
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enzyme-accelerated CO2 capture and storage (CCS) using paper and pulp residues as co-sequestrating agents†
Ayanne
De Oliveira Maciel
,
Paul
Christakopoulos
 ,
Ulrika
Rova
and
Io
Antonopoulou
*
,
Ulrika
Rova
and
Io
Antonopoulou
*
Biochemical Process Engineering, Division of Chemical Engineering, Department of Civil, Environmental and Natural Resources Engineering, Luleå University of Technology, SE-97187 Luleå, Sweden. E-mail: io.antonopoulou@ltu.se
First published on 20th February 2024
Abstract
In the present work, four CaCO3-rich solid residues from the pulp and paper industry (lime mud, green liquor sludge, electrostatic precipitator dust, and lime dregs) were assessed for their potential as co-sequestrating agents in carbon capture. Carbonic anhydrase (CA) was added to promote both CO2 hydration and residue mineral dissolution, offering an enhancement in CO2-capture yield under atmospheric (up to 4-fold) and industrial-gas mimic conditions (up to 2.2-fold). Geological CO2 storage using olivine as a reference material was employed in two stages: one involving mineral dissolution, with leaching of Mg2+ and SiO2 from olivine; and the second involving mineral carbonation, converting Mg2+ and bicarbonate to MgCO3 as a permanent storage form of CO2. The results showed an enhanced carbonation yield up to 6.9%, when CA was added in the prior CO2-capture step. The proposed route underlines the importance of the valorization of industrial residues toward achieving neutral, or even negative emissions in the case of bioenergy-based plants, without the need for energy-intensive compression and long-distance transport of the captured CO2. This is a proof of concept for an integrated strategy in which a biocatalyst is applied as a CO2-capture promoter while CO2 storage can be done near industrial sites with adequate geological characteristics.
Introduction
Carbon capture and storage (CCS) plays an important role in decreasing greenhouse gas (GHG) emissions and mitigating climate change. Industries, such as mining, cement, and pulp and paper, produce alkaline wastes, which have proven capabilities to work as CO2-sequestrating agents.1–3 Yearly, more than 7 billion tons of such residues are produced worldwide, representing a potential to capture and store 2.9–8.5 million tons of CO2 in the long term.4 Mineral weathering is a possible route to utilize alkaline residues in CO2 capture and can bring some benefits since it is a wet route, which can enhance the kinetics of the capture reaction without requiring high, or even any, additional power inputs.5,6 Carbonate rocks (such as limestone, CaCO3 and dolomite, CaMg(CO3)2) are abundant in nature and play an important role as natural carbon sinks, since they react with atmospheric CO2 (weathering). Nevertheless, the process happens in a geological timeframe, which is not quick enough for reaching the worldwide goals targeted for CCS by 2050.7 Consequently, carbon capture utilizing aqueous rich carbonate solutions and concentrated CO2 streams, such as industrial flue gases, has been gaining increasing attention as a path for accelerated weathering in the last decades.Recently, a demonstration carbon-capture plant installed in a coal-fired power unit and using limestone as a co-sequestrant agent revealed a CO2-capture efficiency of up to 55%.8 The mechanism involved in calcium carbonate weathering (acidic conditions) can be explained by two distinct reactions, the first reaction being CO2 hydration (eqn (1)) and the second mineral dissolution (eqn (2)), of which the final targeted product is the formation of bicarbonates (HCO3). The carbonate equilibrium, which includes free CO2 or carbonic acid, bicarbonate ions, and carbonate ions, is highly pH dependent. At pH values <8.3, bicarbonates are predominant rather than carbonate ions. Alkali metal oxides are also relevant for CCS applications since in aqueous solutions these react with CO2 to also produce bicarbonates (eqn (3)). The weathering or mineral dissolution reaction can be favored at acidic conditions (pH < 7.5) for carbonates9,10 and oxides (pH < 8.4).11–13 The CO2 hydration reaction supplies the H+ protons for mineral dissolution and is the limiting step in the process of weathering since it has slow kinetics.
| CO2(g) + H2O ↔ HCO3− + H+ | (1) |
| MCO3 + H+ ↔ M2+ + HCO3− | (2) |
| M(OH)2 + 2H+ + 2CO2(g) ↔ M2+ + 2HCO3− | (3) |
Carbonic anhydrase (CA) is a metalloenzyme that catalyzes the limiting step in mineral weathering (CO2 hydration reaction (eqn (1))). It is one of the fastest existing enzymes with a turnover rate up to 107 s−1, and has demonstrated potential for enhancing the yield in carbon capture, utilization, and storage (CCUS) applications,14–16 and also in accelerating the weathering of carbonates.17–19 Therefore, CA utilization could bring about enhanced mineral dissolution and a higher productivity of bicarbonates, which could then be converted further into a thermodynamically stable form (i.e., mineral carbonation).
Silicate-rich mineral rocks have been demonstrated to be able to store CO2 permanently, in particular, Mg-silicates, such as olivine ((Mg,Fe)2SiO4), since their dissolution/carbonation also might occur under mild temperature and pressure conditions. The direct use of bicarbonate solutions, obtained from a CO2-capture process step, in replacement of the injection of pressurized CO2 gas in the bedrock, could be an alternative to overcome some issues such as gas leakage from storage sites,20 thus enabling safer geological carbon storage.
In contact with solutions rich in bicarbonates, under neutral to acidic conditions, silicates react with the H+ proton, releasing a metallic ion, such as Mg2+ in the case of forsterite (Mg2SiO4). This step is referred to as mineral dissolution or leaching of the mineral rock during the CO2 storage (eqn (4)). The leached Mg2+ ions can react with (bi)carbonate, according to the pH of the aqueous solution, and then form magnesium carbonates (MgCO3), which is referred to as carbonation (eqn (5)). The formed solid carbonates serve as a permanent storage form of CO2.
| M2SiO4 + 4H+ ↔ 2 M2+ + H4SiO4 | (4) |
| M2+ + CO32− ↔ MCO3(s) | (5) |
Fig. 1 proposes a path to combine a process of carbon capture and storage for the pulp and paper industry. The synergies in respect with the logistics, energy efficiency, and the share of common industrial facilities could be a great asset to facilitate the development of CCS units in industrial sites. In Sweden alone, about 130![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons of waste generated from the pulp and paper sector was sent to landfills in 2019, which was subject to tax for waste generation (517 SEK per ton waste).21 Thus, the valorization of such residues could offer an alternative route for promotion of both CCS practices and the circular economy.
000 tons of waste generated from the pulp and paper sector was sent to landfills in 2019, which was subject to tax for waste generation (517 SEK per ton waste).21 Thus, the valorization of such residues could offer an alternative route for promotion of both CCS practices and the circular economy.
 | ||
| Fig. 1 Proposed concept for an in situ CCS strategy utilizing solid residues from the pulp and paper industry. | ||
Aiming to integrate biotechnology to enhance CCS processes, this current study aimed to evaluate the application of industrial residues from the pulp and paper sector, composed majorly of CaCO3, as co-sequestrating agents for a CA-accelerated aqueous carbon-capture process. The utilized residues did not receive any prior treatment (acid, thermal, or mechanical activation22) to potentialize the carbon capture, which could imply lower costs. As a result of the CO2-capture step, a bicarbonate-rich solution was obtained by enzyme-accelerated CO2 hydration and mineral dissolution of the industrial solid residues, which was further used for mineral dissolution and the carbonation of olivine, a highly abundant mineral on earth, in order to understand its potential for the permanent storage of CO2.
For the expected outcomes, it would be desirable not only to accelerate the CO2-capture rate by use of an enzyme, but also to achieve the highest concentration of bicarbonates and the lowest pH value to further promote olivine dissolution and carbonation as a CO2-storage step. A CA mutant with high tolerance to alkaline environments was selected due to the nature of the tested waste. Research on CA application to boost both accelerated mineral weathering and increased bicarbonate production using alkaline wastes is still limited in the literature and has been tested only with well-known materials, such as limestone and brucite. Consequently, the proposed utilization of residues that might contain impurities, such as sulfates, phosphors, and metals, in CA-assisted CO2 capture will contribute to expanding the knowledge in this field.
Materials and methods
Enzyme production
The DNA sequence of an ultrastable CA mutant from Desulfovibrio vulgaris (DvCA8.0)23,24 was incorporated with 6xHis-tag and inserted between the NdeI and XhoI restriction sites of the pET22b(+) vector and transformed in Escherichia coli BL21. The pre-cultures were prepared using Luria Bertani medium containing 100 μg mL−1 ampicillin and incubated overnight at 37 °C. Cultures were done in an auto-induction medium containing lactose (ZYP-5052 without trace elements) and 100 μg mL−1 ampicillin and incubated overnight at 32 °C. The cells were harvested, resuspended in 100 mM pH 8.0 Tris–HCl buffer and lysed in a homogenizer at 700 bar. The produced lysate was filtered to 0.2 μm, freeze-dried, and resuspended in water to create a concentrated enzyme stock solution. Finally, the lysate was dialyzed in 20 mM pH 8.0 Tris–HCl buffer to minimize the buffer interference in the further reactions. Expressions were verified by a His-Tag purification step, SDS-PAGE, and by measuring the CA activity using standard assays (p-nitrophenyl acetate and Wilbur–Anderson assays) in lysates from transformed and non-transformed E. coli cultured cells.Characterization of the co-sequestrating agents
Four distinct industrial solid residues from the Kraft process – lime mud (R1), green liquor sludge (R2), electrostatic precipitator dust (R3), and lime dregs (R4) – were kindly provided by the paper and pulp industry firm Billerud (Karlsborg, Norrbotten, Sweden). X-Ray fluorescence (XRF, Hitachi) and X-ray diffraction (XRD, PANalytical) techniques were utilized for identifying the chemical composition and the phases of each material. The average particle size of the samples was estimated through a particle size and shape analyzer (Camsizer XT, Retsch Technology). The moisture content was determined using a moisture analyzer scale (Sartorius MA30). Due to the irregularity of R4, the material was ground in a roll mill to reach an average particle size <25 μm prior to the particle-size measurements. Scanning electron microscopy (SEM) analysis was conducted to observe the surfaces of the residues.CO2-capture trials
Experiments were carried out in a 250 mL reactor at room temperature, using 167 mL working solution containing 0.4% w/w of industrial solid residue (DM basis) suspended in ultrapure water (18.2 MΩ cm−1). Next, 2.5 mL of concentrated enzyme lysate or 20 mM pH 8.0 Tris–HCl was added to the CA-added or control reactions, respectively. The DvCA8.0 concentration in the concentrated lysate was 27 mg mL−1 with a specific activity of 6690 WAU per mg. In the case of CO2 capture from the open air, the reactor used was a flat beaker left open to allow contact with atmospheric air. In the case of CO2 capture from synthetic gas (20% CO2:80% N2, Linde Gas, Luleå, Sweden) simulating industrial exhaust gases, a three-neck round-bottom flask was used instead. The gas was bubbled in the flask at a flow rate of 7.8 mL min−1. The reactors were agitated using a magnetic stirrer during the reaction. The pH value (pHenomenal, VWR, 1100L) and temperature were monitored over time, and at certain time intervals, aliquots were collected and filtered to 0.45 μm for determination of the Ca2+, Mg2+, and SO42− concentrations (Spectroquant ® test kits, Supelco). At the end of the reaction, the supernatant was filtered to 1.2 μm, and the amount of bicarbonate formed during the reaction was estimated from a modified biomineralization method.25 In short, a solution of 1.3 M Tris and 6.0% w/w CaCl2·2H2O was added to the filtered supernatant in a ratio of 1:4 v/v and agitated at 37 °C and 300 rpm for at least 12 h. The formed solid was filtered, washed, and then oven-dried at 60 °C for 24 h. The final weight of the dried formed matter was measured, and the material was subjected to XRD analysis. Quantification of the bicarbonates or total CO2 captured attributed to combined CO2 hydration (eqn (1)) and mineral dissolution (eqn (2) and (3)) was performed according to Section 1 of the ESI.† The unreacted industrial solid residues recovered by filtration were oven-dried at 50 °C and subjected to XRD analysis in order to identify the chemical composition and phases after the CO2-capture reaction. An overview of the CO2 capture set-up performed in the laboratory is presented in Fig. 2.
 | ||
| Fig. 2 Experimental set-up for CCS in the laboratory. M: Metal. | ||
CO2 storage using olivine as a reference material
Tests were performed according to the scheme process illustrated in Fig. 2. A solution rich in bicarbonate ions obtained in a CO2-capture step was recovered by filtration and utilized for mineral dissolution and carbonation. The supernatant was mixed with high purity milled and dried olivine (Ward's Sciences) at a concentration of 150 g L−1 in a 500 mL Duran bottle, pressurized at 3.0 bar with N2, and left to react under stirring at 600 rpm and room temperature. Two systems were evaluated: one containing the supernatant from a CA-added CO2-capture reaction and one was the control, containing the supernatant from a CO2-capture reaction without CA. Every 24 h, an aliquot was withdrawn, filtered, and the concentrations of Ca2+, Mg2+, alkalinity (CO32−/HCO3−), and SiO2 measured to monitor the dissolution of the olivine minerals over time (eqn (4)). The samples were also analyzed for Fe, Si, and Ni by ICP-SFMS according to SS-EN ISO 17294-2:2016 (ALS Scandinavia, Luleå, Sweden). After 5 days, the pressure was released from the bottle, and the bottle was sealed and then heated at 35–40 °C overnight to favor the carbonation of leached Mg2+ ions (eqn (5)). At the end of the reaction, the solid olivine residue was recovered through vacuum filtration, oven-dried, and kept for further analysis. Thermal gravimetric analysis (TGA; PerkinElmer TGA 8000; 10 °C min−1, 80 L min−1 N2) and XRD were performed on olivine, prior to and after mineral dissolution/carbonation, to identify the carbonates formed and the differences between the samples before and after the reaction, respectively. The quantification of the total CO2 stored was done according to Section 2 of the ESI.† The milled and dried olivine was characterized for its particle-size distribution by sieving and was then subjected to SEM analysis prior to its use in the CO2-storage experiments.Results and discussions
Characterization of industrial solid residues
Except for R4, which was crushed and ground, all the residues were utilized as received. As shown in Table 1, the average particle size (Psize) varied from 7.3 to 23.1 μm. The particle-size distribution is presented in Fig. 3b. The residues had moisture contents varying from 3.6% to 49% approximately, with R2 having the highest water content.| Lime mud (R1) | Green liquor sludge (R2) | Electrostatic precipitator dust (R3) | Lime dregs (R4) | |
|---|---|---|---|---|
| a N.D: not determined. | ||||
| Element composition (% w/w) | ||||
| Mg | 1.13 ± 0.09 | 4.96 ± 0.08 | 1.39 ± 0.03 | 1.56 ± 0.12 |
| Al | 0.10 ± 0.01 | 0.32 ± 0.01 | 0.10 ± 0.00 | 0.97 ± 0.04 |
| Si | 0.17 ± 0.01 | 0.60 ± 0.01 | 0.20 ± 0.02 | 0.54 ± 0.04 |
| P | 0.53 ± 0.01 | 0.17 ± 0.00 | 0.50 ± 0.00 | 0.30 ± 0.00 |
| S | 0 | 1.62 ± 0.00 | 0.14 ± 0.03 | 0.00 ± 0.00 |
| K | 0 | 0.40 ± 0.00 | 0.00 ± 0 | 0 |
| Ca | 38.42 ± 0.10 | 29.98 ± 0.10 | 37.74 ± 0.53 | 37.8 ± 0.86 |
| Mn | 0.03 ± 0.00 | 1.35 ± 0.02 | <0.2 | <0.2 |
| Fe | 0.03 ± 0.03 | 0.35 ± 0.01 | <0.2 | 0.17 ± 0.00 |
| Ni | <0.2 | <0.1 | <0.1 | <0.2 |
|
||||
| Particle size (μm) | ||||
| P size (μm) | 13.99 | 16.75 | 7.3 | 23.1 |
|
||||
| Chemical composition (% w/w) | ||||
| CaCO3 | 96.1 | 75.0 | 94.0 | 90.8 |
| CaO | N.D | N.D | N.D | 2.1 |
| MgSO4 | N.D | 6.1 | N.D | N.D |
| MgO | N.D | 8.1 | N.D | N.D |
| Other phases | 3.9 (metal oxides, alumino-silicates) | 10.5 (alumino-silicates, metallic oxides, carbonides) | 6.0 (silicates, oxides) | 7.1 (silicates, metal oxides) |
|
||||
| Percentage dry matter (% w/w) | ||||
| 83.4 ± 0.9 | 51.5 ± 1.4 | 96.4 ± 1.1 | 93.2 ± 2.4 | |
 | ||
| Fig. 3 (a) Photos of the representative samples, (b) particle-size distributions, (c) SEM analysis and (d) XRD patterns of R1–R4 (R1: lime mud, R2: green liquor sludge, R3: electrostatic precipitator dust, and R4: lime dregs). | ||
Based on elemental analysis by XRF, all the analyzed materials presented a high concentration of Ca – varying from approximately 30.0% to 38.4% (Table 1). Other relevant elements identified were Mg, Al, Si, and S. Mn was found in significant concentrations (1.35%) for R2, as well as S (1.62%), indicating the possible presence of sulfates. Other elements, such as Cr and V, were found in smaller concentrations of <0.15% in all the materials. The XRD results (Fig. 3d) showed that CaCO3 was the majority phase in all the samples. For R2, the presence of MgO and Mg(SO4)2 was also detected. R4 presented peaks for Ca(OH)2 besides the presence of CaCO3. Despite the identification of Mg in all the residues by XRF, XRD analysis only indicated corresponding magnesium-containing phase peaks for sample R2, possibly because this material presented higher concentration than the others. Small amounts of CaO could be also expected in sample R3, since they shared parts of the same process (lime kiln), as shown in Fig. 1. Other materials, such as aluminum silicates or silica, appeared in smaller amounts; however, they were not detected during the XRD analysis. In order to estimate the concentrations of the existing phases, a mass balance was performed according to the methodology described in Section 3 of the ESI.†
Fig. 3a presents the distinct materials after drying and milling. SEM analysis indicated the presence of rhombohedral calcite in all the analyzed residues (Fig. 3c). The R1 and R3 samples had high homogeneity and contained mostly CaCO3. R2 showed the presence of flower-like structures, possibly due to the presence of Mg(OH)2. R4 had the most irregular structure and bigger grain size when compared to the other residues, indicating the presence of other phases, such as silicates and Ca(OH)2.
CO2 capture from open-air reactions
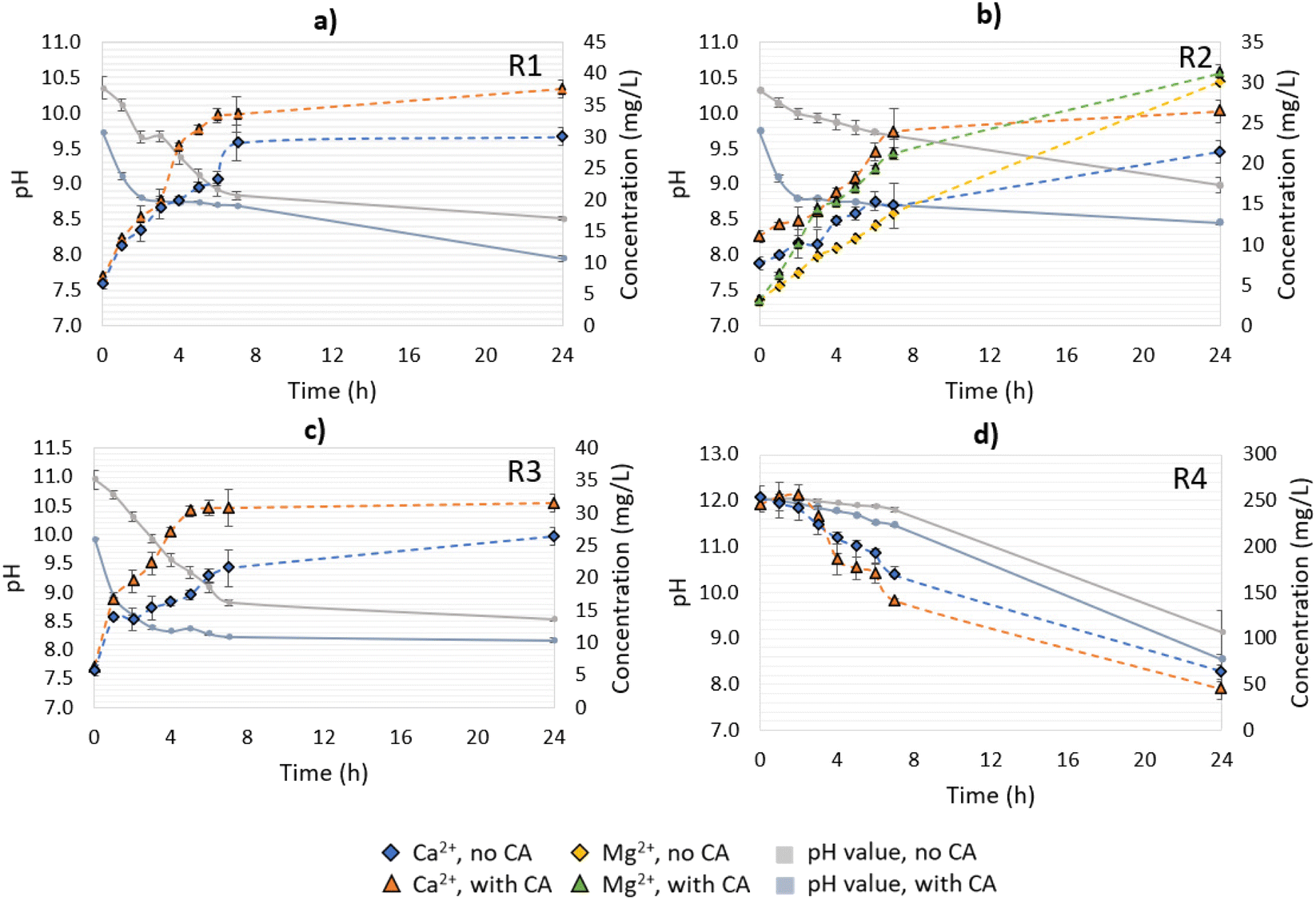 | ||
| Fig. 4 pH variation and ions (Ca2+, Mg2+) release over time during CA-accelerated and non-CA-accelerated CO2 capture in open-air reactions. (a) R1: lime mud, (b) R2: green liquor sludge, (c) R3: electrostatic precipitator dust, and (d) R4: lime dregs. | ||
Differently from the other materials, R4 had a higher Ca2+ concentration at time 0 of the reaction, which decreased over time (Fig. 4d). This can be explained because of the presence of Ca(OH)2, which will form CaCO3 when in contact with the available CO2, and precipitate in the solution. Since the solution pH was higher than 11 over the first 7 h of the CO2-capture experiment, the formation of carbonate ions predominantly occurred over bicarbonates, enabling the free Ca2+ to react with them, leading to precipitation. Between 7 < pH < 10, bicarbonate and carbonate ions co-existed, and the former were favored, leading to a great reduction in the CaCO3-precipitation rate.27 At the end of experiment (24 h), the no- and CA-added R4 reactions presented pH values between 8.6–9.1, which were slightly higher than the other residues, which varied from pH 8.0–8.5. Note that between 7 and 24 h, the system containing R4 had greater pH value variation (2.7–2.9) than the other materials (0.1–0.7), indicating that R4 might require more time to react with the CO2 until reaching a stable pH value. As a general observation, the addition of CA resulted in a faster drop in the pH value over time for all the materials in comparison to the no-CA-added reactions, demonstrating the enhancement effect of the enzyme in accelerating the CO2 capture, and in the formation of H+ ions, as has been already observed in previous studies using limestone and dolomite and bovine carbonic anhydrase (BCA) for accelerated CO2 capture.19
Other works involving the mineral dissolution of materials rich in calcium carbonate under atmospheric CO2 concentration proved that CA played an important role in increasing the Ca2+ release, ranging from 2.3–11.7-fold in comparison to their control samples.17,18,28 In contrast, the present study, performed under non-optimized conditions, presented an increase in Ca2+ leaching of up to 1.25-fold. As a possible improvement, the utilization of buffered solutions, a higher speed of agitation, and a longer time of reaction (for R4) could result in higher CO2 mass transfer to the solution, and consequently higher Ca2+ leaching and bicarbonates production. Also, the residues employed in our tests did not receive any treatment to minimize any existent impurities, such as organics and heavy metals, which can impair the enzyme activity and mineral dissolution. Nevertheless, CA presented a positive effect on the results, and since we aimed at the utilization of enriched CO2 streams (i.e., industrial flue gases), reactions using gas with an enriched CO2 concentration were performed as well. More details about the pH value variation as well as the Ca2+ and/or Mg2+ concentrations for R1–R4 are presented in Fig. 4.
:3.9 to 9:1 (Fig. 5a). Under given conditions and based on the CA-accelerated reaction, the highest amount of total CO2 captured was achieved in descending order for: R4 > R1 > R2 > R3. In particular, the addition of CA in R1 increased the CO2 capture 4.0-fold, in R2 2.3-fold, in R3 1.8-fold, and in R4 1.5-fold, as determined at the end point (24 h). The maximum concentration of bicarbonates (HCO3−) measured in solution for R1 in the CA-accelerated reaction at the end point was 0.48 g L−1 (corresponding to 0.35 g L−1 captured CO2). For R4, 53% of the total amount of CO2 sequestrated was captured in the form of carbonates that were not present in the aqueous solution, and 47% was captured in the form of soluble bicarbonates (Fig. 5d). Thus, the concentration of bicarbonates available in solution after the end of the reaction for R4 in the CA-accelerated reaction was 0.32 g L−1 (corresponding to 0.23 g L−1 captured CO2).
 | ||
| Fig. 5 Quantification of the captured CO2 at the end of the open-air reactions (24 h). (a) R1: lime mud, (b) R2: green liquor sludge, (c) R3: electrostatic precipitator dust, and R4: (d) lime dregs. | ||
Despite the Ca2+ release being lower in R2, the dissolution of Mg(OH)2 contributed to the total bicarbonate concentration in this system, reaching a similar concentration as observed with R1 (Fig. 5b). With R4, part of the CO2 from the capture was destined for the carbonation of Ca(OH)2, which was evident by the decrease in Ca2+ in the solution over time, so the final concentration of bicarbonate measured did not reflect the total amount of CO2 captured. There was an additional amount of captured CO2 that precipitated toward CaCO3, thus the total amount of CO2 captured could be attributed to CO2 hydration and Ca(OH)2 carbonation. Of course, there was also an ongoing reaction of mineral dissolution for CaCO3 (Ca2+ increase); however, this was not evident due to the predominant reaction of Ca(OH)2 carbonation (Ca2+ precipitation, decrease). Thus, it is important to underline that the attribution of the CO2-capture step to different reactions (hydration, dissolution, carbonation) was only apparent and depended on the components detected over time in the aqueous working solution.
Subsequently, notwithstanding the pH drop over time leading to mineral dissolution, the precipitation of carbonates could also occur in the opposite direction in small proportions for R1–R3, as the carbonation of CaCO3 (and MgCO3 for R2) is favored in neutral to alkaline pH values. Besides an improved CO2 capture, a high bicarbonate availability is essential in the mineral carbonation step, so both outcomes should be balanced when selecting which material has the highest potential to be applied in an industrial process.
CO2 capture from CO2-rich gas
 | ||
| Fig. 6 pH variation and ions (Ca2+, Mg2+) release over time during CA-accelerated and non-CA-accelerated CO2 capture from synthetic gas (20% CO2). (a) R1: lime mud, (b) R2: green liquor sludge, (c) R3: electrostatic precipitator dust, and (d) R4: lime dregs. | ||
The CA-added system for R1 reached a pH value of 7.0 in 22.5 min of reaction; without the enzyme, instead, almost three times as much time was required to reach a similar condition (Fig. 6a). Since the reaction was performed until 90 min, the residence time of the non-CA-added system with R2 in acidic conditions was also reduced, leading to a lower dissolution of Mg(OH)2, as evidenced during the time interval between 65–75 min of the reaction. For the CA-added system, the pH value at 65 min was 6.68 while it was 6.99 for the no-CA-added system. The pH plays an important role in the dissolution of Mg(OH)2/MgO, and the two are exponentially related,12 which could explain the boost during the referred to timeframes. Also, the release of Ca2+ was considerably lower when compared to the other residues, possibly influenced by the Mg2+ inhibition effect on CaCO3, as also observed in the open-air reactions (Fig. 6b). The concentration of sulfates was measured for the systems added to R2; however, there was no significant variation of their concentration during the time of the reaction (data not shown).
The reactions with R3 revealed a similar trend to those with R1 in terms of Ca2+ release; however, they had a slightly higher pH value at time 0, possibly due to the presence of small contents of CaO, causing a “delay” in the pH drop in the first minutes of the reaction when compared to R1. Additionally, an improvement of 1.21-fold was verified in comparison to the no-CA-added reaction, at the time of 90 min (R3) (Fig. 6c). Similar results in the Ca2+ leaching enhancement were found in a system using bacterial CA and limestone, which exhibited a boost of 1.22–1.61-fold in comparison to the control reaction.30 In other works utilizing CA for accelerated weathering, the enzyme had a positive effect on the mineral dissolution, enhancing it in a range of 1.6–4.0-fold when compared to the control reactions.28,31,32
For R4, a longer time was required to give the system time to reach neutral to acidic conditions (pH below 7.3), as happened with the other materials (Fig. 6d). For the control reaction, a decrease in Ca2+ concentration was observed in the first 120 min of the reaction (pH > 7.3), which was attributed to Ca2+ precipitation in the carbonation of Ca(OH)2. The results indicated that when the solution reached pH 11 almost 80% of the dissolved Ca2+ ions had already precipitated, and the free Ca2+ comprised less than 8.0% of the initial concentration in the range of 10 < pH < 8. From 120 min (pH 7.3) until the end of the reaction at 180 min, Ca2+ started getting released again due to the mineral dissolution of CaCO3. Interestingly, in the CA-accelerated reaction, a decrease in Ca2+ concentration was not observed at all, as was observed for the respected open-air reaction. The Ca2+ concentration was instead rather stable and even increased slightly, indicating that there was an ongoing competitive reaction between Ca(OH)2 carbonation and mineral dissolution.
R4 and R2 presented the highest leaching improvements because of the presence of the enzyme (Ca2+ and/or Mg2+), despite R2 having a considerably lower absolute concentration of Ca2+ compared with the other materials. Therefore, it was also relevant to evaluate the rate of ions dissolution over time as a metric reaction development. With R1 and R3, the highest dissolution rates were observed at 40 min – meaning the dissolution rate had reached its peak at that time or before – and the addition of the enzyme increased their dissolution rate 1.47- and 1.31-fold compared to the no-CA-added reaction, respectively. For R2, the dissolution rate reached the peak enhancement at 55 min, corresponding to an 1.8-fold increase compared to the system without the enzyme. R4 had the highest dissolution rate at 150 min, with a CA-boosting effect of 2.2-fold in comparison with the non-enzyme assisted reaction. Table 2 displays the Ca2+ dissolution rate results for the distinct materials tested.
| Dissolution rate ([ion] per min) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Time (min) | Lime mud (R1) | Green liquor sludge (R2) | Electrostatic precipitator (R3) | Lime dregs (R4) | ||||
| No CA | With CA | No CA | With CA | No CA | With CA | No CA | With CA | |
| 40 | 1.53 ± 0.21 | 2.26 ± 0.15 | 1.27 ± 0.11 | 1.68 ± 0.0 | — | — | — | — |
| 45 | — | — | — | — | 0.21 ± 0.02 | 0.25 ± 0.01 | — | — |
| 50 | 1.27 ± 0.13 | 1.74 ± 0.11 | 1.11 ± 0.13 | 1.38 ± 0.03 | 0.17 ± 0.01 | 0.21 ± 0.02 | — | — |
| 65 | — | — | — | — | 0.19 ± 0.02 | 0.35 ± 0.01 | — | — |
| 70 | 1.24 ± 0.15 | 1.51 ± 0.07 | 1.01 ± 0.09 | 1.22 ± 0.04 | — | — | — | — |
| 75 | — | — | — | — | 0.27 ± 0.02 | 0.40 ± 0.03 | — | — |
| 90 | 1.07 ± 0.08 | 1.26 ± 0.10 | 0.87 ± 0.10 | 1.06 ± 0.02 | 0.34 ± 0.02 | 0.49 ± 0.02 | — | — |
| 135 | — | — | — | — | — | — | 0.73 ± 0.03 | 0.93 ± 0.06 |
| 150 | — | — | — | — | — | — | 0.61 ± 0.05 | 1.34 ± 0.02 |
| 180 | — | — | — | — | — | — | 0.53 ± 0.03 | 0.55 ± 0.04 |
Industrial residues rich in oxides33 and hydroxides34–37 have high CO2 affinity and have proven their potential for carbon capture;38,39 however, materials rich in carbonates without previous treatment (i.e., calcination, acidification) have been not widely exploited in the literature in direct carbonation studies, since they are not as reactive as their decarbonated forms. CO2 hydration is known to benefit from alkaline pH values, so the accelerated weathering of carbonates (also alkaline) is also an alternative for CO2 capture, without requiring extreme process conditions (high temperature and pressure). In this present work, utilizing residues as received from the industry and working under ambient temperature and pressure conditions, the CO2 capture was improved in the presence of CA, confirming the potential of this biotechnological route for application in carbon capture. Table 3 summarize studies on accelerated weathering with and without CA.
| Without CA | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Co-sequestrating agent | Concentration (g L−1) | Temperature (°C) | Pressure (bar) | Volume of reaction | CO2 intake | Released ions concentration (mg L−1) | CO2 uptake | Reaction time | Ref. |
| a Value of total alkalinity. b Considered as the maximum Mg2+ concentration increase before nucleation – baseline 4.11 mg L−1. c Based on dissolved inorganic carbon (DIC) concentration – includes both carbonates and bicarbonates. d Total concentration of bicarbonates measured – includes the dissolution of the minerals. e Values include measurements from the supernatant and mycelia. f N.Q.: not quantified. | |||||||||
| Steel slag – Ca–K silicates | 33.3 | 25 and 90 | Ambient pressure | 0.2 L | Saturated CO2 solution with injection 30 mL min−1 | ∼105–200 mg L−1 (Ca2+) | N.Q. | 240 h | 39 |
| Red mud – NaOH | 100 | Room temperature | Ambient pressure | 0.2 L | 15.0%, 5 mL min−1 | 330 mg L−1 (Na+) and 173 mg L−1 (Al) | N.Q. | 24 h | 37 |
| Coal fly ashes – alkaline oxides | 100–200 | Room temperature | Ambient pressure | 0.5–1.0 L | Atmospheric | 503–1073 mg L−1 (Ca2+) | N.Q. | 24 h | 40 |
| Calcinated oil shale limestone – CaO rich | 10–100 | Up to 950 | Ambient pressure | ∼0.25 L | 16%, 150 mL min−1 | ∼2820 mg L−1 (Ca2+) | N.Q. | 1 h | 41 |
| Brucite – Mg(OH)2 | 7 | Room temperature | 15 atm | 0.3 L | 100% CO2, 15 atm | 14.7–17.2 g L−1 (Mg2+) | 65.9–71.0 g L−1 | 2.25 h | 42 |
| Limestone – CaCO3 | 5 | Room temperature | 1.5 bar | 5 columns of 0.16 m3–30 m3 h−1 (continuous flow) | ∼10% CO2, 50–200 m3 h−1 | N.Q. | 11 mMa | — | 8 |
| With CA | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Co-sequestrating agent | Concentration (g L−1) | Enzyme | Temperature (°C) | Pressure (bar) | Reactor size | CO2 intake | Max released ions concentration/CA enhancement | CO2 uptake | Reaction time | Ref. |
| Brucite ore – Mg(OH)2 | 50 | Bovine CA (BCA) | Room temperature | Ambient pressure | 0.3 L | 10%, 54–2700 mL min−1 | ∼3.7 mg L−1b | 12.5 mg C per Lc | 3–11 days | 34 |
| Wollastonite – CaSiO4 | 2.3 | CA from Bacillus mucilaginosus | 35 | Ambient pressure | 0.05 L | Atmospheric and 3.9% CO2 | ∼25–66 (Ca2+), 1.19–1.22-fold | N.Q. | 8 h | 31 |
| Limestone and dolomite – CaCO3 and CaMgCO3 | — | CA from Bacillus cereus | Room temperature | Ambient pressure | 30 × 3.0 cm diameter – 3 mL min−1 continuous flow | 0.035–100%, 190 mL min−1 | ∼55–250 (Ca2+), 1.62–2.44-fold | ∼160–850 mg L−1d | 25 min | 30 |
| Wollastonite – CaSiO4 | 10 | CA from Aspergillus nidulans, whole cells | 37 | Ambient pressure | 0.1 L | Atmospheric and 3% CO2 | Up to 550 mg L−1e, up to 18-fold | N.Q. | 2–10 days | 43 |
| Limestone – CaCO3 | 5 | Extracellular CA from Aspergillus penicillium, whole cells | 28 | Ambient pressure | 0.4 L | Atmospheric | Up to ∼135 mg L−1, ∼2.3-fold | N.Q. | 72 h | 18 |
| Limestone – CaCO3 | 0.2 | Extracellular CA from Bacillus sp. GLRT102Ca | Room temperature | Ambient pressure | 0.4 L | Atmospheric | 9.7 mg L−1, 2.4–11.7-fold | N.Q. | 9 h | 17 |
| Limestone – CaCO3 | 10 | CA from Bacillus mucilaginosus – lysate and whole cells | 32 | Ambient pressure | 0.01 L | Atmospheric | Up to 1130 mg L−1–4.0–6.0-fold | N.Q. | 6 days | 28 |
| Pulp and paper residues – CaCO3 and alkaline oxides | 4.0 | Evolved CA from Desulfovibrio vulgaris (DvCA8.0) | Room temperature | Ambient pressure | 0.25 L | 20% CO2, 7.8 mL min−1 | Up to 132 (Ca2+) and 62 (Mg2+), 1.2–2.2-fold | 0.84–1.05 g HCO3− per L or 0.15–0.18 g CO2 per g residuea | 1.5–3 h | This study (not optimized) |
| Pulp and paper residues – CaCO3 and alkaline oxides | 4.0 | Evolved CA from Desulfovibrio vulgaris (DvCA8.0) | Room temperature | Ambient pressure | 0.25 L | Atmospheric | Up to 35 mg L−1 (Ca2+), 2.2–3.1-fold | 0.28–0.48 g HCO3− per L or 0.024–0.042 g CO2 per g residuea | 24 h | This study (not optimized) |
During the first part of the reaction (until the pH reached ∼7.3), the bicarbonate formation had contributions both from the CO2 hydration reaction and the mineral dissolution. As the reaction approached its end (90 min for R1–R3 and 180 min for R4), the CaCO3 and Mg(OH)2 dissolution rates were reduced, and CO2 hydration played a bigger role in the bicarbonate formation. Longer reaction times could also boost the mineral dissolution, however, at slower rates. Overall, the enzyme presence increased the yield of CO2 captured for all the materials. In contrast to the open-air reactions, the addition of CA had a varying response in regards to the extent of the CO2 hydration ratio over mineral dissolution as a percentage of the total CO2 capture. For R1 and R3, the addition of CA did not significantly increase the ratio of CO2 hydration over mineral dissolution neither at 40 nor at 90 min (Fig. 7a and c). For R2, the addition of CA did not significantly affect the ratio of CO2 hydration at 40 min and at 90 min, however the percentage of dissolved Mg(OH)2 over the total CO2 capture increased (Fig. 7b). Interestingly, at 90 min, the addition of CA led to a decrease in the CO2 hydration extent from 80% to 64% and a significant increase in the Mg(OH)2 dissolution, from 14% to 29%. For R4, the control reaction showed predominant Ca2+ precipitation, as in the open-air reactions. Interestingly, the addition of CA significantly accelerated the reaction, leading to apparent Ca2+ dissolution (Fig. 7d). This implies that there was ongoing competing Ca(OH)2 carbonation and CaCO3 dissolution, where CaCO3 dissolution was more predominant.
 | ||
| Fig. 7 Quantification of CO2 captured at 40 and 90 min (end of the reaction) when synthetic gas (20% CO2) was used as the feedstock: (a) R1: lime mud, (b) R2: green liquor sludge, (c) R3: electrostatic precipitator dust, and (d) R4: lime dregs. | ||
Notwithstanding the positive obtained results, the yield of CO2 capture and bicarbonate production could be optimized. It is important to emphasize that the current study was carried out at room temperature and pressure and the conditions were not optimized; thus, there is a window of opportunity for future improvements that could positively influence the CO2 capture and storage reactions. Increasing the partial CO2 pressure could lead to an enhancement in Ca2+ and Mg2+ leaching and a higher concentration of final bicarbonates. The temperature effect also needs to be analyzed since it promotes the kinetics of mineral dissolution, although high temperatures also decrease CO2 solubility, and could lead the bicarbonate ions to decompose. Finally, a higher concentration of co-sequestrant materials and increased CO2 flow rate are to be tested in further studies.
Also, no other CaCO3 crystalline forms were identified than calcite in all the analyzed samples, which could imply that the carbonation of Ca(OH)2 led to the same crystalline phases already present in the sample before the reaction or to amorphous/not detected CaCO3 phases. MgCO3 formation was not detected in the non-reacted solid from R2, excluding the presence of a ‘competitive’ Mg2+ precipitation reaction during CO2 capture.
CO2 storage using olivine as a reference material
| Element | Mg | Si | Fe | Ca | Cr | Mn | Al | Ni | V |
|---|---|---|---|---|---|---|---|---|---|
| a Reported values are for elements with concentration >0.1%. | |||||||||
| Composition (% w/w) | 24.01 ± 0.19 | 18.92 ± 0.03 | 7.65 ± 0.02 | 0.98 ± 0.01 | 0.62 ± 0.01 | 0.12 ± 0.01 | 0.42 ± 0.02 | 0.335 ± 0.02 | 0.200 ± 0.001 |
| Chemical formula | (Mg1.57Fe0.21)SiO4 | CaCO3 | (Mg,Al)6(Si,Al)4O10(OH)8 | Other phases |
|---|---|---|---|---|
| Name of the mineral | Forsterite | Calcium carbonate | Clinochlore | Silicates and oxides |
| Composition (% w/w) | 91.2 | 2.6 | 1.3 | 4.9 |
 | ||
| Fig. 8 (a) Uncrushed olivine; (b) olivine crushed to a powder; (c) SEM analysis of the crushed olivine. | ||
 | ||
| Fig. 9 (a) Captured CO2 concentration, (b) pH, (c) Mg2+ concentration, (d) Mg2+ increase, (e) Ca2+ concentration, (f) Ca2+ decrease, (g) Si concentration, and (h) Si decrease over time during olivine dissolution. ΔM is defined as Mt − Mt = 0. | ||
In the reaction with bicarbonate formed from R1 and CA, the measured release of Mg2+ was higher compared to the control after 120 h of reaction, which was expected since the solution had a higher availability of H+, thus favoring dissolution of the olivine (Fig. 9c). At the end of 120 h, the Mg2+ ions release was 1.49-fold higher for the CA-added system compared to the no-CA-added one (Fig. 9d). The initial Mg2+ and Ca2+ concentration in the supernatant obtained from the reaction with the enzyme was approximately 20% higher than the one without CA, as observed at time = 0 of the dissolution reaction. Interestingly, the Ca2+ concentration dropped over time in both systems, indicating the precipitation of Ca2+. The available CO2 concentration was mostly stable over time in the solution (Fig. 9a). As suggested in a previous study, olivine dissolution leads to the formation of orthosilicic acid and other polymeric silica forms, which can potentially adsorb Ca2+via SiOH bonds.45 This phenomenon could be a possible explanation for the Ca2+ concentration decrease in our study, as it could be attributed to Ca2+ bonding onto the silicates surface (Fig. 9e) or the formation of other identified salts, such as CaSiO4. Nevertheless its formation did not seem to be likely since the reaction conditions, at slightly alkaline and with the presence of HCO3−, should have a contrary effect on the CaSiO4 precipitation rate.46 Besides, the Ca2+ concentration decline was more accentuated in the reaction using bicarbonate solution without the use of CA in the CO2-capture reaction (Fig. 9f). Such a reaction appeared to be fast in both systems, as the consumption of Ca2+ reached a plateau and stabilized after only 48 h. The Si concentration in the solution decreased over time in both systems, reaching a plateau at 48 h, and presented a decreasing concentration of soluble Si corresponding to 0.76–0.20 of the expected stoichiometry, indicating its precipitation to other insoluble phases (such as SiO2(s)) (Fig. 9g and h), and possible interactions with Ca2+ ions, reducing its concentration over time. Another case is that Ca2+ ions could have precipitated to CaCO3, which could have benefited from the higher pH values. Based on the carbonate concentration data (Fig. 9a), some CaCO3 could be formed in the CA-added system, since the total alkalinity concentration decreased by 5.7% at the end of 120 h. However, based on the stoichiometry, if all carbonate had reacted to form CaCO3, this could account for a maximum of only 68% of the total Ca2+ consumption. Except for Ni, which presented a concentration up to 31.7–74.1 μg L−1, the samples were tested for other elements at the ppm level, but no other mineral traces were found in the aqueous phase. Despite Fe accounting for approximately 7.7% of the total mass content of the olivine, an Fe concentration <0.02 mg L−1 was detected at the end of the mineral dissolution (120 h), evidencing that the forsterite (Mg2SiO4) underwent predominant dissolution when compared to the fayalite (FeSiO4) and agreeing with previous studies that indicated that preferential Fe dissolution benefits from acidic environments instead of alkaline conditions.47
A correlation between a lower pH value, dissolved CO2 (bicarbonate), and ions leaching was expected. In the bicarbonate solutions from CA-accelerated and non-CA CO2 capture and after mixing with olivine powder, the initial pH values were 7.55 and 7.88, respectively, with a higher alkalinity (bicarbonates dissolved) measured for the first system, which promoted higher Mg leaching. Other studies utilizing metal alkali silicates have confirmed a similar trend. In a batch CO2 pressurized system (2–10 atm), utilizing a pulp of serpentine at 150 g L−1, 19.4–32.7% leaching of the total magnesium of the mineral was recorded,48 demonstrating the higher the pCO2, the higher the HCO3−, thus the greater leaching of Mg2+. Similarly, a positive correlation between the HCO3− concentration and the release of Ca2+ ions was demonstrated in a system containing Wollastonite – a Ca-silicate mineral (CaSiO3) – in a near neutral pH range of 7–8.46
According to our results, the leached Mg represented only 0.4% of the total amount contained in the olivine sample. The supernatant's bicarbonate concentration was estimated to be not more than 0.02 M, which could impact the dissolution of the forsterite due to the limitation of H+ protons available. Besides, a favored leaching of Mg2+ was demonstrated to occur under acidic to slightly alkaline conditions (pH < 8). As proposed in other studies, the forsterite dissolution rate could be controlled by breaking the layers of the polymerized Si dimer precursor47 (>Si2OH+), whereby H+ protons would be exchanged by Mg2+ ions, and we hypothesized that a similar behavior could be observed in our experiments As the pH of the system increased, a different dissolution mechanism dominated, which could be highly inhibited by the presence of carbonate ions.49 Agreeing with this, the highest Mg leaching was observed in the first 24 h for both the no- and CA-added systems, when their pH values were the lowest, meaning the dissolution rate could be affected as the pH increased over the progress of the experiment. Besides, the formation of Si-rich layers in the mineral could impede the H+ from reaching the surface of the solid, impairing its dissolution.
In another example study that performed the aqueous carbonation of magnesium silicates, a Mg leaching of 17.3% was found.50 Nevertheless, the experiments were conducted under multiple cycles of absorption and with a higher CO2 partial pressure, which facilitated the dissolution of the mineral. Also, temperature plays a role in the kinetics of forsterite dissolution, as demonstrated previously under ambient pressure conditions and acidic conditions.51 In the present study, the average temperature for conducting the experiments was kept between 20–22 °C. As a suggestion for improvement in future works, tests with more concentrated bicarbonate solutions and high temperature should be included.
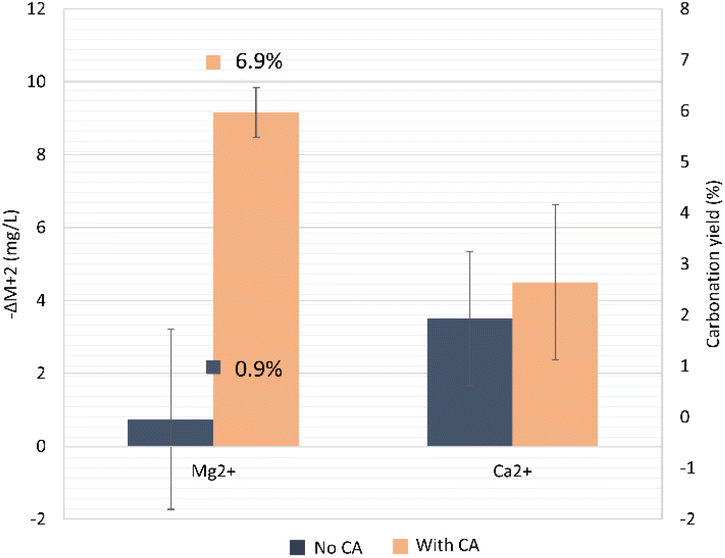 | ||
| Fig. 10 Carbonation yield for the conversion of Mg2+ ions in the formation of MgCO3, at the end of the mineral carbonation. The decrease in concentration of Mg2+ and Ca2+ during the carbonation step is also shown. ΔM is defined as Mt − Mt=0. | ||
The targeted product in the olivine carbonation step in order to confirm the CCS concept is the formation of MgCO3. To quantify the carbonation yield, we looked into the decrease in available Mg2+ in the solution during the carbonation step. As a starting point, we considered the end of olivine dissolution step. In the reaction where bicarbonate from a CA-accelerated CO2 capture step was used, the precipitation into carbonates was 7.1-fold higher when compared to the no-CA-added system. Possibly, the higher concentration of bicarbonates and Mg2+ helped achieve a higher yield from Mg carbonation, which was 6.9% for the CA-added system compared to the non-CA system (0.9%). In the latter system, the variation of the Mg2+ concentration was virtually ΔM ∼ 0, and would probably lead to a high standard deviation, since the used Mg2+ quantification method is not highly sensitive at very low concentrations. It is possible that the carbonation in the absence of CA had not reached equilibrium and more time was needed. The presence of calcite has been proven to promote Mg carbonation in Mg-bearing silicates,56 which could also have happened here with the olivine. Furthermore, mineral carbonation benefits from an increased ionic strength of the solution, which was slightly higher here in the system containing the enzyme. Yet, no additives (salts) to enhance the solution ionic strength were added in the reactions performed in this study. The direct effect of CA in improving the CO2 storage set of reactions could be debated, but there was a clear indirect effect. First, CA boosted the CO2-capture reaction, producing higher amounts of bicarbonate and H+ protons, which further boosted the olivine dissolution step, causing an increased availability of Mg2+ ions. At the end of the carbonation reaction, 43.4 ± 3.0% of the available bicarbonate was consumed from the supernatant for the no-CA-added system and 61.4 ± 6.0% by the CA-added system, which could be explained by the formation of insoluble carbonate salts and the degassing that happened during the heating.
As mentioned in the previous section, studies showed that at conditions of mild temperature and pressure, and low or zero concentration of added salts, mineral carbonation was verified to be controlled by the ions diffusion through a Si-rich layer formed around the olivine core,57 which limits the H+ attack on the unreacted mineral, impairing the mineral dissolution, and consequently leading to a low carbonation yield, as also observed in this study. In addition, the use of bovine CA (BCA) for the mineral carbonation of Wollastonite was shown to bring a faster pH increase in the system, and therefore higher carbonation rates when compared to the control reaction.58 However, since the activity of the CA used in this system was not measured during the mineral dissolution and carbonation, it could not be confirmed that the increased carbonation was related with the presence of the enzyme.
Other studies using Ca/Mg-silicates for CCS have reported carbonation yields as high as 100%;59 however, they were generally performed under conditions such as high pressure, temperature, and CO2 concentration, which could play a role in improving the carbonation extent.60,61 The addition of salts (like NaHCO3) that can buffer the solutions was also suggested could keep the pH value controlled and increase the ionic strength to favor the carbonation,62 a strategy also adopted by some works to optimize the carbon sequestration.63,64 TGA of olivine before and after the CO2-storage reactions was performed. A maximum difference of 0.9% in total mass loss was observed between them. Pure olivine contains CaCO3, while small amounts of MgCO3 (and CaCO3) were possibly formed during the carbonation reaction. The first mass loss happened between 30–120 °C (5.0% of total mass loss), which was due to the moisture content in the analyzed samples. Between 350–600 °C, a notable proportion of the total mass variation was observed, ranging from 37.5%–41.3% for the analyzed samples, which could be attributed to the decomposition of other silica-based materials, such as serpentine.65 About 50% of the total mass loss occurred between 600–900 °C, linked to the thermal decomposition of CaCO3. It is important to note that between 300–350 °C, a slight mass loss of 0.9% between the unreacted and reacted olivine was noted, which could indicate the formation of nesquehonite (MgCO3·3H2O).66 Based on the quantification of the carbonation yield during our experiment, it is expected that the amount of MgCO3 formed during the mineral carbonation would be very small, which can explain why no significant mass loss in the full range of MgCO3 thermal decomposition was found. Besides, the XRD analysis of the olivine showed no detectable differences between before and after the reaction. The XRD analysis results and TGA curves are presented in Fig. S1 and S2 in the ESI.†Table 6 list some distinct studies on the mineral carbonation of silicate materials.
| Main mineral | Concentration (g L−1) | Temperature (°C) | Pressure | Volume of reaction (L) | CO2 intake | % leaching | Carbonation yield and CO2 fixed | Residence time | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a MgCO3 yield. Serpentine: X3Si2O5(OH)4, where X = Mg2+, Fe2+, Ni2+, Mn2+, Zn2+, olivine: mainly (Mg,Fe)2SiO4, lizardite: Mg3(Si2O5)(OH)4, wollastonite: CaSiO3. N.Q.: not quantified. | |||||||||
| Serpentine | 150 | Room temperature | 8 atm | ∼6.2 | 14–18% CO2 | 17.3 (Mg) | 17.9%, 0.215 g CO2 per g solid | 6 h | 48 |
| Olivine | 100 | 185 | 6.5 MPa | 0.045 | 18.2% CO2 | 34 (Mg) | 74.8%, N.Q. | Up to 6 h | 63 |
| Serpentine/lizardite | 50–150 | 22 | 10.5 atm | 0.3 (reactor size) | 18.2% CO2 | N.Q. | 30%, 0.55 g CO2 per g solid | 2.25 h | 61 |
| Lizardite | 150 | 100–150 | 20–150 | ∼0.3 | 100% CO2 | N.Q. | 30%,a N.Q. | Up to 6 h | 64 |
| Lizardite | 150 | Room temperature | 0.4–1.6 bar | ∼5.0 | 14–18% CO2 | ∼15.8% | 33.3%, 0.08 g CO2 per g solid | 6 h | 60 |
| Olivine | ∼200 | Up to 190 | 40–100 bar | 0.01 | 100% CO2 | N.Q. | 100%, N.Q. | Up to 6 h | 59 |
| Rich Mg-bearing silicates | 100 | 185 | 20.7–38.6 bar | 0.6 (reactor size) | 100% CO2 | N.Q. | 71%, N.Q. | 5 h | 57 |
| Olivine | 150 | Room temperature (mineral dissolution) – 35–40 °C (carbonation) | 3 bar (dissolution) – ambient pressure (carbonation) | 0.17 | 1 g per L HCO3− | 0.4% (Mg) | 6.9%, N.Q. | 5 days (mineral dissolution) + 12 h (carbonation) | This study (not optimized) |
| Wollastonite | 2 | Room temperature | 2 bar | 2 | 100% CO2 | N.Q. | N.Q., 0.14 g CO2 per g solid | 22 days | 68 |
As possibilities for improvement, we suggest longer residence times and/or higher temperature as means to increase the yield of carbonation, since the latter could lead to an enhancement in the kinetics of both the olivine dissolution and the carbonation.67 Furthermore, the carbonation experiments were conducted in non-pressurized vessels, which are more likely to be affected by degassing, decreasing the amount of CO2 (bicarbonates) over time, especially under heating, and therefore the utilization of reactors under pressure are recommended. Increasing the supernatant's bicarbonate concentration is another important parameter to be tested in olivine dissolution, which should be a topic of further studies to optimize the conditions for CO2 capture.
Conclusions
Investigation of the routes to valorize alkaline residues is valuable, and their utilization for CCS tackles both CO2 mitigation and waste repurposing. Using CaCO3-rich residues generated in the Kraft process, a route for CCS was proposed in the present study and showed encouraging results. The utilization of CA improved significantly up to 5-fold the CO2 capture and almost 2-fold the mineral dissolution of olivine, when compared to a reaction without addition of the enzyme.Some limitations on the mineral dissolution and carbonation were observed, probably because of the formation of a passivation rich Si-layer around the core of unreacted olivine impeding H+ from attacking the mineral, decreasing its dissolution rate. In addition, the formation of carbonates during the olivine dissolution step with the pH increase over time can strongly inhibit the mineral dissolution rate, leading to a low concentration of Mg2+ leached. The carbonation extent of Mg2+ ions was 7.1-fold higher using bicarbonate from a CA-added capture/dissolution reaction compared to the no-CA-added case, which could potentially be improved in future studies. A carbonation yield of 6.9% was achieved. As recommendations, the utilization of pressurized vessels for mitigating the effects of degassing, longer residences times, and more concentrated bicarbonate solutions could lead to a higher availability of the leached metals, accelerating the carbonation, and so should be considered in future studies. Nevertheless, we successfully demonstrated a proof-of-concept CCS strategy for the paper and pulp industry, which could be achieved even at relatively low pressures (3 bar). Tests at higher pressures will be part of future work in order to simulate geological conditions during the injection of bicarbonate in the bedrock (>100 bar), which will certainly benefit the carbonation yields.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank David Nilsson (Billerud, Karlsborg, Norrbotten, Sweden) for the contribution of the residues utilized in this study. This work is supported by the Swedish Energy Agency project ‘Utilization of industrial residues for an efficient geological BECSS’ – INSURANCE (grant number ID: 2020-019943).References
- F. W. K. Khudhur, J. M. MacDonald, A. Macente and L. Daly, The Utilization of Alkaline Wastes in Passive Carbon Capture and Sequestration: Promises, Challenges and Environmental Aspects, Sci. Total Environ., 2022, 823, 153553, DOI:10.1016/j.scitotenv.2022.153553.
- W. Liu, L. Teng, S. Rohani, Z. Qin, B. Zhao, C. C. Xu, S. Ren, Q. Liu and B. Liang, CO2 Mineral Carbonation Using Industrial Solid Wastes: A Review of Recent Developments, Chem. Eng. J., 2021, 416, 129093, DOI:10.1016/j.cej.2021.129093.
- S.-Y. Pan, E. E. Chang and P.-C. Chiang, CO2 Capture by Accelerated Carbonation of Alkaline Wastes: A Review on Its Principles and Applications, Aerosol Air Qual. Res., 2012, 12(5), 770–791, DOI:10.4209/aaqr.2012.06.0149.
- P. Renforth, The Negative Emission Potential of Alkaline Materials, Nat. Commun., 2019, 10(1), 1401, DOI:10.1038/s41467-019-09475-5.
- N. Zhang, Y. E. Chai, R. M. Santos and L. Šiller, Advances in Process Development of Aqueous CO2 Mineralisation towards Scalability, J. Environ. Chem. Eng., 2020, 8(6), 104453, DOI:10.1016/j.jece.2020.104453.
- R. Zevenhoven, M. Slotte, J. Åbacka and J. Highfield, A Comparison of CO 2 Mineral Sequestration Processes Involving a Dry or Wet Carbonation Step, Energy, 2016, 117, 604–611, DOI:10.1016/j.energy.2016.05.066.
- IPCC. Global Warming of 1.5°C. An IPCC Special Report on the Impacts of Global Warming of 1.5°C above Pre-Industrial Levels and Related Global Greenhouse Gas Emission Pathways, in the Context of Strengthening the Global Response to the Threat of Climate Change, IPCC – Sr15, 2018, vol. 2, pp. 17–20 Search PubMed.
- J. S. Kirchner, A. Berry, F. Ohnemüller, B. Schnetger, E. Erich, H.-J. Brumsack and K. A. Lettmann, Reducing CO 2 Emissions of a Coal-Fired Power Plant via Accelerated Weathering of Limestone: Carbon Capture Efficiency and Environmental Safety, Environ. Sci. Technol., 2020, 54(7), 4528–4535, DOI:10.1021/acs.est.9b07009.
- E. L. A. Sjöberg, Fundamental Equation for Calcite Dissolution Kinetics, Geochim. Cosmochim. Acta, 1976, 40(4), 441–447, DOI:10.1016/0016-7037(76)90009-0.
- P. Cubillas, S. Köhler, M. Prieto, C. Chaïrat and E. H. Oelkers, Experimental Determination of the Dissolution Rates of Calcite, Aragonite, and Bivalves, Chem. Geol., 2005, 216(1–2), 59–77, DOI:10.1016/j.chemgeo.2004.11.009.
- M. Back, M. Kuehn, H. Stanjek and S. Peiffer, Reactivity of Alkaline Lignite Fly Ashes towards CO2 in Water, Environ. Sci. Technol., 2008, 42(12), 4520–4526, DOI:10.1021/es702760v.
- O. S. Pokrovsky and J. Schott, Experimental Study of Brucite Dissolution and Precipitation in Aqueous Solutions: Surface Speciation and Chemical Affinity Control, Geochim. Cosmochim. Acta, 2004, 68(1), 31–45, DOI:10.1016/S0016-7037(03)00238-2.
- D. A. Vermilyea, The Dissolution of MgO and Mg(OH)[Sub 2] in Aqueous Solutions, J. Electrochem. Soc., 1969, 116(9), 1179, DOI:10.1149/1.2412273.
- A. de Oliveira Maciel, P. Christakopoulos, U. Rova and I. Antonopoulou, Carbonic Anhydrase to Boost CO2 Sequestration: Improving Carbon Capture Utilization and Storage (CCUS), Chemosphere, 2022, 134419, DOI:10.1016/j.chemosphere.2022.134419.
- E. Sapountzaki, U. Rova, P. Christakopoulos and I. Antonopoulou, Renewable Hydrogen Production and Storage Via Enzymatic Interconversion of CO2 and Formate with Electrochemical Cofactor Regeneration, ChemSusChem, 2023, 16(17), e202202312, DOI:10.1002/cssc.202202312.
- I. Antonopoulou, U. Rova and P. Christakopoulos, CO2 to Methanol: A Highly Efficient Enzyme Cascade, Methods Mol. Biol., 2022, 317–344, DOI:10.1007/978-1-0716-2269-8_19.
- W. Li, L. Yu, Y. Wu, L. Jia and D. Yuan, Enhancement of Ca2+ Release from Limestone by Microbial Extracellular Carbonic Anhydrase, Bioresour. Technol., 2007, 98(4), 950–953, DOI:10.1016/j.biortech.2006.03.021.
- W. Li, P.-P. Zhou, L.-P. Jia, L.-J. Yu, X.-L. Li and M. Zhu, Limestone Dissolution Induced by Fungal Mycelia, Acidic Materials, and Carbonic Anhydrase from Fungi, Mycopathologia, 2009, 167(1), 37–46, DOI:10.1007/s11046-008-9143-y.
- Z. Liu, D. Yuan and W. Dreybrodt, Comparative Study of Dissolution Rate-Determining Mechanisms of Limestone and Dolomite, Environ. Geol., 2005, 49(2), 274–279, DOI:10.1007/s00254-005-0086-z.
- K. Mortezaei, A. Amirlatifi, E. Ghazanfari and F. Vahedifard, Potential CO2 Leakage from Geological Storage Sites: Advances and Challenges, Environ. Geotech., 2021, 8(1), 3–27, DOI:10.1680/jenge.18.00041.
- T. Jarnerud, Application of Wastes from Pulp and Paper Industries for Steelmaking Processes, 2021 Search PubMed.
- J. Sun, W. Liu, M. Li, X. Yang, W. Wang, Y. Hu, H. Chen, X. Li and M. Xu, Mechanical Modification of Naturally Occurring Limestone for High-Temperature CO2 Capture, Energy Fuel., 2016, 30(8), 6597–6605, DOI:10.1021/acs.energyfuels.6b01131.
- O. Alvizo, L. J. Nguyen, C. K. Savile, J. A. Bresson, S. L. Lakhapatri, E. O. P. Solis, R. J. Fox, J. M. Broering, M. R. Benoit, S. A. Zimmerman, S. J. Novick, J. Liang and J. J. Lalonde, Directed Evolution of an Ultrastable Carbonic Anhydrase for Highly Efficient Carbon Capture from Flue Gas, Proc. Natl. Acad. Sci. U.S.A., 2014, 111(46), 16436–16441, DOI:10.1073/pnas.1411461111.
- M. Sjöblom, I. Antonopoulou, I. G. Jiménez, A. De Oliveira Maciel, S. G. Khokarale, J. P. Mikkola, U. Rova and P. Christakopoulos, Enzyme-Assisted CO2Absorption in Aqueous Amino Acid Ionic Liquid Amine Blends, ACS Sustain. Chem. Eng., 2020, 8(36), 13672–13682, DOI:10.1021/acssuschemeng.0c03497.
- P. Mirjafari, K. Asghari and N. Mahinpey, Investigating the Application of Enzyme Carbonic Anhydrase for CO2 Sequestration Purposes, Ind. Eng. Chem. Res., 2007, 46(3), 921–926, DOI:10.1021/ie060287u.
- R. G. Compton and C. A. Brown, Inhibition of Calcite Dissolution/Precipitation: Mg2+ Cations, J. Colloid Interface Sci., 1994, 445–449, DOI:10.1006/jcis.1994.1248.
- S.-J. Han, M. Yoo, D.-W. Kim and J.-H. Wee, Carbon Dioxide Capture Using Calcium Hydroxide Aqueous Solution as the Absorbent, Energy Fuels, 2011, 25(8), 3825–3834, DOI:10.1021/ef200415p.
- Z. Zhang, B. Lian, W. Hou, M. Chen, X. Li and Y. Li, Bacillus Mucilaginosus Can Capture Atmospheric CO 2 by Carbonic Anhydrase, Afr. J. Microbiol. Res., 2011, 5(2), 106–112, DOI:10.5897/AJMR10.690.
- T. Tenno, K. Uiga, A. Mashirin, I. Zekker and E. Rikmann, Modeling Closed Equilibrium Systems of H2O-Dissolved CO2-Solid CaCO3, J. Phys. Chem. A, 2017, 121(16), 3094–3100, DOI:10.1021/acs.jpca.7b00237.
- T. Shen, W. Li, W. Pan, S. Lin, M. Zhu and L. Yu, Role of Bacterial Carbonic Anhydrase during CO2 Capture in the CO2-H2O-Carbonate System, Biochem. Eng. J., 2017, 123, 66–74, DOI:10.1016/J.BEJ.2017.04.003.
- L. Xiao, B. Lian, J. Hao, C. Liu and S. Wang, Effect of Carbonic Anhydrase on Silicate Weathering and Carbonate Formation at Present Day CO2 Concentrations Compared to Primordial Values, Sci. Rep., 2015, 5(1), 7733, DOI:10.1038/srep07733.
- T. Shen, W. Li, W. Pan, S. Lin, M. Zhu and L. Yu, Role of Bacterial Carbonic Anhydrase during CO2 Capture in the CO2-H2O-Carbonate System, Biochem. Eng. J., 2017, 123, 66–74, DOI:10.1016/j.bej.2017.04.003.
- G. Gadikota and A. A. Park, Accelerated Carbonation of Ca- and Mg-Bearing Minerals and Industrial Wastes Using CO2, in Carbon Dioxide Utilisation: Closing the Carbon Cycle, 1st edn, 2015, pp. 115–137, DOI:10.1016/B978-0-444-62746-9.00008-6.
- I. M. Power, A. L. Harrison and G. M. Dipple, Accelerating Mineral Carbonation Using Carbonic Anhydrase, Environ. Sci. Technol., 2016, 50(5), 2610–2618, DOI:10.1021/acs.est.5b04779.
- Ö. Cizer, E. Ruiz-Agudo and C. Rodriguez-Navarro, Kinetic Effect of Carbonic Anhydrase Enzyme on the Carbonation Reaction of Lime Mortar, Int. J. Architect. Herit., 2018, 12(5), 779–789, DOI:10.1080/15583058.2017.1413604.
- L. Zhao, L. Sang, J. Chen, J. Ji and H. H. Teng, Aqueous Carbonation of Natural Brucite: Relevance to CO 2 Sequestration, Environ. Sci. Technol., 2010, 44(1), 406–411, DOI:10.1021/es9017656.
- D. Bonenfant, L. Kharoune, S. Sauvé, R. Hausler, P. Niquette, M. Mimeault and M. Kharoune, CO2 Sequestration by Aqueous Red Mud Carbonation at Ambient Pressure and Temperature, Ind. Eng. Chem. Res., 2008, 47(20), 7617–7622, DOI:10.1021/ie7017228.
- E. R. Bobicki, Q. Liu, Z. Xu and H. Zeng, Carbon Capture and Storage Using Alkaline Industrial Wastes, Prog. Energy Combust. Sci., 2012, 38(2), 302–320, DOI:10.1016/j.pecs.2011.11.002.
- Q. Zhao, X. Chu, X. Mei, Q. Meng, J. Li, C. Liu, H. Saxén and R. Zevenhoven, Co-Treatment of Waste From Steelmaking Processes: Steel Slag-Based Carbon Capture and Storage by Mineralization, Front. Chem., 2020, 8, 571504, DOI:10.3389/fchem.2020.571504.
- H. Y. Jo, J. H. Kim, Y. J. Lee, M. Lee and S.-J. Choh, Evaluation of Factors Affecting Mineral Carbonation of CO2 Using Coal Fly Ash in Aqueous Solutions under Ambient Conditions, Chem. Eng. J., 2012, 183, 77–87, DOI:10.1016/j.cej.2011.12.023.
- S. K. Puthiya Veetil, K. Rebane, C. R. Yörük, M. Lopp, A. Trikkel and M. Hitch, Aqueous Mineral Carbonation of Oil Shale Mine Waste (Limestone): A Feasibility Study to Develop a CO2 Capture Sorbent, Energy, 2021, 221, 119895, DOI:10.1016/J.ENERGY.2021.119895.
- L. Zhao, L. Sang, J. Chen, J. Ji and H. H. Teng, Aqueous Carbonation of Natural Brucite: Relevance to CO2 Sequestration, Environ. Sci. Technol., 2010, 44(1), 406–411, DOI:10.1021/es9017656.
- Q. Sun and B. Lian, The Different Roles of Aspergillus Nidulans Carbonic Anhydrases in Wollastonite Weathering Accompanied by Carbonation, Geochim. Cosmochim. Acta, 2019, 244, 437–450, DOI:10.1016/j.gca.2018.10.022.
- L. Zhang and J. R. Smyth, Crystal Chemistry of Metal Element Substitution in Olivine and Its High-Pressure Polymorphs: Implications for the Upper-Mantle and the Mantle Transition Zone, Earth-Sci. Rev., 2022, 232, 104127, DOI:10.1016/j.earscirev.2022.104127.
- R. K. Iler, The Chemistry of Silica: Solubility, Polymerization, Colloid and Surface Properties, and Biochemistry, John Wiley and Sons Ltd, New York, 1979, DOI:10.1016/0160-9327(80)90074-5.
- S. V. Golubev, O. S. Pokrovsky and J. Schott, Experimental Determination of the Effect of Dissolved CO2 on the Dissolution Kinetics of Mg and Ca Silicates at 25 °C, Chem. Geol., 2005, 217(3–4), 227–238, DOI:10.1016/j.chemgeo.2004.12.011.
- O. S. Pokrovsky and J. Schott, Kinetics and Mechanism of Forsterite Dissolution at 25 °C and PH from 1 to 12, Geochim. Cosmochim. Acta, 2000, 64(19), 3313–3325 CrossRef CAS.
- N. Kemache, L. C. Pasquier, E. Cecchi, I. Mouedhen, J. F. Blais and G. Mercier, Aqueous Mineral Carbonation for CO2 Sequestration: From Laboratory to Pilot Scale, Fuel Process. Technol., 2017, 166, 209–216, DOI:10.1016/j.fuproc.2017.06.005.
- O. S. Pokrovsky and J. Schott, Kinetics and Mechanism of Forsterite Dissolution at 25°C and PH from 1 to 12, Geochim. Cosmochim. Acta, 2000, 64(19), 3313–3325, DOI:10.1016/S0016-7037(00)00434-8.
- N. Kemache, L. C. Pasquier, E. Cecchi, I. Mouedhen, J. F. Blais and G. Mercier, Aqueous Mineral Carbonation for CO2 Sequestration: From Laboratory to Pilot Scale, Fuel Process. Technol., 2017, 166, 209–216, DOI:10.1016/J.FUPROC.2017.06.005.
- J. J. Rosso and J. D. Rimstidt, A High Resolution Study of Forsterite Dissolution Rates, Geochim. Cosmochim. Acta, 2000, 64(5), 797–811, DOI:10.1016/S0016-7037(99)00354-3.
- J.-H. Bang, S. C. Chae, S.-W. Lee, J.-W. Kim, K. Song, J. Kim and W. Kim, Sequential Carbonate Mineralization of Desalination Brine for CO2 Emission Reduction, J. CO2 Util., 2019, 33, 427–433, DOI:10.1016/j.jcou.2019.07.020.
- M. Hänchen, V. Prigiobbe, R. Baciocchi and M. Mazzotti, Precipitation in the Mg-Carbonate System—Effects of Temperature and CO2 Pressure, Chem. Eng. Sci., 2008, 63(4), 1012–1028, DOI:10.1016/j.ces.2007.09.052.
- I. M. Power, P. A. Kenward, G. M. Dipple and M. Raudsepp, Room Temperature Magnesite Precipitation, Cryst. Growth Des., 2017, 17(11), 5652–5659, DOI:10.1021/acs.cgd.7b00311.
- R. S. Arvidson, M. Collier, K. J. Davis, M. D. Vinson, J. E. Amonette and A. Luttge, Magnesium Inhibition of Calcite Dissolution Kinetics, Geochim. Cosmochim. Acta, 2006, 70(3), 583–594, DOI:10.1016/j.gca.2005.10.005.
- M. Chen, Q. Zhang, Z. Li, H. Hu and C. Wang, Insights into the Mechanochemical Interfacial Interaction between Calcite and Serpentine: Implications for Ambient CO2 Capture, J. Cleaner Prod., 2023, 401, 136715, DOI:10.1016/j.jclepro.2023.136715.
- F. Wang, D. Dreisinger, M. Jarvis and T. Hitchins, Kinetics and Mechanism of Mineral Carbonation of Olivine for CO2 Sequestration, Miner. Eng., 2019, 131, 185–197, DOI:10.1016/j.mineng.2018.11.024.
- F. Di Lorenzo, C. Ruiz-Agudo, A. Ibañez-Velasco, R. Gil-San Millán, J. A. R. Navarro, E. Ruiz-Agudo and C. Rodriguez-Navarro, The Carbonation of Wollastonite: A Model Reaction to Test Natural and Biomimetic Catalysts for Enhanced CO2 Sequestration, Minerals, 2018, 8(5), 209, DOI:10.3390/min8050209.
- E. Eikeland, A. B. Blichfeld, C. Tyrsted, A. Jensen and B. B. Iversen, Optimized Carbonation of Magnesium Silicate Mineral for CO 2 Storage, ACS Appl. Mater. Interfaces, 2015, 7(9), 5258–5264, DOI:10.1021/am508432w.
- I. Mouedhen, N. Kemache, L.-C. Pasquier, E. Cecchi, J.-F. Blais and G. Mercier, Effect of PCO2 on Direct Flue Gas Mineral Carbonation at Pilot Scale, J. Environ. Manage., 2017, 198, 1–8, DOI:10.1016/j.jenvman.2017.04.048.
- L. C. Pasquier, G. Mercier, J. F. Blais, E. Cecchi and S. Kentish, Parameters Optimization for Direct Flue Gas CO2 Capture and Sequestration by Aqueous Mineral Carbonation Using Activated Serpentinite Based Mining Residue, Appl. Geochem., 2014, 50, 66–73, DOI:10.1016/J.APGEOCHEM.2014.08.008.
- W. K. O'Connor, D. C. Dahlin, G. E. Rush, S. J. Gerdemann, L. R. Penner and D. N. Nilsen, Aqueous Mineral Carbonation: Mineral Availability, Pretreatment, Reaction Parametrics, and Process Studies, 2005, DOI:10.13140/RG.2.2.23658.31684.
- J. Li, A. D. Jacobs and M. Hitch, Direct Aqueous Carbonation on Olivine at a CO2 Partial Pressure of 6.5 MPa, Energy, 2019, 173, 902–910, DOI:10.1016/J.ENERGY.2019.02.125.
- E. Benhelal, M. I. Rashid, M. S. Rayson, G. F. Brent, T. Oliver, M. Stockenhuber and E. M. Kennedy, Direct Aqueous Carbonation of Heat Activated Serpentine: Discovery of Undesirable Side Reactions Reducing Process Efficiency, Appl. Energy, 2019, 242, 1369–1382, DOI:10.1016/j.apenergy.2019.03.170.
- T. M. McCollom, F. Klein, B. Moskowitz, T. S. Berquó, W. Bach and A. S. Templeton, Hydrogen Generation and Iron Partitioning during Experimental Serpentinization of an Olivine–Pyroxene Mixture, Geochim. Cosmochim. Acta, 2020, 282, 55–75, DOI:10.1016/j.gca.2020.05.016.
- G. Jauffret, J. Morrison and F. P. Glasser, On the Thermal Decomposition of Nesquehonite, J. Therm. Anal. Calorim., 2015, 122(2), 601–609, DOI:10.1007/s10973-015-4756-0.
- P. Bénézeth, G. D. Saldi, J.-L. Dandurand and J. Schott, Experimental Determination of the Solubility Product of Magnesite at 50 to 200°C, Chem. Geol., 2011, 286(1–2), 21–31, DOI:10.1016/j.chemgeo.2011.04.016.
- J. C.-S. Wu, J.-D. Sheen, S.-Y. Chen and Y.-C. Fan, Feasibility of CO 2 Fixation via Artificial Rock Weathering, Ind. Eng. Chem. Res., 2001, 40(18), 3902–3905, DOI:10.1021/ie010222l.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra06927c |
| This journal is © The Royal Society of Chemistry 2024 |