
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew fluorene-based bipolar charge transporting materials†
Aistė Jegorovė*a,
Marytė Daškevičienėa,
Kristina Kantminienė b,
Vygintas Jankauskasc,
Romualdas Jonas Čepasc,
Alytis Gruodisc,
Vytautas Getautisa and
Kristijonas Genevičiusc
b,
Vygintas Jankauskasc,
Romualdas Jonas Čepasc,
Alytis Gruodisc,
Vytautas Getautisa and
Kristijonas Genevičiusc
aDepartment of Organic Chemistry, Kaunas University of Technology, Radvilėnų pl. 19, Kaunas, 50254, Lithuania
bDepartment of Physical and Inorganic Chemistry, Kaunas University of Technology, Radvilėnų pl. 19, Kaunas, 50254, Lithuania
cInstitute of Chemical Physics, Vilnius University, Saulėtekio al. 3, Vilnius 10257, Lithuania. E-mail: romualdas.cepas@ff.vu.lt
First published on 18th January 2024
Abstract
Air-stable and solution-processable fluorene-based bipolar charge transporting materials (CTMs) were designed, synthesized, and analyzed. These CTMs feature anthraquinone, 9-fluorenone, and 9-dicyanofluorenylidine groups and exhibit good film formation properties for solvent processing. Quantum chemistry simulations and optical absorption measurements proved that several stable conformers and charge transfer complexes form inside the molecules. Hole mobilities in CTMs were around 10−4 to 10−5 cm2 V−1 s−1, while electron mobility in compounds with anthraquinone and 9-dicyanofluorenylidine groups was approximately one order of magnitude lower.
Introduction
The innovative design of organic compounds and the tuning of their practical properties has become an important tool of materials science used to address major contemporary global issues, including the energy crisis and environmental impact. Therefore, the synthesis of new organic functional molecules and their adjustment for high performance materials has been gaining considerable attention in recent years. In the last few decades, the development of organic compounds as active components of electronic and optoelectronic devices, such as organic solar cells (OSCs),1,2 dye-sensitized solar cells (DSCs),3 organic field-effect transistors (OFET),4–6 organic light-emitting diodes (OLEDs),7,8 and organic photorefractive devices9 has contributed to a significant breakthrough in their technological applications and expanded fundamental research knowledge that could be used for future scientific and technological advancement.Electronic or optoelectronic devices that employ organic materials as active elements benefit from the important advantages of organic materials, such as their potentially low cost and light mass as well as the ability to form thin-film large-surface-area flexible devices. The first industrial-scale application of organic semiconductors was xerography.10 Since then, charge transporting materials have played a significant role in many of the technologies that have evolved from xerography.11 Although an extensive data on hole- or electron-transporting materials have been reported in scientific literature, molecules capable of transporting both holes and electrons have received much less attention.12
Different strategies have been implemented to achieve bipolar behaviour of organic materials for electronic and optoelectronic applications. Methods used to ensure bipolarity include layered heterostructure, interpenetrating network, and charge transfer complex creation.13–16 Several disadvantages are associated with the first two of these methods, such as the increased complexity of the manufactured device; the higher probability of occurrence of defect points or nonuniformity of the layer owing to formed defective charge transfer complexes, and the possible long-term instability. In addition, it is well known that the formation of charge transfer complexes significantly reduces the charge mobility of one of the components used,11,17 so achieving balance between the mobility of electrons and holes in the layer becomes a challenging issue. Therefore, a more reasonable approach toward bipolarity of organic materials is to design the bipolar charge transporting material (CTM) as a single compound. Thus, utilization of a bipolar material reduces both the number of mixed materials and helps maintain device stability. Additionally, it is technologically advantageous for such materials to be air-operable and soluble in common organic solvents since low-cost organic electronic or optoelectronic devices require material deposition via solution processes instead of expensive vacuum thermal deposition.
However, the reported number of single-component air-stable and soluble bipolar CTMs capable of transporting both carrier types is limited despite the obvious advantages of such molecules. Aso et al. achieved bipolar transport by attaching a hole-transporting oligothiophene unit to the fullerene core.18 Several other fullerene derivatives bearing, for example, dendritic oligothiophene side chains,19 oligophenyleneethynylenes,20 thioaxanthenes,21 hydrazones,22,23 or hexa-peri-hexabenzocoronene24 have also been shown to exhibit bipolar behaviour, though with moderate charge-carrier mobilities. These compounds have quite complex structures and require expensive starting materials and/or elaborate multistep syntheses. Fullerene derivatives widely used as acceptors in OSCs are associated with poor absorption, high cost, and insufficient stability. As an alternative, bipolar small molecule acceptors based on the fused terthieno[3,2-b]thiophene or benzodithiophene donor core and various electron-withdrawing end groups have been tested in OSCs.25,26
Various small molecule donor materials incorporating diketopyrrolopyrrole fragment in their structure have been designed and synthesized for solution-processed optoelectronic devices.27–33 Imide derivatives are another successful class of materials used for bipolar CTMs.34,35 The carbazole moiety, because of its electron donating ability, is a convenient donor component of bipolar CTMs, while triazine, cyano, pyridine, phosphine oxide, etc. are used as acceptor fragments.36 Velasco et al. investigated stable long-lived organic carbazole-based radical adducts as an electron donor–acceptor system with bipolar charge transport behaviour. High electron and hole mobilities up to 10−2 and 10−3 cm2 V−1 s−1, respectively, were achieved.37,38 Kim et al. reported the synthesis of the bipolar hosts bearing 9,9-bis(9-alkylcarbazole)thioxanthene-S,S-dioxide moiety for application in white organic light emitting diodes. The thioxanthene-S,S-dioxide moiety possesses good electron-transporting ability, while the bis-ethylhexyl-, octyl-, dodecyl- or heptadecanylcarbazole moieties have good hole-transporting ability.39
Recently, we designed and synthesized air-stable and solution-processable tetracarboxydiimide-based bipolar CTMs, bearing electron-transporting 1,4,5,8-naphthalene- or 3,4,9,10-perylenetetracarboxy-diimide cores with attached 1-phenyl-1,2,3,4-tetrahydroquinoline moieties as end-capping hole-transporting groups in a straightforward and simple manner without the need for advanced synthetic procedures, expensive catalysts or sensitive reagents.40
Generally, soluble and air-stable bipolar CTMs have quite complex structures and require expensive starting materials and/or elaborate multistep synthesis. In this work, we have designed and synthesized air-stable and solution-processable fluorine-based bipolar materials V1393, V1421, V1457, V1458, V1484, and V1485 (Fig. 1). These CTMs were obtained from inexpensive starting materials through simple reactions and possess electron transporting anthraquinone, 9-fluorenone, or 9-dicyanofluorenylidine with attached carbazolyl electron-donating moieties. The optical, thermal, and charge carrier transport properties of the synthesized CTMs were studied. The existence of possible conformers and excitation energies were calculated by the means of quantum chemistry simulations.
 | ||
| Fig. 1 Chemical structures of the synthesized bipolar compounds bearing different structural units. | ||
Results and discussion
Target bipolar molecules were synthesized via the synthetic route depicted in Scheme 1. The precursors (side-amines) were synthetized by coupling 3-bromo-9-ethylcarbazole with 2-aminoantraquinone and 2-amino-9H-fluorenone. The target compounds V1393, V1421, V1457, and V1458 were synthesized under Buchwald–Hartwig amination reaction conditions using a central fluorene-based core as the starting material. The dicyanofluorenylidine derivatives V1484 and V1485 were synthesized by modifying the keto group to malononitrile by Knoevenagel condensation in basic medium.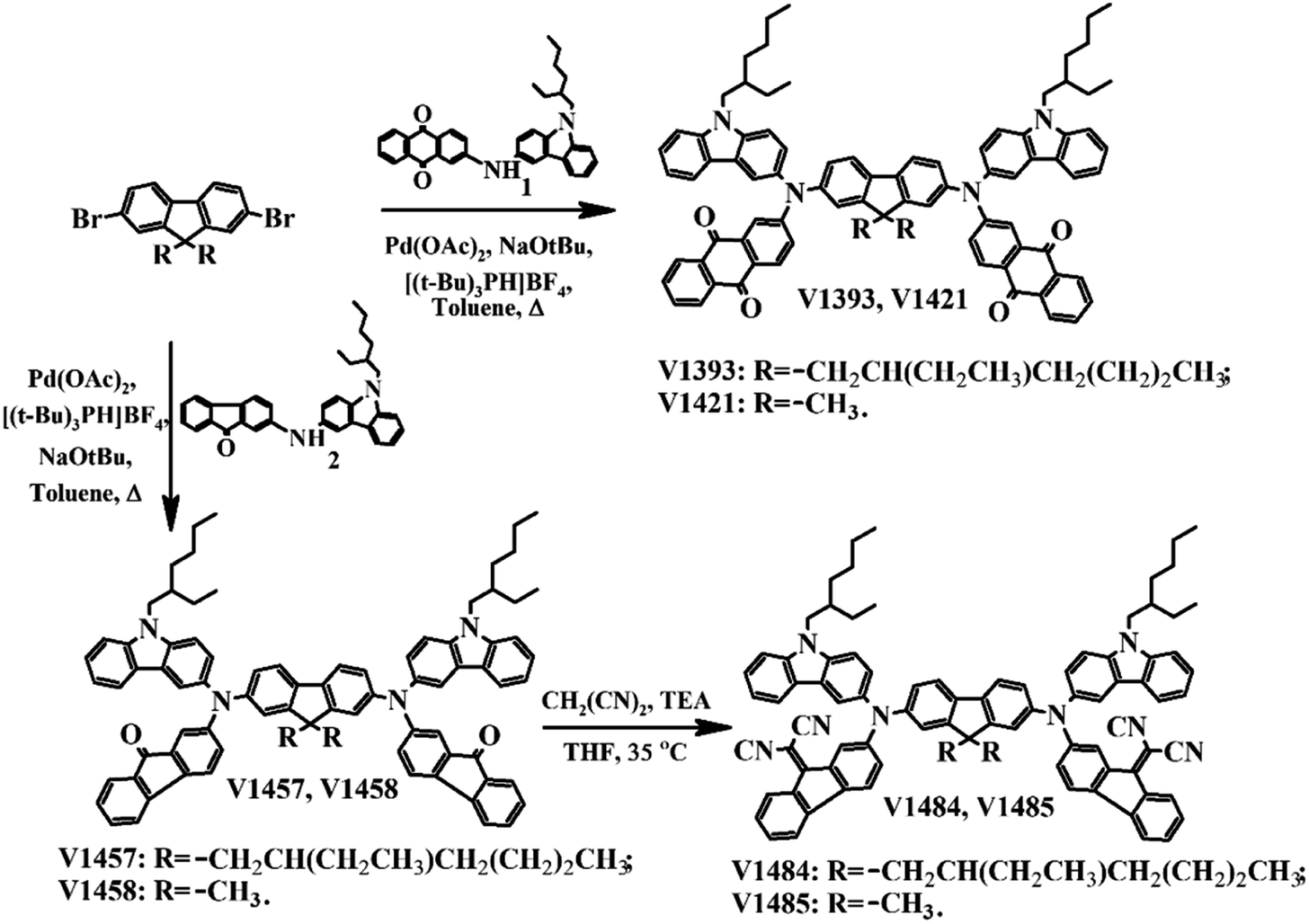 | ||
| Scheme 1 Synthesis of new fluorene-based bipolar compounds V1393, V1421, V1457, V1458, V1484, and V1485. | ||
The chemical structures of the synthesized compounds were verified by NMR spectroscopy, mass spectrometry, elemental analysis, and infrared spectroscopy data. A detailed description of the synthetic procedures and analysis is presented in the ESI.†
Thermal stability of new bipolar molecules was evaluated by thermogravimetric analysis (TGA) in nitrogen atmosphere (Fig. 2). As seen from the data presented in Table 1 and Fig. 2, all bipolar compounds are thermally stable at temperatures higher than 400 °C that is well above operating temperatures of most electronic devices.
 | ||
| Fig. 2 TGA curves of new bipolar molecules. | ||
| Compound | Tga [°C] | Td5a [°C] | λabsb [nm] | Ipc [eV] | HOMOd [eV] | LUMOe [eV] | μ0 holef [cm2 V−1 s−1] | μ0 elec.g [cm2 V−1 s−1] |
|---|---|---|---|---|---|---|---|---|
| a Glass transition (Tg) and 5% weight loss (Td5) temperatures recorded from DSC and TGA, respectively (10 °C min−1, N2 atmosphere).b UV-vis spectra were measured from thin layers.c Ionization energies of the films were measured using PESA.d HOMO energy levels were estimated from the CV measurements.e LUMO energy levels were estimated from the CV measurements.f Hole mobility value at zero field strength.g Electron mobility value at zero field strength. | ||||||||
| V1393 | 109 | 428 | 311, 355, 512 | 5.18 | 5.38 | 3.85 | 2.6 × 10−7 | 4.6 × 10−9 |
| V1421 | 153 | 464 | 311, 352, 514 | 5.21 | 5.51 | 4.0 | 1.4 × 10−7 | 1.1 × 10−9 |
| V1457 | 92 | 423 | 378, 534 | 5.02 | 5.34 | 3.97 | 3.9 × 10−7 | — |
| V1458 | 133 | 485 | 377, 537 | 5.02 | 5.46 | 4.09 | 9.2 × 10−8 | — |
| V1484 | 112 | 406 | 337, 346, 760 | 5.18 | 5.54 | 4.16 | 1.2 × 10−7 | 1.0 × 10−8 |
| V1485 | 161 | 417 | 337, 750 | 5.16 | 5.42 | 4.09 | 2.8 × 10−8 | 3.5 × 10−9 |
The results of differential scanning calorimetry (DSC) measurements are shown in Fig. 3. According to the DSC scans, the glass transition temperatures (Tg) were observed at 109 and 153 °C for V1393 and V1421, respectively. Notably, the Tg of V1457 and V1458 are lower than those of their analogues with anthraquinone moieties (92 °C for V1457 and 133 °C for V1458). The glass transition temperatures increased when the cyano groups were incorporated into the molecules (112 °C for V1484 and 161 °C for V1485). Furthermore, as expected, all materials with longer alkyl side chains demonstrated lower glass transition temperatures than those of the ones with shorter alkyl chains. No endothermic peaks were observed during two heating cycles, what proves that all new materials are amorphous. It is preferable that new bipolar molecules would have an amorphous state in order to obtain good quality films.
 | ||
| Fig. 3 DSC curves for the second run of the bipolar molecules bearing (a) carbonyl groups (V1393, V1421, V1457 and V1458) and (b) cyanoylidene moieties (V1484 and V1485) (heating rate of 10 °C min, N2 atmosphere). | ||
For obtaining more information about electronic structure, quantum chemistry simulations of the molecular structure and several most probable conformers as well as dimers were performed using Gaussian 16 software.41 The density functional (DFT) Cam-B3LYP method and the 6-31G (d) basis set supplemented with polarization functions (d,p) were used for ground state optimization. Solvation effects were not considered in all cases. The list of two most probable molecular conformations is presented in the ESI, Table S1† and xy projections are in Fig. S2–S4.† Generally, total molecular symmetry is absent in the analyzed structures, but in several cases partial symmetry (or quasi-symmetry according to central fluorene unit) could be established (for example, V1421b, V1458b, and V1485b). Substituents are oriented in a chaotic manner, and large number of different conformers could be obtained. The population of low-lying excited molecular states S1 and S2 could be realized using the partially allowed transitions S0 → Sn, n = 1, 2 (Table S2†). The parameters of transition between MO (which are related to the population of “spectroscopic” states Sn, n = 1, 2) are presented in the ESI, Table S3.† In all cases, the dominant and most significant electron jump is provided between HOMO and LUMO. Spatial distributions of electron density (for the HOMO-1, HOMO, LUMO and LUMO+1 of each compound) are presented in the ESI, Tables S4–S6.† Generally, electron transition between HOMO and LUMO corresponds to the charge redistribution between the core (fluorene) and substituents (for example, anthraquinone). In case of both conformers of V1421 and V1393, by population of the lowest excited state, charge redistribution could be named as the closest to that of pure CT. Substituents are present in the role of electron acceptor (charge donor) (Table S4†). The tendencies for both conformers of V1458 and V1457 are the same. The HOMO orbital is localized in the central fluorene unit and in well-oriented substituents (Table S5†). Small changes occur for both conformers of V1485 and V1484. The chaotic orientation of the 2-ethylhexyl- fragment related to the central fluorene unit predetermines the strong non-symmetric displacement of the substituents (Table S6†). Due to that, only the left part of V1484a is involved in significant charge redistribution by excitation.
To understand the nature of electronic excitations, we decided to provide the natural transition orbitals (NTO) analysis using Gaussian16 routine: Density=(Check, Transition=1,2) Pop=(NTO, SaveNTO) for first and second transitions. HOMO and LUMO NTO (which correspond the electronic densities of hole and electron NTO pairs) for allowed first transition are presented in ESI, Tables S7–S9.†
List of several most probable molecular dimers is presented in ESI, Table S10.† Two typical structures in most informative projections are presented in Fig. S23.† First structure of open V-shaped type (dimmer Eh0, V1421b + V1421b) presents amorphous behaviour of low density condensed phase. Intermolecular structure could be organized using weak van-der Waals forces, which relates two molecules through substituents. Second structure of core-to-core type (dimmer Eh2, V1458b + V1458b) represents amorphous behaviour of high density condensed phase. Intermolecular structure could be organized overlap of central core unit of different molecules. Additional relations of two molecules are organized through the substituents. Orientation of two core units is not parallel, but closed to perpendicular (dehidral angle −20 deg).
A comparison of the UV-visible absorption spectra of the new HTMs recorded in THF solution and from solid films is shown in Fig. 4, and the corresponding data are listed in Table 1.
 | ||
| Fig. 4 UV-vis spectra of the target compounds from (a) THF solutions (c = 10−4 mol l−1); (b) solid films. | ||
In general, the spectra of all target compounds display not less than three absorption bands. As expected, the same group molecules, which structures differ in the length of the alkyl chains, present similar absorption profiles from 250 to 900 nm. The spectra of all new compounds display an intensive π–π* absorption bands at 270–450 nm, while the weak absorption band with maximum in the visible region has n–π* nature. While comparing spectra of V1393, V1421, and V1457, V1458 the red shifted absorption peaks can be detected. Stronger absorption for V1393, V1421 around 510 nm is attributed to the presence of additional oxygen atoms. The same phenomenon was observed when dicyanoylide groups were added to the molecule.
When compared to the solution samples, the absorption of film samples exhibits a minimal red shift (∼3 nm) (Fig. 4b), suggesting a significant degree of intermolecular interactions in the solid state. Additionally, this shift supports the presence of aggregates even in low-concentration solutions. Quantum chemistry calculations further validated these findings, indicating that such intermolecular transitions were only feasible in dimer structures. The photoluminescence (PL) measurements were conducted on the solutions and solid layers of materials, but no observable emission was detected.
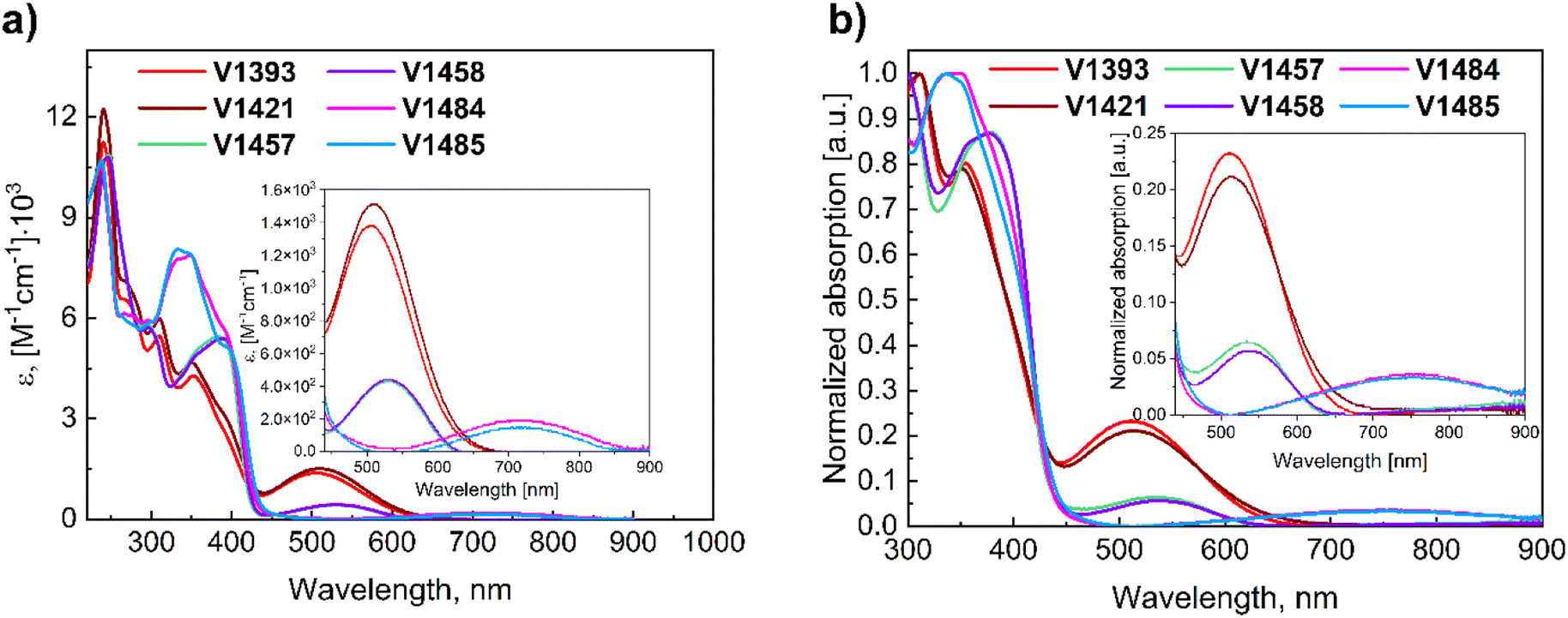
To assess the energy levels of novel bipolar molecules, their oxidation (Eox) and reduction (Ered) potentials in solution and ionization potential (Ip) values in the solid-state were determined using cyclic voltammetry (CV) and photoelectron spectroscopy in air (PESA) techniques, respectively. The experimental results are depicted in Fig. 5, S19,† and Table 1. Materials with anthraquinone moieties have the highest Ip values (5.18 and 5.21 eV for V1393 and V1421, respectively). As expected, the alkyl chain attached to the 9-fluorenyl core practically does not affect Ip. The comparison between molecules containing anthraquinone fragment and molecules containing fluoren-9-one chromophores has revealed compelling evidence that a decrease in the number of electron-withdrawing keto groups within the molecule leads to a reduction in the ionization potential. The opposite effect, i.e. increase of the Ip values, was recorded when cyanoylidene fragments were present in the molecule instead of the keto groups. The inclusion of additional electron-acceptor fragments or strengthening them in this concept resulted in an increase in the ionization potential values.
 | ||
| Fig. 5 Photoemission in air spectra of new compounds with (a) carbonyl groups (V1393, V1421, V1457, and V1458) and (b) cyanoylidene substituents (V1484 and V1485). | ||
The CV measurements (Fig. S19†) have clearly indicated that materials containing anthraquinone exhibit two oxidation peaks and two reduction peaks in a positive region, while compounds incorporating the fluoren-9-one and cyanoylidene groups display three peaks corresponding to oxidation and reduction processes. Comparison of V1393, V1421 with V1457, V1458 has apparently revealed that reducing the number of oxygen atoms in the molecule results in a slight decrease in the oxidation potential (Eox) value. Other modifications in the structures have a minimal effect on the Eox values.
The CV of the analyzed compounds has demonstrated reversible reduction during the scanning process in a negative region, indicating the electrochemical stability of the synthesized materials. It is important to note that these values do not represent absolute electron affinity or ionization energies in the solid state, but they can be utilized to compare different compounds relative to each other. The estimated ranges for the LUMO and HOMO energy levels of the novel compounds were found to be between −3.85 eV and −4.16 eV, and between −5.02 eV and −5.21 eV, respectively.
The hole and electron mobility is a fundamental parameter when bipolar molecules are evaluated. To analyze the charge transport properties of novel compounds, time-of-flight measurements (XTOF) were employed; the transient signals are presented in the ESI, Fig. S24–S29.† These measurements were conducted in films of the pure compounds, and the obtained mobility dependencies on electrical field are presented in Fig. 6.
 | ||
| Fig. 6 Electric field dependencies of the new materials containing (a) anthraquinone and fluorenone functional groups (V1393, V1421, V1457, and V1458) and (b) cyanoylidene group (V1484 and V1485). | ||
The results have clearly indicated that molecules containing anthraquinone fragments (V1393, V1421) and cyanoylidene substituents (V1484, V1485) exhibit both negative and positive charge transport capabilities with hole mobility reaching values of one order of magnitude higher than those of electrons. However, no electron-clear transients were obtained in compounds V1457 and V1458 bearing 9-fluorenone chromophores as electron acceptors. The only noticeable effect of longer alkyl chains (V1393, V1457, and V1484) was a slight improvement of hole mobilities at low electric fields, thus a case could be made that longer alkyl chains may be beneficial for more optimal molecular arrangement for charge transport. Replacement of the keto groups with cyanoylidene groups resulted in decreased hole drift mobility and increased electron mobility. The most balanced charge mobility has been recorded in the bipolar molecules V1484 and V1485, where the hole mobility is 1.2 × 10−7 cm2 V−1 s−1, and the electron mobility is 1.0 × 10−8 V−1 s−1 at near zero electric fields.
Photocurrent decay measurements were conducted to gain deeper insights into charge transport within the layers. The applied electrical field was insufficient to facilitate the extraction of charge carriers from the layer, causing photocurrent decay to be predominantly influenced by charge carrier trapping, detrapping, and recombination processes.42,43
The resulting profiles were normalized to compare the photocurrent tail regions of decay (ranging from 2 × 10−5 to 1 × 10−4 s), which primarily were attributed to deep traps and recombination mechanisms. In layers featuring anthraquinone fragments (Fig. 7b), the photocurrent decay profiles for both holes and electrons exhibit similar kinetics with minimal observable impact from additional alkyl groups. On the contrary, the most significant alterations in the slower decay region were observed when altering the length of the alkyl chains in compounds containing fluorenone groups (V1458, V1457) (Fig. 7a). The lengthening of the alkyl chains resulted in a reduced influence of trapping and detrapping on hole transport and increased recombination, as evidenced by the significantly lower hole mobilities observed in V1458. However, electron photocurrent decay seemed to exhibit only marginal sensitivity to the same alkyl chain modifications. Compounds featuring cyanoylidine fragments (V1484, V1485) (Fig. S30†) displayed a similar improvement when additional alkyl chains were introduced. This led to slower electron photocurrent decay alongside higher electron mobilities. These findings underscore the importance of additional alkyl groups in optimizing the molecular arrangement within the layer, subsequently enhancing charge carrier transport. Notably, the discernible differences in hole and electron current decays corroborate the discrepancies found in charge carrier mobilities.
 | ||
| Fig. 7 Photocurrent decay profiles of compound films containing (a) fluorenone functional groups (V1457 and V1458) and (b) anthraquinone groups (V1393 and V1421). | ||
Conclusions
New amorphous bipolar fluorine-based materials featuring anthraquinone, 9-fluorenone, or 9-dicyanofluorenylidine groups along with attached carbazolyl electron-donating moieties have been developed and synthesized via a straightforward synthesis. These amorphous materials exhibit homogeneous, cohesive, and continuous film-formation properties for solvent processing. Optical absorption and quantum chemistry simulations have confirmed the formation of a CT complex within the molecule, along with the emergence of a few of the most probable dimer structures, specifically V-type and core-to-core type structures. According to quantum chemistry calculations, several stable conformers are possible due to a complex geometry of the molecules.The hole mobility in all materials is around 10−4-10−5 cm2 V−1 s−1, while the electron mobility in compounds with anthraquinone and 9-dicyanofluorenylidine groups is approximately one order of magnitude lower. Electron transport in compounds with 9-fluorenone was highly dispersive and clear kink in photocurrent was not observed.
Author contributions
Aistė Jegorovė: writing – original draft, synthesis and characterization of materials; Marytė Daškevičienė: conceptualization, synthesis and characterization of materials; Kristina Katminienė: writing – original draft; Vygintas Jankauskas: project administration, supervision, investigation; Romualdas Jonas Čepas: investigation, formal analysis, visualization, writing – original draft; Alytis Gruodis: formal analysis, investigation; Vytautas Getautis: conceptualization, supervision; Kristijonas Genevičius: investigation, formal analysis, writing – review & editing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was funded by a grant (No. S-MIP-22-8) from the Research Council of Lithuania. Computations were performed on resources at the High Performance Computing Center “HPC Saulėtekis” (Faculty of Physics, Vilnius University, Lithuania). Dr E. Kamarauskas is acknowledged for ionisation potential measurements.References
- A. Bessette and G. S. Hanan, Chem. Soc. Rev., 2014, 43, 3342–3405 RSC.
- A. Wadsworth, M. Moser, A. Marks, M. S. Little, N. Gasparini, C. J. Brabec, D. Baran and I. McCulloch, Chem. Soc. Rev., 2019, 48, 1596–1625 RSC.
- A. B. Muñoz-García, I. Benesperi, G. Boschloo, J. J. Concepcion, J. H. Delcamp, E. A. Gibson, G. J. Meyer, M. Pavone, H. Pettersson, A. Hagfeldt and M. Freitag, Chem. Soc. Rev., 2021, 50, 12450–12550 RSC.
- M. Chen, L. Yan, Y. Zhao, I. Murtaza, H. Meng and W. Huang, J. Mater. Chem. C, 2018, 6, 7416–7444 RSC.
- H. Jiang, S. Zhu, Z. Cui, Z. Li, Y. Liang, J. Zhu, P. Hu, H.-L. Zhang and W. Hu, Chem. Soc. Rev., 2022, 51, 3071–3122 RSC.
- Y. Weng, Z. Yu, T. Wu, L. Liang and S. Liu, New J. Chem., 2023, 47, 5086–5109 RSC.
- A. J. Shahnawaz, S. S. Swayamprabha, M. R. Nagar, R. A. K. Yadav, S. Gull, D. K. Dubey and J.-H. Jou, J. Mater. Chem. C, 2019, 7, 7144–7158 RSC.
- M. Sheokand, Y. Rout and R. Misra, J. Mater. Chem. C, 2022, 10, 6992–7017 RSC.
- K.-L. Wang, J.-C. Jiang, C.-H. Jhu, S. Wada, T. Sassa and M. Horie, J. Mater. Chem. C, 2020, 8, 13357–13367 RSC.
- D. S. Weiss, J. Imaging Sci. Technol., 2016, 60, 1–24 CrossRef.
- D. S. Weiss and M. Abkowitz, Chem. Rev., 2010, 110, 479–526 CrossRef CAS PubMed.
- Y. Zhang, Y. Wang, C. Gao, Z. Ni, X. Zhang, W. Hu and H. Dong, Chem. Soc. Rev., 2023, 52, 1331–1381 RSC.
- E. J. Meijer, D. M. de Leeuw, S. Setayesh, E. van Veenendaal, B.-H. Huisman, P. W. M. Blom, J. C. Hummelen, U. Scherf and T. M. Klapwijk, Nat. Mater., 2003, 2, 678–682 CrossRef CAS PubMed.
- C. Rost, D. J. Gundlach, S. Karg and W. Rieß, J. Appl. Phys., 2004, 95, 5782–5787 CrossRef CAS.
- T. D. Anthopoulos, D. M. de Leeuw, E. Cantatore, S. Setayesh, E. J. Meijer, C. Tanase, J. C. Hummelen and P. W. M. Blom, Appl. Phys. Lett., 2004, 85, 4205–4207 CrossRef CAS.
- A. Dodabalapur, H. E. Katz, L. Torsi and R. C. Haddon, Science, 1995, 269, 1560–1562 CrossRef CAS PubMed.
- Y. Yamaguchi, T. Fujiyama, H. Tanaka and M. Yokoyama, Denshi Shashin Gakkaishi, 1991, 30, 274–281 CAS.
- Y. Kunugi, K. Takimiya, N. Negishi, T. Otsubo and Y. Aso, J. Mater. Chem., 2004, 14, 2840–2841 Search PubMed.
- N. Negishi, Y. Ie, H. Tada, T. Kaneda and Y. Aso, Chem. Lett., 2007, 36, 544–545 Search PubMed.
- J.-F. Nierengarten, T. Gu, T. Aernouts, W. Geens, J. Poortmans, G. Hadziioannou and D. Tsamouras, Appl. Phys. A, 2004, 79, 47–49 Search PubMed.
- S. Amriou, A. Mehta and M. R. Bryce, J. Mater. Chem., 2005, 15, 1232–1234 CAS.
- S. Urnikaite, T. Malinauskas, V. Gaidelis, V. Jankauskas and V. Getautis, Chem.–Asian J., 2012, 7, 614–620 CrossRef CAS PubMed.
- S. Urnikaite, T. Malinauskas, V. Gaidelis, R. Maldzius, V. Jankauskas and V. Getautis, Carbon, 2011, 49, 320–325 Search PubMed.
- W. W. H. Wong, D. Vak, Th. B. Singh, S. Ren, C. Yan, D. J. Jones, I. I. Liaw, R. N. Lamb and A. B. Holmes, Org. Lett., 2010, 12, 5000–5003 Search PubMed.
- Y. Chen, T. Liu, H. Hu, T. Ma, J. Y. L. Lai, J. Zhang, H. Ade and H. Yan, Adv. Energy Mater., 2018, 8, 1801203 Search PubMed.
- L. Gao, Z.-G. Zhang, H. Bin, L. Xue, Y. Yang, C. Wang, F. Liu, T. P. Russell and Y. Li, Adv. Mater., 2016, 28, 8288–8295 CrossRef CAS PubMed.
- Y. Zhang, C. Kim, J. Lin and T.-Q. Nguyen, Adv. Funct. Mater., 2012, 22, 97–105 CrossRef CAS.
- L. Wang, X. Zhang, H. Tian, Y. Lu, Y. Geng and F. Wang, Chem. Commun., 2013, 49, 11272–11274 Search PubMed.
- A. Riaño, P. M. Burrezo, M. J. Mancheño, A. Timalsina, J. Smith, A. Facchetti, T. J. Marks, J. T. L. Navarrete, J. L. Segura, J. Casado and R. P. Ortiz, J. Mater. Chem. C, 2014, 2, 6376–6386 RSC.
- T. Bura, N. Leclerc, R. Bechara, P. Lévêque, T. Heiser and R. Ziessel, Adv. Energy Mater., 2013, 3, 1118–1124 CrossRef CAS.
- Y. Lin, L. Ma, Y. Li, Y. Liu, D. Zhu and X. Zhan, Adv. Energy Mater., 2013, 3, 1166–1170 CrossRef CAS.
- H. Bai, P. Cheng, Y. Wang, L. Ma, Y. Li, D. Zhu and X. Zhan, J. Mater. Chem. A, 2013, 2, 778–784 RSC.
- H. Shi, W. Fu, M. Shi, J. Ling and H. Chen, J. Mater. Chem. A, 2015, 3, 1902–1905 RSC.
- Q. Wu, L. Li, J. Hai, X. Zhang, Z. Lu, J. Yang, Y. Liu, L. Zhang and C. Zhan, Dyes Pigm., 2016, 132, 41–47 CrossRef CAS.
- G. Balaji, T. S. Kale, A. Keerthi, A. M. Della Pelle, S. Thayumanavan and S. Valiyaveettil, Org. Lett., 2011, 13, 18–21 Search PubMed.
- Y. Wang, J. H. Yun, L. Wang and J. Y. Lee, Adv. Funct. Mater., 2021, 31, 2008332 CrossRef CAS.
- M. Reig, C. Gozálvez, V. Jankauskas, V. Gaidelis, J. V. Grazulevicius, L. Fajarí, L. Juliá and D. Velasco, Chem.–Eur. J., 2016, 22, 18551–18558 CrossRef CAS PubMed.
- A. Bobet, A. Cuadrado, L. Fajarí, I. Sirés, E. Brillas, M. P. Almajano, V. Jankauskas, D. Velasco and L. Juliá, J. Phys. Org. Chem., 2019, 32, e3974 CrossRef.
- K. S. Kim, D. U. Kim, K. S. Joung and J.-W. Yu, Thin Solid Films, 2020, 696, 137781 CrossRef.
- M. Daskeviciene, S. Urnikaite, G. Planciunaite, T. Malinauskas, A. Gruodis, M. Viliunas, E. Kamarauskas, V. Gaidelis, V. Jankauskas and V. Getautis, Dyes Pigm., 2018, 158, 157–164 CrossRef CAS.
- Citation | Gaussian.com, https://gaussian.com/citation/, (accessed October 26, 2023) Search PubMed.
- S. Valouch, M. Nintz, S. W. Kettlitz, N. S. Christ and U. Lemmer, IEEE Photonics Technol. Lett., 2012, 24, 596–598 CAS.
- C. R. McNeill, I. Hwang and N. C. Greenham, J. Appl. Phys., 2009, 106, 024507 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra07583d |
| This journal is © The Royal Society of Chemistry 2024 |