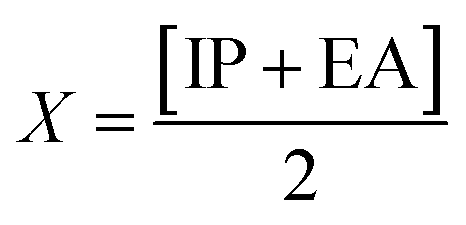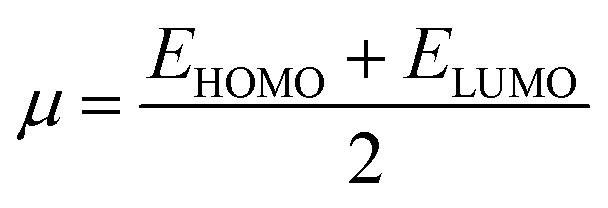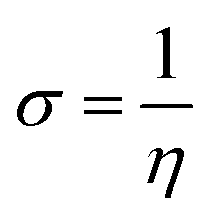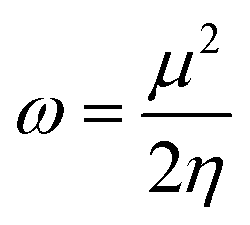DOI:
10.1039/D3RA06817J
(Paper)
RSC Adv., 2024,
14, 12841-12852
Use of benzothiophene ring to improve the photovoltaic efficacy of cyanopyridinone-based organic chromophores: a DFT study†
Received
7th October 2023
, Accepted 8th April 2024
First published on 19th April 2024
Abstract
The benzothiophene based chromophores (A1D1–A1D5) with A–π–A configuration were designed via end-capped tailoring with benzothiophene type acceptors using reference compound (A1R). Quantum chemical calculations were accomplished at M06/6-311G(d,p) level to probe optoelectronic and photophysical properties of designed chromophores. Therefore, frontier molecular orbitals (FMOs), binding energy (Eb), open circuit voltage (Voc), transition density matrix (TDM), density of state (DOS) and UV-Vis analyses of A1R and A1D1–A1D5 were accomplished. The designed compounds (A1D1–A1D5) exhibited absorption values in the visible region as 616.316–649.676 nm and 639.753–665.508 nm in gas and chloroform phase, respectively, comparing with reference chromophore. An efficient charge transference from HOMO towards LUMO was found in A1D1–A1D5 chromophores which was further supported by TDM and DOS analyses. Among all chromophores, A1D2 exhibited unique characteristics such as reduced band gap (2.354 eV), higher softness (σ = 0.424 eV), lower exciton binding energy (0.491 eV) and maximum value of open circuit voltage (Voc = 1.981 V). Consequently, A1D2 may be considered as potential candidate for the development of optoelectronic devices. These analyses revealed that the studied compounds exhibited promising findings. They may be utilized in the realm of organic solar cells.
Introduction
Addressing the urgent energy crisis requires embracing efficient, durable, and environmentally friendly sources as exhaustible energy reserves deplete.1–3 Wind, solar, biomass, and hydro-power have emerged as crucial alternatives for sustainable economic growth and combating energy scarcity.4 Solar cells, considered the most compelling option, offer a safe, clean, and abundant source, contributing significantly to global efforts for net-zero carbon emissions. With clean production processes and increasing accessibility, solar energy is poised to surpass fossil fuels, potentially replacing coal entirely by 2050 and becoming the predominant global energy source by the end of the century.5–7 Inorganic silicon-based solar cells have gained considerable interest owing to their notable power conversion efficiencies (PCEs), exceptional charge transfer capabilities, robust thermal stability, low toxicity, and abundant natural availability.8,9 However, organic solar cells (OSCs) have emerged as a viable alternative, replacing conventional counterparts due to their capacity to overcome limitations such as brittleness, non-tunable energy levels, and high manufacturing cost.10,11 Over the past few decades, fullerene derivatives, particularly [6,6]-phenyl-C61-butyric acid methyl ester (PCBM), have been the dominant electron acceptors in OSCs.12,13 However, the inherent limitations of fullerene-based systems, such as low absorption coefficients, limited tunability, and batch-to-batch variations, have spurred a quest for alternative acceptor materials.14 In recent years, non-fullerene acceptors (NFAs) have emerged as a transformative paradigm in the field of organic photovoltaics, offering a versatile and tunable platform for enhancing the efficiency and stability of OSCs.15,16 The shift towards non-fullerene based systems has been driven by their unique advantages, including broader absorption spectra,17 tunable energy levels,18 improved charge transport properties,19 and enhanced morphological control.20 This paradigm shift not only addresses the drawbacks associated with fullerene-based acceptors but also opens new avenues for the development of high-performance OSCs.21 The structure–property relationship (architecture and the nature of electron with drawing/donating groups) played a significant role in tunning the photovoltaic properties of NFAs.22,23 The NFAs with central donating core and terminal with electron withdrawing units are reported with significant photovoltaic properties such as reduction of HOMO/LUMO energy gap,24 significant charge transfer,17 thermal stability25,26 and wide-range of spectrum.27,28 The economic manufacturability, effective tunability of energy levels, wide optical absorption range, and high morphological consistency render NF-SMAs as potential candidates over fullerene-based acceptors, establishing them as outstanding candidates for efficient OSCs. Consequently, significant research efforts have been directed towards addressing the challenges and advancing the understanding of NF-SMAs in the context of OSCs.29 Molecular engineering plays a crucial role in optimizing the photovoltaic properties of non-fullerene acceptors by precisely tailoring molecular structures to enhance absorption spectra, charge mobility, and energy levels, thereby improving the overall performance of OSCs.30–32 Literature data elucidates that incorporating benzothiophene (BT) based acceptors, coupled with electron-withdrawing moieties, enhances charge transfer toward acceptor regions.33,34 This leads to increased values of short-circuit current (Jsc) and open-circuit voltage (Voc) in NFAs. This effect is attributed to the selective positioning of a lower-lying LUMO, while the HOMO remains nearly unchanged. Additionally, the absorption band is effectively broadened, contributing to these improvements.35 Currently, density functional theory (DFT) has played a pivotal role in elucidating the electronic and optical properties of non-fullerene-based OSCs.36 By employing DFT simulations, researchers gain valuable insights into the molecular interactions, charge transport mechanisms, and energetics of these innovative materials. This computational approach not only aids in the rational design of efficient NFAs but also facilitates a deeper understanding of the complex interplay between molecular structures and device performance, ultimately paving the way for the development of high-performance OSCs.37–39 Keeping in view the importance of NFAs based OSCs, in current study a series of cyanopyridinone (CP) based chromophores (A1D1–A1D5) with A–π–A framework was designed from synthesized BFA1 chromophore.40 These compounds were designed by molecular engineering with benzothiophene based acceptors in order to improve the photovoltaic properties. From literature study, we found out that the CP based chromophores with benzothiophene acceptors have not been reported yet. Quantum chemical calculations were employed to investigate the key electronic, optic and photovoltaic characteristics of A1D1–A1D5 at M06/6-311G(d,p) functional of DFT approach. It is anticipated that newly designed derivatives may be synthesized by experimentalists as proficient photovoltaic OSCs in future.
Methodology
In order to investigate the photovoltaic properties of A1R and A1D1–A1D5 chromophores, quantum chemical calculations were accomplished with the aid of Gaussian 09 software,41 at TD-DFT/TDF approaches. At first step, the structures of studied compounds were optimized at M06 functional,42 in conjunction with the 6-311G(d,p) basis set.43 After obtaining true minima geometries, various analyses such as frontier molecular orbitals (FMOs), global reactivity parameters (GRPs), density of states (DOS), transition density matrix (TDM), open circuit voltage (Voc), and UV-Vis absorption were accomplished at abovementioned functional, in order to investigate the impact of benzothiophene based acceptors on the designed chromophores. To interpret the data from outputs, different software: Avogadro,44 Chemcraft,45 PyMOlyze 2.0,46 Gaussum,47 Multiwfn 3.7,48 and Gauss View 6.0.16 (ref. 49) were utilized. The Voc of A1R and A1D1–A1D5 chromophores was determined with the aid of eqn (1).50| | |
Voc = (|EHOMOD| − |ELUMOA|) − 0.3
| (1) |
Here, 0.3 is the empirical constant, while E stands for the energy of molecular orbitals.
Results and discussion
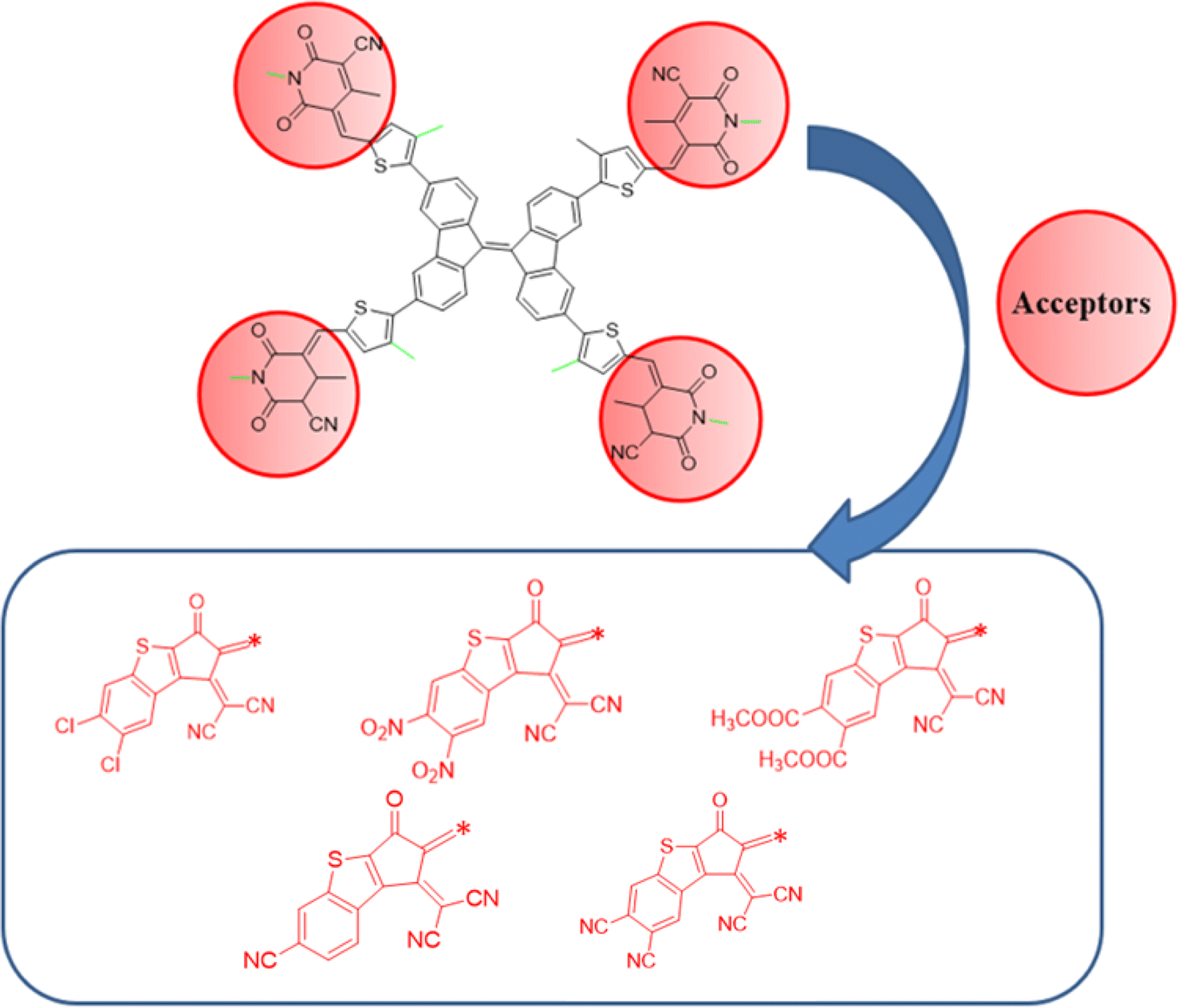
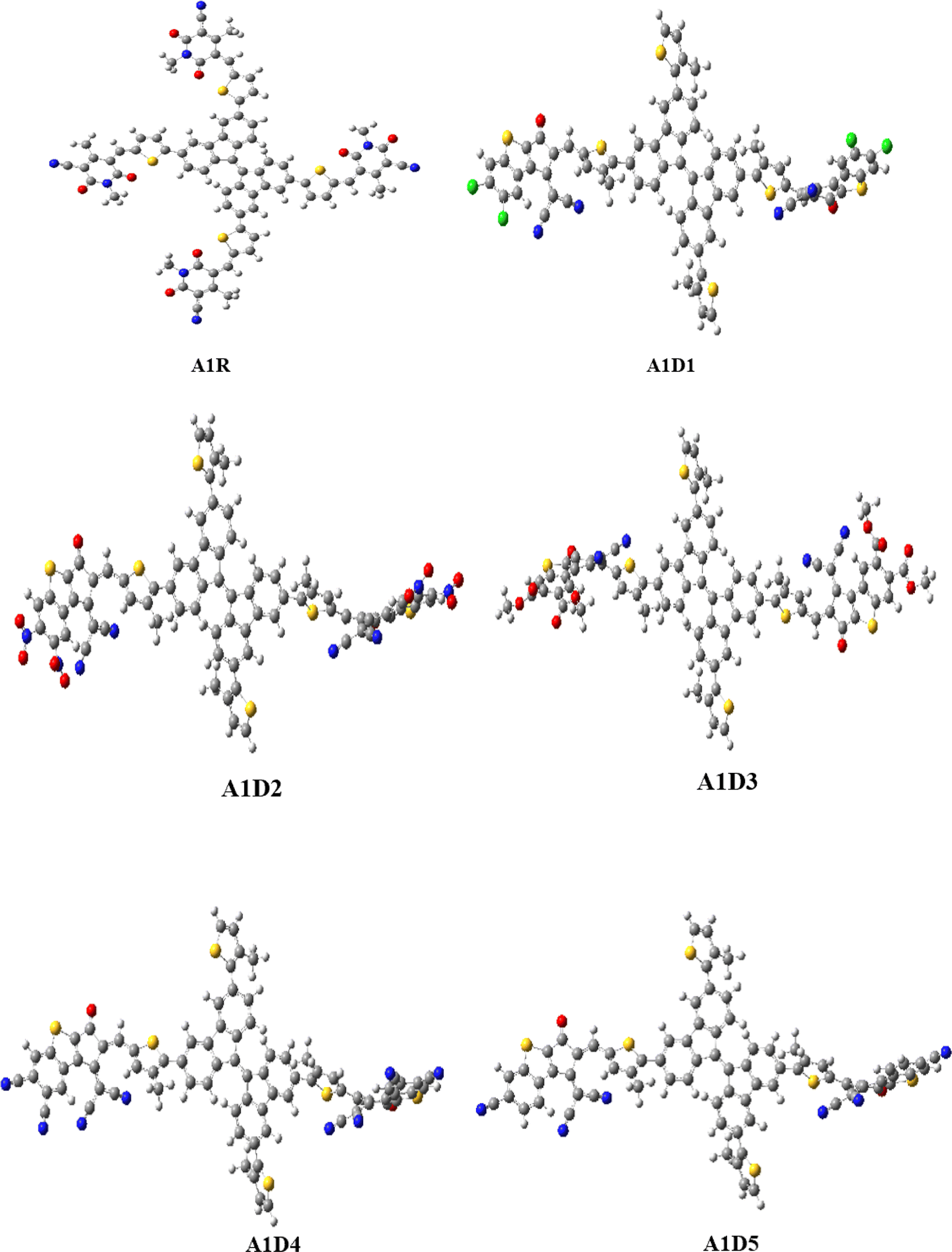
In this study, a series of CP based chromophores (A1R and A1D1–A1D5) with A–π–A framework was designed from a reported synthesized BFA1 compound.40 This compound consisted on a cyanopyridinone-based (CP) central core that acts as a π-spacer, and 1,4-dimethyl-5-methylene-2,6-dioxo-1,2,5,6-tetrahydropyridine-3-carbonitrile terminal acceptor. To reduce the computational cost, we substituted long alkyl groups, such as hexyl (–C6H13) and butyl (–C4H9), with methyl (–CH3) groups in BFA1 and named as A1R, as illustrated in Fig. 1. Literature review revealed that modifying end-capped acceptors is helpful in achieving significant photovoltaic responses in organic chromophores.51 The incorporation of BT acceptors, coupled with electron-withdrawing groups, enhances CT towards acceptor components, resulting in increased Jsc and Voc values in OSCs. This is attributed to the selectively lower-lying LUMO with minimal alteration in the HOMO, combined with effective broadening of the absorption band.52 Therefore, in current study, we designed A1D1–A1D5 by structural tailoring with benzothiophene (BT) based acceptor, to tune the optoelectronic and photovoltaic properties of these compounds depicted in Fig. 2. The influence of BT acceptors was explored through DFT approach. Literature data explained that the reaction with negative value of ΔrG° elucidates that the reaction is spontaneous with significant formation of products.53,54 The negative values of ΔrG° are obtained using eqn (S1)† for studied compounds as can be seen in Table S1† which indicated the feasibility of product formation (A1D1–A1D5) and their stability. Fig. 3 presents the optimized geometries of investigated molecules, while their cartesian coordinates are illustrate in Tables S1–S6.†
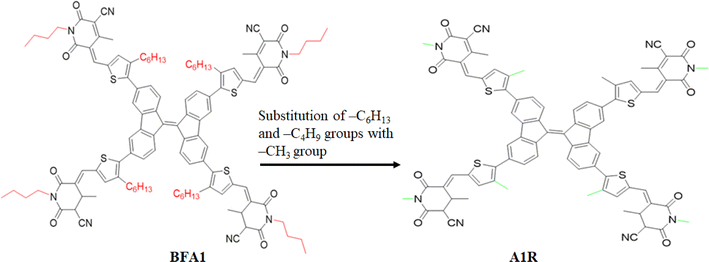 |
| | Fig. 1 Modification of BFA1 into A1R via replacing bulky groups with –CH3 group. | |
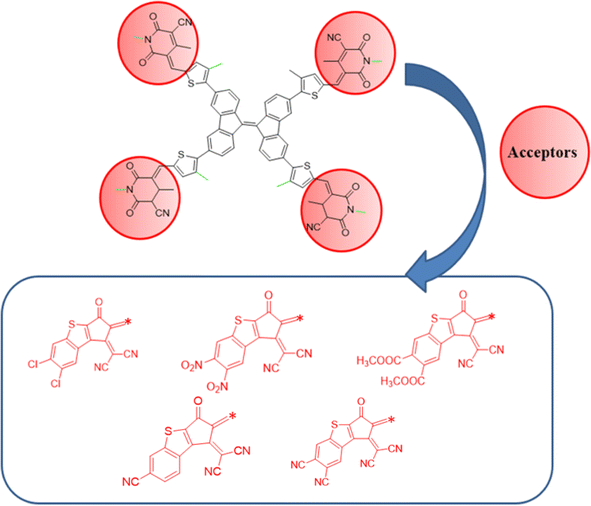 |
| | Fig. 2 Schematic representation of designing of A1D1–A1D5 from A1R. | |
 |
| | Fig. 3 Optimized geometries of A1R and A1D1–A1D5. | |
Frontier molecular orbitals (FMO) analysis
The FMO analysis is used to determine light absorption capacity, reactivity and optoelectronic properties of OSCs.55 The energy difference between molecular orbitals predicts the properties like reactivity,56 stability,26 hardness, softness,57 and intramolecular charge transfer (ICT) of chromophores.58 Therefore, HOMO/LUMO energies and ΔE are calculated for A1R and A1D1–A1D5 and results are tabulated in the Table 1.
Table 1 Computed EHOMO, ELUMO and energy gap of studied compounds in eV
| Compounds |
EHOMO |
ELUMO |
Band gap |
| A1R |
−6.302 |
−3.557 |
2.745 |
| A1D1 |
−5.623 |
−3.136 |
2.487 |
| A1D2 |
−5.684 |
−3.330 |
2.354 |
| A1D3 |
−5.616 |
−3.130 |
2.486 |
| A1D4 |
−5.672 |
−3.263 |
2.409 |
| A1D5 |
−6.149 |
−3.158 |
2.991 |
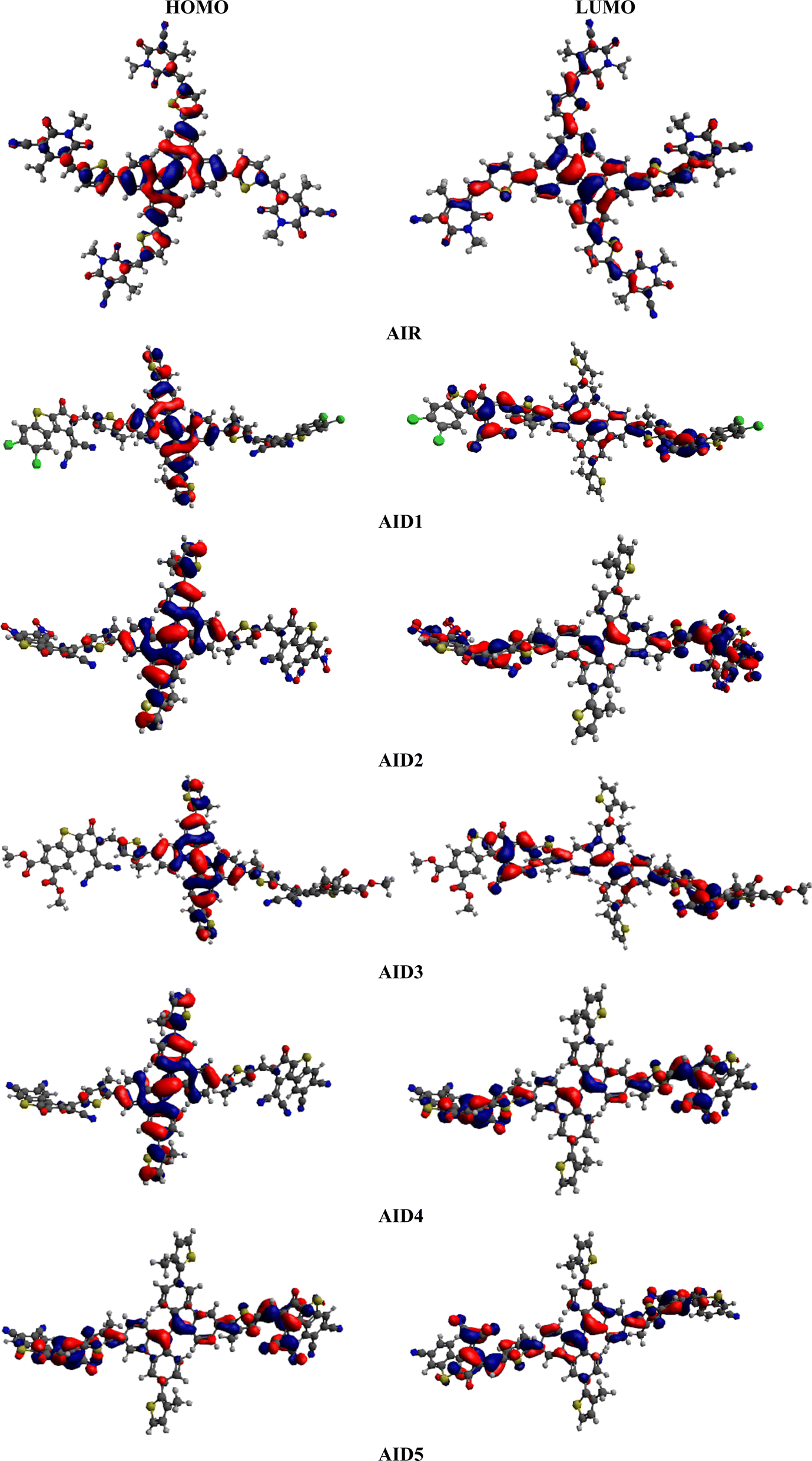
The computed HOMO/LUMO (−6.302/−3.557 eV) values of reference chromophore showed harmony with reported experimental values (−5.45/−3.85 eV).40 This close harmony illustrated the suitable selection of functional for current investigations. The data in Table 1 exploits a comparable band gap in derivatives when compared with reference compound. Among all benzothiophene based acceptor derivatives, the least band is investigated in A1D2 (2.354 eV) and. A1D4 (2.409 eV). This minute reduction in energy gap might be due to the presence of strong electron withdrawing –CN and –NO2 units on terminal acceptors. As literature study revealed that presence of strong electron withdrawing unit over the terminal acceptor unit, lower the LUMO and reduced the band gap.59,60 The values of the energy gaps (ΔE) reduce in the following order: A1D5 > A1R > A1D1 > A1D3 > A1D4 > A1D2. The different molecular orbital energies and the intramolecular charge transfer (ICT) process in the chromophores can also be studied through FMOs analysis.61,62 The red and blue colors in Fig. 4 illustrate the presence of charge densities over the different moieties in entitled chromophores. In A1R, the greater charge density is located over the central π-spacer CP core and smaller charge is concentrated at terminal units in both HOMO and LUMO. This charge is efficiently moved towards the end-capped acceptors from CP core in derivatives when tailored with BT based acceptors. In Fig. 4, a more pronounced electronic cloud is situated in terms of red and blue surfaces over the CP π-spacer in the HOMO and BT acceptors in the LUMO orbitals for all derivatives (A1D1–A1D5). Same phenomena of CT with in A1R and A1D1–A1D5 chromophores is examined in other molecular orbitals (HOMO−1/LUMO+1 and HOMO−2/LUMO+2) as illustrated in Fig. S1 and Table S7.† This efficient ICT in derivatives increase the polarizability in them which might improve the optoelectronic properties of studied chromophores.
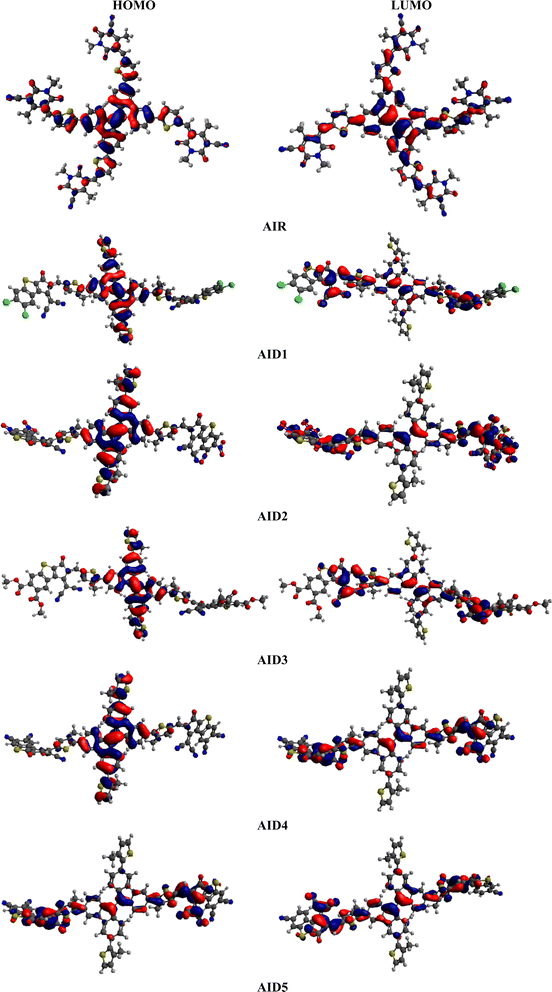 |
| | Fig. 4 Charge distribution pattern on HOMO–LUMO of A1R and A1D1–A1D5 chromophores. | |
Global Reactivity Parameters
The GRPs are employed to compute reactivity, stability and electron withdrawing/donating capabilities of chromophores.63,64 The energy gap is utilized to calculate electron affinity (EA), ionization potential (IP), global softness (σ), electronegativity (X), hardness (η), global electrophilicity index (ω), chemical potential (μ)65 and ΔNmax (ref. 66) which are estimated with the aid of eqn (2)–(9).| |
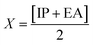 | (4) |
| |
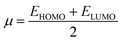 | (6) |
| |
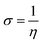 | (7) |
| |
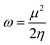 | (8) |
| |
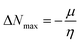 | (9) |
The findings in Table S11† elucidate that all derivatives showed comparable values of IP = 5.623–6.149 eV, EA = 3.136–3.330 eV and X = 4.379–4.653 eV with reference chromophore. These results explained their greater tendency to accept electron owing to the presence of robust terminal acceptors.67 The global hardness (η) and softness (σ) are used to estimate the reactivity and stability of compounds.68 Compounds which possess higher softness and lower hardness are consider as more reactive and polarize compounds that promote the charge transfer in an organic solar cell.69 All the designed compounds except A1D5 expressed lower value of global hardness (η = 1.243–1.177 eV) with higher softness values (σ = 0.424–0.402 eV−1) than parent molecule (η = 1.372 eV and σ = 0.364 eV−1), manifested that tailored chromophores might have the higher reactivity along with greater rate of polarizability that boosted charge transfer and the photovoltaic characteristics of derivatives. The decreasing order of softness values is computed as: A1D2 > A1D4 > A1D1 = A1D3 > A1DR > A1D5 which is inversely related with hardness. The A1D2 showed the minimum band gap (2.354 eV) and lowest hardness (1.177 eV) along with higher softness (0.424 value eV−1) among all the reported compounds (see Table S11 and Fig. S2†). These unique characteristics in A1D2 might be due to the presence of strong electron withdrawing –NO2 units on terminal benzothiophene based acceptor.
Density of state (DOS) analysis
To support the electronic charge distribution pattern over the different parts (CP π-spacer and BT acceptor units) of A1R and A1D1–A1D5 as studied by the FMOs investigation, DOS analysis has been carried out. For this purpose, the investigated compounds (A1R and A1D1–A1D5) are divided into two portions, i.e., π-spacer and end-capped acceptor units as shown in graphs (in Fig. 5) by red and green lines, respectively. On terminal acceptors the electronic cloud in HOMO orbital is founds as: A1R = 17.5, A1D1 = 3.6, A1D2 = 3.1, A1D3 = 3.4, A1D4 = 3.1 and A1D5 = 3.4%, while for LUMO this charge distribution pattern is studied as A1R = 18.0, A1D1 = 47.2, A1D2 = 51.9, A1D3 = 46.9, A1D4 = 54.9, and A1D1 = 43.7 (see Table S8†). Similarly, in π-spacer the charge contributed for HOMO is 82.5, 96.4, 96.9, 96.6, 96.9, and 96.6%, whereas 82.0, 52.8, 48.1, 53.1, 45.1 and 56.3% is examined in LUMO for A1R and A1D1–A1D5, respectively. These percentages provide insights into the population of electrons at different energy levels within a molecule. From these values it was examined that efficient charge transfer is found from central unit in HOMO towards acceptor in LUMO. Further, the DOS graphs in Fig. 5 also described that the highest charge density is located over π-spacer (green color) for HOMO and LUMO, the greatest charge density is distributed over acceptor moiety (red red) which supported the FMOs findings as illustrated in Fig. 4.
 |
| | Fig. 5 DOS maps of A1R and A1D1–A1D5 compounds. | |
UV-Vis analysis
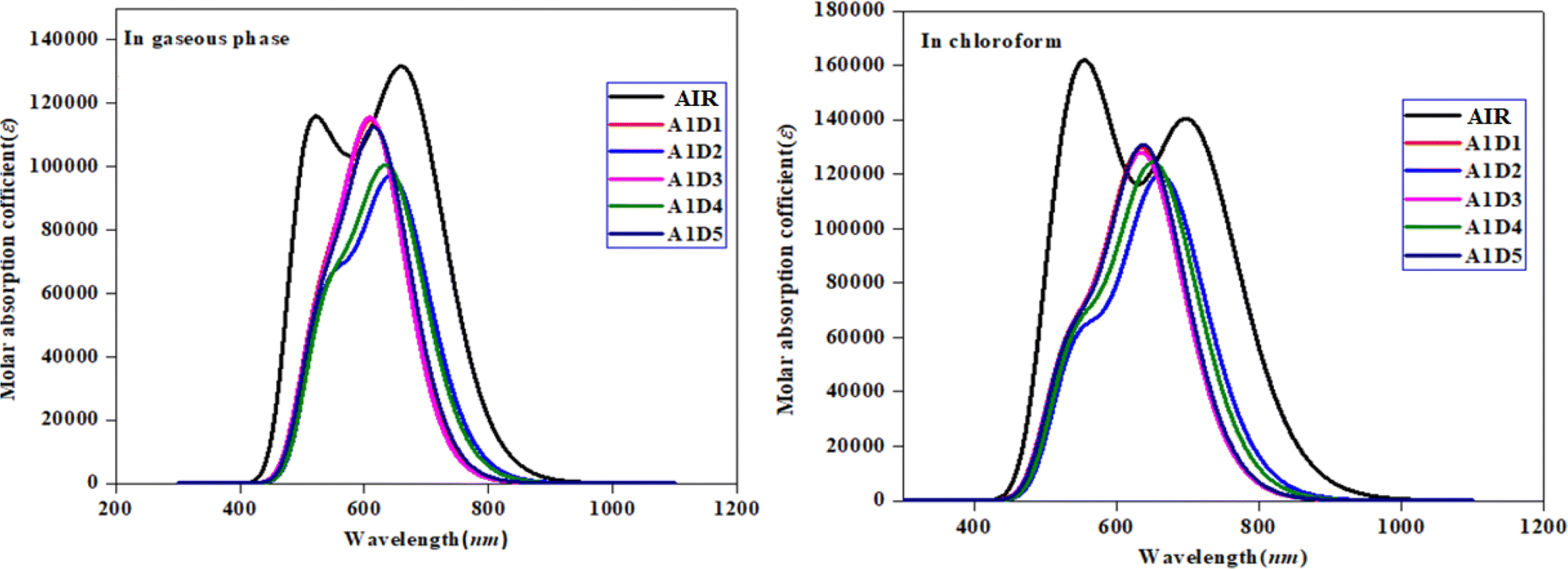
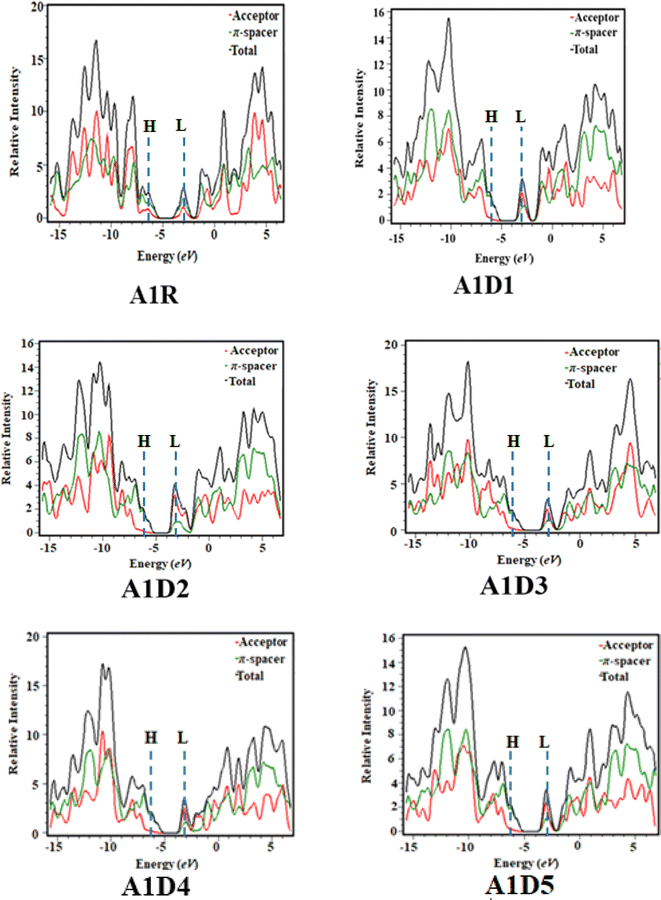
UV-Vis analysis was performed in chloroform and gaseous phase in order to determine the impact of benzothiophene based acceptors on absorption properties of A1D1–A1D5 chromophores. Various parameters like, excitation energy (E), oscillator strength (fos), maximum absorption wavelength (λmax) and molecular orbital contributions were calculated and presented in Tables 2 and 3, while other transitions are shown in the Tables S9 and S10.† Moreover, the absorption spectra of A1D1–A1D5 chromophores are displayed in Fig. 6 in both gaseous and solvent phase.
Table 2 The wavelength, excitation energy and oscillator strength of A1R and A1D1–A1D5 in gaseous phase
| Compounds |
λ (nm) |
E (eV) |
fos |
Major MO contributions |
| A1R |
671.164 |
1.847 |
1.656 |
H → L (97%) |
| A1D1 |
618.468 |
2.005 |
1.448 |
H → L (89%) |
| A1D2 |
649.676 |
1.908 |
1.243 |
H → L (92%) |
| A1D3 |
616.316 |
2.012 |
1.473 |
H → L (90%) |
| A1D4 |
644.141 |
1.925 |
1.217 |
H → L (73%) |
| A1D5 |
623.412 |
1.989 |
1.420 |
H → L (89%) |
Table 3 The wavelength, excitation energy and oscillator strength of A1R and A1D1–A1D5 in solvent phasea
| Compounds |
λ (nm) |
E (eV) |
fos |
Major MO contributions |
| MO = molecular orbital, H = HOMO, L = LUMO, fos = oscillator strength, wavelength = λ (nm). |
| A1R |
704.816 |
1.759 |
1.852 |
H → L (96%) |
| A1D1 |
639.753 |
1.938 |
1.705 |
H → L (81%) |
| A1D2 |
665.508 |
1.863 |
1.589 |
H → L (79%) |
| A1D3 |
640.150 |
1.937 |
1.675 |
H → L (81%) |
| A1D4 |
656.696 |
1.888 |
1.615 |
H → L (76%) |
| A1D5 |
641.707 |
1.932 |
1.725 |
H → L (83%) |
 |
| | Fig. 6 UV-Vis spectra of the compounds in gaseous and solvent phase. | |
The results of Tables 2 and 3 show that the values of maximum absorption wavelength (λmax) for all the compounds (A1R and A1D1–A1D5) are shifted towards bathochromic in chloroform solvent due to solvent effect as compared to gaseous phase. The observed λmax ranges are found as 640.150–665.508 nm for A1R and A1D1–A1D5 in chloroform solvent, while 616.316–649.676 nm in gaseous phase, correspondingly. Among all the designed compounds (A1D1–A1D5), A1D2 exhibited the maximum value of absorption wavelength in gaseous (649.676 nm) as well as in solvent phase (665.508 nm). The reason for greater value might be due to the strong electron-withdrawing nitro group (–NO2) which effectively withdraws electrons from the π-spacer towards acceptor moiety. Whereas, A1D1 having chloro (–Cl) group in their acceptor portion showed the minimum λmax in both solvent (639.753 nm) and gaseous phase (618.468 nm). Moreover, other chromophores including: A1D3–A1D5 displayed the values of 640.150, 656.696 and 641.707 nm in the solvent phase and 616.316, 644.141 and 623.412 nm in the gaseous phase, correspondingly. The absorption spectra of all studied compounds are shown by Fig. 6 in gaseous as well as solvent. The observed increasing order of λmax in all the derivatives in solvent phase is: A1D1 < A1D3 < A1D5 < A1D4 < A1D2 and A1D3 < A1D1 < A1D5 < A1D4 < A1D2 in gaseous phase. The above analysis revealed that narrowed band gap along with highest λmax in A1D2 resulted in maximum charge transference which make it suitable candidate to be utilized as non-fullerene-based material in solar cells to obtain excellent performance.
Open circuit voltage
The open circuit voltage (Voc) analysis is a vital parameter to determine the efficiency of solar cells.70 It provides the maximum electronic current that can be drawn from any optical device. The highest possible voltage that any device can produce at zero current is referred as Voc.71 It is affected by a number of variables including charge carrier recombination, electrode work function, fluorescence proficiency, light intensity, light source, OSC device temperature and many other variables.72 Some scaling metrics have been proposed for calculating Voc and can be utilized to precisely estimate the Voc of a solar cell. The HOMO of the donor material can be tuned using the LUMO of an acceptor material.73 The Voc is substantially influenced by the band gap between the donor and acceptor.74 For open circuit voltage measurements, the polymer molecule i.e., PBDB-T (EHOMO = −5.401 eV and ELUMO = 2.328 eV) is selected as donor and the reference (A1R) and designed molecules (A1D1–A1D5) are considered as acceptors. An approximated approach developed by Scharber et al.75 can be utilized to evaluate the results of Voc comprehensively by using the eqn (1).
Table 4 shows that the values of Voc for A1R and A1D1–A1D5 related to the energy gap of PBDB-THOMO and acceptor PBDB-TLUMO are calculated to be 1.201, 1.965, 1.981, 1.971, 1.838 and 1.943 V, respectively. All our compounds displayed a voltage which is comparable to the voltage of parent chromophore. Among all the tailored compounds, A1D2 exhibited the greatest Voc value of 1.981 V with reference to the energy difference of HOMOdonor–LUMOacceptor. The Voc findings of all the studied chromophores found in following descending order: A1D2 > A1D3 > A1D1 > A1D5 > A1D4 > A1D6 > A1R. As previously mentioned, the values of HOMO and LUMO influence the Voc. Improved optoelectronic characteristics and a higher Voc are produced by lower acceptor LUMO. A low-lying LUMO of acceptor increases the transport of electrons from HOMO of donor molecules, directly enhancing the optoelectronic properties. Additionally, PCE values are amplified by the band gap between acceptor and donor units. The molecular orbital energy diagram of the chromophores in relation to PBDB-T can be seen in Fig. 7 which demonstrates that the acceptor chromophores possess a more substantial LUMO level than the donor polymer PBDB-T. As a result, the studied molecular optoelectronic properties are improved due to the effective transference of electrons from the donor to the acceptor part.
Table 4 The energy and open circuit voltage of A1R and A1D1–A1D5
| Compounds |
ΔE (eV) |
Voc (V) |
| A1R |
0.901 |
1.201 |
| A1D1 |
2.265 |
1.965 |
| A1D2 |
2.071 |
1.981 |
| A1D3 |
2.271 |
1.971 |
| A1D4 |
2.138 |
1.838 |
| A1D5 |
2.243 |
1.943 |
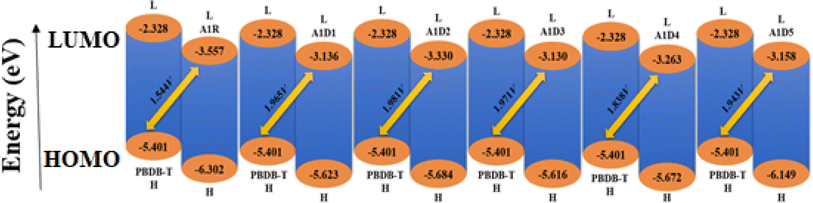 |
| | Fig. 7 Graphical representation of Voc for designed compounds with PBDB-T. | |
Transition density matrix (TDM) analysis
The TDM investigation is extensively utilized for the evaluation of degree of transitions in organic solar cells.76 From TDM computations, the interaction among acceptor–donor moieties, electron–hole localization and electron excitation can be estimated.77 Herein, TDM analysis was accomplished at M06/6-311G(d,p) functional between S0 → S1 state in A1R and A1D1–A1D5 chromophores. Owing to the little contribution of hydrogen atoms in transitions, their effect has been ignored. The electrons are uniformly distributed and diagonal charge transference has been observed by the bright portion of TDM pictographs for A1R and A1D1–A1D5. Fig. 8 reveals an efficacy in the transfer of charge density from the central CP π-spacer towards the peripheral BT acceptors in derivatives, as evidenced by bright color against a dark blue background. This phenomenon facilitates efficient charge transfer and supported the FMOs and DOS investigations.
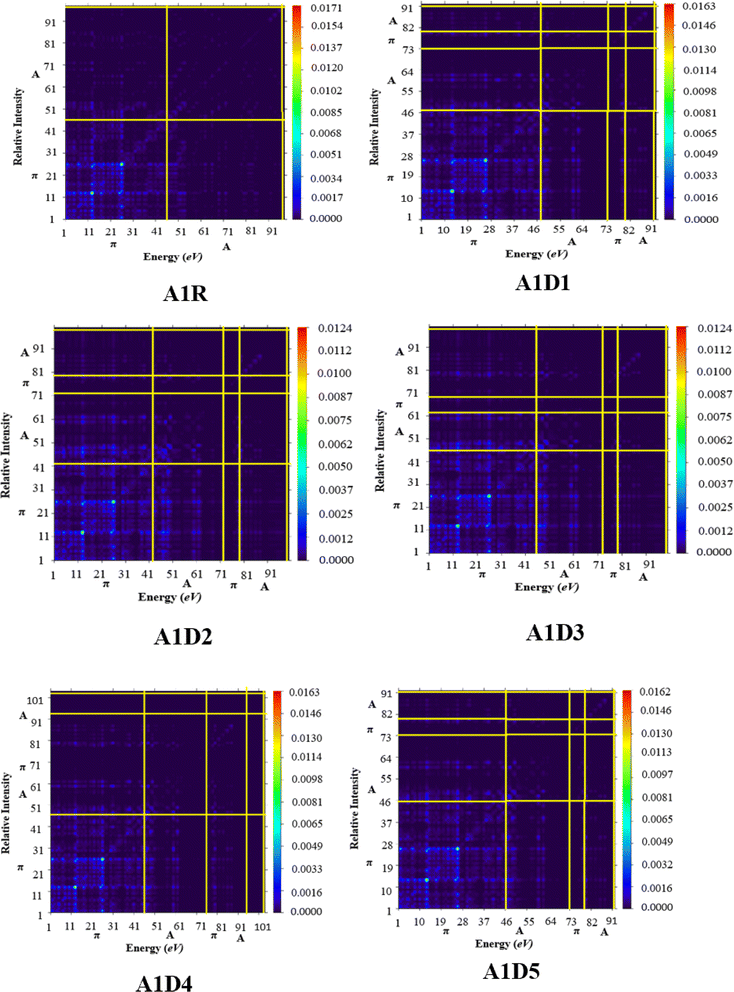 |
| | Fig. 8 TDM heat maps of A1R and A1D1–A1D5 illustrating the charge transfer from CP central core towards terminal BT acceptors. | |
Exciton binding energy investigations
Exciton binding energy (Eb) is another substantial parameter to evaluate photovoltaic characteristics of the compounds (A1R and A1D1–A1D5). The Eb in NF-OSCs materials refers to the energy required to dissociate an electron–hole (exciton) pair that is generated upon absorption of a photon.78 In the context of organic solar cells, this binding energy is a parameter that influences the efficiency of charge separation and subsequent charge carrier transport.79 The Eb of the chromophores was computed by using the eqn (10).where Eb is the energy difference, Eopt is the first singlet–singlet excitation energy. The computed values of Eb are exhibited in Table 5.
Table 5 The computed binding energies of A1R and A1D1–A1D5 in eV
| Compounds |
EH–L |
Eopt |
Eb |
| A1R |
2.745 |
1.759 |
0.986 |
| A1D1 |
2.487 |
1.938 |
0.549 |
| A1D2 |
2.354 |
1.863 |
0.491 |
| A1D3 |
2.486 |
1.937 |
0.549 |
| A1D4 |
2.409 |
1.888 |
0.521 |
| A1D5 |
2.991 |
1.932 |
1.059 |
From results summarized in Table 5, a comparable trend to the FMO is noted in Eopt. Furthermore, the values of Eb for A1R and A1D1–A1D5 are computed to be 0.986, 0.549, 0.491, 0.549, 0.521 and 1.059 eV, respectively. The declining trend of Eb is found as follows: A1D5 > A1R > A1D1 = A1D3 > A1D4 > A1D2. The lowest value of Eb (0.491 eV) is observed in A1D2 which depicted that it has the maximum capacity of exciton separation and Jsc.
Conclusion
In this study, five cyanopyridinone based compounds with A-π-A framework were designed from A1R via structural modification of terminal moieties with benzothiophene based acceptors. The primary focus in present study is to investigate the impact of benzothiophene based acceptors on electronic, photovoltaic and photophysical properties along with cyanopyridinone based core. All the designed compounds (A1D1–A1D5) displayed comparable band gap (2.354–2.991 eV) along with high bathochromic shift (640.150–665.508 nm) as compared to A1R (ΔE = 2.745 eV, λmax = 704.816 nm). A significant charge is transferred from central cyanopyridinone based core towards BT acceptors in derivatives as illustrated in FMOs, DOS and TDM investigations. Moreover, open circuit voltage values are computed in the range of 1.838–1.981 V for the designed compounds. The exciton binding energy (0.491–1.059 eV) of A1D1–A1D4 compounds is found to be smaller than A1R except A1D5 which represented their greater exciton dissociation rate. Among all the derivatives, A1D2 displayed the maximum Voc (1.981 V) value owing to it unique properties such as narrowest band gap (2.354 eV), highest λmax in both the gaseous (649.676 nm) as well as solvent phase (665.508 nm) and lowest binding energy (0.491 eV). These characteristics indicated that A1D2 might be considered as potential chromophore for OSCs with efficient photovoltaic properties. Consequently, the introduction of benzothiophene acceptors with electron-withdrawing groups improved the charge transfer, leading to improve values of Jsc and Voc for OSCs.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported and funded by the Deanship of Scientific Research at Imam Mohammad Ibn Saud Islamic University (IMSIU) (grant number IMSIU-RP23109).
References
- M. J. B. Kabeyi and O. A. Olanrewaju, Front. energy res., 2022, 9, 45 CrossRef.
- A. M. Omer, Renewable Sustainable Energy Rev., 2008, 12, 2265–2300 CrossRef CAS.
- W. Fang, Z. Liu and A. R. S. Putra, Renewable Energy, 2022, 194, 1142–1152 CrossRef.
- O. Ellabban, H. Abu-Rub and F. Blaabjerg, Renewable Sustainable Energy Rev., 2014, 39, 748–764 CrossRef.
- J. Ajayan, D. Nirmal, P. Mohankumar, M. Saravanan, M. Jagadesh and L. Arivazhagan, Superlattices Microstruct., 2020, 143, 106549 CrossRef CAS.
- I. Jamil, H. Lucheng, S. Habib, M. Aurangzeb, E. M. Ahmed and R. Jamil, Energy Rep., 2023, 9, 1501–1534 CrossRef.
- T. Bradford, Solar Revolution: the Economic Transformation of the Global Energy Industry, MIT Press, 2008 Search PubMed.
- W. Tress, Adv. Energy Mater., 2017, 7, 1602358 CrossRef.
- R. Liu and B. Sun, Acta Chim. Sin., 2015, 73, 225 CrossRef CAS.
- M. Khalid, I. Shafiq, M. Zhu, M. U. Khan, Z. Shafiq, J. Iqbal, M. M. Alam, A. A. C. Braga and M. Imran, J. Saudi Chem. Soc., 2021, 25, 101305 CrossRef CAS.
- D. Wöhrle and D. Meissner, Adv. Mater., 1991, 3, 129–138 CrossRef.
- H.-S. Lin and Y. Matsuo, in Handbook of Fullerene Science and Technology, ed. X. Lu, T. Akasaka and Z. Slanina, Springer Nature Singapore, Singapore, 2022, pp. 851–888 Search PubMed.
- V. A. Brotsman, V. A. Ioutsi, A. V. Rybalchenko, V. Yu. Markov, N. M. Belov, N. S. Lukonina, S. I. Troyanov, I. N. Ioffe, V. A. Trukhanov, G. K. Galimova, A. A. Mannanov, D. N. Zubov, E. Kemnitz, L. N. Sidorov, T. V. Magdesieva, D. Yu. Paraschuk and A. A. Goryunkov, Chem.–Asian J., 2017, 12, 1075–1086 CrossRef CAS PubMed.
- A. Wadsworth, Z. Hamid, J. Kosco, N. Gasparini and I. McCulloch, Adv. Mater., 2020, 32, 2001763 CrossRef CAS PubMed.
- M. N. Arshad, I. Shafiq, M. Khalid and A. M. Asiri, ACS Omega, 2022, 7, 11606–11617 CrossRef CAS PubMed.
- L. Ye, W. Ye and S. Zhang, J. Semicond., 2021, 42, 101607 CrossRef CAS.
- M. U. Khan, F. Shafiq, S. S. Al Abbad, J. Yaqoob, R. Hussain, Z. H. Alsunaidi, G. Mustafa and S. Hussain, Molecules, 2023, 28, 3625 CrossRef CAS PubMed.
- L. Xie, Y. Zhang, W. Zhuang, S. Y. Jeong, Q. Bian, H. Li, J. Cao, W. Liu, H. Tan and H. Y. Woo, Chem. Eng. J., 2022, 427, 131674 CrossRef CAS.
- W.-C. Wang, Y.-W. Lin, S.-H. Peng, C.-T. Chuang, C.-C. Chang and C.-S. Hsu, Org. Electron., 2020, 86, 105899 CrossRef CAS.
- F. Shen, J. Xu, X. Li and C. Zhan, J. Mater. Chem. A, 2018, 6, 15433–15455 RSC.
- Y.-Q.-Q. Yi, H. Feng, N. Zheng, X. Ke, B. Kan, M. Chang, Z. Xie, X. Wan, C. Li and Y. Chen, Chem. Mater., 2019, 31, 904–911 CrossRef CAS.
- Q. He, P. Ufimkin, F. Aniés, X. Hu, P. Kafourou, M. Rimmele, C. L. Rapley and B. Ding, SusMat, 2022, 2, 591–606 CrossRef CAS.
- Y. Zhang, Y. Lang and G. Li, EcoMat, 2023, 5, e12281 CrossRef CAS.
- X. Zhang, J. Yao and C. Zhan, Sci. China: Chem., 2016, 59, 209–217 CrossRef CAS.
- D. Luo, C. J. Brabec and A. K. K. Kyaw, Nano Energy, 2023, 108661 CrossRef CAS.
- Y. Wang, J. Lee, X. Hou, C. Labanti, J. Yan, E. Mazzolini, A. Parhar, J. Nelson, J. Kim and Z. Li, Adv. Energy Mater., 2021, 11, 2003002 CrossRef CAS.
- Q. Wei, W. Liu, M. Leclerc, J. Yuan, H. Chen and Y. Zou, Sci. China: Chem., 2020, 63, 1352–1366 CrossRef CAS.
- I. Shafiq, M. Khalid, M. Muneer, M. A. Asghar, R. Baby, S. Ahmed, T. Ahamad, S. F. de Alcântara Morais and A. A. Braga, Mater. Chem. Phys., 2023, 308, 128154 CrossRef CAS.
- A. Kuzmich, D. Padula, H. Ma and A. Troisi, Energy Environ. Sci., 2017, 10, 395–401 RSC.
- Y. Pan and G. Sun, ChemSusChem, 2019, 12, 4570–4600 CrossRef CAS PubMed.
- Q. Qi, X. Guo, B. Zhu, P. Deng, H. Zhan and J. Yang, Org. Electron., 2020, 78, 105616 CrossRef CAS.
- A. Mahmood, J. Hu, A. Tang, F. Chen, X. Wang and E. Zhou, Dyes Pigm., 2018, 149, 470–474 CrossRef CAS.
- S.-L. Chang, K.-E. Hung, F.-Y. Cao, K.-H. Huang, C.-S. Hsu, C.-Y. Liao, C.-H. Lee and Y.-J. Cheng, ACS Appl. Mater. Interfaces, 2019, 11, 33179–33187 CrossRef CAS PubMed.
- H. Cao, N. Bauer, C. Pang, J. Rech, W. You and P. A. Rupar, ACS Appl. Energy Mater., 2018, 1, 7146–7152 CrossRef CAS.
- A. Mahmood, J. Cluster Sci., 2019, 30, 1123–1130 CrossRef CAS.
- S.-L. Chang, K.-E. Hung, F.-Y. Cao, K.-H. Huang, C.-S. Hsu, C.-Y. Liao, C.-H. Lee and Y.-J. Cheng, ACS Appl. Mater. Interfaces, 2019, 11, 33179–33187 CrossRef CAS PubMed.
- M. R. S. A. Janjua, J. Photochem. Photobiol., A, 2023, 444, 115003 CrossRef CAS.
- S. B. Numbury, Oxford Open Mater. Sci., 2022, 2, itac002 CrossRef.
- W. Taouali, K. Alimi, A. Sindhoo Nangraj and M. E. Casida, J. Comput. Chem., 2023, 44, 2130–2148 CrossRef CAS PubMed.
- C. Wei, T. Zhang, Y. Zhao, Y. Zhou, M. Hailin and T. Yu, New J. Chem., 2021, 45, 7637–7646 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, et al., Gaussian 09, Revision A. 02, Gaussian Inc., Wallingford
CT, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- M. P. Andersson and P. Uvdal, J. Phys. Chem. A, 2005, 109, 2937–2941 CrossRef CAS PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- G. A. Zhurko, 2014.
- A. L. Tenderholt, Pymolyze, https://sourceforge.net/ Search PubMed.
- N. M. O'boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
- T. Lu, Software Manual, Version Search PubMed.
- R. D. Dennington, T. A. Keith and J. M. Millam, GaussView 5.0, Gaussian Inc., Wallingford, 2008, p. 20 Search PubMed.
- M. N. Arshad, I. Shafiq, M. Khalid, M. Asad, A. M. Asiri, M. M. Alotaibi, A. A. C. Braga, A. Khan and K. A. Alamry, Polymers, 2023, 15, 1508 CrossRef CAS PubMed.
- P. Mahalingavelar, Energy Fuels, 2022, 36, 2095–2107 CrossRef CAS.
- S.-L. Chang, K.-E. Hung, F.-Y. Cao, K.-H. Huang, C.-S. Hsu, C.-Y. Liao, C.-H. Lee and Y.-J. Cheng, ACS Appl. Mater. Interfaces, 2019, 11, 33179–33187 CrossRef CAS PubMed.
- F. Colmenero, A. M. Fernández, J. Cobos and V. Timón, J. Phys. Chem. C, 2018, 122, 5268–5279 CrossRef CAS.
- J. Murillo-Gelvez, K. P. Hickey, D. M. Di Toro, H. E. Allen, R. F. Carbonaro and P. C. Chiu, Environ. Sci. Technol., 2019, 53, 5816–5827 CrossRef CAS PubMed.
- M. Khalid, I. Shafiq, M. Zhu, M. U. Khan, Z. Shafiq, J. Iqbal, M. M. Alam, A. A. C. Braga and M. Imran, J. Saudi Chem. Soc., 2021, 25, 101305 CrossRef CAS.
- C. T. Tsapi, S. N. Tasheh, N. K. Nkungli, A. D. T. Fouegue, C. I. L. Alongamo and J. N. Ghogomu, J. Chem., 2023, 5287422 CAS.
- M. Irfan, H. A. Khan, S. Bibi, G. Wu, A. Ali, S. G. Khan, N. Alhokbany, F. Rasool and K. Chen, Sci. Rep., 2024, 14, 2732 CrossRef CAS PubMed.
- A. Mahmood, J.-Y. Hu, B. Xiao, A. Tang, X. Wang and E. Zhou, J. Mater. Chem. A, 2018, 6, 16769–16797 RSC.
- M. Khalid, H. M. Lodhi, M. U. Khan and M. Imran, RSC Adv., 2021, 11, 14237–14250 RSC.
- C. B. Nielsen, S. Holliday, H.-Y. Chen, S. J. Cryer and I. McCulloch, Acc. Chem. Res., 2015, 48, 2803–2812 CrossRef CAS PubMed.
- C. Liu, Y. Li, M. Takao, T. Toyao, Z. Maeno, T. Kamachi, Y. Hinuma, I. Takigawa and K. Shimizu, J. Phys. Chem. C, 2020, 124, 15355–15365 CrossRef CAS.
- M. Jarończyk, P. F. Lipiński, J. C. Dobrowolski and J. Sadlej, Chem. Pap., 2017, 71, 1429–1443 CrossRef.
- A. Mahmood, S. U.-D. Khan, U. A. Rana, M. R. S. A. Janjua, M. H. Tahir, M. F. Nazar and Y. Song, J. Phys. Org. Chem., 2015, 28, 418–422 CrossRef CAS.
- A. Mahmood, A. Irfan, F. Ahmad and M. R. S. A. Janjua, Comput. Theor. Chem., 2021, 1204, 113387 CrossRef CAS.
- H. Chermette, J. Comput. Chem., 1999, 20, 129–154 CrossRef CAS.
- D. A. Zainuri, M. Abdullah, S. N. A. M. Nizar, S. N. F. A. Rahman, N. Zahari, M. F. Zaini, M. A. Abu Bakar, S. Arshad and A. R. Ibrahim, Polycyclic Aromat. Compd., 2023, 1–24 Search PubMed.
- M. Khalid, M. Zafar, S. Hussain, M. A. Asghar, R. A. Khera, M. Imran, F. L. Abookleesh, M. Y. Akram and A. Ullah, ACS Omega, 2022, 7, 23532–23548 CrossRef CAS PubMed.
- R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef CAS.
- I. Shafiq, I. Amanat, M. Khalid, M. A. Asghar, R. Baby, S. Ahmed and S. M. Alshehri, Synth. Met., 2023, 297, 117410 CrossRef CAS.
- M. Khalid, W. Anwer, M. Adeel, Z. Shafiq, A. A. C. Braga, M. A. Assiri, M. Imran and A. Ullah, RSC Adv., 2022, 12, 29010–29021 RSC.
- M. Usman Khan, J. Iqbal, M. Khalid, R. Hussain, A. A. Carmo Braga, M. Hussain and S. Muhammad, RSC Adv., 2019, 9, 26402–26418 RSC.
- M. Khalid, I. Shafiq, M. Zhu, M. U. Khan, Z. Shafiq, J. Iqbal, M. M. Alam, A. A. C. Braga and M. Imran, J. Saudi Chem. Soc., 2021, 25, 101305 CrossRef CAS.
- M. Haroon, T. Akhtar, M. Khalid, H. Mehmood, M. Adnan Asghar, R. Baby, R. Orfali and S. Perveen, RSC Adv., 2023, 13, 7237–7249 RSC.
- A. Irfan and A. Mahmood, J. Cluster Sci., 2018, 29, 359–365 CrossRef CAS.
- D. Mühlbacher, M. Scharber, M. Morana, Z. Zhu, D. Waller, R. Gaudiana and C. Brabec, Adv. Mater., 2006, 18, 2884–2889 CrossRef.
- A. Mahmood, J.-Y. Hu, B. Xiao, A. Tang, X. Wang and E. Zhou, J. Mater. Chem. A, 2018, 6, 16769–16797 RSC.
- A. Irfan and A. Mahmood, J. Cluster Sci., 2018, 29, 359–365 CrossRef CAS.
- A. Dkhissi, Synth. Met., 2011, 161, 1441–1443 CrossRef CAS.
- K. Zheng, Q. Zhu, M. Abdellah, M. E. Messing, W. Zhang, A. Generalov, Y. Niu, L. Ribaud, S. E. Canton and T. Pullerits, J. Phys. Chem. Lett., 2015, 6, 2969–2975 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Gul Mariaab,
Nadeem Razac,
Ataualpa A. C. Braga
*ab,
Gul Mariaab,
Nadeem Razac,
Ataualpa A. C. Braga