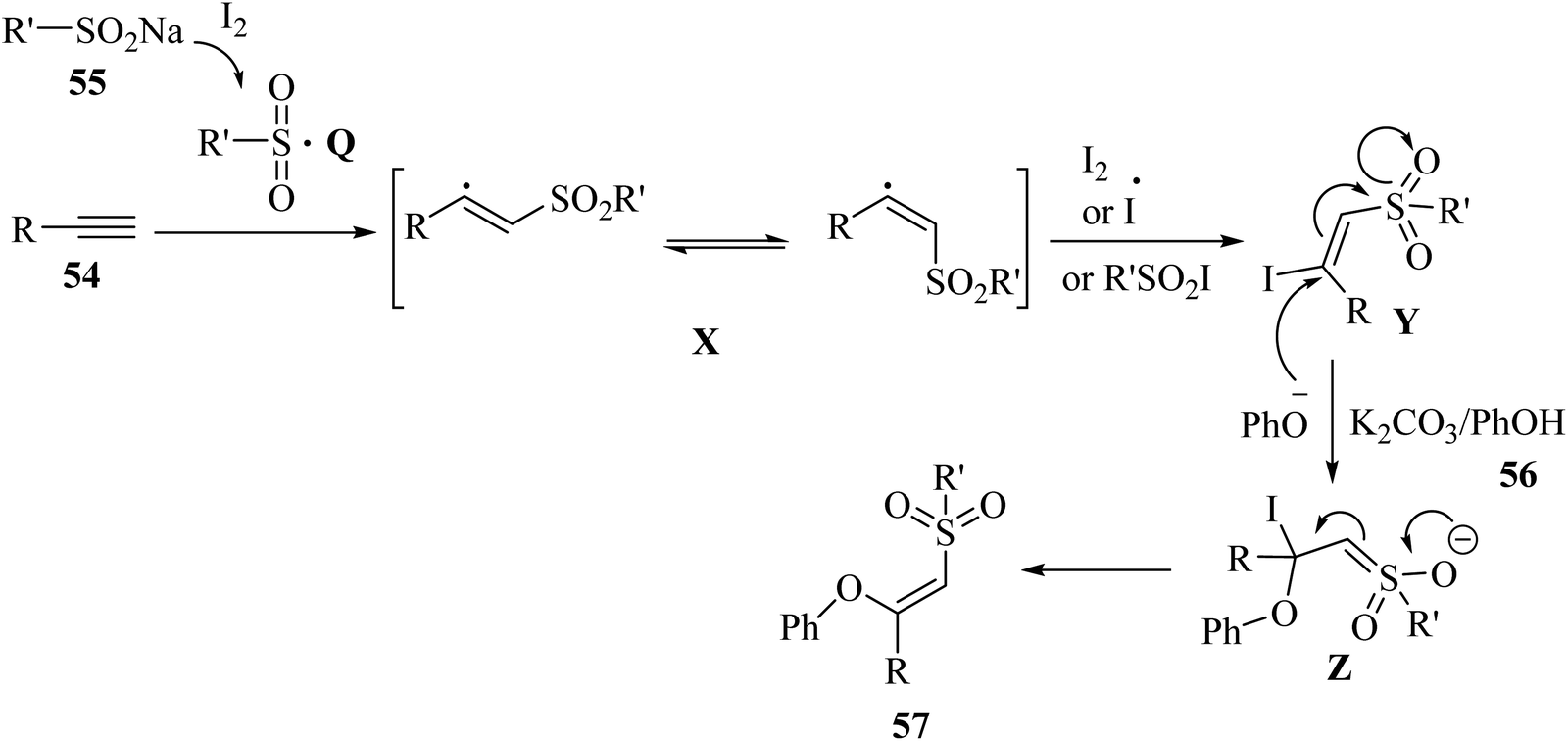
Initially, sulfonyl radical Q was generated via the reaction between I2 and 55. Subsequently, radical Q reacted with 54 and produced vinyl sulfone radicals X. After that, the vinyl sulfone radical was attacked by R'SO2I, I2, or I˙ and generated β-iodovinylsulfone intermediate Y. Next, intermediate Z was generated via the nucleophilic addition of PhO− to intermediate Y. Finally, intermediate Z produced the desired product via the elimination of iodide (Scheme 24).
 | ||
| Scheme 24 Possible reaction mechanism for the synthesis of (Z)-β-phenoxy vinylsulfones 57.49 | ||
The desired products were obtained in 73% to 87% yield. According to the results, the electron-deficient groups (Cl and CF3) on the phenyl ring gave lower reactivity than electron-rich groups (Me and t-Bu). In addition, the internal alkynes and aliphatic terminal alkynes produced 57 in 72–77% yield.
Chen et al.50 constructed C–S and C–Se bonds and produced unexpected β-sulfonylvinylselane structures 60 via the reaction among alkyne derivatives 58, SO2, and diselenides 59 in high selectivity for the E configuration (Scheme 25).
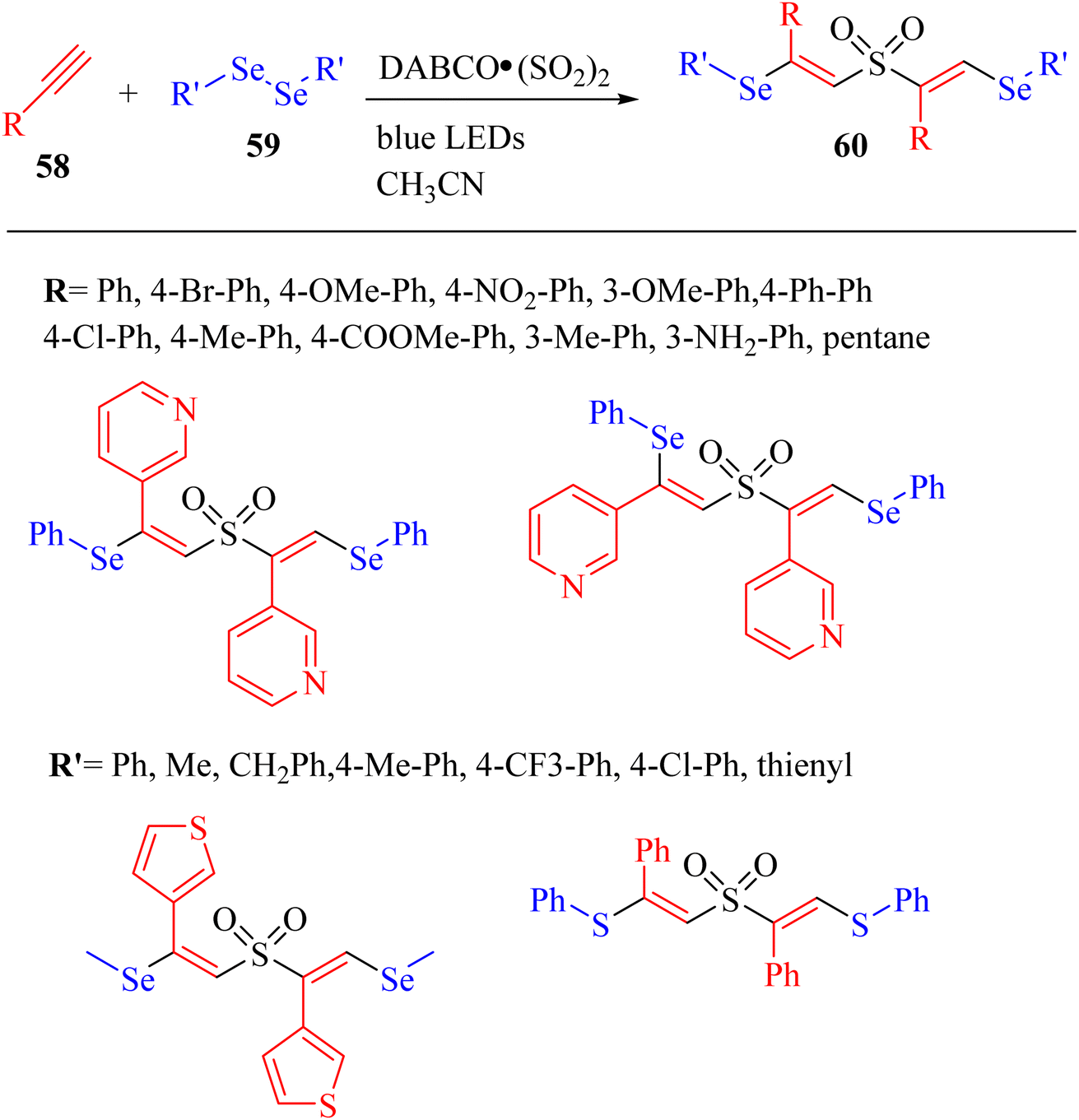 | ||
| Scheme 25 Synthesis of β-sulfonylvinylselane 60 via the visible-light-mediated multicomponent reaction.50 | ||
Initially, 59 under blue light irradiation generated selenium radical O. Then, 58 reacted with O and produced vinylic radical intermediate P, which after being captured by SO2, produced sulfonyl radical intermediate Q. After that, vinylic radical intermediate R was generated via the attack of radical intermediate Q to 59 and trapped by 59. Finally, 60 was obtained (Scheme 26).
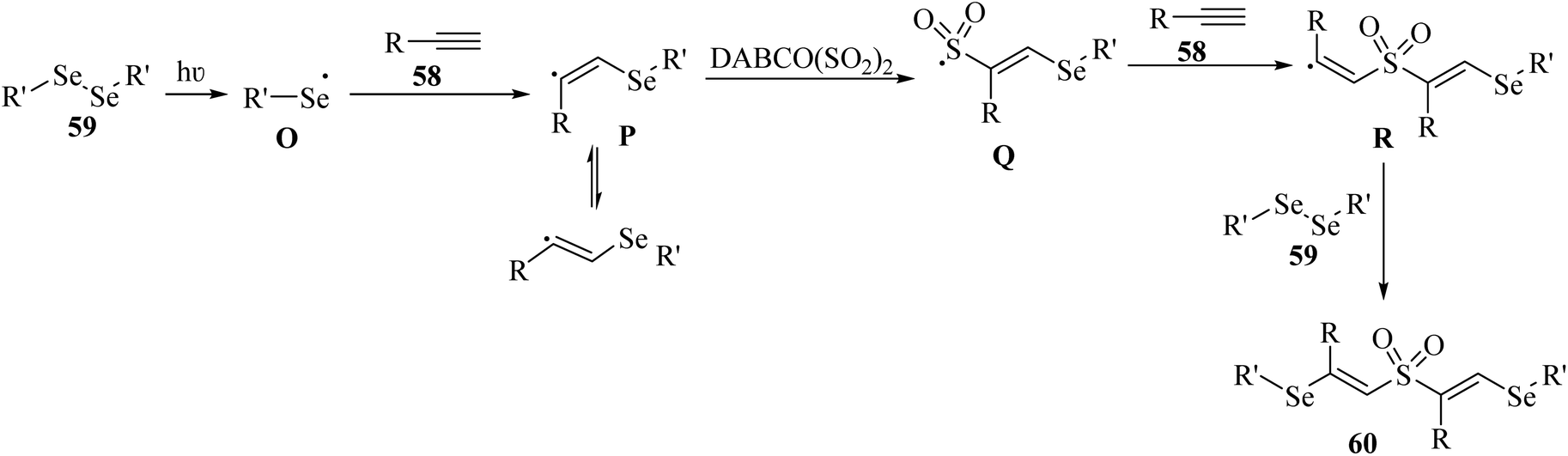 | ||
| Scheme 26 Possible reaction mechanism for the synthesis of unexpected β-sulfonylvinylselanes 60.50 | ||
The starting materials were mixed in CH3CN under blue LEDs. The unexpected β-sulfonylvinylselanes were produced in good yield (up to 71%). According to the results, when various substrate alkynes 58 on the phenyl ring were used, the desired unexpected β-sulfonylvinylselanes were produced in 35–71% yield and when 4-ethynyl-1,1′-biphenyl was used, the unexpected β-sulfonylvinylselane was obtained in 54% yield. Also, 3-ethynylpyridine and its isomer were investigated and the corresponding unexpected β-sulfonylvinylselanes were obtained in moderate to low yield (42% and 3%, respectively). In contrast, alkyl acetylene was unsuitable and the corresponding product not obtained. In the case of alkyl diselenides, the tandem reactions of 1,2-dimethyldiselane with 3- ethynylthiophene, 4-methylphenylacetylene, phenylacetylene, and 4-bromophenylacetylene produced the corresponding unexpected β-sulfonylvinylselanes in good yield of 47% to 64%.
Zhu and co-workers51 synthesized stereo-defined olefins 64 via single electron/triplet energy transfer and nickel catalysis. This strategy is a good method for producing tri-substituted alkenes. The reaction proceeded via the reaction among aryl halide derivatives 61, alkyne derivatives 62, and sodium sulfonate 63 in the presence of an Ru photocatalyst, NiCl2·dme, and 4,4′,4′′-tri-tert-butyl-2,2':6′,2′′-terpyridine as a ligand in DMF at room temperature for 24 h under blue LED irradiation (7.4 W) (Scheme 27).
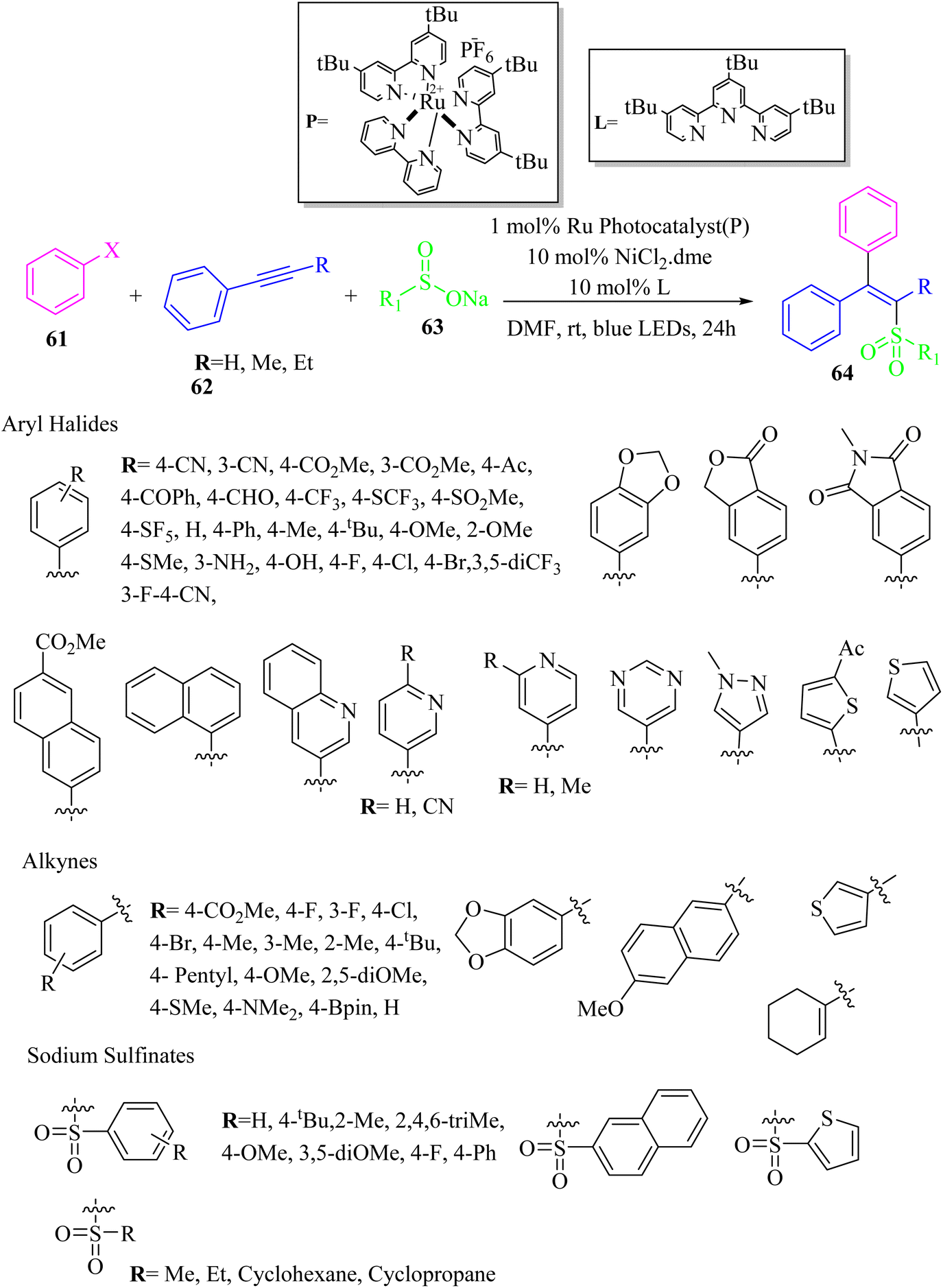 | ||
| Scheme 27 Synthesis of tri-substituted alkenes via nickel catalysis.51 | ||
Initially, the RuII or IrIII complex absorbed visible light in the photocatalytic cycle and generated *IrIII or *RuII triplet excited state. Then, the radical precursor was oxidized to the corresponding radical and formed RuI and IrII reductants. The desired radical was added to a NiI complex and entered the Ni catalytic cycle. Subsequently, it underwent reduction and 1,2 migratory insertion of alkyne, producing A. Then, intermediate B was produced by intermediate A under Ni-assisted anti/syn isomerization. Next, intermediate C was produced by intermediate B via the oxidative addition of the aryl halide. Subsequently, 64 was generated from intermediate C via reductive elimination (Scheme 28).
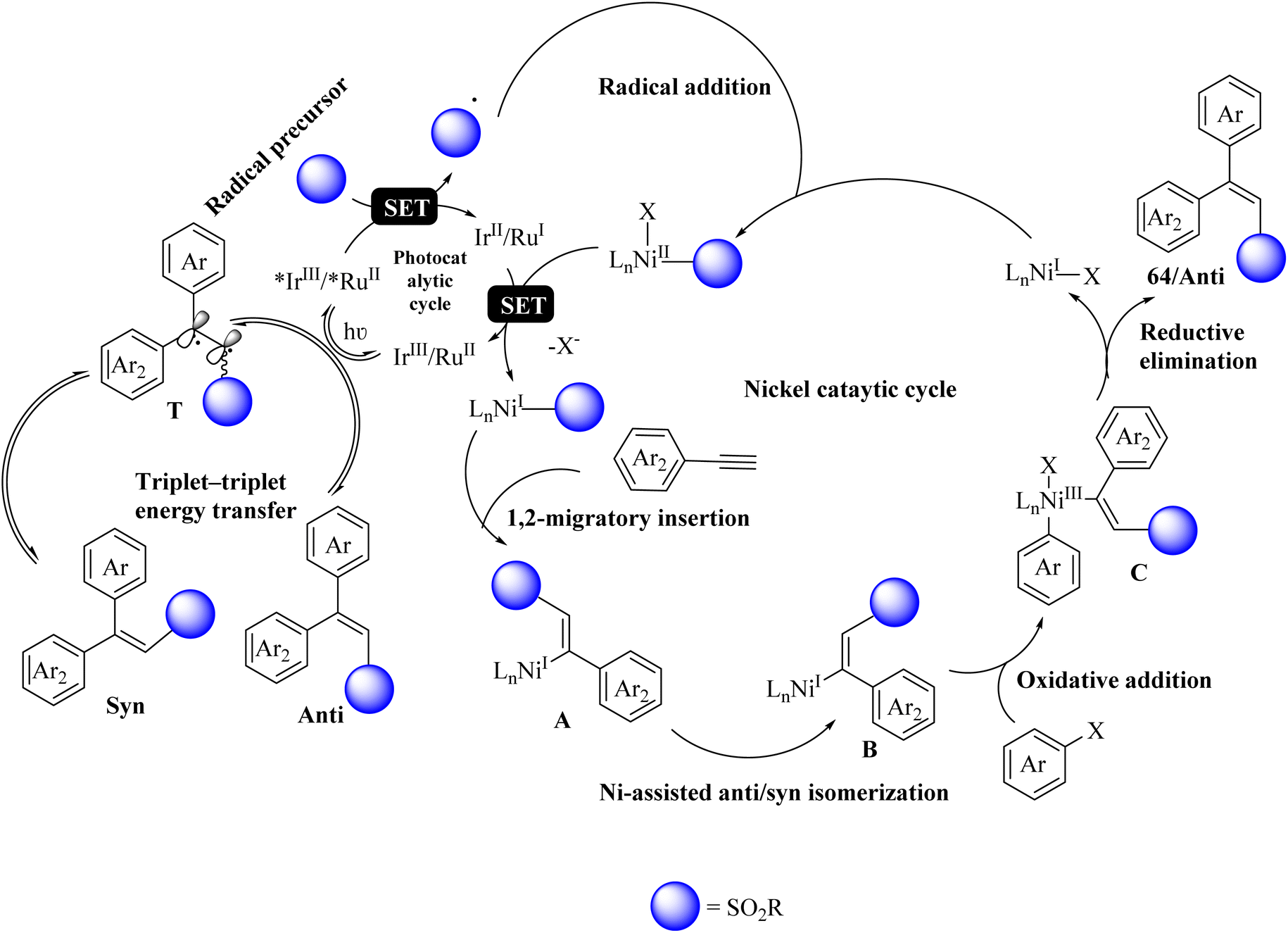 | ||
| Scheme 28 Possible reaction mechanism for the synthesis of stereo-defined olefins.47 | ||
According to the results, various alkynes with various functional groups were used (boronic ester, acetal, amine, bromide, ester, fluoride, chloride, alkyl, methoxy, and methylthio groups) and produced the desired olefins in 29–99% yields. Furthermore, 3-ethynylthiophene produced the corresponding olefin in 76% yield and cyclohexenyl alkyne and internal alkyne derivatives were suitable substrates for producing the desired products (29–60%).
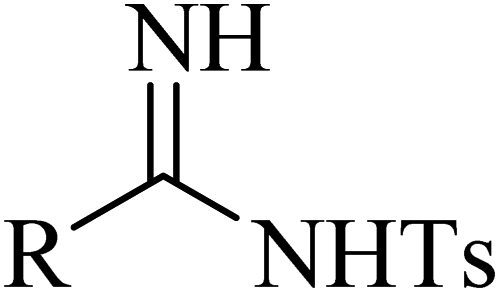
Yi et al.52 synthesized arylamidines 69 via the four-component reaction of sulfonyl azide derivatives 65, terminal alkyne derivatives 66, water 67, and oxime halide 68 by [3 + 2] cycloaddition in the presence of triethylamine and Cu(I) in DCM at room temperature for 12 h under open air (Scheme 29).
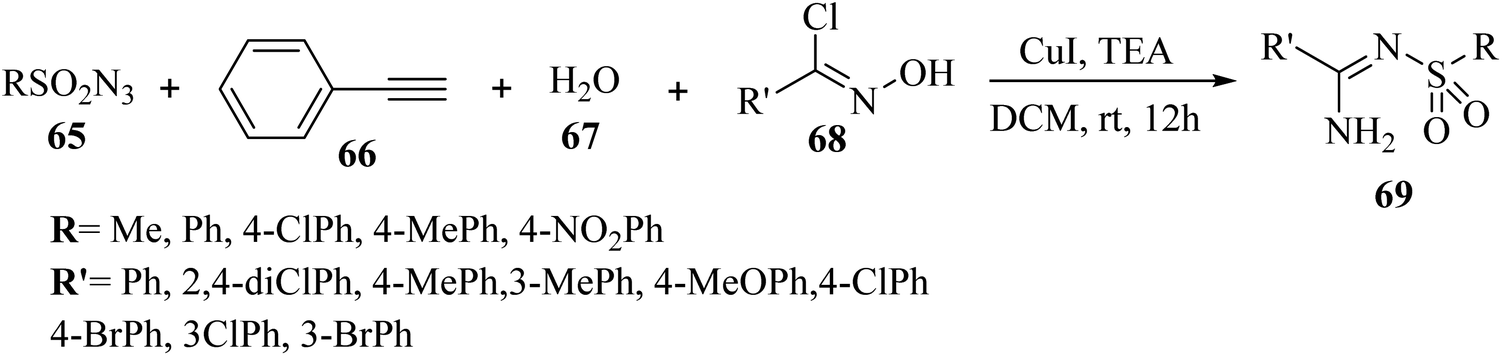 | ||
| Scheme 29 Synthesis of arylamidines 69 from oxime chlorides.52 | ||
Initially, ketenimine intermediate F was generated via the reaction between 66 and TsN3 in the presence of Et3N and CuI. Then, oxime halide 68 generated nitrile oxide G under Et3N. Subsequently, G reacted with intermediate F and produced intermediate H via the formal [3 + 2] cycloaddition reaction. Next, intermediate I was generated via the electronic ring opening of intermediate H with the help of OH− or H2O. Intermediate I reacted with OH− or H2O and produced intermediate J, accompanied by the formation of 66. Finally, 69 was obtained from intermediate J in the presence of OH− or H2O, accompanied by the release of O2. It should be noted that the role of H2O in the reaction is important, which provides the OH− anion and the source of proton, and also may promote the cleavage of the N–O and C–O bonds (Scheme 30).
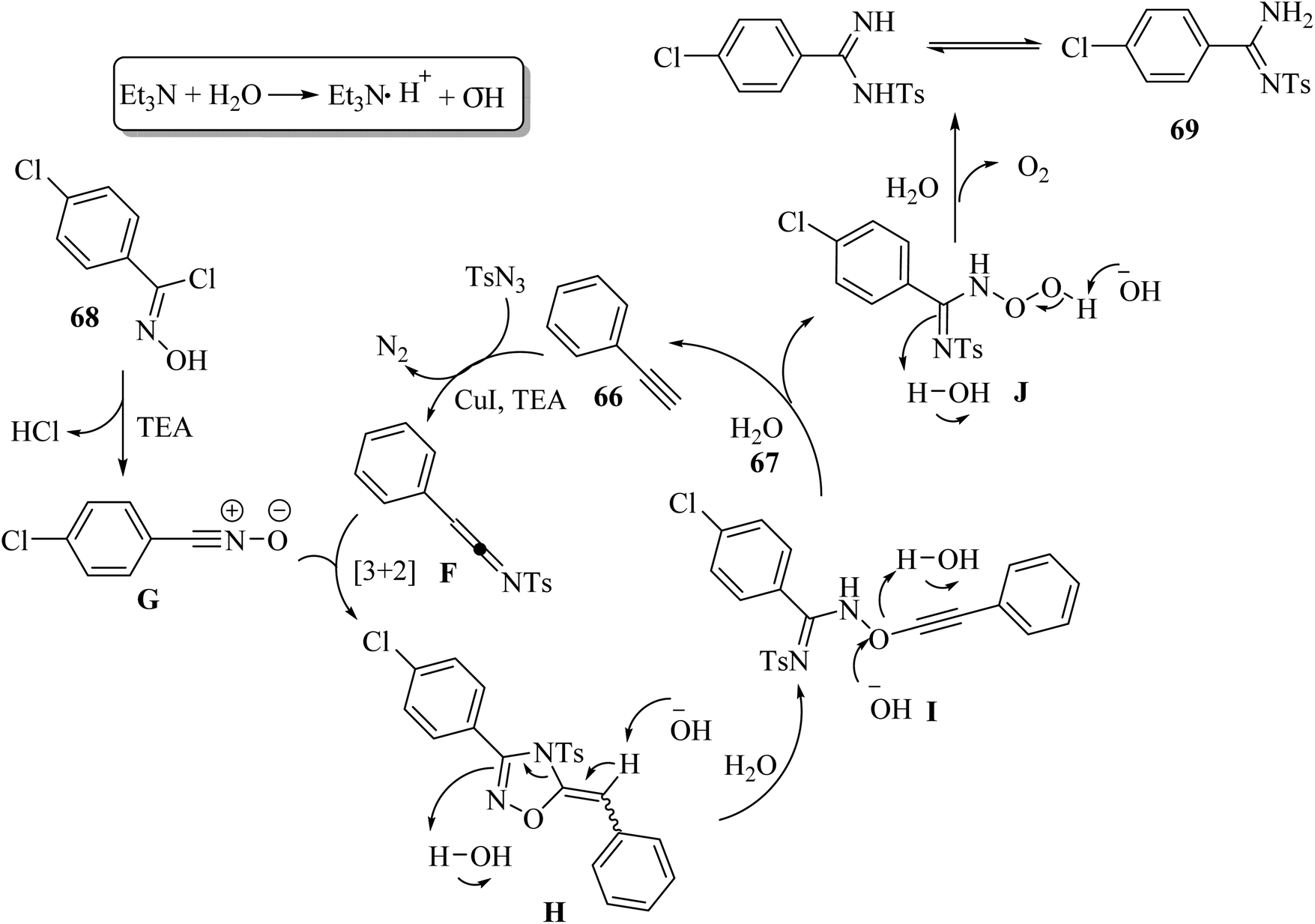 | ||
| Scheme 30 Possible reaction mechanism for the synthesis of arylamidines 69.52 | ||
According to the results, arylamidines were produced in 68–92% yield. Importantly, oxime chloride derivatives 68 as the carbon source and water 67 played a critical role in the reaction. In addition, terminal alkyne derivatives, which can lead to high yields of corresponding products using lower amounts of reactant, may play a catalytic role.
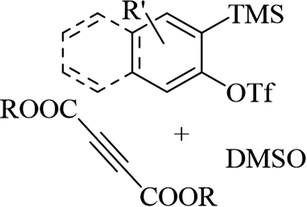
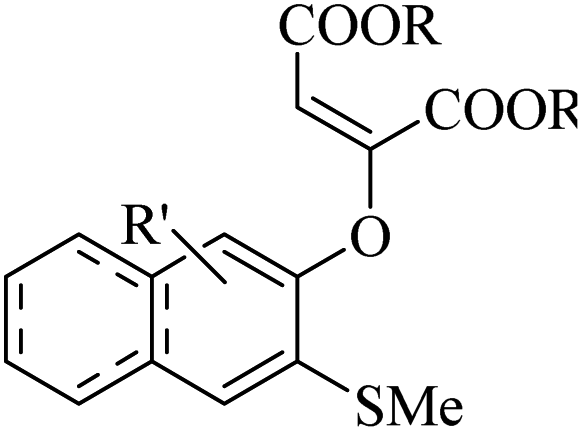
Hazarika et al.53 synthesized 2-[(ortho-methylthio)aryloxy] substituted dialkyl maleates 73 via the reaction among alkyne derivatives 70, DMSO 72, and aryne derivatives 71 in the presence of KF at 50 °C (Scheme 31).
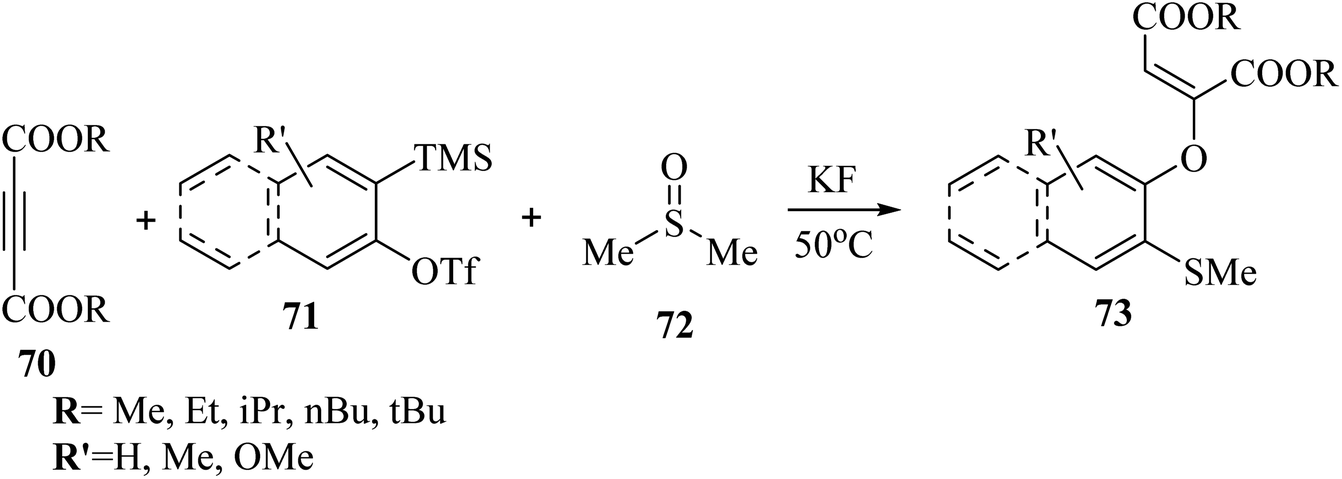 | ||
| Scheme 31 Synthesis of dialkyl maleates 3 from activated alkyne.53 | ||
Initially, four-membered ring intermediate E was formed via the reaction between benzyne, which was produced in situ, and DMSO. Then, intermediate E underwent ring opening due to the ring strain, and o-quinone intermediate F was generated. Zwitterion G was the resonance form of F. G underwent nucleophilic attack to form the activated alkyne, and also demethylation, which led to the generation of anion H. Finally, anion H produced 73 in the presence of H2O (Scheme 32).
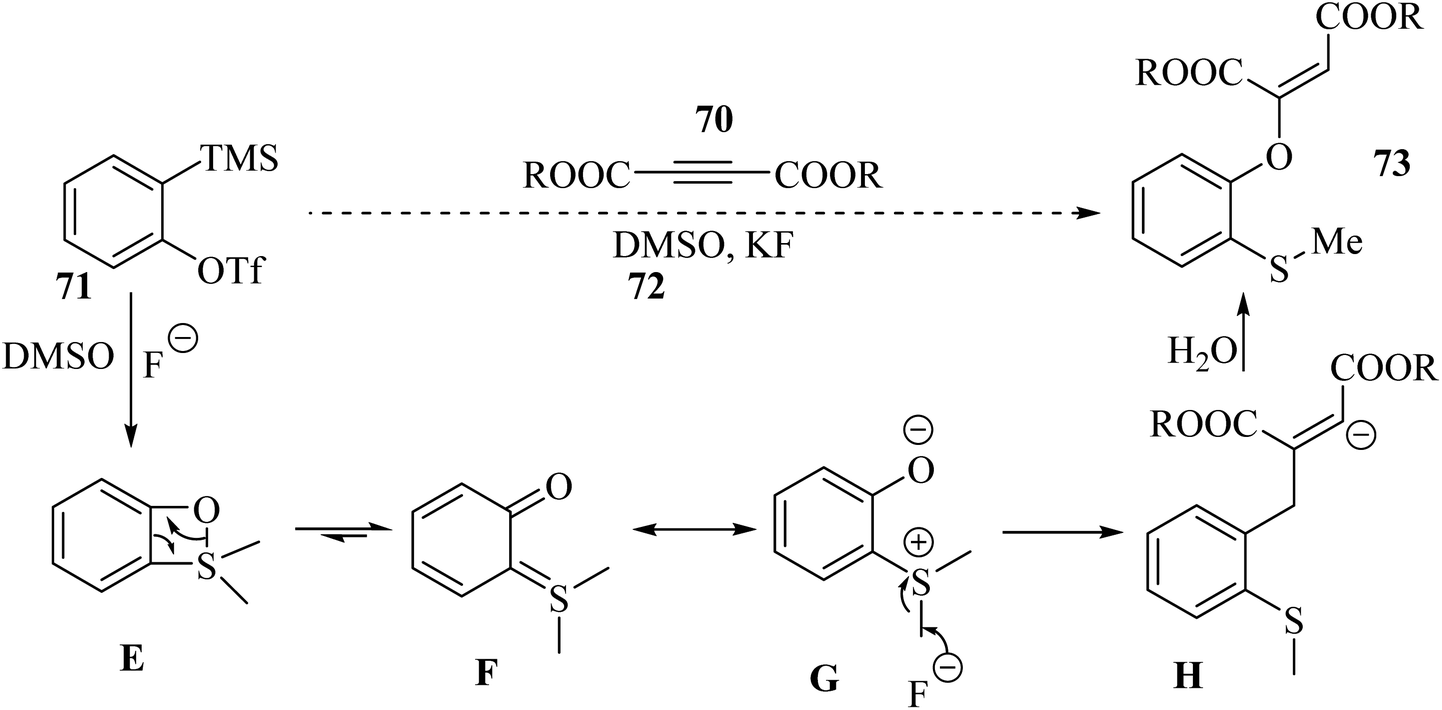 | ||
| Scheme 32 Possible reaction mechanism for the synthesis of 2-[(o-methylthio)aryloxy]-substituted dialkyl maleate.53 | ||
This strategy produces trisubstituted vinyl ethers with remarkable stereospecificity. According to the results, different types of activated alkyne derivatives treated with o-silyl aryl triflate produced the corresponding products in 63–81% yield. Notably, bulky alkyl group-substituted activated alkyne derivatives as a comparison to small alkyl group-substituted activated alkyne derivatives showed lower yields. Investigation of symmetrical aryne precursors showed that the desired products were produced in 59–77% yield. Interestingly, the symmetrical aryne precursors 3-(trimethylsilyl)-2-naphthyl trifluoromethanesulfonate, and 4,5-dimethoxy-2-(trimethylsilyl)phenyl trifluoromethanesulfonate produced a small amount of the corresponding products, which could be due to steric hindrance. Also, unsymmetrical aryne precursors such as 4-methoxy-2-(trimethylsilyl)phenyltrifluoromethanesulfonate and 2-methyl-6-(trimethylsilyl)phenyltrifluoromethanesulfonate were evaluated and produced the desired products in 66–69% yield. When the unsymmetrical aryne precursor 4-methyl-2-(trimethylsilyl)phenyltrifluoromethanesulfonate was treated with activated alkyne derivatives such as di-methylacetylenedicarboxylate and di-ethylacetylenedicarboxylate, regioisomer mixtures, they gave 69% and 63% yield, respectively.
In 2019, indolizine thione derivatives 76 were synthesized via the formation of C–N, C–C and C–S bonds via the reaction among ynals 75, elemental sulfur, and 2-alkylpyridines 74 was reported by Chen and co-workers (Scheme 34).54 Two conditions were investigated for the synthesis of indolizine thione derivatives 76.
Firstly, 74 and 75 reacted and generated enyne intermediate U via Knoevenagel condensation. Intermediate V was produced from intermediate U via the intramolecular nucleophilic attack and activation of S. Then, the cleavage of the disulfide bond produced 76 (Scheme 33a). Also, W generated intermediate Z via the elimination process and electrophilic addition. Next, intermediate Z produced 76 via aminothiation with elemental sulfur (Scheme 33b).
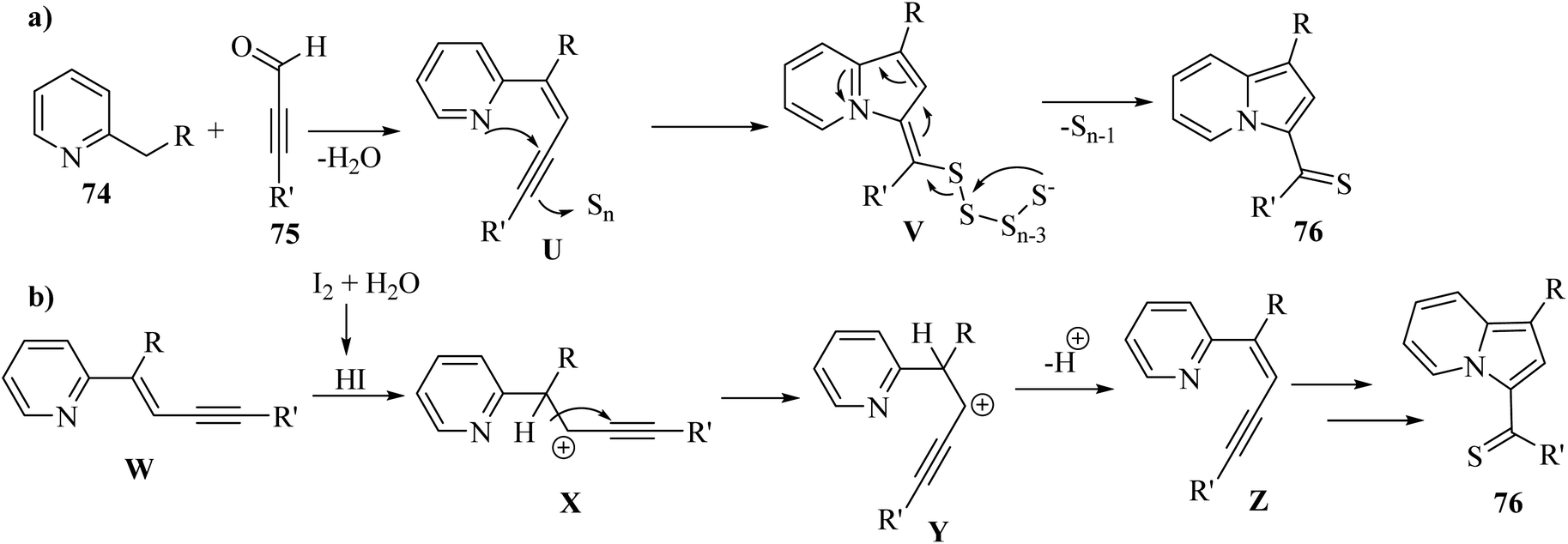 | ||
| Scheme 33 Possible reaction mechanism for the synthesis of (a) indolizine thione derivatives without I2 and (b) indolizine thione derivatives with I2.54 | ||
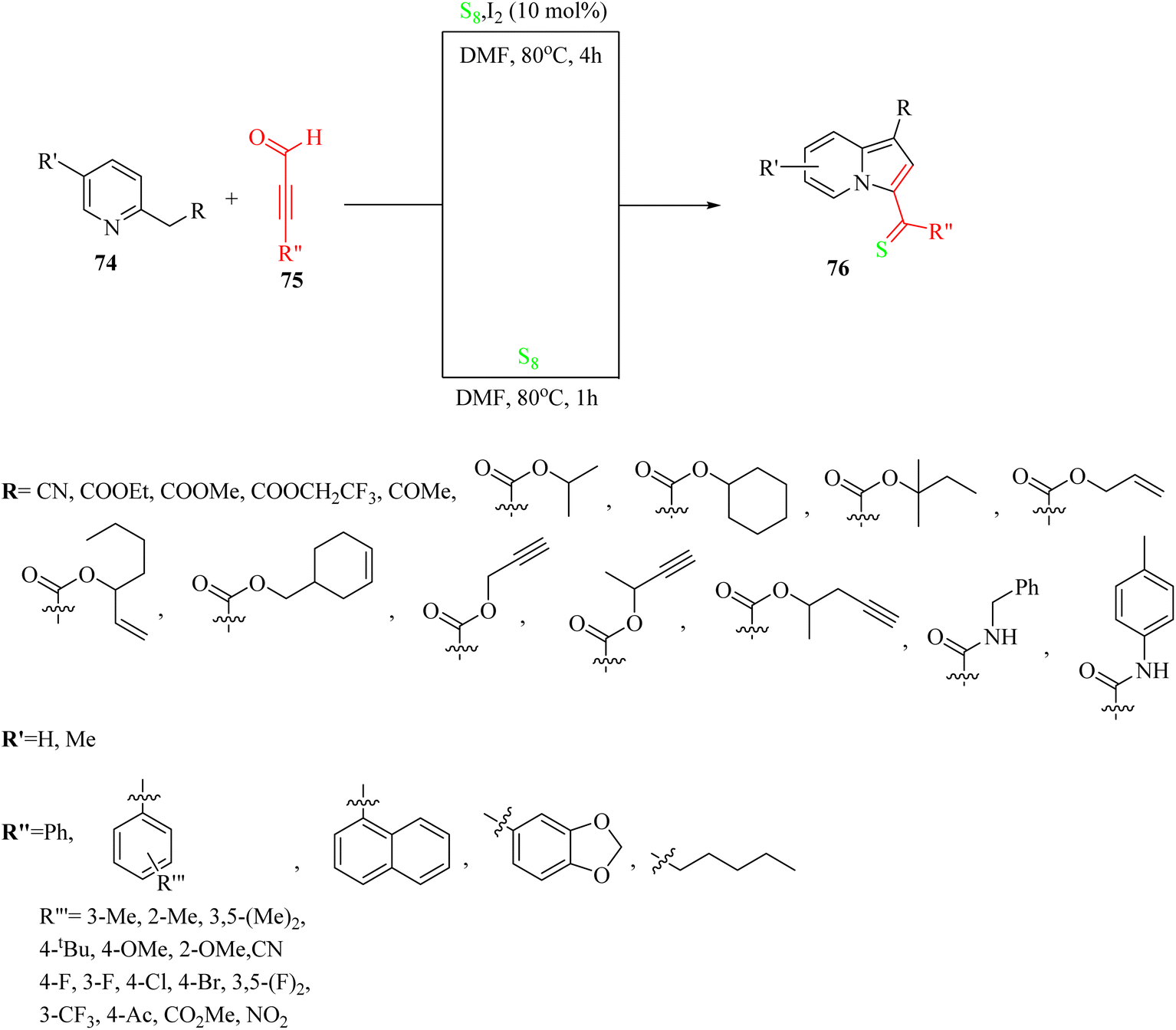 | ||
| Scheme 34 Synthesis of indolizine thiones 76 from ynals.54 | ||
The desired products were synthesized once in 50–88% yield in DMF under argon at 80 °C for 1 h, and again the reaction process for the synthesis of the corresponding products was carried out in the presence of iodine as a catalyst and under the same conditions for 4 h and products were obtained in 52–93% yield. According to the results without the catalyst, ynals 75 with different groups (electron-donating and -withdrawing) produced the corresponding indolizine thiones in 50–86% yield. Me or OMe at the o-position of the benzene ring had a low impact on the efficiency of the reaction. In addition, the disubstituted substrates (3,5-(Me)2 and 3,5-(F)2) produced the desired indolizine thiones in 70% and 52% yield, respectively. Ynals with strong and weak electron-poor substitutes reacted smoothly. In contrast, aliphatic ynal did not produce the corresponding indolizine thione. According to the results from the I2-catalyzed aminothiation of 2-alkylpyridines, the ynal bearing a naphthalene moiety produced the desired indolizine thione in 93% yield. Interestingly, the heterocycle ynal produced the desired indolizine thione in 53% yield.
Jin and co-workers55 synthesized 2,3-dihydro-1H-imidazo[1,2-a]indole derivatives 80 via the reaction among allylamine derivatives 79, sulfonyl azide derivatives 78, and alkyne derivatives 77 (Scheme 35). In the first step, triethylamine (Et3N) as a base and CuI as a catalyst were added to the mixture in DMSO at room temperature for 1 h. After that, L1, K2CO3, and CuI were added to the mixture and heated at the temperature of 80 °C for 6 h.
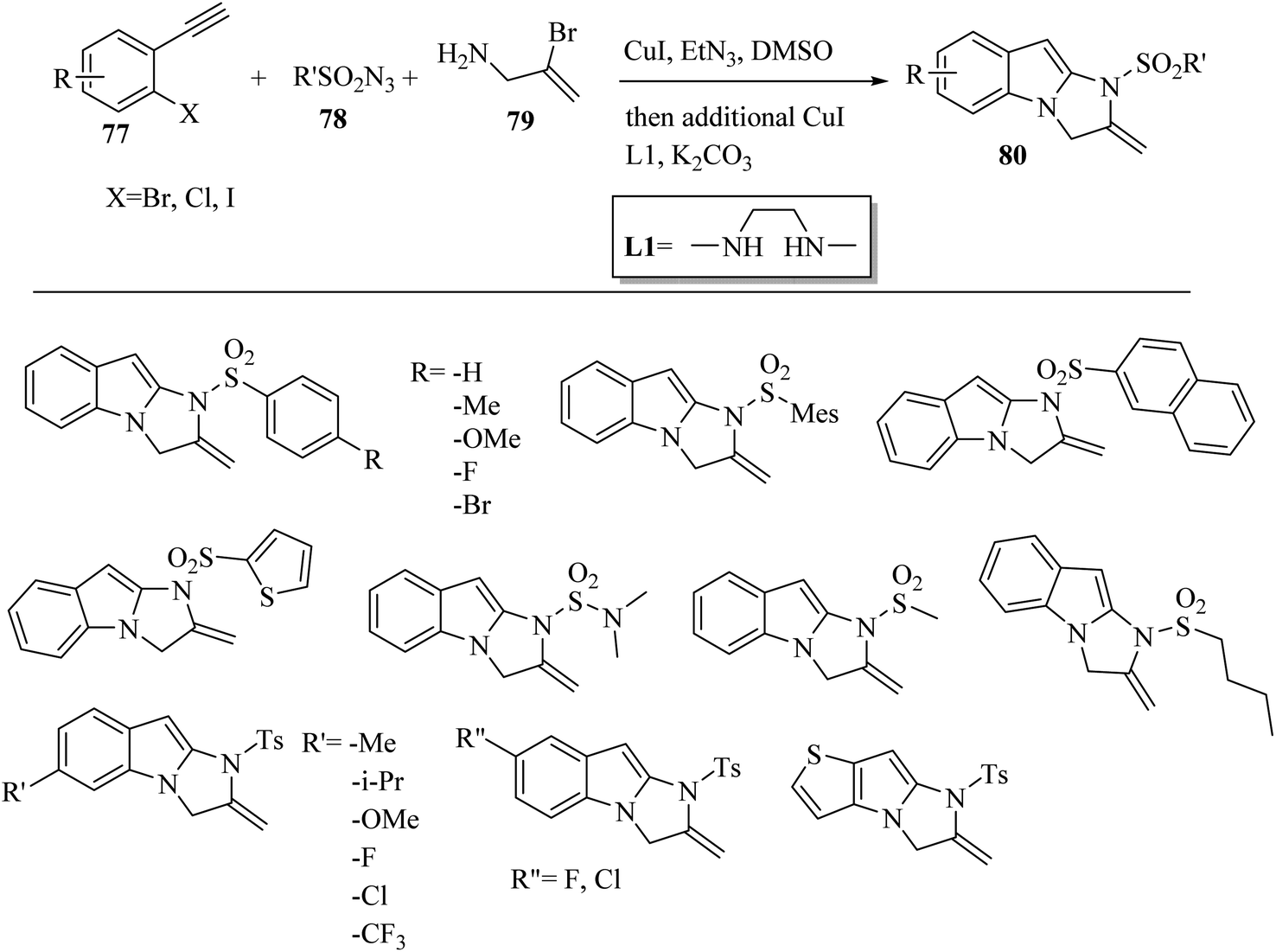 | ||
| Scheme 35 Synthesis of 2,3-dihydro-1H-imidazo[1,2-a]indole derivatives 80.55 | ||
Initially, 77 and 78 generated intermediate G via a Cu-catalyzed azide–alkyne cycloaddition reaction. Then, intermediate G produced ketenimine H by the extrusion of N2, and carboxamidine I and/or its tautomer I′ was generated via the nucleophilic addition of 79 to intermediate H. After that, 80 was produced via the Cu-catalyzed C–N coupling reaction (Scheme 36).
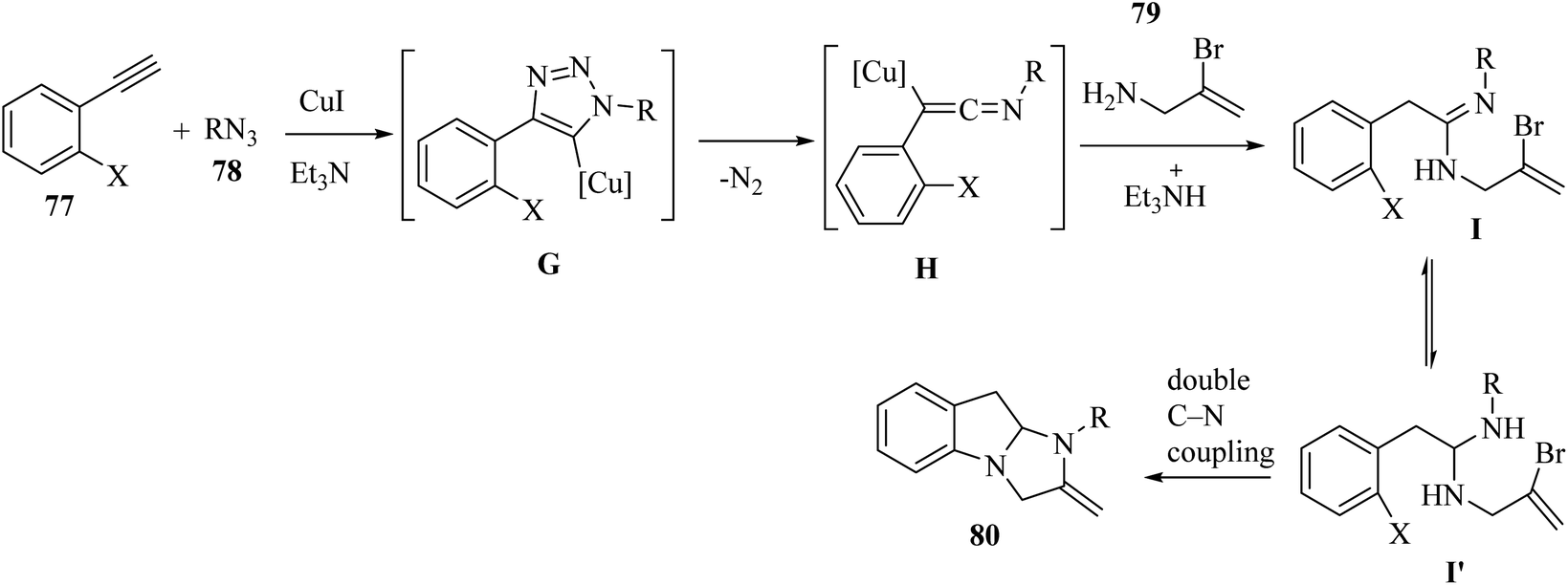 | ||
| Scheme 36 Possible reaction mechanism for the synthesis of 2,3-dihydro-1H-imidazo[1,2-a]indole derivatives 80.55 | ||
The corresponding products were produced with in isolated yield of 16–77%. According to the results, 1-bromo-2-ethnylbenzene demonstrated higher reactivity compared to the iodo- and chloro-substituted substrates. In addition, aromatic alkyne derivatives with different groups reacted with para-toluenesulfonyl azide.
Xu et al.56 synthesized α,α-bis-sulfonyl arylketone derivatives 84 via the reaction among H2O 83, bromoalkyne derivatives 81, and sulfinic acid derivatives 82 at room temperature under visible-light irradiation in DMSO solvent and N2 atmosphere for 12 h (Scheme 37).
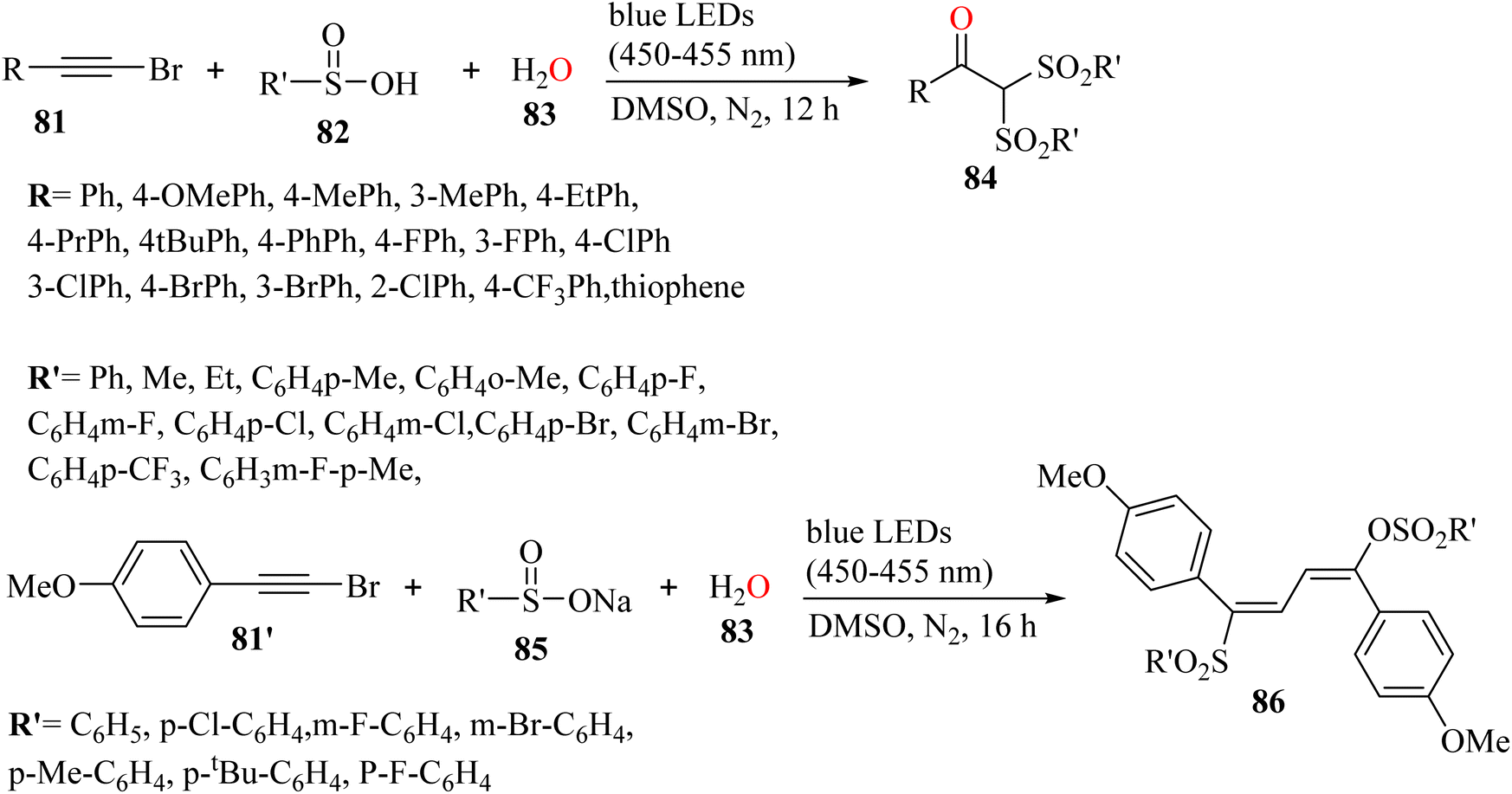 | ||
| Scheme 37 Synthesis of α,α-bis-sulfonyl arylketones under visible-light irradiation.56 | ||
Initially, sulfinyl sulfones B were produced via the condensation of A, which decomposed to radical C. Then, radical C was added to 81 and generated intermediate D, which removed the bromide radical (Br˙). After that, surplus sulfonyl C reacted with E to afford intermediate F via conjugate addition. Next, the hydroxyl radical (˙OH) coupled with intermediate F and produced intermediate G, which was tautomerized to 84. It should be noted that ˙OH may be generated via the reaction between H2O and possible radical intermediates (Scheme 38a). Also, E was afforded via a similar procedure, except intermediate H was produced via the 1,4-addition reaction of E and extra A′, and also an initial hydrolysis. In addition, Br˙ and alkynyl radical were generated via the homolytic cleavage of 81. Next, intermediate I was produced via the coupling of intermediate H with an alkynyl radical. The important point about intermediate I was that it can be unstable. Finally, R'SO3H reacted with intermediate I via electrophilic addition and generated 86 (Scheme 38b).
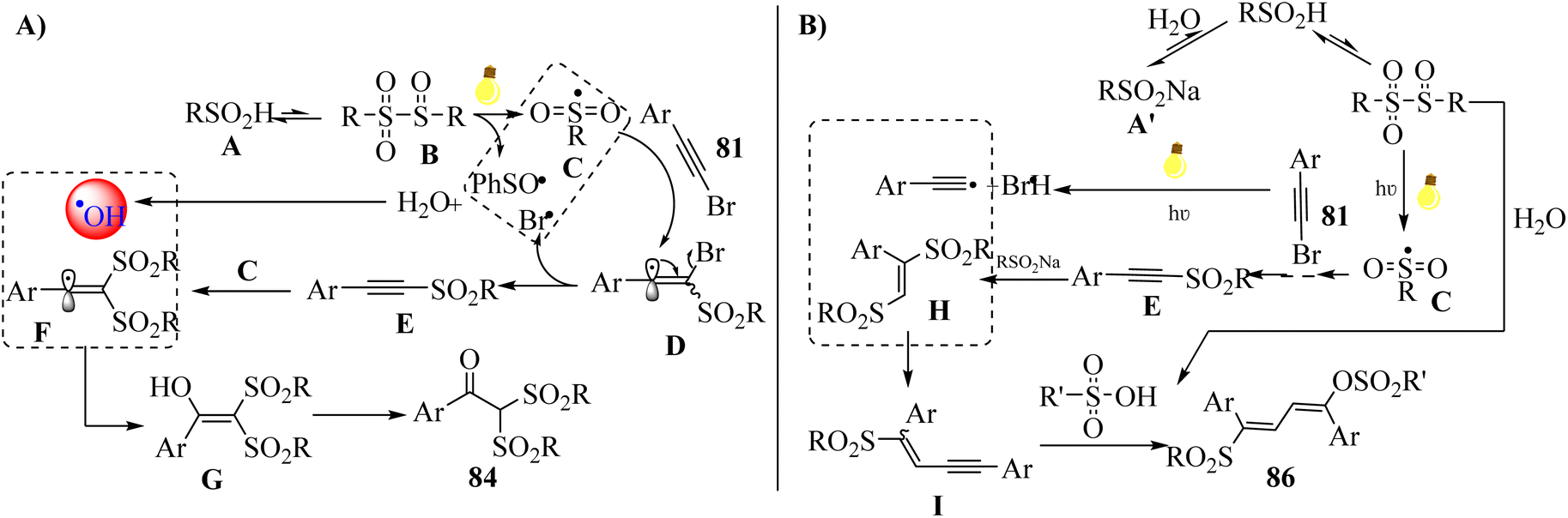 | ||
| Scheme 38 Possible reaction mechanism for the synthesis of α,α-bis-sulfonyl arylketones.56 | ||
In the case of 81, aromatic bromoalkynes bearing para-Me, meta-Me, para-Et, para-Pr, and para-tBu were reacted with benzenesulfonic acid and produced the corresponding arylketone derivatives in 76% to 89% yield. A phenyl substituent at the para-position on the benzene ring of bromoalkyne produced the corresponding arylketone in 94% yield. Bromoalkyne derivatives with a halogen-substituent at the meta- and para-position were tested and produced the corresponding arylketone derivatives in 72% to 82% yield. In contrast, substrates with Cl at the o-position did not react with benzenesulfonic acid to obtain the corresponding product. CF3 and thiophene substituents on the alkynyl bromide were also reacted and produced the corresponding arylketone derivatives in 68% and 53% yield, respectively. Importantly, the aliphatic bromoalkyne of 1-bromohex-1-yne was not transformed in this reaction, and no desired product was produced. Toluenesulfonic acid was tested and produced the corresponding arylketone in 60% yield. In contrast, when o-toluenesulfonic acid was employed, no reaction occurred. Furthermore, sulfonic acid derivatives with halogen substituents on the benzene ring produced the desired arylketone derivatives in 64% to 84% yield. An CF3 group on the benzene ring was reacted and produced the desired product in 57% yield. In addition, benzenesulfonic acid with disubstituted groups of Me and F produced the corresponding product in 43% yield. Also, ethanesulfonic and methylsulfonic acid were tolerated and the desired arylketone derivatives obtained in 85% and 81% yield, respectively.
Bhat and Lee57 synthesized 3-arylsulfonylated thioflavones 90 via the three-component reaction of aryl diazonium salts 88, DABSO 89, and alkyl-substituted alkynones 87 in CH3CN under N2 at 23 °C for 4 h (Scheme 39).
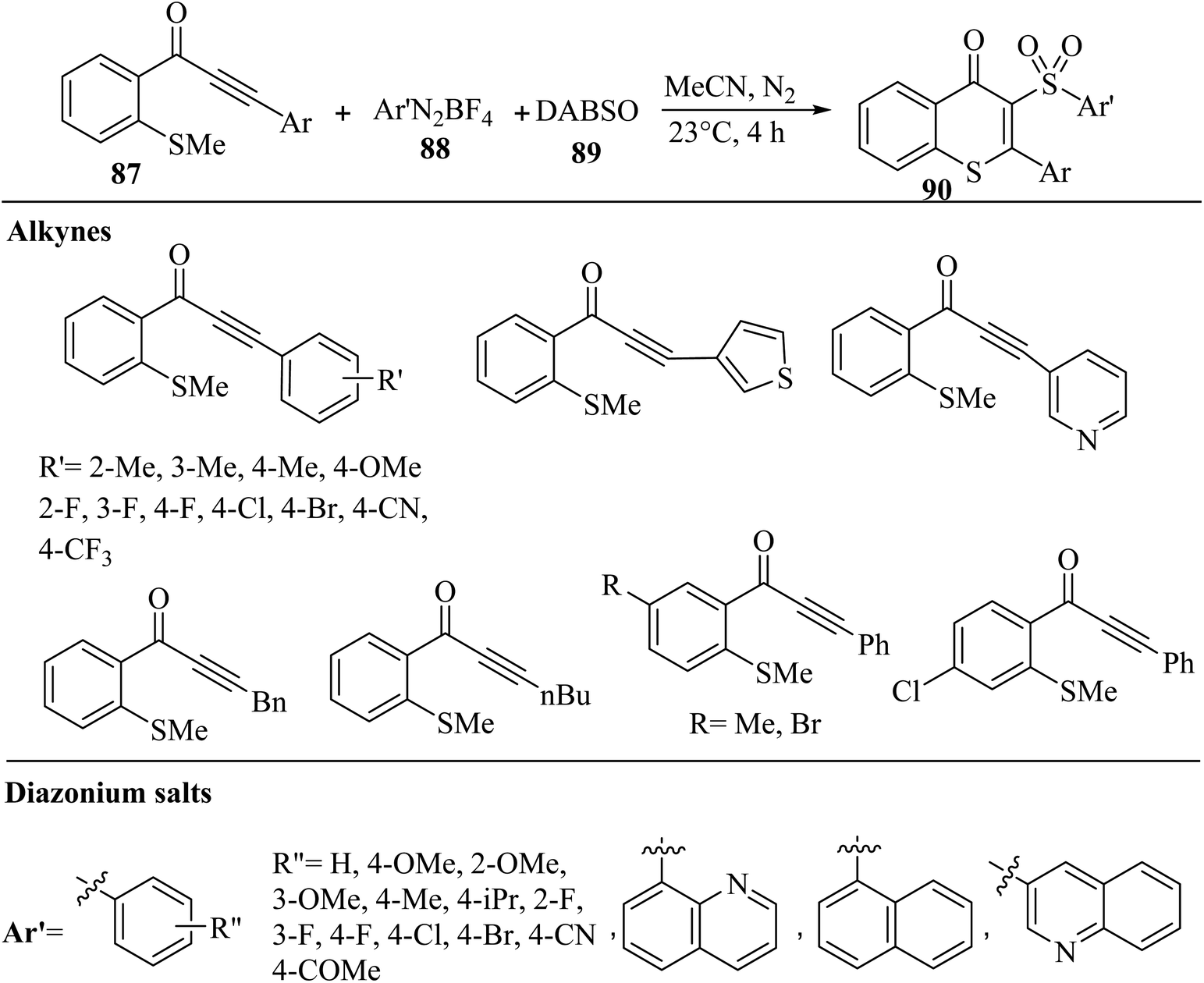 | ||
| Scheme 39 Synthesis of 3-arylsulfonylated thioflavone derivatives 90 by alkyl-substituted alkynone derivatives.57 | ||
Initially, complex B was produced via the electrostatic interaction between A and DABSO. Then, complex B produced C and D via homolytic cleavage of the N–S bond and concomitant single-electron transfer. After that, a radical intermediate was generated via the reaction between E and 87. Finally, via intramolecular radical cyclization, 90 was produced. It should be noted that by-product S was observed in this reaction, which was produced via the reaction between 87 and the methylsulfonyl radical (Scheme 40).
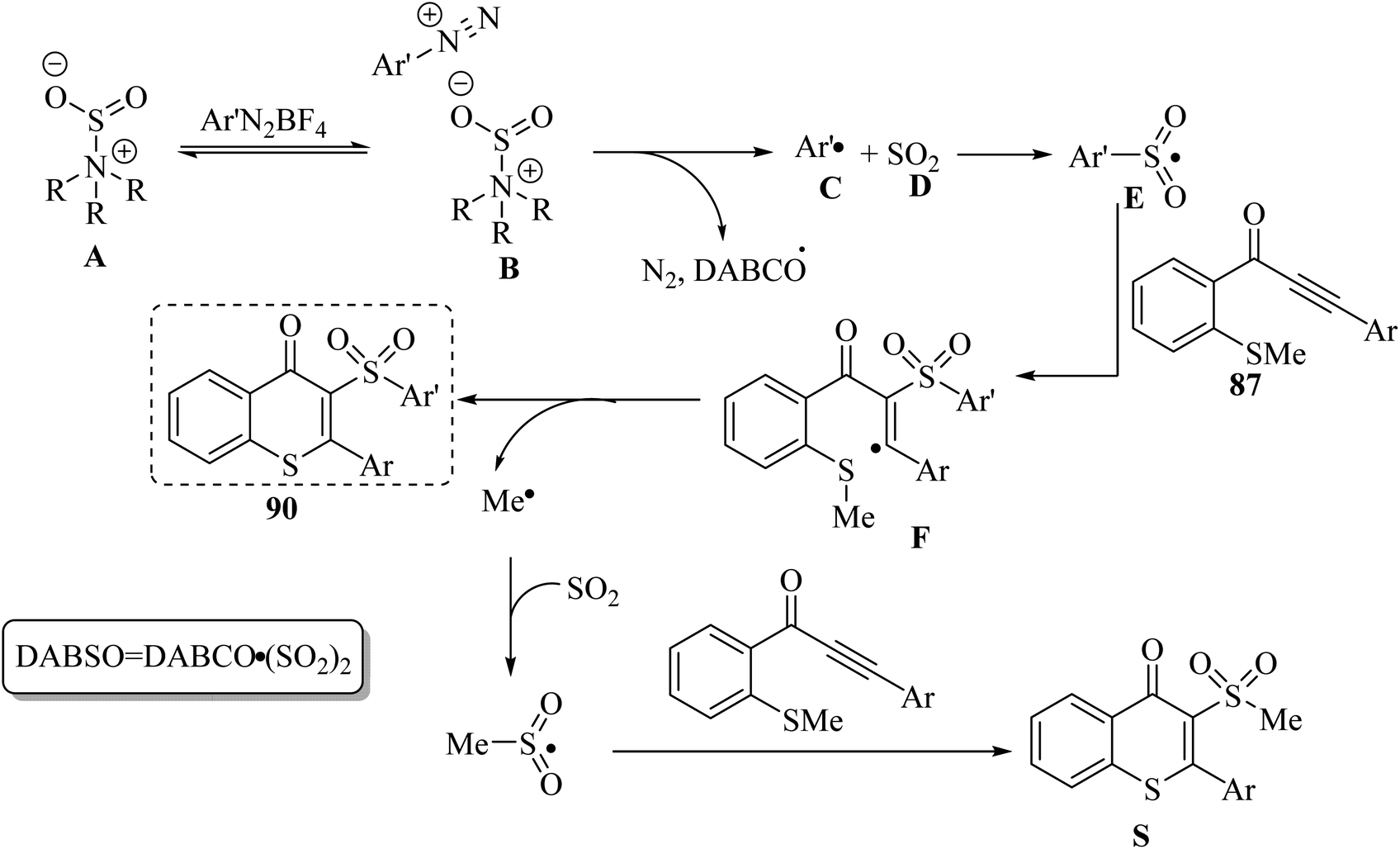 | ||
| Scheme 40 Possible reaction mechanism for the synthesis of 3-arylsulfonylated thioflavones.57 | ||
In the case of 88, the corresponding products in the presence of different substituents produced in 40–97% yield. No obvious electronic effects of these substituents were observed. Moreover, the p-, m-, and o-substituted aryl diazonium salt derivatives produced the desired -arylsulfonylated thioflavones in 87–97% yield. Heteroaromatic and naphthyl substituents were tested, although lower yields were shown with quinoline-substituted diazonium salt derivatives. In the case of 87, alkyne derivatives with electron-donating and -withdrawing substituents produced the corresponding 3-arylsulfonylated thioflavones in 52–91% yield. No significant differences in reactivity were observed for the ortho, meta, and para substituents. Also, thienyl or pyridyl groups produced the corresponding 3-arylsulfonylated thioflavones in 57–96% yield. Unfortunately, the use of 1-(2-(methylthio)phenyl)hept-2-yn-1-one or 1-(2-(methylthio)phenyl)-4-phenylbut-2-yn-1-one as the alkyl-substituted alkynone was not successful and did not produce the corresponding products.
In 2022, Kalari and co-workers58 synthesized β-acyl allyl sulfone 94 via the reaction among alkynes 91, DMSO 93, and sodium sulfinate derivatives 92 in the presence of Selectfluor at 110 °C for 12 h (Scheme 41).
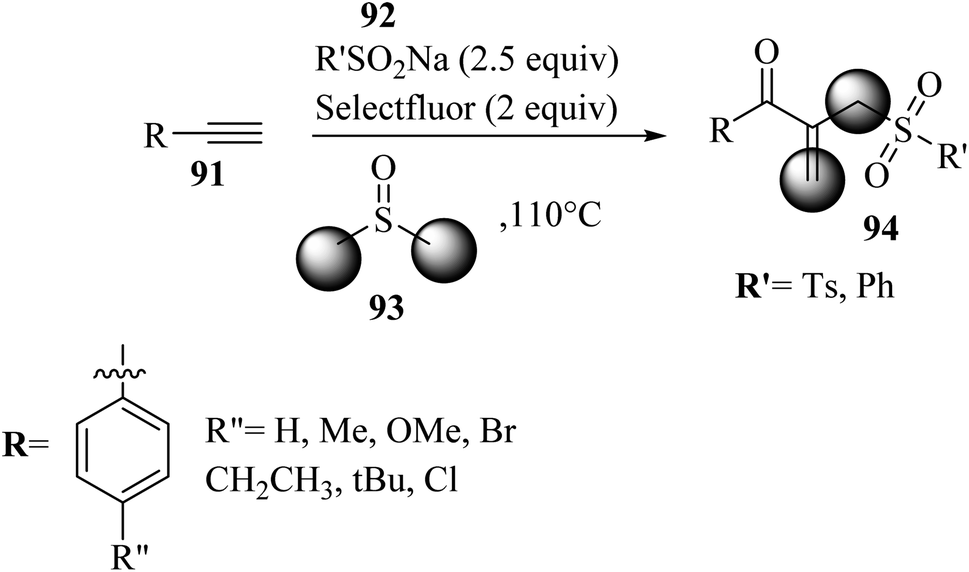 | ||
| Scheme 41 Synthesis of β-acyl allyl sulfone 94 by Selectfluor.58 | ||
Initially, 93 was activated by Selectfluor to afford E (radical cation). Then, E reacted with F− and generated key intermediate F. Intermediate G was produced via the reaction between 91 and intermediate F, which reacted with additional F to afford intermediate H. After that, I was generated via the demethylthiolation of intermediate H. Finally, 94 was obtained via the reaction between 92 and I (Scheme 42).
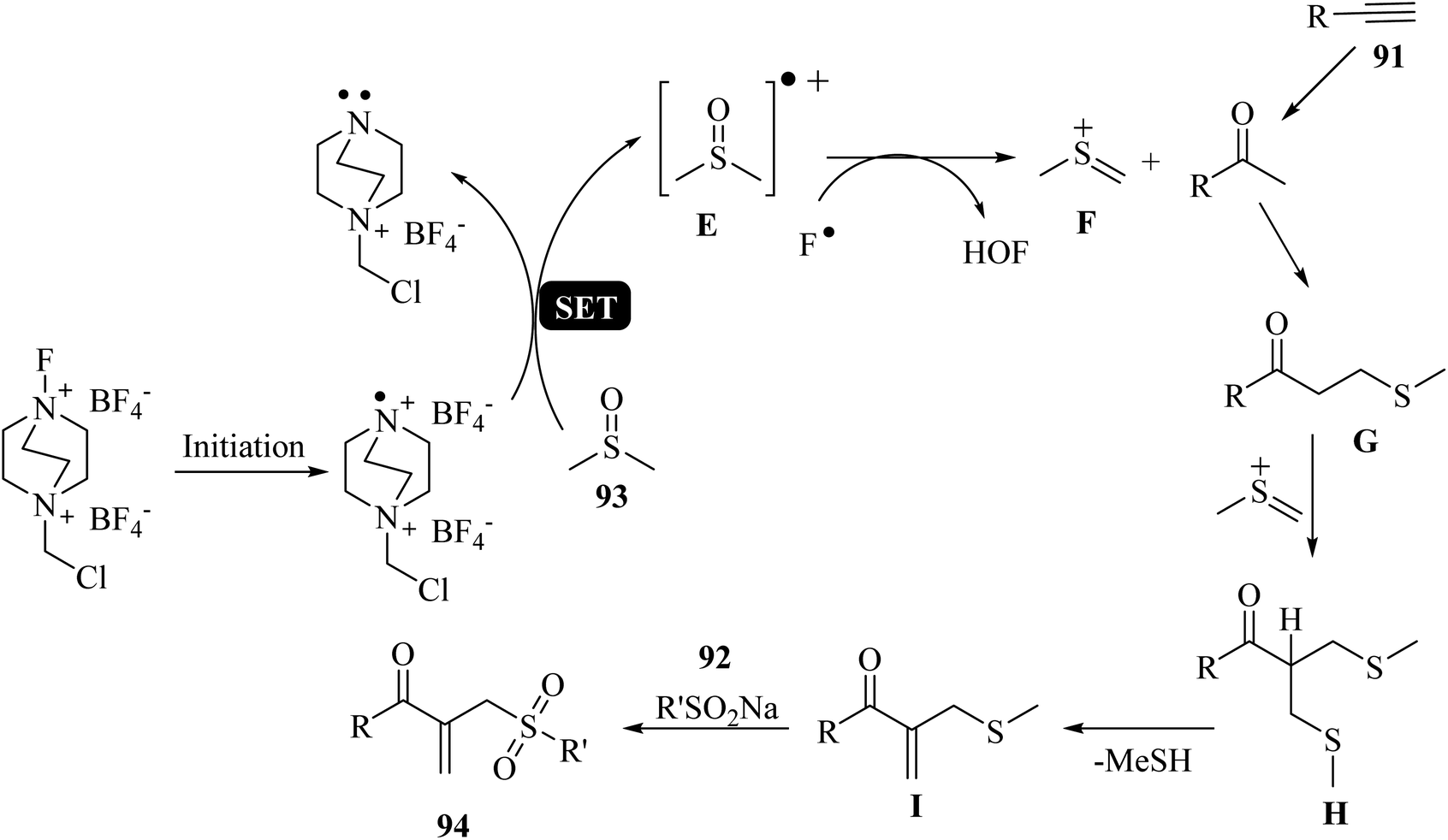 | ||
| Scheme 42 Possible reaction mechanism for the synthesis of β-acyl allyl sulfones 94.58 | ||
In the above-mentioned reaction, DMSO acts as a dual-carbon synthon. Phenyl acetylene without substrates and phenyl acetylenes bearing Me-, OMe-, tBu-, ethyl-, Br-, and Cl-afforded the desired products in 41–66% yield. It should be noted that the investigation of aliphatic alkyne ethynylcyclohexane or 1-ethynylcyclohex-1-ene showed that no desired product was synthesized.
A summary of the reaction of alkynes with some of sulfur-containing structures, and also the limitations and progress of reactions are presented in Table 2.
| Reactants | Product | Catalyst | Solvent | Method employed | Reaction time | Yield | Limitations or progress | Ref. |
|---|---|---|---|---|---|---|---|---|
 |
 |
Ag3PO4 | DMSO | H2O, 70 °C | 6 h | Up to 74% | (1) New transformation of alkynes | Tang et al.59 |
| (2) C–C, C–N, and C–S bonds formed. | ||||||||
| (3) Excellent FG tolerance | ||||||||
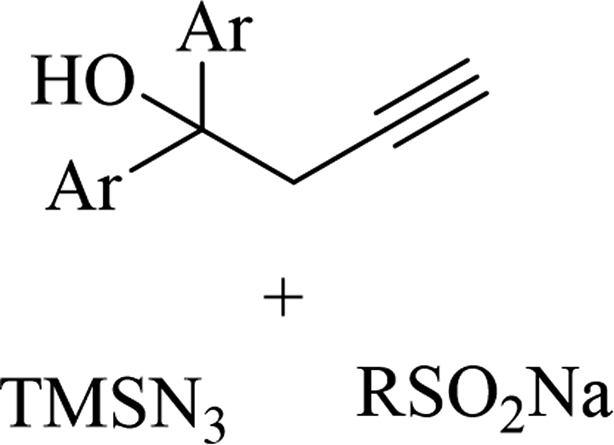 |
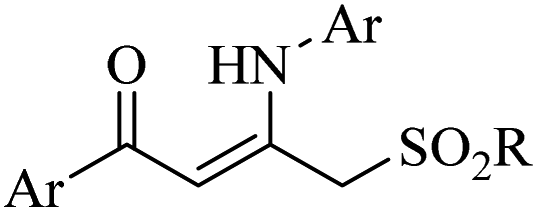 |
Ag3PO4 | DMSO | H2O, 70 °C | 4 h | Up to 75% | Efficient C to N aryl migration of homopropargylic alcohol | Ning et al.60 |
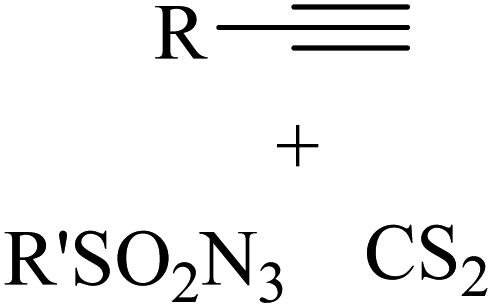 |
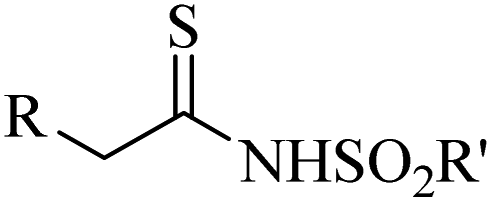 |
CuBr·SMe2 | Toluene | BDU, 100 °C | 22 h | 53–97% | Electronic and steric variations had not remarkable effect on the reaction | Khalaj et al.61 |
 |
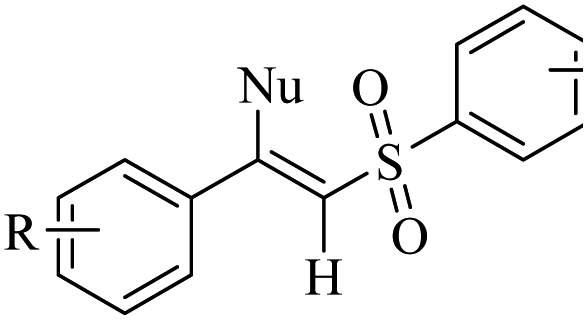 |
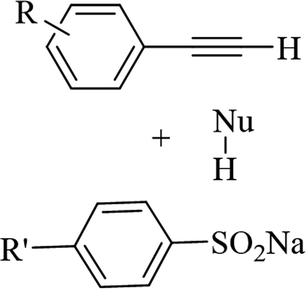
Eosin Y | EtOH | I2, K2CO3, visible light, RT | 48 h | 52–80% | (1) Exclusive Z-selectivity | Sahoo et al.62 |
| (2) C–S and C–O bond formation | ||||||||
 |
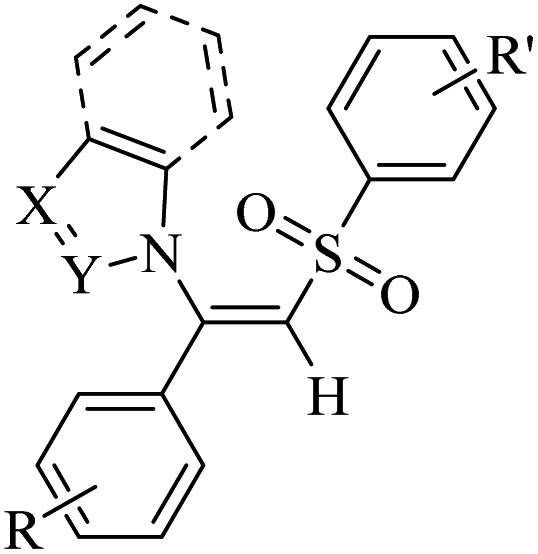 |
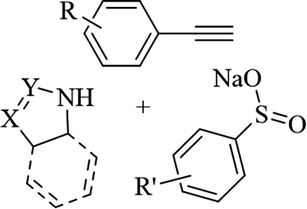
— | DMF | I2, K2CO3, 70 °C | 5 h | Up to 85% | (1) C–S and C–N bond formation | Ansari et al.63 |
| (2) Excellent stereoselectivity | ||||||||
| (3) Mild operation | ||||||||
 |
 |
— | — | KF, 50 °C | 5 h | Up to 81% | (1) Stereoselective | Hazarika et al.64 |
| (2) Transition-metal-free | ||||||||
| (3) Multiple bond cleavage and bond formation |
2.3 Reaction of acids
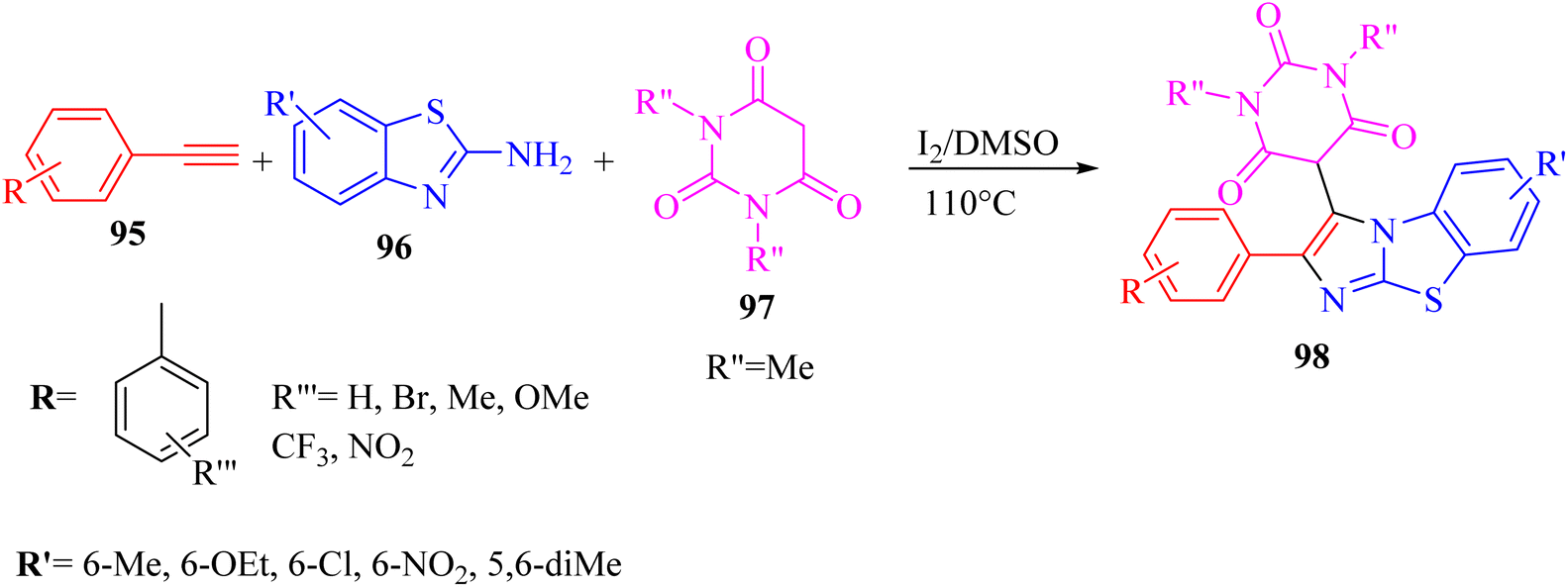 | ||
| Scheme 43 Synthesis of 2-arylbenzo[d]imidazo[2,1-b]thiazole derivatives 98.65 | ||
Initially, 95 reacted with I2 in DMSO and generated B via intermediate A. Then, electron-deficient alkene C was produced via Knoevenagel condensation between B and 97. After that, D was produced via the aza-Michael addition of 96 to C. Finally, 98 was prepared via the intramolecular cyclization of D under acidic conditions together with H2O removal (Scheme 44).
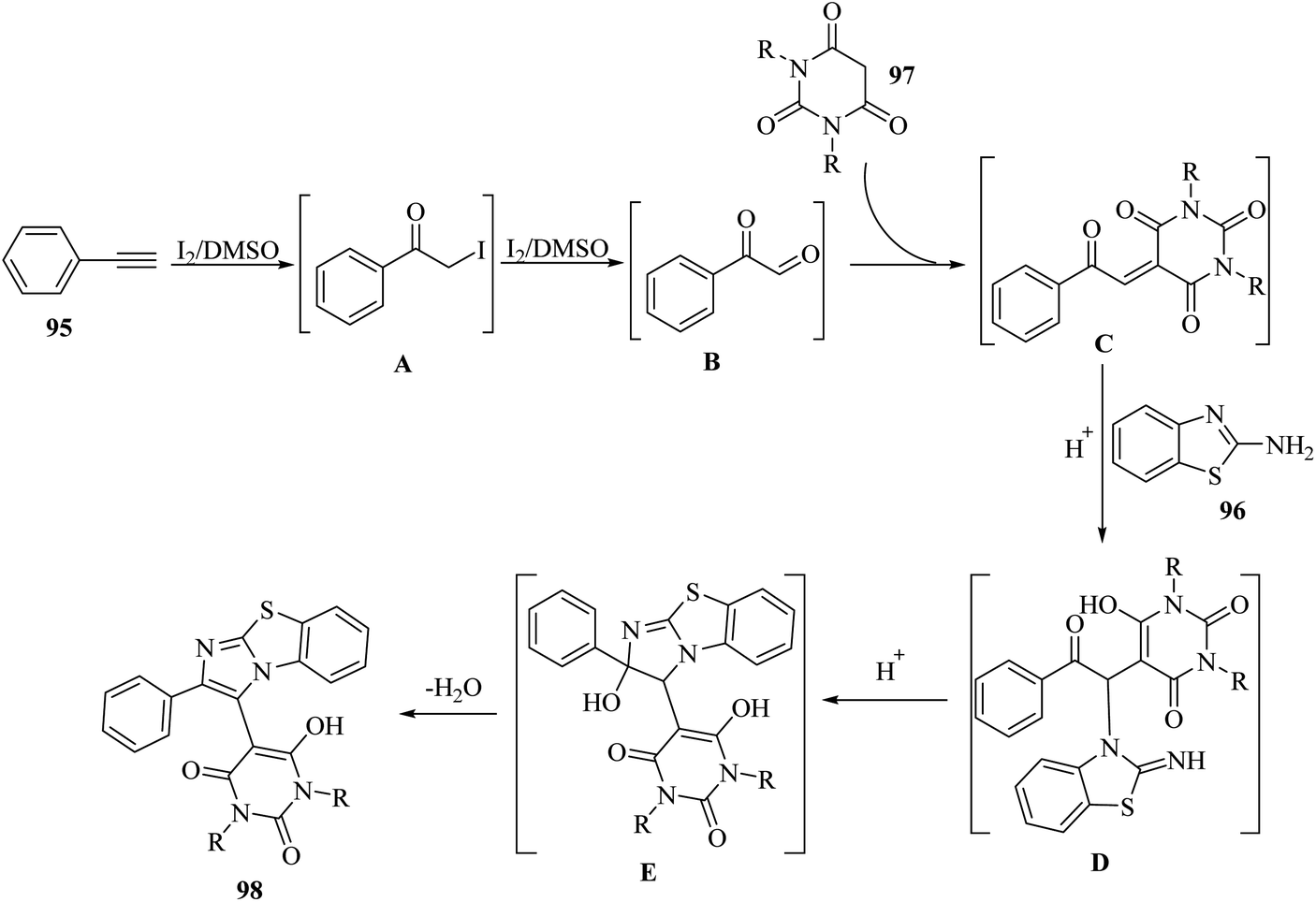 | ||
| Scheme 44 Possible reaction mechanism for the synthesis of 2-arylbenzo[d]imidazo[2,1-b]thiazole derivatives 98.65 | ||
According to the results, 2-aminobenzothiazoles 96 with a variety of groups such as 6-NO2, 6-Me, 5,6-dimethyl, 6-OEt and 6-Cl produced the desired products in 74%,78%, 80%, 83%, and 76% yield, respectively. Moreover, phenyl acetylene derivatives 95 with either electron-donating or -withdrawing groups (Br, Me, OMe, CF3, and NO2) produced the desired products in 79%, 85%, 70%, 85%, and 84% yield, respectively.
Panday et al.66 synthesized pyrimidine-linked naphthoquinone-fused pyrrole derivatives 102 via iodine-mediated one-pot multicomponent reactions (Scheme 45). The desired product was obtained via the reaction among 2-amino-1,4-naphthoquinone 101, barbituric acid derivatives 100, and terminal aryl alkynes 99 in DMSO and molecular iodine at 110 °C for 4–5 h.
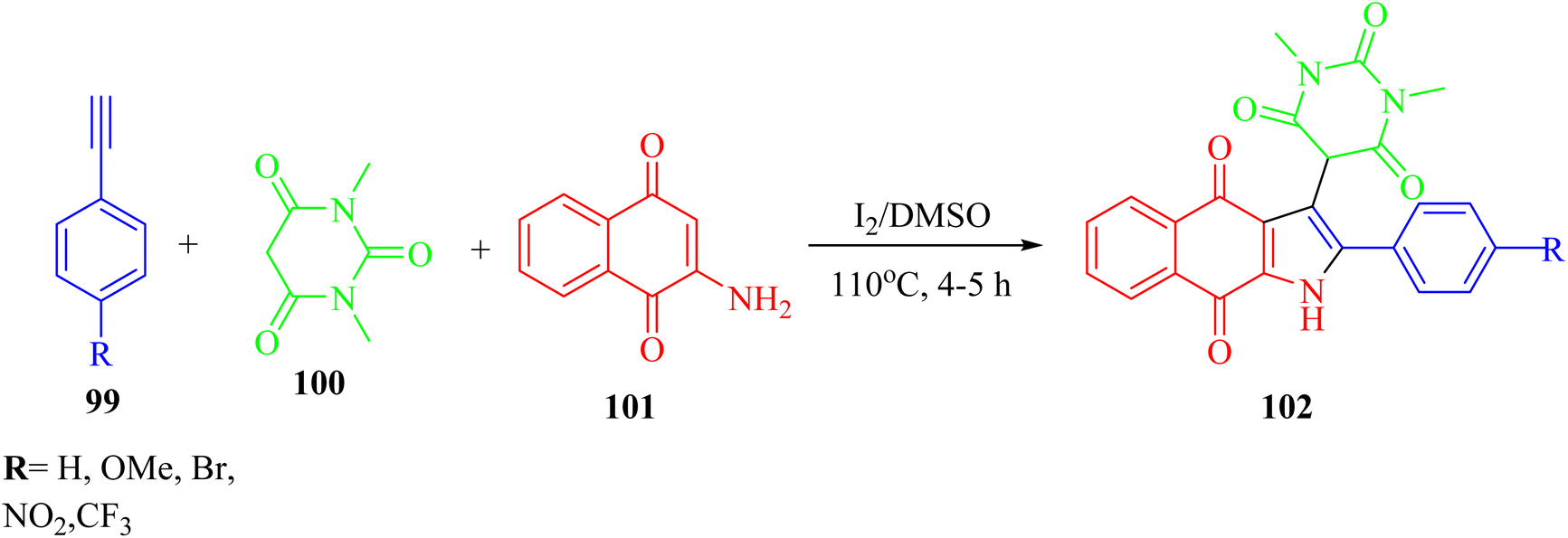 | ||
| Scheme 45 Synthesis of naphthoquinone-fused pyrroles 102.66 | ||
2-Iodoacetophenone C was generated via the reaction between 99 and I2. Next, C oxidized to phenylglyoxal T in the presence of DMSO. Then, alkene compound U was produced via the reaction between T and 100. After that, trisubstituted methane intermediate V was obtained via the 1,4 addition of reaction between U and 101. Finally, 102 was obtained via intramolecular cyclization together with H2O removal (Scheme 46).
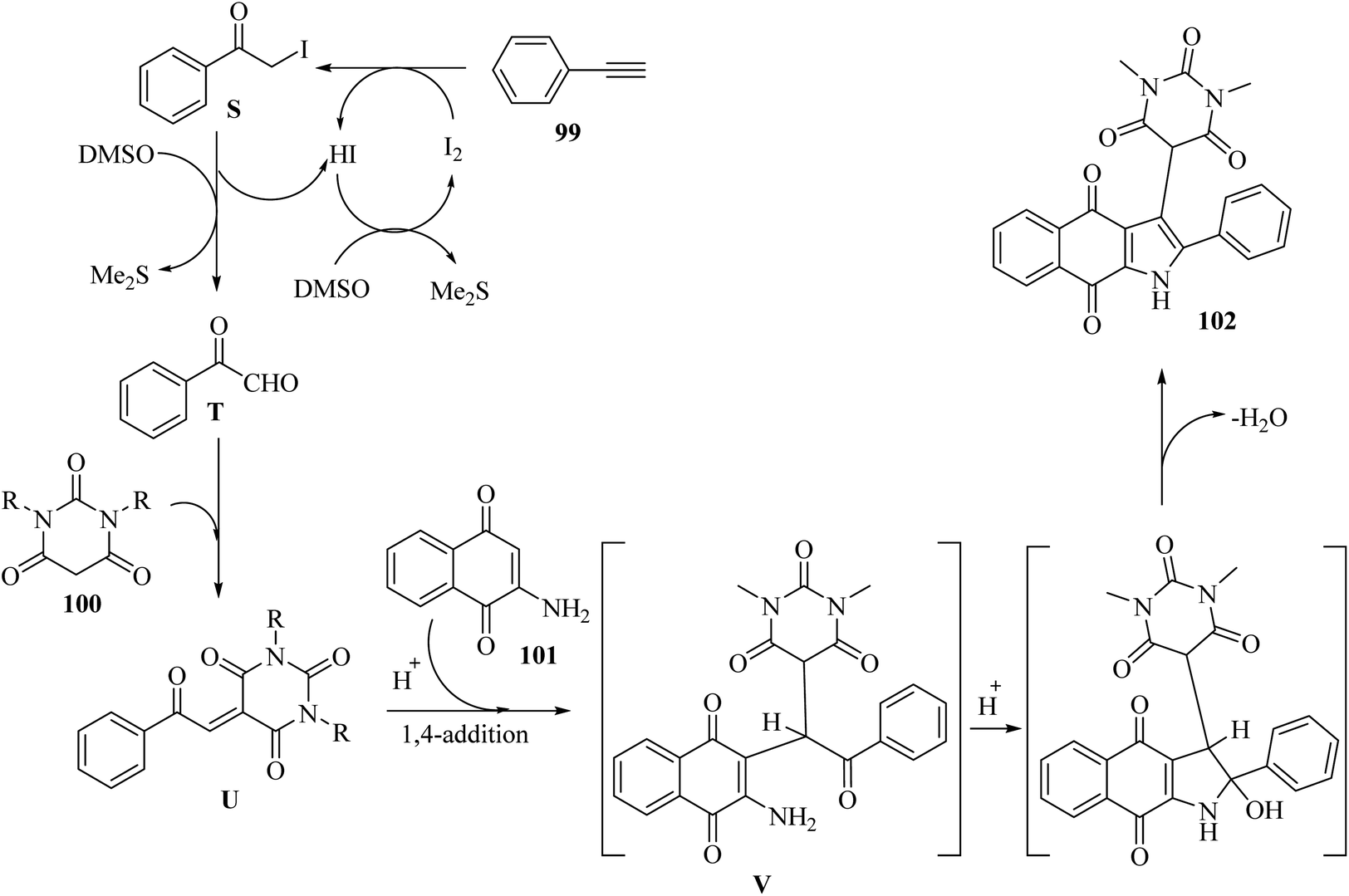 | ||
| Scheme 46 Possible reaction mechanism for the synthesis of pyrimidine-linked naphthoquinone-fused pyrrole derivatives 102.66 | ||
Various alkynes (4-OMe, 4-Br, 4-NO2, and 4-CF3 phenyl acetylenes) produced the desired products with an isolated yield of 60% to 78%.
In 2022, Bhuyan et al.67 synthesized pyrano[2,3-d]pyrimidine-2,4(3H,5H)-dione derivatives 106 via the reaction among aromatic aldehydes 104, N,N-dimethyl barbituric acid 103, and terminal alkyne derivatives 105 in the presence of ZnCl2 in HFIP under ultrasonic irradiation at 50 °C for 1 h (Scheme 47).
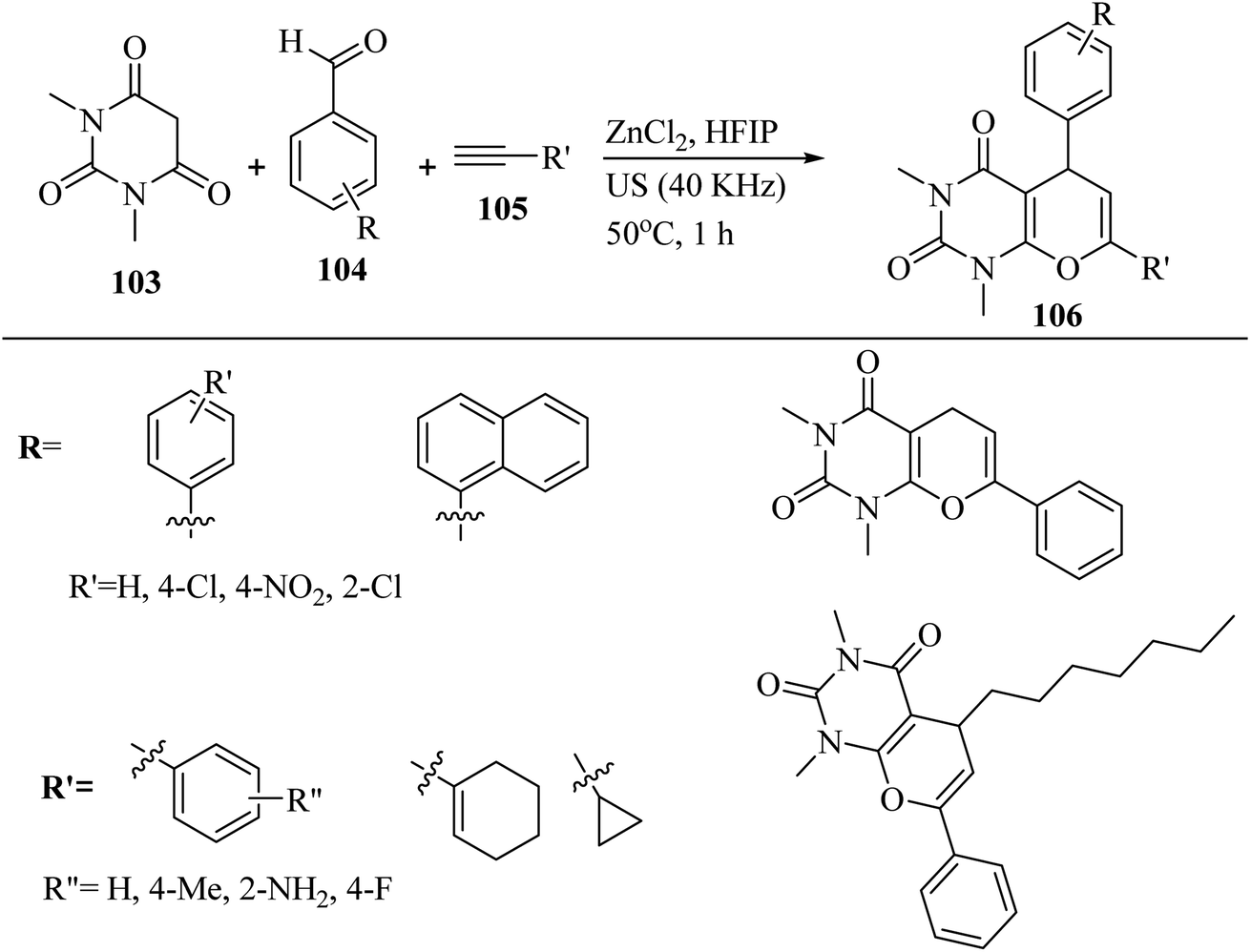 | ||
| Scheme 47 ZnCl2-catalyzed synthesis of pyrano[2,3-d]pyrimidine-2,4(3H,5H)-dione derivatives 106.67 | ||
Firstly, 103 reacted with 104 via Knoevenagel condensation and generated G. Simultaneously, ZnCl2 reacted with 105 and produced zinc(II) alkynilide H. After that, H underwent conjugative alkynylation with G and produced the intermediate I. Finally, the alkynylated product 106 was produced through 6-endo-dig cyclization (Scheme 48).
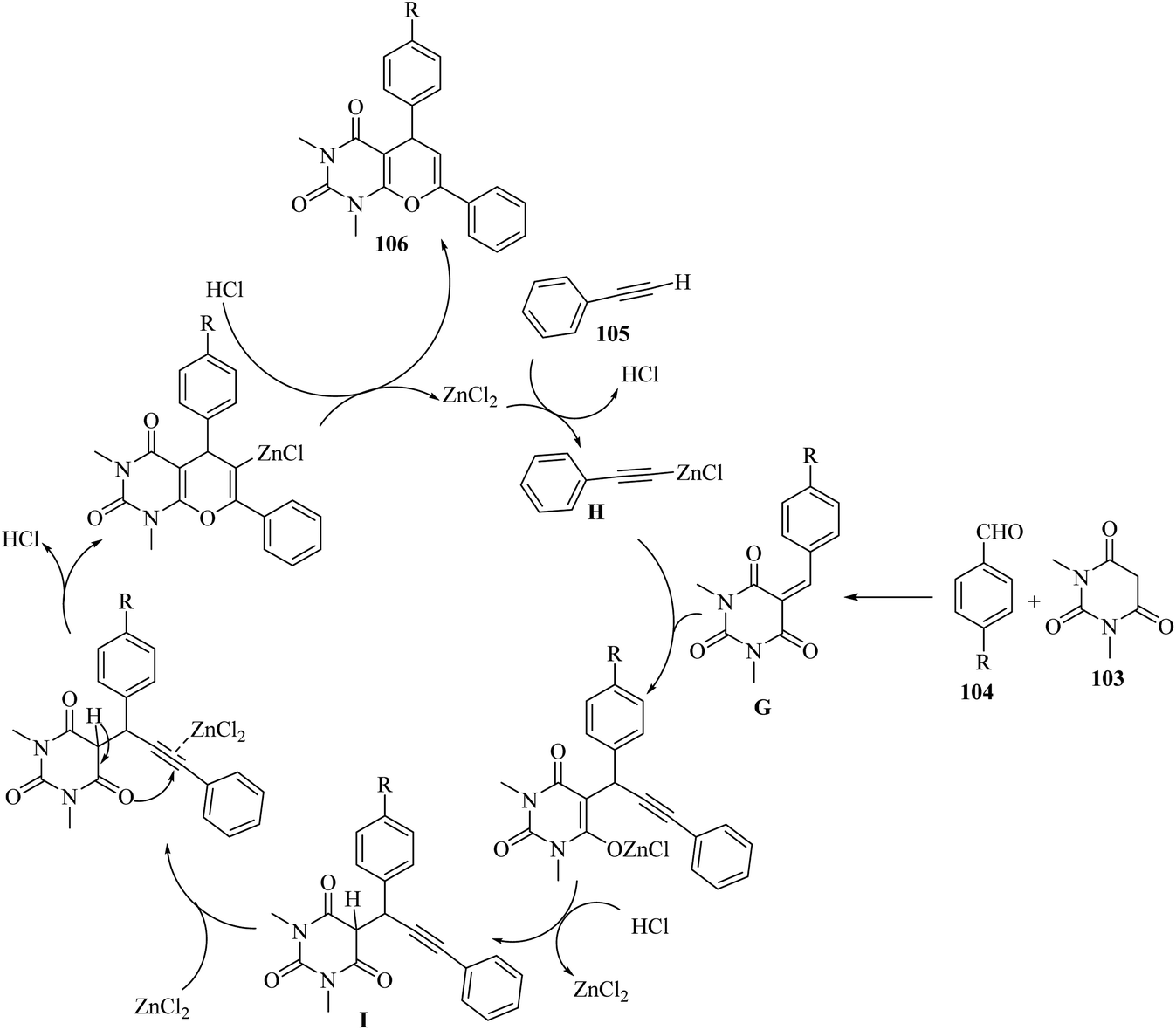 | ||
| Scheme 48 Possible reaction mechanism for the synthesis of pyrano[2,3-d]pyrimidine-2,4(3H,5H)-dione derivatives 106.67 | ||
Various electronic functional groups such as –F, –NO2, –NH2, –Me, and –Cl present on both aldehyde derivatives 104 and alkyne derivatives 105 were shown to tolerate the reaction conditions. However, although all terminal alkynes performed well, aromatic alkynes (yield: 79–95%) gave higher yields than aliphatic alkynes (yield: 68–73%). Electronic factors play a role among aromatic alkynes, given that those with electron-donating groups such as –NH2 and CH3 were observed to produce slightly lower products than their unsubstituted counterparts. Also, the electron-withdrawing –F group has little impact on the product yield. Among the various groups on the aromatic ring of aldehydes, the electron-withdrawing groups -Cl and –NO2 in the para position were found to support the reactions when aromatic alkyne derivatives were used as one of the reaction partners. However, when using aliphatic alkynes, this trend was not observed, but the effect was minimal. Interestingly, the –Cl group placed in the ortho position in aromatic aldehydes had the opposite effect, with lower yields being obtained in these cases. The aromatic bicyclic aldehyde also reacted well and gave a good yield of product (70%). n-Octanal provided the desired product in trace amounts, as identified by HRMS. All other aliphatic aldehydes did not give suitable products under the reaction conditions.
Asghari and co-workers68 synthesized spiro 1,3-oxazines 110 via the reaction among N,N′-disubstituted parabanic acid derivatives 109, imine derivatives 107, and dialkyl acetylenedicarboxylate derivatives (DAAD) 108 in dry dichloromethane without catalyst at room temperature for 1–6 h (Scheme 49).
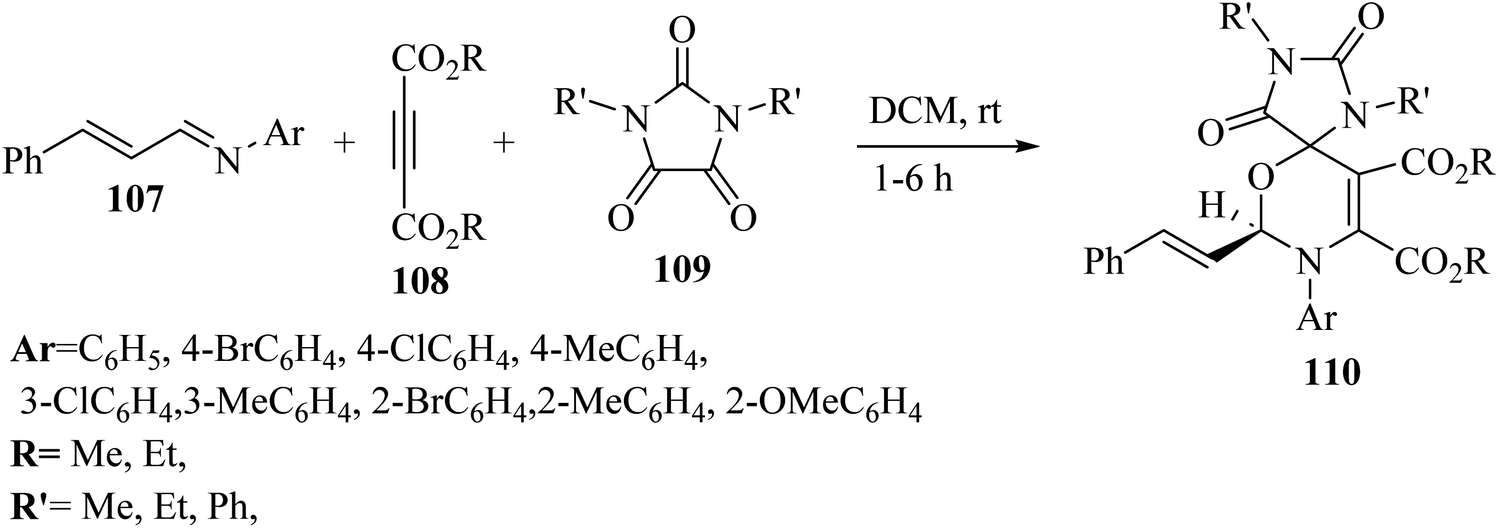 | ||
| Scheme 49 Synthesis of spiro 1,3-oxazine derivatives 110.68 | ||
Initially, 107 reacted with 108 and produced 1,4-dipolar intermediate J. After that, zwitterionic intermediate K was generated via attack of J to the carbonyl group of 109. Finally, 110 was obtained via the intramolecular reaction between iminium and alkoxide group of intermediate K (Scheme 50).
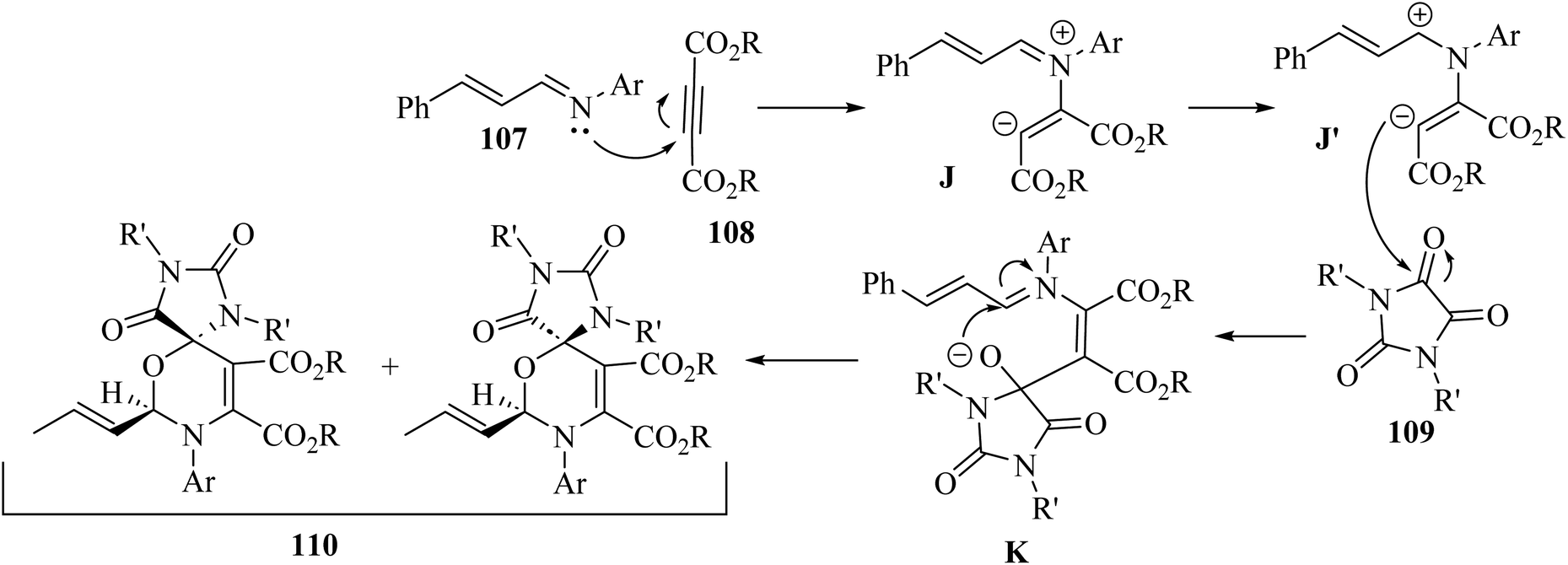 | ||
| Scheme 50 Possible reaction mechanism for the synthesis of spiro 1,3-oxazines 110.68 | ||
The desired products in the above-mentioned method were produced in 44–98% yield. According to the results, the different groups (withdrawing and donating) at the para position of the N-arylaldimine derivatives as nucleophiles were effective and the desired 1,3-oxazines obtained in 84–98% yield. The reaction yields decreased when using electron-withdrawing groups at the meta position in the N-arylaldimine derivatives (65–84%) but the yield of the reaction in the presence of a methyl group increased, which could be due to the inductive effect at the meta position (96%). In addition, when 2-substituted N-arylaldimines were tested, the reaction yields decreased, which could be because of the hindrance effect at the ortho position (44–76%).
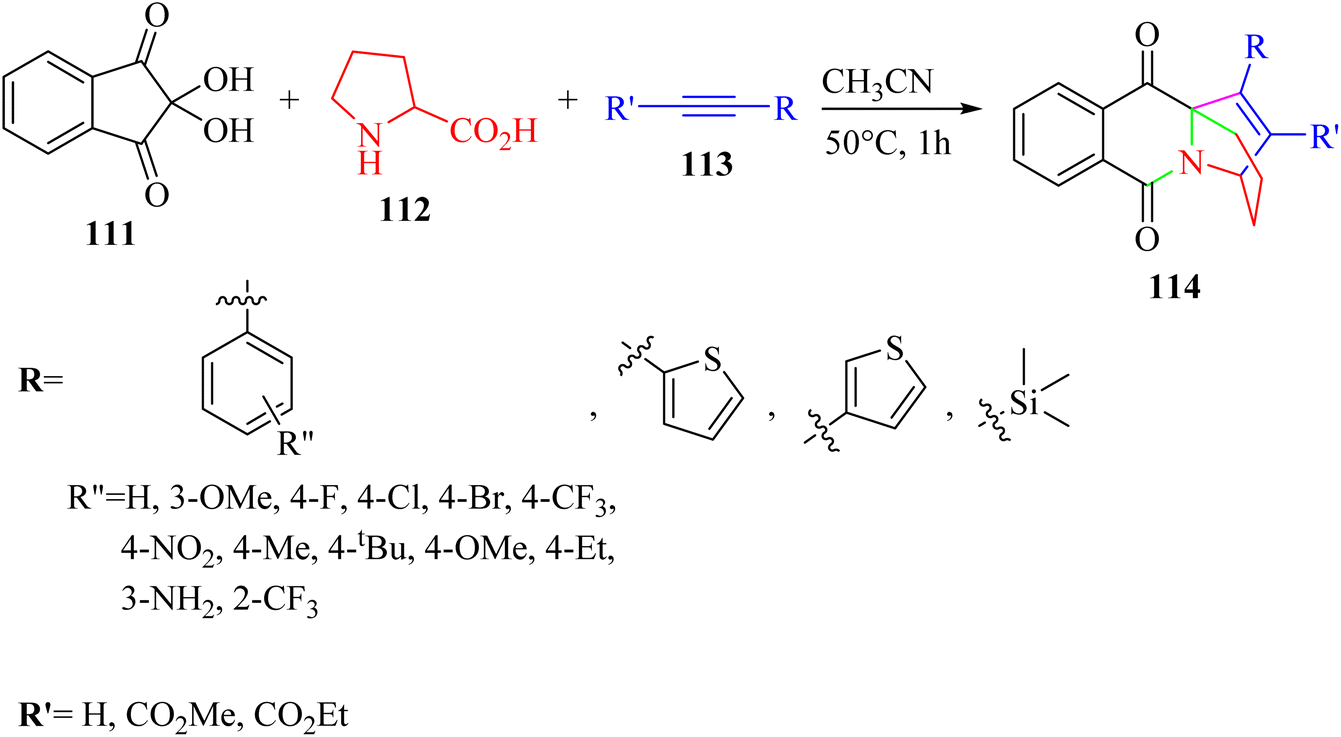 | ||
| Scheme 51 Synthesis of pyrido [1,2-b] isoquinoline derivatives 114.69 | ||
Initially, intermediate K was generated via the reaction between ninhydrin and 112. After that, aziridinium cation intermediate L was produced via the attack of amine on the ninhydrin carbonyl carbon. Next, intermediate L underwent ring expansion and produced intermediate M. After that, intermediate N was obtained via the nucleophilic attack of the enol carbon on the proline carbon. It should be noted that the authors could not isolate intermediate N. In the next step, intermediate O was produced from intermediate N by the loss of H2O, followed by decarboxylation. Next, intermediate O was converted into isoquinolinium ylide O′, probably. Finally, 114 was produced via the reaction between O′ and 113 by [3 + 2]-cycloaddition reaction (Scheme 52).
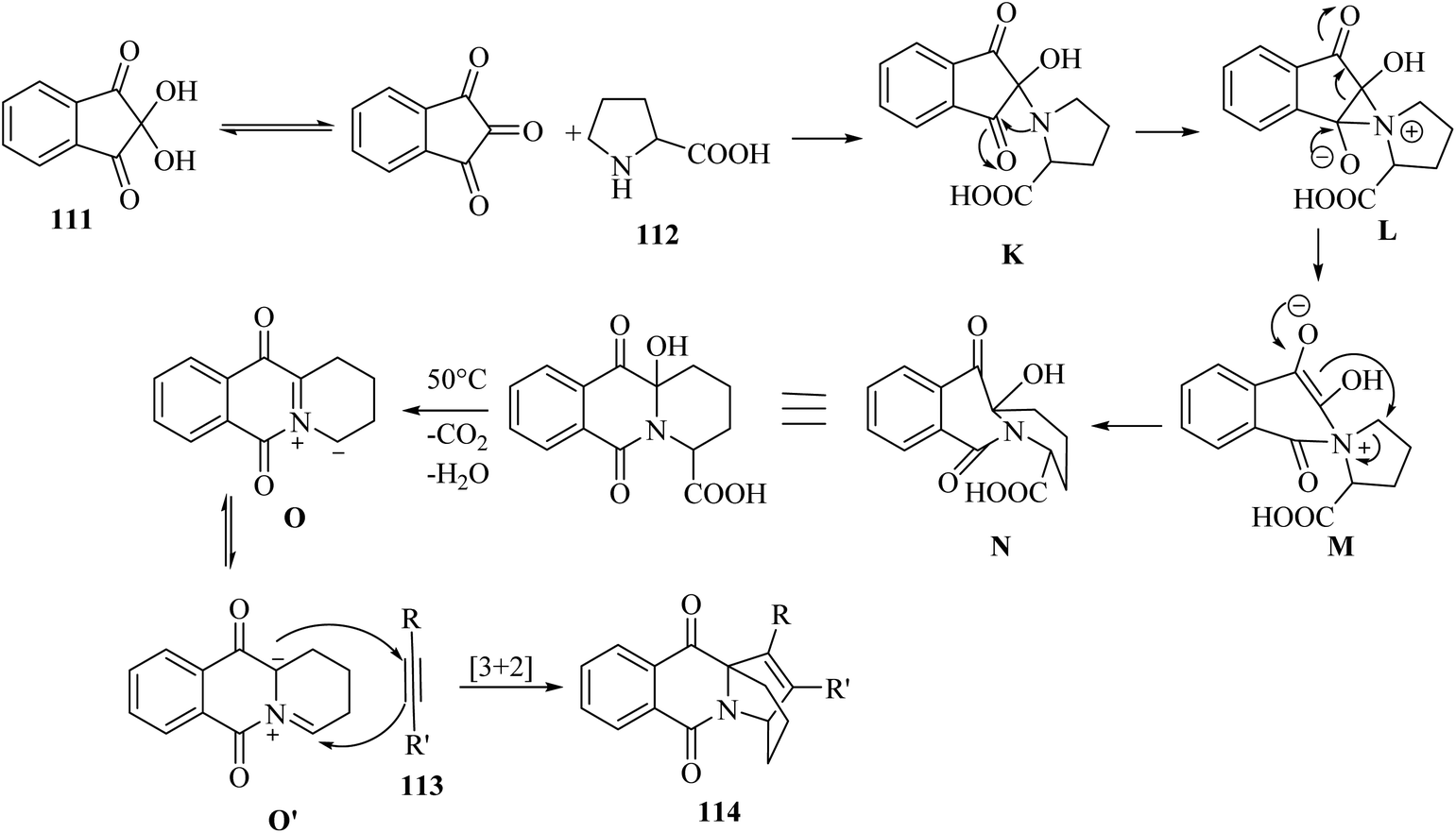 | ||
| Scheme 52 Possible reaction mechanism for the synthesis of ethanopyrido[1,2-b]isoquinolines 114.69 | ||
According to the results, various aromatic alkyne derivatives containing different types of groups at different positions on the aromatic ring produced the desired of ethanopyrido derivatives in 67–90% yield. Heteroaryl alkyne (thiophene) also produced the desired ethanopyrido derivatives in 75–80% yield. The above-mentioned method was extended with ethyl phenylpropiolate derivatives and produced the corresponding ethanopyrido derivatives in 70–90% yield. Furthermore, unactivated terminal alkynes produced the desired products in 70–88% yield. Importantly, terminal aromatic alkynes containing free amine group also produced the corresponding of ethanopyrido[1,2-b]isoquinoline in 88% yield. Moreover, TMS acetylene was also treated with isoquinolinium ylide and produced the corresponding ethanopyrido[1,2-b]isoquinoline in 73% yield. However, the reaction did not proceed with 1-butyne and cyclohexylacetylene.
Thakur et al.70 synthesized 3-substituted 2-quinolone derivatives 118 via the reaction among terminal alkynes 116, 2-iodoaniline derivatives 115, and oxalic acid 117 as the C1 source in the presence of polystyrene-supported palladium (Pd@PS) nanoparticle catalyst, and TBAI as a base in DMF at 135 °C under MW irradiations for 1 h (Scheme 53).
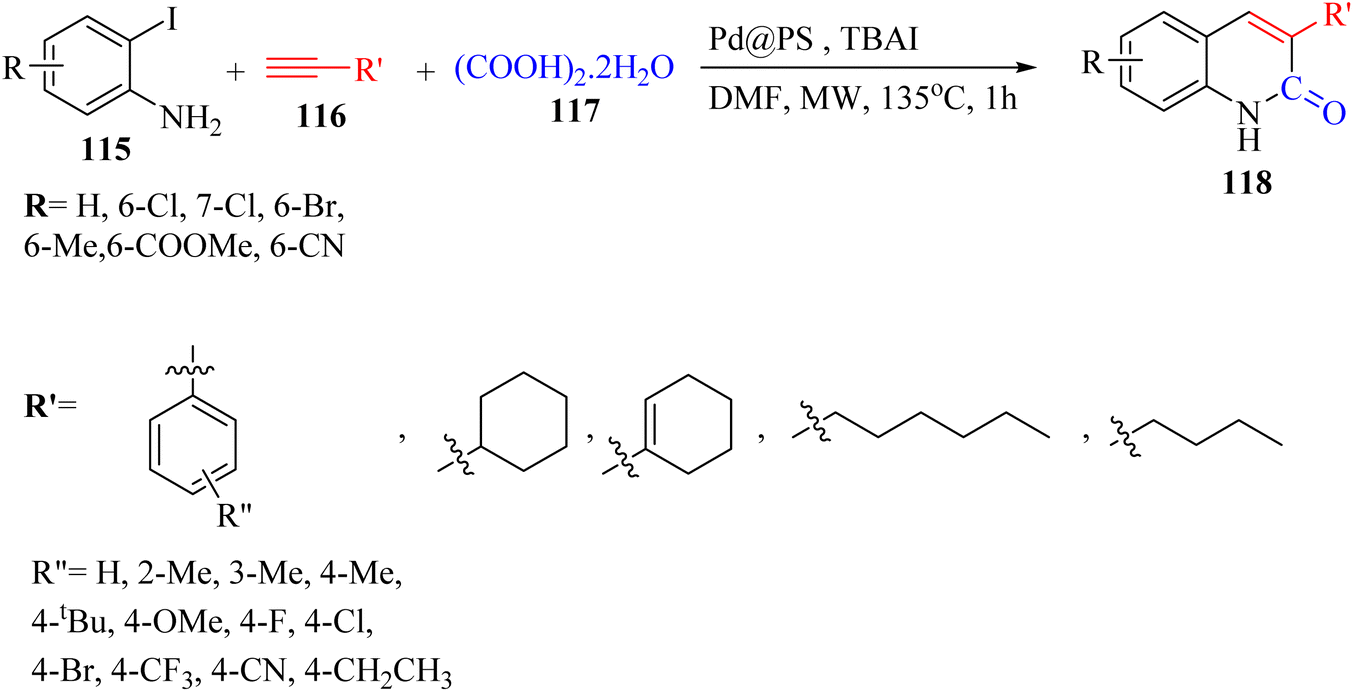 | ||
| Scheme 53 Synthesis of 3-substituted 2-quinolone derivatives 118 by oxalic acid.70 | ||
Initially, 115 and Pd@PS reacted and generated arylpalladium intermediate A via oxidative addition. Then, vinylpalladium intermediate B was obtained via the insertion of 116 in intermediate A. When intermediate B is produced, two pathways (A or B) may be followed. In the case of pathway A, intermediate C was produced via the interaction of CO with Pd metal. Next, acylpalladium intermediate D was obtained via the insertion reaction. After that, 118 was generated via the nucleophilic attack of the amine group on the carbonyl of intermediate D. In this pathway for synthesis of the 118, another minor pathway exists, where intermediate E was obtained via the formylation of intermediate B. Then, intermediate E formed 118. In the case of pathway B, intermediate F was produced via the reaction between CO2 and the amine group of intermediate B. Next, intermediate F produced intermediate G in the presence of base. Finally, intermediate G generated 118 via the nucleophilic attack of the vinyl group at the carbon of the isocyanate group together with the regeneration of Pd@PS (Scheme 54).
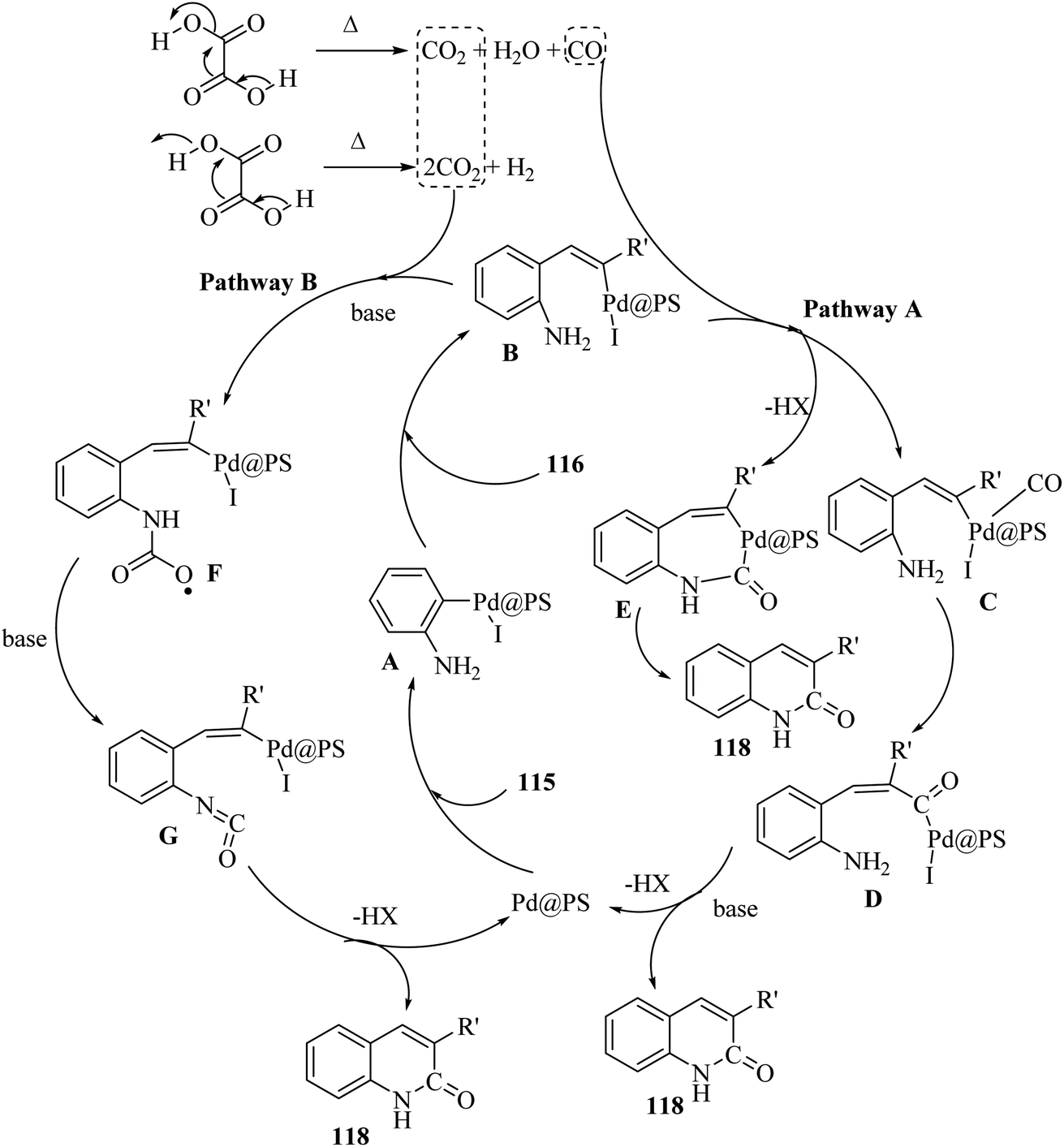 | ||
| Scheme 54 Possible reaction mechanism for the synthesis of 3-substituted 2-quinolone derivatives 118.70 | ||
All the 4-CH3, 4-COOCH3, 4-CN, 4-Cl, 5-Cl, 4-Br, and 4-F substituted 2-iodoanilines reacted well with 4-ethynyltoluene and phenylacetylene to produce the desired 2-quinolone derivatives in 43–75% yield. The relatively lower yield of 2-ethynyltoluene compared to 3-ethynyltoluene could be due to the steric hindrance at the ortho position of the alkyne. The reaction of 2-iodoaniline with 4-tert-butylphenylacetylene led to the formation of the corresponding 3-substituted 2-quinolone in 64% yield. Moreover, the C2H5/4-OCH3-substituted alkynes produced the desired 2-quinolone derivatives in 56% and 58% yield, respectively. The halogen-substituted aromatic alkynes also produced the corresponding products in 43–55%. The aromatic alkyne derivatives containing electron-withdrawing groups (–CF3 and –CN) produced the desired 2-quinolone derivatives in 42% and 55% yield, respectively. In addition, alicyclic alkynes (1-ethynylcyclohexene and 1-ethynylcyclohexane) also produced the desired 2-quinolone derivatives in 52% and 49% yield, respectively. The aliphatic alkynes were tested and produced the desired 2-quinolone derivatives in 54–57% yield.
Zeng and co-workers71 synthesized α-carbonyloxy ester derivatives 122 via a two-stage process. In the first stage, carboxylic acids 119, and ynol ethers 120 reacted in the presence of Ag2O in dioxane at 100 °C for 5 h and produced alpha-alkoxy enol ester intermediate (1), and then 3-chloroperoxybenzoic acid 121 added to the mixture and stirred at room temperature for 12 h (Scheme 55).
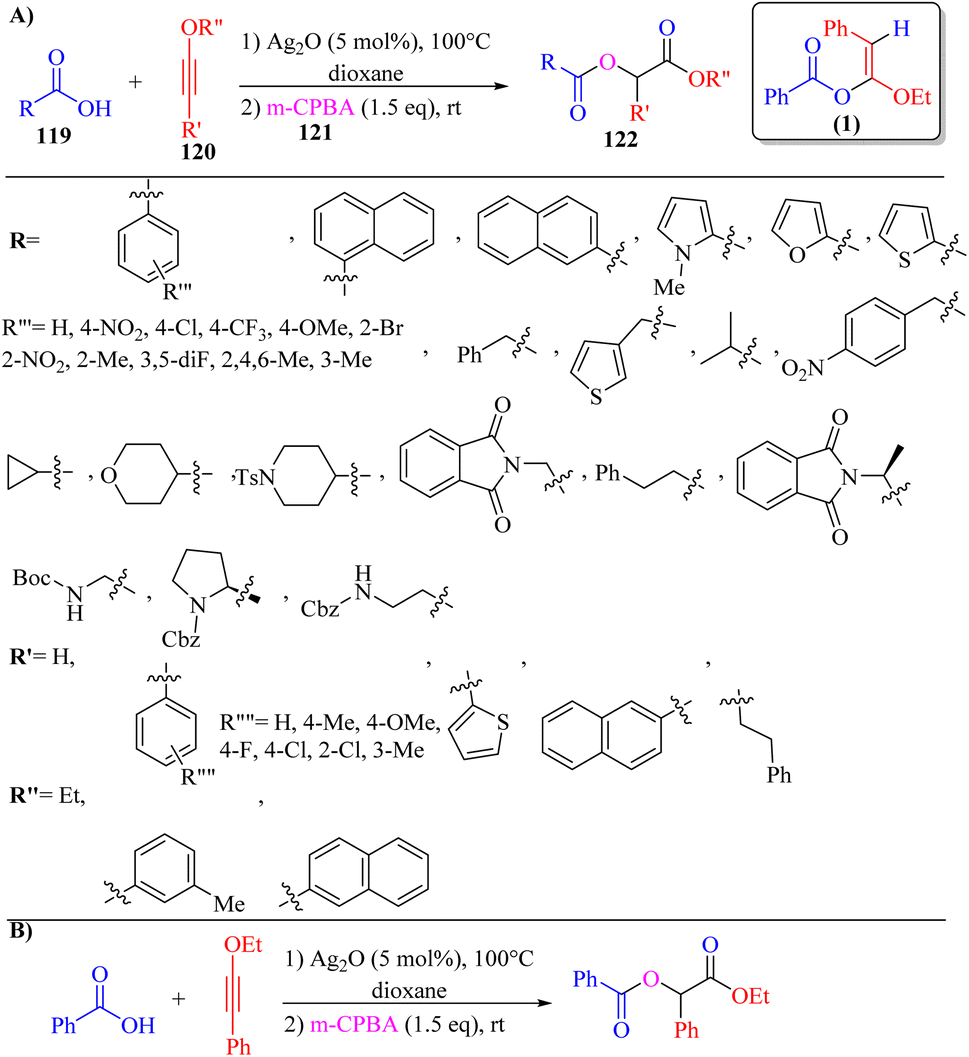 | ||
| Scheme 55 (A) Ag2O catalyzed synthesis of α-carbonyloxy esters 122. (B) Gram-scale reaction for the synthesis of α-carbonyloxy ester; 1: α-alkoxy enol ester intermediate synthesized via the reaction between benzoic acid and ynol ether.71 | ||
Firstly, 119 and 120 reacted and produced α-alkoxy enol ester K under catalytic Ag2O promotion. After the synthesis of K, two pathways may exist. In pathway A, intermediate L was generated via the reaction between 121 and the carbonyl group of K. Next, 122 was produced from intermediate L via fragment cascade and Baeyer–Villiger-type reaction. In pathway B, intermediate M was generated via the epoxidation of K by 121. Finally, 122 was obtained from M (Scheme 56).
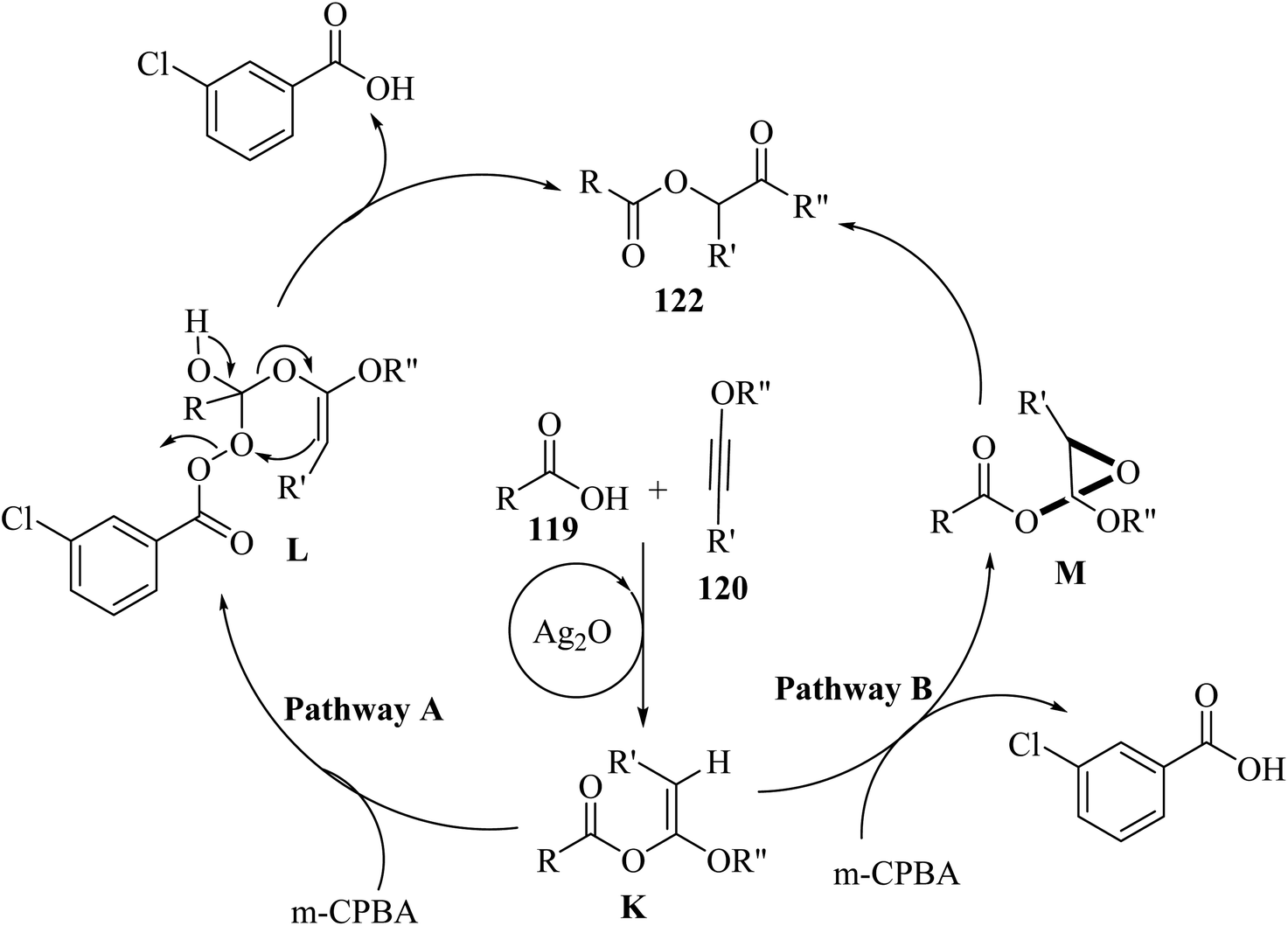 | ||
| Scheme 56 Possible reaction mechanism for the synthesis of α-carbonyloxy ester derivatives 122.71 | ||
When various carboxylic acid derivatives 119 and ynol ether derivatives 120 were evaluated, the desired α-carbonyloxy ester derivatives were produced in 45–91% yields. In the case of 119, various groups produced the corresponding products in 69–83% yield. Also, heterocyclic carboxylic acid derivatives (thiophene, pyrrole, and furan) and naphthyl carboxylic acids produced the corresponding product in 91%, 57%, 53%, 86% and 70% yield, respectively. Moreover, aliphatic carboxylic acids also produced the corresponding products in 49–79% yield. Also, N-protected amino acid derivatives produced the corresponding products in 72–81% yield. Importantly, the products generated from L-proline and L-alanine showed dr values of 54:46 and 53:47, respectively. In the case of 120, various substituted groups in the benzene ring such as para-methoxy, para-fluoro, para-chloro, ortho-chloro, meta-methyl, and para-methyl produced the corresponding products in 49–83% yield. In addition, the aliphatic and terminal ynol ether in the above-mentioned method engaged and produced the desired α-carbonyloxy ester derivatives in 45% and 49% yield, respectively.
Wang et al.72 synthesized β-amino amides 127 with high diastereoselectivity via the four-component reaction of triazenyl alkynes 123, carboxylic acids 125, aldehydes 124 and anilines 126 in the presence of Sc(OTf)3 and 4 Å MS in DCE at 80 °C (Scheme 57). In the above-mentioned method, the alkyne scaffold serves as a C2 fragment, which can lead to high diastereoselectivity.
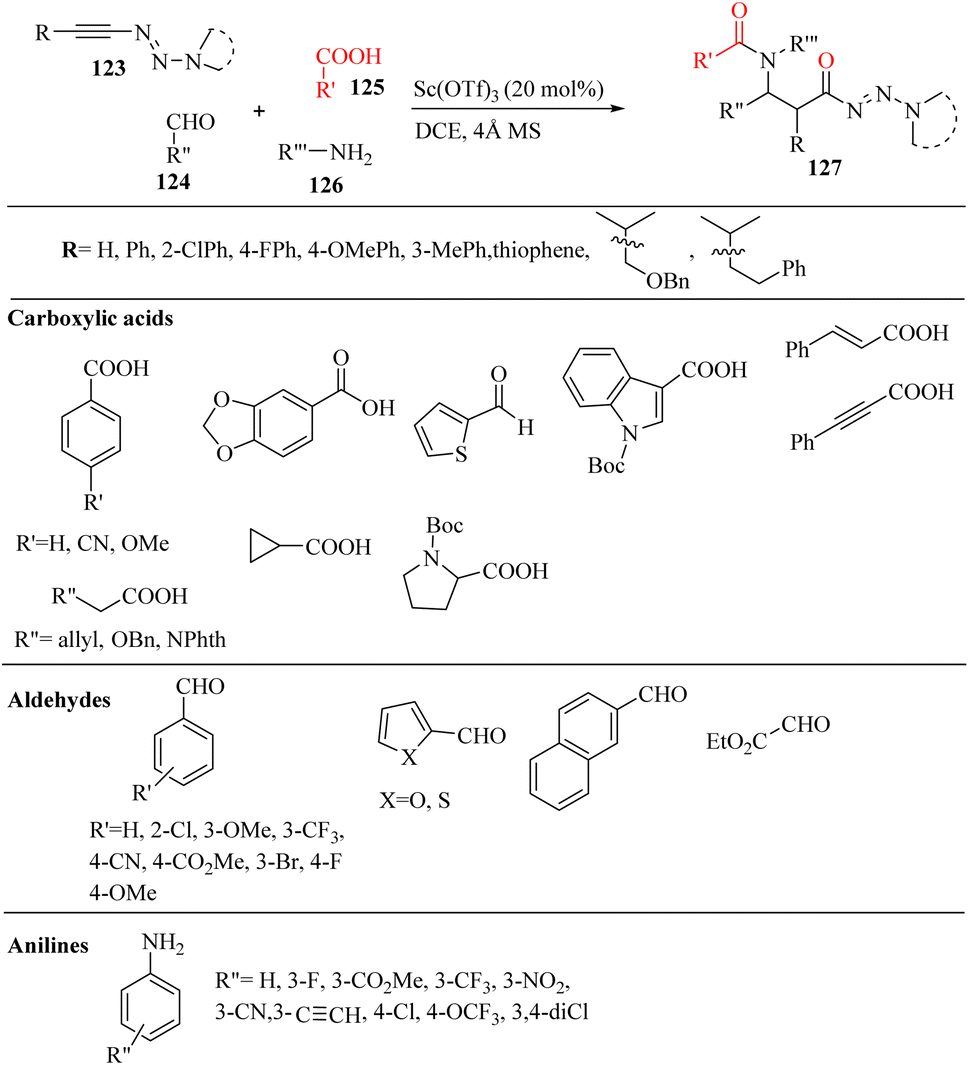 | ||
| Scheme 57 Synthesis of β-amino amide derivatives 127 using Sc(OTf)3.72 | ||
Initially, 124 and 126 reacted and produced imine R, probably, and also probably 123 reacted with 125 and generated E-enol ether Q. After that, a Zimmerman–Traxler chair-like transition state was formed. It should be noted that Sc(OTf)3 could be coordinated with the nitrogen atom of the imine and triazene group. Next, intermediate T was obtained via intramolecular addition of the enol ether to R. Finally, 127 was produced via the Mumm rearrangement together with the regeneration of the catalyst. The important point about this reaction was that 123 acted as a bifunctional building block (Scheme 58).
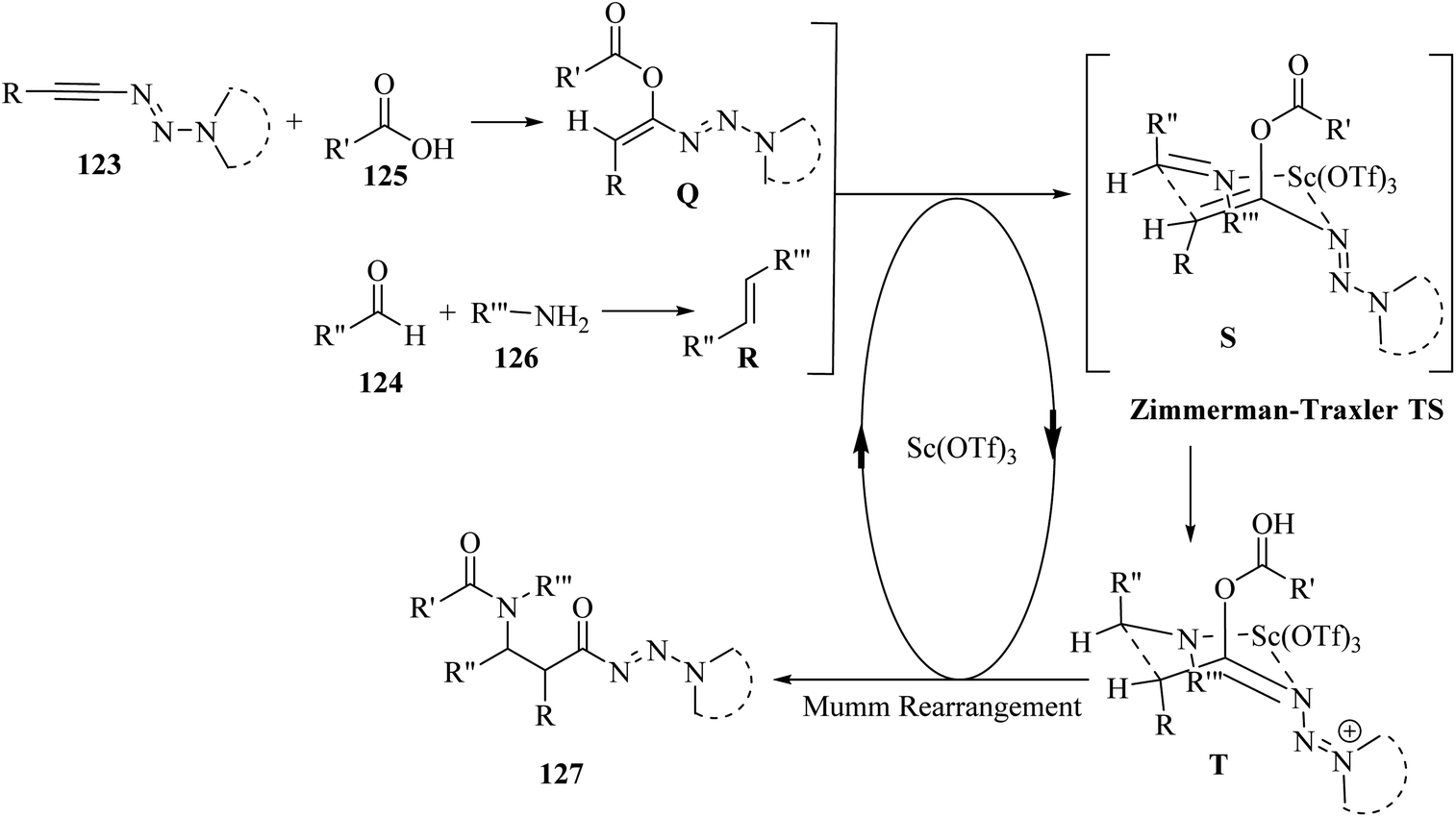 | ||
| Scheme 58 Possible reaction mechanism for the synthesis of β-amino amides 127.72 | ||
In the case of 123, various substitutions in the aromatic ring such as F, Cl, Me, and OMe were tolerated and produced the corresponding products in 81–87% yield with excellent diastereoselectivity (d.r = up to >20:1). It should be noted that the substitution position did not have a remarkable effect on the reaction yields. The thiophene-2-yl-substituted triazenyl alkyne also produced the corresponding β-amino amide in 90% yield (d.r = >20:1). The alkyl-substituted triazenyl alkyne derivatives such as benzyloxymethyl and phenylethyl produced the corresponding β-amino amides in 87% and 86% yield (d.r = 5:3 and >20:1), respectively. The benzyloxymethyl-substituted triazenyl alkyne produced the corresponding beta-amino amide with moderated d.r, which may be due to the O atom of the benzyloxymethyl group coordinating with Sc(OTf)3. In addition, the trimethylsilyl-substituted triazenyl alkyne produced the desilylation beta-amino amide product in 70% yield, and when piperidine-substituted triazenyl alkyne was tested, the desired β-amino amide could be formed in 76% yield (d.r = >20:1). In the case of 125, aldehydes, and anilines, various substituted benzoic acids such as methoxy, [1,3]dioxole, and cyano were tested in the reaction and produced the corresponding β-amino amides in 89%, 90%, and 80% yield, respectively with excellent d.r (d.r = >20:1). The heterocyclic carboxylic acids bearing indole and thiophene produced the corresponding β-amino amides in 82% and 90% yield, respectively. Unsaturated carboxylic acids such as 3-phenylpropiolic acid and cinnamic acid also produced the corresponding beta-amino amides in 80% and 90% yield, respectively. Aliphatic carboxylic acids (allyl, protected alcohol, amino, and cyclopropyl) were well engaged in the reaction and produced the corresponding β-amino amide derivatives in 92%, 90%, 90%, and 87% yield, respectively. Also, N-Boc L-proline produced the desired product in 80% yield. Next, a variety of benzaldehydes with the substitution of OMe, Cl, F, Br, CF3, CN, and CO2Me was tolerated and produced the corresponding beta-amino amide derivatives in 80% to 95% yield. In addition, anisaldehyde produced the desired product in 63% yield. The heterocyclic aldehyde derivatives were tested and the desired beta-amino amide derivatives obtained in 62% to 73% yield. Ethyl oxoacetate also produced the desired β-amino amide in 70% yield. Finally, various aniline derivatives 126 were tested and anilines with differential substituted positions and functionalities (F, Cl, CF3, CO2Me, NO2, CN, ethyne, and OCF3) were tolerated and the desired β-amino amide derivatives obtained in 71–89% yield. Importantly, aliphatic aldehydes and alkylamines failed in the reaction and did not produce the desired β-amino amides.
2.4 Synthesis of heterocyclic structures via the reaction among alkynes, aldehydes and amines
Fernández and co-workers73 synthesized pyrano[2,3,4-ij]isoquinoline derivatives 131 (132 was generated in a molar ratio of 2.5:1:1) via the reaction among dimethyl acetylenedicarboxylate 133, ortho-alkynylsalicylaldehydes 135 and anilines 134 (molar ratio: 1:1:1) in the presence of AgOTf in THF under reflux (Scheme 59).
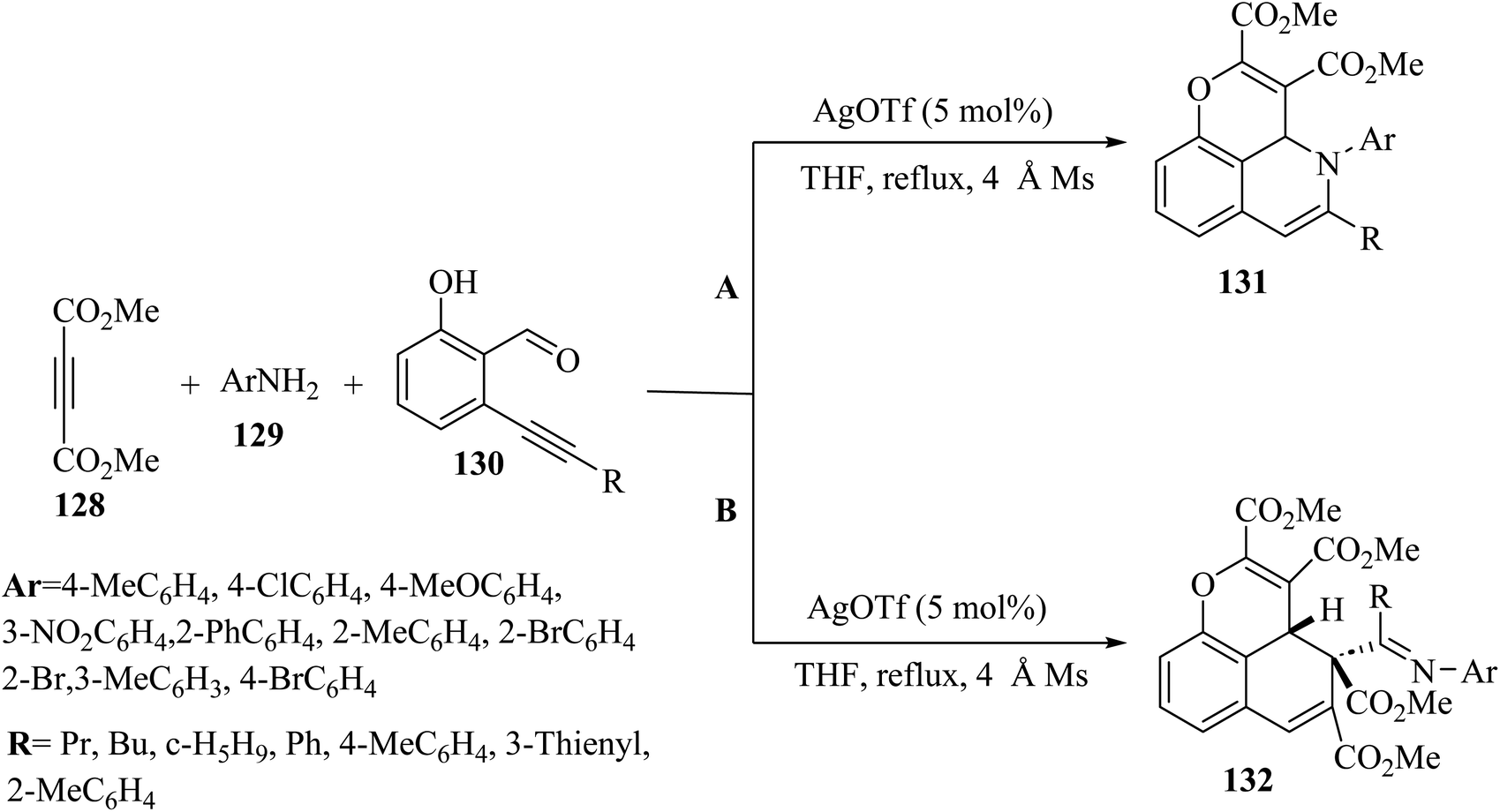 | ||
| Scheme 59 (A) Synthesis of pyrano[2,3,4-ij]isoquinoline derivatives (molar ratio = 1:1:1) and (B) synthesis of benzo[de]chromene derivatives (molar ratio = 2.5:1:1).73 | ||
Initially, intermediate L was generated via the coordination of AgOTf to the triple bond of K, which was prepared by the reaction between 129 and 130. After that, intermediate M was obtained via the intramolecular addition of the nitrogen of the imine to the alkyne. Subsequently, 8-isoquinolinones N was produced via the intramolecular protodemetalation reaction together with the regeneration of the silver catalyst. Next, intermediate N via [4 + 2] cycloaddition reactions with 128 generated the first desired product 131. In addition, when an excess of 128 was used, derivatives 131 reacted with it and produced cyclobutenes O via [2 + 2] cycloaddition reactions. After that, the cyclobutene underwent electrocyclic ring opening and produced novel tricyclic intermediates P. Then, P may produce the bicyclic intermediates Q via another ring-opening process. Finally, 132 was obtained via the electrocyclic ring-closing process of intermediates Q (Scheme 60).
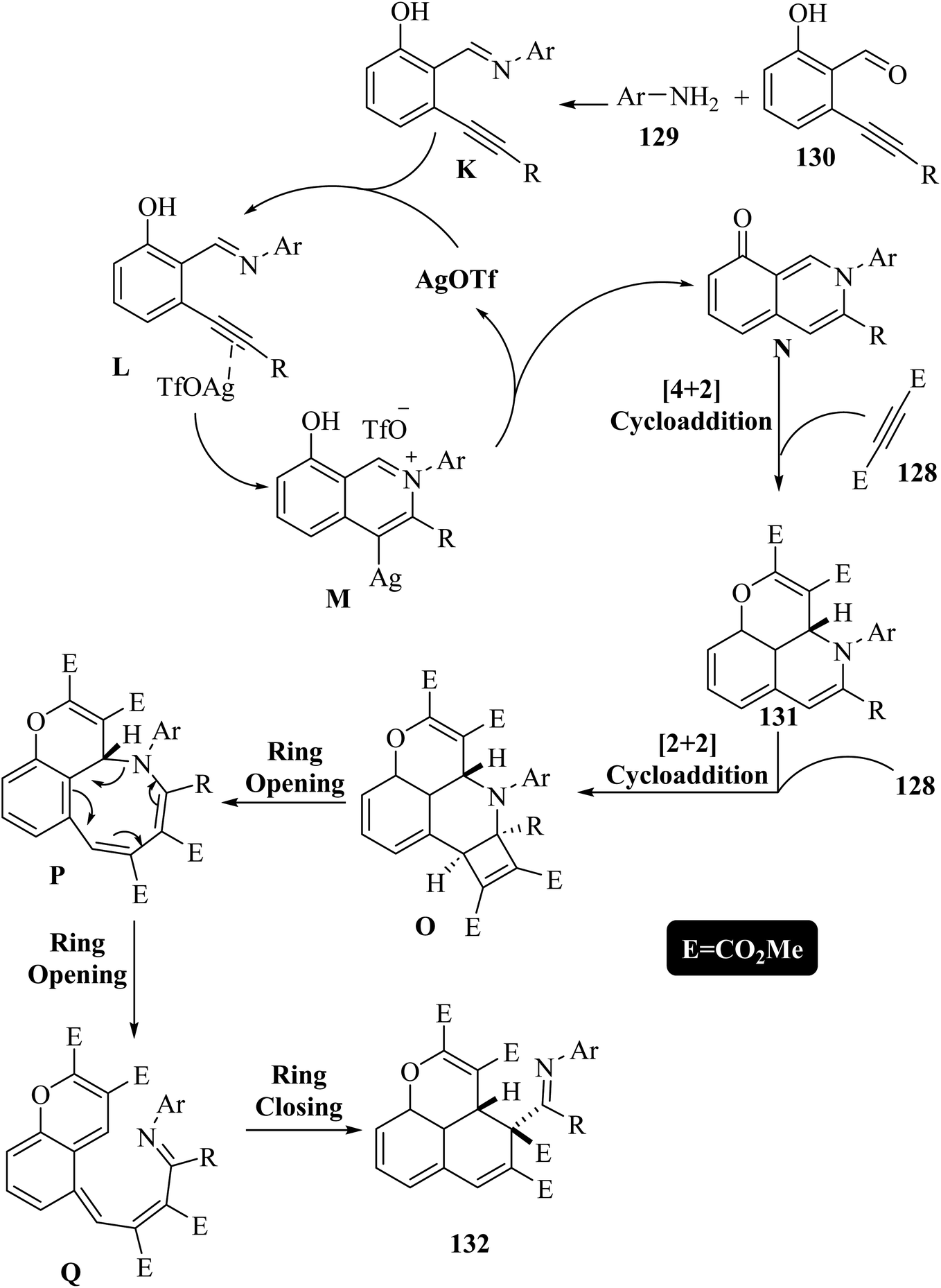 | ||
| Scheme 60 Possible reaction mechanism for the synthesis of benzo[de]chromenes and pyrano[2,3,4-ij]isoquinolines.73 | ||
The desired products were produced in moderate yields. According to the results, when an excess of dimethyl acetylenedicarboxylate (molar ratio: 2.5) was used under the same condition, benzo[de]chromene derivatives 137 were synthesized instead of pyrano[2,3,4-ij]isoquinoline derivatives 136. The corresponding products were obtained in moderate yield (40–48%).
Das et al.74 synthesized 1,3-disubstituted tetrahydroisoquinolines 141 and isoindolines 142 via the reaction among aldehyde derivatives 138, aniline derivatives 140 and alkyne derivatives 139 with high yield (up to 92%), diastereoselectivity (up to 9:1), and enantioselectivity (up to 99%) (Scheme 61). Imination–alkynylation occurred at room temperature in C6H5CH3, but the desired products were produced in the presence of an equimolar solution of lithium hexamethyldisilyl amide in THF at 0 °C. According to the results, due to higher yield and enantioselectivity using 4-methoxyaniline, the above-mentioned reaction was extended to various terminal alkyne derivatives with this amine. When different substituents were used on the aromatic ring of the alkyne, no remarkable changes in selectivities and yields were shown and the corresponding products obtained in high yields (up to 86%), and ee (up to 96%). Trimethylsilyl acetylene in this reaction was unable to produce the desired product. In addition, aliphatic terminal alkynes were investigated and produced the corresponding products with enantioselectivities of 87–93% and high yields of 84–86%.
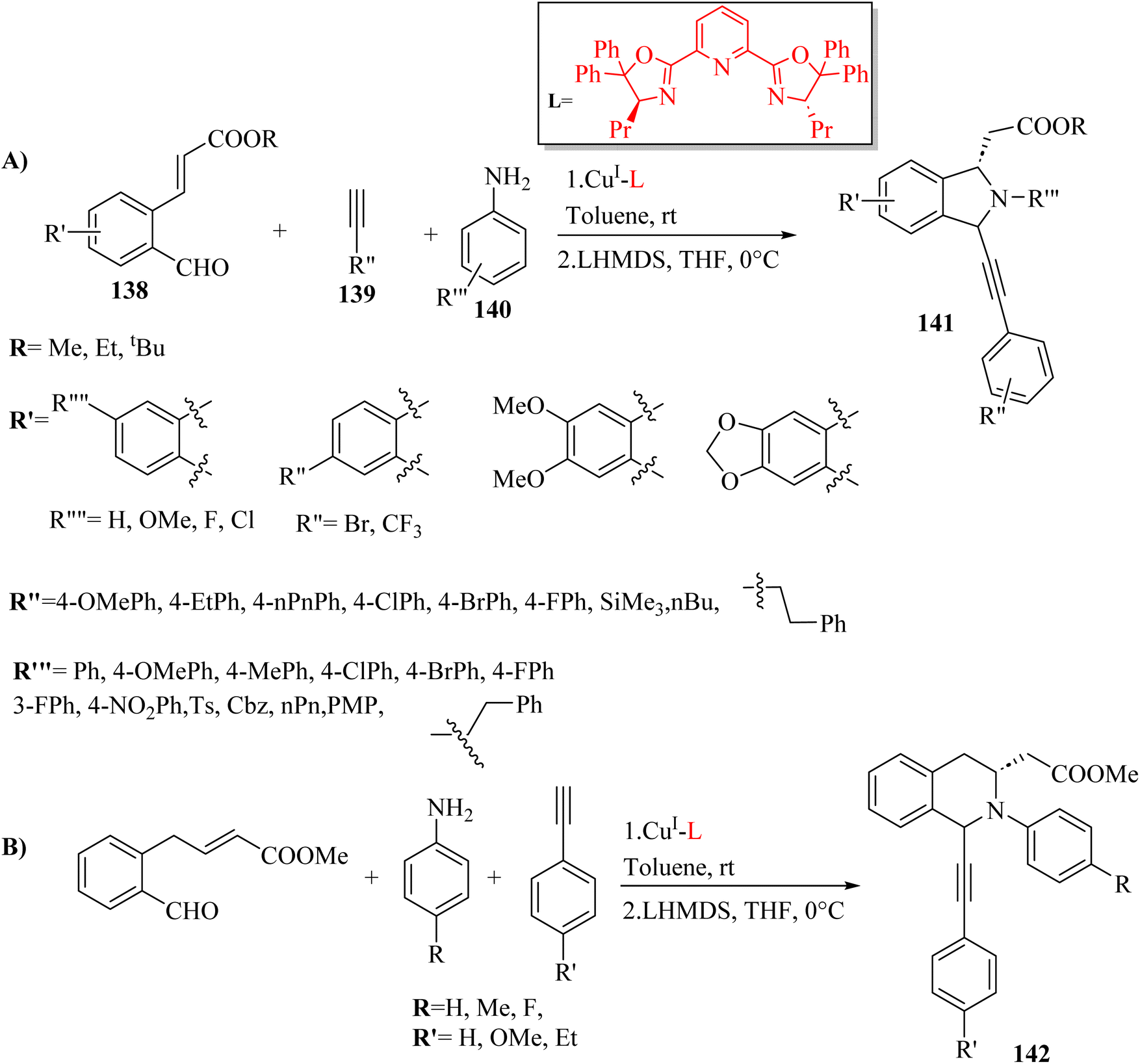 | ||
| Scheme 61 (A) Synthesis of 1,3-disubstituted tetrahydroisoquinolines and (B) isoindolines.74 | ||
Chandra et al.75 synthesized 4-arylated quinolines 146 via the reaction among paraformaldehyde 145, anilines 143, and alkynes 144 through [4 + 2] cycloaddition of alkyne derivatives and imine in the presence of (±) camphor-10-sulfonic acid (CSA) in TFE under microwave irradiation at 90 °C for 20 to 30 min (Scheme 62).
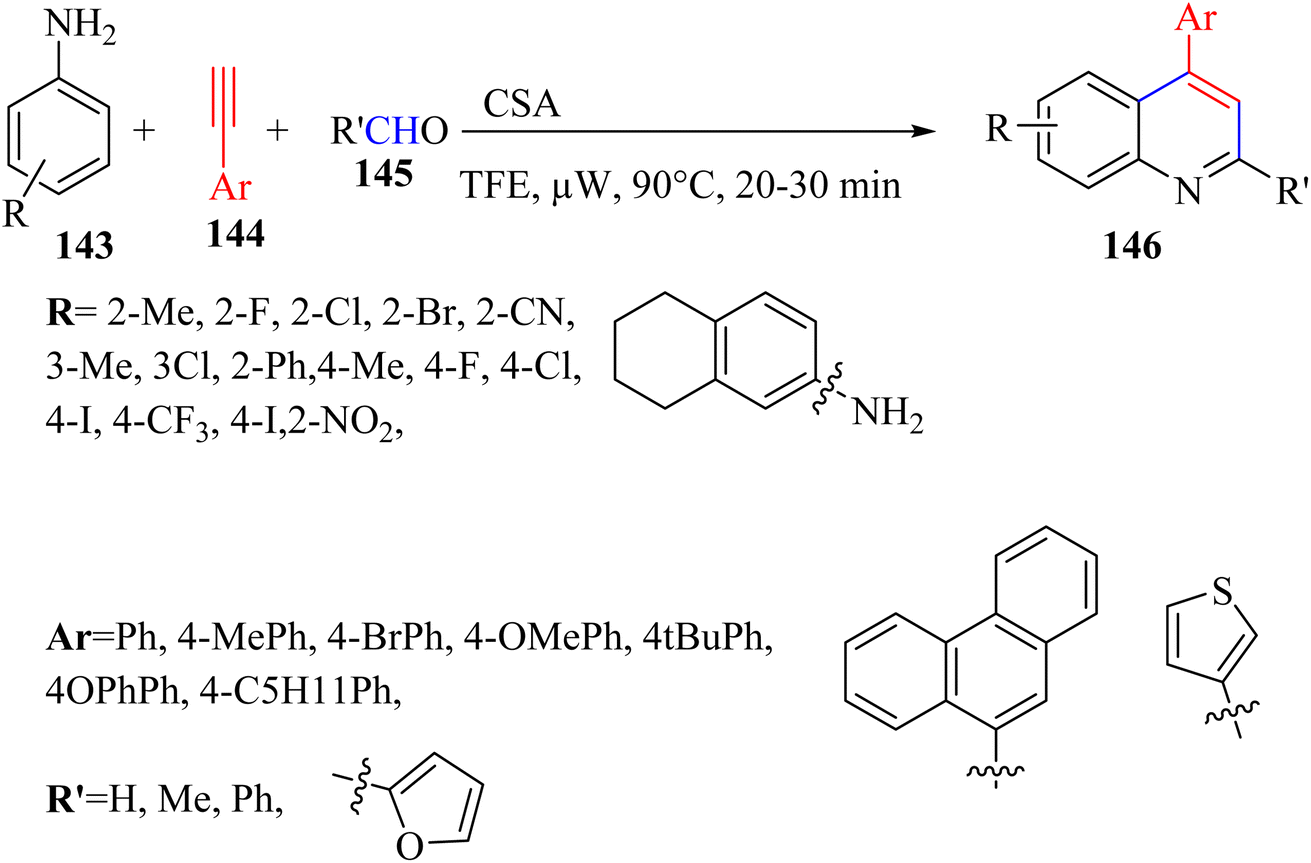 | ||
| Scheme 62 Synthesis of 4-arylated quinoline derivatives 146.75 | ||
Initially, the reaction between 145 and 143 may in situ produce intermediate A. After that, intermediate D was generated from [4 + 2] cycloaddition of 142 with intermediate B via a Povarov-type multicomponent reaction. Finally, 146 was produced via the spontaneous oxidation of intermediate D (Scheme 63).
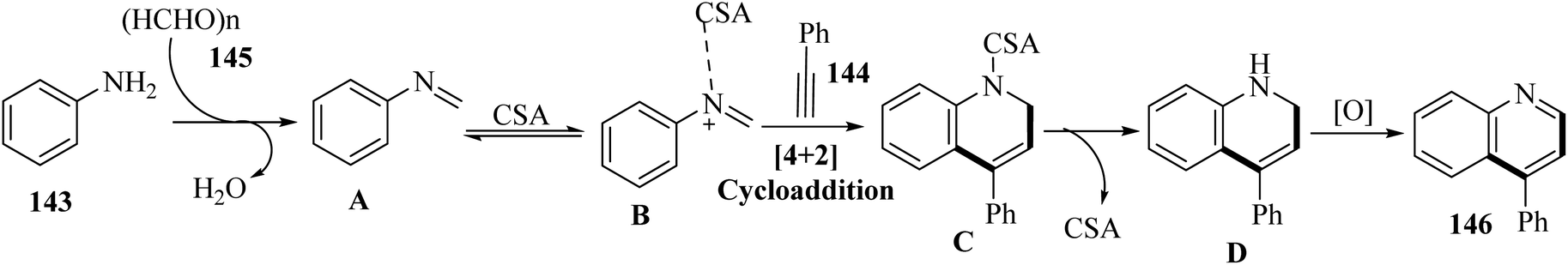 | ||
| Scheme 63 Possible reaction mechanism for the synthesis of 4-arylated quinolones 146.75 | ||
According to the results, the corresponding products with different paraformaldehyde, aniline, and alkyne derivatives were produced in excellent yields (up to 90%). In the case of 144, various alkyne derivatives were reacted with ortho-toluidine and different para-substituted phenyl acetylenes produced the corresponding quinolines in 31–76% yield. Interestingly, polyaromatic alkyne was tolerated and the desired quinoline was produced in 68% yield. In addition, 3-ethynylthiophene produced the desired quinoline in 48% yield. In contrast, internal alkyne did not react.
In 2019, 2,4-disubstituted quinolones 150 were synthesized via the reaction among alkynes 148, aldehydes 147, and amines 149 in toluene at 110 °C (Scheme 64). When different alkynes 148, aldehydes 147, and amine derivatives 149 were used, the desired quinolines were produced in 46–92% yield. Also, 3-thienylacetlyene, 1-hexyne, and 1-ethynylcyclohexene produced the desired quinolone derivatives in 91%, 55%, and 86% yield, respectively.76
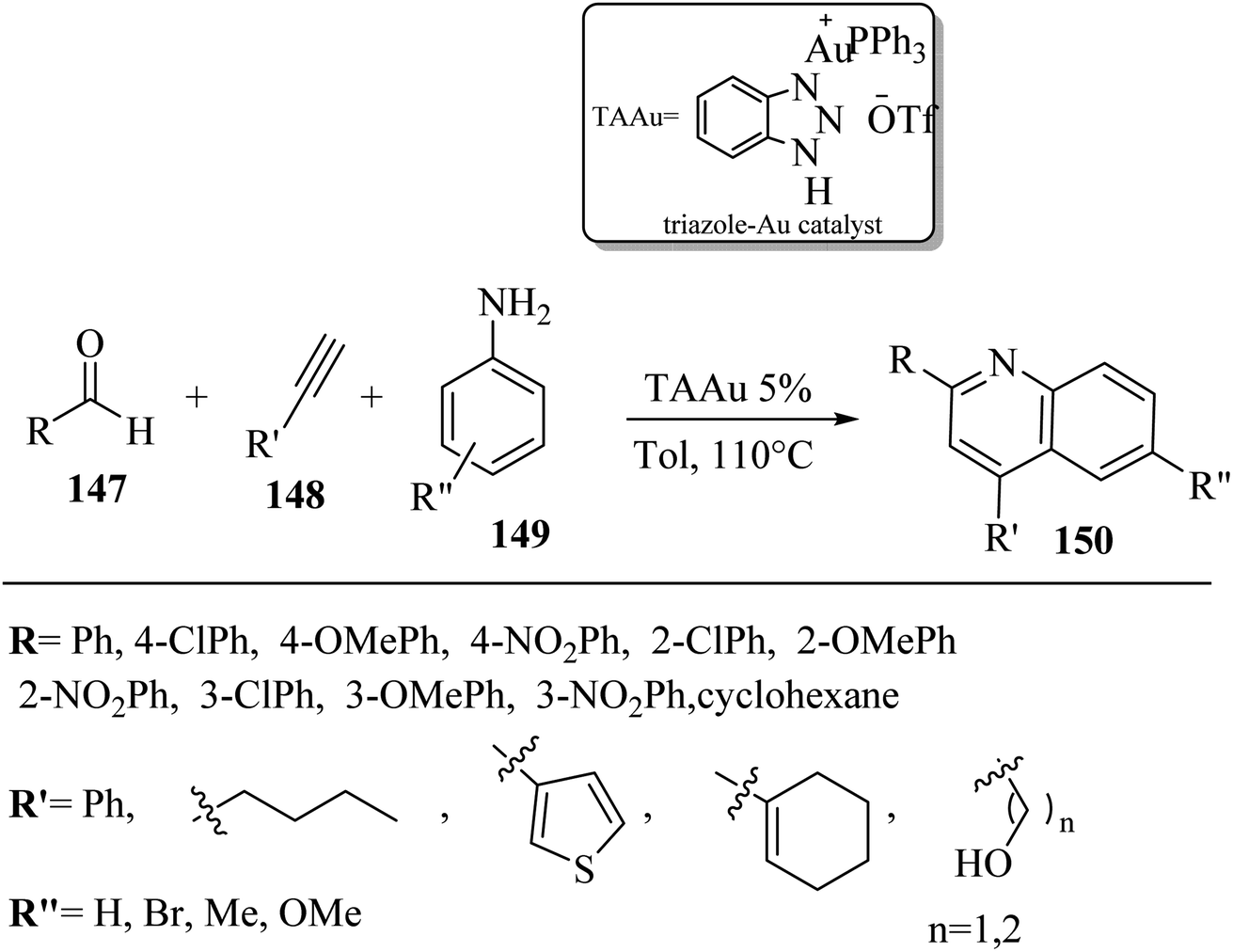 | ||
| Scheme 64 Synthesis of 2,4-disubstituted quinoline derivatives 150 via the triazole–gold catalyst.76 | ||
Yu and co-workers77 synthesized spirofuran-hydropyrimidinone derivatives 154 via the three-component Biginelli-like tandem reaction of alkynol 153 as an enolizable carbonyl equivalent, aromatic aldehydes 151, and (thio)urea 152 in the presence of co-catalyst of palladium chloride (transition metal) and trifluoroacetic acid (Brønsted acid) in dioxane at 50 °C for 12 h (Scheme 65).
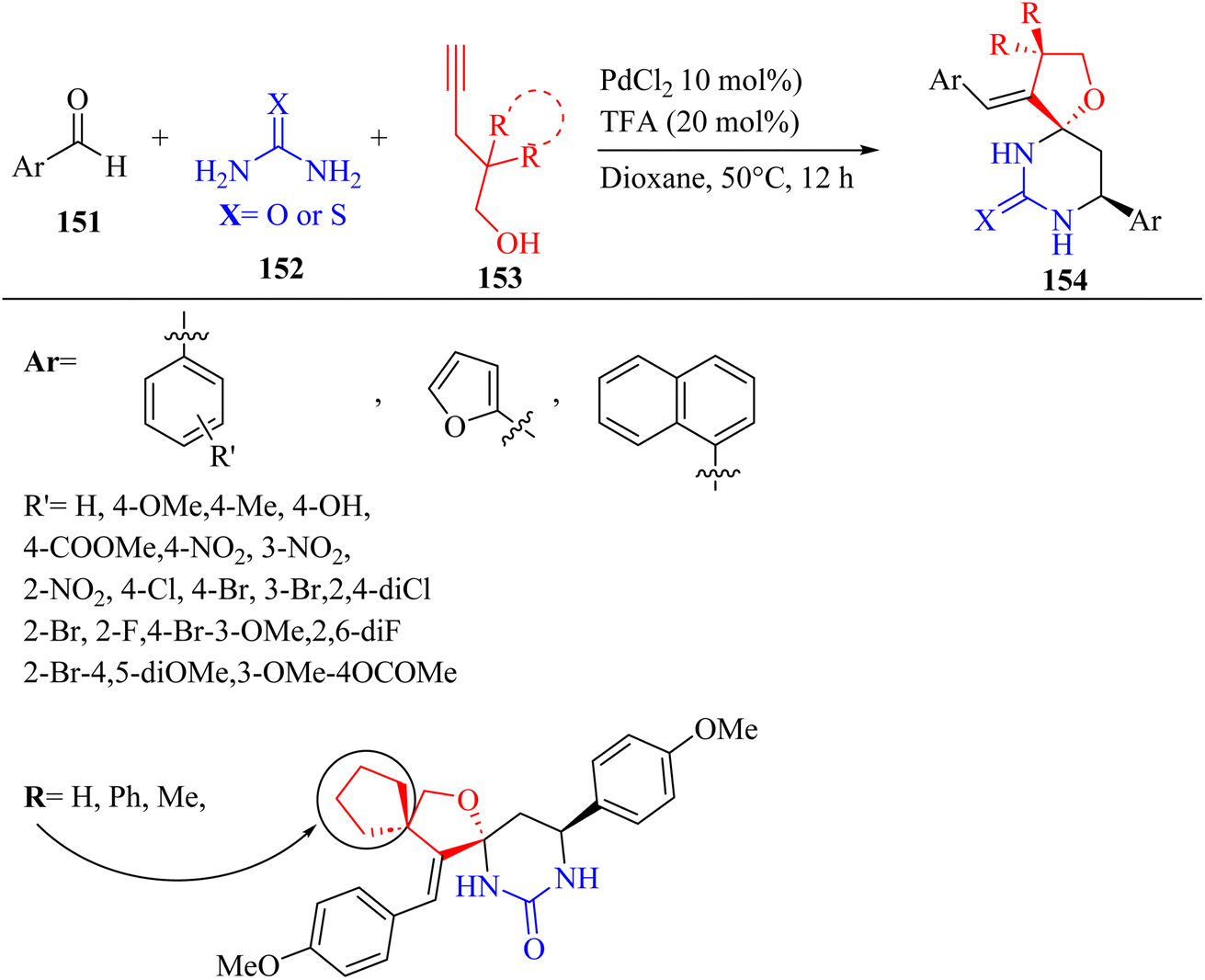 | ||
| Scheme 65 Synthesis of spirofuran-hydropyrimidinones 154 through a Biginelli-like tandem reaction with co-catalyst of PdCl2 and TFA.77 | ||
Initially, N-acyliminium intermediate G was generated via the condensation of 151 and 152 in the TFA. After that, vinylpalladium species H was obtained via intramolecular addition of the OH group to the electron-deficient alkyne, where it should be mentioned that the coordination of PdCl2 with 153 increased the electrophilicity of the triple bond. Next, intermediate I was produced via the attack of H to intermediate G. Then, endocyclic enol ether J was generated via the isomerization of the exocyclic double bond in the presence of the Pd catalyst. Next, oxonium intermediate K was obtained via the nucleophilic attack of the enol ether to a second aldehyde. Finally, the catalyst was regenerated and 154 was obtained via intramolecular cyclization (Scheme 66).
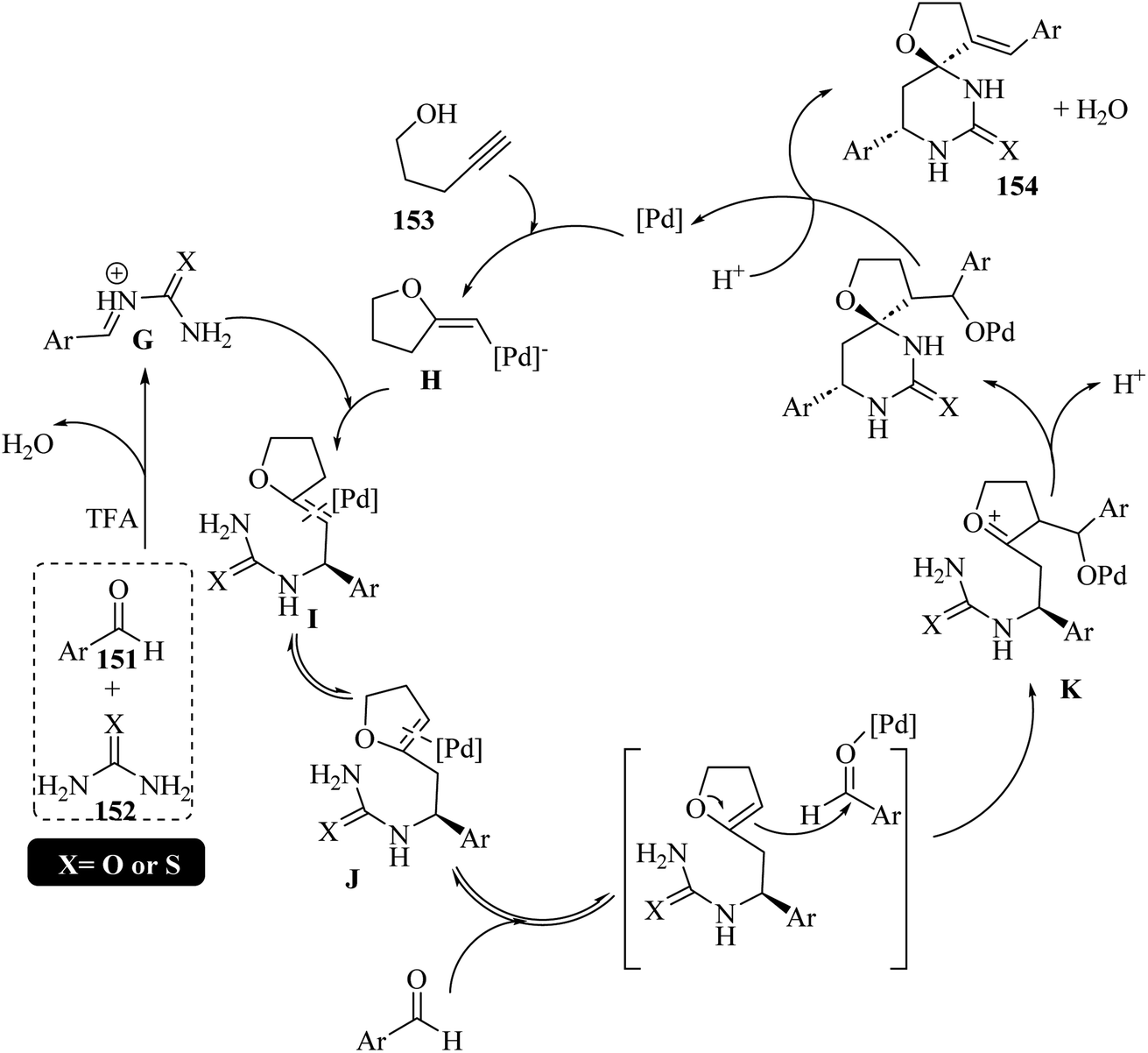 | ||
| Scheme 66 Possible reaction mechanism for the synthesis of spirofuran-hydropyrimidinone derivatives 154.77 | ||
The desired spirofuran-hydropyrimidinone derivatives were produced in 45–87% yield and diastereoselectivity. In the case of 151, some groups such as ester, carbonyl, nitro, halide, and hydroxyl were amenable in this catalytic system. Generally, benzaldehyde with electron-withdrawing substituents produced the corresponding spirofuran-hydropyrimidinone derivatives in worse yields compared to that with donating groups (71–78% and 80–87% yield). Notably, the reaction of benzaldehyde with a substituent at the meta position proceeded worse than that at the para position. Also, di- and tri-substituted benzaldehydes produced the corresponding spirofuran-hydropyrimidinone derivatives in 77–85% yield. Other aldehydes such as furfural and 1-naphthaldehyde produced the corresponding spirofuran-hydropyrimidinone derivatives in 73% and 81% yield, respectively but no desired product could be produced with 2-pyridine formaldehyde. When urea was replaced with thiourea, the corresponding spirofuran-hydropyrimidinone derivatives were produced in 45–57% yield. The scope of 153 also was investigated and the desired products were produced in 62–77% yield.
Purohit and co-workers78 via the reaction among aldehyde derivatives 156, aminopyridines 155 and alkynes 157 synthesized substituted imidazopyridines 158 in the presence of PW-CIS500 nano-catalyst at 100 °C under neat condition (Scheme 67). The reaction proceeded smoothly when used various alkynes 157, aldehydes 156, and amines 155. According to the study, the desired imidazopyridines were produced with an isolated yield of 82–95%. Also, various aryl-alkynes/aldehydes with electron-donating and electron-withdrawing groups produced the desired imidazopyridines with an isolated yield of 82% to 95%.
 | ||
| Scheme 67 Synthesis of imidazopyridine derivatives 158.78 | ||
Xian et al.79 synthesized 2,4-diphenylpyrimido[1,2-b]indazole derivatives 162 via the reaction among alkyne derivatives 161, 1H-indazol-3-amine 159, and aldehydes 160 under solvent-free conditions in the presence of FeCl3 at 110 °C (Scheme 68).
 | ||
| Scheme 68 Synthesis of 2,4-diphenylpyrimido[1,2-b]indazole derivatives 162 under solvent-free conditions.79 | ||
Initially, the condensation reaction between 159 and 160 led to the formation of imine A, which released an H2O molecule through the reaction. Then, iron complex intermediate B was obtained via the attack of iron acetylide complex to A, which isomerized to intermediate C. After that, intermediate D was obtained after regioselective attack of the intramolecular N–H bond activated the triple bond through 6-endo-dig-cyclization. Finally, 162 was generated via the autoxidation and demetalation of intermediate D (Scheme 69).
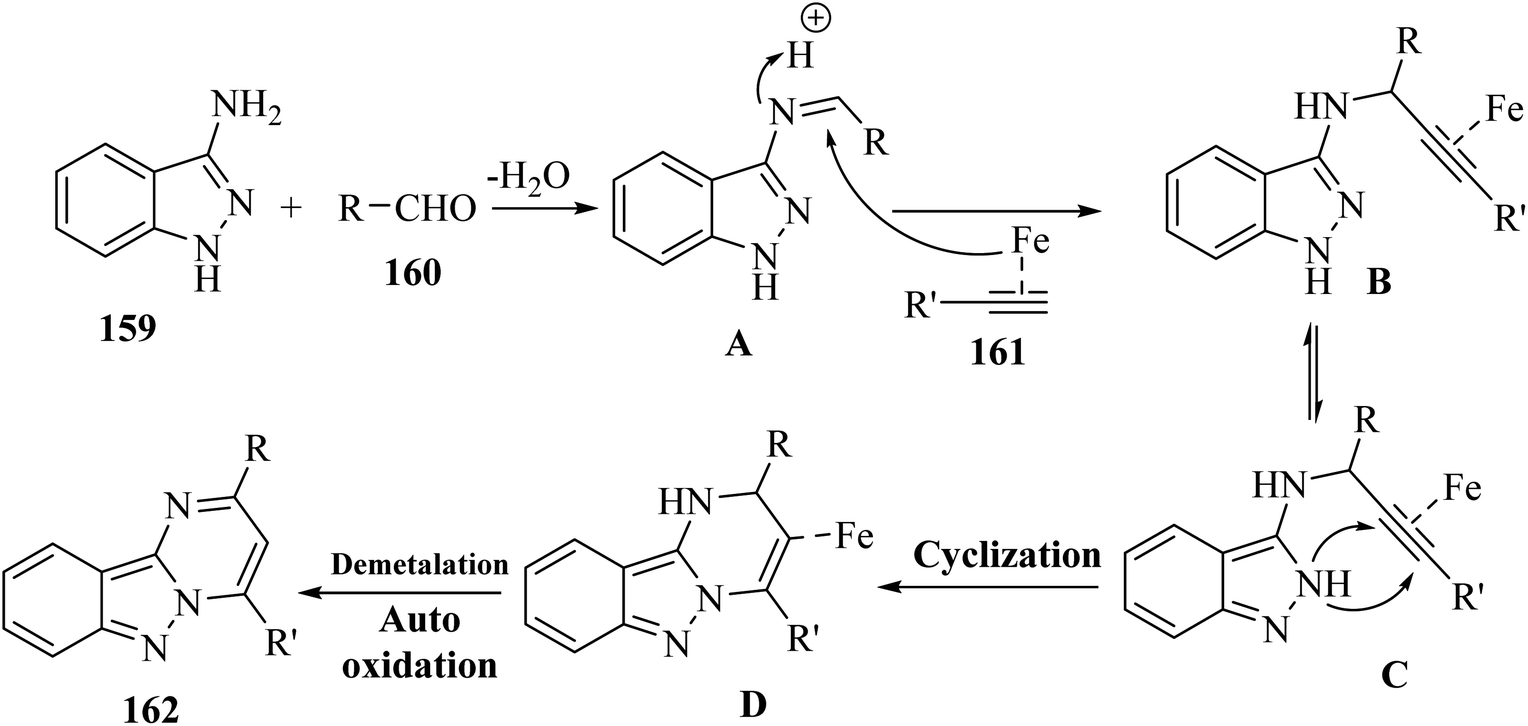 | ||
| Scheme 69 Possible reaction mechanism for the synthesis of 2,4-diphenylpyrimido[1,2-b]indazole derivatives 162.79 | ||
The desired indazole derivatives were produced in 69% to 93% yield. Alkynes with different groups gave the desired indazole derivatives in 83–85% yield. Under the same conditions, substituted aromatic alkynes and substituted aromatic aldehydes were evaluated and produced the corresponding products with an isolate yield of 73% to 79%. This method has advantages such as the use of available materials and an environmentally friendly catalyst. Also, the above-mentioned method is a good way to produce pyrimidine derivatives.
Yuan et al.80 synthesized γ-carbonyl-α-amino acids 166 via the reaction among amines 165, α-carbonyl aldehyde derivatives 163, and propargylic alcohols 164 in the presence of AgNO3 in CH2Cl2 at room temperature for 24 h (Scheme 70).
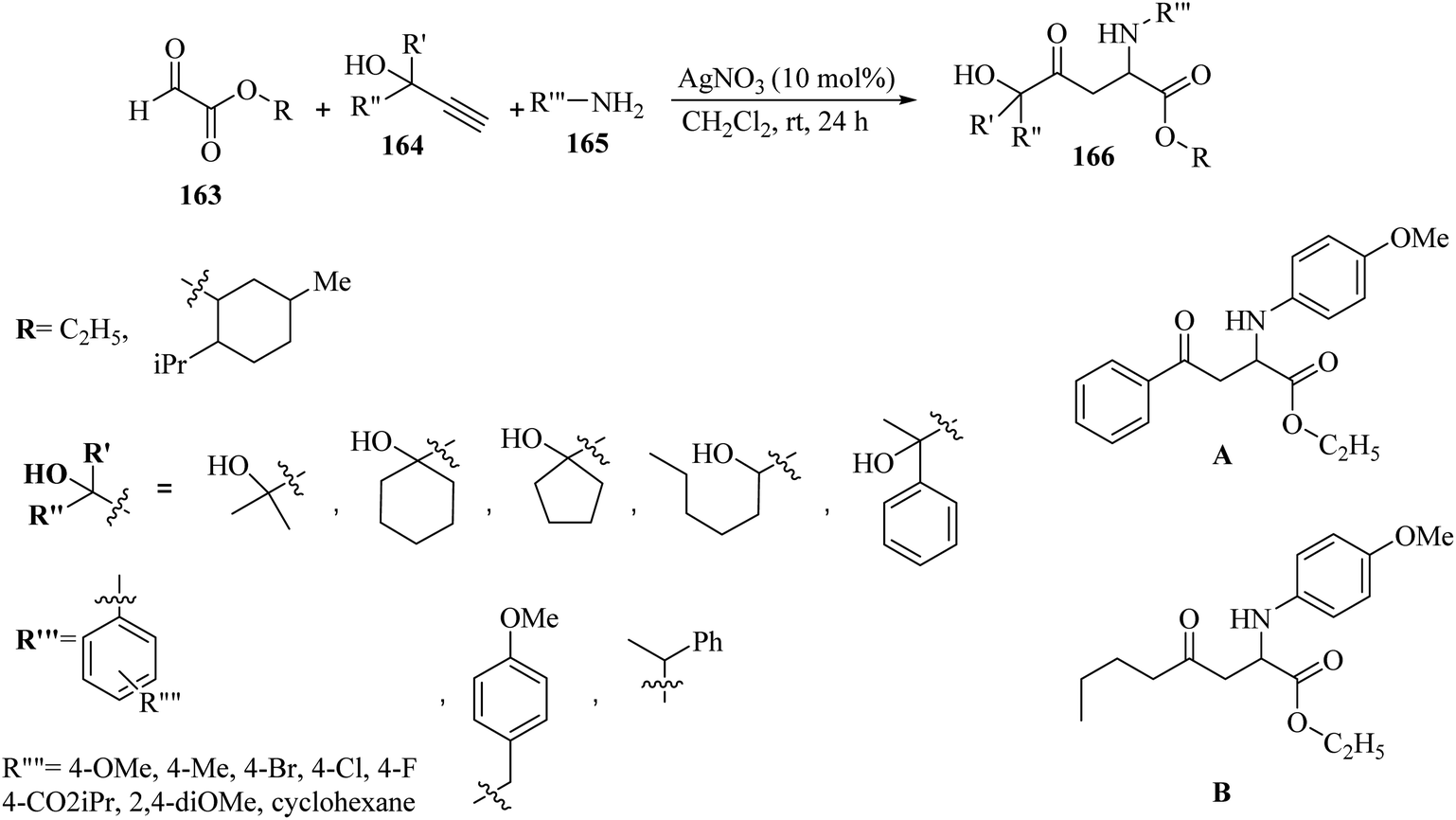 | ||
| Scheme 70 AgNO3-catalyzed synthesis of γ-carbonyl-α-amino acid derivatives 166.80 | ||
Initially, the reaction among 164, 165, and glyoxylate in the presence Ag catalyst generated product A via a Mannich-type reaction with intermediate B. In this section, intermediate B acted as a nucleophile (cycle Z). Subsequently, intermediate silver–alkyne complex C was produced via the coordination of intermediate A with Ag+. Next, intermediate D was obtained via the reaction between intermediate C and H2O. In the last step, enol E was generated via the protonation of intermediate D, and then E was tautomerized to 166 (cycle X) (Scheme 71).
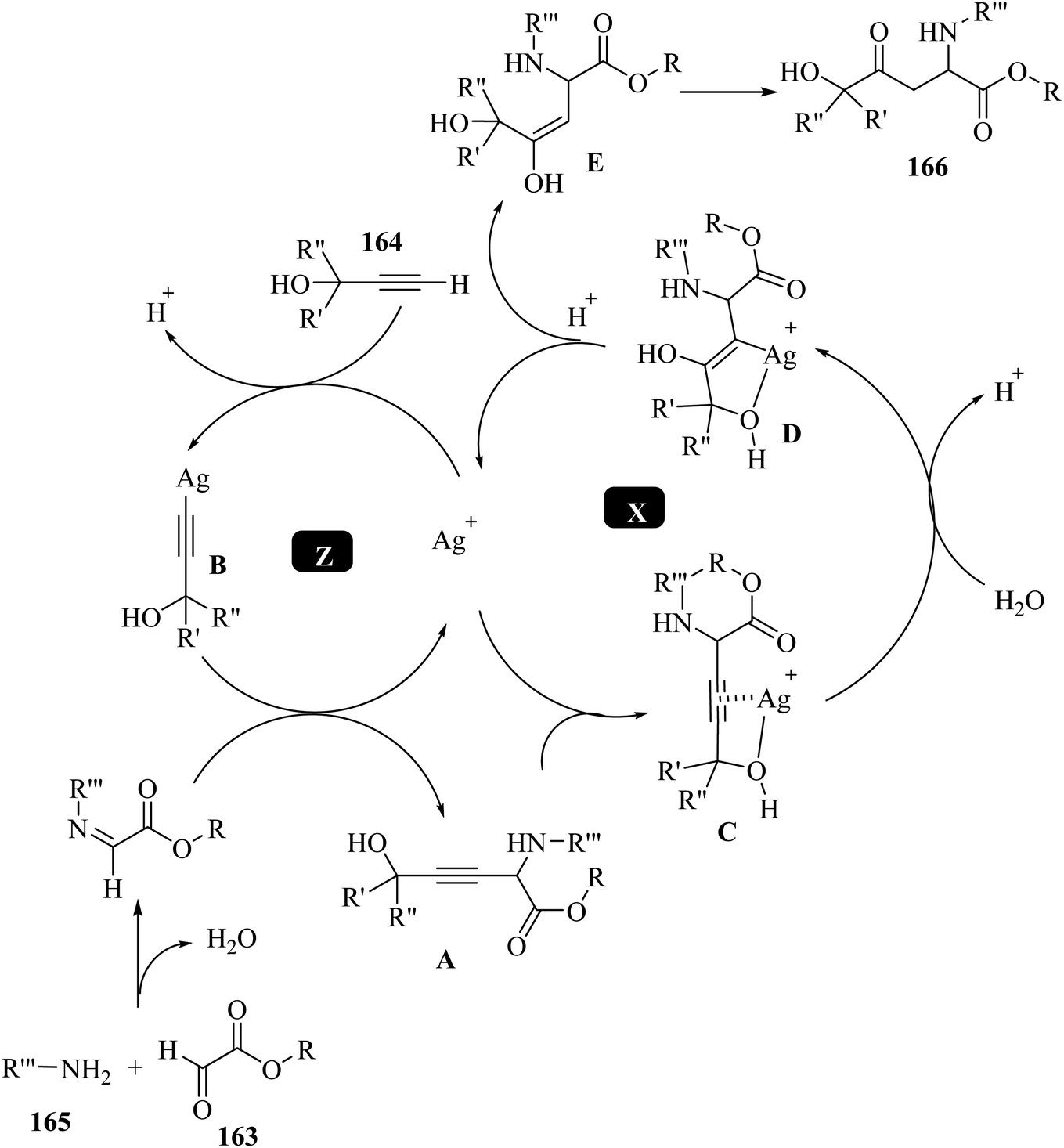 | ||
| Scheme 71 Possible reaction mechanism for the synthesis of γ-carbonyl-α-amino acids 166.80 | ||
According to the results, γ-carbonyl-α-amino acids were produced in 71% to 91% yield. Aromatic amine derivatives containing either electron-withdrawing or -donating groups on the aromatic ring produced the corresponding γ-carbonyl-α-amino acids in 78–90% yield. Aliphatic amine derivatives were also compatible and produced the corresponding γ-carbonyl-α-amino acids in 71–87% yield. In the above-mentioned method, when morpholine was used, none of the desired γ-carbonyl-α-amino acids were detected. In addition, a variety of secondary and tertiary propargyl alcohol derivatives including acyclic and cyclic substituted propargyl alcohol derivatives produced the desired products. However, general aliphatic and aromatic alkynes such as 1-hexyne and phenylacetylene were used, and none of the corresponding products were detected (B and A, respectively). In this transformation, the alcohol OH group of propargylic alcohol played an essential role.
Mohammod and co-workers81 synthesized 3-aminoindolysines 170 via the reaction among pyridine-2-carbaldehyde 167, secondary amine derivatives 168, and terminal alkyne derivatives 169 in the presence of the CuCN/[bmim][PF6] system under an N2 atmosphere at 70 °C (Scheme 72).
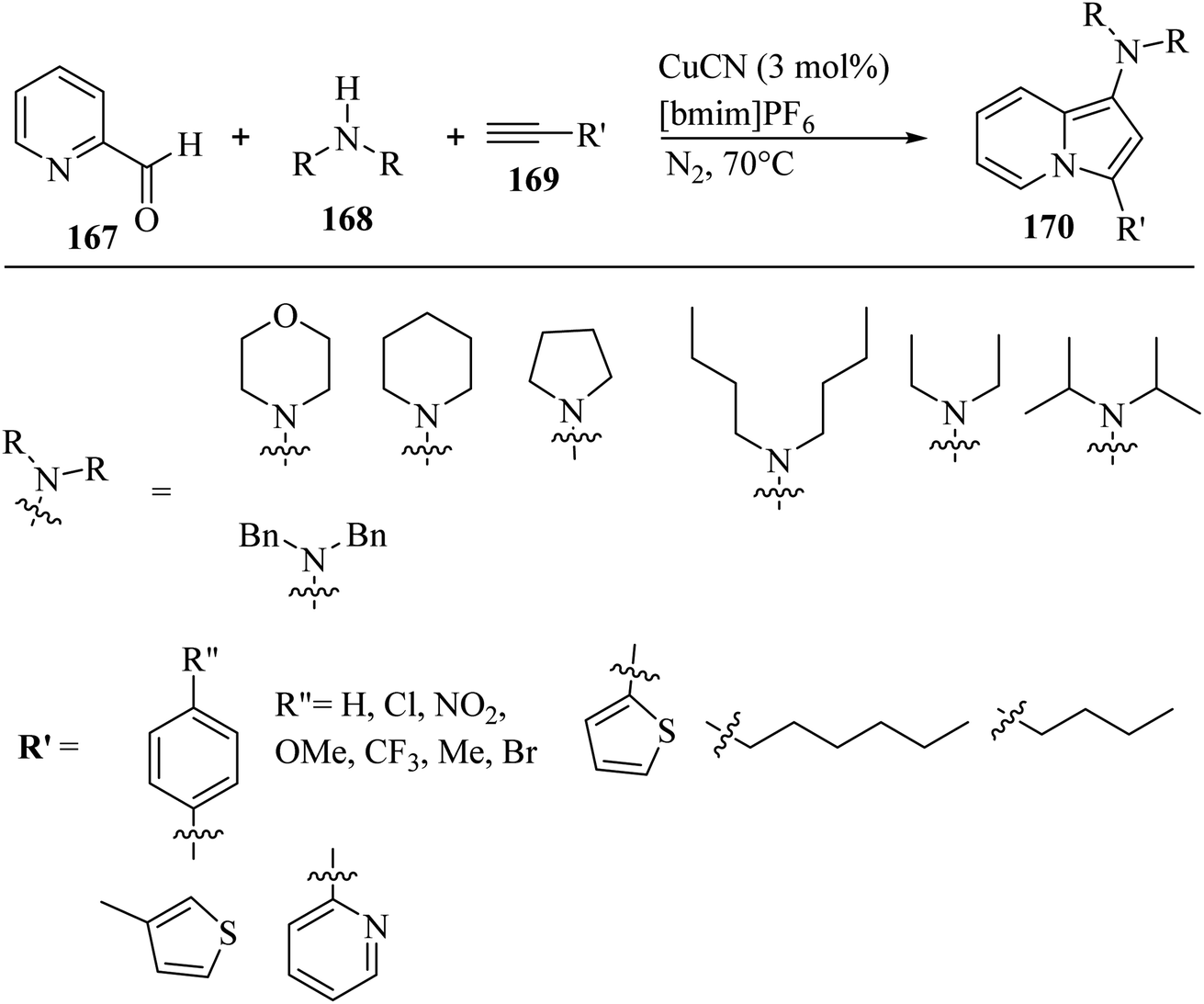 | ||
| Scheme 72 Synthesis of 3-aminoindolysines 170 in the presence of the CuCN/[bmim][PF6] system.81 | ||
Firstly, Cu-acetylide intermediate G was generated via the activation of 169 by CuCN. Then, N-propargylamine amine intermediate H was obtained via the reaction between intermediate G and iminium ion F. In this stage, the electrophilicity of the internal alkyne increased because the triple bond of 169 was coordinated with CuCN. After that, cationic heterocyclic intermediate I was produced via the nucleophilic attack of the N lone pair to the activated triple bond of alkyne, which proceeded via intramolecular cyclization. Next, intermediate J was generated through the deprotonation of intermediate I. Finally, the desired product 170 was produced together with the regeneration of catalyst via the protonation and demetallation of intermediate J (Scheme 73).
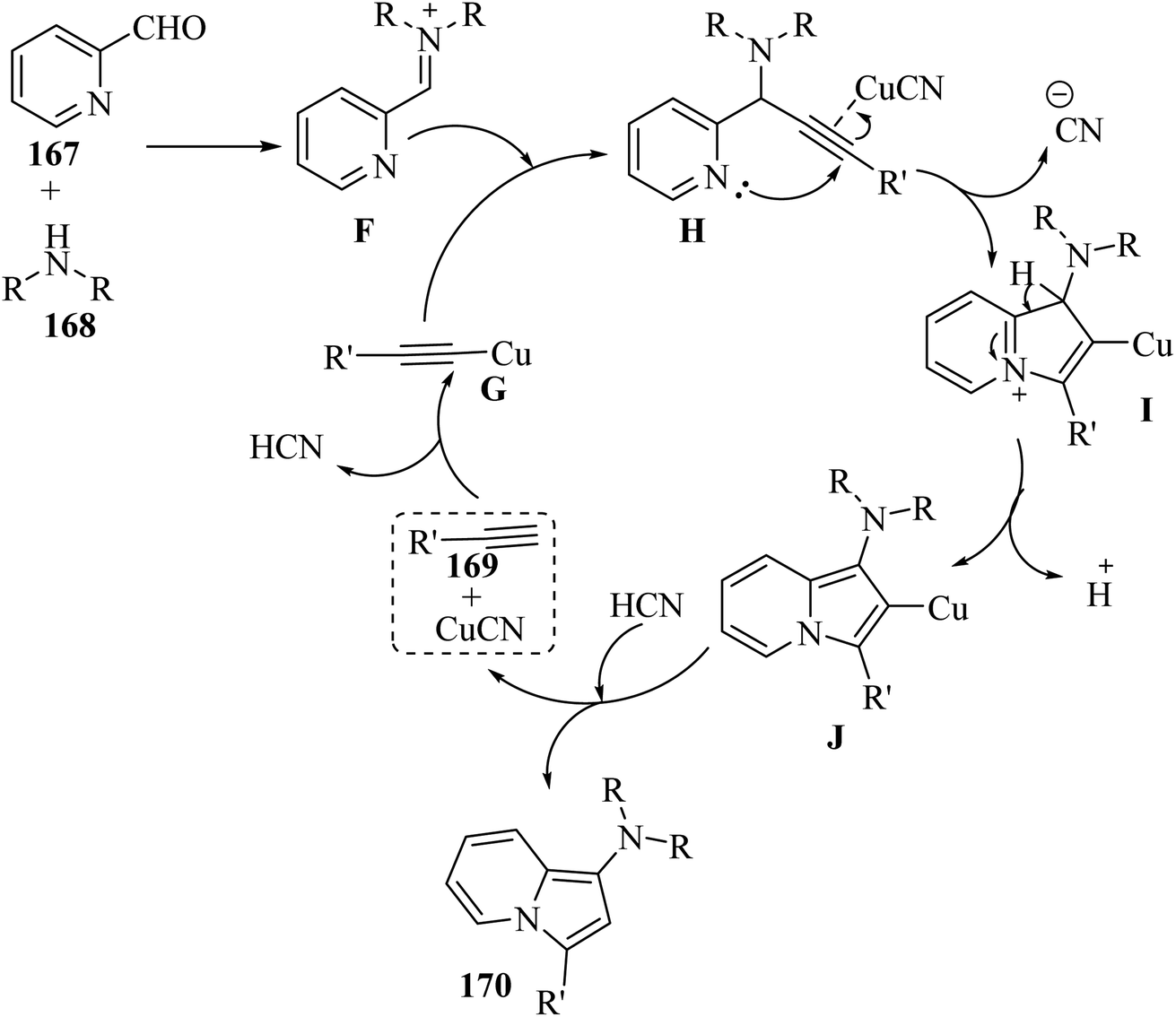 | ||
| Scheme 73 Possible reaction mechanism for the synthesis of 3-aminoindolysines 170.81 | ||
It should be noted that the CuCN/[bmim][PF6] system was reused six times with no noticeable change in its performance. In the case of 169, phenylacetylene and phenylacetylene derivatives with various substituents such as 4-Me, 4-OMe, 4-CF3, 4-Br, 4-Cl, and 4-NO2 provided the corresponding 3-aminoindolizines in 67–89% yield. However, only modest yields of the corresponding 3-aminoindolysines were isolated with aliphatic alkynes (70–71%), which is possibly due to the instability of the products, where they rapidly degraded during column chromatography. In addition, the use of heteroaryl alkynes was effective to obtain the corresponding 3-aminoindolizines in 82–84% yield. In the case of 168, moderate yields of the resulting 3-aminoindolizine derivatives were obtained with aliphatic acyclic amine derivatives (67–72%).
Cui et al.82 synthesized pyrrolo[2,1-a]isoquinolines 174 via the reaction among terminal alkyne derivatives 173, aldehydes 172, and tetrahydroisoquinolines 171 in the presence of CuCl2 and PhCOOH (condensation promoter) in DMF at 130 °C (Scheme 74).
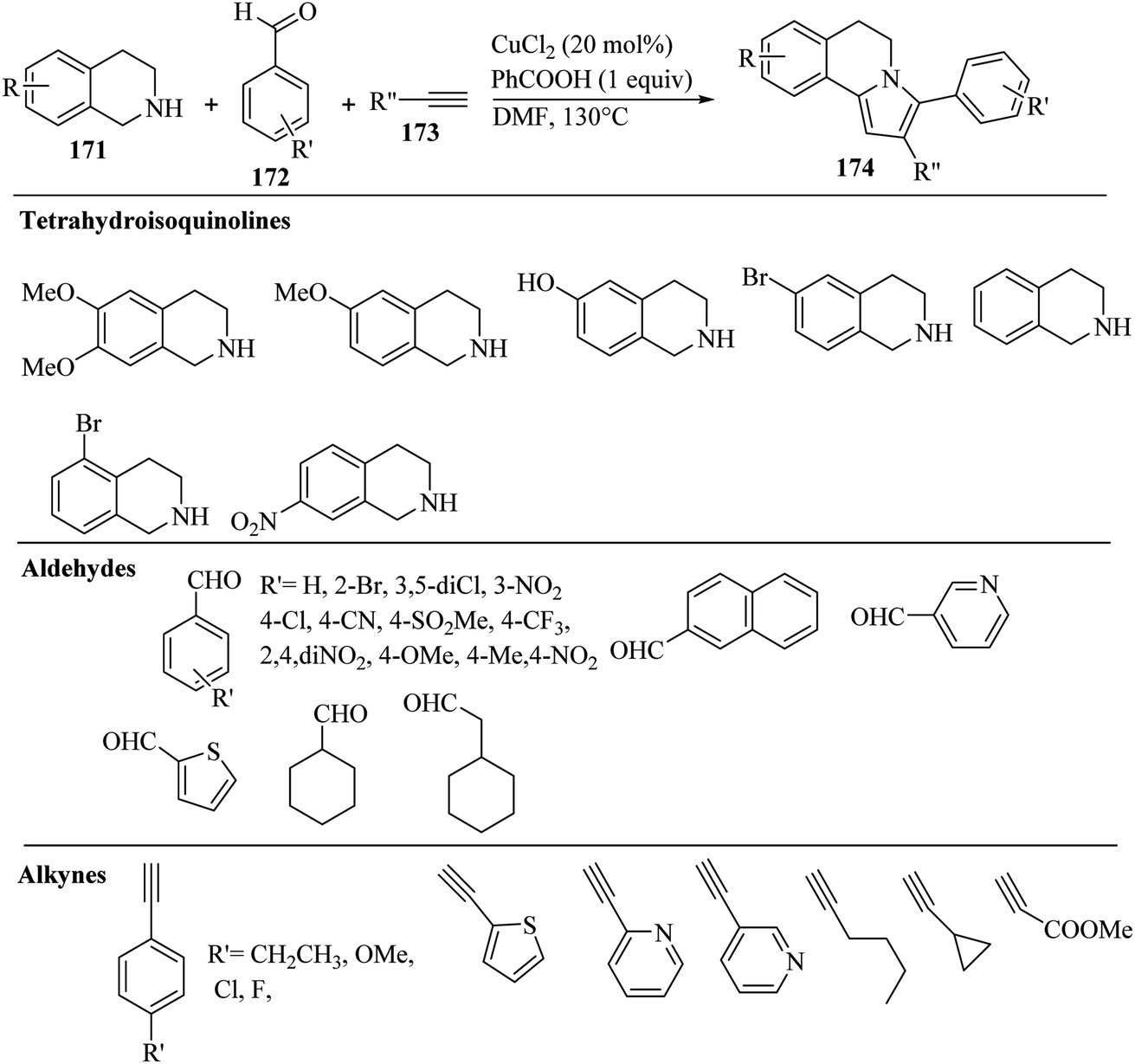 | ||
| Scheme 74 CuCl2-catalyzed synthesis of pyrrolo[2,1-a]isoquinolines 174.82 | ||
Initially, the Cu catalyst reacted with 173 and produced F. Meanwhile, two imine ions G and G′ were generated via the reaction between 171 and 172. Then, amine H was obtained via the Mannich-type addition of activated alkyne F to ion G′, which is more stable than the other ion. After that, H underwent oxidation in the presence of the Cu catalyst, and then its product was deprotonated to produce azomethine ylide. After that, intermediate I was generated via the 5-endo-dig cyclization of azomethine ylide. Lastly, 174 was obtained via the final proton transfer together with the release of the Cu catalyst (Scheme 75). It should be noted that the Cu catalyst may have played three roles in the reaction, as follows: (1) activation of 173 for Mannich-type addition, (2) producing an iminium ion via the oxidation of the tertiary amine, and (3) 5-endo-dig cyclization via the activation of propargyl amine.
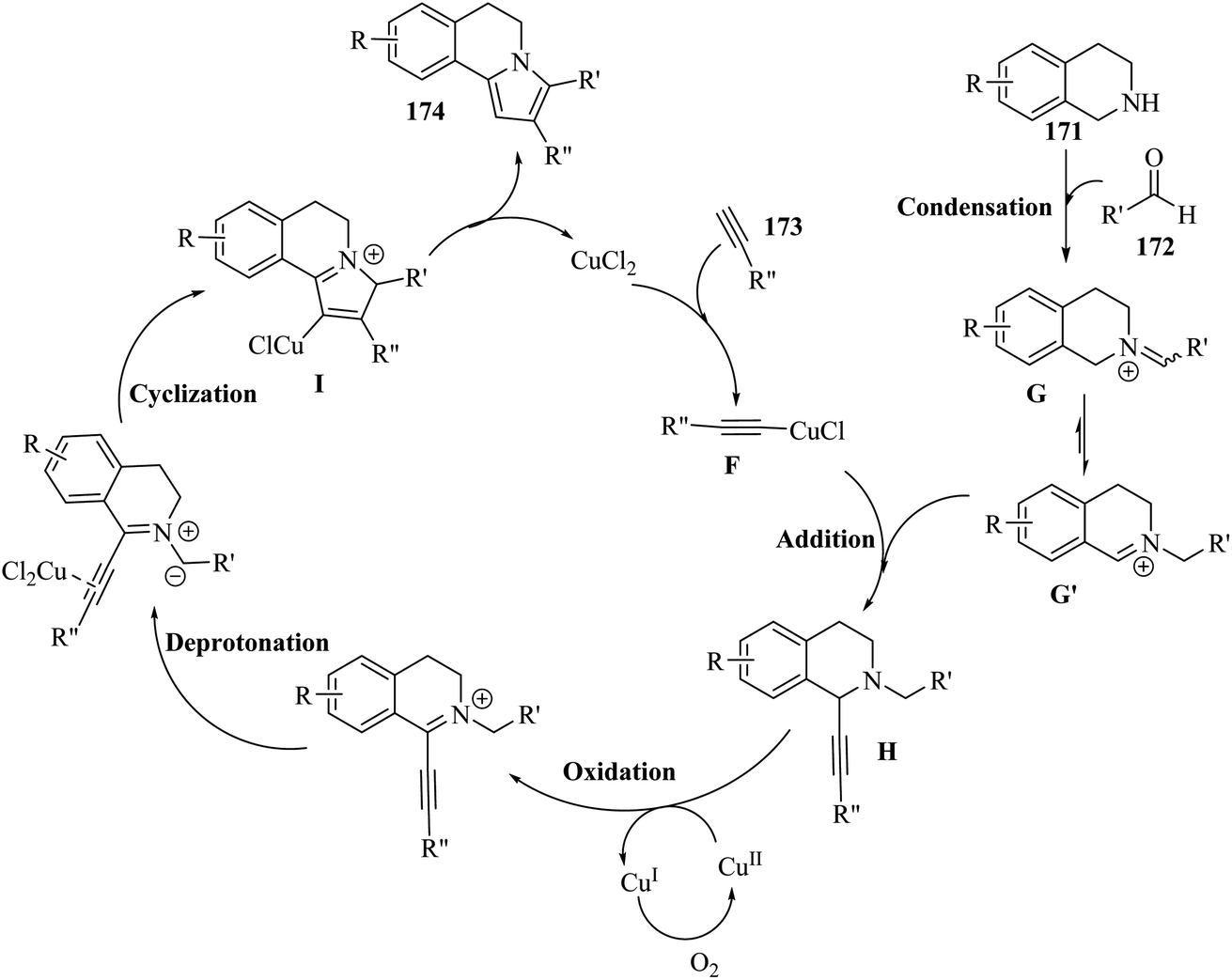 | ||
| Scheme 75 Possible reaction mechanism for the synthesis of pyrrolo[2,1-a]isoquinolines 174.82 | ||
According to the results, the corresponding isoquinoline derivatives were produced in 17–69% yield via a condensation/Mannich-type addition/oxidation/cyclization cascade sequence. In the case of 171, with the exception of the NO2 group, the usual electron-withdrawing and -donating groups can be compatible under the optimal conditions at the tetrahydroisoquinoline moiety such as MeO, OH, Br, and Cl (28−66% yield). The NO2 group dramatically deactivated tetrahydroisoquinoline and did not produce the corresponding product. In the case of 172, various substituted benzaldehydes were tested in the above-mentioned reaction and produced the desired isoquinoline derivatives in 17–69% yield. In general, aldehyde derivatives with electron-withdrawing groups performed better in terms of reaction yield, which is likely due to the easier condensation and better stability of the intermediates generated in situ. Also, when using 2,4-dinitrobenzaldehyde, no corresponding product was detected, which is probably due to steric hindrance. All pyridyl, thienyl, and naphthyl substituents could be incorporated in the pyrrole[2,1-a]isoquinoline backbone in reasonable yields (31–33%). Moreover, a trace amount of the corresponding isoquinoline derivative was detected when using aliphatic aldehyde and the corresponding intermediate (37%). In the case of 173, the electron-rich and -deficient terminal alkynes were compatible in the present reaction (53–66%). Heterocyclic alkynes such as 2-pyridyl, 3-pyridyl, and 2-thienyl also were tolerated and produced the corresponding isoquinoline derivatives in 31%, 46%, and 51% yield, respectively. In addition, alkyl-substituted terminal alkynes were tested and produced the corresponding products in 19–35% yield. Unfortunately, ethyl propiolate did not give the corresponding product, which may be related to the high reactivity of this alkyne.
2.5 Reaction of α-diazoesters or ketones
He et al.83 synthesized seven-membered 1,4-oxazepine structures 180 and five-membered indolizine derivatives 178 in 34–93% yield via the rhodium-catalyzed regiodivergent [5 + 2] and [3 + 2] cycloaddition of quinolinium ylide derivatives 175 with alkyne derivatives (176 or 176′), respectively, in CH2Cl2 for [5 + 2] cycloaddition and xylenes for [3 + 2] cycloaddition at room temperature (Scheme 76).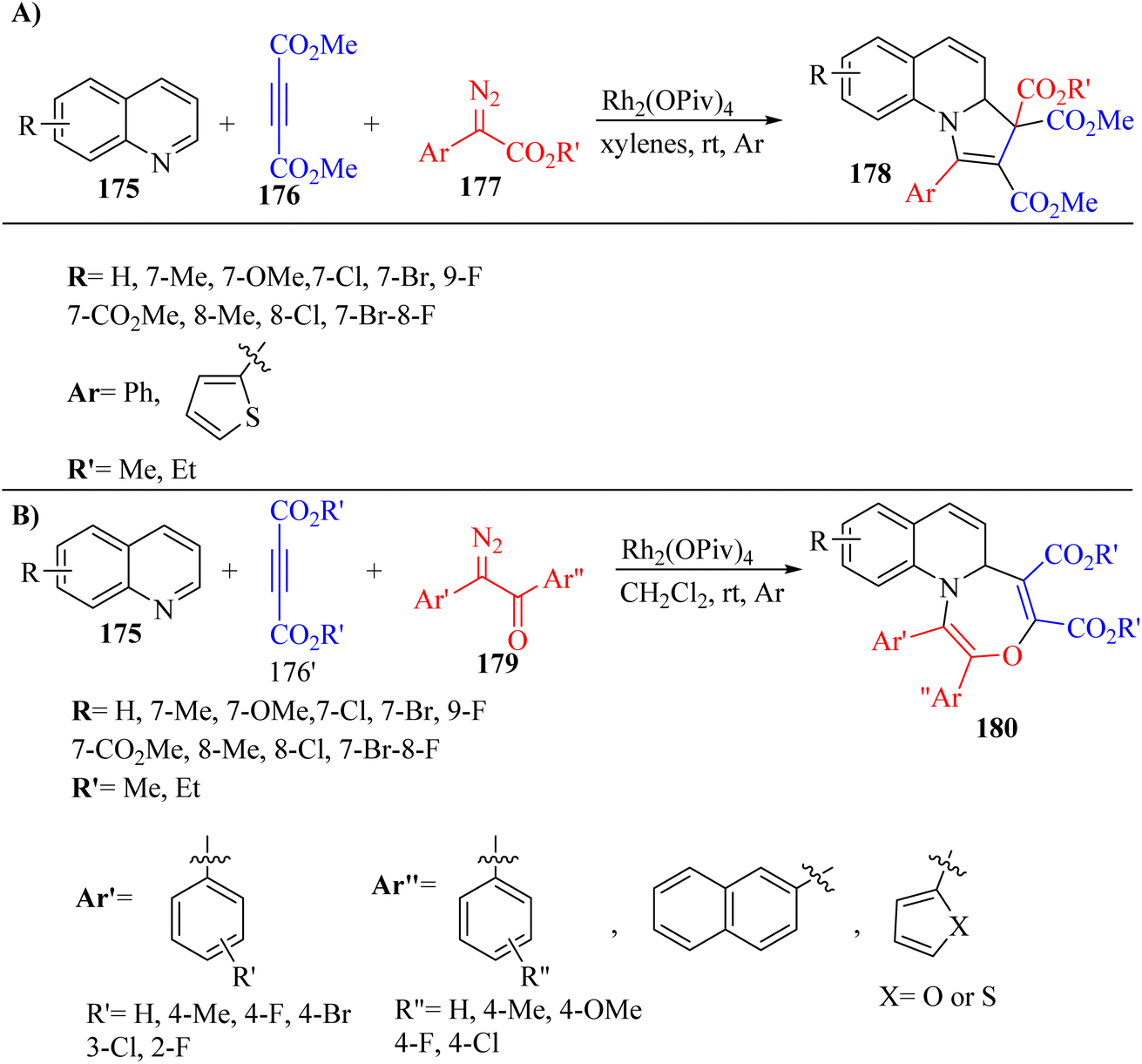 | ||
| Scheme 76 (A) Synthesis of five-membered indolizine (scope for [3 + 2] cycloaddition) and (B) synthesis of seven-membered 1,4-oxazepine (scope for [5 + 2] cycloaddition).83 | ||
Initially, rhodium carbene A was produced via the reaction between diazo compound 177 and Rh complex together with N2 removal. Then, intermediate B was generated via the nucleophilic addition of 175 to A. After that, intermediates C and C′ were produced via the dissociation of the rhodium salt. Next, intermediate D was obtained via the reaction between an alkyne and intermediate C, and this reaction was processed via 1,3-dipolar [3 + 2] cycloaddition. Subsequently, 178 was produced via a metal-assisted 1,3-ester migration process. In addition, the reaction between an alkyne and intermediate C′ led to 180, which proceeded via 1,5-dipolar [5 + 2] cycloaddition (Scheme 77).
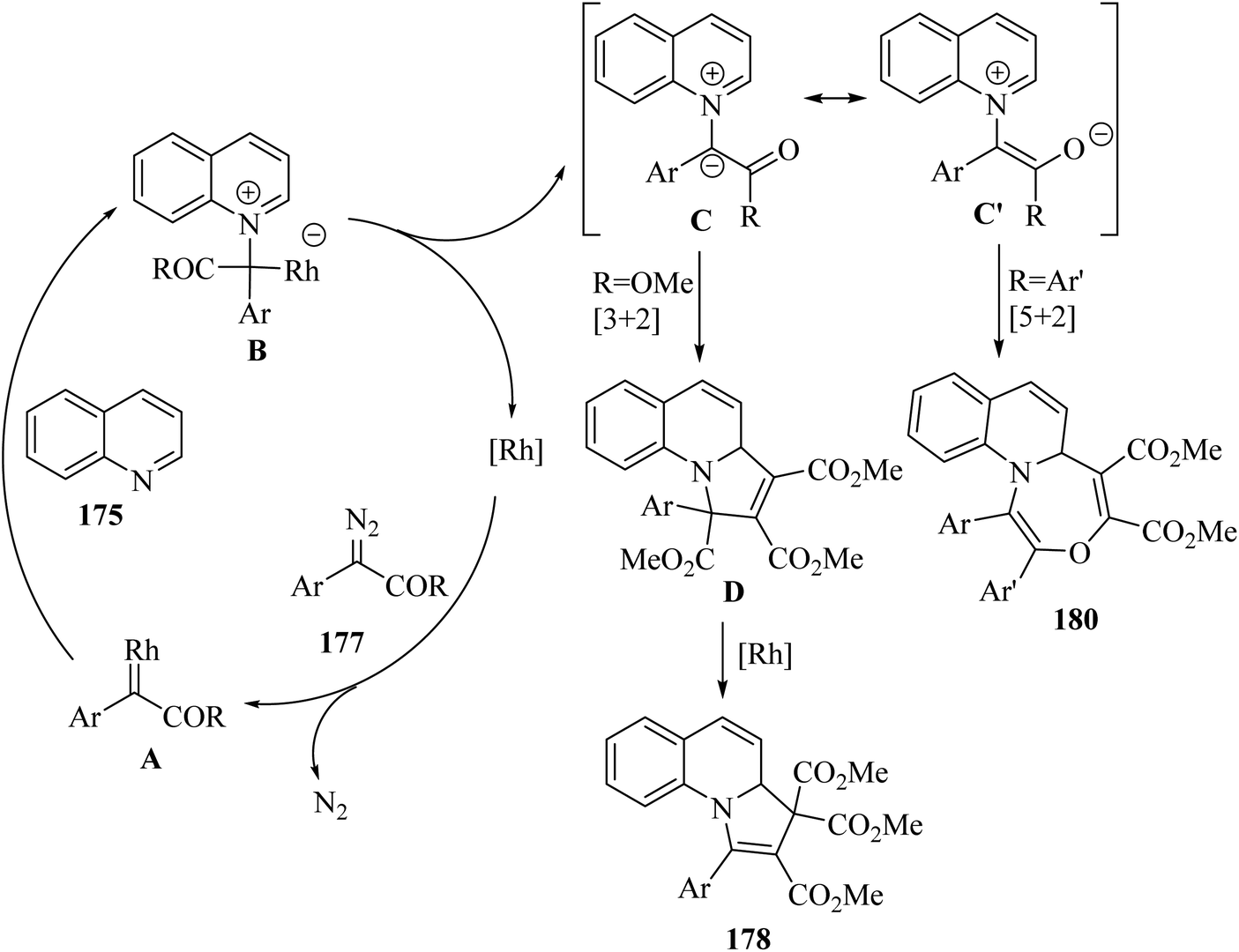 | ||
| Scheme 77 Possible reaction mechanism for the synthesis of seven-membered 1,4-oxazepines 180 and five-membered indolizines 178.83 | ||
In the case of 180, various α-diazoketone derivatives were investigated and produced the desired oxazepines in 51% to 96% yield. The types of groups (electron-donating and -withdrawing) on both aryl rings of the α-diazoketone derivatives produced the desired oxazepines in 51–96% yield. In addition, diazoketone derivatives containing a bulky 2-naphthyl group, thiophene, and furan gave the desired products in 61%, 56%, and 68%, yield, respectively. Diethyl acetylenedicarboxylate also produced the corresponding oxazepine in 87% yield. A variety of quinoline derivatives was evaluated, and the desired oxazepines were obtained in 34–93% yield, except for 8-fluoroquinoline, which could not produce the corresponding product. In the case of 178, various aryl diazoacetates were tested and produced the corresponding indolizines in 71–87% yield. Moreover, a diazo compound with thiophene was compatible in the transformation and produced the corresponding indolizine in 38% yield. Next, a variety of quinolines was tested and quinolone derivatives with various 6- and 7-substituents produced the corresponding indolizines in 58% to 92% yield. Notably, 6-Br-7-fluoroquinoline produced the corresponding indolizine in 68% yield, but 8-fluoroquinoline did not produce the corresponding product in the above-mentioned method.
Li et al.84 synthesized multi-substituted 1-arylaziridine-2-carboxylates via the reaction among alkyne derivatives 181, α-diazoester derivatives 183 and nitrosoarenes 182 and the mixture was stirred in 1,4-dioxane at 50 °C for 24 h under catalyst-free conditions (Scheme 78).
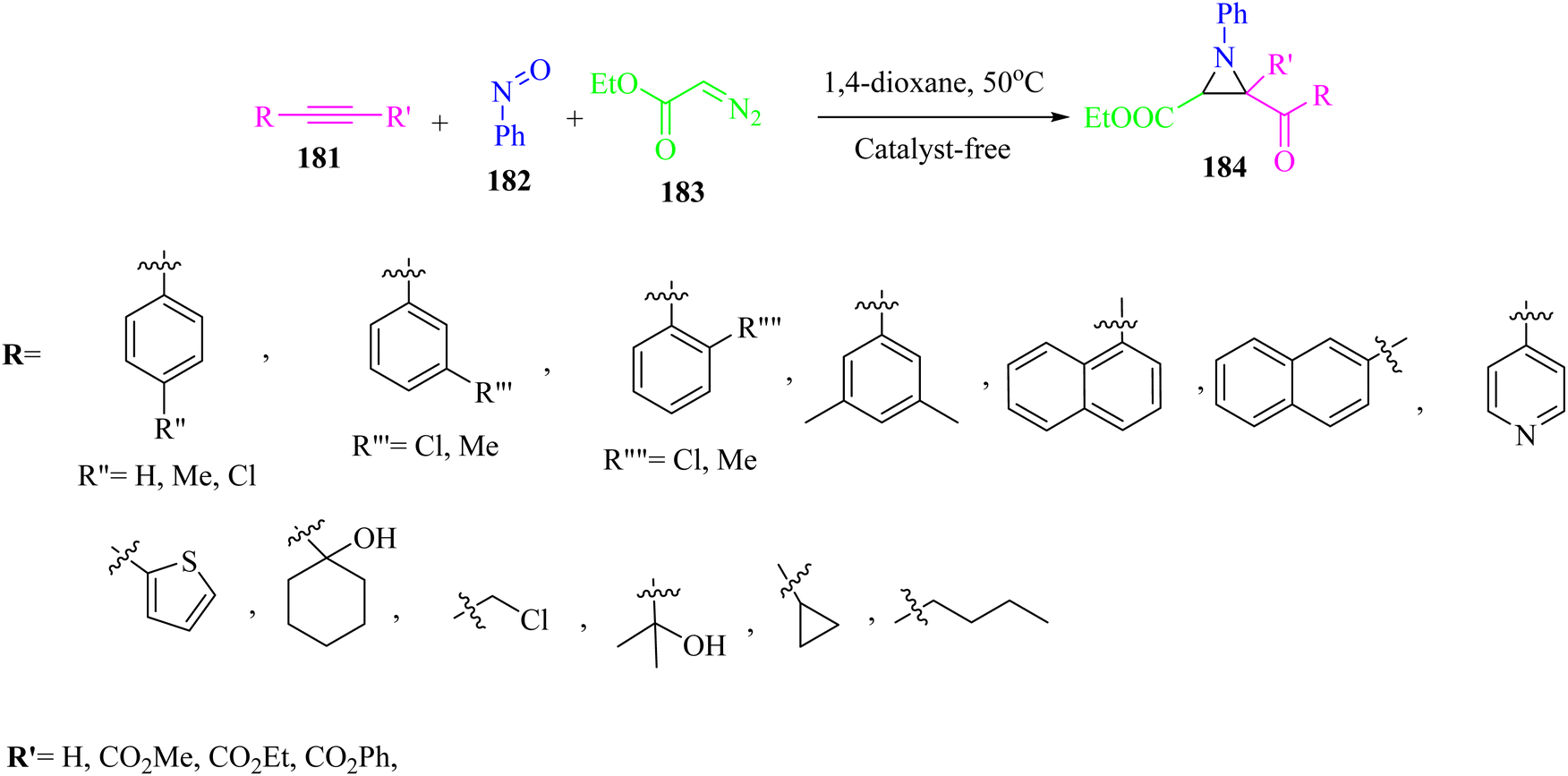 | ||
| Scheme 78 Synthesis of 1-arylaziridine-2-carboxylates 184.84 | ||
Firstly, intermediate K was formed from 183. Then, intermediate L was generated via the reaction between 182 and intermediate K. After that, intermediate M was produced via the removal of N2 from intermediate L. Next, isoxazoline N was prepared from the reaction between M and 181 via [3 + 2] cycloaddition. Finally, the desired product cis-184 or trans-184 was generated via a Baldwin rearrangement. The important point about this reaction is that the use of an ester group instead of R′ and an aryl group instead of R led to the production of cis′-184 due to steric hindrance in transition state O. Also, when using an H atom instead of R′, two transition states (P and Q) were formed, where the cis form was dominant (Scheme 79).
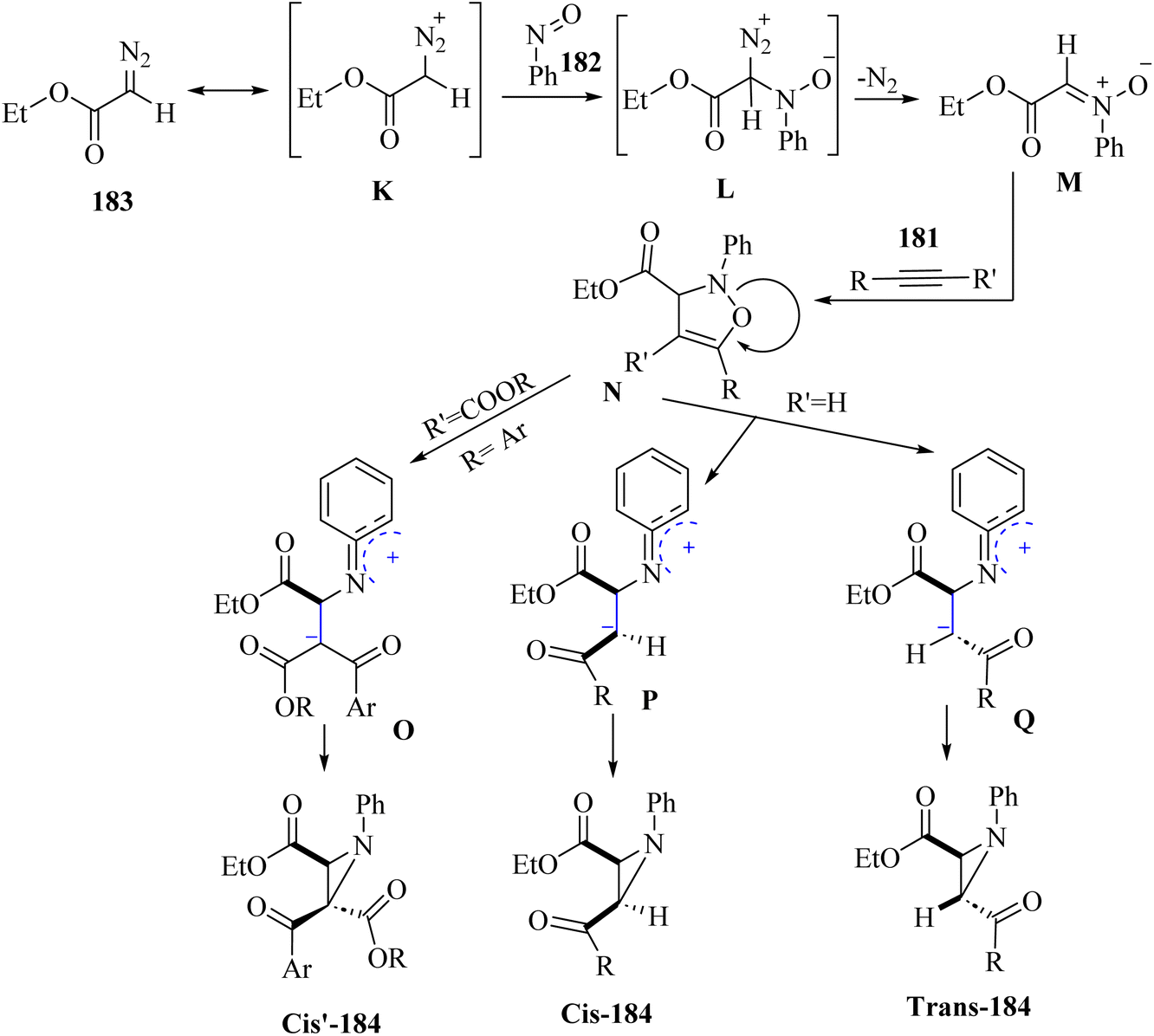 | ||
| Scheme 79 Possible reaction mechanism for the synthesis of multi-substituted 1-arylaziridine-2-carboxylates 184.84 | ||
The above-mentioned method provides mild conditions for the synthesis of different types of multi-functionalized aziridine derivatives in up to 91% yield. Also, this method has advantages such as the use of catalyst-free strategy and readily accessible and cheap starting materials. According to the screened internal and terminal alkynes, they all produced the desired products in 56–91% yield. Internal alkynes produced the corresponding products (as single diastereomeric) exclusively in 71–91% yield. The evaluation of ester moieties of phenylpropiolic acid ester (Me, Et, and Ph) demonstrated that steric hindrance affected the yield, where sterically hindered aryl group provided relatively lower yields (89%, 87%, and 73%, respectively). Also, ethyl 3-phenylpropiolate derivatives with an electron-donating group (Me at ortho, meta, and para position) resulted in higher yields than that having an electron-withdrawing group such as Cl (84–91% and 71–80% yield). In addition, the structure with two substituents (3,5-dimethyl) on the phenyl ring gave the corresponding product produced in 75% yield, which showed that steric hindrance affected the yield. Furthermore, internal alkyne derivatives with a naphthyl group gave the corresponding products in 83% to 91% yield. Moreover, aliphatic terminal alkyne derivatives were evaluated and produced the corresponding cis isomers as the major products (56–82% yield) with moderate diastereoselectivity (up to 71:29 cis/trans). Unfortunately, ethynylcyclopropane and 2-ethynylthiophene gave the corresponding products in 78% and 83% yield with very low diastereoselectivity (50:50 cis/trans and 52:48 cis/trans), respectively.
Zhou et al.85 synthesized tetrasubstituted pyrrole 188 via the reaction among alkynes 187, 1-substituted benzimidazoles 186, diazoacetates 185, and [Fe(TPP)Cl] in 1,4-dioxane for 12 h under an Ar atmosphere at room temperature (Scheme 80).
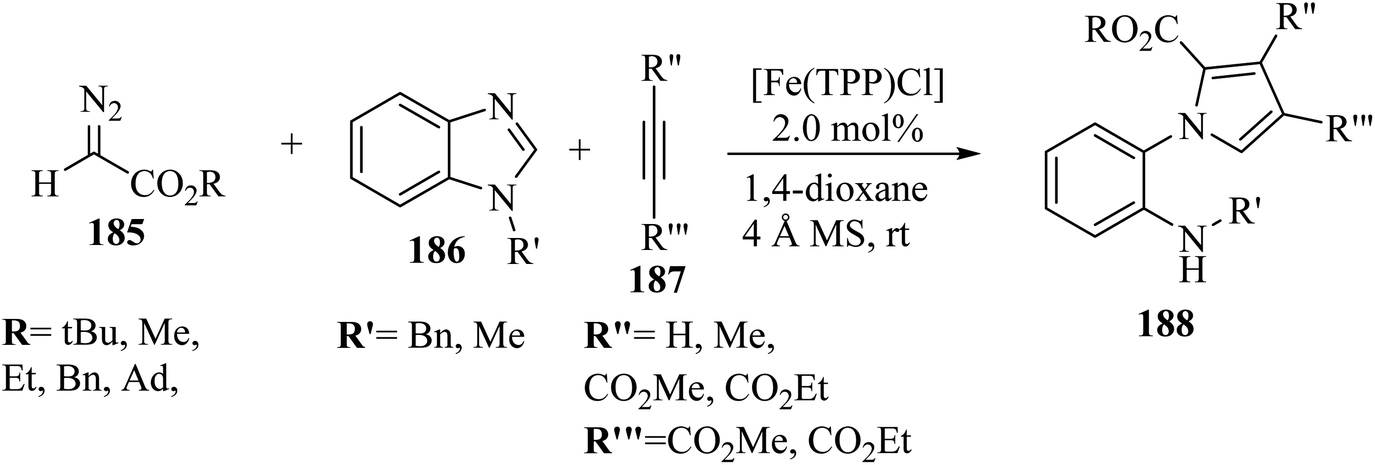 | ||
| Scheme 80 Synthesis of multi-substituted pyrroles 188.85 | ||
Initially, the reaction between 185 and 186 produced benzimidazolium N-ylide intermediate A in the presence of an iron catalyst. Then, intermediate B was produced via the reaction between 187 and intermediate. Finally, 188 was generated via fast isomerization of intermediate B, which was a thermodynamically favorable ring-opening aromatization process (Scheme 81).
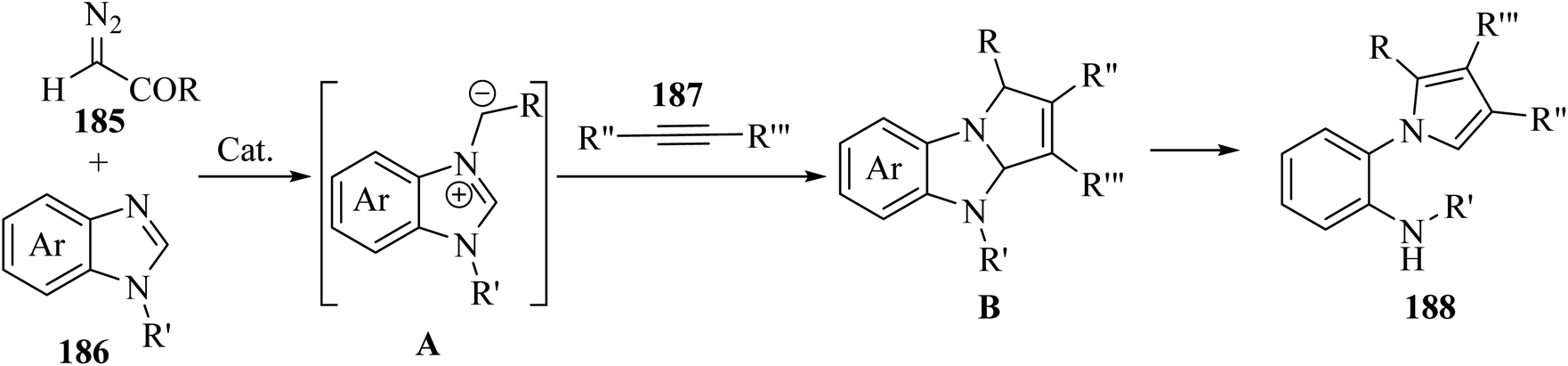 | ||
| Scheme 81 Possible reaction mechanism for the synthesis of tetra-substituted pyrrole 188.85 | ||
The desired products were produced with an isolated yield of 70–90%. The above-mentioned method has advantages such as the use of mild reaction conditions, available starting materials, operational simplicity, and inexpensive catalyst.
Lv and co-workers86 synthesized (E)-beta-monofluoroalkyl-β,γ-unsaturated esters (192 and 195) via the reaction among 1,1-alkylmonofluoroalkylation of terminal alkyne derivatives 189, 2-fluoro-1,3-dicarbonyl compounds (191 and 194), and diazo compounds (190 and 193) in the presence of CuI and K2CO3 in acetone at 80 °C for 2 h (Scheme 82).
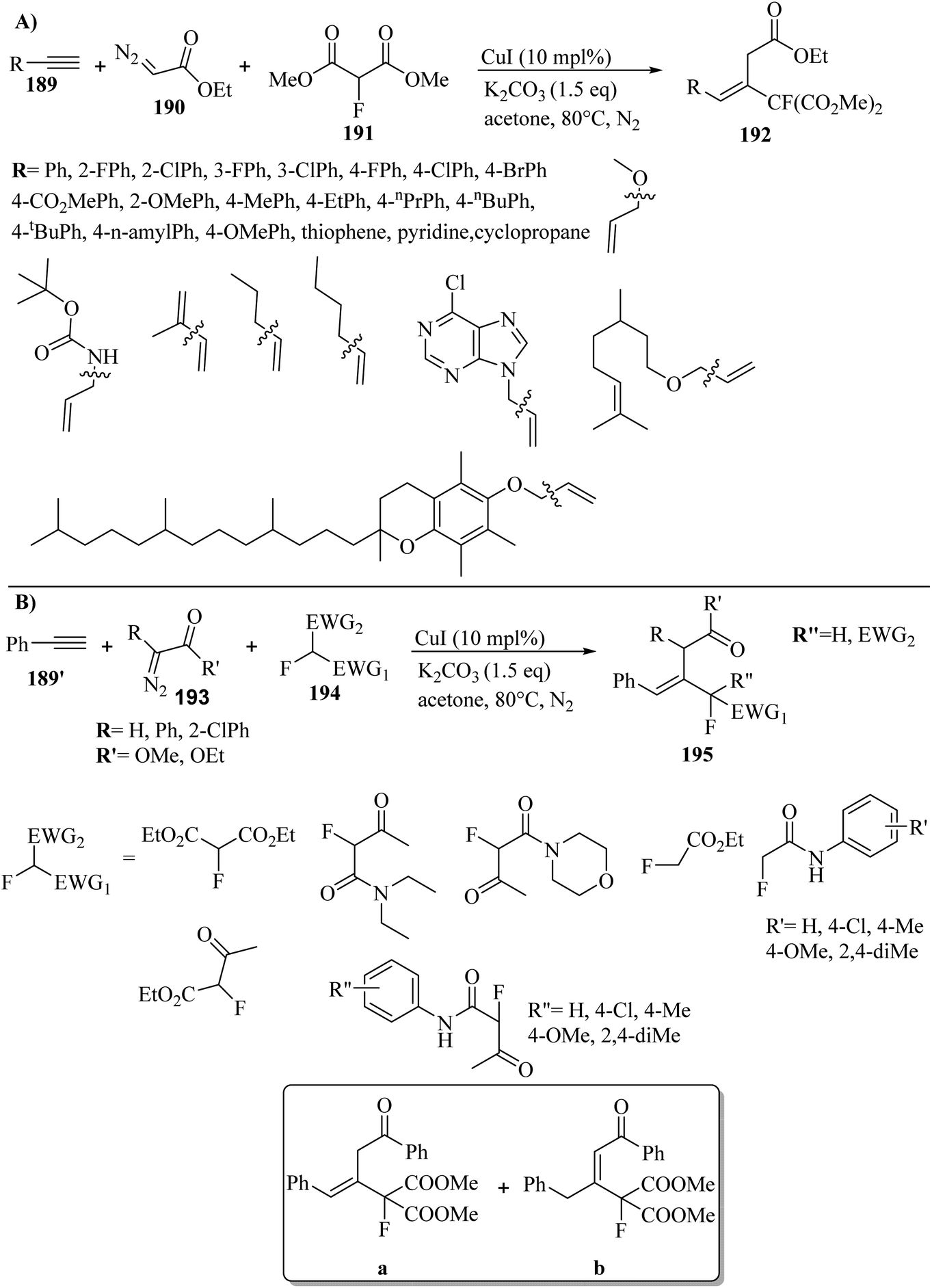 | ||
| Scheme 82 CuI-catalyzed synthesis of (E)-β-monofluoroalkyl-β,γ-unsaturated esters or ketones: (A) scope of alkynes in the 1,1-alkylmonofluoroalkylation and (B) scope of diazo compounds and monofluoroalkylation reagents in 1,1-alkylmonofluoroalkylation.86 | ||
Initially, Cu acetylide was produced via the reaction between 189 and CuI in the presence of K2CO3. Then, copper carbene K was generated via the reaction between 190 and Cu acetylide. Next, intermediate L was obtained from the migratory insertion of the alkynyl group into the K, followed by protonation of intermediate L to produce intermediate M. After that, intermediate N was produced by the rearrangement of intermediate M. Finally, the desired product 192 was produced via the reaction between intermediate N and dimethyl 2-fluoromalonate in the presence of K2CO3. It should be noted that the nucleophilic attack of the monofluoroalkyl group to the Ph from the opposite side was preferred to avoid steric interactions (Scheme 83).
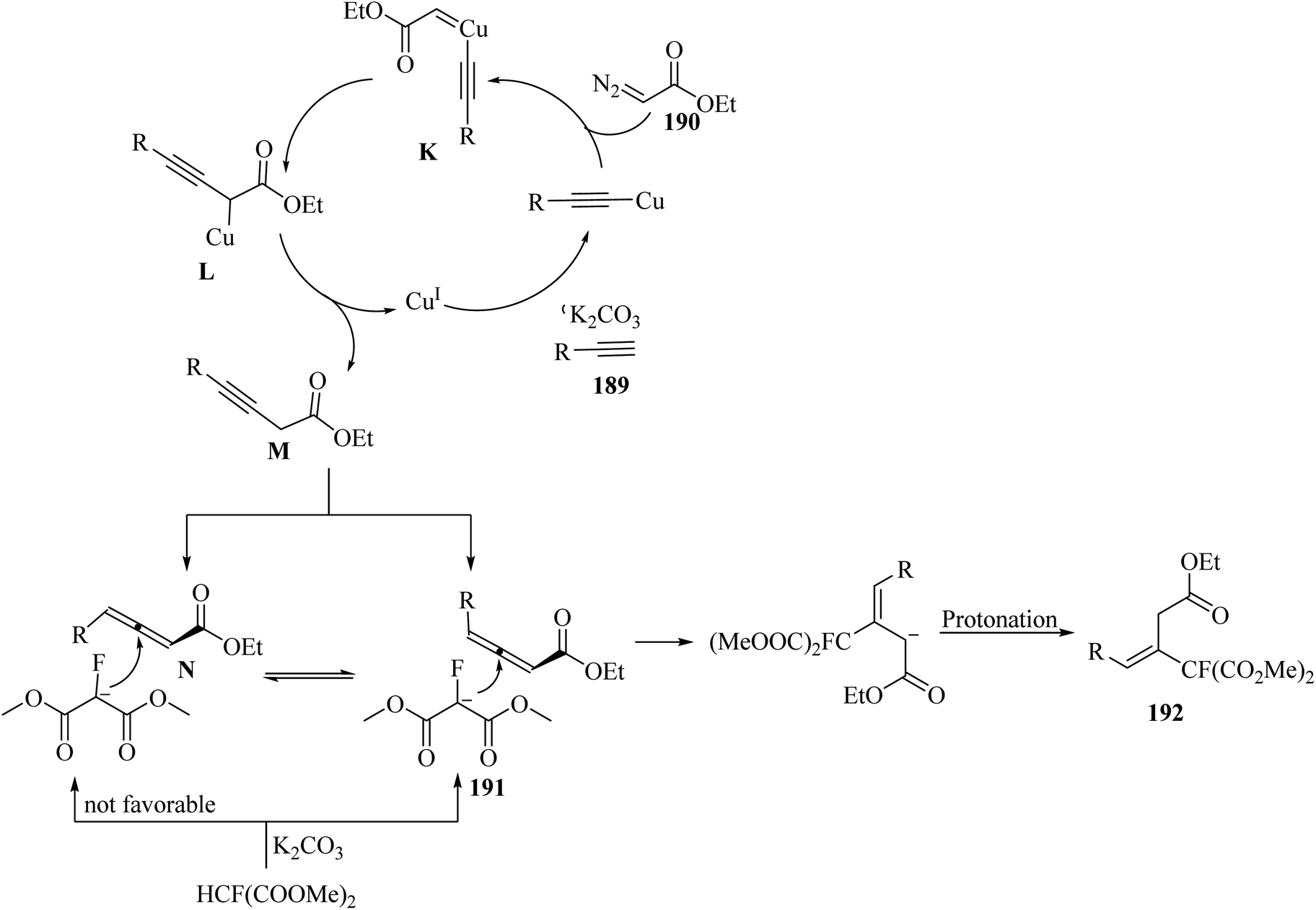 | ||
| Scheme 83 Possible reaction mechanism for the synthesis of (E)-beta-monofluoroalkyl-β,γ-unsaturated esters.86 | ||
In the case of 189, the reaction of dimethyl 2-fluoromalonate, 2-diazoacetate, and various terminal alkynes gave the corresponding β,γ-unsaturated products in 35−91% yield. Also, para, ortho, and meta-fluoro, chloro ethynylbenzenes under the 1,1-alkylmonofluoroalkylation reaction produced the desired products in the same yields, which showed that steric hindrance had no effect. 4-Br and 4-COOMe ethynylbenzenes were reacted and produced the corresponding products in 76% and 70% yield, respectively. In addition, alkyne derivatives with electron-donating groups on the aromatic rings gave the corresponding β,γ-unsaturated products in 70% to 85% yield. Alkynes with a pyridine or thiophene moiety gave the desired β,γ-unsaturated in 45–77% yield. N-propargyl amides, conjugated enynes, and alkyl propargyl ethers gave the desired products in 70–75% yield. Moreover, the non-activated aliphatic alkynes produced the corresponding β,γ-unsaturated products in 35% to 40% yield. Further, the citronellol-based alkyne derivative, D,L-α-tocopherol-based alkyne derivative, and purine-based alkyne derivative produced the desired β,γ-unsaturated products in 85%, 70%, and 81% yield, respectively. Diethyl 2-fluoromalonate produced the desired product in 78% yield. Also, 2-F-1-morpholinobutane-1,3-dione and N,N-diethyl-2-F-3-oxobutanamide were tested and produced the desired products in 70% and 56% yield, respectively. Interestingly, ethyl 2-fluoro-3-oxobutanoate reacted with ethynylbenzene and ethyl 2-diazoacetate and produced the desired β,γ-unsaturated product in 70% yield. In addition, 2-F-acetoacetamides with different groups on the aromatic rings were converted to the desired β,γ-unsaturated products in 68% to 81% yield. Importantly, when K2CO3 was replaced with K3PO4, the corresponding products were produced in 52% to 78% yield. The reaction of ethyl 2-diazoacetate compared to 2-diazo-1-phenylethanone and methyl aryldiazoacetates was higher under identical conditions. When acetone was replaced with THF, methyl aryldiazoacetates were suitable substrates and produced the desired β,γ-unsaturated products in 39–40% yield. Finally, 2-diazo-1-phenylethanone was transformed into the 1,1-alkylmonofluoroalkylation products (Scheme 82B(a) and (b)) in 1,4-dioxane with a total yield of 35%.
2.6 Reaction of isocyanides
Pelliccia et al.87 synthesized iminofurans 199 via an isocyanide-based three-component reaction with the reactants of diethyl bromomalonate 196 and phenylacetylene derivatives 197 in the presence of fac-Ir(ppy)3 as the photoredox catalyst and Na2CO3 as the base in MeCN under irradiation with a 30 W blue LED at room temperature for 20 h (Scheme 84).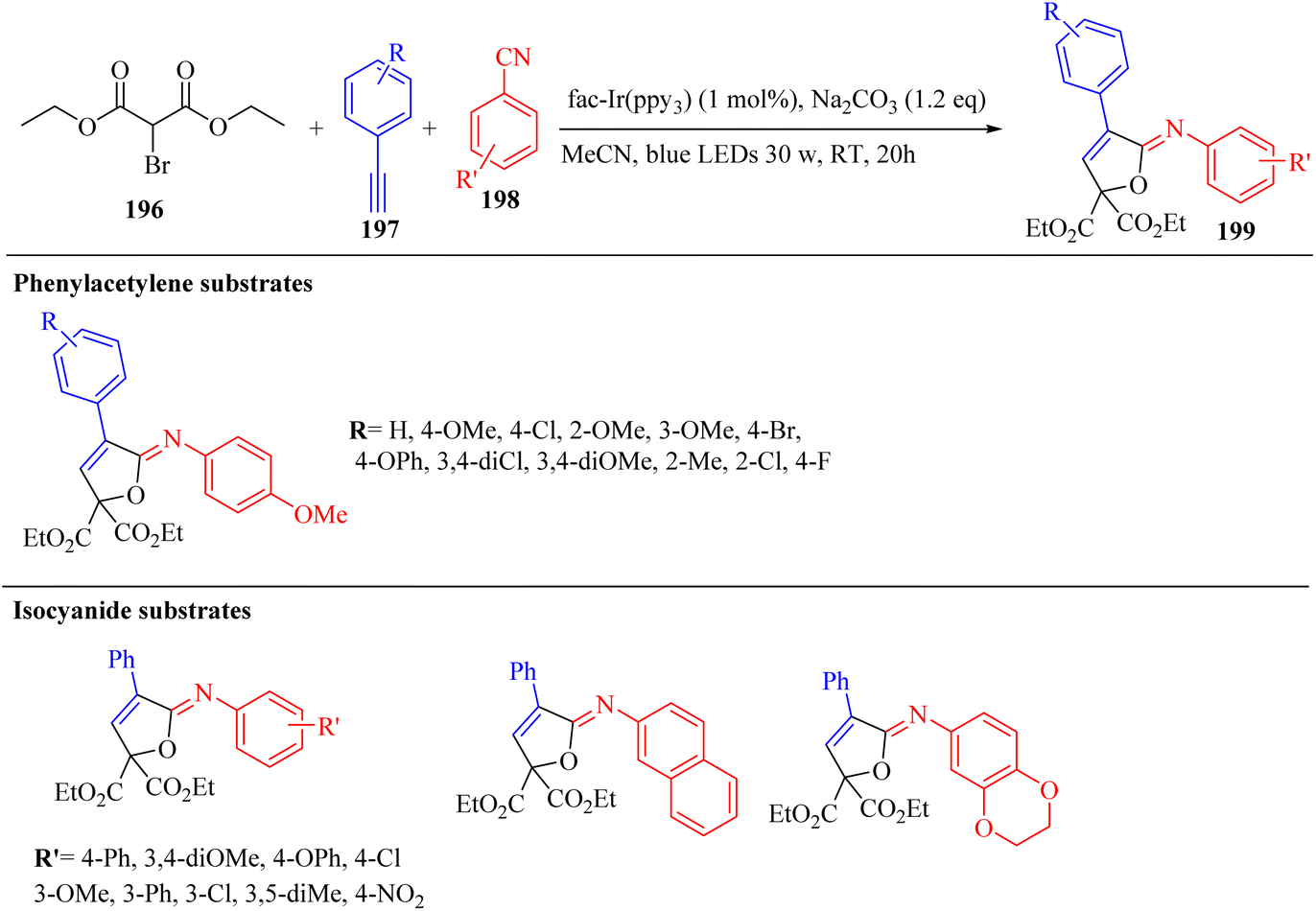 | ||
| Scheme 84 Fac-Ir(ppy)3-catalyzed synthesis of iminofuran derivatives 199.87 | ||
Initially, the Ir3+* photocatalyst was generated from fac-Ir(ppy)3, which underwent metal-to-ligand charge transfer under visible-light irradiation. Then, alkyl radical A was formed via single-electron transfer (SET) between Ir3+* and 196. Next, vinyl radical B was generated via the reaction between A and 197, followed by the reaction of 198 with B, which led to the formation of imidoyl radical intermediate C. After that, Ir4+ oxidized this key species to nitrilium ion D. Next, amide E was prepared from D in the presence of H20. In the following step, 199 was generated from E, which underwent oxidative cyclization (Scheme 85).
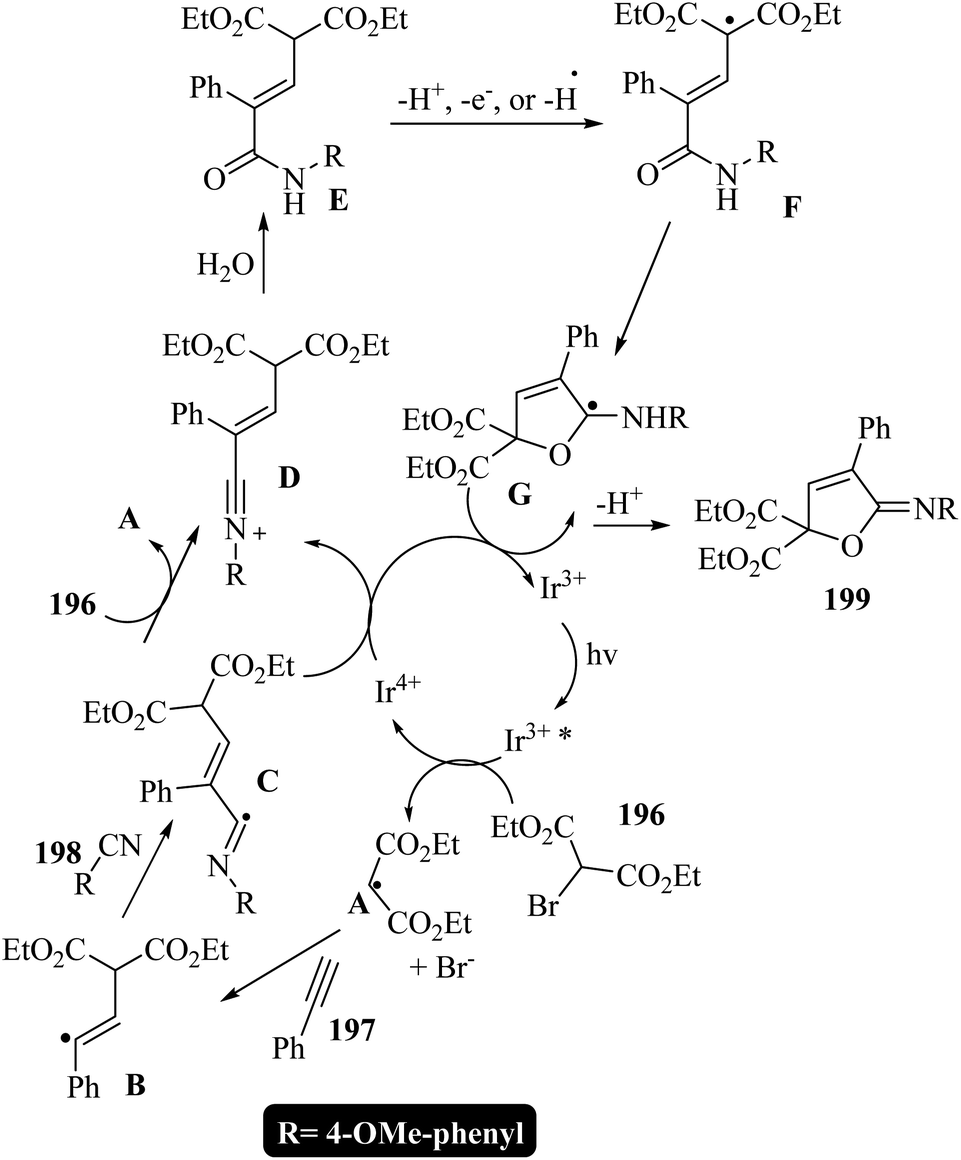 | ||
| Scheme 85 Possible reaction mechanism for the synthesis of iminofurans 199.87 | ||
Investigation of isocyanides in the above-mentioned reaction demonstrated that a variety of groups on the phenyl ring could produce the corresponding products in 35–54% yield. In contrast, aliphatic isocyanides such as cyclohexylisocyanide and tert-butylisocyanide did not produce the corresponding products. Moreover, the evaluation of phenylacetylene derivatives showed that neither the position of the aromatic ring substituents nor their electronic nature significantly affected the reaction yield (23–60%) except when bromo phenylacetylene was tested, where the reaction yield dramatically decreased (23%). In the case of aliphatic derivatives of alkynes, the use of these derivatives was not possible due to them not being reactive at all.
Gao and co-workers88 synthesized captodative olefins (203 or 207) via the palladium-catalyzed (Pd(OAc)2) three-component reaction of alkynes (200 or 204), carboxylic acids (206 or 202), and isocyanides (201 or 205) in the presence of Ag2O as a silver salt, and P(o-tol)3 as a ligand in PhCl under N2 at 30–50 °C for 5 h (or 70 °C for 5–15.5 h) (Scheme 86).
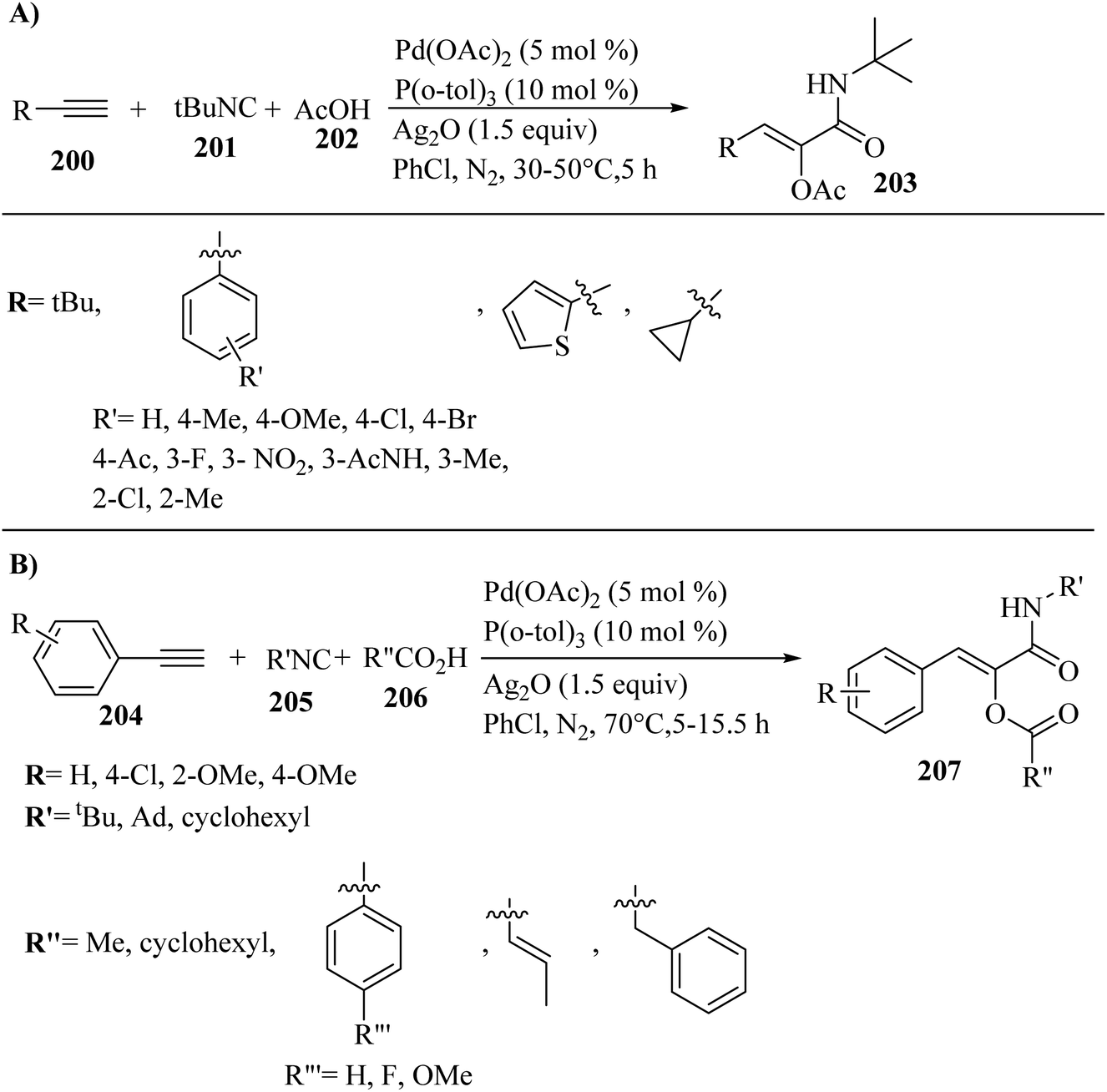 | ||
| Scheme 86 Synthesis of captodative olefins: (A) substrate scope of alkyne derivatives and (B) substrate scope of isocyanides, alkynes, and carboxylic acids.88 | ||
Initially, the reaction among 200, 206, and Ag2O produced silver acetylide K together with the formation of silver carboxylate. Then, complex L was obtained via the attack of the carboxylic anion on K, which subsequently caused the transmetalation process with Pd(II). Then, imidoyl palladium(II) intermediate N was generated via the isocyanide migratory insertion reaction to vinyl palladium complexes M. Finally, via the successive reductive elimination process and treatment with H2O 270 or 203 was produced (Scheme 87).
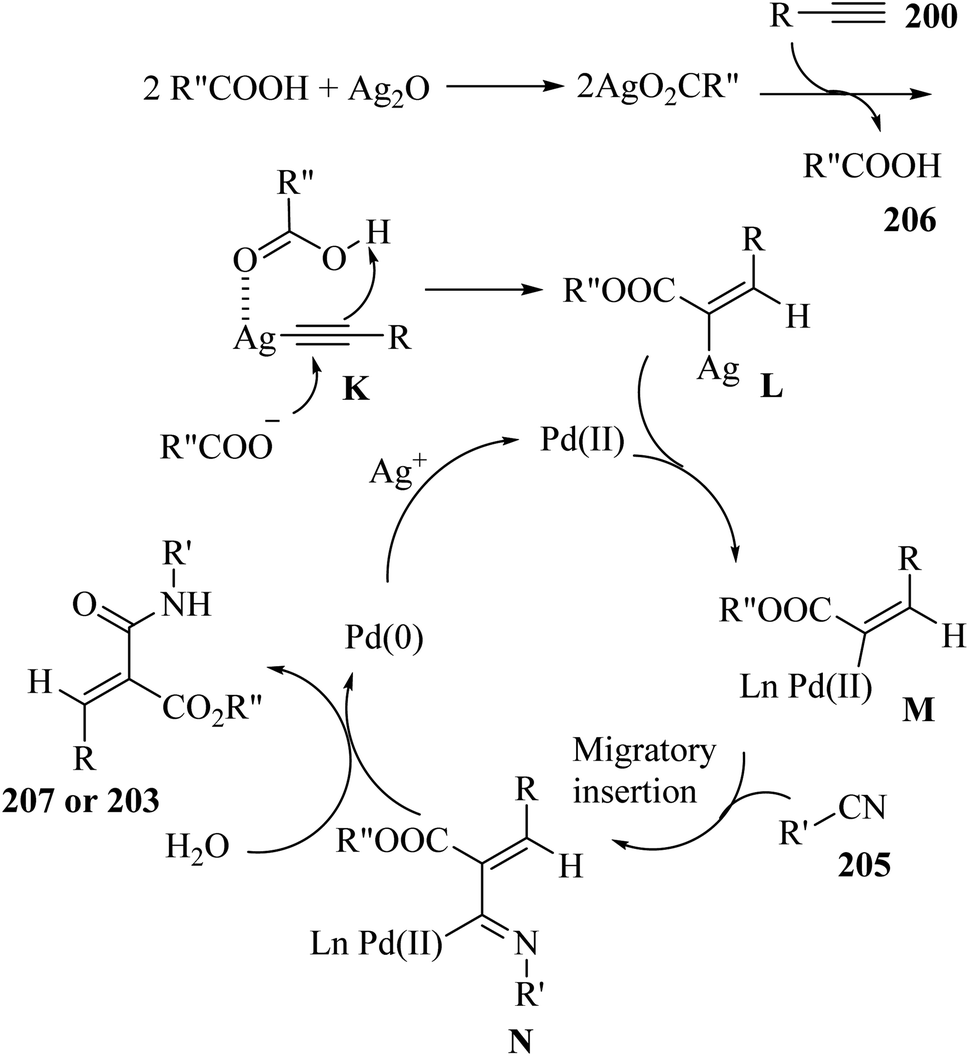 | ||
| Scheme 87 Possible reaction mechanism for the synthesis of captodative olefins.88 | ||
A variety of alkyne, carboxylic acid, and isocyanide derivatives was investigated. According to the results, aryl acetylenes bearing different substitutions (alkyl, alkoxy, halides, acyl, nitro, and amide) produced the corresponding olefins in 48% to 90% yield. Interestingly, when phenylpropiolic acid was used instead of phenylacetylene, the corresponding olefin was produced in 87% yield. In addition, thienyl alkyne gave the corresponding olefin in 44% yield, and the enyne substrate also could be transformed into the corresponding olefin in 57% yield. Also, alkyl acetylenes (tBu and cyclopropane) produced the corresponding olefins in 70% and 67% yields, respectively (Scheme 86A). A variety of acids and isocyanides was tested in the above-mentioned reaction. It was found that for other carboxylic acids, higher temperature was more favorable and produced the corresponding olefins in 47% to 86% yield (Scheme 86B). Moreover, 1-adamantyl and cyclohexyl isocyanide were tested and produced the corresponding olefins in 44% to 79% yield, respectively. Interestingly, when α-acidic isocyanides such as isocyanoacetates were used, no desired products could be observed.
Singh and co-workers89 reported a novel method for the synthesis of tetrahydroquinoline-fused tetracyclic derivatives 2a. This method was performed in two steps. In the first step, aniline derivatives 209, benzaldehyde derivatives 208, propiolic acid derivatives 211, and isocyanide derivatives 210 were stirred in MeOH at room temperature and produced derivatives 1a. Then derivatives 1a were reacted with molecular iodine (electrophile) and produced the corresponding derivatives 2a (Scheme 88).
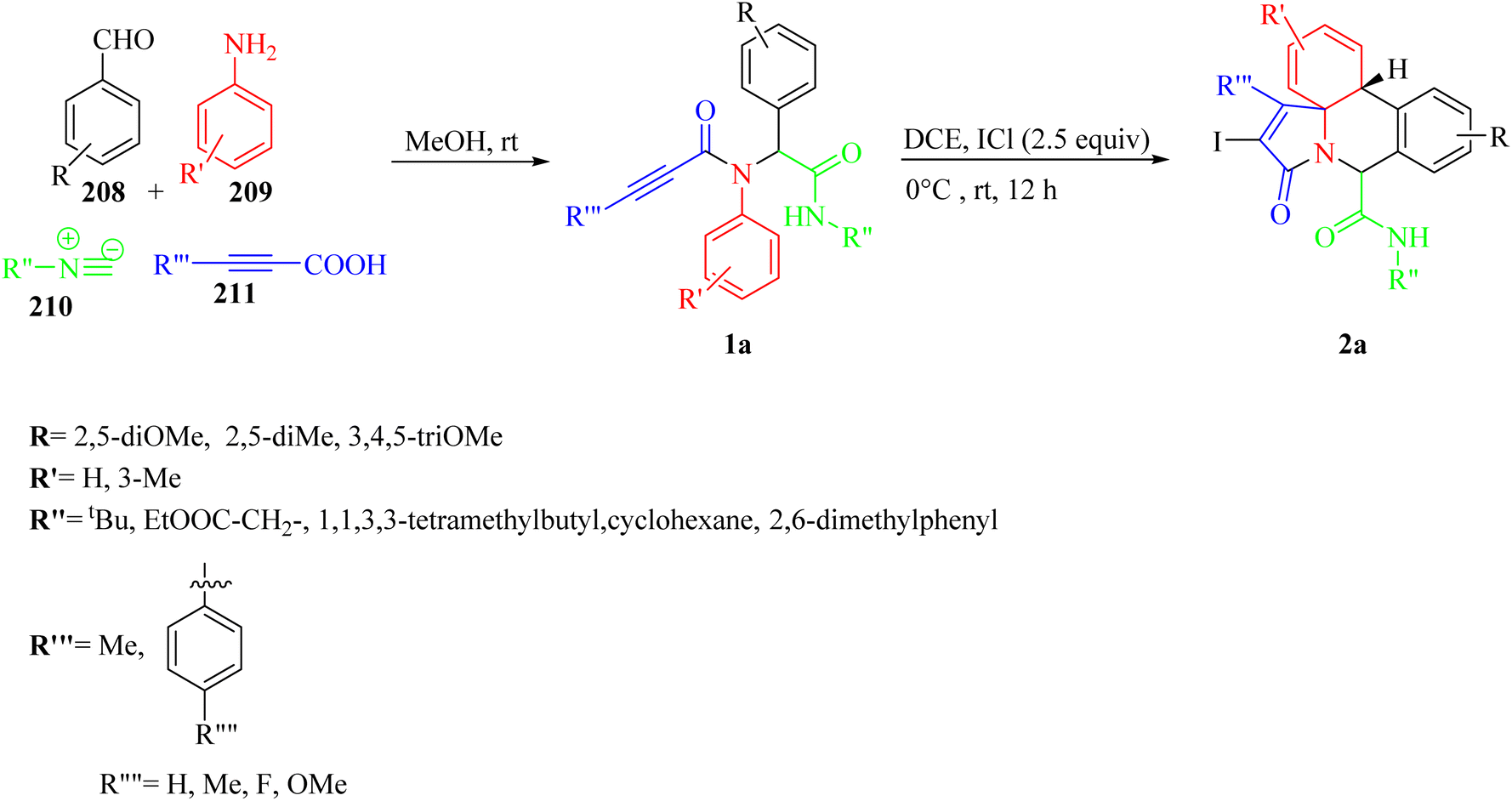 | ||
| Scheme 88 Synthesis of tetrahydroquinoline-fused tetracyclic heterocycles 2a.89 | ||
Initially, intermediate A was generated via the activation of a triple bond of alkyne (π activation) by the iodonium (I+) ion. After that, the regioselective spirocyclic carbocation intermediate B was obtained via the ipso nucleophilic attack of the aniline to the activated triple bond. In this step, it should be noted that this process was done via a 5-endo-dig method. Finally, 2a was produced via the immediate trapping of the carbocation by the intramolecular nucleophile. In this step, it should be noted that this process was done through the Friedel–Crafts-type cyclization (Scheme 89).
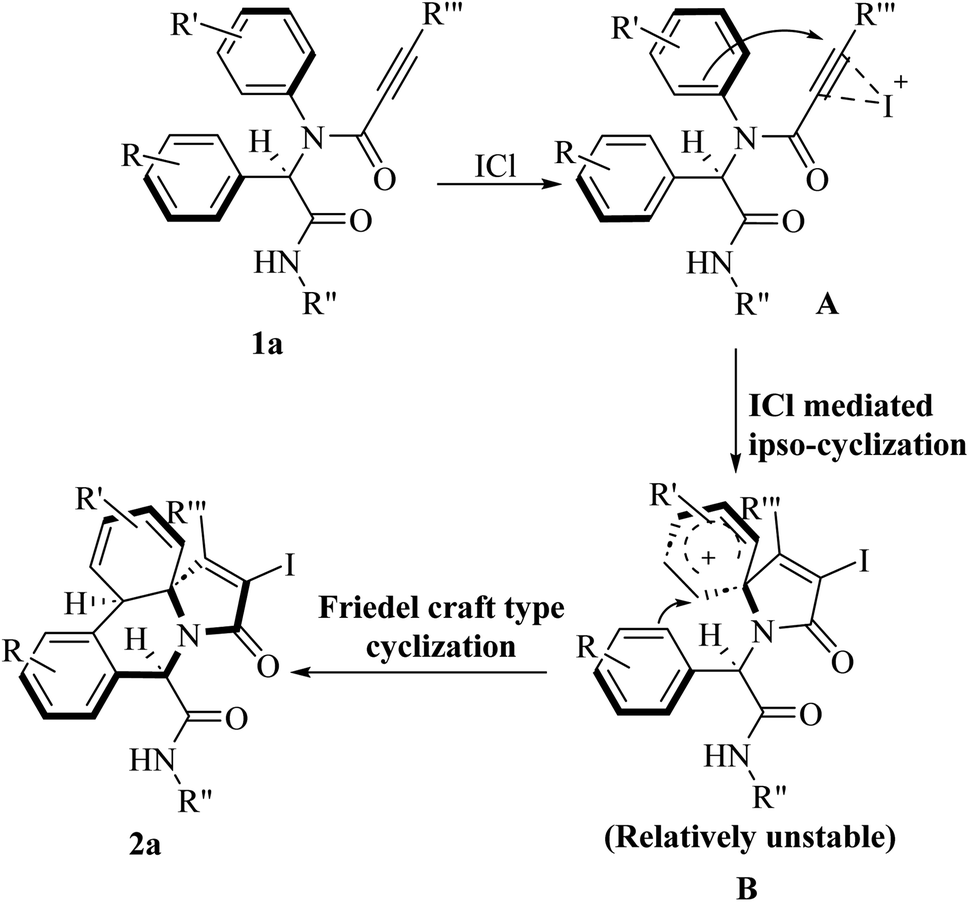 | ||
| Scheme 89 Possible reaction mechanism for the synthesis of tetrahydroquinoline-fused tetracyclic derivatives 2a.89 | ||
The Ugi adducts were produced in 67–91% yield and were compatible with substituted phenyl propiolic acid derivatives, which transformed to the corresponding tetrahydroquinolines in 64% to 87% yield. Also, in this transformation, substituent electronic factors had an influence on the phenyl propiolic acid. The presence of an electron-donating group on phenyl propiolic acid produced the required products in better yields (71–87%) compared to fluorine-substituted adducts (58–64%). The Ugi adducts derived from 2,5-dimethoxy and 3,4,5-trimethoxy aldehyde reacted with the various isocyanide derivatives and produced the corresponding products in 71% to 76% yield. Furthermore, functionalized phenyl propiolic acid, aldehyde and cyclohexylisocyanide-derived Ugi adduct produced the corresponding products in 58% to 77% yield. It should be noted that the aliphatic acid derivatives having Ugi adducts transformed the corresponding products in 69% to 71% yield. In addition, a variety of aromatic and aliphatic substituted isocyanides was tested and produced the corresponding cyclized products in 52% to 83% yield. Among the aliphatic isocyanide derivatives, only n-pentyl isonitrile showed the minimum yield. Moreover, the aromatic isonitrile-based Ugi adduct produced the corresponding products in 56% and 62% yield.
Yang and co-workers90 synthesized perfluoroalkyl-containing pyrano[3,4-c]pyrroles 215 via the reaction among methyl perfluoroalk-2-ynoate derivatives 214, 3-aroyl (or heteroyl) acrylic acid derivatives 212, and isocyanide derivatives 213 in MeCN at room temperature for 12 h (Scheme 90).
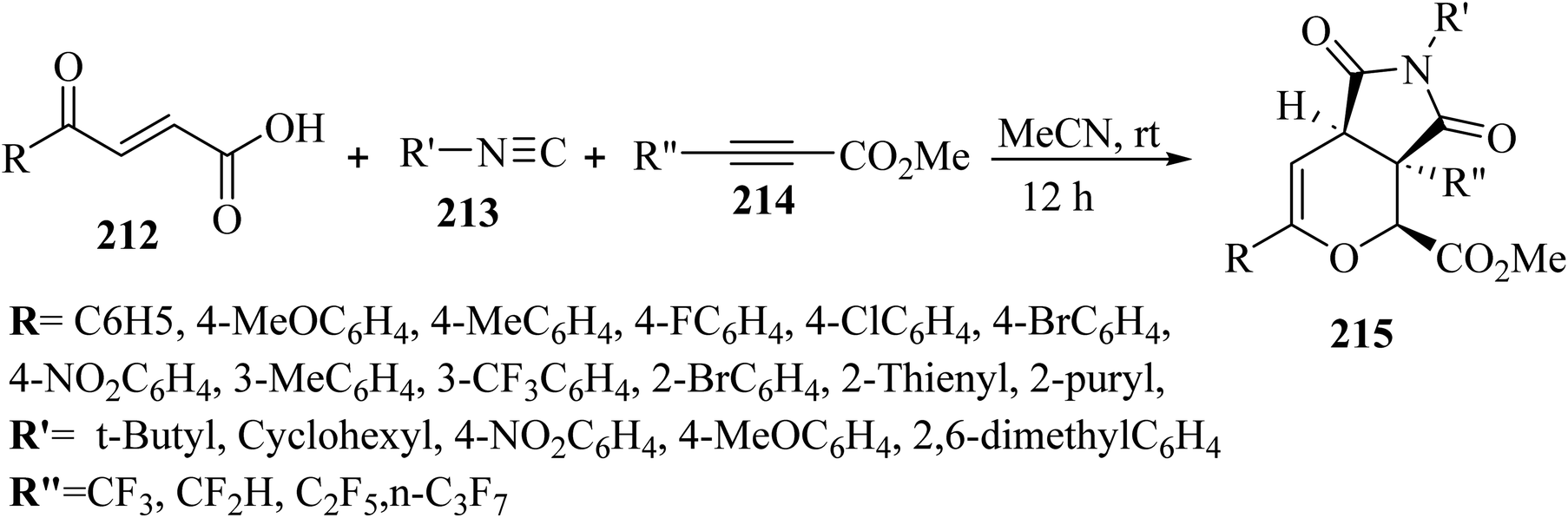 | ||
| Scheme 90 Synthesis of perfluoroalkyl derivatives 215 without a catalyst.90 | ||
Initially, zwitterion J was obtained via the Michael addition of 213 to 214. Then, J was protonated by 212 to give aminium K and carboxyl anion L. After that, isoimide M was generated from the reaction between intermediate K and L via a Passerini-type reaction. Next, M produced conformers N and N′ via the Mumm rearrangement. Finally, the exo isomer was generated conformer N as the major diastereoisomer via the intramolecular oxo-Diels–Alder reaction. It should be noted that the endo isomer was obtained as the minor product due to the steric hindrance between CO2Me and the R group in conformer N′ (Scheme 91).
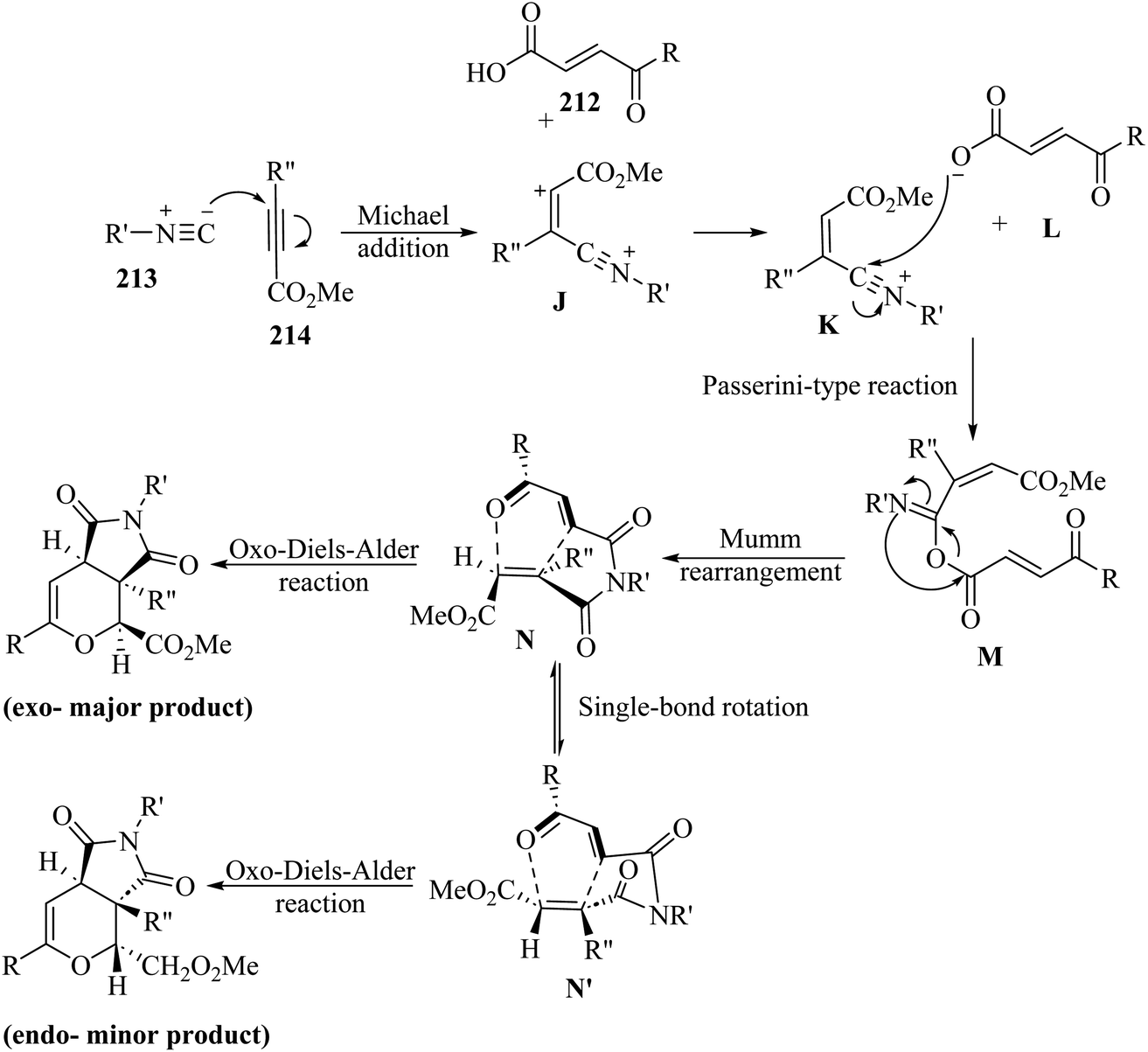 | ||
| Scheme 91 Possible reaction mechanism for the synthesis of perfluoroalkyl-containing pyrano[3,4-c]pyrroles.90 | ||
Various R-substituted acrylic acid derivatives were evaluated and acrylic acid derivatives with different types of heteroaryl or aryl groups at the terminal position produced the desired perfluoroalkyls in 45% to 97% yield with high diastereoselectivity (up to >99:1 dr). Generally, an electron-withdrawing group on the acrylic acid phenyl ring afforded the corresponding perfluoroalkyls in higher yields compared to an electron-donating substituent at the same position. Substrates with n-C3F7, CF2H, and C2F5 were tested and produced the corresponding perfluoroalkyls in 45% to 93% yield, which showed little detrimental effect on the yields and had no adverse influence on the diastereoselectivity. Next, various isocyanides were investigated and the corresponding perfluoroalkyls produced in 32% to 98% yield (up to >92:1 dr). A summary of the reaction of alkynes with isocyanide derivatives, and also the limitations and progress of the reactions are presented in Table 3.
| Reactants | Product | Catalyst | Solvent | Method employed | Reaction time | Yield | Limitations or progress | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Investigation of scope of arylacetylenes under N2 (UV and LED method = 24 h). | ||||||||
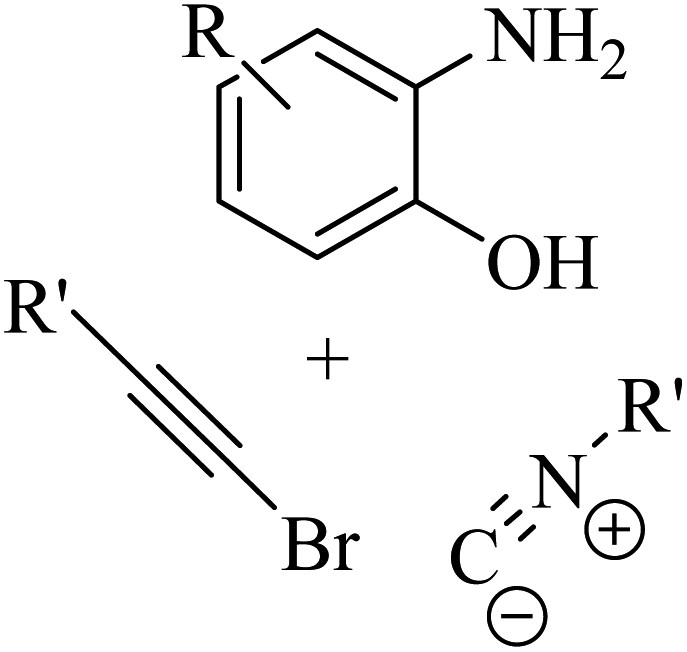 |
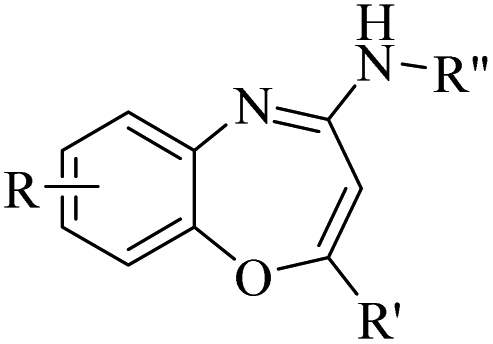 |
Pd(PPh3)2Cl2 | 1,4-Dioxane | PPh3, Cs2CO3, 80 °C | 2 h | 47–94% | 2-Amino-3-pyridinol and 2-aminothiophenol did not react | Liu et al.94 |
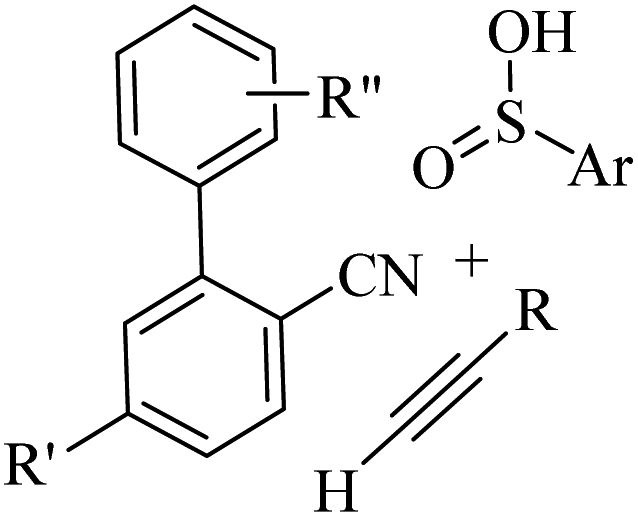 |
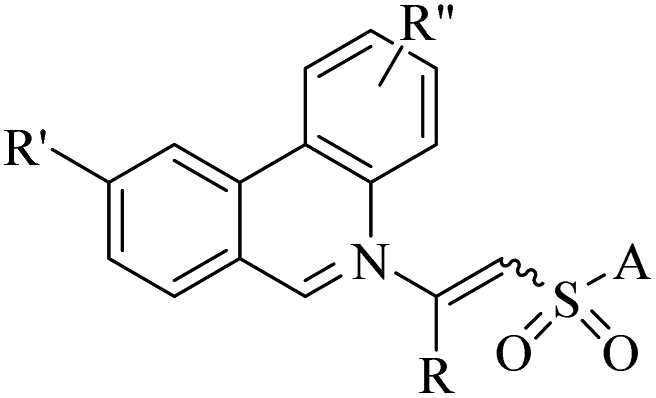 |
— | DMSO/H2O | Method 1: pyridine, LED, O2a, RT | Method 1: 16 h | Up to 73% | (1) High selectivity | Li et al.95 |
| Method 2: pyridine, UV, O2a, RT | Method 2: 24 h | (2) Made new EDA complexes from arylsulfinate anions | ||||||
The reaction among isocyanides, alkynes and fullerenes was reviewed by Li et al.91–93 Investigating the types of alkynes in this type of multicomponent reaction requires a more detailed investigation to discover a way to carry out the reaction of alkynes that are not involved in the reaction. Even checking the position of C–H alkynes and activating this part to be involved in the reaction need to be investigated. The yield of the obtained products is also low, which is a limitation in this field and requires more extensive research in this type of reaction. Some recent developments in this field are shown in Table 4.
| Reactants | Product | Catalyst | Solvent | Method employed | Reaction time | Yield | Limitations or progress | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Used in the reaction between fullerenes, alkyl isocyanides, and DMAD.b Used in the reaction between fullerenes, alkyl isocyanides, and unsymmetric alkyne substrates. | ||||||||
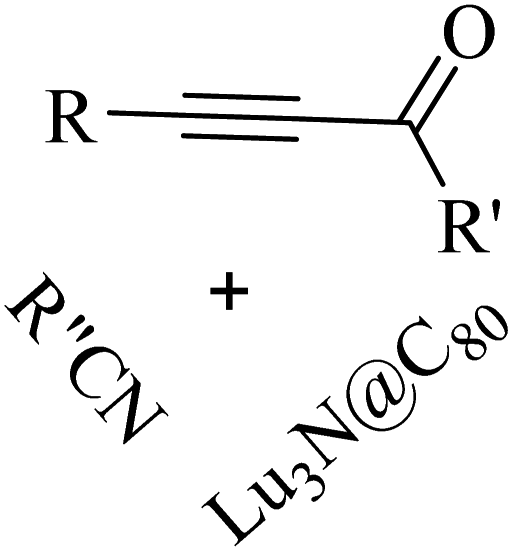 |
 |
o-Dichlorobenzene | Air, 120 °C | 1 h | 27–30% | (1) One-step reaction for synthesis of a static solid-state cluster in an NCF | Li et al.91 | |
| (2) This research proposed a vision about potential regioselectivity in further reactions and advances in quantum information materials | ||||||||
 |
 |
— | o-Dichlorobenzene | N2, 80 °Ca or 120 °Cb | 1 h | 4–36% | (1) Alkynes containing a substituent and an ester group on one side provided resonance and inductive effects on the other side of the triple bond (ester group has key role) | Li et al.92 |
| (2) The 4-NO2-Ph substituent at both ends of the CC triple bond of did not show similar results |
||||||||
3 C–H activation
3.1 Synthesis of propargylamines
Yan and co-workers96 reported a green method for the synthesis of propargylamine derivatives 219 via the reaction among alkynes 218, aldehyde derivatives or ketone derivatives 216, and amines 217 in the presence of PS–PEG-BPy–CuBr2 as a catalyst and N2 at 110 °C under solvent-free conditions (Scheme 92). The desired propargylamines were produced in 66–97% yield. Several alkynes were investigated and the results showed that aromatic alkynes with p-trifluoromethyl or p-methyl substituents produced the corresponding products in 99% and 98% yield, respectively. When the aromatic alkyne has a methyl substitution on the ortho position, it gave the corresponding product in 94% yield. Furthermore, aliphatic acetylenes were used and the desired propargylamines were produced in yields of 79% and 74%.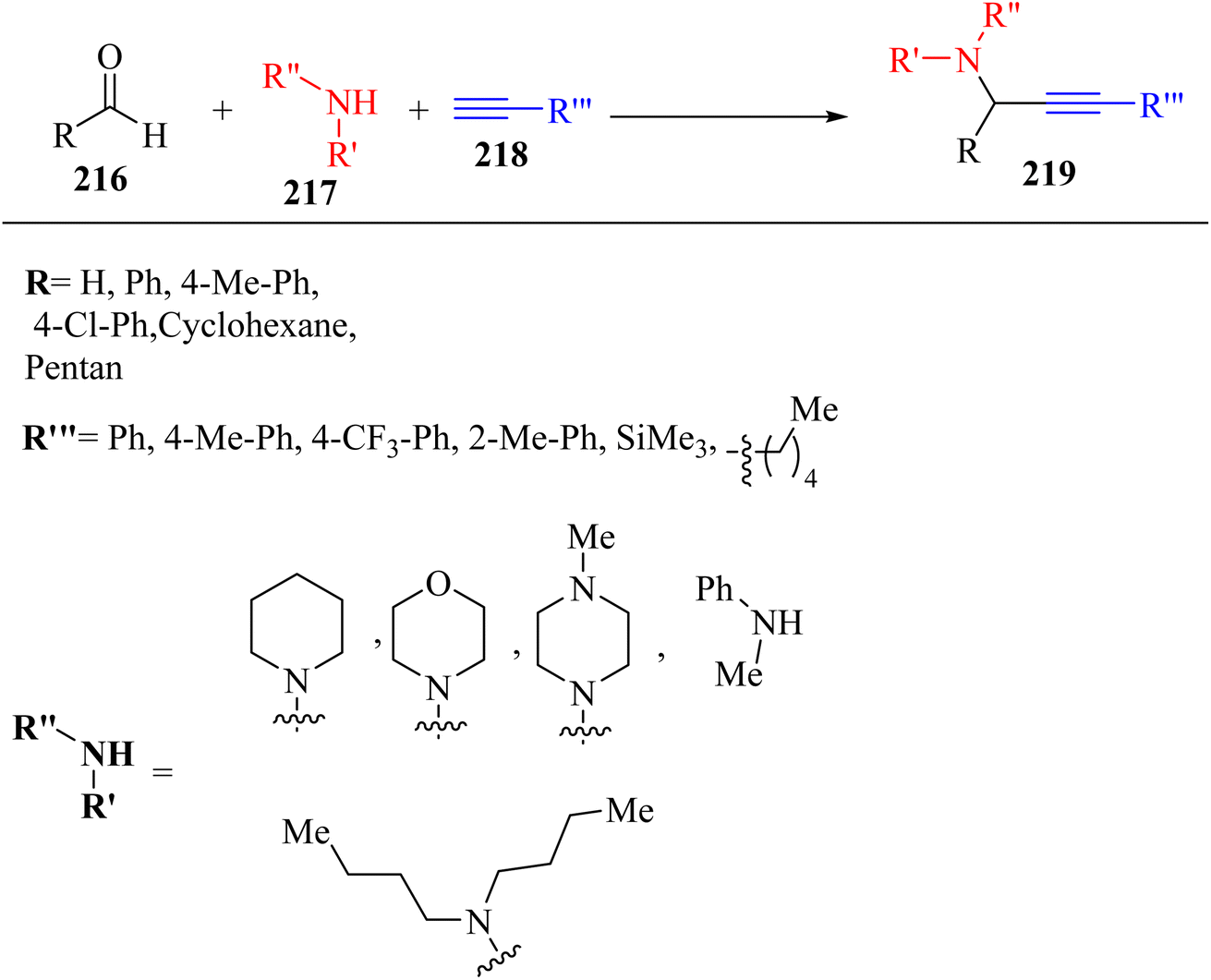 | ||
| Scheme 92 Green synthesis of propargylamine derivatives 219 via A3 coupling reactions.96 | ||
Propargylamines 223 were synthesized by Dhanasekaran and co-workers97 via the reaction among 2-ethynyl anilines 222, amines 221, and substituted benzaldehydes 220 in the presence of a CuI-iPrpyboxdiPh complex and N-Boc-(L)-proline as the catalyst system in DCE at room temperature (Scheme 93). According to the results, N-substituted 2-ethynyl aniline derivatives gave the desired propargylamines in high yields (up to 87%). N-Tosyl-2-ethynyl aniline produced the products in low enantioselectivities with respect to N-substituted 2-ethynylaniline derivatives. Also, 2-ethynyl anilines with various substituents on the aromatic ring produced the desired products with high levels of enantioselectivities (up to 95% ee) in good yields (up to 87%).
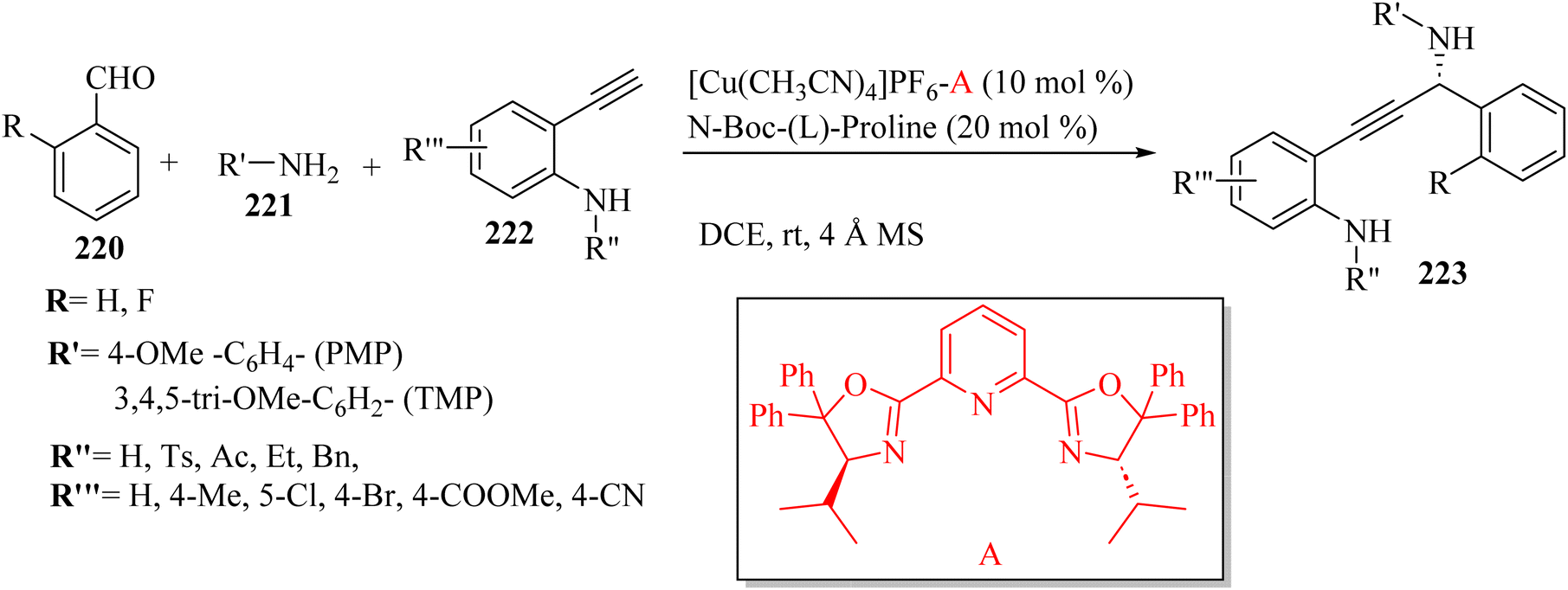 | ||
| Scheme 93 Synthesis of propargylamines 223 via a chiral CuI–box complex with 2-ethynyl anilines.97 | ||
Milen et al.98 synthesized propargylamines 227 via the reaction among acetylenes 225, aldehydes 224, and secondary amines 226, where they studied twelve readily available copper-containing mineral derivatives. It should be noted that the reaction was performed in the presence of chrysocolla, brochantite, azurite, and malachite in PhMe under reflux for 20 h and the corresponding propargylamines were obtained in 36% to 96% yield (Scheme 94). Firstly, various benzaldehyde derivatives with phenylacetylene and morpholine were tested and the results showed that aldehydes with either electron-donating or -withdrawing groups on the phenyl ring produced the desired propargylamine derivatives in 21% to 92% yield. Also, the reaction of 9H-fluorene-2-carbaldehyde and naphthalene-2-carbaldehyde produced the desired propargylamines in 87% and 91% yield, respectively. Furthermore, heteroaromatic carbaldehyde derivatives with a furanyl, thiazolyl, pyridinyl or thiophenyl moiety produced the corresponding propargylamines in 78% to 94% yield. When 1-methylpyrrol-2-carboxaldehyde and 5-fluoro-1-benzothiophene-2-carbaldehyde were used as the starting materials, lower yields were obtained (36% and 51%, respectively). In addition, aliphatic aldehyde heptanal produced the corresponding propargylamine in 88% yield. Subsequently, the reaction of benzaldehyde and morpholine with 4-substituted phenylacetylenes was evaluated. 4-Methylphenylacetylene showed good reactivity and produced the corresponding propargylamine in 93% yield. However, 4-nitrophenylacetylene gave the corresponding product in 47% yield. Furthermore, the reaction could be performed with aliphatic acetylene derivatives (dec-1-yne and oct-1-yne) and produced the corresponding propargylamines in 77% and 83% yield, respectively. Next, secondary amine derivatives were also investigated and produced the corresponding propargylamines in 59% to 96% yield. Dibutylamine was also reacted with phenylacetylene and benzaldehyde to produce the corresponding propargylamine in 71% yield. Importantly, an attempted extension of the reaction to aniline, with phenylacetylene and benzaldehyde, led to the formation of a Schiff base.
 | ||
| Scheme 94 Synthesis of propargylamine derivatives via Cu-containing minerals.98 | ||
Thakur and Khare99 synthesized stereoselective chiral propargylamines 231 via the reaction among heterocyclic amine derivatives 229, aromatic aldehyde derivatives 228, deoxy sugar based alkyne 230 in the presence of CuI, and L-proline (L) in CH3C6H5 under reflux (Scheme 95).
 | ||
| Scheme 95 CuI-catalyzed synthesis of deoxy sugar-based chiral propargylamine derivatives.99 | ||
Initially, complex A was generated via the reaction between CuI and 230. Then, 228 and 229 reacted and produced intermediate aminal B via the Aldol-type condensation. Next, A and B reacted together and produced the complex C. Then, the coordinated alkyne in the complex C was deprotonated together with the removal of H2O. After that, complex D was produced via the ligation of L-proline (L) with complex C, which gave Cu-acetylide with a coordinated iminium ion at the end. Finally, 231 was generated via the addition of Cu-acetylide with a coordinated iminium ion together with the regeneration of the catalyst and L (Scheme 96).
 | ||
| Scheme 96 Possible reaction mechanism for the synthesis of stereoselective chiral propargylamines.99 | ||
According to the results, when ortho/meta-substituted benzaldehyde derivatives reacted with 3,4,6-tri-O-acetyl-2-deoxy propargyl-O-α-D-glucopyranoside/3,4,6-tri-O-acetyl-2-deoxy propargyl-O-α-D-galactopyranoside and piperidine/morpholine/pyrrolidine the desired propargylamine derivatives were produced in 68% to 86% yield. Under similar conditions, 4-hydroxy benzaldehyde compounds decreased the percentage yield of the corresponding products in comparison with other synthesized compounds (30–65%).
Martinez-Amezaga100 synthesized 1-substituted propargylic tertiary amines 235 via the three-component reaction of alkynes 234, amines 233, and aldehydes or 2-oxoacid derivatives 232 in the presence of Cu(OTf)2 in toluene at 120 °C for 6 h (Scheme 97). When the reaction condition for the synthesis of 235 was done under microwave heating for 1 h, a mixture of 235 and 1,3-disubstituted allene A was observed (Scheme 97).
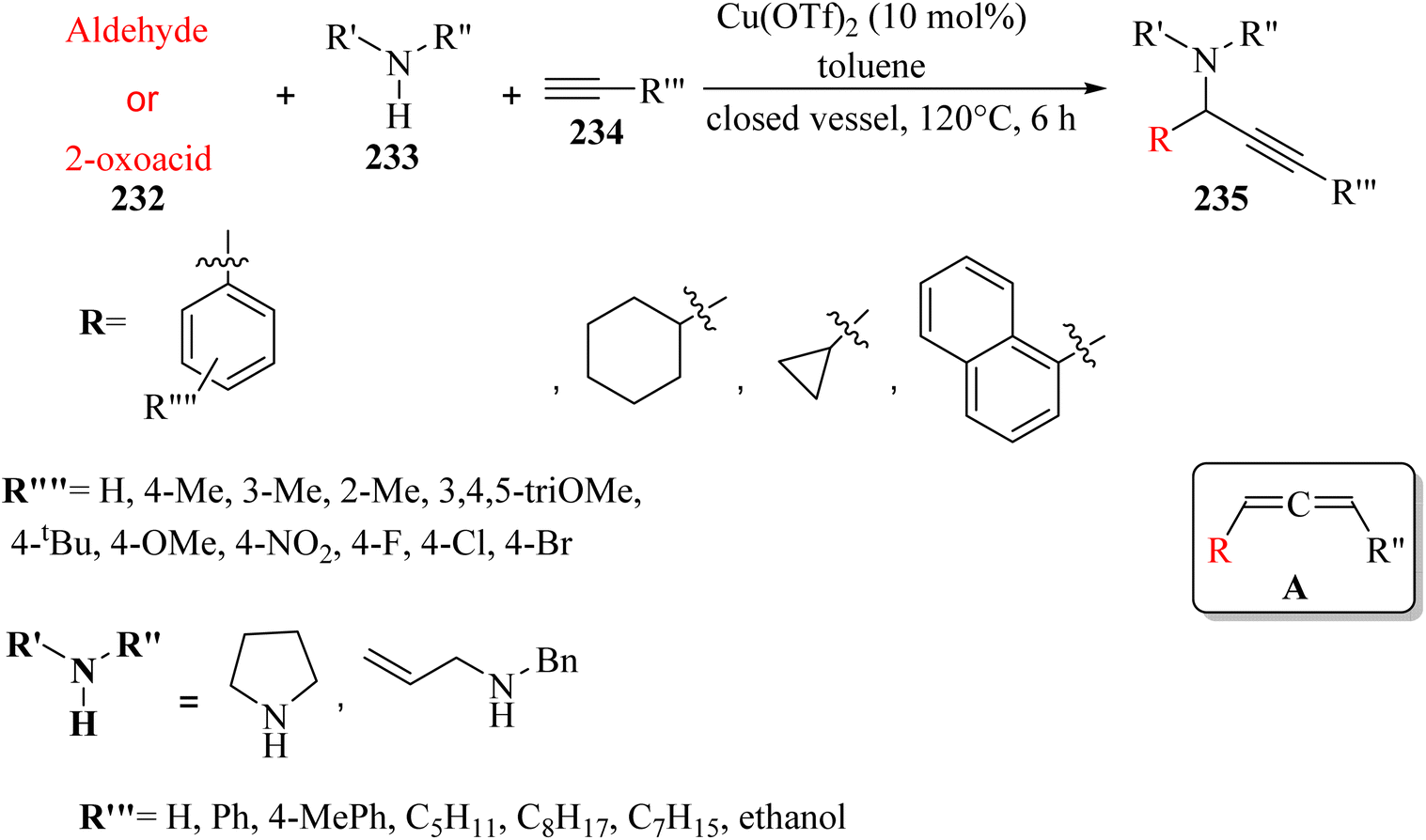 | ||
| Scheme 97 Cu(OTf)2-catalyzed synthesis of propargylamine derivatives.100 | ||
The synthesis mechanism of A could have occurred as follows: initially, 235 was generated, followed by the coordination of Cu to the triple bond. After that, this process caused 1,5-hydride transfer and β-elimination, which led to the synthesis of A (Scheme 98).
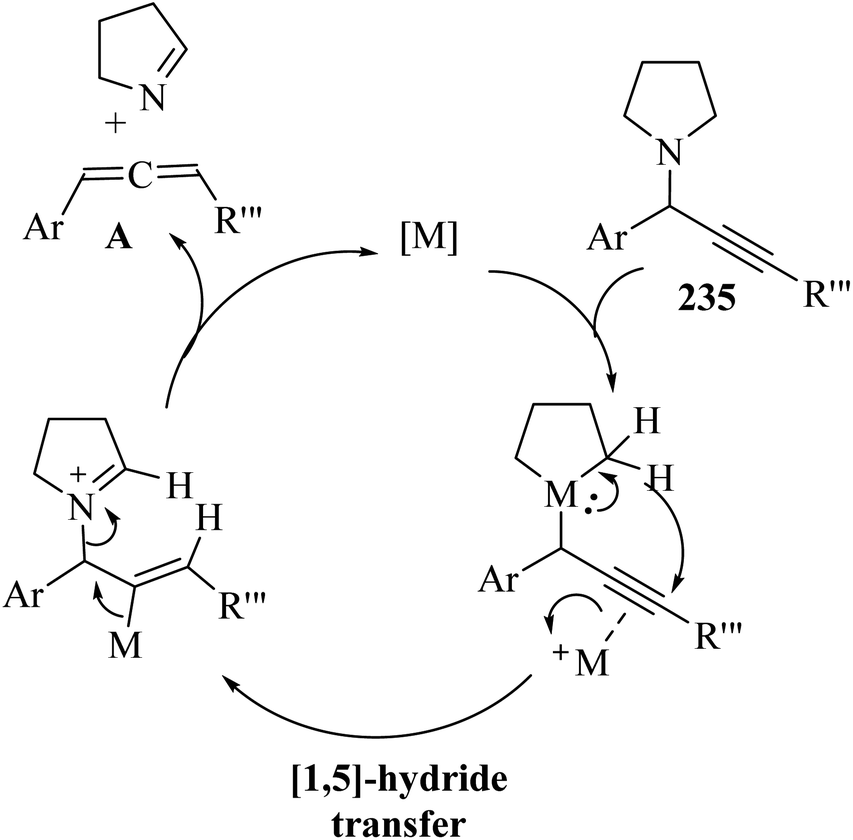 | ||
| Scheme 98 Possible reaction mechanism for the synthesis of 1-substituted propargylic tertiary amines.100 | ||
According to the study, propargylamines were obtained in 9% to 99% yield. In addition, propargylamines were obtained in 50% to 65% yield using phenylglyoxylic acid. Also, ethynyltrimethylsilane as the alkyne component in the above-mentioned method gave the corresponding propargylamine in 49% yield. Also, Sonogashira coupling was performed using tetrakis(triphenylphosphine)palladium(0) as the catalyst and aryl iodide, giving the corresponding product in 79% yield.
Neofotistos and co-workers101 synthesized tetrasubstituted propargylamines 239 via the reaction among amine derivatives 237, alkyne derivatives 238, and ketone derivatives 236 in the presence of MnBr2 as a metal source under neat conditions at 130 for 20 h (Scheme 99). In the case of 236, the mixture of cyclopentanone, piperidine, and phenylacetylene produced the corresponding propargylamine in 67% yield. Cycloheptanone produced the corresponding product in 73% yield. In addition, when cyclohexanone or cyclopentanone was reacted with pyrrolidine produced, the desired propargylamines were obtained in 91% and 75% yield, respectively. In contrast, when cyclohexane-1,2-dione was used, the corresponding propargylamine was not obtained. Linear ketones such as 3-pentanone, 2-pentanone, and 4-decanone decreased the yields (48%, 38%, and 68%, respectively). Also, 4-methoxy-acetophenone and 4-chloro-acetetophenone produced the corresponding propargylamines in 34% and 42% yield, respectively. When benzophenone was used, the corresponding propargylamine was not observed, which may be due to the increased steric hindrance imparted from the two phenyl moieties in the alpha-position of the carbonyl group. In the case of 238, cyclohexanone with pyrrolidine or piperidine was coupled with a variety of terminal alkyne derivatives and the corresponding propargylamines obtained in 57% to 89% yield. 3-Ethynyl-toluene was coupled with piperidine and cyclohexanone and produced the corresponding propargylamine in 81% yield. p-Methoxyphenylacetylene reacted was with piperidine and cyclohexanone and produced the corresponding propargylamine in 87% yield. Interestingly, the reaction and nornicotine, 2-methylbut-3-yn-2-ol, and cyclohexanone gave the corresponding product in 81% yield, which was produced as a single diastereoisomer. In addition, 4-chloro-ethynylbenzene was reacted with cyclohexanone and pyrrolidine and produced the corresponding propargylamine in 77% yield, and when piperidine replaced pyrrolidine, the corresponding product was produced in 64% yield. 2-Br-phenylacetylene was coupled with piperidine and cyclohexanone and produced the corresponding propargylamine in 57% yield. Furthermore, when p-trifluomethyl-phenylacetylene was used, the corresponding product was obtained in low yield. Finally, 1-octyne reacted well with piperidine and cyclohexanone and produced the corresponding propargylamine in 70% yield. In the case of 237, various amine derivatives were reacted with cyclohexanone and phenylacetylene. When morpholine was used, the corresponding product was obtained in 94% yield. Also, benzylamine produced the corresponding propargylamine in 46% yield. In addition, when p-methoxybenzylamine was used, it gave the corresponding propargylamine in 43% yield. Aliphatic amines were also investigated. N-Octylamine reacted with phenylacetylene and cyclohexanone and produced the desired propargylamine in 67% yield. Also, cyclohexylamine gave the corresponding propargylamine in 58% yield. In addition, secondary amine di-n-propylamine produced the desired propargylamine in 61% yield. Finally, the corresponding product of p-methoxy-aniline decomposed when exposed to light.
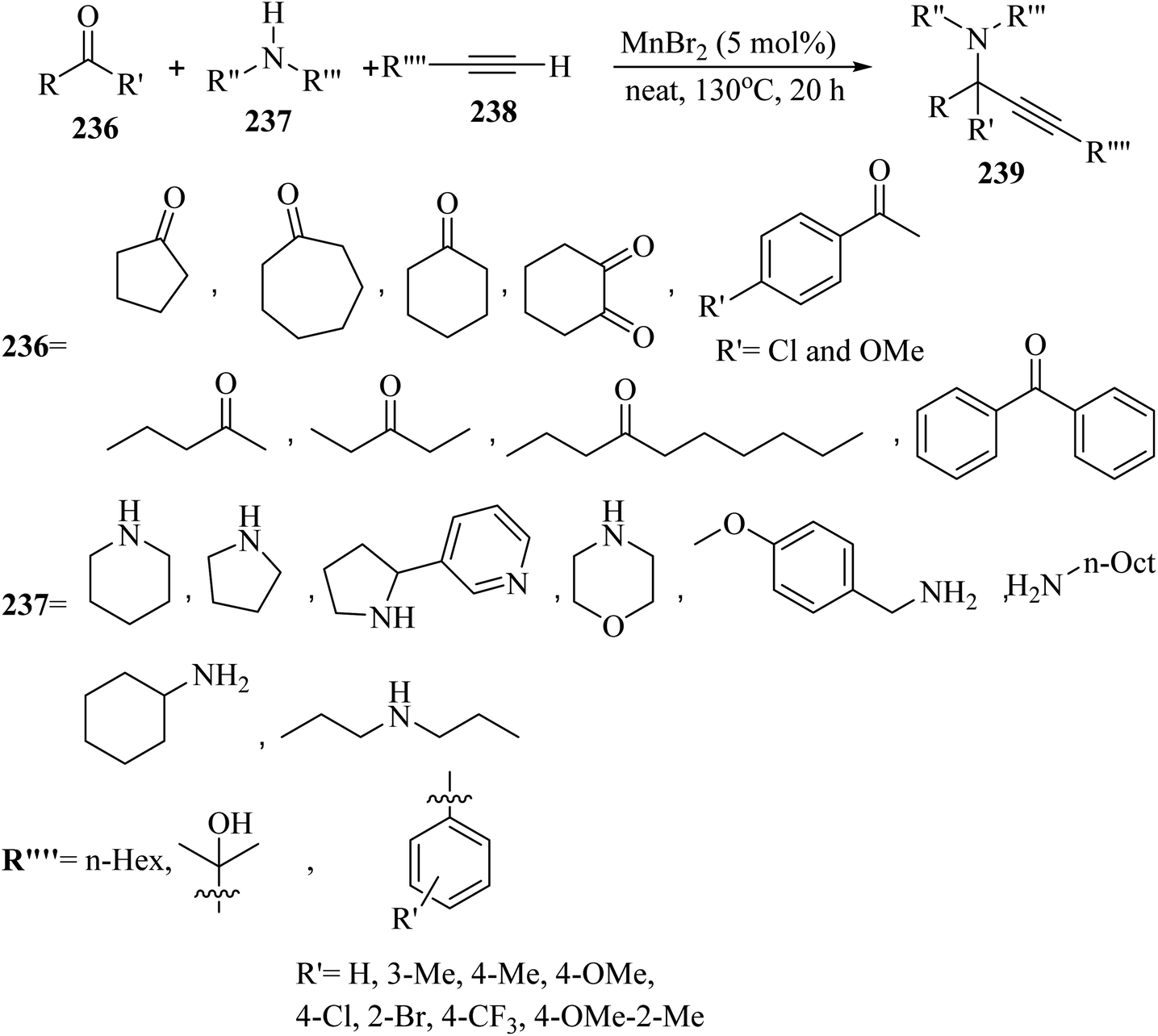 | ||
| Scheme 99 MnBr2-catalyzed synthesis of propargylamine derivatives.101 | ||
Huang et al.102 synthesized propargylamines 243 via the reaction among alkyne derivatives 242, glyoxylic acid monohydrate 241, and secondary amine derivatives 240 in DCE at 95 °C for 12 h (Scheme 100).
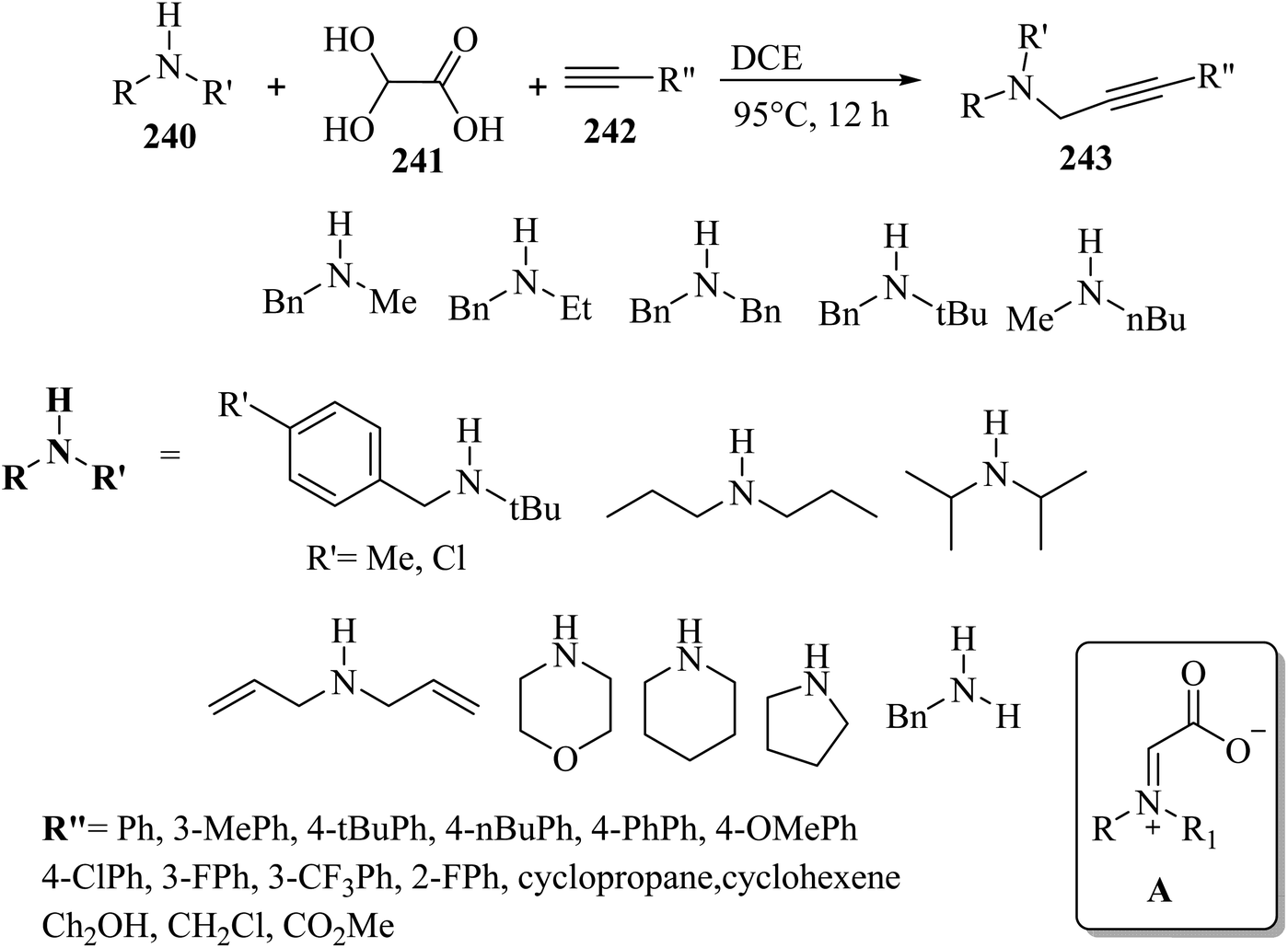 | ||
| Scheme 100 Synthesis of propargylamine derivatives using glyoxylic acid monohydrate.102 | ||
Initially, iminium salt species A was generated via the reaction between 240 and 241 together with H2O removal. Then, the sp carbon of alkyne was activated by the carboxyl group of intermediate A and produced the coupled product C via the subsequent Michael addition. Finally, 243 was produced via the further conversion of C (Scheme 101).
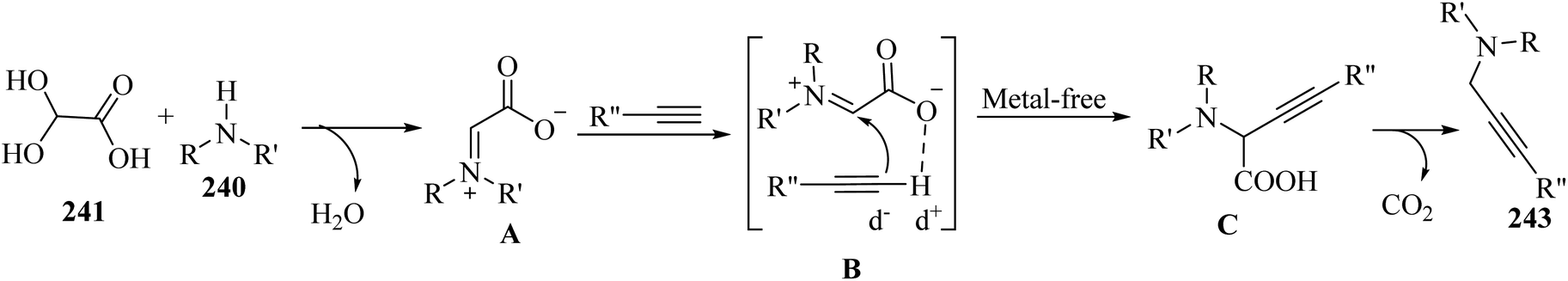 | ||
| Scheme 101 Possible reaction mechanism for the synthesis of propargylamines.102 | ||
In the case of 240 and 242, the yield increased when the steric bulk on the amine substrate increased, which may be due to intermediate A. All N-alkyl substituted benzylamine derivatives were tested and produced the desired propargylamines in 78–93% yield except for the product NH(Bn)2 (21%). N-tert-Butyl benzylamines containing various groups on the phenyl moiety were reacted and produced the corresponding propargylamines in 78% and 93% yield, respectively. Moreover, secondary aliphatic amine derivatives were also reacted and produced the corresponding propargylamines in 54–79% yield. When the primary and cyclic amine derivatives were tested, only morpholine gave a low yield of the corresponding propargylamine (13%). Next, phenylacetylenes bearing electron-neutral groups such as phenyl and electron-donating substituents such as Me, tBu, and OMe produced the corresponding propargylamines in 89%, 72%, 78%, and 82% yield, respectively. Also, phenylacetylene derivatives with electron-withdrawing groups such as CF3, Cl, and F produced the corresponding propargylamines in 65–69% yield. In addition, aliphatic alkyne derivatives were investigated and produced the corresponding propargylamines in 72% to 83% yield. Importantly, methyl propiolate and prop-2-yn-1-ol, 3-chloroprop-1-yne were tested, but no desired products were produced.
A summary of the synthesis of propargylamine derivatives under metal-free conditions, and also the limitations and progress of the reactions are presented in Table 5.
| Reactants | Product | Catalyst | Solvent | Method employed | Reaction time | Yield | Limitations or progress | Ref. |
|---|---|---|---|---|---|---|---|---|
 |
 |
Nano-colloidal silica@APTPOSS | Toluene | Microwave irradiation, 50 °C | 20–25 min | Up to 96% | (1) Efficient catalyst | Safaei-Ghomi et al.103 |
| (2) Recoverability of the catalyst. | ||||||||
| (3) Low catalyst loading | ||||||||
| (4) Short reaction times. | ||||||||
| (5) A simple procedure | ||||||||
| (6) High atom economy | ||||||||
 |
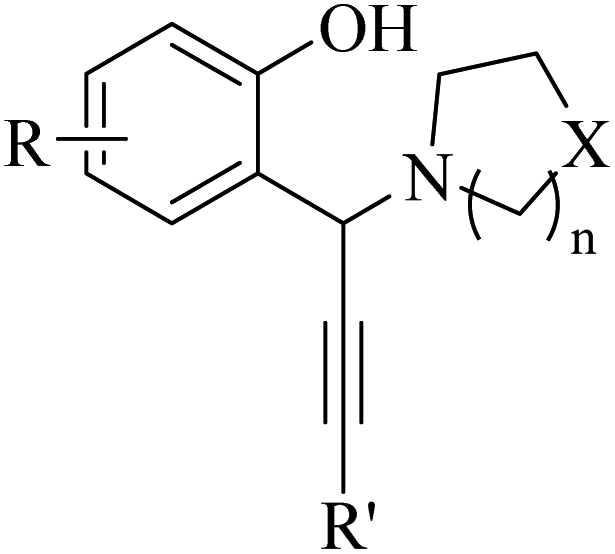 |
— | Solvent-free | 80–90 °C | 4–8 h | 82–90% | (1) Metal-catalyst-free | Ghosh et al.104 |
| (2) Solvent-free conditions | ||||||||
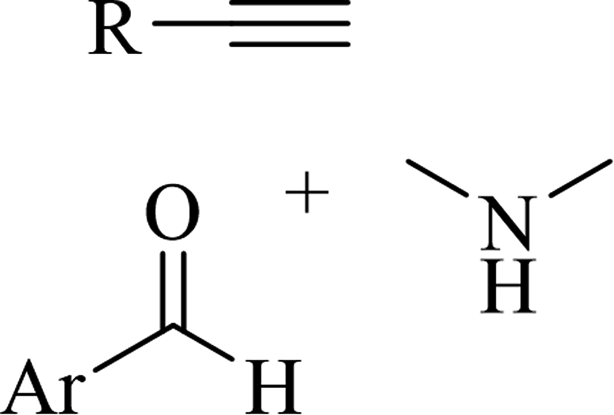 |
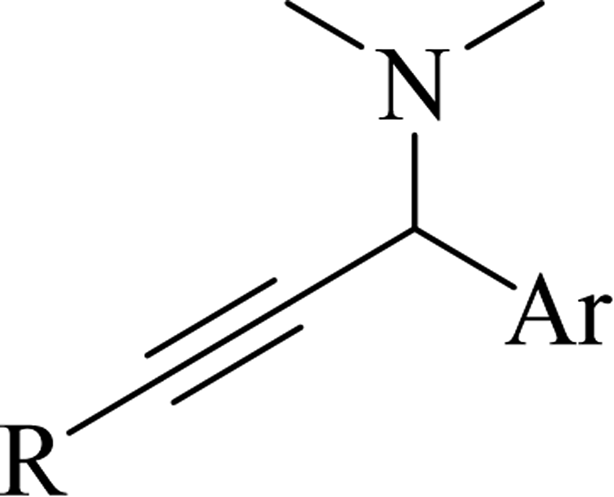 |
— | Magnetized water | MW, 60 °C | 110–160 min | 65–90% | (1) Simplicity | Bakherad et al.105 |
| (2) Low cost | ||||||||
| (3) Short reaction time | ||||||||
| (4) High reaction yield | ||||||||
| (5) Easy work-up | ||||||||
| (6) Absence of hazardous organic solvents | ||||||||
 |
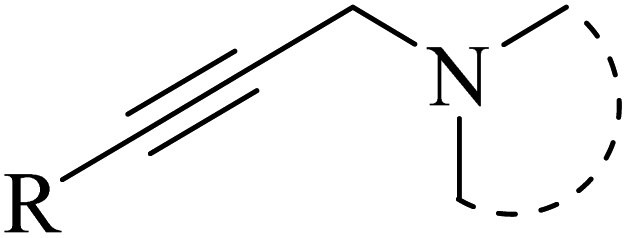 |
— | DCM (play solvent and reactant role) | 70 °C | 12 h | (1) Simplicity of operation | Rawat et al.106 | |
| (2) Mild conditions | ||||||||
| (3) High functional group tolerance |
3.2 Reaction of CO2
Guo et al.107 synthesized alkyl 2-alkynoates 247 (24–96%) via the reaction among terminal alkyne derivatives 245, CO2 244, and alkyl halides 246 in the presence of Ag2WO4 (cat.) and Cs2CO3 (base) in DMF at room temperature for 24 h (Scheme 102).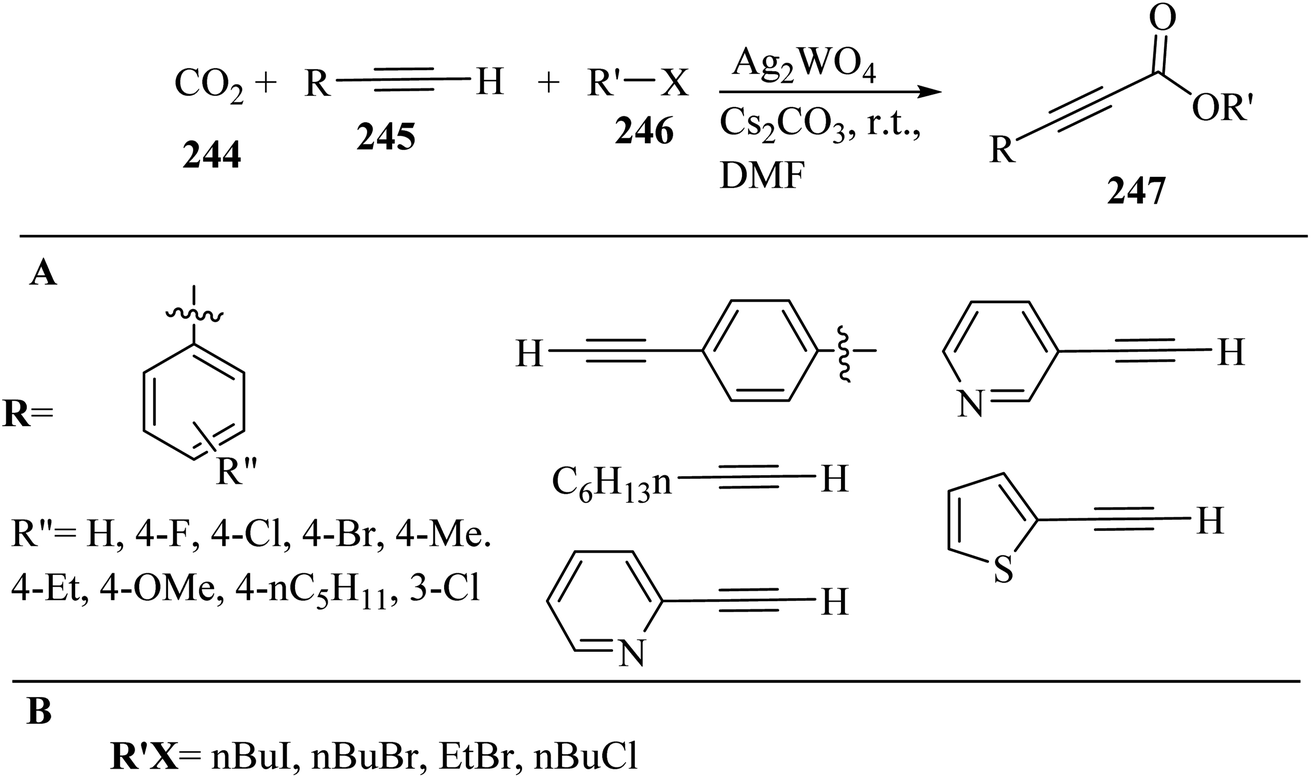 | ||
| Scheme 102 Synthesis of alkyl 2-alkynoates by Ag2WO4: (A) scope of alkynes and (B) scope of alkyl halides.107 | ||
Initially, Ag-acetylide A was generated via the coordination of Ag and 245 in the presence of the base. Then, simultaneously WO42−-CO2 adduct B was obtained via CO2 activation by tungstate. After that, Ag-phenylpropiolate C was produced via the insertion of B into the sp-hybridized Ag–C bond together with the release of WO42−. Finally, 247 was generated from C using n-BuI (Scheme 103). It should be noted that the type of solvent in this transformation played an important role, where DMF showed the best result among the solvents.
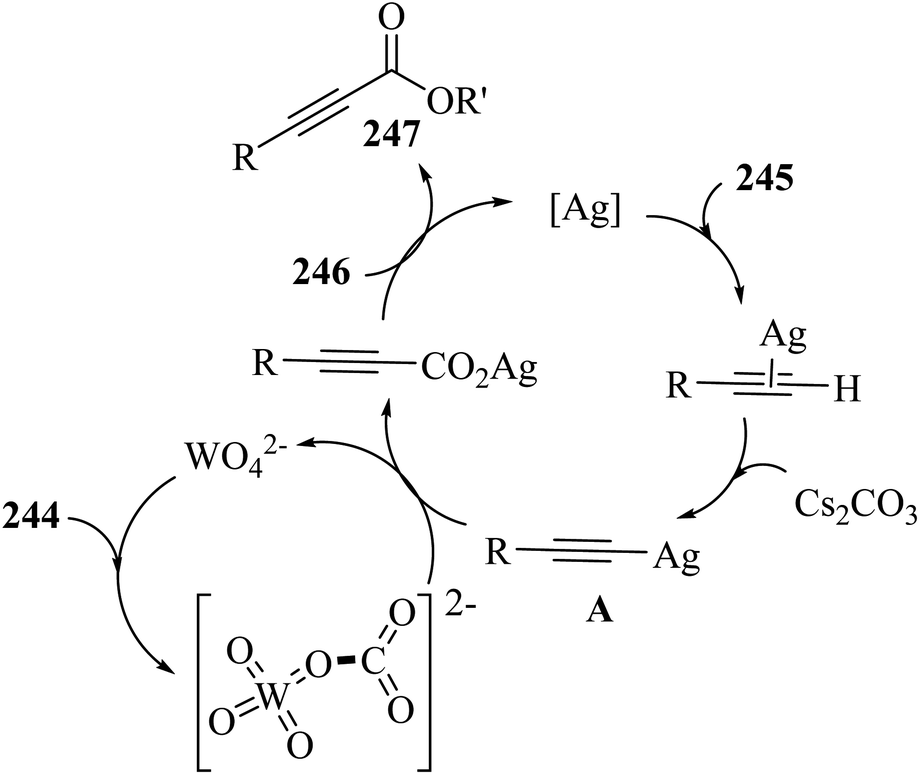 | ||
| Scheme 103 Possible reaction mechanism for the synthesis of alkyl 2-alkynoates.107 | ||
Phenylacetylenes containing electron-withdrawing and electron-donating groups were carboxylated and produced the desired alkyl 2-alkynoate derivatives in 75–91% yield. 1,4-Diethynylbenzene generated the biscarboxylative and monocarboxylative products. Between them, the biscarboxylative product was the major product and the monocarboxylative product was the minor product (64% and 24% yield, respectively). In this transformation, heteroaromatic acetylenes and aliphatic alkyne derivatives also were tolerated and produced the desired alkyl 2-alkynoate derivatives. This method has advantages such as use of a halogen-free catalyst and simple preparation of the catalyst.
Xie et al.108 synthesized alkynoates 251 via the reaction among terminal alkyne derivatives 249, CO2 248, and n-BuI 250 in the presence of L (cat.) and Cs2CO3 in DMF at room temperature for 12 h (Scheme 104).
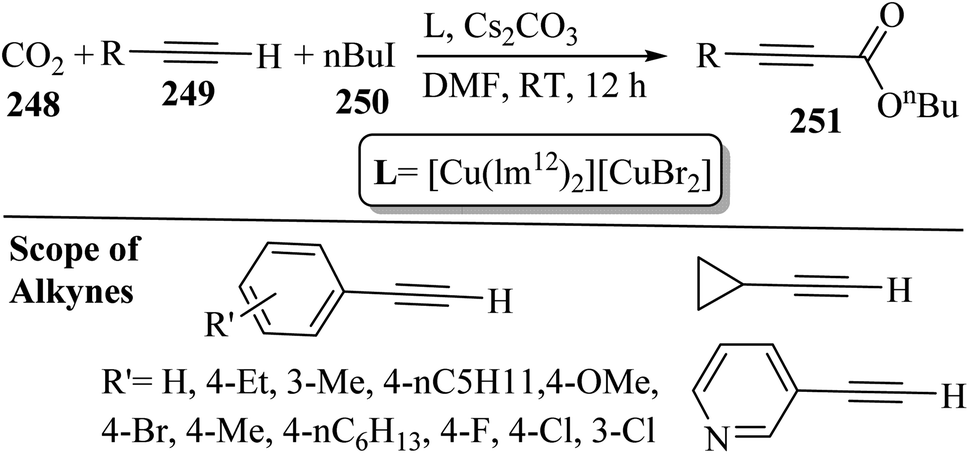 | ||
| Scheme 104 Synthesis of alkynoates.108 | ||
Initially, Cu-acetylide B was generated from activated alkyne A, in the presence of Cs2CO3. Then, propynoate intermediate C was produced from B via the insertion of CO2 into the sp-hybridized Cu–C. Finally, 251 was obtained from intermediate C in the presence of 250. According to this report, the ligand causes the reaction to proceed under mild conditions, and also advances the efficiency of the catalyst (Scheme 105).
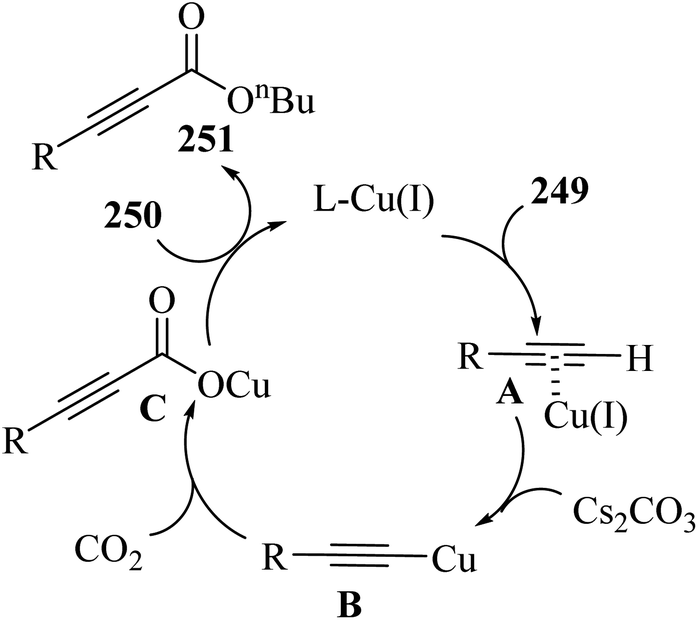 | ||
| Scheme 105 Possible reaction mechanism for the synthesis of alkynoates.108 | ||
Phenylacetylene derivatives 249 containing electro-rich groups at the para or meta positions generated the desired 251 in 88–96% yield. Also, the substrate with Br produced the desired 251 in 85% yield. In addition, aliphatic alkynes generated the desired 251 in up to 97% yield. Generally, the yield of the reaction decreased when using a strong electron-withdrawing group.
Zhang et al.109 synthesized 2-alkynoate derivatives 255 via the reaction among CO2 252, nBuI 254, and terminal alkyne derivatives 253 in the presence of Ag–NHC and Cs2CO3 in DMF at 40 °C for 48 h (Scheme 106).
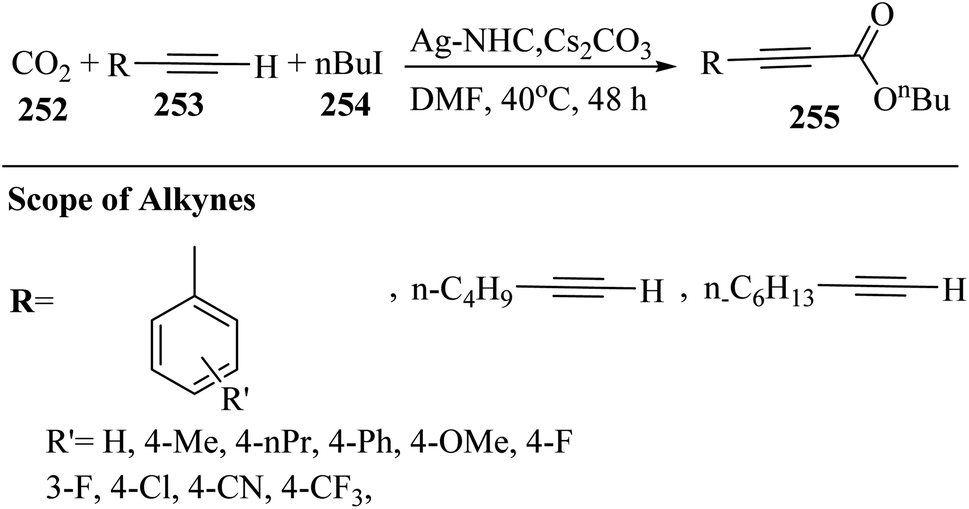 | ||
| Scheme 106 Synthesis of 2-alkynoate derivatives.109 | ||
Initially, 253 coordinated with Ag–NHC and produced intermediate A. Then, intermediate B was produced from intermediate A in the presence of Cs2CO3. After that, Ag-acetylide C was obtained from intermediate B together with the elimination of CsHCO3. Next, Ag-propiolate intermediate D was produced via the insertion of CO2 into the Ag–C bond of intermediate C. Finally, 255 was generated via the reaction between nBuI and intermediate D together with the regeneration the catalyst (Scheme 107).
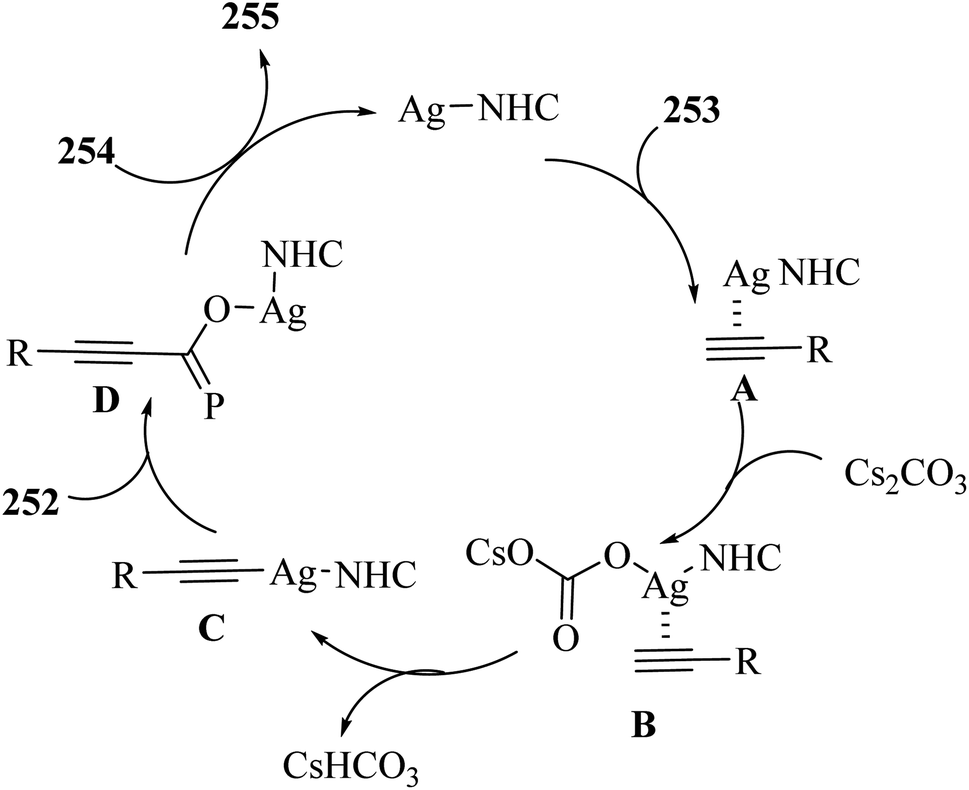 | ||
| Scheme 107 Possible reaction mechanism for the synthesis of 2-alkynoate derivatives.109 | ||
Derivatives of 253 containing electron-donating groups gave the desired 2-alkynoate derivatives 255 in 77–94% yield. In addition, halo derivatives of 253 generated the desired 2-alkynoate derivatives 255 in 82–86% yield. Derivatives of 253 containing a CN or CF3 group produced the desired 2-alkynoate derivatives 255 in 84% and 81% yield, respectively. Furthermore, linear alkyl terminal alkyne derivatives 253 were tolerated and produced the desired 2-alkynoate derivatives 255 in 80–82% yield. It should be noted that the selectivity of the reaction was good due to the production of only one isolated product.
Guo et al.110 synthesized benzyl 2-alkynoate derivatives 259 via the reaction among CO2 256, aryl/alkyl terminal alkyne derivatives 257, and benzyl halide derivatives 258 in the presence of Cs2CO3 and AgI in CH3CN under ligand-free conditions at 40 °C for 48 h (Scheme 108).
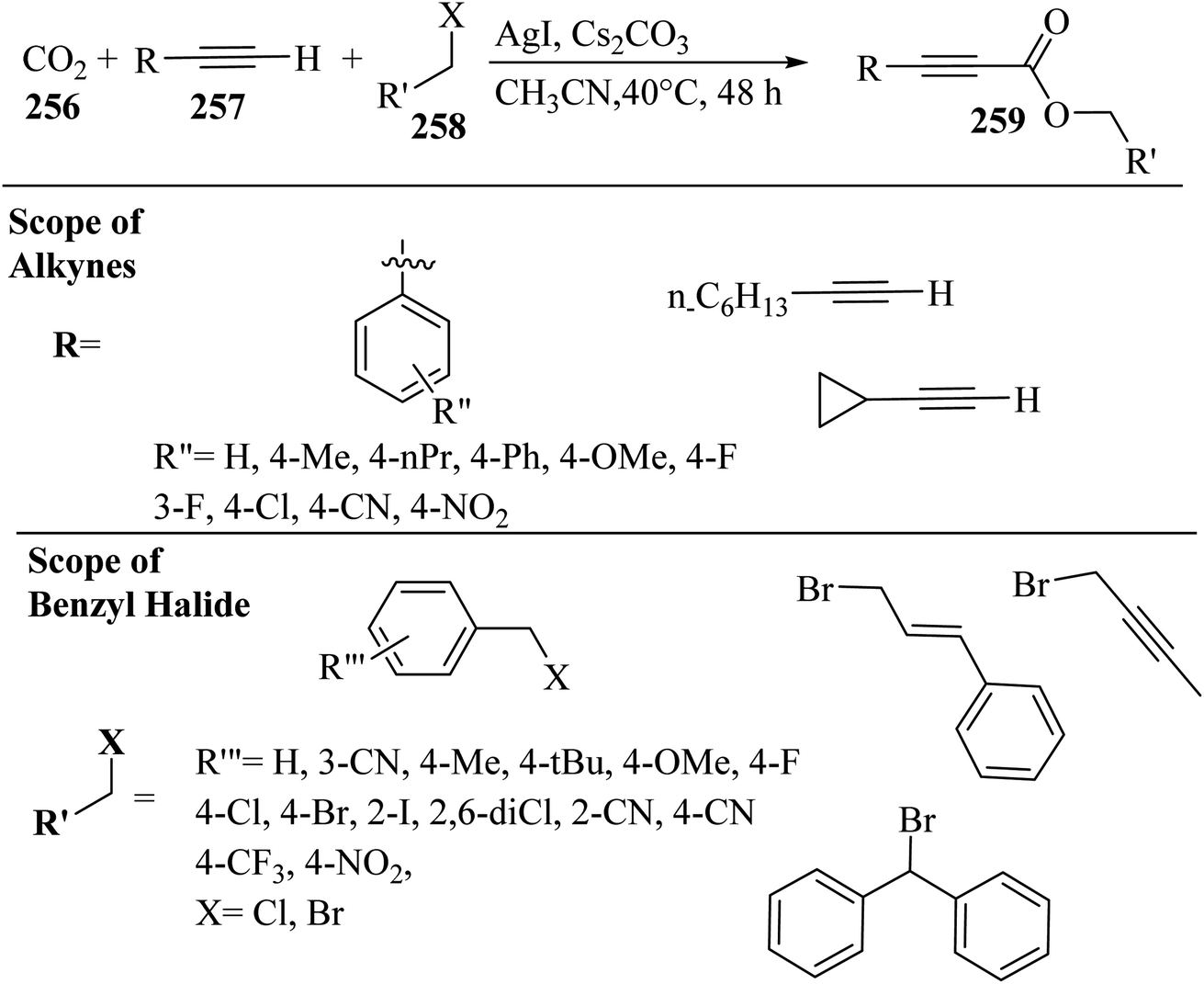 | ||
| Scheme 108 Synthesis of benzyl 2-alkynoate derivatives.110 | ||
Initially, intermediate K was obtained via the coordination of 257 and Ag(I). Then, Ag(I)-acetylide L was generated via the deprotonation of K by Cs2CO3 together with the release of CsHCO3. After that, Ag-propiolate intermediate M was produced via the insertion of CO2 in the C–Ag bond. Finally, 259 was obtained via the reaction between M and 258 together with the release of Ag(I) (Scheme 109).
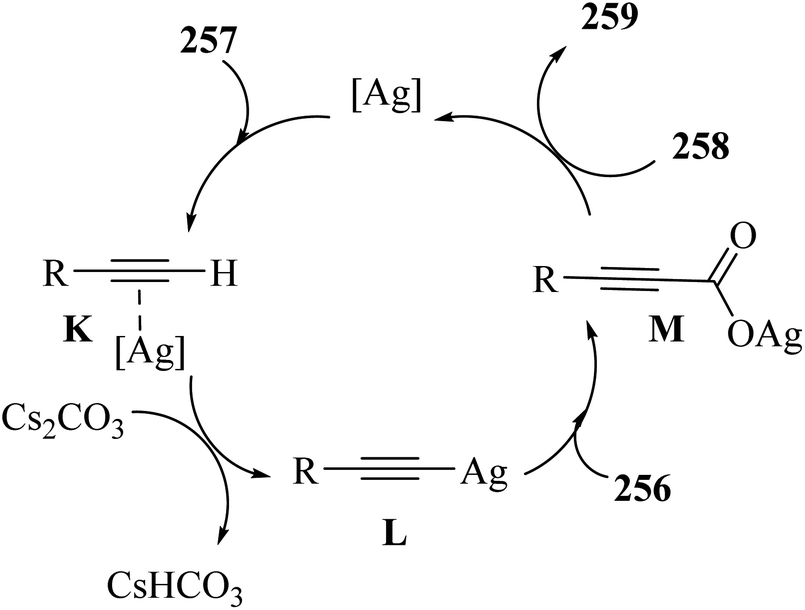 | ||
| Scheme 109 Possible reaction mechanism for the synthesis of benzyl 2-alkynoate derivatives.110 | ||
The electronic effect of the groups on the phenyl ring of aryl alkyne derivatives were demonstrated to have an important effect on the reactivity. In the case of 257, derivatives of 257 containing electron-donating groups produced the desired 259 in 66–73% yield. Also, derivatives of 257 containing F and Cl groups were produced the desired 259 in 65–75% yield. In contrast, aryl alkyne derivatives containing CN or NO2 group produced the desired 259 in 45% and 33% yield, respectively. Also, aliphatic alkyne derivatives 257 generated desired 2-alkynoate derivatives 259 in 38–50% yield. The low product yields by aliphatic alkynes may be due to the low activity of the activation of their sp C–H bond. In the case of the benzyl halide derivatives 258, aryl chlorides or bromides 258 containing electron-donating groups generated the desired 2-alkynoate derivatives 259 in 71–85% yield. Also, halo derivatives of 258 gave the desired 2-alkynoate derivatives 259 in 62–91% yield. Aryl halide derivatives containing CF3, NO2, and CN gave the desired 2-alkynoate derivatives 259 in 63–87% yield. In addition, allylic/propargylic halide derivatives produced the desired 2-alkynoate derivatives 259 in 42–47% yield. It should be noted that secondary benzyl halide-(bromomethylene)dibenzene was tested and produced the desired 2-alkynoate 259 in 8% yield.
Papastavrou et al.111 synthesized propargylic esters 263 via the reaction among organochlorides 262, CO2 260, and terminal alkyne derivatives 261 in the presence of K2CO3 and NHC in DMF at 60 °C for 20 h (Scheme 110). In the case of 262, 2-chloroacetate esters generated the desired 263 in 25–28% yield. Organochlorides containing a Cl atom on the carbonyl carbon did not give the desired 263. In contrast, allylic chlorides showed good activity and produced the desired 263 in 42–97%. The low yield of chloropropene and chlorobutene may be related to their low boiling points, which were lower than the reaction temperature. Similarly, the yield of crotyl chloride was high, which has a boiling point of 85 °C. In addition, benzyl chloride derivatives produced the desired ester 263 in 59–84% yields. In contrast, picolyl chloride did not generate the desired ester 263, which may be due to the presence of a pyridine moiety in its structure. It should be noted that organobromide derivatives did not give the desired esters 263. Furthermore, benzyl chlorides containing electron-poor and electron-rich groups produced the desired esters 263 in 59–84% yield. In the case of 261, alkyl-substituted terminal alkyne derivatives and a hydroxyl-bearing alkyne did not generate the desired esters 263. Phenyl acetylene bearing p-NO2 and p-CF3 produced the desired esters in trace and 91% yield, respectively. The result showed that electron-poor groups could produce the desired esters 263 but the NO2 group was not very consistent. Phenylacetylene derivatives containing an Me or OMe group on the aromatic ring, and also containing no aromatic substituents produced the desired esters 263 in 64–97% yield.
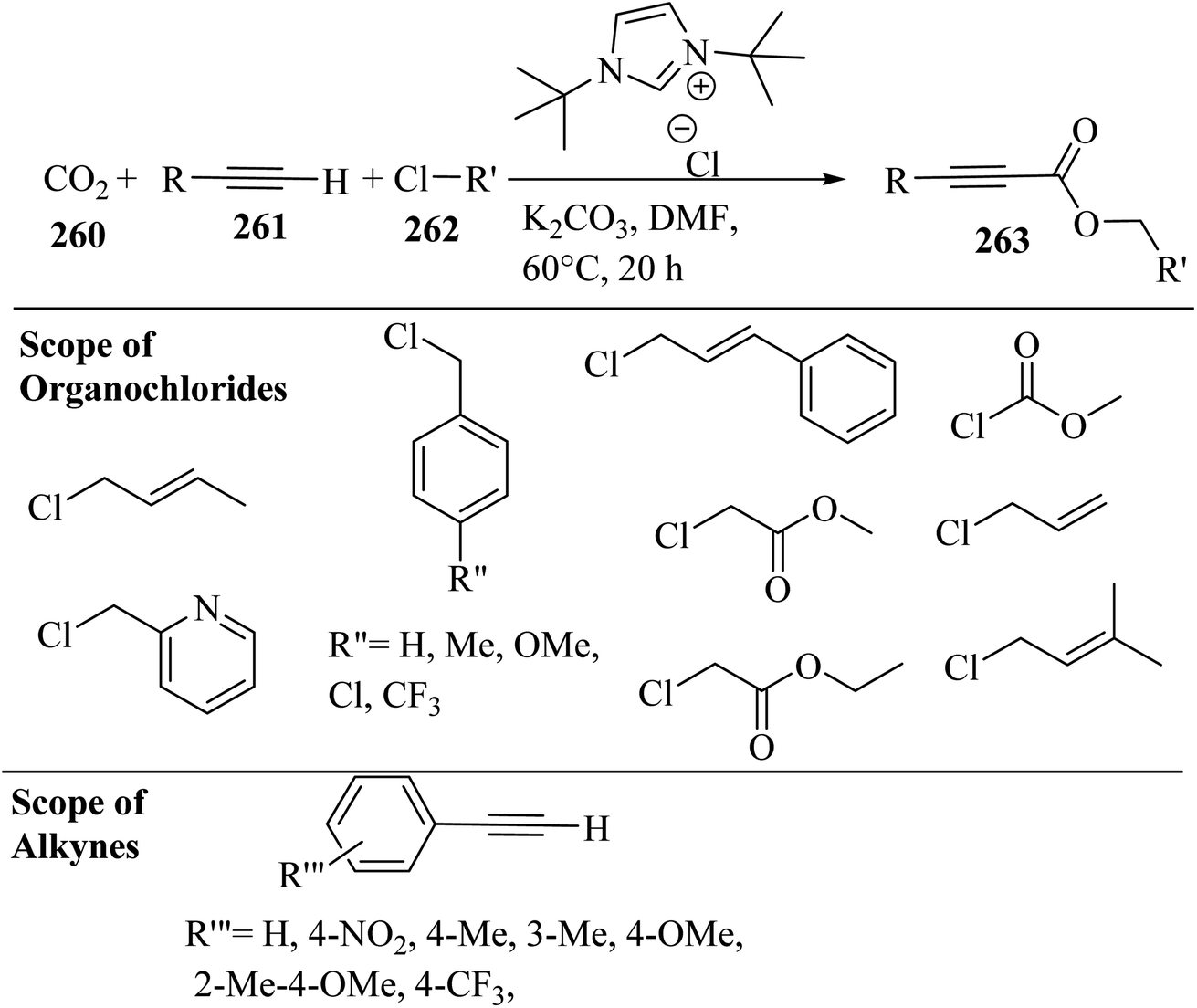 | ||
| Scheme 110 Synthesis of propargylic esters.111 | ||
Zhang et al.112 synthesized 4-pentenyl 2-alkynoate derivatives 267 and 5-hexenyl 2-alkynoate derivatives 268 via the reaction among CO2 264, terminal alkyne derivatives 265, and 5-bromopentene or 6-bromohexene derivatives 266 in the presence of AgI and Cs2CO3 in DMF at 60 °C for 24 h (Scheme 111). The selectivity of the reaction was high and no direct coupling by-products were observed. Alkyne derivatives containing electron-withdrawing and -donating groups on the phenyl ring produced the desired carboxylative products (267 and 268) in 69–91% yield. Alkyl-substituted terminal alkynes in comparison to aryl-substituted substrates showed low yields (59–66%). These results may be due to the higher stability of the aryl-substituted Ag(I) acetylide with respect to the alkyl-substituted Ag(I) acetylide intermediate.
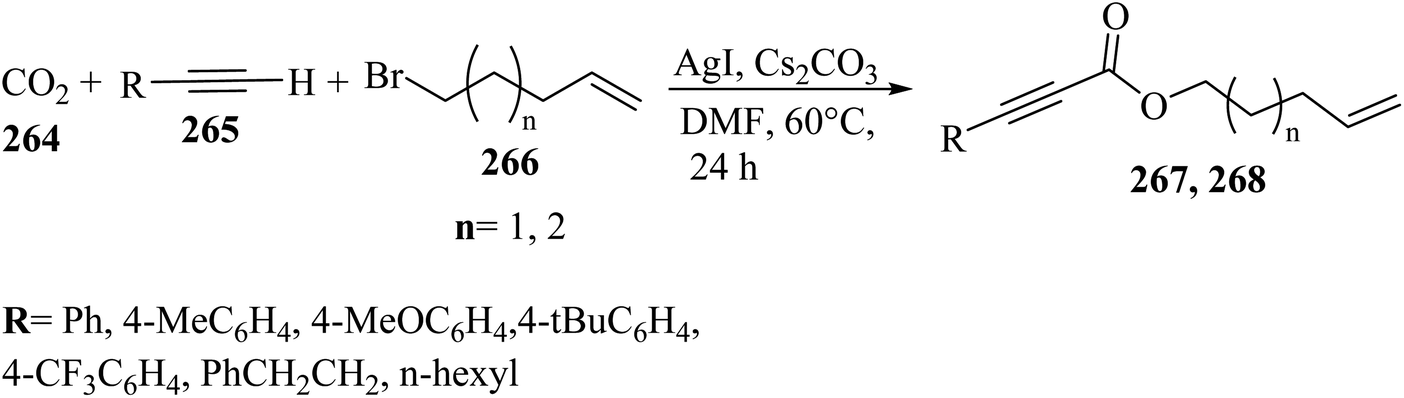 | ||
| Scheme 111 Synthesis of 4-pentenyl 2-alkynoate derivatives.112 | ||
3.3 Reaction of CO
Yang et al.113 synthesized a-chirality ketones 272 via the reaction among terminal alkyne derivatives (271 and 271′), CO 269, and enallene derivatives (270 and 270′) in the presence of BQ, Pd(OAc)2, and chiral phosphoric acid (CPA) X in PhCl at 0 °C for 24 h (Scheme 112).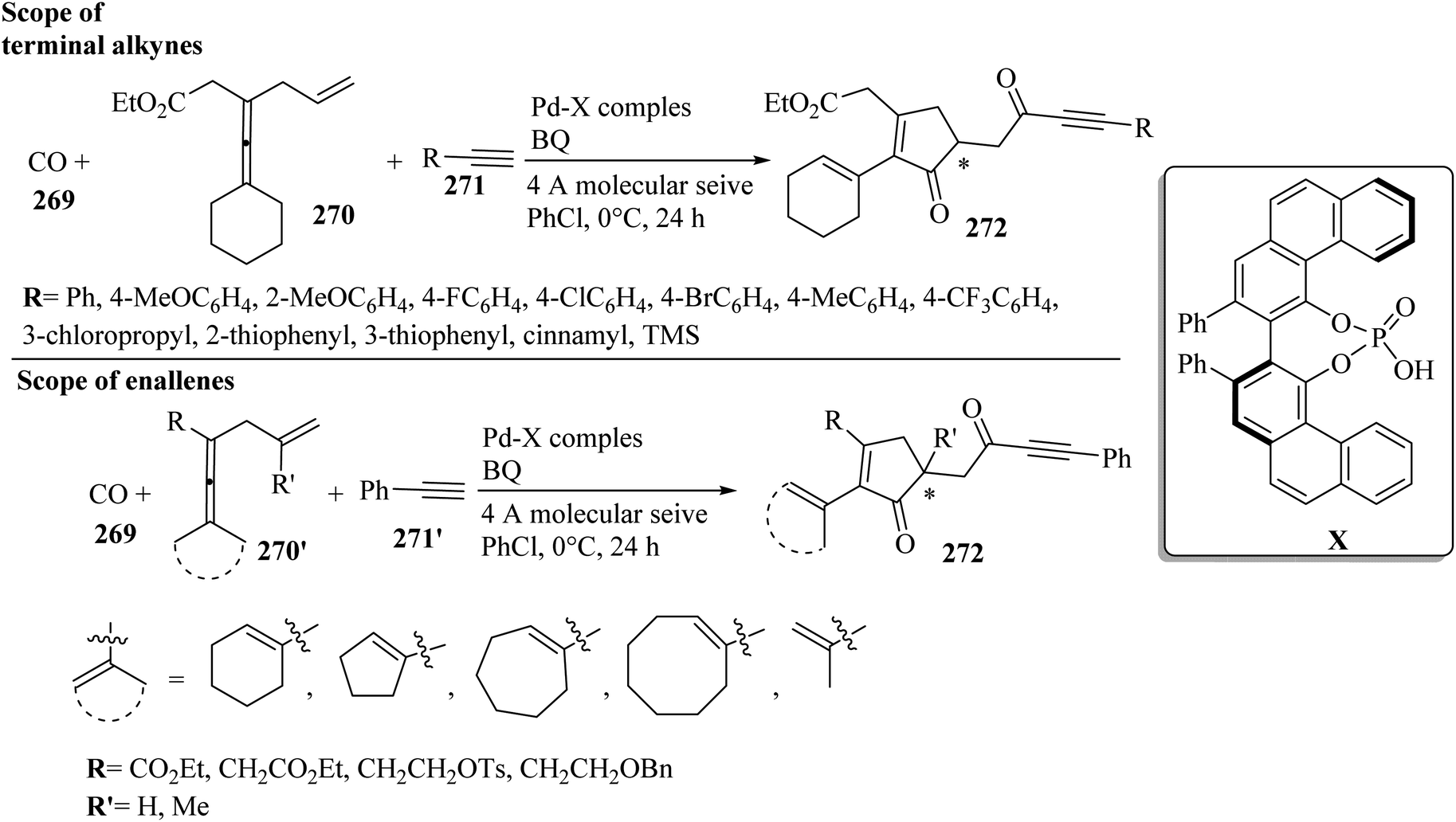 | ||
| Scheme 112 Synthesis of a-chirality ketones 272.113 | ||
Initially, intermediate A was generated via the coordination of 270′ to PdII. Then, carbonyl PdII intermediate B was obtained via the insertion of CO and the subsequent attack of 270 on intermediate A. After that, olefin inserted into the C–Pd bond via enantioselective migration and produced intermediate C, which caused chirality at the alpha-position of the ketone. Lastly, 272 was generated via the carbonylative alkynylation of intermediate C together with the release of Pd0. To close the catalytic cycle, BQ reoxidized the released Pd0 to PdII (Scheme 113).
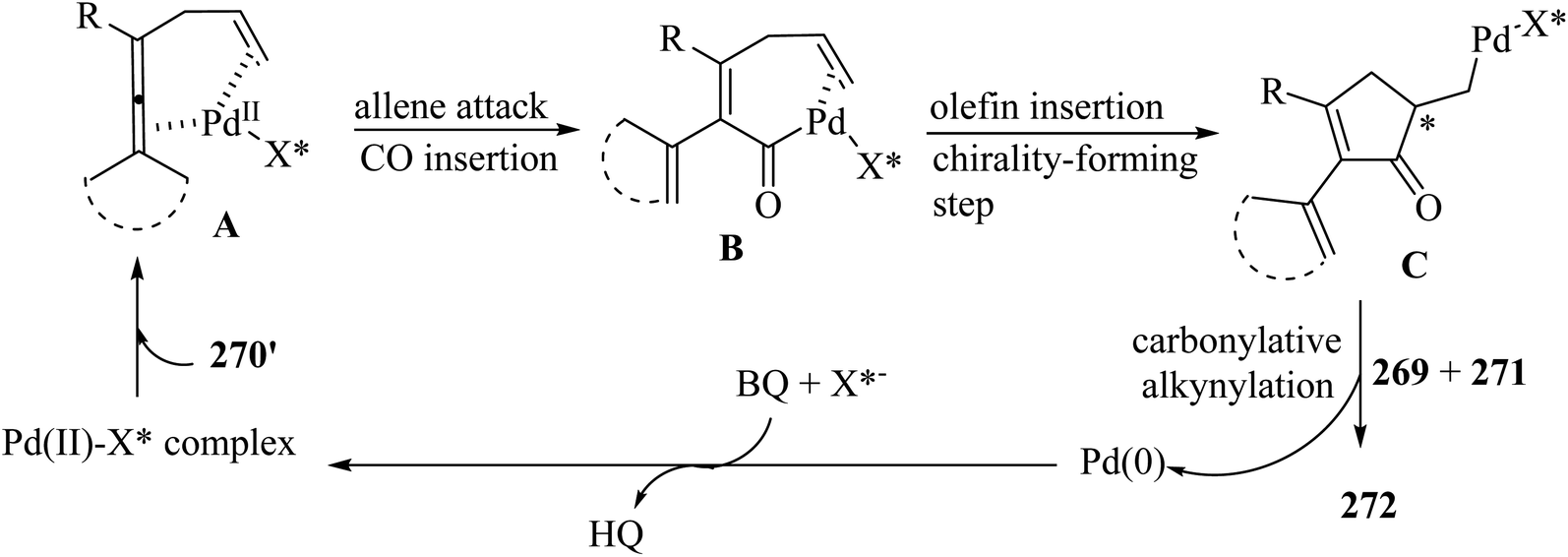 | ||
| Scheme 113 Possible reaction mechanism for the synthesis of a-chirality ketones 272.113 | ||
In the case of 271, phenylacetylenes containing electron-donating and -withdrawing groups generated the desired 272 in 48–81% yield and high er (up to 95.5:4.5). Aliphatic acetylene derivatives, as well as heteroaryl acetylene derivatives, were tolerated and produced the desired 272 in 64–73% yield. Importantly, alkyne containing TMS generated the desired 270 in 5% yield and poor er (56.5:43.5), and the reaction time for the preparation of this desired compound was 60 h. Poor enantioselectivity of the desired 272 was obtained when slow alkyne-quenching of the key Pd species existed. In the case of 270′, when two Me were replaced with a terminal cycloalkyl group, the desired 272 was generated with a slight decrease in the enantiomeric ratio. The size of the ring was tested and the results showed that all rings could generate the desired 272 but had slightly lower er values compared to the six-membered rings. When an internal Me group on the olefin moiety was used, the desired 272 was generated in a lower er. Substituents on the allene moiety had a remarkable effect on the reactivity, as well as enantioselectivity. 2,3-Allenoate had lower er and yield with respect to 3,4-allenoate. Functional groups of sulfonyl ester and ether were suitable in the reaction and produced the desired 272 in 66% and 70% yield (93.5:6.5 and 95:5 er), respectively.
Zhu et al.114 synthesized ynone derivatives 276 via the reaction among CO 273, terminal alkyne derivatives 275, and enallene derivatives 274 in the presence of Pd(TFA)2 and BQ in CH3Ph at room temperature (Scheme 114).
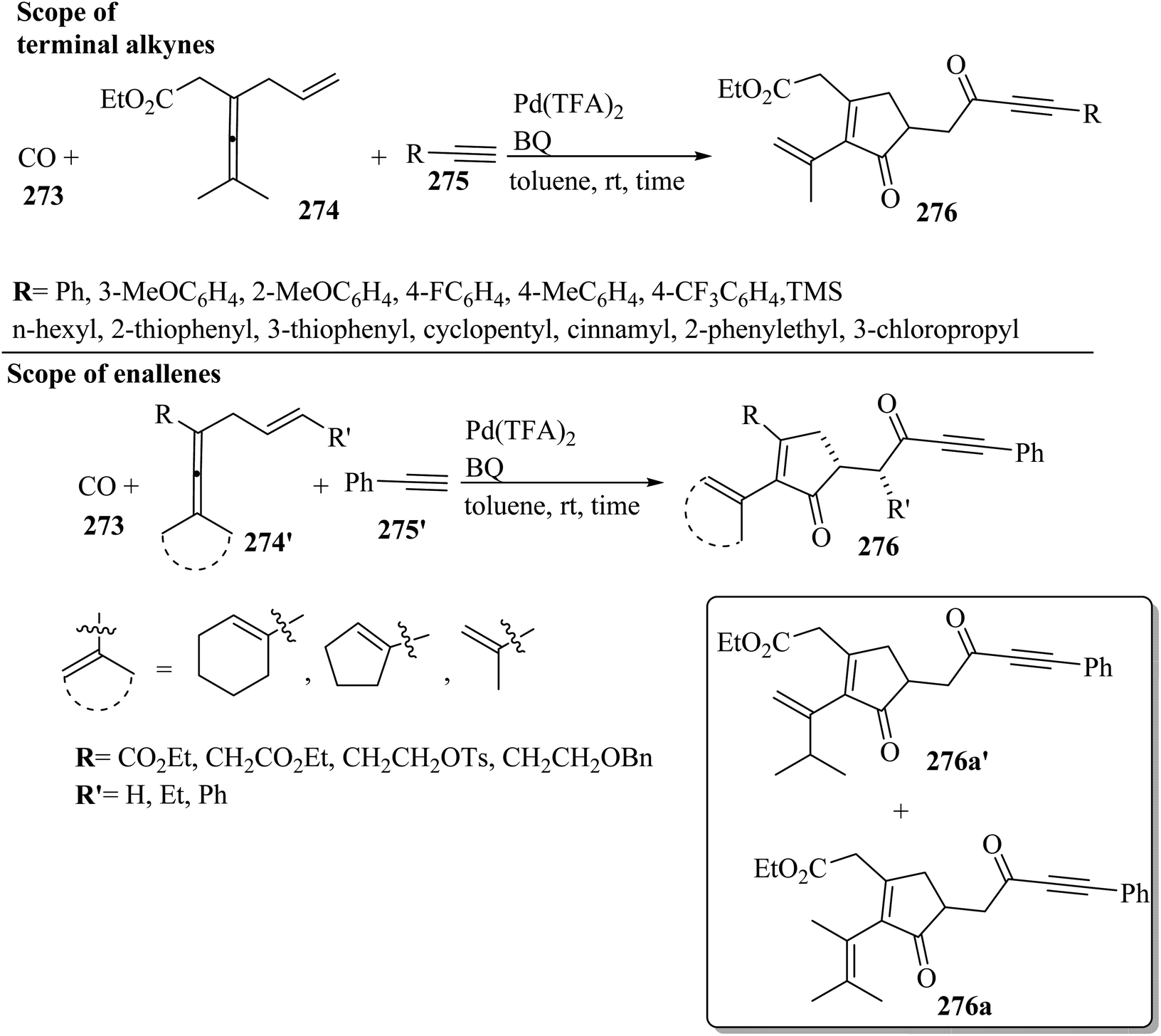 | ||
| Scheme 114 Synthesis of ynone derivatives 276.114 | ||
Initially, intermediate A was produced via the coordination of 247′ to Pd(II). Then, intermediate B was generated from intermediate A via allene attack, where the additional coordination of the olefin to Pd(II) was vital. After that, intermediate C was obtained via the selective insertion of CO in the C–Pd bond of intermediate B. In this step, olefin insertion could occur, which led to the formation of intermediate F. Next, intermediate C produced intermediate E via olefin and carbon monoxide insertion reactions. Lastly, intermediate G was generated via the reaction between 275′ and Pd in intermediate E, which after reductive elimination, gave the desired product together with the release of Pd(0). The released product was reoxidized to Pd(II) by p-benzoquinone (Scheme 115).
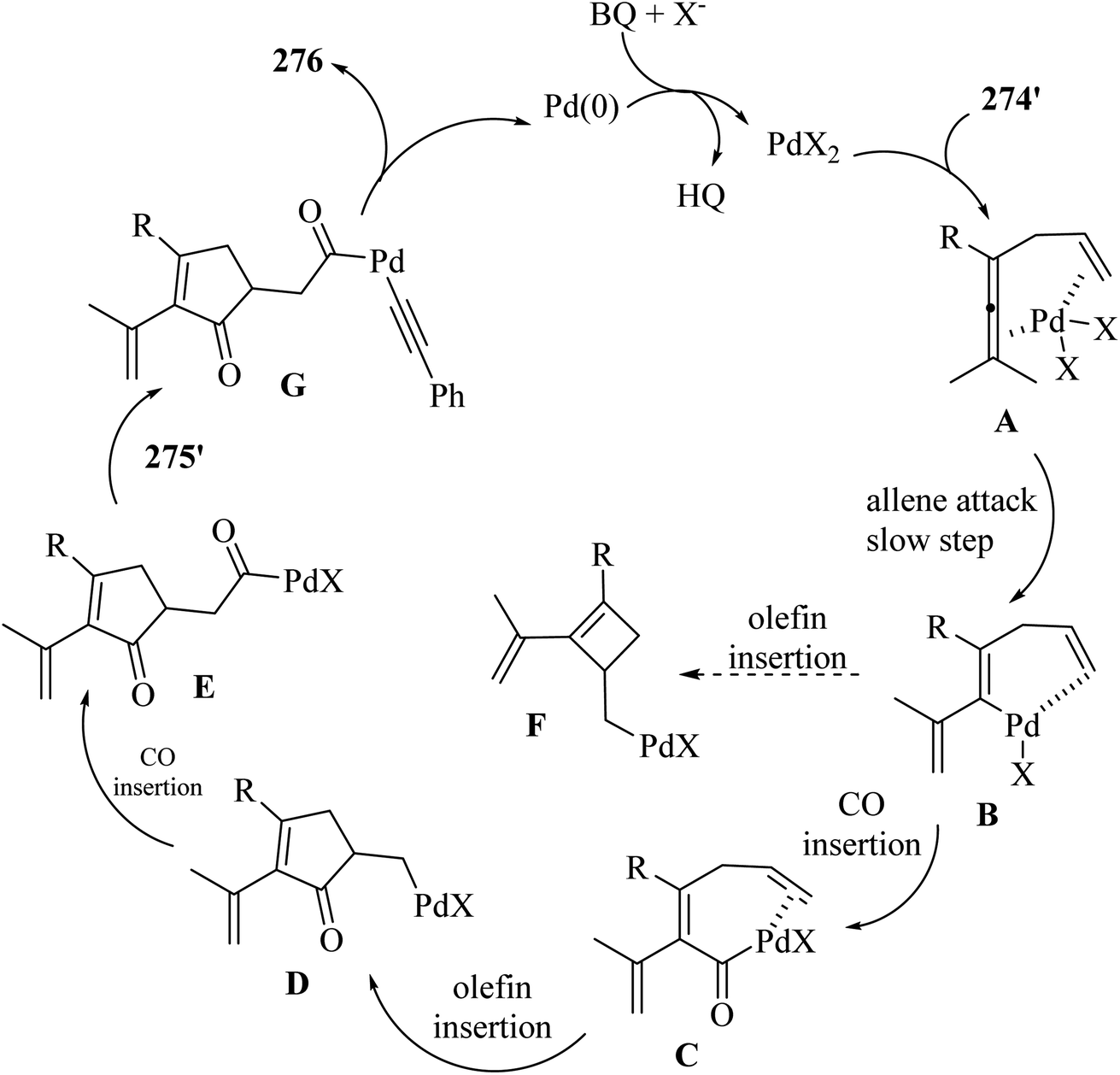 | ||
| Scheme 115 Possible reaction mechanism for the synthesis of ynone derivatives 276.114 | ||
In the case of 275, phenylacetylene derivatives containing electron-withdrawing and -donating groups generated the desired 276 in 70–81% yield. Also, heteroaryl acetylenes produced the desired 276 in 66–75% yield. It should be noted that aliphatic cyclic and acyclic terminal alkynes were tolerated well and produced the desired 276 in 74–83% yield. In addition, functional groups of TMS and Cl were tested and produced the desired 276 in 73% and 72% yield, respectively. In the case of 274′, cyclohexylidene enallene produced the desired 276 in 73% yield but cyclopentylidene enallene gave the desired 276 in a lower yield (58% yield). Tosyl and benzyl 3,4-dienol were tested and produced the desired 276 in 76% and 75% yield, respectively. It should be noted that the use of Ph or Et substituent rather than R′′ substituent on the olefin had a great effect on the reaction (41% and 40% yield, respectively). Also, the dissymmetric allene containing i-Pr and Me gave the desired 276a and 276a′ in 40% and 30% yield, respectively. It should be noted that selective allenic C–H cleavage gave 276a and 276a′.
Qiu et al.115 synthesized spiro[3.4]octene derivatives 280 and spiro[4.4]nonene derivatives 281 via the reaction among dienallene derivatives 278, CO 277, and alkyne derivatives 279 in the presence of Pd(TFA)2 and BQ. For the preparation of 280, CH3CN was used as the solvent at 80 °C, whereas DCE was used as the solvent for the preparation of 281 at room temperature (Scheme 116).
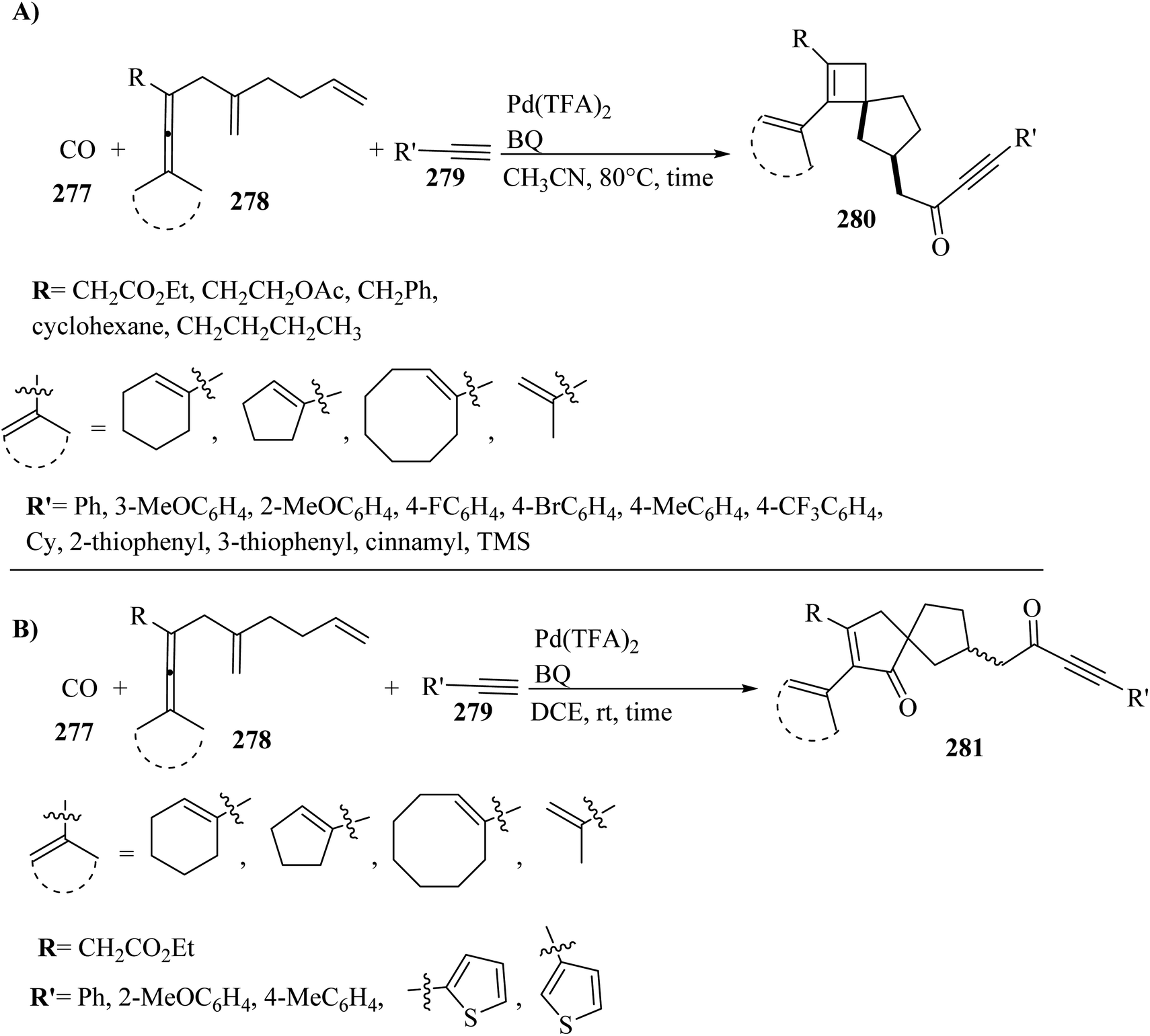 | ||
| Scheme 116 Synthesis of (A) spiro[3.4]octene derivatives 280 and (B) spiro[4.4]nonene derivatives 281.115 | ||
Initially, intermediate A was generated from the reaction between Pd(TFA)2 and 278 via allene attack involving allenic C–H bond cleavage. Then, intermediate B was produced from intermediate A via olefin insertion. After that, intermediate D was obtained from intermediate C via olefin and CO insertions. Lastly, intermediate E was generated via the reaction between intermediate D and 279, which gave the desired 280 after reductive elimination. Alternatively, intermediate F was obtained from intermediate A via carbonylation. Then, intermediate I was generated from intermediate G and H via olefin–olefin–CO insertion. Finally, the desired 281 was generated from the reaction between intermediate I and 279 via intermediate J (Scheme 117). It should be noted that CH3CN choose the pathway that was favorable for the production of intermediate B instead of intermediate F. This solvent suppressed CO coordination, and hence insertion.
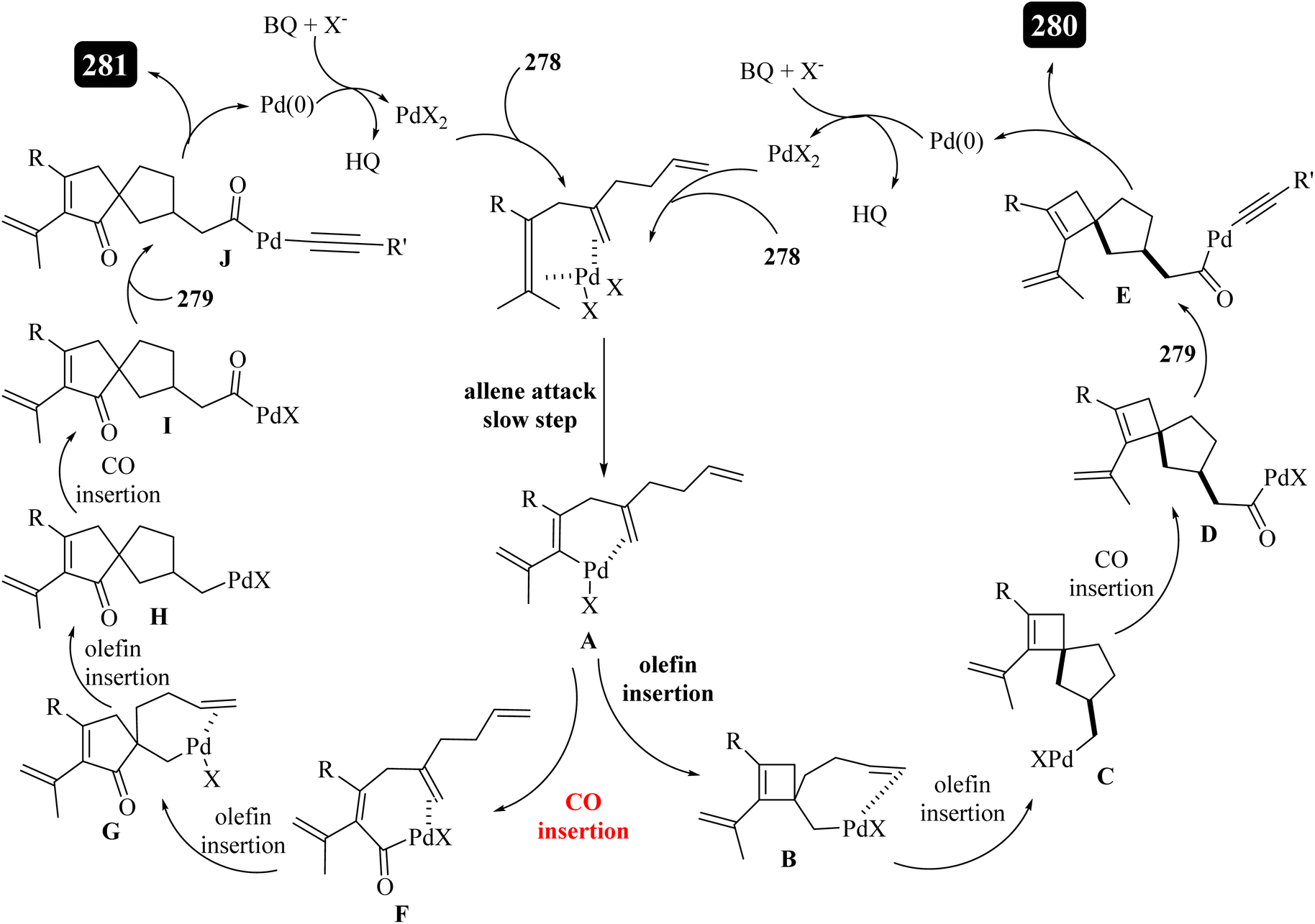 | ||
| Scheme 117 Possible reaction mechanism for the synthesis of spiro[3.4]octene derivatives 280 and spiro[4.4]nonene derivatives 281.115 | ||
In the case of 279 for the formation of 280, arylalkyne derivatives with electron-donating and -withdrawing groups generated the desired 280 in 64–79% yield. Also, heteroaryl acetylene derivatives were tolerated well and produced the desired 280 in 71–83% yield. Aliphatic terminal alkyne derivatives were tested and produced the desired 280 in 66–70% yield. Importantly, the TMS group reacted well and generated the desired 280 in 79% yield, which could be utilized for further functionalization after desilylation. In the case of 278 for the formation of 280, cyclooctylidene, cyclopentylidene, and cyclohexylidene produced the desired 280 in 80%, 71%, and 66% yield, respectively. In addition, the unsymmetric allene containing methyl and phenyl groups generated the desired 280 in 62% yield. It should be noted that when using n-butyl, cyclohexyl, and benzyl groups instead of R, the desired 280 was obtained in 55%, 60%, and 63% yield, respectively. In the case of the formation of 281, substrates of Me, cyclopentylidene, cyclohexylidene, or cyclooctylidene on the dienallene moiety were tolerated well and generated the desired 281 in 90%, 82%, 93%, and 89% yield, respectively. Arylacetylenes containing o-MeO and p-Me groups produced the desired product in 80% and 87% yield, respectively. Furthermore, heteroaryl acetylene derivatives were tested and the desired 281 obtained in 72–78% yield.
Volla and Backvall116 reported an oxidative carbocyclization–carbonylation–alkynylation reaction for the synthesis of ynone derivatives 285 via the reaction among alkyne derivatives 283, CO 282, and enallene derivatives 284 in the presence of Pd(TFA)2 and 1,4-BQ in ClCH2CH2Cl at room temperature for 12 h (Scheme 118).
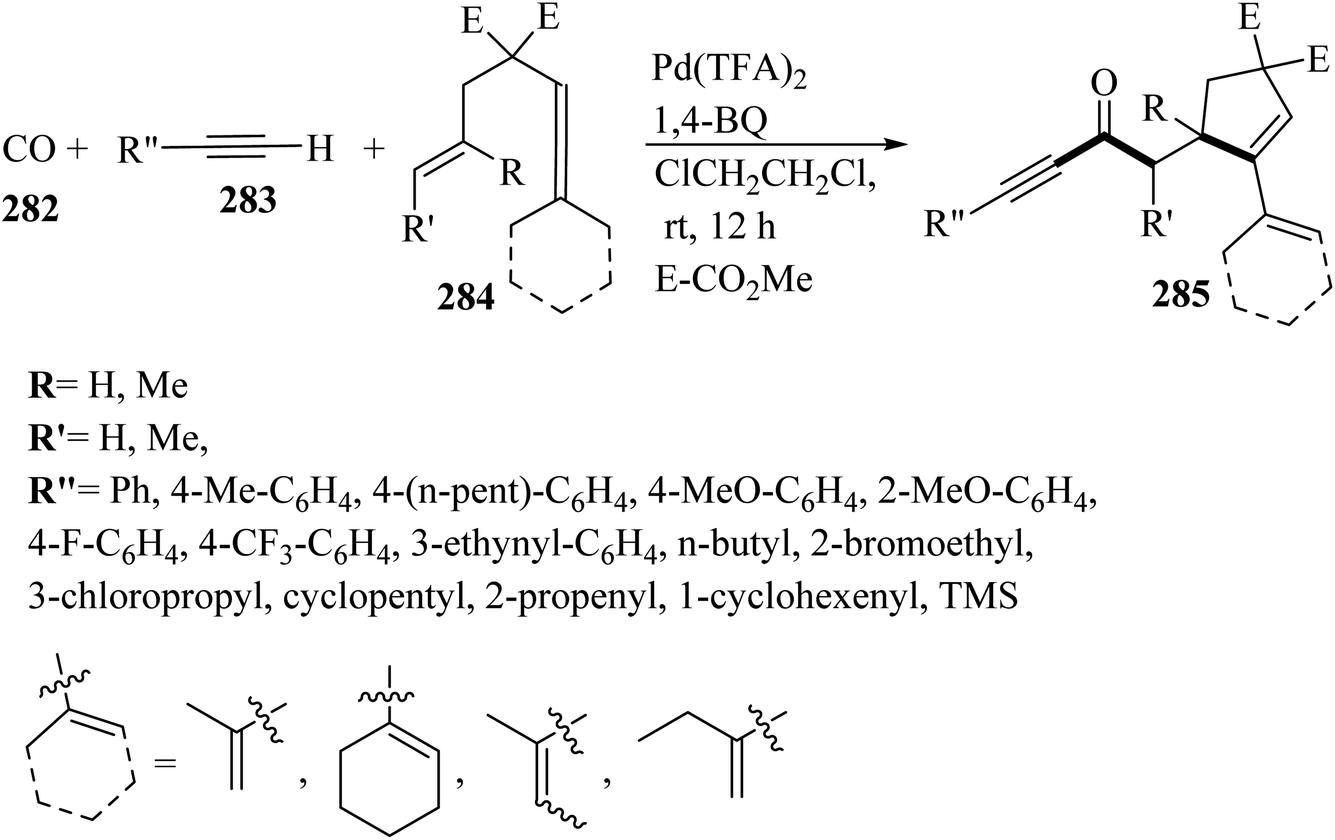 | ||
| Scheme 118 Synthesis of ynone derivatives 285.116 | ||
Initially, π-complex A was generated with alkene and allene units by Pd. Then, intermediate B was obtained from intermediate A via the attack of the allene moiety on Pd(II), which was followed by the production of intermediate C via carbocyclization. After that, acylpalladium(II) species D was obtained from intermediate C via the insertion of CO in the Pd–C bond of intermediate C. Next, intermediate E was generated via the subsequent transfer of an alkynyl moiety. Finally, 285 was produced from E via reductive elimination together with the release of Pd(0), and Pd(0) was converted to Pd(II) via oxidization (Scheme 119).
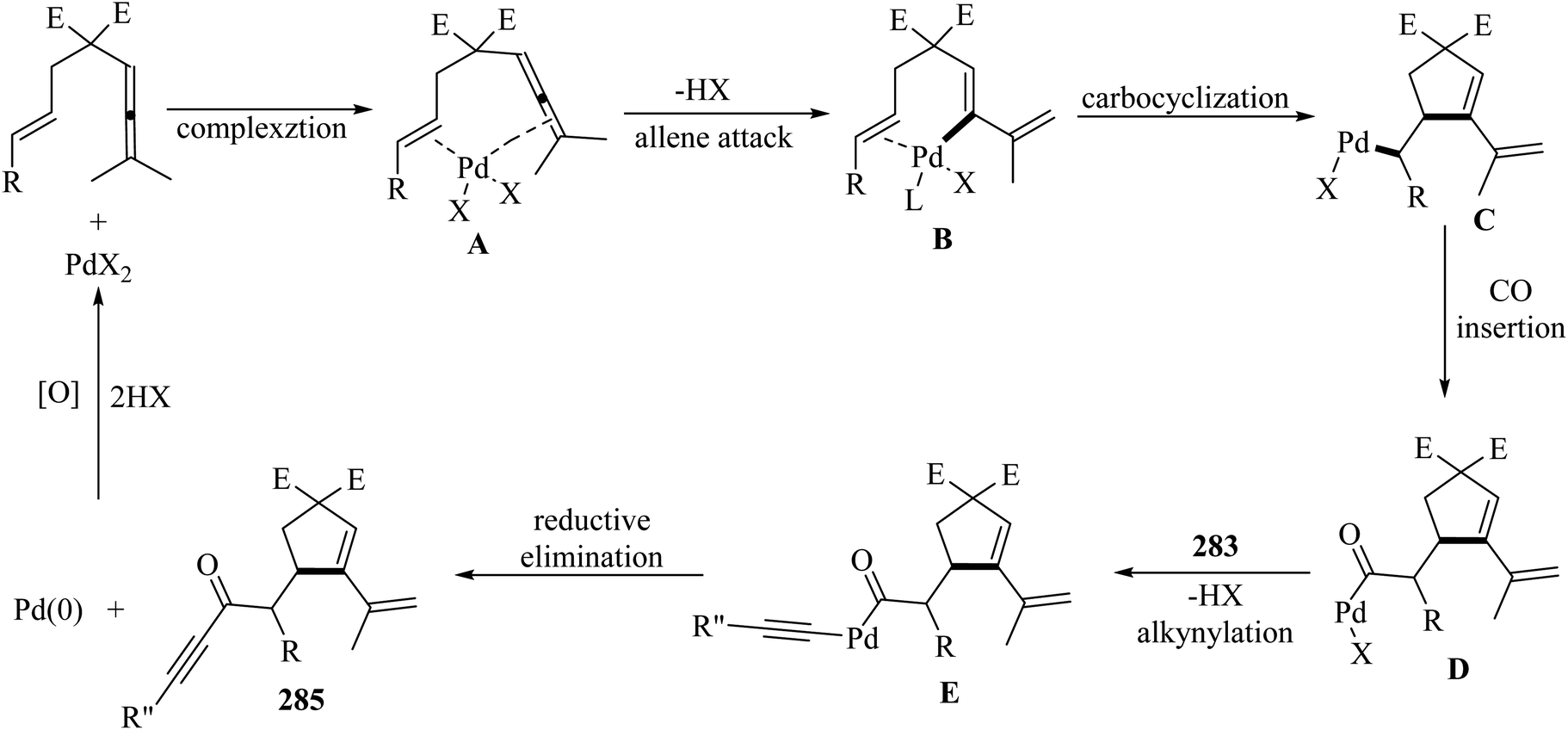 | ||
| Scheme 119 Possible reaction mechanism for the synthesis of ynone derivatives 285.116 | ||
In the case of 283, phenylacetylene derivatives possessing electron-donating and -withdrawing groups produced the desired 285 in 72–87% yield. It should be noted that steric and electronic factors on the aromatic ring did not show sensitivity. 1,3-Diethynylbenzene, which has two terminal alkyne units, produced monoalkynylation 285 without reaction of the second alkyne group during the reaction. Also, aliphatic cyclic and acyclic terminal alkyne derivatives generated the desired 285 in 54–94% yield. Importantly, TMS was tolerated well in the reaction and produced the desired TMS-product 285 in 81% yield, which could be utilized for further functionalization after desilylation. In the case of 284, when the dimethyl groups of the allene unit were replaced with methylethyl or pentamethylene units, the temperature was increased to 40 °C, and the desired 285 was produced in 88% and 76% yield, respectively. In addition, enallene, which contains a methyl group on the olefin unit, gave the desired 285 with an all-carbon quaternary center via the cascade carbocyclization–carbonylation–alkynylation in 73% yield at 80 °C. It should be noted that 1,6-enallene X did not generate the desired 258 even at high temperatures.
Chandrasekhar et al.117 synthesized 3-aryl-4-(arylethynyl)-2H-chromen-2-one derivatives 289 via the reaction among CO 286, terminal alkyne derivatives 287, and 2-iodoaryl 2-arylacetate derivatives 288 in the presence of Pd(PPh3)2(Cl)2 and Et3N in toluene at 80 °C for 3–10 h (Scheme 120A). According to the results, various substituents were evaluated on the three aryl rings, and all groups such as Cl, NO2, Br, Me, OMe, and tBu were tolerated well in the reaction and produced the desired 289 in 70–95% yield (Scheme 120a). In addition, trimethylsilylacetylene 291 generated the desired 292 in 78% yield, and the reaction time for the preparation was increased to 12 h (Scheme 120B). For more evaluations, 2-iodo-3,5-dimethylphenyl 2-phenylacetate 293 was investigated under the standard reaction conditions and produced just Sonogashira coupling product 295 in 8% yield after 12 h and did not produce the desired 289 (Scheme 120C). Also, 1-iodo-2-naphthyl acetate 296 under the standard reaction conditions generated just Sonogashira coupling product 297 in 98% after 4 h and did not produce the desired 289 or other products (Scheme 120D). A summary of the recent reaction of alkynes with CO and also limitations and progress of the reactions have been shown in Table 6.
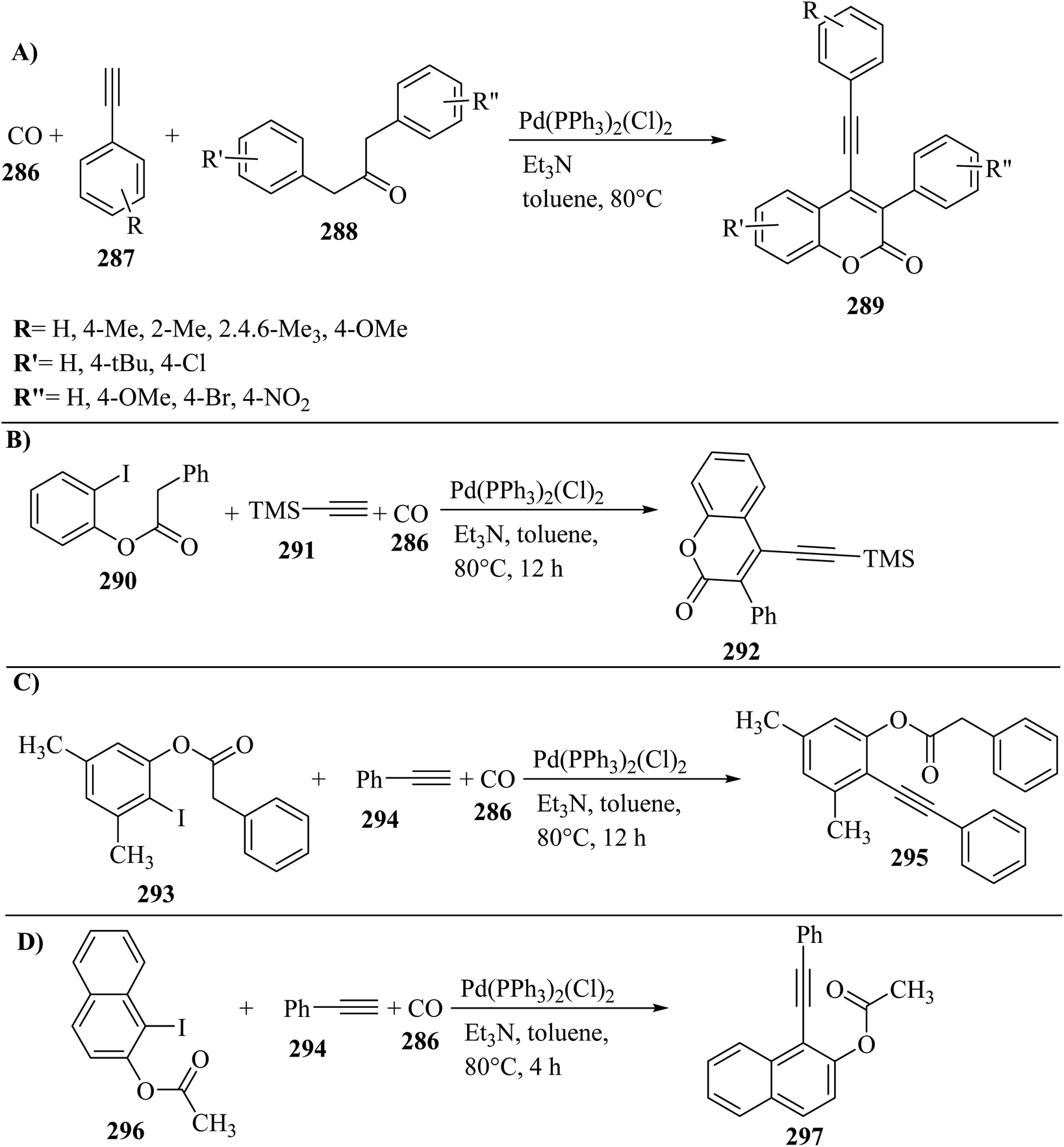 | ||
| Scheme 120 (A) Synthesis of 3-aryl-4-(arylethynyl)-2H-chromen-2-one derivatives 289, (B) 3-phenyl-4-((trimethylsilyl)ethynyl)-2H-chromen-2-one 292, (C) 3,5-dimethyl-2-(phenylethynyl)phenyl 2-phenylacetate 295, (D) 1-(phenylethynyl)naphthalen-2-yl acetate 297.117 | ||
| Reactants | Product | Catalyst | Solvent | Method employed | Reaction time | Yield | Limitations or progress | Ref. |
|---|---|---|---|---|---|---|---|---|
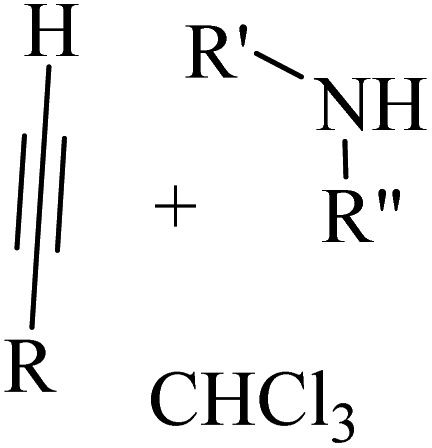 |
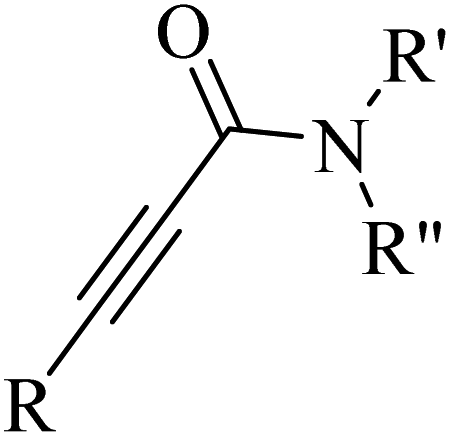 |
PdCl2(PPh3)2–cataCXium A | 1,4-Dioxane | CsOH·H2O, 80 °C, microwave | 50 min | 35–70% | (1) CHCl3 as in situ CO surrogate | Rathod and Jain118 |
| (2) Short reaction time | ||||||||
| (3) No prefunctionlization needed | ||||||||
| (4) Bioactive alk-2-ynamides | ||||||||
| (5) Catalytic system was new | ||||||||
| (6) Substrate scope was broad | ||||||||
| (7) 2-Ethynylpyridine could not produce the desired product | ||||||||
| (8) Trace amount of product was produced by trimethylsilylacetylene | ||||||||
| (9) Di-n-propylamine did not produce the desired product | ||||||||
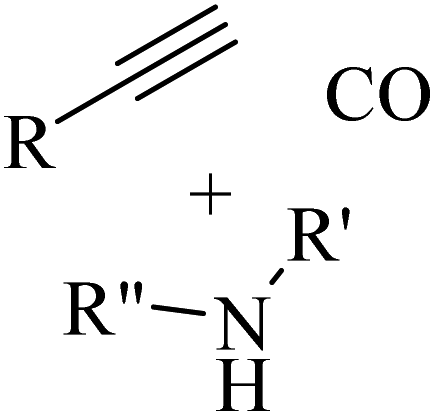 |
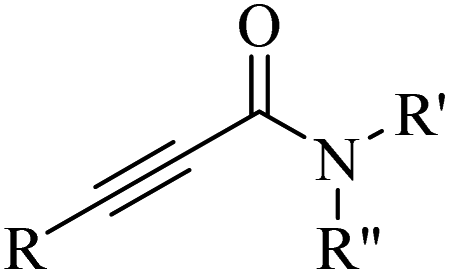 |
IPr-Pd-Peppsi-Cl2 | Dioxane | KI, K3PO4, 100 °C | 18 h | 50–95% | (1) Mild conditions | Zhang et al.119 |
| (2) High atom efficiency | ||||||||
| (3) Alkyl and aryl of the alkynes could be efficiently | ||||||||
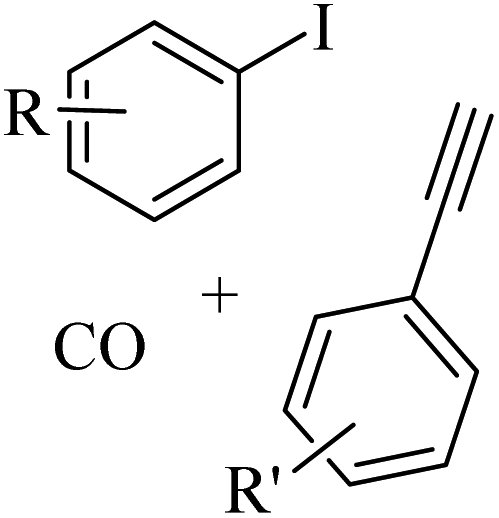 |
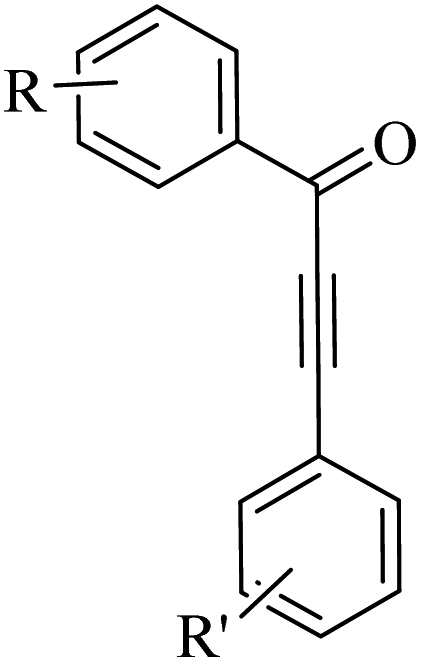 |
NiCl2·6H2O | DMF | dppb, K2CO3 130 °C | 7–12 h | 20–76% | (1) First time (CO2H)2 was used as in situ CO surrogate, bench stable, ex situ solid, and easy to handle | Shaifali et al.120 |
| (2) First report for carbonylative Sonogashira coupling by using CO-surrogate under Ni catalyst | ||||||||
 |
 |
Pd(OAc)2 | EtOAc | [NBu4]I, 80 °C, O2 in N2 | 6 h | 52–92% | (1) The method avoids the use of dangerous | Hughes et al.121 |
| 1,4-Dioxane | ||||||||
| (2) Used O2 as the terminal oxidant | ||||||||
| (3) Does not require the use of ligands | ||||||||
| (4) Develop synthesis of 2-ynamides through oxidative carbonylation | ||||||||
| (5) A variety of alkynes and amines can be used | ||||||||
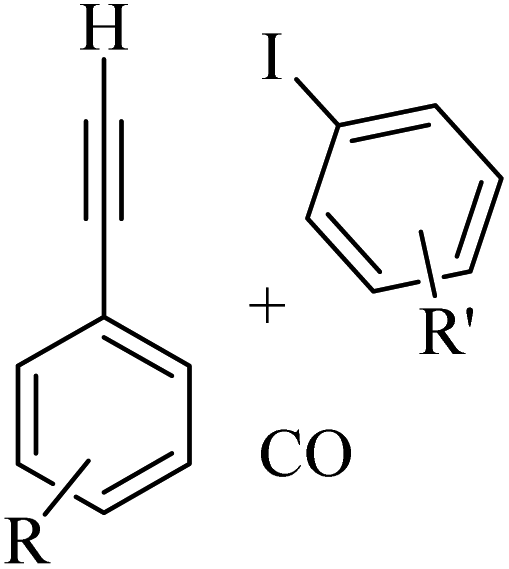 |
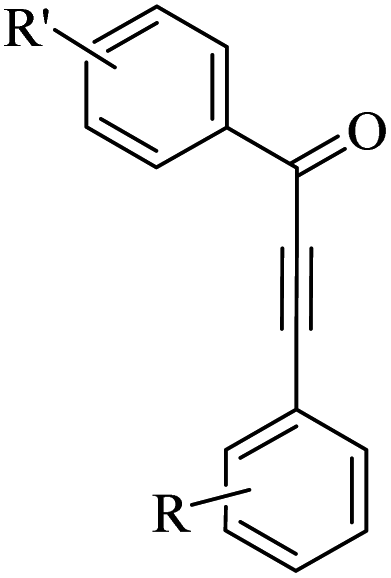 |
Pd(II)APTES@K10 | DME | NEt3 80 °C | 7 h | 60–92% | (1) Method gave easy and simple access toward dibenzoylmethane | Chavan et al.122 |
| (2) The electron withdrawing aryl halides can be efficiently used as an electrophile | ||||||||
| (3) Recovery of catalyst by simple filtration | ||||||||
 |
 |
Pd/C | Toluene | Et3N, 130 °C | 4 h | 63–97% | (1) Mild conditions | Liu et al.123 |
| (2) No need to add toxic phosphine ligands | ||||||||
| (3) Easy catalyst recycling | ||||||||
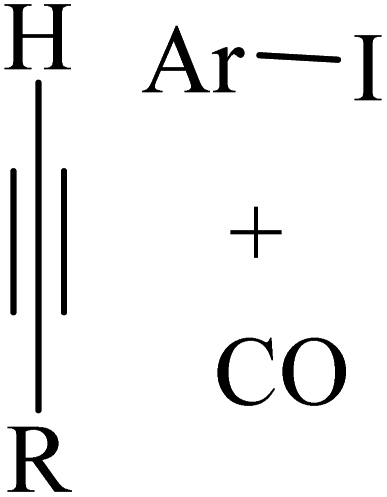 |
 |
P(DVB-IL)-Pd | H2O | Et3N, 130 °C | 6 h | 27–95% | (1) The catalyst is easily separated from the products. | Wang et al.124 |
| (2) Reuse of catalyst without activity loss | ||||||||
| (3) Environmental synthesis of α, β-alkynyl ketones via the heterogeneous carbonylation reaction | ||||||||
| (4) The method avoids the use of toxic phosphine ligands | ||||||||
 |
 |
Pd/Fe3O4 | Toluene | Et3N, 130 °C | 4 h | 50–95% | (1) Highly efficient catalyst for the carbonylative Sonogashira coupling reaction | Liu et al.125 |
| (2) Catalyst can be reused with sustained selectivity and activity | ||||||||
| (3) Recoverability of catalyst | ||||||||
| (4) Catalyst could be reused seven times without remarkable loss in activity and selectivity |
4 Conclusion
The need to improve the reaction conditions in the synthesis of heterocyclic compounds is an important and constant issue, and thus researchers are devoting their efforts to finding optimal and better methods and trying to design methods that solve the related problems. Among them, alkynes, which are a very important and widely use groups in chemistry, are used in important multi-component reactions given that they possess two active sites to carry out the reaction. Due to this characteristic of alkynes, it is challenging for researchers to design and activate a specific position. Meanwhile, researchers use ligands, catalysts, solvents, and even different methods such as microwave heating, which is a priority to perform the desired reactions using environmentally friendly conditions because both the reaction time is short and a low temperature is required, and also non-toxic materials are used, which help to improve the reaction conditions. We present our vision for improving the CC and C–H activation of alkynes in the various reactions that lead to a variety of important structures.
The amount of catalyst used in the investigated reactions showed that the amount of catalyst was very effective in the reactions and the alkyne used acted selectively. Furthermore, the presence of electron-withdrawing groups in alkynes (most of the reactions) reduces the efficiency of the reactions, and also aromatic alkynes participate in reactions better than aliphatic alkynes and have higher efficiency, although it should be noted that in some reactions aliphatic alkynes did not react at all. Also, the type of solvent is decisive in the reactions and plays a key role, especially in the reaction of alkynes with CO2 or CO. In addition, the used molar ratios of alkynes are effective in the synthesis of compounds and different products are created by using different molar ratios.
This article highlighted some applications of the CC and C–H positions of alkynes in reactions with other important compounds in multicomponent reactions for the synthesis of important compounds and heterocycles and focused on recent advances in the use of alkynes. Also, the articles highlighted in this review are impressive and extensive and provide valuable insight into the activation of CC and C–H alkynes.
We hope that this review will encourage researchers to do more research in this field and cover the gaps in previous research, as well as offer insight into the design of new reactions and new chemical transformations of alkynes to prepare important structures.
4.1 Future remarks
In the selectivity of alkynes, the use of a suitable catalyst is very important, and thus we suggest the use of efficient catalysts in this field, such as nanocatalysts, photocatalysts as well as enzymes. Also, in this case, the used ligands are very effective, and the use of a suitable ligand and catalyst combination helps to optimize the reactions, the selectivity of alkynes and also helps to carry out the reaction of all types of alkynes. Therefore, we suggest using or synthesizing ligands and catalysts that can activate the CC or C–H position with the highest selectivity.
Conflicts of interest
There are no conflicts to declare.References
- R. Salvio, M. Moliterno and M. Bella, Asian J. Org. Chem., 2014, 3(4), 340–351 CrossRef CAS.
- For reviews on alkyne synthesis, see: (a) D. Habrant, V. Rauhala and A. M. P. Koskinen, Chem. Soc. Rev., 2010, 39, 2007–2017 RSC; (b) J. P. Brand and J. Waser, Chem. Soc. Rev., 2012, 41, 4165–4179 RSC.
- (a) Acetylene Chemistry. Chemistry, Biology and Material Science, ed. F. Diederich, P. J. Stang and R. R. Tykwinski, WILEY-VCH, Weinheim, 2005 Search PubMed; (b) R. Gleiter and D. B. Werz, Alkynes Between Main Group Elements: From Dumbbells via Rods to Squares and Tubes, Chem. Rev., 2010, 110, 4447–4488 CrossRef CAS PubMed.
- X. Ren and Z. Lu, Chin. J. Catal., 2019, 40(7), 1003–1019 CrossRef CAS.
- C. Hu, J. Mena and I. V. Alabugin, Nat. Rev. Chem, 2023, 7(6), 405–423 CrossRef PubMed.
- T. Long, C. Zhu, L. Li, L. Shao, S. Zhu, M. Rueping and L. Chu, Nat. Commun., 2023, 14(1), 55 CrossRef CAS PubMed.
- L. Zheng, X. Guo, Y. C. Li, Y. Wu, X. S. Xue and P. Wang, Angew. Chem., Int. Ed., 2023, 62(5), e202216373 CrossRef CAS PubMed.
- P. Zhou, H. Jiao, K. Niu, H. Song, Y. Liu and Q. Wang, ACS Sustainable Chem. Eng., 2023, 11(6), 2607–2612 CrossRef CAS.
- Y. Q. Zhang, L. Hu, L. Yuwen, G. Lu and Q. W. Zhang, Nat. Catal., 2023, 1–8 Search PubMed.
- W. Li, S. Chen, J. Xie, Z. Fan, K. Yang and Q. Song, Nat. Synth., 2023, 2(2), 140–151 CrossRef.
- X. Li, M. Jiang, J. Zuo, X. Song, J. Lv and D. Yang, Sci. China: Chem., 2023, 66(3), 791–798 CAS.
- X. Xu, A. Gao, W. Chen, X. Xu, J. Li and C. Cui, ACS Catal., 2023, 13(6), 3743–3748 CrossRef CAS.
- S. Tao, Y. Wang, Q. Pan, J. Zhao, Q. Bu, F. Chen, J. Liu, B. Dai, D. Wei and N. Liu, Green Chem., 2023, 25(17), 6704–6716 RSC.
- P. Sivaguru, S. Cao, K. R. Babu and X. Bi, Acc. Chem. Res., 2020, 53(3), 662–675 CrossRef CAS PubMed.
- S. K. Banjare, P. S. Mahulkar, T. Nanda, B. V. Pati, L. O. Najiar and P. C. Ravikumar, Chem. Commun., 2022, 58(74), 10262–10289 RSC.
- C. de Graaff, E. Ruijter and R. V. Orru, Chem. Soc. Rev., 2012, 41(10), 3969–4009 RSC.
- B. B. Touré and D. G. Hall, Chem. Rev., 2009, 109, 4439–4486 CrossRef PubMed.
- R. Kakuchi, Angew. Chem., Int. Ed., 2014, 53, 46–48 CrossRef CAS PubMed.
- C. Lamberth, A. Jeanguenat, F. Cederbaum, A. De Mesmaeker, M. Zeller, H.-J. Kempf and R. Zeun, Bioorg. Med. Chem., 2008, 16, 1531–1545 CrossRef CAS PubMed.
- (a) E. Ruijter and R. V. A. Orru, Drug Discovery Today: Technol., 2013, 10, e15–e20 CrossRef PubMed; (b) A. Dömling, W. Wang and K. Wang, Chem. Rev., 2012, 112, 3083–3135 CrossRef PubMed; (c) I. V. Magedov and A. Kornienko, Chem. Heterocycl. Compd., 2012, 48, 33–38 CrossRef CAS PubMed; (d) P. Slobbe, E. Ruijter and R. V. A. Orru, Med. Chem. Commun., 2012, 3, 1189–1218 RSC; (e) C. Kalinski, M. Umkehrer, L. Weber, J. Kolb, C. Burdack and G. Ross, Mol. Diversity, 2010, 14, 513–522 CrossRef CAS PubMed; (f) I. Akritopoulou-Zanze, Curr. Opin. Chem. Biol., 2008, 12, 324–331 CrossRef CAS PubMed.
- A. Domling, W. Wang and K. Wang, Chem. Rev., 2012, 112(6), 3083–3135 CrossRef CAS PubMed.
- H. Zheng, Y. J. Mei, K. Du, X. T. Cao and P. F. Zhang, Molecules, 2013, 18(11), 13425–13433 CrossRef CAS PubMed.
- S. Hosseininezhad and A. Ramazani, Arabian J. Chem., 2023, 16, 105234 CrossRef CAS.
- C. Cimarelli, Molecules, 2019, 24(13), 2372 CrossRef CAS PubMed.
- R. C. Cioc, E. Ruijter and R. V. Orru, Green Chem., 2014, 16(6), 2958–2975 RSC.
- B. Jiang, T. Rajale, W. Wever, S. J. Tu and G. Li, Chem.–Asian J., 2010, 5, 2318–2335 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- M. G. Finn and V. V. Fokin, Chem. Soc. Rev., 2010, 39(4), 1231–1232 RSC.
- J. Hou, X. Liu, J. Shen, G. Zhao and P. G. Wang, Expert Opin. Drug Discovery, 2012, 7(6), 489–501 CrossRef CAS PubMed.
- Click Chemistry for Biotechnology and Materials Science, ed. J. Lahann, John Wiley & Sons, 2009 Search PubMed.
- P. Bao, H. Yue, N. Meng, X. Zhao, J. Li and W. Wei, Org. Lett., 2019, 21(18), 7218–7222 CrossRef CAS PubMed.
- P. Bao, N. Meng, Y. Lv, H. Yue, J. S. Li and W. Wei, Org. Chem. Front., 2019, 6(24), 3983–3988 RSC.
- X. He, R. Li, M. Xie, J. Duan, Q. Tang and Y. Shang, New J. Chem., 2020, 44(28), 12266–12273 RSC.
- A. A. Saikia, R. N. Rao, S. Das, S. Jena, S. Rej, B. Maiti and K. Chanda, Tetrahedron Lett., 2020, 61(36), 152273 CrossRef CAS.
- L. L. Zhang, M. T. Li, L. L. Shen and Q. P. Wu, Synthesis, 2020, 52(02), 304–310 CrossRef CAS.
- A. A. Khandar, A. Sheykhi, M. Amini, A. Ellern and L. K. Woo, Polyhedron, 2020, 188, 114698 CrossRef.
- Z. Liu, L. Wang, T. Yu, Y. Sun, H. Chen, W. Gao and B. Tang, Org. Chem. Front., 2020, 7(18), 2628–2633 RSC.
- C. Wang, Q. Li, S. Wang, G. Zhu, A. Zhu and L. Li, RSC Adv., 2021, 11(60), 38108–38114 RSC.
- V. Camberlein, N. Kraupner, N. B. Karroum, E. Lipka, R. Deprez-Poulain, B. Deprez and D. Bosc, Tetrahedron Lett., 2021, 73, 153131 CrossRef CAS.
- D. Yuan, S. Wang, G. Zhu, A. Zhu and L. Li, Tetrahedron, 2021, 81, 131911 CrossRef CAS.
- X. X. Wang, B. X. Sun, Z. W. Zhao, X. Chen, W. J. Xia, Y. Shen and Y. M. Li, Adv. Synth. Catal., 2022, 364(1), 165–171 CrossRef CAS.
- B. X. Sun, X. N. Wang, T. G. Fan, Y. J. Hou, Y. T. Shen and Y. M. Li, J. Org. Chem., 2023, 88(7), 4528–4535 CrossRef CAS PubMed.
- F. Wang, N. Zhu, P. Chen, J. Ye and G. Liu, Angew. Chem., Int. Ed., 2015, 54(32), 9356–9360 CrossRef CAS PubMed.
- H. Xiong, N. Ramkumar, M. F. Chiou, W. Jian, Y. Li, J. H. Su, X. Zhang and H. Bao, Nat. Commun., 2019, 10(1), 122 CrossRef PubMed.
- J. Chen, T. Liang, H. Zhao, C. Lin, L. Chen and M. Zhang, Org. Biomol. Chem., 2019, 17(19), 4843–4849 RSC.
- B. Liu, Y. Ning, M. Virelli, G. Zanoni, E. A. Anderson and X. Bi, J. Am. Chem. Soc., 2019, 141(4), 1593–1598 CrossRef CAS PubMed.
- X. He, R. Li, M. Xie, J. Duan, Q. Tang and Y. Shang, New J. Chem., 2020, 44(28), 12266–12273 RSC.
- K. P. S. Cheung and G. C. Tsui, Org. Lett., 2017, 19, 2881–2884 CrossRef CAS PubMed.
- M. Y. Ansari, N. Kumar and A. Kumar, Org. Lett., 2019, 21(11), 3931–3936 CrossRef CAS PubMed.
- H. Chen, R. Ding, H. Tang, Y. Pan, Y. Xu and Y. Chen, Chem.–Asian J., 2019, 14(19), 3264–3268 CrossRef CAS PubMed.
- C. Zhu, H. Yue, B. Maity, I. Atodiresei, L. Cavallo and M. Rueping, Nat. Catal., 2019, 2(8), 678–687 CrossRef CAS.
- F. Yi, Q. Sun, J. Sun, C. Fu and W. Yi, J. Org. Chem., 2019, 84(11), 6780–6787 CrossRef CAS PubMed.
- H. Hazarika, K. Neog, A. Sharma, B. Das and P. Gogoi, J. Org. Chem., 2019, 84(9), 5846–5854 CrossRef CAS PubMed.
- Z. Chen, P. Liang, F. Xu, Z. Deng, L. Long, G. Luo and M. Ye, J. Org. Chem., 2019, 84(19), 12639–12647 CrossRef CAS PubMed.
- H. Jin, D. Liu, B. Zhou and Y. Liu, Synthesis, 2020, 52(09), 1417–1424 CrossRef CAS.
- H. Xu, R. Ye, Z. Li, M. Y. Han and L. G. Meng, Adv. Synth. Catal., 2021, 363(10), 2670–2675 CrossRef CAS.
- V. S. Bhat and A. Lee, Adv. Synth. Catal., 2022, 364(17), 3088–3093 CrossRef CAS.
- S. Kalari, U. B. Karale and H. B. Rode, J. Org. Chem., 2022, 87(5), 2435–2445 CrossRef CAS PubMed.
- J. Tang, P. Sivaguru, Y. Ning, G. Zanoni and X. Bi, Org. Lett., 2017, 19(15), 4026–4029 CrossRef CAS PubMed.
- Y. Ning, A. Mekareeya, K. R. Babu, E. A. Anderson and X. Bi, ACS Catal., 2019, 9(5), 4203–4210 CrossRef CAS.
- M. Khalaj, Monatsh. Chem., 2020, 151, 79–85 CrossRef CAS.
- A. K. Sahoo, A. Dahiya, B. Das, A. Behera and B. K. Patel, J. Org. Chem., 2021, 86(17), 11968–11986 CrossRef CAS PubMed.
- M. Y. Ansari, S. Swarnkar and A. Kumar, Chem. Commun., 2020, 56(66), 9561–9564 RSC.
- H. Hazarika, K. Neog, A. Sharma, B. Das and P. Gogoi, J. Org. Chem., 2019, 84(9), 5846–5854 CrossRef CAS PubMed.
- A. Jana, P. Bhaumick, A. K. Panday, R. Mishra and L. H. Choudhury, Org. Biomol. Chem., 2019, 17(21), 5316–5330 RSC.
- A. K. Panday, D. Ali and L. H. Choudhury, Org. Biomol. Chem., 2020, 18(26), 4997–5007 RSC.
- P. Bhuyan, P. Bhorali, S. Kataky, S. J Bharali, A. K. Guha and L. Saikia, Sustainable Chem. Pharm., 2022, 30, 100852 CrossRef CAS.
- S. Asghari, D. Alizadeh, H. Younesi and G. Firouzzadeh Pasha, Polycyclic Aromat. Compd., 2022, 42(9), 6303–6319 CrossRef CAS.
- S. S. Shinde, S. Laha, D. K. Tiwari, B. Sridhar and P. R. Likhar, Org. Biomol. Chem., 2019, 17(16), 4121–4128 RSC.
- V. Thakur, A. Sharma, N. Sharma and P. Das, Adv. Synth. Catal., 2019, 361(3), 426–431 CrossRef CAS.
- L. Zeng, H. Sajiki and S. Cui, Org. Lett., 2019, 21(16), 6423–6426 CrossRef CAS PubMed.
- C. Wang, Z. Lai, H. Xie and S. Cui, Angew. Chem., Int. Ed., 2021, 60(10), 5147–5151 CrossRef CAS PubMed.
- P. Fernández, C. Valdés, F. J. Fañanás and F. Rodríguez, J. Org. Chem., 2019, 84(6), 3184–3191 CrossRef PubMed.
- B. G. Das, S. Shah and V. K. Singh, Org. Lett., 2019, 21(13), 4981–4985 CrossRef CAS PubMed.
- D. Chandra, A. K. Dhiman, R. Kumar and U. Sharma, Eur. J. Org Chem., 2019, 2019(16), 2753–2758 CrossRef CAS.
- F. Zhang, Q. Lai, X. Shi and Z. Song, Chin. Chem. Lett., 2019, 30(2), 392–394 CrossRef CAS PubMed.
- S. Yu, J. Wu, H. Lan, L. Gao, H. Qian, K. Fan and Z. Yin, Org. Lett., 2019, 22(1), 102–105 CrossRef PubMed.
- G. Purohit, A. Kharkwal and D. S. Rawat, ACS Sustain. Chem. Eng., 2020, 8(14), 5544–5557 CrossRef CAS.
- L. Xian, C. T. Ma, Y. G. Ouyang, J. Q. Di and Z. H. Zhang, Appl. Organomet. Chem., 2020, 34(11), e5921 CrossRef CAS.
- C. Yuan, X. Zhao and G. Nan, Tetrahedron Lett., 2020, 61(39), 152318 CrossRef CAS.
- M. Mohammod, V. Guguloth, C. S. Vasam and N. S. Thirukovela, Synth. Commun., 2022, 52(9–10), 1254–1267 CrossRef CAS.
- H. L. Cui and X. H. Chen, J. Org. Chem., 2022, 87(22), 15435–15447 CrossRef CAS PubMed.
- M. He, N. Chen, J. Wang and S. Peng, Org. Lett., 2019, 21(13), 5167–5171 CrossRef CAS PubMed.
- X. Li, T. Feng, D. Li, H. Chang, W. Gao and W. Wei, J. Org. Chem., 2020, 85(15), 9538–9547 CrossRef CAS PubMed.
- K. Zhou, M. Bao, J. Huang, Z. Kang, X. Xu, W. Hu and Y. Qian, Org. Biomol. Chem., 2020, 18(3), 409–414 RSC.
- Y. Lv, W. Pu, X. Zhu, C. Chen and S. Wang, J. Org. Chem., 2021, 86(15), 10043–10054 CrossRef CAS PubMed.
- S. Pelliccia, A. I. Alfano, P. Luciano, E. Novellino, A. Massarotti, G. C. Tron, D. Ravelli and M. Giustiniano, J. Org. Chem., 2019, 85(4), 1981–1990 CrossRef PubMed.
- M. Gao, M. Zou, J. Wang, Q. Tan, B. Liu and B. Xu, Org. Lett., 2019, 21(6), 1593–1597 CrossRef CAS PubMed.
- K. Singh, B. K. Malviya, P. K. Jaiswal, V. P. Verma, S. S. Chimni and S. Sharma, Org. Lett., 2019, 21(17), 6726–6730 CrossRef CAS PubMed.
- S. Yang, L. Yao, Z. Fan, J. Han, J. Chen, W. He, H. Deng, M. Shao, H. Zhang and W. Cao, J. Fluorine Chem., 2021, 243, 109723 CrossRef CAS.
- Y. Li, T. J. Emge, A. Moreno-Vicente, W. P. Kopcha, Y. Sun, I. F. Mansoor, M. C. Lipke, G. S. Hall, J. M. Poblet, A. Rodríguez-Fortea and J. Zhang, Angew. Chem., 2021, 133(48), 25473–25477 CrossRef.
- Y. Li, W. P. Kopcha, T. J. Emge, Y. Sun and J. Zhang, Org. Lett., 2021, 23(22), 8867–8872 CrossRef CAS PubMed.
- Y. Li, W. P. Kopcha, A. Rodriguez-Fortea and J. Zhang, Synlett, 2022, 33(10), 907–912 CrossRef CAS.
- B. Liu, Y. Li, M. Yin, W. Wu and H. Jiang, Chem. Commun., 2012, 48(93), 11446–11448 RSC.
- Y. Li, T. Miao, P. Li and L. Wang, Org. Lett., 2018, 20(7), 1735–1739 CrossRef CAS PubMed.
- S. Yan, S. Pan, T. Osako and Y. Uozumi, ACS Sustain. Chem. Eng., 2019, 7(10), 9097–9102 CrossRef CAS.
- S. Dhanasekaran, V. K. Kannaujiya, R. G. Biswas and V. K. Singh, J. Org. Chem., 2019, 84(6), 3275–3292 CrossRef CAS PubMed.
- M. Milen, G. Györke, A. Dancsó and B. Volk, Tetrahedron Lett., 2020, 61(10), 151544 CrossRef CAS.
- K. Thakur and N. K. Khare, Carbohydr. Res., 2020, 494, 108053 CrossRef CAS PubMed.
- M. Martinez-Amezaga, R. A. Giordano, D. N. P. Gori, C. P. Squizatto, M. V. Giolito, O. G. Scharovsky, V. R. Rozados, M. J. Rico, E. G. Mata and C. M. Delpiccolo, Org. Biomol. Chem., 2020, 18(13), 2475–2486 RSC.
- S. P. Neofotistos, N. V. Tzouras, M. Pauze, E. Gómez-Bengoa and G. C. Vougioukalakis, Adv. Synth. Catal., 2020, 362(18), 3872–3885 CrossRef CAS.
- L. Huang, Y. Xie, P. Ge, J. Huang and H. Feng, Eur. J. Org Chem., 2021, 2021(17), 2448–2451 CrossRef CAS.
- J. Safaei-Ghomi, S. H. Nazemzadeh and H. Shahbazi-Alavi, Main Group Met. Chem., 2017, 40, 129–135 CAS.
- S. Ghosh, K. Biswas, S. Bhattacharya, P. Ghosh and B. Basu, Beilstein J. Org. Chem., 2017, 13, 552–557 CrossRef CAS PubMed.
- M. Bakherad, F. Moosavi, R. Doosti, A. Keivanloo and M. Gholizadeh, New J. Chem., 2018, 42, 4559–4566 RSC.
- V. S. Rawat, T. Bathini, S. Govardan and B. Sreedhar, Org. Biomol. Chem., 2014, 12, 6725–6729 RSC.
- C. X. Guo, B. Yu, J. N. Xie and L. N. He, Green Chem., 2015, 17(1), 474–479 RSC.
- J. N. Xie, B. Yu, Z. H. Zhou, H. C. Fu, N. Wang and L. N. He, Tetrahedron Lett., 2015, 56(50), 7059–7062 CrossRef CAS.
- Z. Z. Zhang, R. J. Mi, F. J. Guo, J. Sun, M. D. Zhou and X. C. Fang, J. Saudi Chem. Soc., 2017, 21(6), 685–690 CrossRef CAS.
- F. J. Guo, Z. Z. Zhang, J. Y. Wang, J. Sun, X. C. Fang and M. D. Zhou, Tetrahedron, 2017, 73(7), 900–906 CrossRef CAS.
- A. T. Papastavrou, M. Pauze, E. Gómez-Bengoa and G. C Vougioukalakis, ChemCatChem, 2019, 11(21), 5379–5386 CrossRef CAS.
- L. Zhang, W. Zhang, S. H. I. Linglong, R. E. N. Xiang and L. U. Xiaobing, Chin. J. Catal., 2013, 34(6), 1179–1186 CrossRef CAS.
- B. Yang, Y. Qiu, T. Jiang, W. D. Wulff, X. Yin, C. Zhu and J. E. Bäckvall, Angew. Chem., 2017, 129(16), 4606–4610 CrossRef.
- C. Zhu, B. Yang and J. E. Backvall, J. Am. Chem. Soc., 2015, 137(37), 11868–11871 CrossRef CAS PubMed.
- Y. Qiu, B. Yang, C. Zhu and J. E. Bäckvall, J. Am. Chem. Soc., 2016, 138(42), 13846–13849 CrossRef CAS PubMed.
- C. M. Volla and J. E. Backvall, Org. Lett., 2014, 16(16), 4174–4177 CrossRef CAS PubMed.
- A. Chandrasekhar, V. Ramkumar and S. Sankararaman, Eur. J. Org Chem., 2016, 2016(23), 4041–4049 CrossRef CAS.
- G. K. Rathod and R. Jain, J. Org. Chem., 2023, 88(11), 7219–7227 CrossRef CAS PubMed.
- C. Zhang, J. Liu and C. Xia, Catal. Sci. Technol., 2015, 5(10), 4750–4754 RSC.
- S. Ram, V. Thakur and P. Das, Org. Biomol. Chem., 2019, 17(29), 7036–7041 RSC.
- N. L. Hughes, C. L. Brown, A. A. Irwin, Q. Cao and M. J. Muldoon, ChemSusChem, 2017, 10(4), 675–680 CrossRef CAS PubMed.
- S. P. Chavan, G. B. B. Varadwaj, K. Parida and B. M. Bhanage, Appl. Catal., A, 2015, 506, 237–245 CrossRef CAS.
- J. Liu, J. Chen and C. Xia, J. Catal., 2008, 253(1), 50–56 CrossRef CAS.
- Y. Wang, J. Liu and C. Xia, Tetrahedron Lett., 2011, 52(14), 1587–1591 CrossRef CAS.
- J. Liu, X. Peng, W. Sun, Y. Zhao and C. Xia, Org. Lett., 2008, 10(18), 3933–3936 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |