
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConductive imprinted polymeric interfacially modified electrochemical sensors based on covalently bonded layer-by-layer assembly of Gr/Au with flower-like morphology for sensitive detection of 2,4,6-TCP†
Ziang Xu‡
ab,
Xiangying Jin‡b,
Yuqing Lib,
Manwen Zhangb,
Wenhua Yinb,
Yanyan Yangb,
Wenchao Jia *b and
Danping Xieb
*b and
Danping Xieb
aCollege of Chemistry and Environmental Science, Hebei University, Baoding 071002, China
bState Environmental Protection Key Laboratory of Environmental Pollution Health Risk Assessment, South China Institute of Environmental Sciences, Ministry of Ecology and Environment, Guangzhou, 510655, China. E-mail: jiawenchao1988@gmail.com
First published on 25th January 2024
Abstract
Polymeric membrane sensors based on molecular imprinted polymers (MIPs) have been attractive analytical tools for detecting organic species. However, the MIPs in electrochemical sensors developed so far are usually prepared by in situ polymerization of pre-polymers and non-covalent adsorption on the surface of the working electrode. Meanwhile, the MIPs in the electrochemical sensors developed are typically made of a non-conductive polymer film. This results in a relatively low current due to the lack of electron transfer. Additionally, the smoothness of the traditional electrochemical substrate results in a low specific surface area, which reduces the sensitivity of the electrochemical sensor. Here, we describe a novel electrochemical sensor with a conductive interface and MIPs modification. The electrochemical sensor was modified by covalent coupled layer by layer self-assembly with the imprinted polymer film. The incorporation of these two conductive functional materials improves the conductivity of the electrodes and provides interface support materials to obtain high specific surface area. By using 2,4,6-trichlorophenol as the model, the sensitivity of the developed conductive sensor was greatly improved compared to that of the traditional MIPs sensor. We believe that the proposed MIPs-based sensing strategy provides a general and convenient method for making sensitive and selective electrochemical sensors.
Introduction
Molecularly imprinted polymers (MIPs) are of great interest to researchers because of their highly selective recognition properties for targets like antibodies, as well as their simple synthesis, low preparation cost and stability in properties. As excellent selective functional materials, they have been used extensively in life sciences,1 monitoring and analysis of contaminants in the environment2 and harmful factors in food.3 Due to their high potential for serving as stable and high-efficiency recognition elements, MIPs have been rapidly integrated into electrochemical sensors. Therefore, combined with electrochemical sensors, they have proved useful for detecting various types of targets (such as organic pollutants,4 disease marker proteins,5 bacteria and viruses,6 etc.) by amperometric,7 potentiometric or impedance measurements. Free radical polymerization is a common method to prepare MIP membranes.8 A MIP polymer membrane was prepared by applying a pre-polymer solution containing template molecules onto the surface of the working electrode using drip-coating or spin-coating techniques. An in situ polymerization reaction was then carried out on the surface of the working electrode to form the MIP polymer membrane.9 Once the polymerization reaction is finished, the polymer is obtained, and the template molecules are removed by either electrochemical oxidation10 or using different elution reagents.11 This process allows for the creation of imprinted cavities within the polymer structure.12Although the preparation technology of the above-mentioned molecular imprinted membranes has been successfully applied in various sensing technologies, there are still many challenging issues.13,14 First of all, the polymer film prepared by the in situ polymerization of the pre-polymerization solution on the surface of the working electrode, due to the heterogeneity between the liquid–solid interface, the adhesion strength between the polymer film and the electrode surface is low and difficult to control.15 Moreover, there is a potential risk that the prepared imprinted polymer film may detach easily from the sensor surface during its usage or when removing the imprinted template molecules,16 which will eventually lead to a low service life of the sensor and low repeatability of the experiment. Therefore, many researchers have tried to overcome the problem of polymerization film falling off by increasing the thickness of the imprinted film to enhance the stability of the polymeric film on the sensor surface.17 This increase in thickness tends to reduce the sensitivity of the sensor surface.18 The covalent coupling between the polymeric film and the sensor interface can effectively improve the adhesion between the polymeric film and the sensors surface, which is an effective means to solve the problem that the polymeric film is easy to fall off. Unfortunately, so far, there have been no relevant reports on the utilization of a covalently immobilized polymer film on the surface of the working electrode for an electrochemical molecular imprinting sensor. Second, due to the poor compatibility of the polymeric membrane with the working electrode interface, the electronic transfer and recombination kinetics of analytes at the interface are limited,19,20 thereby reducing the potential analytical performance.21 This is undesirable for trace analyte detection assays.
In this study, we achieved, for the first time, the in situ polymerization and covalent immobilization of MIP films on the surface of a superconducting working electrode with a large specific surface area. The modification of the electrode utilizes the preparation method of layer-by-layer assembly through covalent coupled. Firstly, the surface of the working electrode containing amino functional groups was treated with glutaraldehyde, the surface of the electrode was modified with aldehyde functional groups, and then the single-layer graphene modified with amino functional groups was covalently coupled with the aldehyde groups on the electrode surface, to realize the assembly and immobilization of the graphene layer. The exposed residual amino groups on the surface of the graphene-modified working electrode provide action sites for in situ electro-reduced gold flower-like (Au-F), ensuring the absorption force required for the immobilization of Au-F. The surface of the working electrode that has not been fully filled with Au-F was further modified by aldehyde groups of glutaraldehyde which can be used as the active site of covalent coupling reaction of amino polymers.22 Graphene based gold composites not only provide a fixed substrate with high specific surface area for imprinted polymers, but also serve as a three-dimensional network for electron conduction with a single layer of super conductive graphene and flower shaped gold particles prepared by in situ electrodeposition reduction. It is worth noting that although there have been a few reports about covalent immobilization of imprinted film on sensor surface,23 the coupled immobilization of MIPs on electrochemical sensing interfaces has rarely been described and has not been applied to fabricate MIP-based electrochemical sensors. Using the proposed MIP of the superconducting substrate as the acceptor, the proposed concept was evaluated and demonstrated for further detection of neutral phenols. 2,4,6-Trichlorophenol (2,4,6-TCP), one of the major endocrine disruptors, was chosen as a model for phenolic compounds. The results show that the polymer film electrode based on superconducting substrate can significantly improve the detection sensitivity of neutral phenols.
Experimental
Apparatus and reagents
The ESI† clearly describe the various instruments, chemicals, and materials used in the experiment, as well as the production of screen printed electrodes.Fabrication of SPCE/Gr/Au-F/MIPs
The SPCE underwent processing using an EPL-YS4060D machine to convert it into PP paper following a predetermined pattern template. A conductive medium of silver ink was then printed and cured at 95 °C for 15 minutes. Working electrodes, with an area of 4.9 mm2, were fabricated using carbon ink and heated at 95 °C for 10 minutes. The reference electrode was created using silver/silver chloride ink, while the counter electrode was formed using carbon ink. An insulating ink was subsequently printed to serve as an insulating layer, outlining the working area. Prior to use, the SPCE was pretreated in 100 mM PBS (pH 7.0), applying a +1.7 V anodic potential to each working electrode for 5 minutes.The electrodes were sequentially washed with ultrapure water and ethanol. Subsequently, a 5% (v/v) aqueous glutaraldehyde solution was carefully applied onto the completely dried working electrode. The solution was left to react for 1 hour before being rinsed with ultrapure water to remove any excess, unreacted glutaraldehyde. A solution of amino-functionalized monolayer graphene (NH2-Gr) at a concentration of 0.5 mg mL−1 was then dropped onto the aldehyde-based modified working electrode surface. The solution was allowed to react for 1 hour at room temperature in order to obtain SPCE/Gr. Finally, similar preparation to previously reported,24 in situ electrochemical reduction deposition of Au-F was performed on the electrochemical sensor electrode using chronoamperometry (CA). In brief, a mixture sample containing 1 mM HAuCl4 and 0.1 M KCl (10 μL) was applied onto the surface of SPCE/Gr. Subsequently, a negative potential (−0.6 V vs. Ag/AgCl) was applied to the working electrode for 5 minutes. Following this, the electrode was washed with ultrapure water to prepare the SPCE/Gr/Au-F.
The molecularly imprinted sensor was prepared by “porogen imprinting technology”. The PBS with pH 5.0 was used as a solvent. To generate the molecularly imprinted polymer, an electropolymerization solution consisting of 1.25 mM 2,4,6-TCP and 5 mM o-PD was sonicated for 30 minutes. Subsequently, the electropolymerization process was conducted on the surface of the SPCE/Gr/Au-F working electrode. This was achieved by applying cyclic voltammetry with a voltage range of −0.5 to 1.2 V at a scan rate of 0.05 V s−1 for 20 cycles in the electropolymerization solution. After completion, and gently rinse the electrode surface with ultrapure water. The electrode was soaked in acetic acid–methanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9, v/v) eluent for 10 min, followed by removal from the eluent and another round of washing with ultrapure water. After elution, an electropolymerized molecularly imprinted membrane that selectively recognized 2,4,6-TCP was obtained (shown in Scheme 1). For comparison, the non-imprinting (NIPs) polymer modified electrode (SPCE/Gr/AuNPs/NIPs) was prepared using the same method, except that no 2,4,6-TCP template molecules were added.
:9, v/v) eluent for 10 min, followed by removal from the eluent and another round of washing with ultrapure water. After elution, an electropolymerized molecularly imprinted membrane that selectively recognized 2,4,6-TCP was obtained (shown in Scheme 1). For comparison, the non-imprinting (NIPs) polymer modified electrode (SPCE/Gr/AuNPs/NIPs) was prepared using the same method, except that no 2,4,6-TCP template molecules were added.
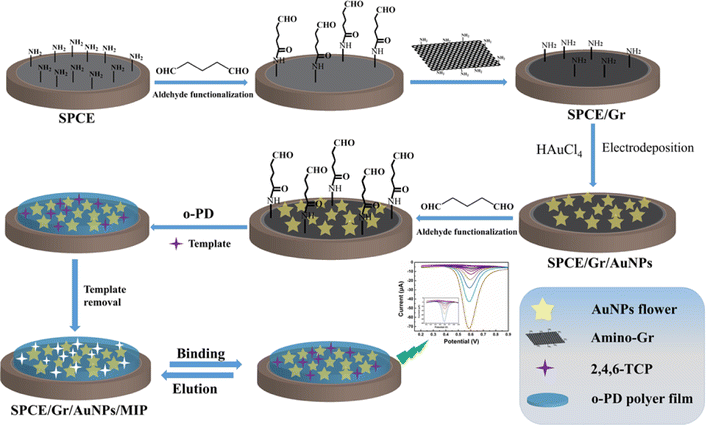 | ||
| Scheme 1 Illustrates the schematic representation of the preparation process for the SPCE/Gr/AuNPs/MIP electrode. | ||
Electrochemical measurements
Unless stated otherwise, a supporting electrolyte of 100 mM PBS (pH 7.0) was utilized. The electrochemical performance of the electrode was assessed through cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS), employing [Fe(CN)6]4−/3− as the redox probe. The quantitative detection of 2,4,6-TCP was accomplished using square wave voltammetry (SWV).CV was carried out within the range of −1.2 to + 1.2 V at a scan rate of 100 mV s−1. SWV measurements were conducted with the following parameters: potential window, 0 to +1.0 V; increment potential, 0.01 V; pulse width, 0.05 V; sample width, 0.0167 s; pulse period, 0.5 s. Following each measurement, the SPCE/Gr/AuNPs/MIPs electrode was thoroughly rinsed with ultrapure water and subjected to five cycles of scanning in 100 mM pH 7.0 PBS.
Detection of 2,4,6-TCP in real water sample
Water samples were collected from the Pearl River (Guangzhou, China) and Haizhu lake (Guangzhou, China), and subsequently pretreated by passing through a 0.45 μm filter membrane. Prior to the electrochemical measurement, the samples were supplemented with an appropriate volume of PBS, and the pH was adjusted to 7.0 by utilizing either HCl or NaOH.Results and discussion
Characterization of morphology and electrochemical properties
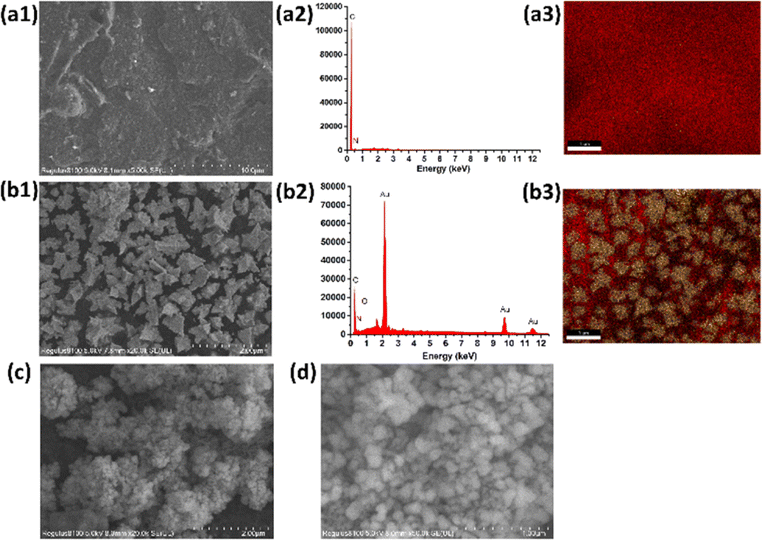 | ||
| Fig. 1 SEM images of (a) SPCE/Gr, (b) SPCE/Gr/Au-F, (c) and (d) SPCE/Gr/Au-F/MIP, the corresponding EDX spectra (a2)–(b2) as well as X-ray mapping images (a3)–(b3). | ||
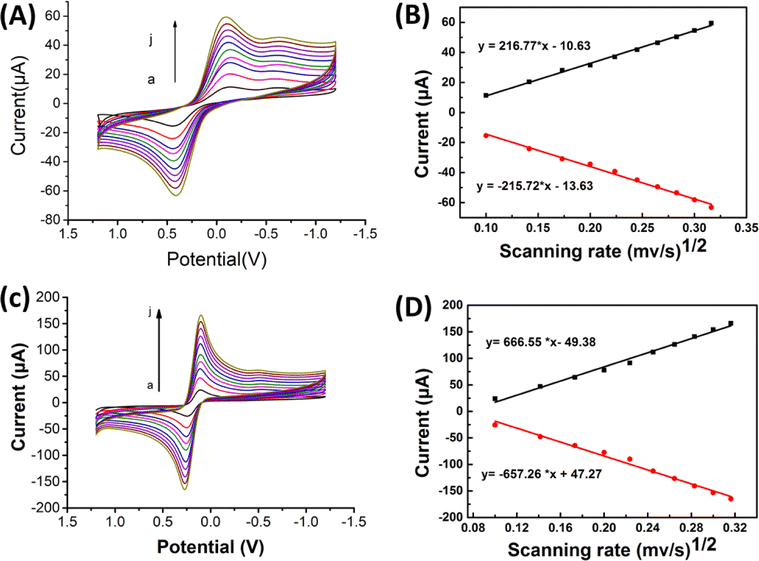 | ||
| Fig. 2 Cyclic voltammetric measurements were conducted using 1:1 mmol L−1 K3Fe(CN)6/K4Fe(CN)6 in 0.1 mol L−1 KCl at various scan rates (a–j, 0.01, 0.02, 0.03, 0.04, 0.05, 0.06 0.07, 0.08, 0.09, 0.1 V s−1). The relation between anodic (■) and cathodic (●) peak currents and the square root of the scan rate (ν1/2) was analyzed for different working electrodes: (A) and (B) SPCE and (C) and (D) treated SPCE/Gr/Au-F. | ||
Fig. 3(A) and (B) presents the analysis of the surface electrochemical properties of the modified electrodes using cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS). In order to illustrate the electrochemical behavior of SPCE/Gr/Au-F/MIP, CV sensorgrams were recorded at each step to assess the alterations occurring on the electrode surface as depicted in Fig. 3(A). The bare SPCE exhibited well-defined CV curves, which corresponded to the peak currents of the reduction/oxidation reaction. These peak currents were approximately equal and reversible. The introduction of graphene onto the SPCE surface led to a remarkable increase in the redox peak current (curve b), which was consistent with literature reports.25,26 The SPCE/Gr/Au-F electrodes exhibited the highest peak current value, indicating a faster electron transfer capability after the introduction of gold flower compared to the Gr-only modified surface (curve c). This suggests that the graphene/Au-F have a good electron transfer capacity.
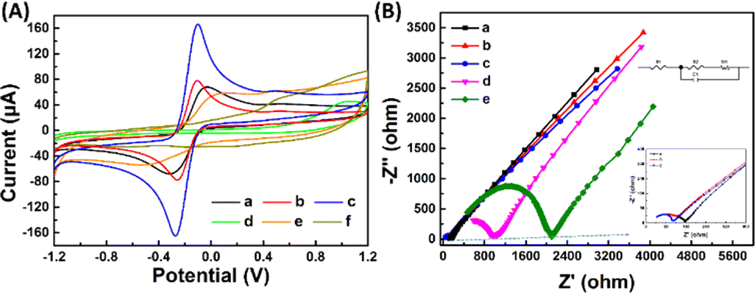 | ||
| Fig. 3 CV curves (A) of SPCE (a), SPCE/Gr (b), SPCE/Gr/Au-F (c), SPCE/Gr/Au-F/MIPs before (d) and after (e) template molecules was removed, SPCE/Gr/Au-F/MIPs after re-identification with 2,4,6-TCP (f) in 5.0 mM [Fe(CN)6]3−/4− solution containing 0.1 M KCl; EIS spectra (B) of SPCE (a), SPCE/Gr (b), SPCE/Gr/Au-F (c), SPCE/Gr/Au-F/MIPs before (e) and after (d) the template molecules was removed, SPCE/Gr/Au-F/MIPs after the subsequent binding of 2,4,6-TCP (f) in 5.0 mM [Fe(CN)6]3−/4− solution containing 0.1 M KCl. | ||
Compared to SPCE/Gr/Au-F (curve c), the peak current measured for the electrode after the electropolymerization step decreased sharply. This indicates the successful deposition of the polymer onto the surface of SPCE/Gr/Au-F, which eventually inhibits the redox reaction of [Fe(CN)6]4−/3− occurring on the electrode surface, leading to no obvious peak current signal (curve d). In the next step, after the removal of template molecules encapsulated in the polymer membrane with the washing solution, imprinted molecular holes appear on the surface of SPCE/Gr/Au-F/MIP, which provide a pathway for [Fe(CN)6]4−/3− to access the electrode surface. Therefore, the redox peak signal appears on the CV image, but the response intensity is significantly lower than that before electropolymerization (curve e). Subsequently, the successfully eluted SPCE/Gr/Au-F/MIP modified electrode is immersed in a 10 μM 2,4,6-TCP solution. In this sense, 2,4,6-TCP is again absorbed by the MIP membrane, plugging the molecular voids, causing the peak current to drop again (curve f). The results of the electrochemical characterization studies confirm that the developed sensor platform is well-suited for detecting 2,4,6-TCP.
Electrochemical impedance spectroscopy (EIS) is a widely employed analytical method that serves as a powerful tool for studying the interface characteristics of an electrode. In Fig. 3(B), the typical Nyquist plots of five modified electrodes are shown in a [Fe(CN)6]3−/4− mixture (1:1) with a 0.1 M KCl solution. SPCE/Gr/Au-F clearly exhibits the smallest semicircle diameter at high frequency, indicating the lowest charge transfer resistance (Rct). After depositing a MIP membrane on the surface of this electrode, the impedance increases significantly (curve e) because the MIP membrane causes less electron transfer. Subsequently, the 2,4,6-TCP template was removed from the MIP membrane by washing solution (curve d). This shows that the charge transfer resistance (Rct) decreases due to the generation of some recognition holes which reduces the resistance to electron transfer. The obtained results are consistent with the CV spectra.
Condition optimization studies
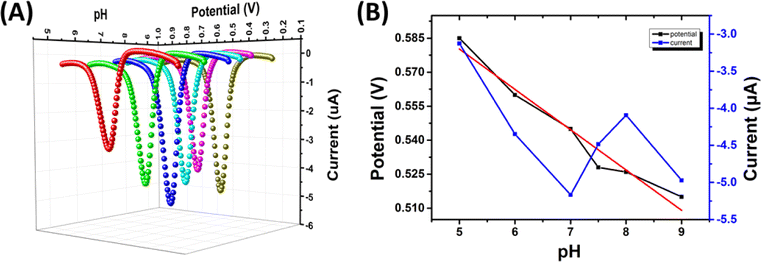 | ||
| Fig. 4 (A) SWV of SPCE/Gr/Au-F/MIP of 2,4,6-TCP. (B) Calibration plots were constructed to illustrate the relationship between the response current and potential with respect to pH. | ||
ν). The linear regression equation was determined as Ep (V) = 0.0201lnν (V s−1) + 0.33, with a coefficient of determination (R2) of 0.986. This observation is consistent with Laviron's theory, which establishes a relationship between the oxidation potential (Ep) and the natural logarithm of the scan rate (lnν) for irreversible anodic reactions. The equation describing this relationship is as follows: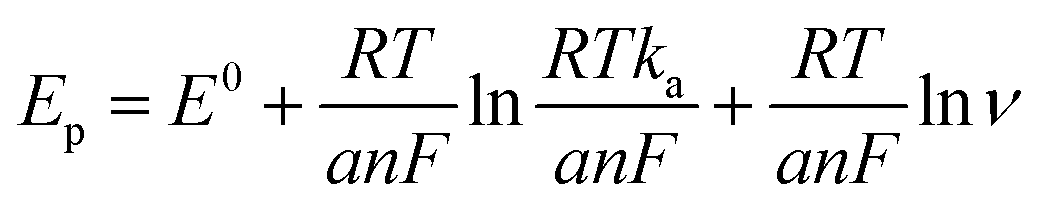
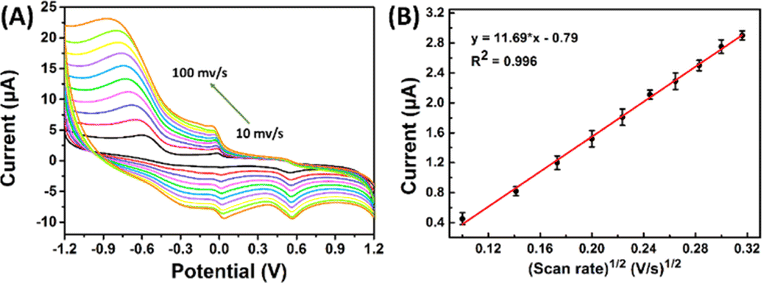 | ||
| Fig. 5 (A) CVs recorded at SPCE/Gr/Au-F/MIP in pH 7.0 PBS containing 5 μM 2,4,6-TCP at the scan rates from 10 to 100 mV s−1; (B) the linear dependence of 2,4,6-TCP on scan rate. | ||
The equation used to describe the relationship between the oxidation potential (Ep) and the natural logarithm of the scan rate (lnν) involves several parameters. These parameters include the electron transfer number (n), the electron transfer coefficient (a), the standard rate constant (ka), the scan rate (ν), and constants such as R, T, and F. In the case of the irreversible electrode process, the value of α is defined as 0.5. By plotting Ep against lnν and performing calculations, the electron transfer number (n) for 2,4,6-TCP was determined to be 2. Hence, the oxidation process of 2,4,6-TCP on the SPCE/Gr/Au-F/MIP electrode involves the transfer of two electrons and the participation of two protons.
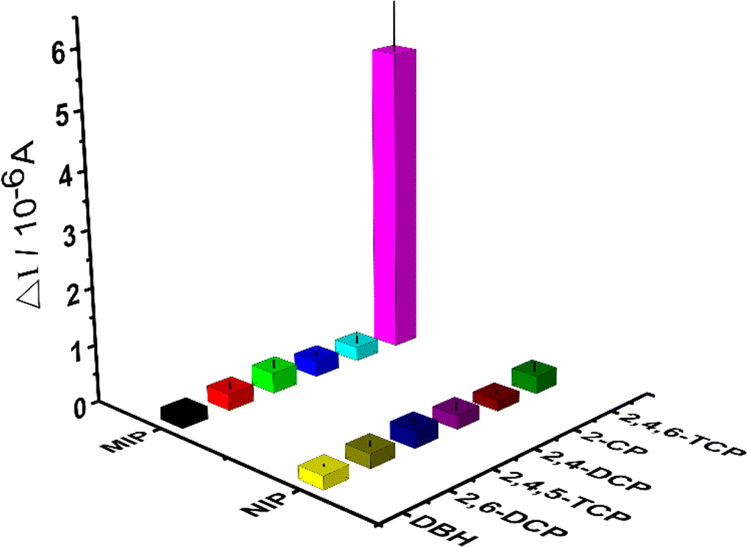 | ||
| Fig. 6 Selectivity studies by different kinds of chlorophenols (2,4,5-TCP, 2,4-DCP and 2-CP) of each interfering compounds on the MIP-coated and NIP-coated electrodes. | ||
The selectivity and relative selectivity coefficients were determined as follows: k = [current value2,4,6-TCP]/[current valuecompetitor] and k′ = kMIP/kNIP. The values of k and k' were summarized in Table 1, respectively. The current values obtained for 2,4,6-TCP imprinted SPCE/Gr/Au-F/MIP electrodes in the presence of 2,4,5-TCP, 2,4-DCP, 2-CP, 2,6-DCP and DBH were 0.36 μA, 0.28 μA, 0.25 μA, 0.29 μA, and 0.22 μA, respectively. The current value of 2,4,6-TCP was 5.35 μA at the same concentration. According to these values, 2,4,6-TCP imprinted electrodes was 14.86, 19.10, 21.40, 18.45, and 26.75 times more selective for 2,4,6-TCP than 2,4,5-TCP, 2,4-DCP, 2-CP, 2,6-DCP, and DBH, respectively. These results indicate that the prepared molecularly imprinted polymer membrane can effectively recognize the steric and chemical structures of 2,4,6-TCP molecules.
| Other compounds | k (MIP) | k (NIP) | k′ |
|---|---|---|---|
| 2-CP | 21.40 | 3.64 | 5.88 |
| 2,4-DCP | 19.10 | 2.04 | 9.36 |
| 2,6-DCP | 18.45 | 2.04 | 9.04 |
| 2,4,5-TCP | 14.86 | 1.7 | 8.74 |
| DBH | 26.75 | 3.4 | 8.74 |
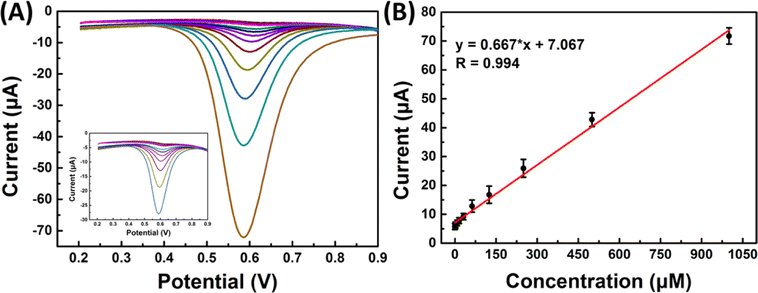 | ||
| Fig. 7 (A) SWV sensorgrams of SPCE/Gr/Au-F/MIP in the 2,4,6-TCP solution at different concentrations; (B) linear calibration graph. | ||
The reproducibility of analytical methods is the most important parameter for evaluating detection methods, four distinct MIP surfaces were prepared using the same method independently, and their corresponding square wave voltammetry (SWV) recordings were compared. The relative standard deviation (RSD) value was determined to be 1.83% (n = 4), indicating a highly reproducible fabrication process for 2,4,6-TCP detection. Furthermore, the repeatability of the signals was assessed by conducting four consecutive measurements on the same sample within a single day, resulting in an RSD value of 2.78%. These findings demonstrate the reproducibility of the sensor surface and its consistent and reusable detection performance.
Furthermore, the stability of the working electrode surface was assessed by a consistent fluid flushing procedure. By enclosing the electrode within a microfluidic chip chamber (shown in Fig. S2†), we observed that the electrode maintained excellent analytical performance for 2,4,6-TCP even after undergoing flushing at varying flow rates (100 μL min−1, 500 μL min−1, 1000 μL min−1, 1500 μL min−1, 2000 μL min−1) (shown in Fig. S3†). This observation underscores the robustness of our prepared molecularly imprinted sensor, demonstrating its remarkable resistance to detachment Table 2.
| Water sample | Addition amount/nM | Measured quantity/nM | Recovery/% | RSD/% |
|---|---|---|---|---|
| Pearl river | 20.00 | 23.24 ± 1.66 | 116.20 | 1.83 |
| 100.00 | 104.06 ± 4.31 | 104.05 | 1.59 | |
| 500.0 | 496.10 ± 7.50 | 99.22 | 0.53 | |
| Haizhu lake | 20.00 | 19.64 ± 1.37 | 98.20 | 1.02 |
| 100.00 | 99.30 ± 2.28 | 99.05 | 0.47 | |
| 500.00 | 504.83 ± 15.60 | 100.97 | 0.55 |
Conclusions
In this study, we have successfully developed a novel electrochemical sensor for the detection and identification of 2,4,6-TCP. The sensor is based on the utilization of molecularly imprinted polymers (MIPs) and incorporates a layer-by-layer modification strategy involving graphene, gold flower-like (Au-F), and MIPs, all covalently bonded onto screen-printed carbon electrodes (SPCE). The three-dimensional structure of the sensor enhances its adsorption capacity and facilitates efficient electron transfer. The presence of graphene and Au-F further enhances the sensor's sensitivity by ensuring good electrical conductivity and a large surface area. Importantly, the prepared MIPs electrodes can still be used for a long time with good performance under the stress condition of strong rinsing. When applied to real water samples, the SPCE/Gr/Au-F/MIP sensor demonstrates reliable and satisfactory performance. It offers several advantages, including cost-effectiveness, ease of preparation, high sensitivity, excellent selectivity, acceptable reproducibility, and stability. Overall, this newly developed electrochemical sensor shows great promise for a wide range of sensing applications.Author contributions
The authors declare that they have no known conflicts of interest or personal relationships that could have influenced the work presented in this paper. The CRediT authorship contribution statement was provided. Ziang Xu: methodology, data curation. Xiangying Jin: resources, data curation. Manwen Zhang: methodology, funding acquisition. Wenhua Yin: writing – original draft. Yanyan Yang: writing – original draft. Wenchao Jia: conceptualization, methodology, writing – review & editing, supervision, funding acquisition. Danping Xie: supervision.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (2019YFC1803900), Guangdong Basic and Applied Basic Research Foundation (Grant No. 2022A1515110111), Guangzhou Science and Technology Innovation Development Special Fund Fundamental and Applied Fundamental Research Project (Grant No. 202102020479), and the Central Government Fund Supporting Non-profit Scientific Institutes for Basic Research and Development (Grant No. PM-zx703-202011-322, PM-zx703-202112-330, PM-zx097-202204-126).Notes and references
- A. M. Bossi, Nat. Chem., 2020, 12, 111–112 CrossRef CAS PubMed.
- S. S. Sandhu, Y. G. Kotagiri, P. U. A. I. Fernando I, M. Kalaj, N. Tostado, H. Teymourian, E. M. Alberts, T. L. Thornell, G. R. Jenness, S. P. Harvey, S. M. Cohen, L. C. Moores and J. Wang, J. Am. Chem. Soc., 2021, 143, 18261–18271 CrossRef CAS PubMed.
- C. C. Villa, L. T. Sánchez, G. A. Valencia, S. Ahmed and T. J. Gutiérrez, Trends Food Sci. Technol., 2021, 111, 642–669 CrossRef CAS.
- S. Wang, M. Pan, K. Liu, X. Xie, J. Yang, L. Hong and S. Wang, Food Chem., 2022, 381, 132225 CrossRef CAS PubMed.
- L. Wang, M. Pagett and W. Zhang, Sens. Actuators, A, 2023, 5, 100153 Search PubMed.
- A. Raziq, A. Kidakova, R. Boroznjak, J. Reut, A. Öpik and V. Syritski, Biosens. Bioelectron., 2021, 178, 113029 CrossRef CAS PubMed.
- C. V. Raju, Y. V. Manohara Reddy, C. H. Cho, H. H. Shin, T. J. Park and J. P. Park, Food Chem., 2023, 428, 136811 CrossRef CAS PubMed.
- M. Daoud Attieh, Y. Zhao, A. Elkak, A. Falcimaigne-Cordin and K. Haupt, Angew. Chem., Int. Ed., 2017, 56, 3339–3343 CrossRef CAS PubMed.
- Y. Wei, H. Liu, S. Wang, K. Yu and L. Wang, The Analyst, 2023, 148, 3851–3859 RSC.
- M. Wei, X. Geng, Y. Liu, H. Long and J. Du, J. Electroanal. Chem., 2019, 842, 184–192 CrossRef CAS.
- J. Chen, X. Huang, L. Wang, C. Ma, S. Wu and H. Wang, Anal. Methods, 2020, 12, 996–1004 RSC.
- D. Lu, D. Z. Zhu, H. Gan, Z. Yao, J. Luo, S. Yu and P. Kurup, Sens. Actuators, B, 2022, 352, 131055 CrossRef CAS.
- L. Carballido, T. Karbowiak, P. Cayot, M. Gerometta, N. Sok and E. Bou-Maroun, Chem, 2022, 8, 2330–2341 CAS.
- A. Ostovan, M. Arabi, Y. Wang, J. Li, B. Li, X. Wang and L. Chen, Adv. Mater., 2022, 34, 2203154 CrossRef CAS PubMed.
- T. Kamra, S. Chaudhary, C. Xu, L. Montelius, J. Schnadt and L. Ye, J. Colloid Interface Sci., 2016, 461, 1–8 CrossRef CAS PubMed.
- M. Roushani and N. Zalpour, React. Funct. Polym., 2021, 169, 105069 CrossRef CAS.
- H. Lu, M. Liu, H. Cui, Y. Huang, L. Li and Y. Ding, Electrochim. Acta, 2022, 427, 140858 CrossRef CAS.
- T. Phi Van, T. P. Nguy and L. T. N. Truong, Anal. Methods, 2022, 14, 2195–2203 RSC.
- B. Deiminiat, G. H. Rounaghi and M. H. Arbab-Zavar, Sens. Actuators, B, 2017, 238, 651–659 CrossRef CAS.
- S. Li, J. Li, Q. Lin and X. Wei, The Analyst, 2015, 140, 4702–4707 RSC.
- J. Völkle, K. Kumpf, A. Feldner, P. Lieberzeit and P. Fruhmann, Sens. Actuators, B, 2022, 356, 131293 CrossRef.
- H. Boyacıoğlu, B. B. Yola, C. Karaman, O. Karaman, N. Atar and M. L. Yola, Appl. Surf. Sci., 2022, 578, 152093 CrossRef.
- P. Maurya and R. Verma, The Analyst, 2023, 148, 1141–1150 RSC.
- A. Huang, H. Li and D. Xu, J. Electroanal. Chem., 2019, 848, 113189 CrossRef CAS.
- H. Maseed, V. M. Reddy Yenugu, S. S. Devarakonda, S. Petnikota, M. Gajulapalli and V. V. S. S. Srikanth, ACS Appl. Nano Mater., 2023, 6, 18531–18538 CrossRef CAS.
- B. Rajeswari, B. Sravani, M. Cheffena, R. Janraj Naik, Y. Veera Manohara Reddy, G. Madhavi, K. V. N. Suresh Reddy and M. Jong Kim, Inorg. Chem. Commun., 2023, 151, 110627 CrossRef CAS.
- M. Nawaz, H. Shaikh, J. A. Buledi, A. R. Solangi, C. Karaman, N. Erk, R. Darabi and M. B. Camarada, Carbon Letters, 2023 DOI:10.1007/s42823-023-00562-8.
- W. Sun, H. Chu, J. Liu, N. Wang and Y. Wang, Sens. Actuators, B, 2023, 393, 134146 CrossRef CAS.
- M. Akilarasan, E. Tamilalagan, S.-M. Chen, S. Maheshwaran, T.-W. Chen, A. M. Al-Mohaimeed, W. A. Al-Onazi and M. S. Elshikh, Microchim. Acta., 2021, 188, 72 CrossRef CAS PubMed.
- W.-L. Chen, T.-W. Lee and C. Chen, Chemosphere, 2023, 335, 139003 CrossRef CAS PubMed.
- K.-Y. Hwa, A. Ganguly, A. Santhan and T. S. Kanna Sharma, Chemosphere, 2021, 282, 130874 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra06668a |
| ‡ These authors contributed equally |
| This journal is © The Royal Society of Chemistry 2024 |