
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoluminescence switching in quantum dots connected with fluorinated and hydrogenated photochromic molecules†
Ephraiem S. Sarabamouna,
Jonathan M. Bietschb,
Pramod Aryalb,
Amelia G. Reidc,
Maurice Curran d,
Grayson Johnsond,
Esther H. R. Tsaie,
Charles W. Machanc,
Guijun Wang*b and
Joshua J. Choi*d
d,
Grayson Johnsond,
Esther H. R. Tsaie,
Charles W. Machanc,
Guijun Wang*b and
Joshua J. Choi*d
aDepartment of Physics, University of Virginia, Charlottesville, VA 22904, USA
bDepartment of Chemistry and Biochemistry, Old Dominion University, Norfolk, VA 23529, USA. E-mail: g1wang@odu.edu
cDepartment of Chemistry, University of Virginia, PO Box 400319, Charlottesville, VA 22904, USA
dDepartment of Chemical Engineering, University of Virginia, Charlottesville, VA 22904, USA. E-mail: jjc6z@virginia.edu
eCenter for Functional Nanomaterials, Brookhaven National Laboratory, Upton, NY 11973, USA
First published on 2nd January 2024
Abstract
We investigate switching of photoluminescence (PL) from PbS quantum dots (QDs) crosslinked with two different types of photochromic diarylethene molecules, 4,4'-(1-cyclopentene-1,2-diyl)bis[5-methyl-2-thiophenecarboxylic acid] (1H) and 4,4'-(1-perfluorocyclopentene-1,2-diyl)bis[5-methyl-2-thiophenecarboxylic acid] (2F). Our results show that the QDs crosslinked with the hydrogenated molecule (1H) exhibit a greater amount of switching in photoluminescence intensity compared to QDs crosslinked with the fluorinated molecule (2F). With a combination of differential pulse voltammetry and density functional theory, we attribute the different amount of PL switching to the different energy levels between 1H and 2F molecules which result in different potential barrier heights across adjacent QDs. Our findings provide a deeper understanding of how the energy levels of bridge molecules influence charge tunneling and PL switching performance in QD systems and offer deeper insights for the future design and development of QD based photo-switches.
Introduction
A photo-switch is a device which can reversibly switch between two discrete states upon illumination. Such systems have been investigated widely for various applications, including both as a potential biomarking and drug delivery system (photo-pharmacology),1–4 and in photonics as optical analogues to transistors.5,6 Recently, the combination of photochromic molecules (PCMs) with quantum dots (QDs) has emerged as a promising approach to developing new photo-switch devices.7–9 These hybrid systems exploit the high quantum yield of quantum dots, and the reversible photo-induced transformation capabilities of photochromic molecules to produce bright, light-controlled switches.Photochromic molecules are a class of compounds that exhibit reversible isomerization between two distinct forms upon irradiation with light of specific wavelengths. These molecules have a wide range of applications, including in data storage/optical memory, bio imaging, and high sensitivity optical switches.10–15 PCMs have also been used extensively in the nanomaterials field where they provide a mechanism to indirectly control the assembly and properties of a nanomaterial system using light which is noninvasive and allows for a high level of remote spatiotemporal resolution.16 PCMs have been used in conjunction with nanoparticles (NPs) to switch a NP catalyst on/off,17 to aggregate NPs and disperse them,18,19 to control the fluorescence levels of NPs between two states (both by using Förster resonance energy transfer (FRET)7,20 and charge tunneling8), to switch a NP system's magnetization,21,22 conductivity,23 and even to turn superconductivity of materials on/off.24 Among the different classes of photochromic molecules, diarylethenes have emerged as particularly interesting due to their good thermal stability, fatigue resistance, and ultra-fast switching speeds,25–28 making them attractive for use as the PCM of choice in photo-switching systems. Quantum dots (QDs), on the other hand, since their original discovery in the 1980s,29 have garnered significant attention due to their high quantum yield and size-dependent optoelectronic properties, making them promising candidates for various applications, including in solar cells, light-emitting diodes, and biosensors.30–38 PbS quantum dots, in particular, have emerged as a versatile material for near-infrared optoelectronics, owing to their tunable bandgaps and exceptional photostability.39
In the literature, QD/PCM photo-switches traditionally employed FRET as the mechanism for switching.7,20,40,41 While FRET has proven effective in inducing photo-switching, its applicability is limited as it requires the alignment of the PCM absorbances with the photoluminescence (PL) of the quantum dots. Recently, Yoon et al. proposed an alternative mechanism for achieving photo-switching by utilizing charge tunneling between PCM bridged PbS QDs.8 For this mechanism, the energy levels of the bridging PCM ligands can be thought of as barriers for charge tunneling between neighboring quantum dots.42 While inter-QD charge tunneling has previously been studied for controlling optical properties36,43 and conductivity23,44,45 in similar systems, this work represents a novel exploration of utilizing charge tunneling between QDs as a mechanism for photo-switching. This switching mechanism is significant since it allows for the easy fabrication of NIR, light controlled photo-switches with non-destructive read-out (UV and visible light are used to prepare the state of the system and NIR light is used to probe the state).
In this paper, we demonstrate that the amount of observed PL switching in a tunneling controlled PCM/QD system can be modified by chemically modulating the energy levels of the bridging molecule. This is accomplished by contrasting the PL switching of the QDs linked by 4,4'-(1-cyclopentene-1,2-diyl)bis[5-methyl-2-thiophenecarboxylic acid] (1H) and 4,4'-(1-perfluorocyclopentene-1,2-diyl)bis[5-methyl-2-thiophenecarboxylic acid] (2F). By fluorinating the cyclopentene ring, we hypothesized that the charge transfer barrier between the QDs could be altered to tune the relative PL switching ratios in our system without affecting the binding between the QDs and the ligands (Fig. 1). Our work demonstrates that the energy level of the bridging molecule can be used as a parameter to modulate the amount of PL switching, and through systematic comparison and analysis, we aim to shed light on the underlying structure–property relationships and provide guidance for future studies and applications in the field of quantum dot-based photo-switchable systems.
 | ||
| Fig. 1 Schematic summarizing the key mechanism investigated in this work. UV and visible light are used to change the configuration of a hydrogenated or a fluorinated photochromic molecule between its open and closed configurations. The difference in energy levels of the molecules results in a greater or lesser tunneling rate thus affecting the amount of PL switching observed in our system. | ||
Results and discussion
For the following experiment, hydrogenated (1H) and fluorinated (2F) dithienylethene molecules (dithienylethenes are a subclass of diarylethenes where the aromatic subunits are substituted with thiophene moieties) were synthesized (see ESI† for details on synthesis and characterization of the molecules Fig. S1–S4†). Both molecules exhibit a ring closure isomerization upon UV illumination and a ring opening isomerization upon visible light illumination. The configuration change of the molecules is accompanied by a color change. Absorbance measurements of the two molecules clearly show the change of configuration after light exposure (Fig. 2b and c). For 1H, exposure at 300 nm resulted in peak formations around 350 nm and 540 nm. After exposure at 540 nm, the 350 nm and 540 nm peaks were suppressed, and a new peak formed around 300 nm. For 2F, 350 nm exposure resulted in peak formations around 380 nm and 590 nm. After exposure at 590 nm, those peaks were suppressed, and a new peak formed around 340 nm. Comparing these absorption peaks with the PL peak of the QDs (Fig. S5†), which occurs around 1060 nm, rules out the FRET based switching mechanism previously reported in the literature, since that mechanism necessitates an overlap of the PL peak of the florescent specie (QD in this case) with the absorption peak of the absorbing specie (the PCMs). Our absorbance results for both molecules, in the free state in solution, show near complete switching at least for the open configuration of the molecules as evidenced by the almost complete disappearance of the absorbance peak in the visible wavelength range after visible light illumination. It is important to note, however, that this does not guarantee that the configuration change will be equally complete for the photostationary states of the PCMs when bound to the QD surface. | ||
| Fig. 2 (a) Molecular structure of the “open” and “closed” configurations of 1H and 2F. Absorbance spectra of (b) 1H and (c) 2F in methanol after UV and visible light illumination. | ||
PbS QDs were synthesized and deposited as thin films on glass in a spin-coating procedure. The absorbance and PL spectra of these QDs are given in the ESI (Fig. S5†). Oleate ligands from the QD synthesis were removed and the QDs were crosslinked with the dithienylethene photochromic molecules through a ligand exchange procedure in methanol (see ESI† for detailed ligand exchange procedure). Though the functionalization efficiency of the two ligands could not be directly quantified in the solid state, since the ligands have identical binding groups, it was assumed that the amounts of ligand exchange for 1H and 2F were comparable.
We also note that since inter-QD tunneling is our mechanism of PL switching, the amount of PL quenching will be dominated by the available nearest neighbor tunneling pathways and will not be significantly affected by adding more tunneling pathways. This implies that the results presented are unlikely to be due to differences in the number of ligands bound to the QD surface.
Absorbance spectra were taken of the QD crosslinked films in both configurations of the PCMs to ensure that film absorbances were not affected by PCM configuration change especially at the excitation wavelength of 750 nm for the QDs (Fig. S6†). PL measurements (Fig. 3a and b) of the PCM linked films were taken after exposing the film to monochromatic UV or visible light. Comparison with the as-synthesized QDs in solution shows that the PL peak of the QDs redshifts by ∼40 nm after the ligand exchange procedure. The crosslinked samples were exposed to UV and visible light to switch the configuration of the ligands between the “open” and the “closed” states respectively. Following each exposure, the photoluminescence (PL) of the quantum dots (QDs) was measured. As predicted, both 1H and 2F crosslinked QD samples showed a significant increase in QD PL after visible light exposure and a decrease in PL after UV exposure. Samples were exposed till they reached the asymptotic maximum and minimum PL intensity (Fig. S7†). This was done to minimize any effect from potential differences in the configuration switching rates of the two ligands. The samples were exposed to two consecutive cycles of UV and visible light exposure. The maximum peak intensity was retained showing that the switching is reversible (Fig. 3a and b). Interestingly, the PL switching effect is larger for the 1H crosslinked film than for 2F. To quantify the difference, we define the “relative PL change” (A) which represents the integrated PL area of the system with the open PCM divided by the integrated PL area of the system with the closed PCM. Using this metric, the average relative PL change for 1H is ∼4.75 whereas the relative PL change for 2F is ∼2.77.
 | ||
| Fig. 3 PL spectra of (a) 1H and (b) 2F crosslinked QD films after successive cycles of UV and visible light illumination. (c) Measured switching amounts of three samples of QDs crosslinked with molecules 1H or 2F. (Raw spectra are presented in Fig. S8 in the ESI†). | ||
Lifetime measurements of both our 1H and 2F crosslinked samples after visible and UV light exposure were also taken (Fig. S9†). Measurements showed shorter excitonic lifetimes in the closed configuration of the PCMs which is consistent with an increase in the charge tunneling rate in the closed configuration of the PCMs. The change in excitonic lifetimes upon configuration change was greater for the hydrogenated than for the fluorinated ligand crosslinked sample. This is consistent with the greater observed PL change for the hydrogenated crosslinked sample seen in Fig. 3c.
In order to further support inter-QD tunneling as the mechanism of PL switching as opposed to some other mechanisms such as energy transfer from QDs to surface ligands, and to demonstrate that crosslinked QDs are necessary for our observed switching effect, a mono-esterified form of molecule 1H was synthesized (Fig. S10†). The absorbance of the free mono-esterified ligand in both its open and closed states (Fig. S10†) were similar to the absorbance of molecule 1H (Fig. 2b). Two solutions were prepared, by adding molecule 1H or the mono-esterified ligand to the QDs in solution as described in the experimental methods section. When measured, the solution with the 1H molecules showed significant switching in PL intensity of QDs (Fig. S11a†) while the solution with the mono-esterified ligand molecules did not show any PL switching (Fig. S11b†). These results suggest that the observed PL switching in this work does require the crosslinking of QDs and is likely not due to energy transfer to surface ligands.
Since the tunneling process is dependent on both changes in barrier width and barrier height, grazing incidence small angle X-ray scattering (GISAXS) measurements were taken of our bridged QDs in both the open and closed configurations to measure the inter-QD distances (Fig. S12 and Table S1†). Our GISAXS measurements show that both 1H and 2F molecules have length of ∼1 nm and change less than 1 Å upon opening and closing of the rings. These results closely match previous values in literature.8,46 The results suggest that barrier width change is minimal and is similar for both molecules thus ruling out change in barrier width as a potential explanation for the difference in PL switching amounts for the two molecules.
To investigate the effects of changes in PCM energy levels on the PL switching behavior of our system, the energy levels of isolated 1H and 2F were experimentally measured using differential pulse voltammetry (DPV) in acetonitrile and calculated using density functional theory (DFT). Focusing on the DPV reduction spectra of 1H and 2F (Fig. 4a and b), new peak(s) was observed to form for both molecules after UV irradiation. The peaks at the more positive potentials are associated with the LUMO level of the molecules. For 1H, an E1/2 of −1.43 V vs. Fc+/Fc is observed (see experimental methods section for more details) which corresponds to a LUMO level of −2.97 eV. This value, taken with the molecular absorbance from Fig. 2, gives a HOMO level of −5.25 eV for the closed state of 1H. For the open state, 1H has a peak around −1.1 V, which gives a LUMO level of −2.14 eV and a HOMO level of −6.26 eV. 1H results match those that have appeared previously in literature.47 For 2F, a peak forms around −1.02 V. This value corresponds to a LUMO level of −3.3 eV. This, taken with the absorption peak data, gives a HOMO level of −5.43 eV for the closed state of 2F. For the open state, 2F has its first peak around −2.22 V, which corresponds to a LUMO level of −2.51 eV and a HOMO level of −6.64 eV.
 | ||
| Fig. 4 Reduction DPV plots for (a) 1H and (b) 2F. (c) Energy level alignment of the system. QD energy was obtained from literature (3.2 nm PbS)48 and energies of 1H and 2F were determined from our DPV and absorbance results. | ||
Oxidation results are given in the ESI (Fig. S13†). To further investigate the relationship between the energy levels and the difference in the amount of PL switching, we can use the results from our DPV measurements and literature values for the energy levels of the quantum dots48 to graph the energy level alignment of our system (Fig. 4c). It is important to note that the given energy levels of the PCMs are only those of the free state ligands as measured using DPV and electronic absorbance spectroscopy, and therefore do not account for any potential energy level shift due to quantum dot surface binding. Since both PCMs have identical binding groups; however, we assume that the magnitude of the energy level shift for both molecules should be similar. This suggests that the relative difference between the energy levels of the two free molecules especially in the regime of small energy change is a useful parameter based on which accurate insights into our system can be made.
One such insight can be obtained by considering the energy level alignment of our system (Fig. 4c) in light of the following equation derived in the mathematical background section of the ESI.†
 | (1) |
Considering the LUMO levels in Fig. 4c, we notice that molecule 2F's Ealignment is 0.2 eV smaller than 1H, while Egap for the two molecules are virtually identical (difference of 0.03 eV between the two). From eqn (1), we see that such an energy alignment would result in a larger switching amount for 2F than 1H regardless of EQD. This suggests that electron tunneling through the LUMO level of the PCMs cannot account for the observed PL switching behavior, where QDs crosslinked with 1H have greater PL switching than those crosslinked with 2F. Considering next the HOMO level alignment of our system, we notice that molecule 1H's Ealignment value is about 0.18 eV smaller than 2F, while Egap is 0.2 eV larger for 2F as compared to 1H. Since, 1H's Ealignment value is smaller, hole tunneling can, in theory, account for the observed PL switching behavior of our system. This suggests that tunneling in our system is likely hole-dominated. Therefore, eqn (1) above must be applied to the HOMO levels of our system with EQD representing the valence band maximum of the QD energies, and EPCM representing the HOMO energies of the PCMs.
Density functional theory (DFT) calculations were run on both the hydrogenated (1H) and the fluorinated (2F) molecules to calculate the energy levels of both PCMs in their open and closed configurations. The electronic structure of the molecules was characterized using a hybrid exchange-correlation potential using the Becke and Hartree–Fock exchange and Lee, Yang, and Parr correlation (B3LYP)26 in the Gaussian03 package.49,50
The calculated absorbance spectra in Fig. 5 show good agreement with the experimentally determined absorbance values in Fig. 2. The orbital diagrams show similar orbital structure for the two molecules with few differences. Most notably, the frontier orbitals of 2F have a greater contribution from the cyclopentene ring due to the incorporation of fluorine, which stabilizes the HOMO and LUMO energies as compared to 1H. The generated DFT energy levels (Table S2†) demonstrated the same relative differences in 1H vs. 2F energy levels as the DPV results.
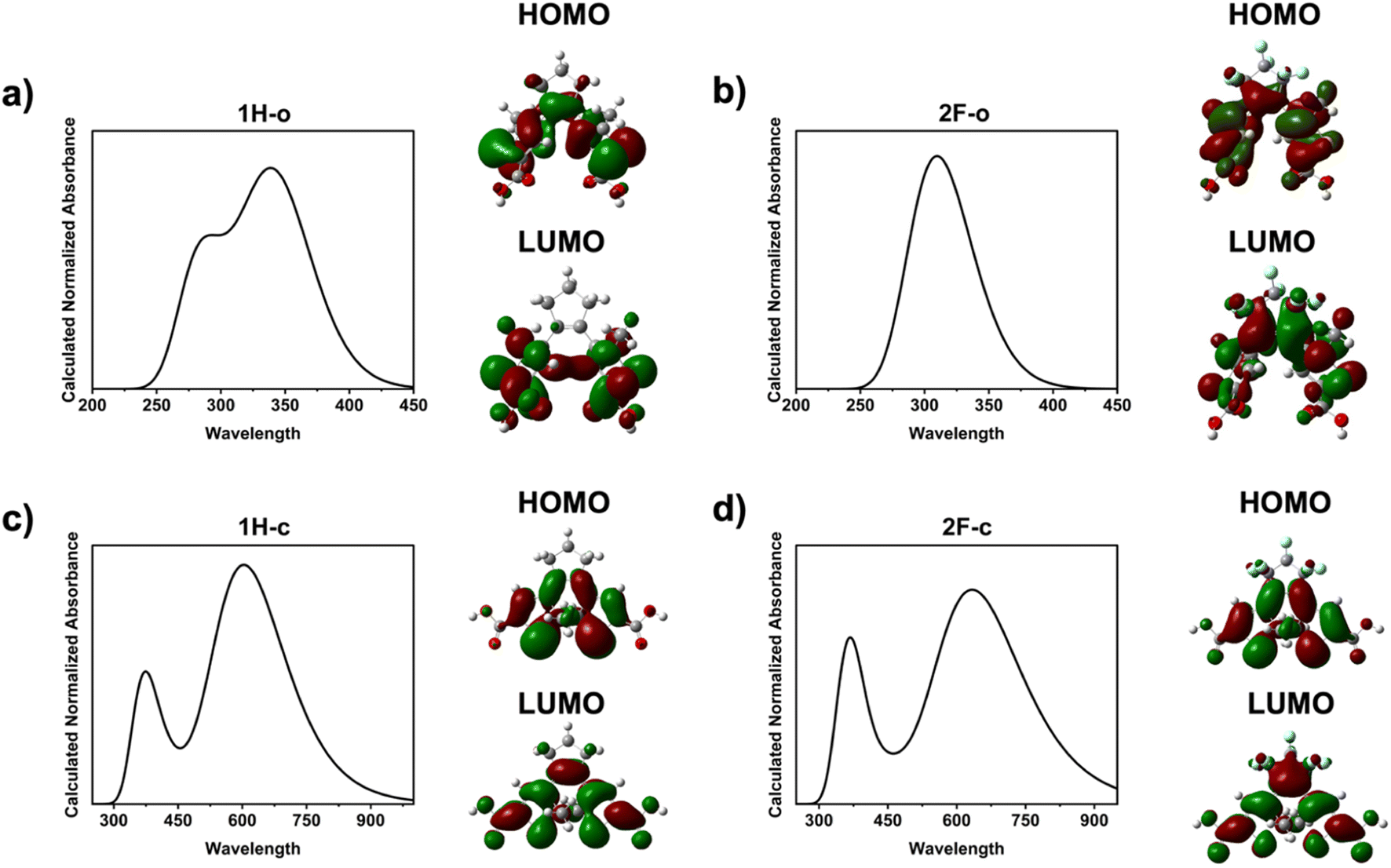 | ||
| Fig. 5 Calculated absorbances and orbitals (both HOMO and LUMO orbitals) for open molecules (a) 1H and (b) 2F, and closed molecules (c) 1H and (d) 2F. | ||
Therefore, eqn (1) can be used as a guide to suggest QD/diarylethene tunneling based photo-switches for future investigation. To maximize the relative switching amount of these photo-switches, QDs and PCMs should be chosen to minimize Ealignment while maximizing Egap. The maximum possible value for A occurs when Ealignment = 0 and Egap is large, in which case A = 1 + γ. γ can be maximized by choosing QDs with smaller sizes but longer excitonic lifetimes (this can be done by improving surface passivation). If the QDs are sufficiently small, however, then Ealignment can become negative in value. In this regime, tunneling will no longer become the mechanism of switching, instead excitonic decay can be dominated by the triplet energy transfer (TET) process. This mechanism was recently demonstrated by Hou et al.51 to provide almost complete PL suppression in small PbS quantum dots with large bandgaps. For switches employing QDs with smaller bandgaps in the deeper NIR regime, however, inter-QD tunneling based switching as demonstrated in this work will be a dominant mechanism.
Conclusion
In this work, we have investigated the photo-switching behavior of two types of photochromic molecules bridging PbS quantum dots. We have shown that the energy levels of the photochromic molecules can play a critical role in the photo-switching properties of the composite system. Our results indicate that the hydrogenated molecule (1H) exhibits a greater photo-switching effect than the fluorinated molecule (2F), which when considered with the DPV and DFT obtained energy levels of the molecules, suggests that the tunneling of our system is likely hole-dominated. Our findings provide a deeper understanding of the energy-level effects on the photo-switching performance of this quantum dot-based system and offer insights for the future design and development of tunneling based hybrid QD/PCM photo-switches.Experimental methods section
General method and procedure for the photochromic molecules' syntheses
All reactions were carried out under anhydrous conditions and under inert atmosphere (argon or nitrogen) utilizing a Schlenk system. Glassware was flame dried and cooled in a desiccator before use. All reagents and solvents were obtained from commercial suppliers and used directly without further purifications. Product purification was carried out via column chromatography with 230–400 mesh silica gel using isocratic or gradient solvent systems. The synthetic procedures are detailed in the ESI.† Reaction monitoring was carried out using thin-layer chromatography (TLC) with fluorescence indicator. TLC was visualized under 254 nm UV light and through PMA staining. Compound characterization was carried out through 1H NMR and proton-decoupled 13C NMR utilizing a Bruker 400 MHz NMR spectrometer in d6-DMSO or CDCl3. The CDCl3/DMSO-d6 peaks were calibrated at 7.26/2.50 ppm and at 77.0/39.5 ppm, in the 1H NMR and 13C NMR respectively. Further structural characterization was carried out via 19F NMR and 2D NMR experiments such as HSQC and COSY.PbS QD synthesis
To synthesize the PbS QD batch needed for this experiment, lead(II) oxide (3 mmol, 99.999%, Alfa Aesar) was mixed with 30 mmol of oleic acid (90%, Sigma Aldrich) in a three-neck flask. An appropriate amount of 1-octadecene (90%, Alfa Aesar) was added to bring the total volume of the solution to 30 mL. The solution was stirred in the flask for 1 hour under vacuum at 110 °C during which time it turned transparent. Under argon gas flow, the temperature was adjusted to 90 °C in preparation for injection. In an N2-filled glovebox, 0.1 M of hexamethyldisilathiane ((TMS)2S) solution was prepared by mixing 378 μL of (TMS)2S in 18 mL of 1-octadecene (ODE). 15 mL of the 0.1 M (TMS)2S solution was taken out of the glovebox and quickly injected into the three-neck flask. After 70 s, the reaction flask was submerged in an ice bath to quickly quench the temperature of the reaction solution. During the purification process, unreacted species were removed by a series of centrifuge steps after cleaning the product with methyl acetate and hexane. The absorbances and PLs of the synthesized batches were measured (ESI†).Ligand exchange mechanism
Firstly, a glass substrate was cleaned by sonication in a sequence of solvents including soapy water (Hellmanex soap and deionized water), deionized water, isopropanol and acetone. The slides were then treated by UV irradiation for 5 min. In an N2-filled glovebox compartment 0.1 mL of a 10 mg mL−1 solution of quantum dots in tetrachloroethylene (TCE) was dispensed on the slide and the slide was spin-coated at 2000 rpm for 60 s. Anhydrous methanol (0.1 mL) was deposited on the slide to minimize the effect of surface tension in preparation for PCM deposition. Next a solution of 3.1 mM PCM in methanol (0.1 mL) was dispensed on the slide for 60 seconds to allow for ligand exchange to occur. This process was repeated twice more to ensure complete ligand exchange. After each application of the PCM solution, excess ligands were spin coated off the substrate at 2000 rpm for 60 s. The films were then encapsulated with a 200 micrometer glass cover slide using epoxy curing. The slides were expose to UV light for 20 min to allow the epoxy to harden, sealing the slide.Absorbance, photoluminescence (PL), and time-resolved photoluminescence (TR-PL)
Absorbance measurements were performed using a PerkinElmer Lambda 950S spectrophotometer equipped with an integrating sphere. PL measurements were taken using a PTI Quantamaster 400 system. Quantum dots were excited at 750 nm. Time-resolved PL was taken with a time correlated single photon counting set-up with a pulsed 633 nm laser diode as the excitation light source. Time-resolved PL measurements were taken at the peak PL emission wavelength of the QD sample (1100 nm).PL measurements of the PCM crosslinked QD thin films
The encapsulated PCM treated QD thin film samples were placed on the stage in the spectrofluorometer. The light wavelength was changed to the appropriate wavelength to induce the configuration change of the PCM. After inducing the configuration change, without changing the position of the sample or any other parts of the setup, the excitation light wavelength was changed to 750 nm to obtain the PL spectra from QDs. All instrumental parameters such as slit widths, detector voltage, stage angle etc. were kept constant throughout the measurements.DFT calculation
Firstly, a ground-state geometry optimization was performed to ensure the molecule structures were optimized and a frequency calculation was performed to validate each stationary point. A TD-DFT energy calculation was performed to compute the first three low-lying excited states. Following each TD-DFT energy calculation, the UV-Vis absorbance spectra was generated and compared to experimental results. The HOMO and LUMO energy values were recorded from the energy calculation outputs. The hybrid functional B3LYP was chosen since it was best suited to calculate the optical properties of our system.49,50 The 6-31G basis set was used and the calculated absorbance spectra matched well with experimental absorbances of the molecules.Solution-state 1H PCM and mono-esterified PCM treated PbS QD sample preparation
10 mg mL−1 concentration of PbS QD in tetrachloroethylene (TCE) and 1.8 mM of PCM or mono-esterified PCM in methanol (MeOH) were prepared and mixed to yield a molar ratio of 1 QD![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 PCM for the mono-esterified ligand or 1 QD:10 PCM for the 1H ligand. The prepared solution was then stirred in a N2 glovebox environment overnight. The solution was transferred to a cuvette for optical measurements.
:100 PCM for the mono-esterified ligand or 1 QD:10 PCM for the 1H ligand. The prepared solution was then stirred in a N2 glovebox environment overnight. The solution was transferred to a cuvette for optical measurements.
GISAXS measurement
GISAXS characterizations were performed at the 11-BM Complex Materials Scattering (CMS) beamline at the National Synchrotron Light Source II (NSLS-II) at Brookhaven National Laboratory. Thin film samples were measured at incident angles from 0.10 to 0.25° with a 200 μm (H) × 50 μm (V) beam at 13.5 keV (wavelength λ = 0.9184 Å). 2D scattering patterns were obtained using Dectris Pilatus 2M, 2 meters downstream of the samples.DPV measurement
All DPV experiments were performed using a Metrohm Autolab PGSTAT302N potentiostat. The glassy carbon working (ϕ = 3 mm) and non-aqueous silver/silver chloride pseudoreference electrode behind a CoralPor frit were obtained from CH Instruments. The pseudoreference electrode was obtained by depositing chloride on bare silver wire in 10% HCl at oxidizing potentials and stored under light-free conditions in 0.1 M tetrabutylammonium hexafluorophosphate/acetonitrile solution prior to use. The counter electrode was a glassy carbon rod (ϕ = 3 mm). All experiments were performed in a modified scintillation vial (20 mL volume) as a single-chamber cell with a cap modified with ports for all electrodes and a sparging needle. Tetrabutylammonium hexafluorophosphate (TBAPF6) was purified by recrystallization from ethanol and dried in a vacuum oven before being stored in a desiccator. All data were referenced to an internal ferrocene standard (ferricenium/ferrocene reduction potential under stated conditions). All voltammograms were corrected for internal resistance during data collection. Experiments were carried out with a solution of 1.0 mM of molecules 1H and 2F in a 0.1 M TBAPF6 acetonitrile (MeCN). The solution was saturated with argon before measurement for molecule 1H. The experiments with molecule 2F were done in a nitrogen atmosphere glovebox. Standard reduction potentials (E1/2) were determined from DPV utilizing the Parry–Osteryoung equation52 where Ep is the peak potential and ΔE is the modulation amplitude:
In all of these experiments, ΔE = 0.025 V, the modulation time is 0.01 s, the interval time is 0.1 s, and the scan rate is 50.354 mV s−1.
Author contributions
J. J. C. and G. W. conceived and designed the research. E. S. S., J. M. B., P. A., A. G. R., E. H. R. T. performed experiments. M. C. performed the DFT calculations. All authors contributed to data analysis. E. S. S., G. J., G. W., and J. J. C. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Science Foundation under Grant No. DMR-2003978 and DMR-2003853. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation. This research used beamline 11BM (CMS) of the National Synchrotron Light Source II and the Center for Functional Nanomaterials (CFN), both of which are U.S. Department of Energy (DOE) Office of Science User Facilities operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704.References
- D. Bléger and S. Hecht, Angew. Chem., Int. Ed., 2015, 54, 11338–11349 CrossRef PubMed.
- S. Samanta, C. Qin, A. J. Lough and G. A. Woolley, Angew. Chem., Int. Ed., 2012, 51, 6452–6455 CrossRef CAS PubMed.
- R. Tong, H. D. Hemmati, R. Langer and D. S. Kohane, J. Am. Chem. Soc., 2012, 134, 8848–8855 CrossRef CAS PubMed.
- A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC.
- F. M. Raymo, Adv. Mater., 2002, 14, 401–414 CrossRef CAS.
- F. M. Raymo and S. Giordani, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4941–4944 CrossRef CAS PubMed.
- Y. Akaishi, A. D. Pramata, S. Tominaga, S. Kawashima, T. Fukaminato and T. Kida, Chem. Commun., 2019, 55, 8060–8063 RSC.
- L. U. Yoon, S. B. Adhikari, E. S. Sarabamoun, J. M. Bietsch, E. H. R. Tsai, G. Wang and J. J. Choi, J. Mater. Chem. C, 2021, 9, 16006–16013 RSC.
- S. Padgaonkar, C. T. Eckdahl, J. K. Sowa, R. López-Arteaga, D. E. Westmoreland, E. F. Woods, S. Irgen-Gioro, B. Nagasing, T. Seideman, M. C. Hersam, J. A. Kalow and E. A. Weiss, Nano Lett., 2021, 21, 854–860 CrossRef CAS PubMed.
- Y. Zhang, K. Zhang, J. Wang, Z. Tian and A. D. Q. Li, Nanoscale, 2015, 7, 19342–19357 RSC.
- C. Yun, J. You, J. Kim, J. Huh and E. Kim, J. Photochem. Photobiol., C, 2009, 10, 111–129 CrossRef CAS.
- I. Yildiz, E. Deniz and F. M. Raymo, Chem. Soc. Rev., 2009, 38, 1859–1867 RSC.
- T. Fukaminato, J. Photochem. Photobiol., C, 2011, 12, 177–208 CrossRef CAS.
- W. Szymański, J. M. Beierle, H. A. V. Kistemaker, W. A. Velema and B. L. Feringa, Chem. Rev., 2013, 113, 6114–6178 CrossRef PubMed.
- J. Zhang, Q. Zou and H. Tian, Adv. Mater., 2013, 25, 378–399 CrossRef CAS PubMed.
- L. Wang and Q. Li, Chem. Soc. Rev., 2018, 47, 1044–1097 RSC.
- L. Zhu, H. Yan, C. Y. Ang, K. T. Nguyen, M. Li and Y. Zhao, Chem.–Eur. J., 2012, 18, 13979–13983 CrossRef CAS PubMed.
- D. Manna, T. Udayabhaskararao, H. Zhao and R. Klajn, Angew. Chem., 2015, 127, 12571–12574 CrossRef.
- K. J. M. Bishop, C. E. Wilmer, S. Soh and B. A. Grzybowski, Small, 2009, 5, 1600–1630 CrossRef CAS PubMed.
- L. Zhu, M.-Q. Zhu, J. K. Hurst and A. D. Q. Li, J. Am. Chem. Soc., 2005, 127, 8968–8970 CrossRef CAS PubMed.
- M. Suda, M. Nakagawa, T. Iyoda and Y. Einaga, J. Am. Chem. Soc., 2007, 129, 5538–5543 CrossRef CAS PubMed.
- R. Mikami, M. Taguchi, K. Yamada, K. Suzuki, O. Sato and Y. Einaga, Angew. Chem., Int. Ed., 2004, 43, 6135–6139 CrossRef CAS PubMed.
- S. J. van der Molen, J. Liao, T. Kudernac, J. S. Agustsson, L. Bernard, M. Calame, B. J. van Wees, B. L. Feringa and C. Schönenberger, Nano Lett., 2009, 9, 76–80 CrossRef CAS PubMed.
- M. Suda, R. Kato and H. M. Yamamoto, Science, 2015, 347, 743–746 CrossRef CAS PubMed.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- M. Irie, Chem. Rev., 2000, 100, 1685–1716 CrossRef CAS PubMed.
- J. C. Owrutsky, H. H. Nelson, A. P. Baronavski, O. K. Kim, G. M. Tsivgoulis, S. L. Gilat and J. M. Lehn, Chem. Phys. Lett., 1998, 293, 555–563 CrossRef CAS.
- J. Owrutsky, H. Nelson, A. Baronavski, O.-K. Kim, G. Tsivgoulis, S. Gilat and J.-M. Lehn, Chem. Phys. Lett., 1998, 293, 555–563 CrossRef CAS.
- A. Ekimov and A. Onushchehko, ZhETF Pisma Redaktsiiu, 1981, 34, 363 CAS.
- P. V. Kamat, J. Phys. Chem. C, 2008, 112, 18737–18753 CrossRef CAS.
- Y. Shirasaki, G. J. Supran, M. G. Bawendi and V. Bulović, Nat. Photonics, 2013, 7, 13–23 CrossRef CAS.
- K. E. Sapsford, T. Pons, I. L. Medintz and H. Mattoussi, Sensors, 2006, 6, 925–953 CrossRef CAS.
- D. V. Talapin, J.-S. Lee, M. V. Kovalenko and E. V. Shevchenko, Chem. Rev., 2010, 110, 389–458 CrossRef CAS PubMed.
- J. Tang and E. H. Sargent, Adv. Mater., 2011, 23, 12–29 CrossRef CAS PubMed.
- J. J. Choi, Y.-F. Lim, M. E. B. Santiago-Berrios, M. Oh, B.-R. Hyun, L. Sun, A. C. Bartnik, A. Goedhart, G. G. Malliaras and H. D. Abruna, Nano Lett., 2009, 9, 3749–3755 CrossRef CAS PubMed.
- L. Sun, J. J. Choi, D. Stachnik, A. C. Bartnik, B.-R. Hyun, G. G. Malliaras, T. Hanrath and F. W. Wise, Nat. Nanotechnol., 2012, 7, 369–373 CrossRef CAS PubMed.
- M. F. Frasco and N. Chaniotakis, Sensors, 2009, 9, 7266–7286 CrossRef CAS PubMed.
- I. L. Medintz, A. R. Clapp, H. Mattoussi, E. R. Goldman, B. Fisher and J. M. Mauro, Nat. Mater., 2003, 2, 630–638 CrossRef CAS PubMed.
- F. W. Wise, Acc. Chem. Res., 2000, 33, 773–780 CrossRef CAS PubMed.
- S. A. Díaz, L. Giordano, T. M. Jovin and E. A. Jares-Erijman, Nano Lett., 2012, 12, 3537–3544 CrossRef PubMed.
- S. M. Emin, N. Sogoshi, S. Nakabayashi, T. Fujihara and C. D. Dushkin, J. Phys. Chem. C, 2009, 113, 3998–4007 CrossRef CAS.
- A. Nitzan, Annu. Rev. Phys. Chem., 2001, 52, 681–750 CrossRef CAS PubMed.
- J. J. Choi, J. Luria, B.-R. Hyun, A. C. Bartnik, L. Sun, Y.-F. Lim, J. A. Marohn, F. W. Wise and T. Hanrath, Nano Lett., 2010, 10, 1805–1811 CrossRef CAS PubMed.
- K. Matsuda, H. Yamaguchi, T. Sakano, M. Ikeda, N. Tanifuji and M. Irie, J. Phys. Chem. C, 2008, 112, 17005–17010 CrossRef CAS.
- G. D. Lilly, A. C. Whalley, S. Grunder, C. Valente, M. T. Frederick, J. F. Stoddart and E. A. Weiss, J. Mater. Chem., 2011, 21, 11492–11497 RSC.
- M. Irie, Photochem. Photobiol. Sci., 2010, 9, 1535–1542 CrossRef CAS PubMed.
- G. Guirado, C. Coudret, M. Hliwa and J.-P. Launay, J. Phys. Chem. B, 2005, 109, 17445–17459 CrossRef CAS PubMed.
- B.-R. Hyun, Y.-W. Zhong, A. C. Bartnik, L. Sun, H. D. Abruña, F. W. Wise, J. D. Goodreau, J. R. Matthews, T. M. Leslie and N. F. Borrelli, ACS Nano, 2008, 2, 2206–2212 CrossRef CAS PubMed.
- F. L. E. Jakobsson, P. Marsal, S. Braun, M. Fahlman, M. Berggren, J. Cornil and X. Crispin, J. Phys. Chem. C, 2009, 113, 18396–18405 CrossRef CAS.
- J. Frisch, M. Herder, P. Herrmann, G. Heimel, S. Hecht and N. Koch, Appl. Phys. A: Mater. Sci. Process., 2013, 113, 1–4 CrossRef CAS.
- L. Hou, R. Ringström, A. B. Maurer, M. Abrahamsson, J. Andréasson and B. Albinsson, J. Am. Chem. Soc., 2022, 144, 17758–17762 CrossRef CAS PubMed.
- E. P. Parry and R. A. Osteryoung, Anal. Chem., 1965, 37, 1634–1637 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Syntheses procedure and characterization of PCMs, PbS QD optical characterization, asymptotic limits of PL switching, raw PL spectra for 1H and 2F samples, GISAXS results of PCM bridged QD assemblies, oxidation DPV spectra, mathematical background, and calculated DFT energy levels for the PCMs. See DOI: https://doi.org/10.1039/d3ra07539g |
| This journal is © The Royal Society of Chemistry 2024 |