
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in the synthesis and utility of thiazoline and its derivatives
Sumit Kumar†
a,
Aditi Arora†
a,
Shivani Sapra
a,
Rajesh Kumar
b,
Brajendra K. Singh
 *a and
Sunil K. Singh
*c
*a and
Sunil K. Singh
*c
aBioorganic Laboratory, Department of Chemistry, University of Delhi, Delhi-110007, India. E-mail: singhbk@chemistry.du.ac.in
bDepartment of Chemistry, R. D. S College, B. R. A. Bihar University, Muzaffarpur, 842002, India
cDepartment of Chemistry, Kirori Mal College, University of Delhi, Delhi-110007, India. E-mail: chem.sunil@kmc.du.ac.in
First published on 2nd January 2024
Abstract
Thiazolines and their derivatives hold significant importance in the field of medicinal chemistry due to their promising potential as pharmaceutical agents. These molecular entities serve as critical scaffolds within numerous natural products, including curacin A, thiangazole, and mirabazole, and play a vital role in a wide array of physiological reactions. Their pharmacological versatility encompasses anti-HIV, neurological, anti-cancer, and antibiotic activities. Over the course of recent decades, researchers have extensively explored and developed analogs of these compounds, uncovering compelling therapeutic properties such as antioxidant, anti-tumor, anti-microbial, and anti-inflammatory effects. Consequently, thiazoline-based compounds have emerged as noteworthy targets for synthetic endeavors. In this review, we provide a comprehensive summary of recent advancements in the synthesis of thiazolines and thiazoline-based derivatives, along with an exploration of their diverse potential applications across various scientific domains.
 Sumit Kumar | Sumit Kumar obtained his Master's Degree in Organic Chemistry from Department of Chemistry, University of Delhi in 2020 and Honours Degree from Kirori mal College, University of Delhi in 2018. He is a third year PhD student under the supervision of Prof. Brajendra K. Singh, Department of Chemistry, University of Delhi. His research interests include design and synthesis of sugar-modified heterocyclic motifs of therapeutic importance. He visited Medgar Evers College, City University of New York (CUNY) in the Chemistry and Environmental Science Department as a Research Scholar. He has published several research articles in various reputed journals such as European Journal of Polymer Chemistry, New Journal of Chemistry, Beilstein Journal of Organic Chemistry, and others. |
 Aditi Arora | Aditi Arora obtained her Honours Degree from Miranda House, University of Delhi in 2018. She completed her Master's in Organic Chemistry from Department of Chemistry, University of Delhi in 2020. She was a Gold Medalist during her Master's. At present, she is pursuing PhD from Department of Chemistry, University of Delhi. Her research interests include design and synthesis of carbohydrate and coumarin modified molecules. She has published several research articles in various reputed journals. |
 Shivani Sapra | Shivani Sapra did her Master's in Chemistry from Chaudhary Charan Singh University, Meerut in 2018. Earlier, she did her graduation from Shyam Lal College, University of Delhi in 2016. After that she worked as a Project fellow in Sri Venkateswara College, University of Delhi. At present, she is working under Prof. BK Singh, Department of Chemistry. Her research interest lies in the field of modification of sugar molecules. |
 Rajesh Kumar | Dr Rajesh Kumar received his Master of Science degree in Chemistry from University of Delhi in 2010. He joined the same Department for a PhD and completed his PhD in 2017 and during his PhD, Dr Kumar visited University of Southern Denmark as a Research Assistant from December 2014 to August 2015. After completion of PhD, he joined as Assistant Professor in Chemistry at B. R. A. Bihar University, India. He has published more than 30 research papers in reputed national and international journals such as Journal of Organic Chemistry, Theranostics, European Polymer Journal etc. His research interest lies in Metal complexes, Biotransformations, Catalysis, Green Chemistry, Nucleoside and Heterocyclic Chemistry. |
 Brajendra K. Singh | Professor Brajendra K. Singh currently holds the position of Professor in the Department of Chemistry, University of Delhi, Delhi, India. He visited the Department of Chemistry, University of Leuven, Leuven, Belgium as International Scholar for the period November 2005–April 2007 and then as post-doctoral fellow till November 2009. His key interest areas are the study and investigation of bio-catalysis and metal-catalysis, C–H activation, specifically in the design, synthesis and evaluation of bioactive compounds and LED mediated synthesis. He has published more than 85 research articles in internationally reputed journals. He has delivered around 30 Invited Lectures in various National and International Conferences at different Universities and Institutes. He has served and serving in different capacities in administration of Delhi university. |
 Sunil K. Singh | Dr Sunil K Singh, received his PhD in 2010 from the Department of Chemistry, University of Delhi, India in the area of synthesis of modified nucleosides and biocatalytic organic transformations. After completing his PhD, he joined Kirori Mal College a constituent College of the University of Delhi as an Assistant Professor in 2011. Recently, he is promoted to Associate Professor in the same Department. He has published more than 31 articles in the Journal of international repute. |
1. Introduction
Heterocyclic compounds play a pivotal role in various domains such as pharmaceuticals, catalytic ligands, fine chemicals, and agrochemicals.1 Among these compounds, thiazolines, sulfur-containing analogs, have received relatively less attention. Thiazolines constitute a specific class of organic compounds characterized by a five-membered ring structure composed of four carbon atoms and one sulfur atom, with the potential for a nitrogen atom substitution (Fig. 1). This heterocyclic ring system is a derivative of thiazole, a structurally similar compound featuring sulfur and nitrogen atoms within the five-membered ring. Thiazolines exhibit diverse applications and attract interest in both organic and medicinal chemistry (Fig. 2). Their broad biological relevance has resulted in their presence in a wide range of synthetic and natural products (Fig. 3), thereby enhancing their significance over time.2 While encountering unsubstituted thiazolines in their pure form is rare, their derivatives are more prevalent, with specific derivatives showcasing bioactivity. A noteworthy aspect of thiazolines is their occurrence in various biologically active molecules, including specific antibiotics and natural products (Fig. 4). Thiazolines can also be identified in certain vitamins and coenzymes, underscoring their importance in biological processes.3 Importantly, thiazolines are synthesized through the conventional post-translational modification of cysteine residues.4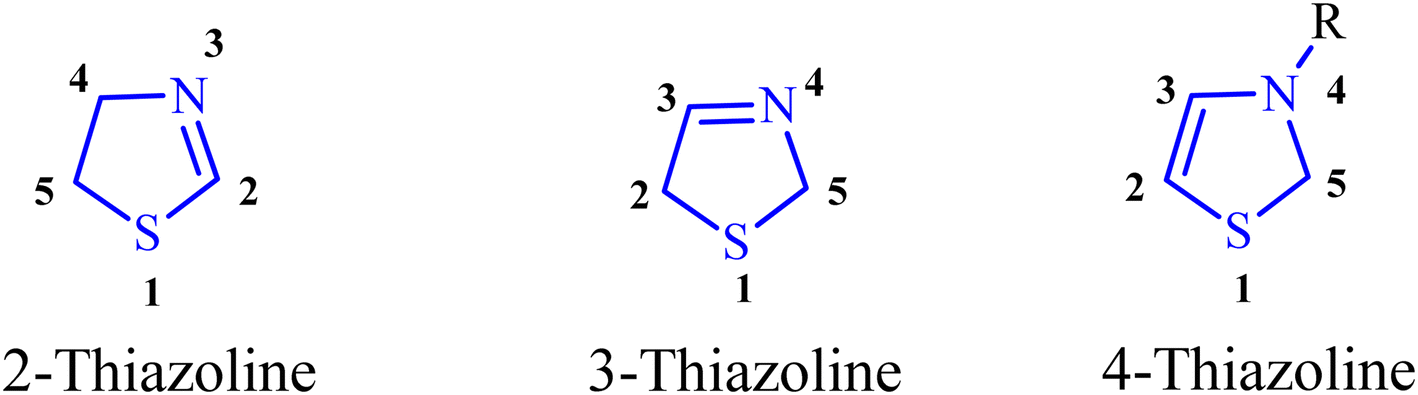 | ||
| Fig. 1 General structure and numbering of the thiazoline heterocycle. | ||
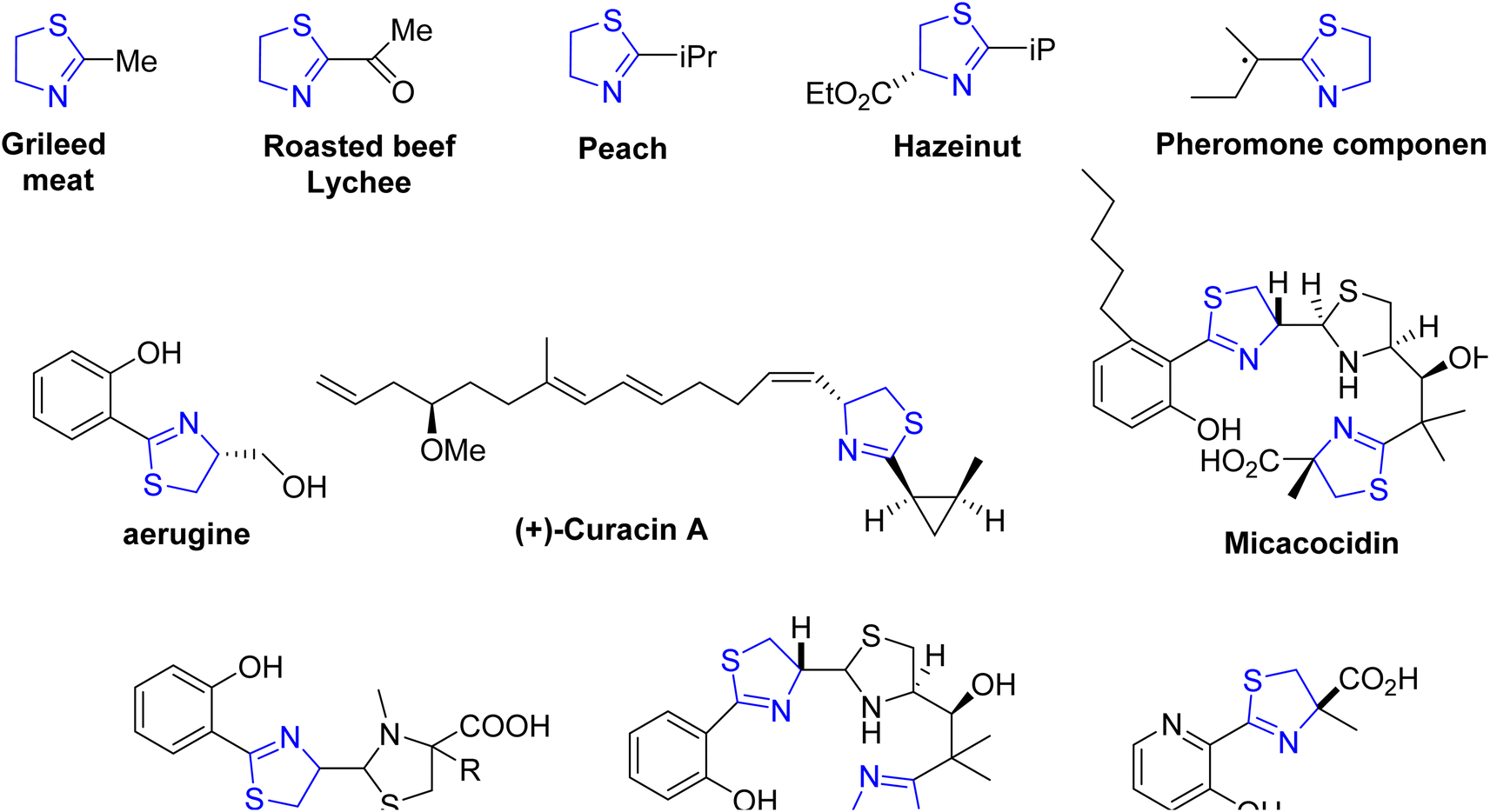 | ||
| Fig. 2 Derivatives of thiazolines sourced from natural origins. | ||
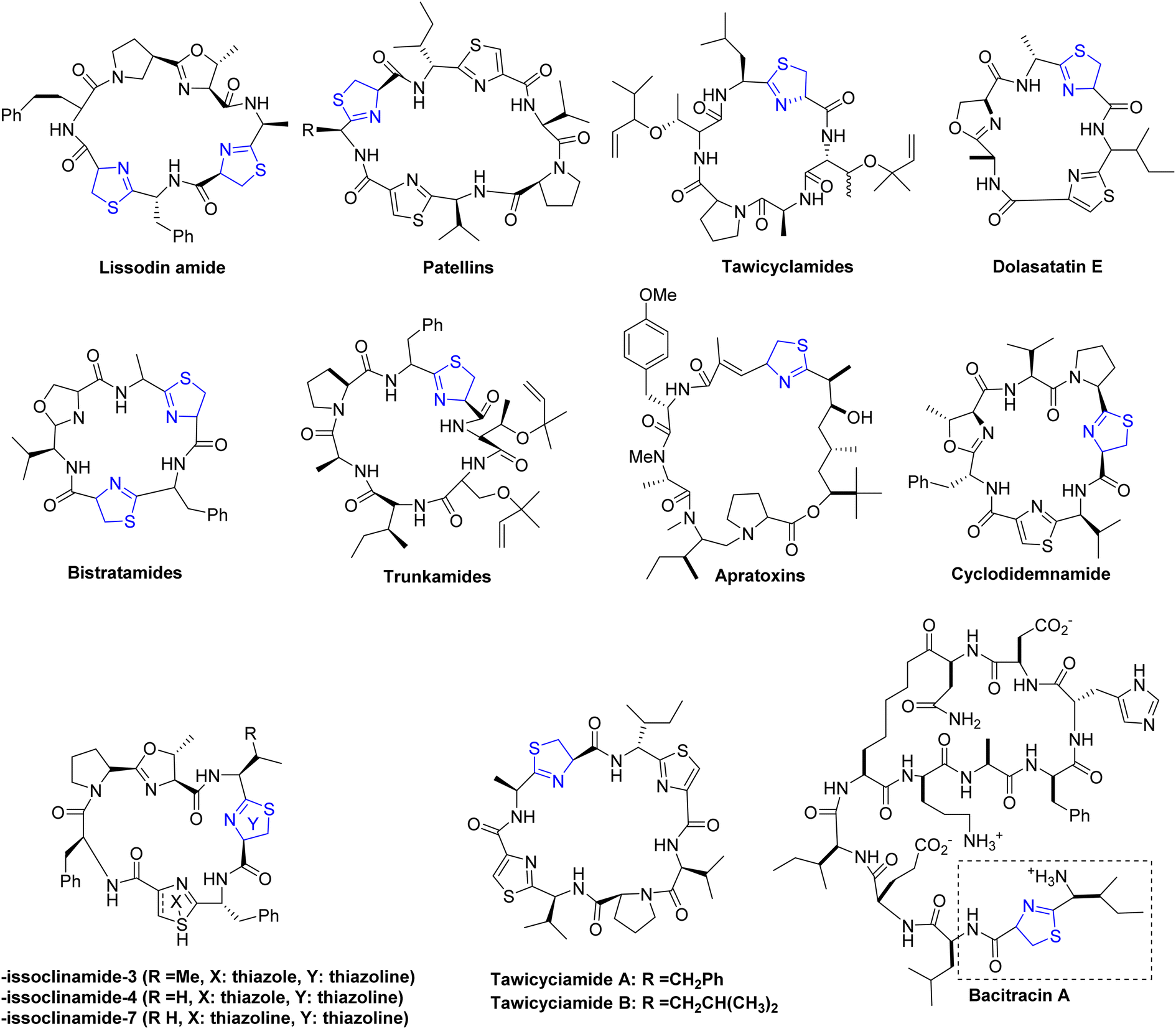 | ||
| Fig. 3 Cyclopeptide alkaloids containing thiazoline as structural segments. | ||
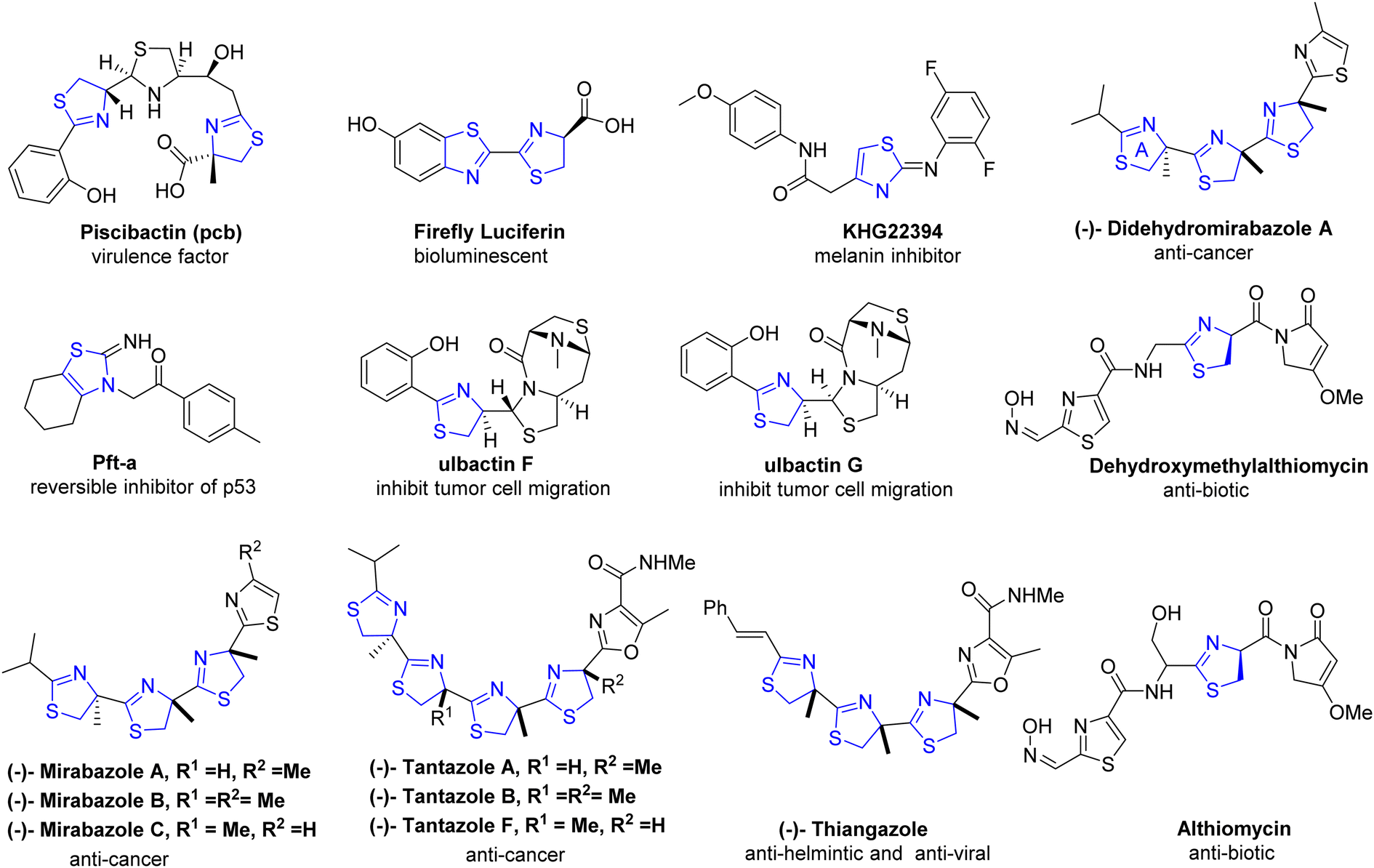 | ||
| Fig. 4 Nature derived biological active scaffolds in relevance of thiazoline. | ||
Within the extensive class of thiazolines, a diverse range of heterocyclic compounds, certain volatile derivatives stand out for their significant role in flavor and food chemistry.5 To date, researchers have identified over 30 distinct thiazoline structures present in food and natural sources,6 with notable examples found in cooked meat7 and specific exotic fruits such as litchis (Fig. 2).8
Thiazoline heterocycles can be found in a multitude of bioactive natural compounds of peptide origin (Fig. 3).9 Thiazolines can be further functionalized to introduce various chemical groups, making them versatile building blocks for the synthesis of diverse organic molecules. The thiazoline ring constitutes a structural component in various compounds, with the apratoxins serving as prominent contemporary illustrations (Fig. 3).10 These compounds were initially isolated from the marine cyanobacterium Lyngbya majuscule by Harvey ex Gomont and subsequently identified by Moore, Paul, and other researchers.11 Thiazoline imparts conformational stability and facilitates binding to proteins, RNA, and DNA by serving as a recognition site. The amide group of the preceding residue undergoes nucleophilic attack by the cysteine thiol group, followed by a dehydration step, resulting in the formation of thiazolines from peptides.12 Although, most chemical processes use serine residues, however, cysteine residues are used in the biosynthetic pathway of thiazolines.13 In processed foods, the Maillard reaction occurs, leading to the generation of these molecules through interactions involving dicarbonyl compounds, aldehydes, ammonia, and hydrogen sulfide.14 The pharmacological attributes of thiazoline have also been investigated. Some thiazoline compounds exhibit noteworthy properties such as anti-HIV15 and anti-cancer16 activities, and they are also capable of inhibiting cell division (Fig. 4).17
In 1909, Richard Willstatter successfully synthesized the first thiazolines by dialkylating thioamides.18 Thiazoles substituted in the industrial setting act as precursors for the synthesis of the amino acid cysteine, wherein 2-aminothiazoline-4-carboxylic acid serves as an intermediate compound during the commercial manufacturing process of L-cysteine.19
Thiazolines play a pivotal role in the synthesis of pharmaceuticals and biologically active natural compounds, exemplified by micacocidin, which exhibits antibacterial properties (Fig. 3).20,21 The role of firefly luciferin in the bioluminescent process of fireflies is extensively documented in scientific literature.22 Numerous fascinating natural compounds, among them curacin A, largazole, and tantazole B, feature thiazoline rings within their molecular structures.23,24 They also serve as ligands in coupling reactions catalyzed by transition metals.25 Due to the distinctive properties of sulfur, there has been a recent surge in research on the chemical attributes of thiazolines. Furthermore, thiazoline derivatives have garnered increased attention as valuable ligands in chemical synthesis and asymmetric catalysis.26 Overall, thiazolines are intriguing compounds with important roles in both natural products and synthetic chemistry. Their unique structural features and diverse applications continue to make them a subject of interest for researchers in various fields. An updated exploration of this field is imperative, given the expanding applications of thiazolines over time. Therefore, our objective is to provide a comprehensive overview of thiazolines, encompassing their chemistry, synthetic methodologies, and applications that have contributed to their increased utilization within the pharmaceutical sector, as ligands in asymmetric catalysis, and in organic synthesis over the past fifteen years. We have exclusively focused on the reports that have not been addressed in prior literature.27,28
2. Recent advances in the synthesis
Jeon et al.29 reported a metal-free oxidative di-functionalization of N-allylthioamides. In this methodology, thiazoline frameworks were synthesized under mild conditions by employing PIDA as the oxidant in conjunction with electron-deficient amines. Various benzothioamides and substrates with differing electron densities were used to produce thiazolines 2a–2f in high yields. Additionally, this process was applicable to amides derived from both thiophene 2h and pyridine 2g, as well as an aliphatic amide 2i, demonstrating compatibility under standard reaction conditions (Scheme 1).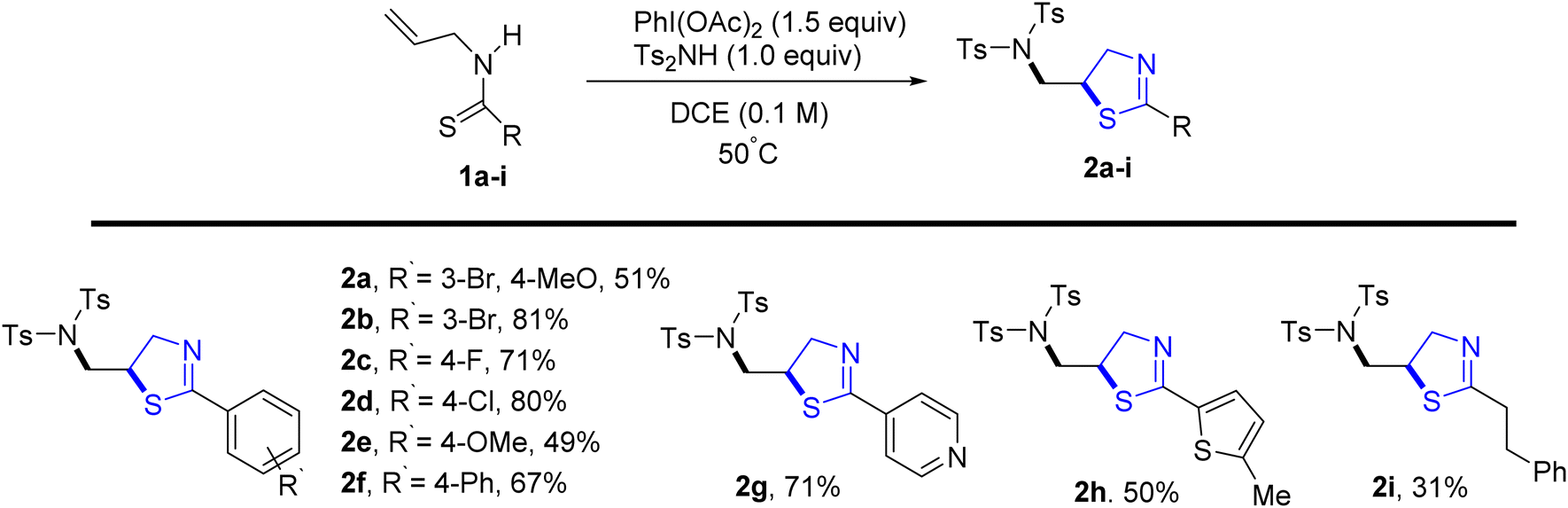 | ||
| Scheme 1 Synthesis of thiazolines 2a–2i. | ||
Multicomponent reactions (MCRs) are gaining growing appeal due to their enhanced efficiency, reduced waste generation, atom economy, and straightforward procedures.30 With the use of microwave technology, Appalanaidu et al.31 developed a novel one-pot technique to obtain the thiazoline analogues following a 3-CR between carbon disulfide, 1°-amine 3 and differently substituted bromo acylketones 4. This methodology enables the synthesis of desired compounds in a single step, yielding excellent yields, rendering it particularly valuable in the fields of synthetic and medicinal chemistry. Various primary amines, including aniline, butylamine, furan-2-ylmethanamine, benzylamine, cyclohexylamine, 2-bromo-1-phenylethanone, and substituted anilines, were subjected to treatment with CS2 to produce the HBr salt of the corresponding thiazoline derivatives. Subsequently, these obtained salts were neutralized using a saturated solution of Na2CO3, yielding the final thiazoline analogs 5a–5j (Scheme 2).
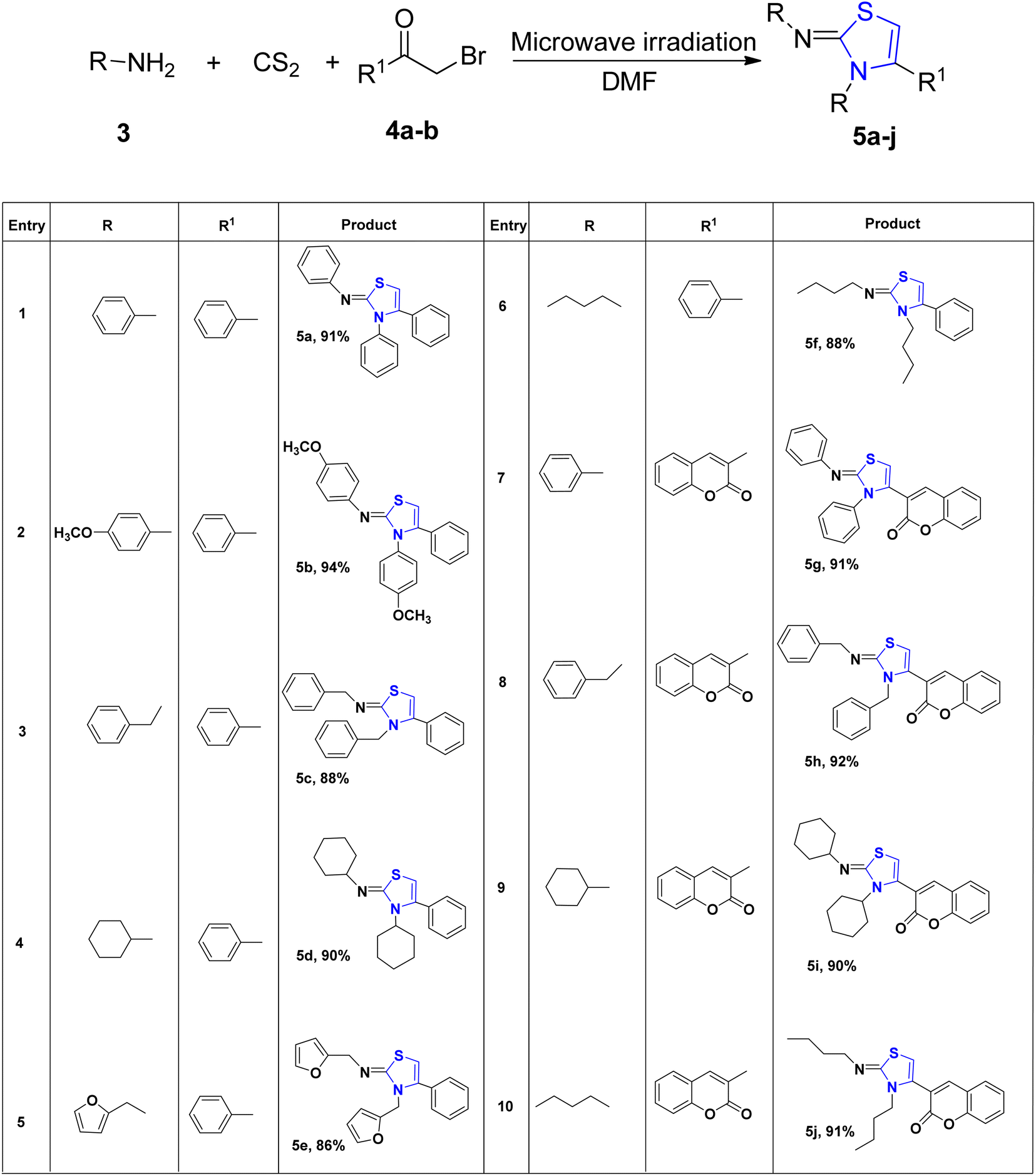 | ||
| Scheme 2 Synthesis of thiazoline derivatives 5a–5j. | ||
Four different research laboratories have used Pattenden's method32 for thiazoline synthesis to achieve the synthesis of largazole, which is isolated from fungi.33–36 Thiazoline–thiazole acid 8a can be obtained in a quantifiable yield through the cyclo-condensation of α-methylcysteine hydrochloride 6a with nitrile 7 under optimized conditions. These conditions involve maintaining a temperature of 70 °C for 2 hours in a phosphate solution with a pH of 5.95 in methanol. However, when conducting the reaction using methyl ester 6b in EtOH at 50 °C for 72 hours with the addition of Et3N, lower yield of the thiazole ester 8b was observed (Scheme 3).
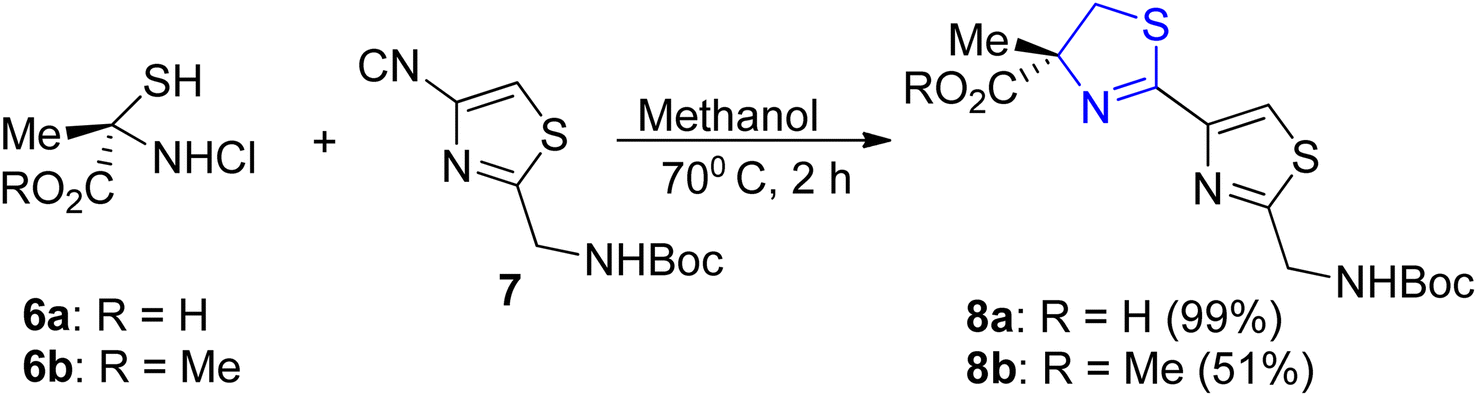 | ||
| Scheme 3 Synthesis of thiazoline thiazole molecule 8a–b. | ||
Padmavathi et al.37 synthesized thiazolines 11a–c through the condensation of Z-styryl sulfonylacetate 10a–c with aminothiol 9, catalyzed by SmCl3 in the presence of n-butyllithium (Scheme 4). The SmCl3-activated carbonyl carbon served as the site of nucleophilic attack by the thiol during the reaction.
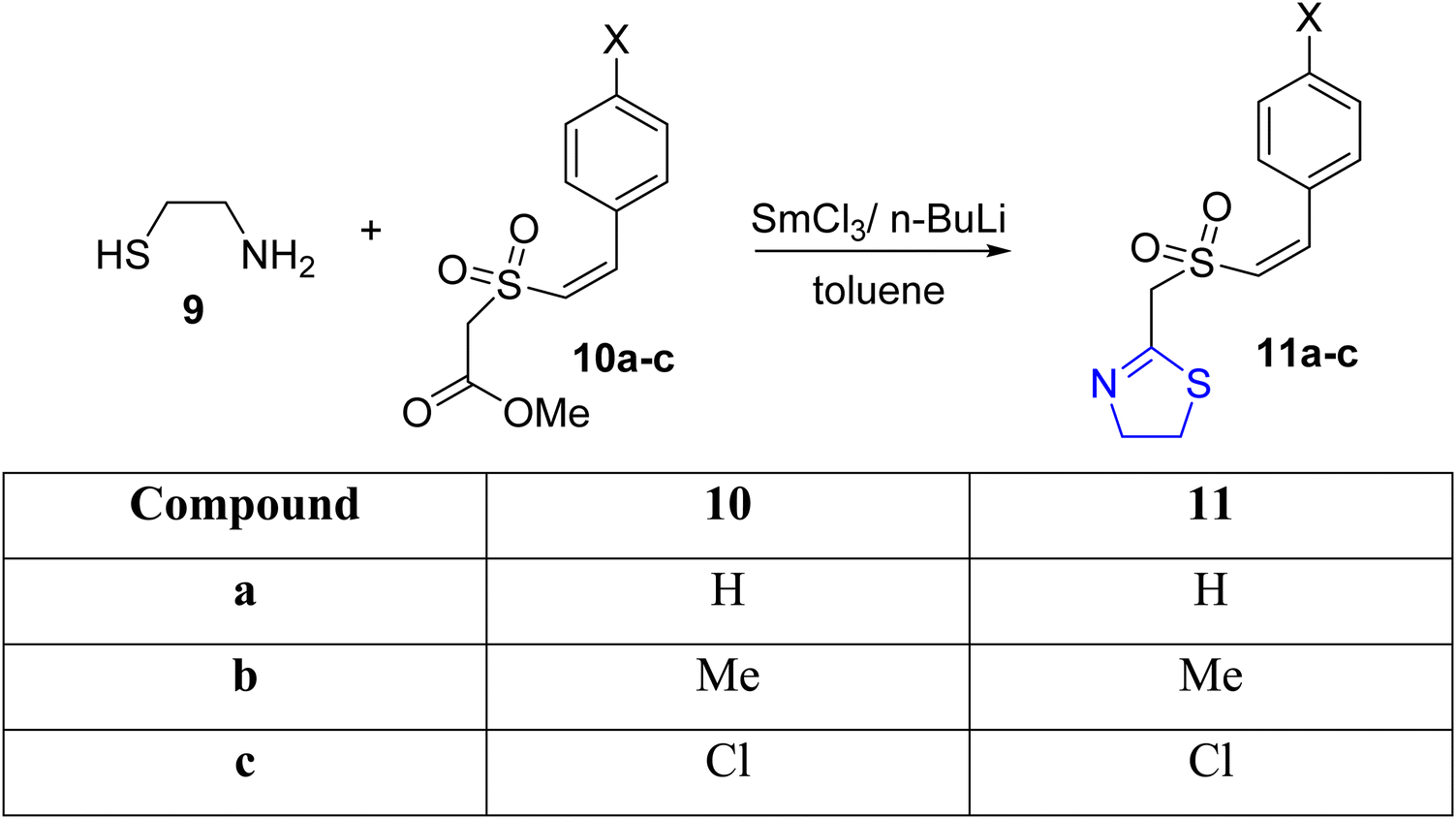 | ||
| Scheme 4 Synthesis of thiazolines 11a–c. | ||
Alom et al.38 have successfully outlined a practical synthetic approach for the important pharmaceutical motif, thiazoline 14 and 15. This method utilizes a straightforward one-pot process involving intermolecular alkene 12 and thioamide 13 substrates that are readily available and straightforward to obtain. Notably, it exhibits compatibility with a diverse array of functional groups, as demonstrated in Scheme 5a and b.
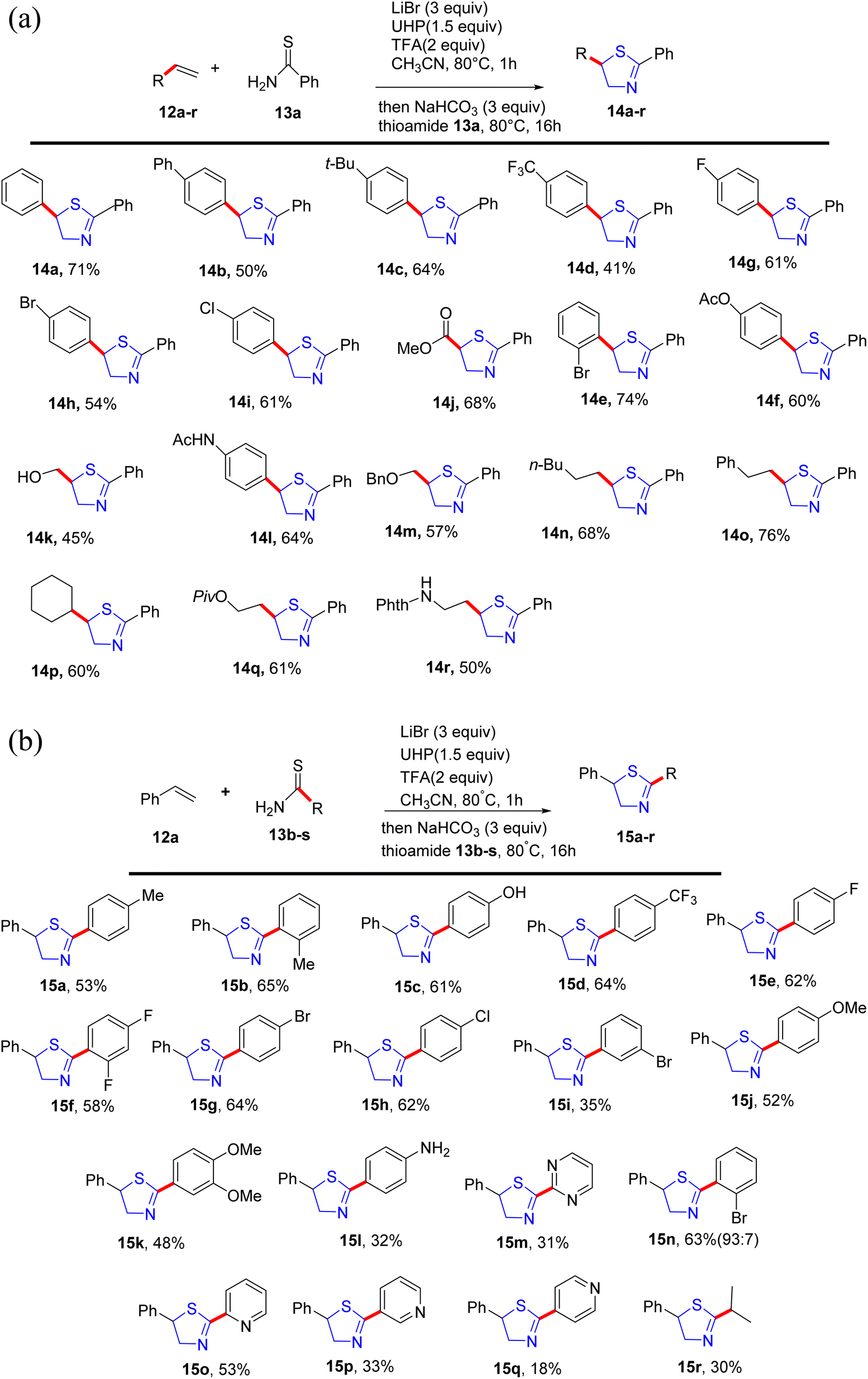 | ||
| Scheme 5 (a) Substrate scope for alkene. (b) Substrate scope for thioamide. | ||
The transformation of amino thiols into thiazolines through the utilization of α,α-difluoroalkylamines gives high yields under favorable conditions (Scheme 6). In accordance with this scheme, Fukuhara et al.39 synthesized phenyl thiazoline 18a and tert-butyl thiazoline 18b through the reaction of an ester 16 with 17a (DFBP) and 17b (DFMPP), respectively. To prevent racemization of the carbon containing the carboxylate group, triethylamine was introduced after the addition of difluoroalkylamine.
 | ||
| Scheme 6 Synthesis of thiazolines 18a–b. | ||
Alsharif et al.40 designed and developed an approach with a primary focus on synthesizing novel thiazolines. However, the current methodologies present challenges in the convenient synthesis of thiazoline derivatives 21a–l and 23a–p, as depicted in Scheme 7a and b. The investigation relied on hexafluoroisopropanol, a solvent known for its strong hydrogen bonding and polar characteristics. Due to its recyclability and recoverability, coupled with the fact that most reactions do not necessitate extensive work-up and rigorous purification, HFIP stands out as an environmentally friendly solvent.41 HFIP also facilitates a wide spectrum of reactions.42
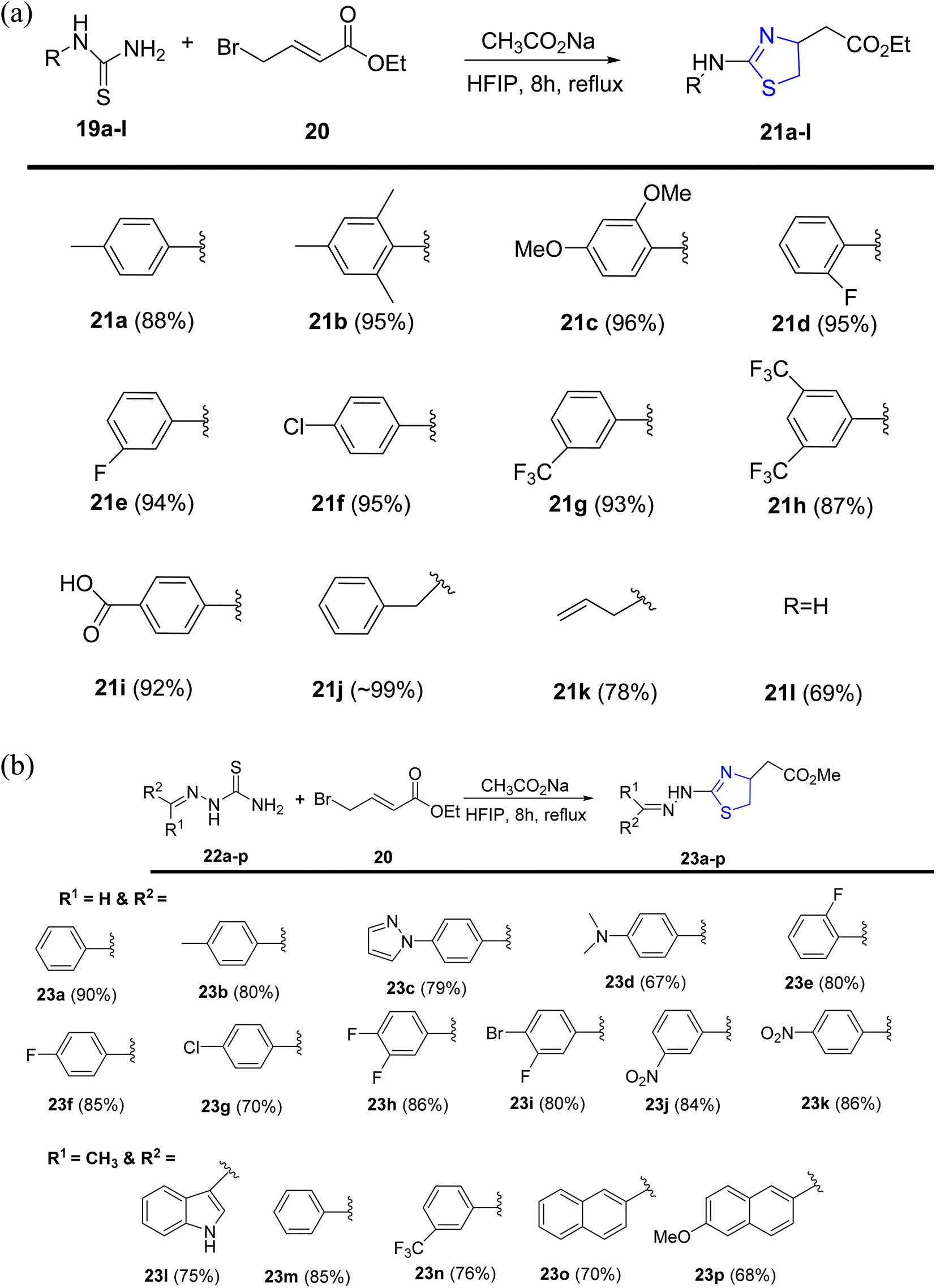 | ||
| Scheme 7 (a) Synthesis of thiazoline derivatives 21a–l. (b) Synthesis of thiazoline derivatives 23a–p. | ||
Kamila et al.43 recently unveiled an efficient approach for the synthesis of arylated thiazolines, utilizing aminothiol 24 and aryl ketonitriles 25a–k as substrates, all achieved without the presence of any solvents (Scheme 8). The incorporation of microwave radiation facilitated the condensation process. The authors outlined a methodology encompassing a thiol nucleophilic attack, water elimination, and the formation of an acrylonitrile derivative. Subsequently, the amino group participated in an intramolecular conjugate attack on the acrylonitrile derivative, resulting in the desired 2-aryl-thiazolines 26a–k, with acetonitrile removal as the final step.
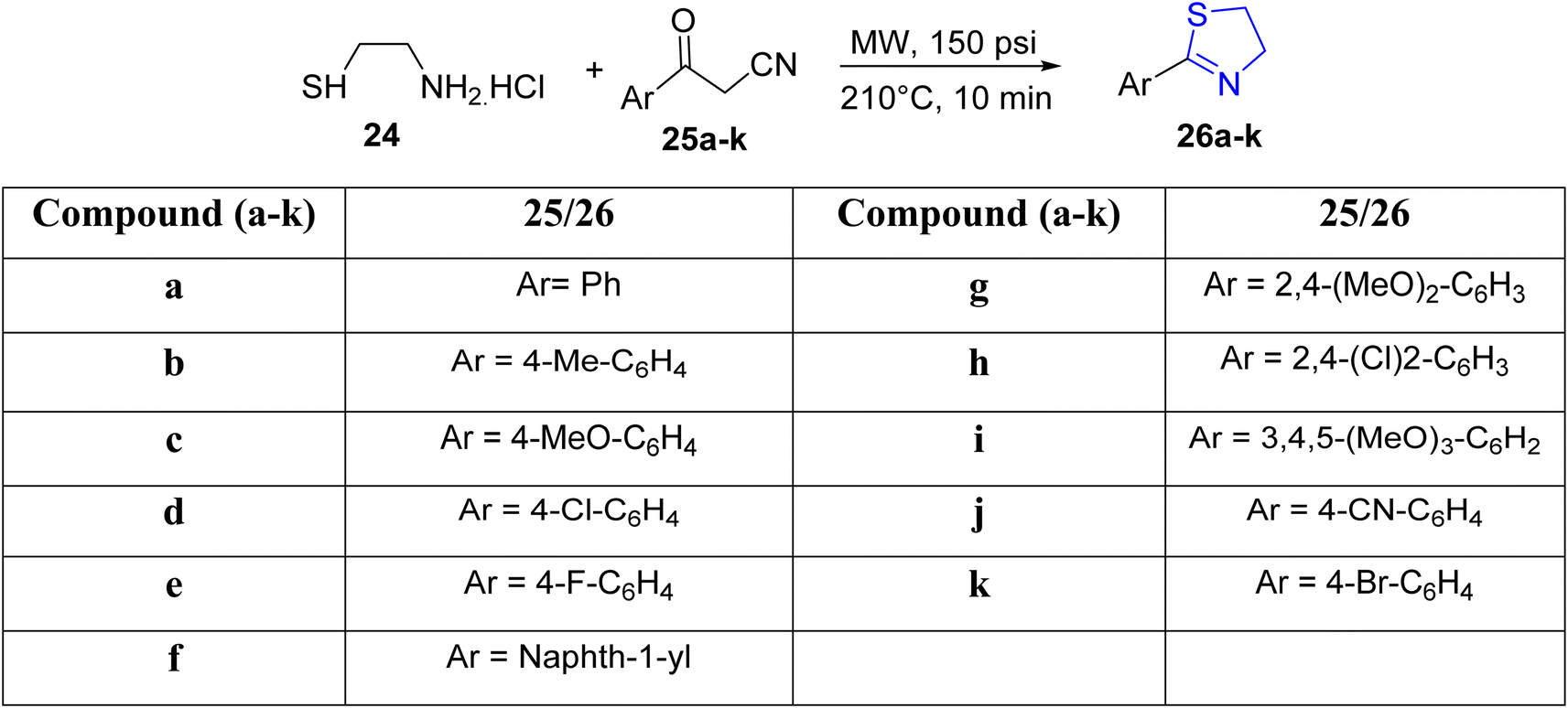 | ||
| Scheme 8 Synthesis of thiazoline derivatives 26a–k. | ||
Sakakura et al.44 reported the synthesis of thiazolines 28a–d through a dehydrative cyclization process of S-unprotected cysteine dipeptide 27 catalysed by molybdenum(VI). Notably, under optimized conditions (Scheme 9), the epimerization of the C-2-exomethine group was observed to be less than 6%.
 | ||
| Scheme 9 Synthesis of thiazolines 28a–d. | ||
Viñuelas-Zahínos et al.45 reported the synthesis of the thiazoline-based ligand ATHTd 31. The synthetic procedure for 31 involved addition of an ethanolic solution of 2-acetyl-2-thiazoline 29 to the ethanolic solution of the hydrochloride salt of (2-thiazolin-2-yl) hydrazine 30, along with potassium acetate. The reaction mixture was subsequently refluxed for 2 hours to yield compound 29 (Scheme 10). Characterization was conducted using various spectroscopic techniques, elemental analysis, and X-ray diffraction. Furthermore, ATHTd 31 was employed for complexation with Ni, Zn, and Cu metal ions, resulting in the corresponding complexes [Ni(ATHTd)2](NO3)2·H2O, [NiCl(ATHTd)(H2O)2]Cl, [ZnCl2(ATHTd)2], and [CuCl2(ATHTd)]. These complexes were also subjected to solid-state characterization using spectroscopic techniques and X-ray diffraction, along with elemental analysis.
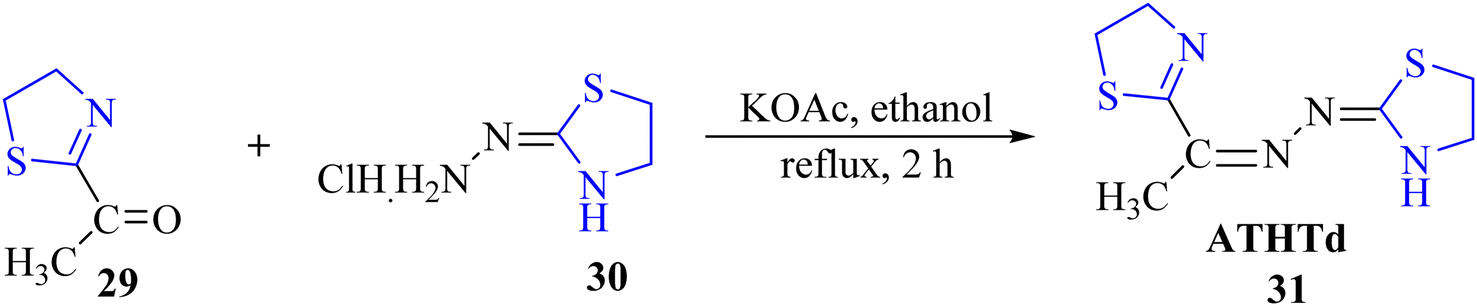 | ||
| Scheme 10 Synthesis of ATHTd 31. | ||
Attanasi et al.46 employed thioamides 33a–b and cycloalkenyl-1-diazenes 32a–c for the synthesis of diverse cycloalkyl-thiazolines 34a–c. Subsequently, these compounds were further converted into fused cycloalkyl–thiazolinepyrazole complexes 35a–c, as depicted in Scheme 11 to synthesize a variety of cycloalkyl–thiazolines 34a–c, which were further transformed into fused cycloalkyl–thiazolinepyrazole complexes 35a–c (Scheme 11).
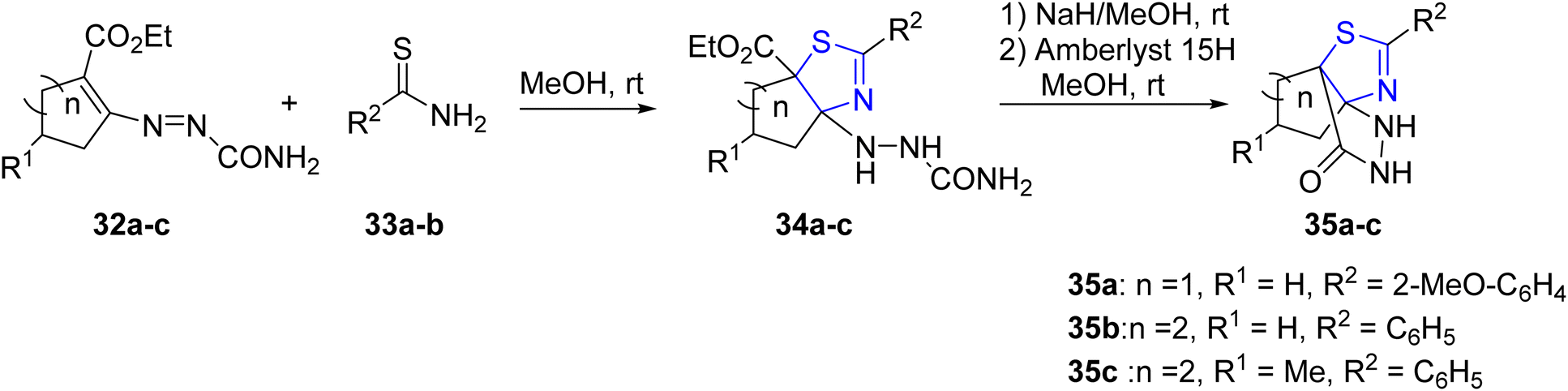 | ||
| Scheme 11 Synthesis of cycloalkyl–thiazolines 34a–c and fused cycloalkyl–thiazolinepyrazole complexes 35a–c. | ||
3. Reactivity and application of thiazolines
Thiazolines exhibit reactivity due to the presence of two nucleophilic centres localized on the nitrogen and sulfur atoms, along with an electrophilic centre on the carbon atom of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond. This versatility makes thiazolines as valuable reagents in the synthesis of diverse compounds, including carbonyls,47,48 β-amino thiols,49,50 thiazoles,51,52 and thiazolinium salts53 (Fig. 5). When treated with a base, the resulting carbanion can engage with various electrophiles, giving rise to a range of functionalized thiazolines. Examples include thiazoline phosphonates, serving as valuable synthetic intermediates in applications such as Horner–Wadsworth–Emmons (HWE) reactions,54 and vinyl thiazolines, which function as Michael acceptors or heterodienes.55 In industrial applications, substituted thiazoles play a crucial role as precursors in the synthesis of the amino acid cysteine. Specifically, 2-aminothiazoline-4-carboxylic acid serves as an intermediate in the commercial manufacturing process of L-cysteine.19
N bond. This versatility makes thiazolines as valuable reagents in the synthesis of diverse compounds, including carbonyls,47,48 β-amino thiols,49,50 thiazoles,51,52 and thiazolinium salts53 (Fig. 5). When treated with a base, the resulting carbanion can engage with various electrophiles, giving rise to a range of functionalized thiazolines. Examples include thiazoline phosphonates, serving as valuable synthetic intermediates in applications such as Horner–Wadsworth–Emmons (HWE) reactions,54 and vinyl thiazolines, which function as Michael acceptors or heterodienes.55 In industrial applications, substituted thiazoles play a crucial role as precursors in the synthesis of the amino acid cysteine. Specifically, 2-aminothiazoline-4-carboxylic acid serves as an intermediate in the commercial manufacturing process of L-cysteine.19
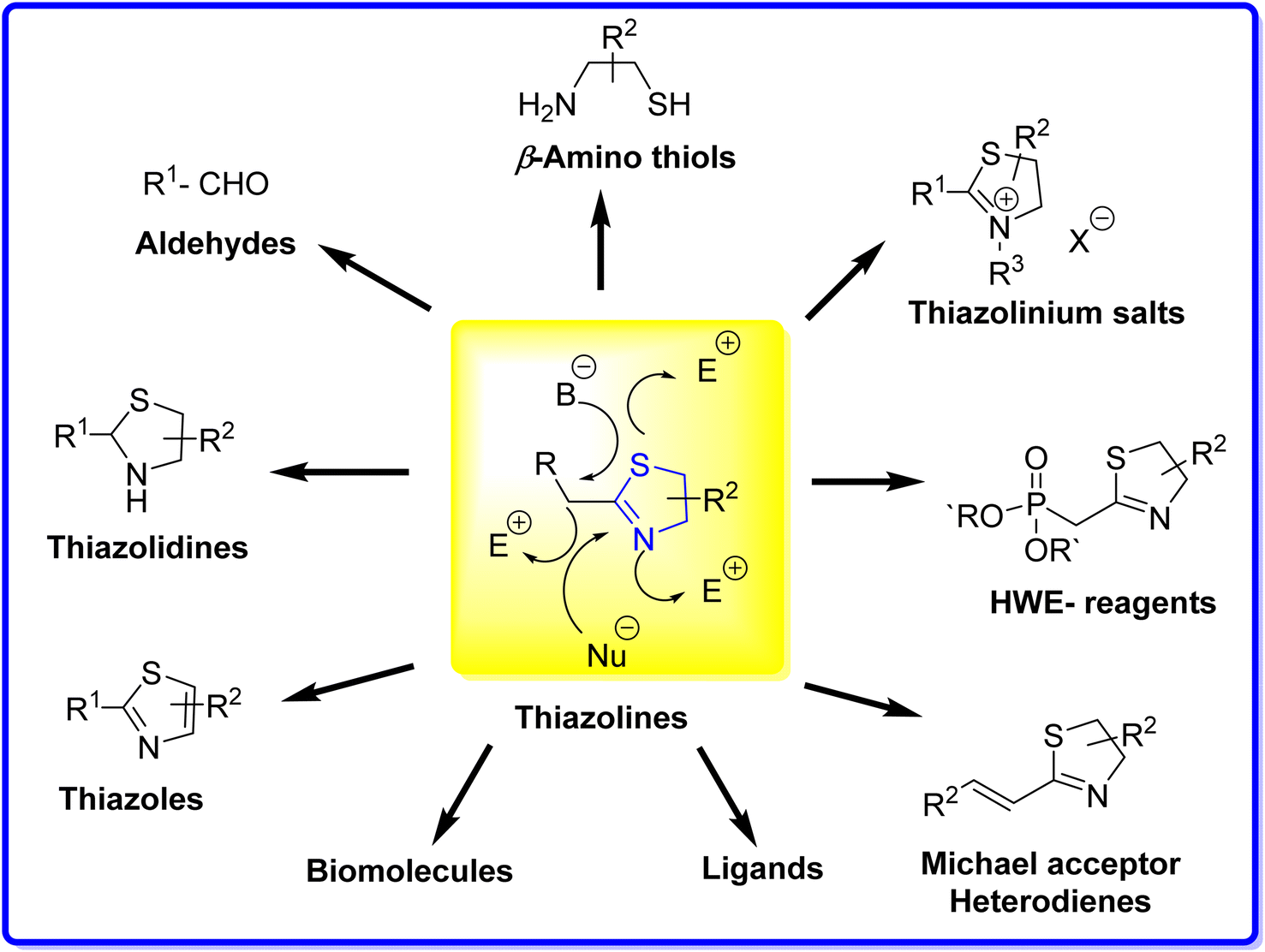 | ||
| Fig. 5 Reactivity and applications of thiazolines. | ||
Furthermore, thiazolines with a chiral centre find applications as chiral auxiliaries or building blocks, enabling the synthesis of more complex chiral structures. These structures include thiazoline-containing biomolecules56,57 and chiral ligands utilized in asymmetric catalysis.58,59 The discussion here encompasses the role of thiazolines in fluorescence, catalysis, pharmacology, and other diverse fields.
3.1. Fluorescence property
Fluorescence-based sensors exhibit several merits, including ease of manipulation and operation, swift response kinetics, exceptional sensitivity, straightforward instrumentation, cost-effectiveness, and the attainment of low detection limits. Consequently, these attributes render them a preferential choice when contrasted with alternative analytical methodologies.60,61 Diverse sensor modalities have been employed for the detection of metal ions, encompassing quantum dots (QDs), nanomaterials, organic molecules, and biopolymers.62,63 Nevertheless, there remains an imperative requirement for the development of highly sensitive and biocompatible near-infrared (NIR) sensors capable of discerning heavy metal ions within biological systems. Recent times have witnessed a notable surge in enthusiasm for fluorescent sensors, owing to their substantial potential in the realm of ion detection within biologically relevant media, characterized by their exceptional selectivity and sensitivity.64,65Even when present at low concentrations, the harmful contaminant mercury (Hg) can exert significant deleterious effects on both the environment and human health. Consequently, there is an acute demand for methodologies that exhibit high sensitivity, efficacy, and precision in detecting mercuric ions within biological matrices. Erdemir et al.66 engineered a fluorescent sensor designated as THI 41, characterized by the inclusion of a Hg2+-sensitive thiazoline moiety, an electron-deficient dicyanovinyl functionality, and an electron-rich diethylamino group. The strong affinity between S2− ions and Hg2+ ions was observed to induce a reversal in the binding interaction between probe 41 and Hg2+ ions. Additionally, probe 41 manifested a positive solvatochromic effect attributable to intramolecular energy transfer, facilitated by the diethylamino functionality, toward the dicyanovinyl group. Moreover, it was found to be highly selective for Hg2+ ions. The synthesis of 4-(bis(2-chloroethyl)amino)benzaldehyde 37 followed a previously established procedure.67 Subsequently, 2-thiazoline-2-thiol 38 was reacted with benzaldehyde 37 in acetonitrile, and the resulting mixture was refluxed for a duration of 72 hours to yield compound 39. In the final step of the synthesis, compound 39 was subjected to reflux with compound 40 in a mixture of piperidine and ethanol for a duration of 12 hours, resulting in the formation of the fluorescent probe THI 41, as depicted in Scheme 12. The determined detection limit for THI 41 was 7.22 mM, and it exhibited a satisfactory linear correlation with varying levels of Hg2+. Furthermore, employing a confocal laser scanning microscope, it was conclusively demonstrated that THI 41 can serve as a proficient fluorescent probe for visualizing Hg2+ ions within living HeLa cells, without any observable adverse effects on cervical cancer and epithelial cells.51
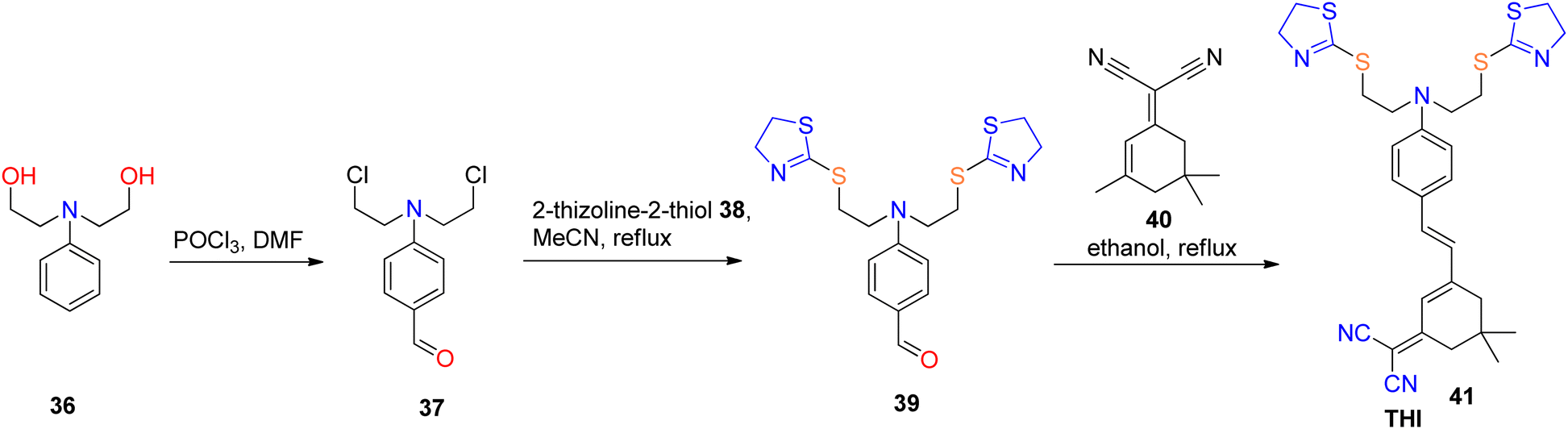 | ||
| Scheme 12 Synthesis of new red-emitting fluorescent probe (THI) 41. | ||
Within the realm of chemistry, the detection of volatile acids represents an economically promising domain. This is primarily attributed to the pivotal role of pH estimation in ascertaining acidity levels, which holds significant importance across a spectrum of applications encompassing chemical reactions, biological processes, the pharmaceutical industry, and environmental monitoring.68,69 Chaudhary et al.70 successfully synthesized six novel compounds, denoted as 43a–e, exhibiting acid-sensitive attributes with exceptional yields ranging from 84% to 95%. These compounds were designed with a thiazoline ring serving as the electron acceptor moiety and a phenothiazine ring acting as the electron-donor unit. The synthesis of thiazoline-based compounds 43a–e, hinging on the phenothiazine-5-oxide ring, was accomplished utilizing a previously established methodology (Scheme 13).
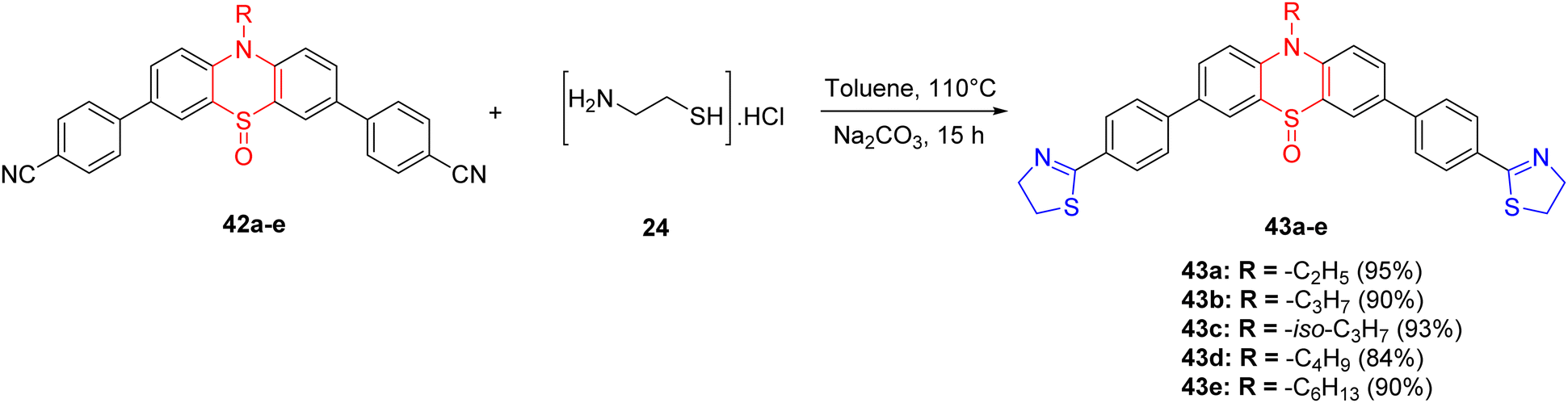 | ||
| Scheme 13 Synthesis of novel “push–pull” compounds 43a–e. | ||
The reaction involved refluxing the precursor molecules 42a–e in the presence of cysteamine hydrochloride 24 and sodium carbonate in toluene under a nitrogen atmosphere at 110 °C for a duration of 15 hours. Notably, compounds 43a–e exhibited robust fluorescence both in solution and in solid states. The incorporation of phenyl rings endowed these compounds with the ability to detect volatile acids, such as trifluoroacetic acid (TFA) and hydrochloric acid (HCl), owing to the presence of a thiazoline unit. In absorption and emission experiments, a distinctive red-shift was observed, attributed to the protonation of the thiazoline ring, induced by potent intramolecular charge transfer (ICT) interactions. The addition of triethylamine (TEA) was found to reverse these spectral alterations. Upon exposure to acids, a distinct color change from colorless to yellow was observed, which could subsequently be reverted to colorless upon the addition of TEA.
Compound 43c exhibited a remarkable minimum detection limit of 0.98 parts per million (ppm) for TFA, while compound 43a demonstrated a noteworthy minimum detection limit of 13.1 parts per billion (ppb) for HCl. Further elucidation of the energy gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of compounds 43a–e, as well as their protonated analogues, was conducted through density functional theory (DFT) studies.55
Copper ions in the form of Cu2+ hold a dual significance in scientific contexts, as they function as both significant metal pollutants and essential micronutrients for all known life forms. In the case of Cu2+ ions, a plethora of fluorescent chemo-sensors are available; however, only a limited subset of these sensors are categorized as “turn-on” sensors, primarily because Cu2+, being paramagnetic, exerts a quenching effect on fluorescence.71 Preferred among these are the “turn-on” fluorescence sensors, as they exhibit a reduced susceptibility to false positive signals and offer enhanced multiplexing capabilities.72 Utilizing a one-pot synthetic approach, Wang et al.73 successfully synthesized a sensor denoted as 45, which incorporated both thiazoline 38 and pyrene moieties 44. This sensor was subsequently employed to enhance the fluorescence emission of pyrene monomers, thereby enabling the development of a highly sensitive and selective detector for Cu2+. The synthetic route to produce sensor 45 is detailed in Scheme 14. Fluorescent titration experiments were conducted, and Job's plots were subsequently employed to ascertain a stoichiometry of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 between sensor 45 and Cu2+. This complexation was corroborated by spectroscopic analysis, density functional theory (DFT) measurements, and Fourier-transform infrared (FTIR) data. In the presence of Hg2+, compound 45 exhibited an “on-off” fluorescence response at 460 nm, although qualitative detection was achieved. Notably, when exposed to a range of investigated metal ions, sensor 45 singularly demonstrated a remarkably sensitive and selective “turn-on” fluorescence response for the detection of Cu2+. Employing statistical deviations and linear regression analyses, the 45-Cu2+ complex was evaluated for its detection limit and association constant. The results indicated the functionality of sensor 45 for Cu2+ detection across a wide pH spectrum, spanning from 2.0 to 11.0.
:1 between sensor 45 and Cu2+. This complexation was corroborated by spectroscopic analysis, density functional theory (DFT) measurements, and Fourier-transform infrared (FTIR) data. In the presence of Hg2+, compound 45 exhibited an “on-off” fluorescence response at 460 nm, although qualitative detection was achieved. Notably, when exposed to a range of investigated metal ions, sensor 45 singularly demonstrated a remarkably sensitive and selective “turn-on” fluorescence response for the detection of Cu2+. Employing statistical deviations and linear regression analyses, the 45-Cu2+ complex was evaluated for its detection limit and association constant. The results indicated the functionality of sensor 45 for Cu2+ detection across a wide pH spectrum, spanning from 2.0 to 11.0.
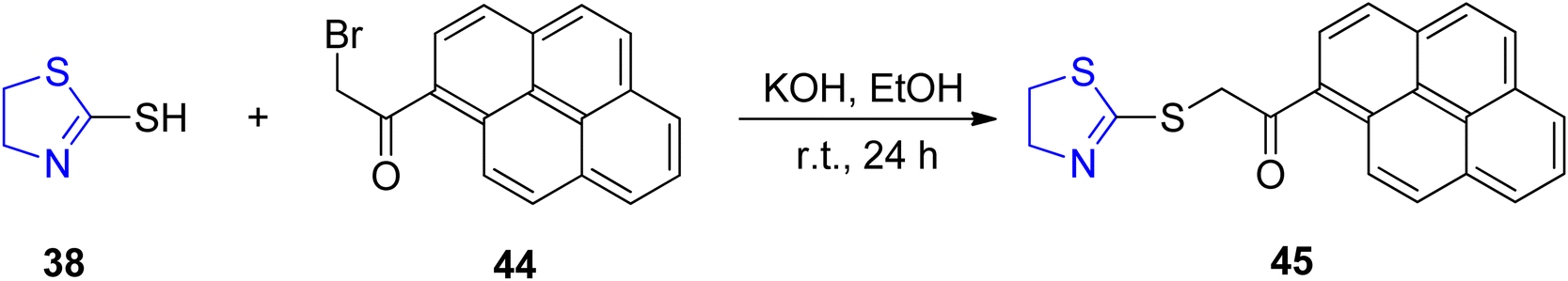 | ||
| Scheme 14 Synthesis of a “turn-on” thiazoline–pyrene sensor 45. | ||
The development of white organic light-emitting diodes (WOLEDs) for forthcoming lighting technologies has garnered substantial interest. Luminescent complexes featuring carbene–metal–amide bonding, incorporating metals like Cu, Au, and Ag, present a compelling alternative to costly metal-based OLEDs employing elements such as Ir and Pt. This preference stems from their notable advantages, including high decay rates and emission efficiency. Ruduss et al.74 reported the synthesis of eight novel Cu(I) complexes, designated as 52–59, featuring an uncharacterized carbene moiety derived from 1,3-thiazoline. These compounds underwent comprehensive evaluation to ascertain their photoluminescent properties and their prospective utility in organic light-emitting diodes (OLEDs). The synthesis of carbene precursors, specifically 46, 48, and 50, was accomplished following established procedures available in the literature.75 Subsequently, these precursors underwent in situ carbene generation to yield the corresponding Cu(I) complexes, specifically 47, 49, and 51. These Cu(I) complexes were then subjected to reactions with various deprotonated carbazolides, including tBuCbz, Cbz, MetBuCbz, and MeCbz, to facilitate the synthesis of amide ligands, as depicted in Scheme 15a–c. The orchestration of both monomer and excimer components was carefully executed to achieve electroluminescence (EL) under optimized emitter structure and mass fraction conditions. This served as the foundational framework for the development of a white organic light-emitting diode (WOLED) characterized by an impressive quantum efficiency of 16.5%, accompanied by a single emission exhibiting a peak brightness exceeding 40000 candelas per square meter (cdm−2). Notably, the broad overlapping emission bands of the monomer and excimer components ensured a color rendering index (CRI) exceeding 80 for the resultant WOLED.
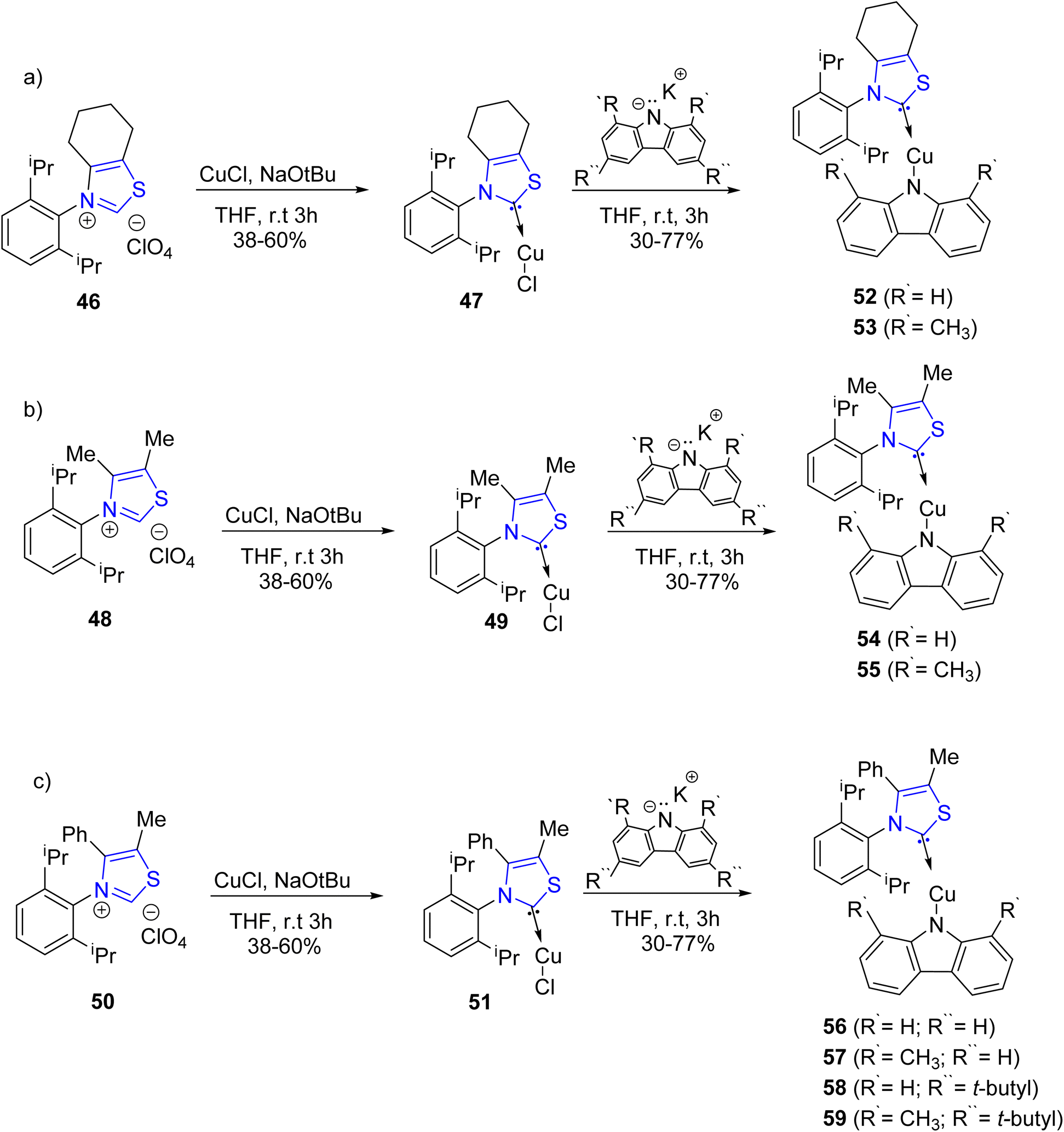 | ||
| Scheme 15 (a–c) Synthesis of CMA emitters 52–59. | ||
3.2. Catalytic property
The versatile chelating capabilities exhibited by multi-donor ligands containing sulfur (S), nitrogen (N), and oxygen (O) atoms in coordination with transition metal ions have garnered significant attention in numerous catalytic reactions.76,77 A representative example of such ligands is Htzol, a naturally occurring compound found in (S) (−)-desferrithiocin. Investigations have revealed that substituting a thiazoline ring with an oxazoline ring induces a profound alteration in the reactivity of the respective ligand.78,79 When contrasted with its oxazoline counterpart, the utility of thiazolines as ligands in coordination and organometallic chemistry is relatively limited. Since Helmchen began researching thiazoline-containing ligands in 1991,80 their use in asymmetric catalysis has decreased. As reported by López-Cortés and collaborators, substituting the oxazoline backbone with a thiazoline counterpart results in an enhancement of the catalytic activity.81 The development of innovative chiral ligands holds paramount significance in scientific research, given the pivotal role of asymmetric metal-catalyzed reactions in synthetic organic chemistry. Recent focus has been directed toward bis(thiazolines) following their synthesis, including various pyridyl bis(thiazolines) and methylene-bridged bis(thiazolines), as established by Masson and Gulea. This renewed attention underscores the emerging importance of thiazoline-containing ligands in contemporary research endeavors.82 The thiazolines, which are sulfur-analogs of oxazolines and amongst the vast range of chiral ligands, are a relatively new family of ligands.83Amini et al.84 provided an account of the catalytic competence of a thiazoline–Cu(II) complex in facilitating cycloaddition reactions without the need for additional redox cofactors. They synthesized three novel complexes, featuring Pd(II), Cu(II), and Co(III), employing Htzol (thiazoline-based compound) 60 as a ligand. Htzol ligand exhibited bidentate thiazoline coordination, serving as an O, N-donor. The standard synthetic approach for the complexes entailed the reaction of the thiazoline ligand with metal acetates in a methanolic solvent medium, as described in Scheme 16. Subsequently, complexes 61–63 were investigated for their catalytic efficacy in promoting the [3 + 2]-cycloaddition reaction between alkyl halide 64, sodium azide 65 and alkyne substrates 66 (Scheme 17).69 Optimization of reaction conditions, including catalyst loading, reaction temperature, and reaction duration, was performed to enhance the efficiency of the azide–alkyne cycloaddition reactions.
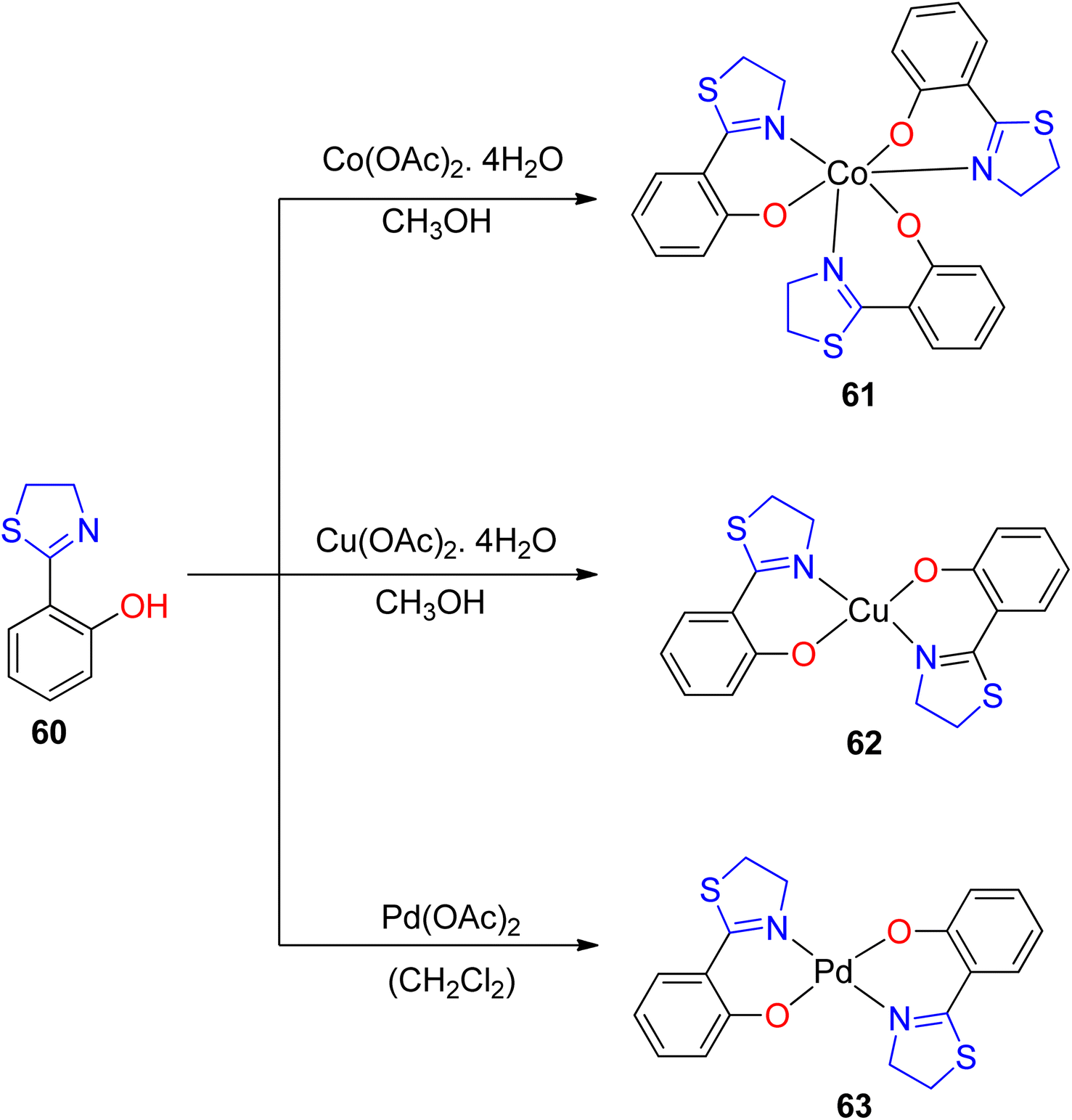 | ||
| Scheme 16 Synthesis of complexes 61–63. | ||
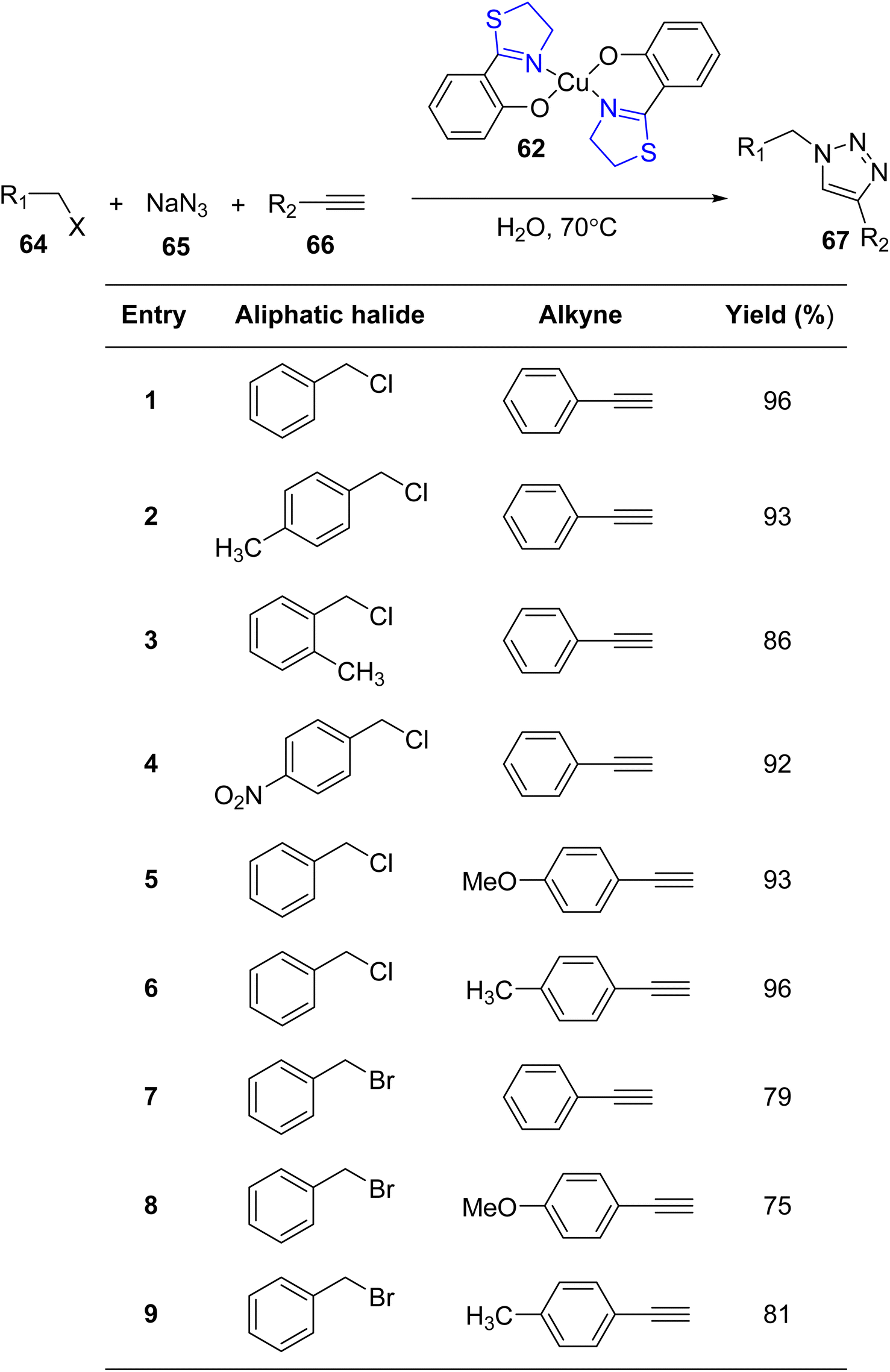 | ||
| Scheme 17 [3 + 2]-Cycloaddition reaction between azide and alkyne substrates. | ||
Irmak et al.85 gave the first synthetic methodology for pyridiyl bis(thiazoline) ligand based on sugar (Scheme 18). Further, it was investigated for its use in asymmetric cyclopropanation processes (Scheme 19). Per-OTMS group was used to protect the amino sugar 68 in order to obtain the desired acetyl-protected bis(amide). The resultant derivative 69 was firstly protected with the OTMS group followed by its reaction with dipicolinic acid chloride to yield 70b. Compound 70b on subsequent desilylation and acetylation gave the bis(amide) 72b. Under the conditions as outlined in the literature,86 bis(amide) 72b when refluxed with Lawesson's reagent produced the bis(thioamide) 73b. Double cyclization could eventually be achieved by extending the reaction period. As a result, target compound 75 was achieved with a 44% total yield (Scheme 18). They conducted experiments using pyridyl bis(thiazoline) 75 as a ligand in metal-catalysed cyclopropanation reactions involving styrene 76 and ethyl diazoacetate 77 under varying conditions. Subsequently, they also explored the reaction with two other alkenes, namely 4-methoxy styrene compound 76b and 1,1-diphenyl ethylene 79 (Scheme 19).
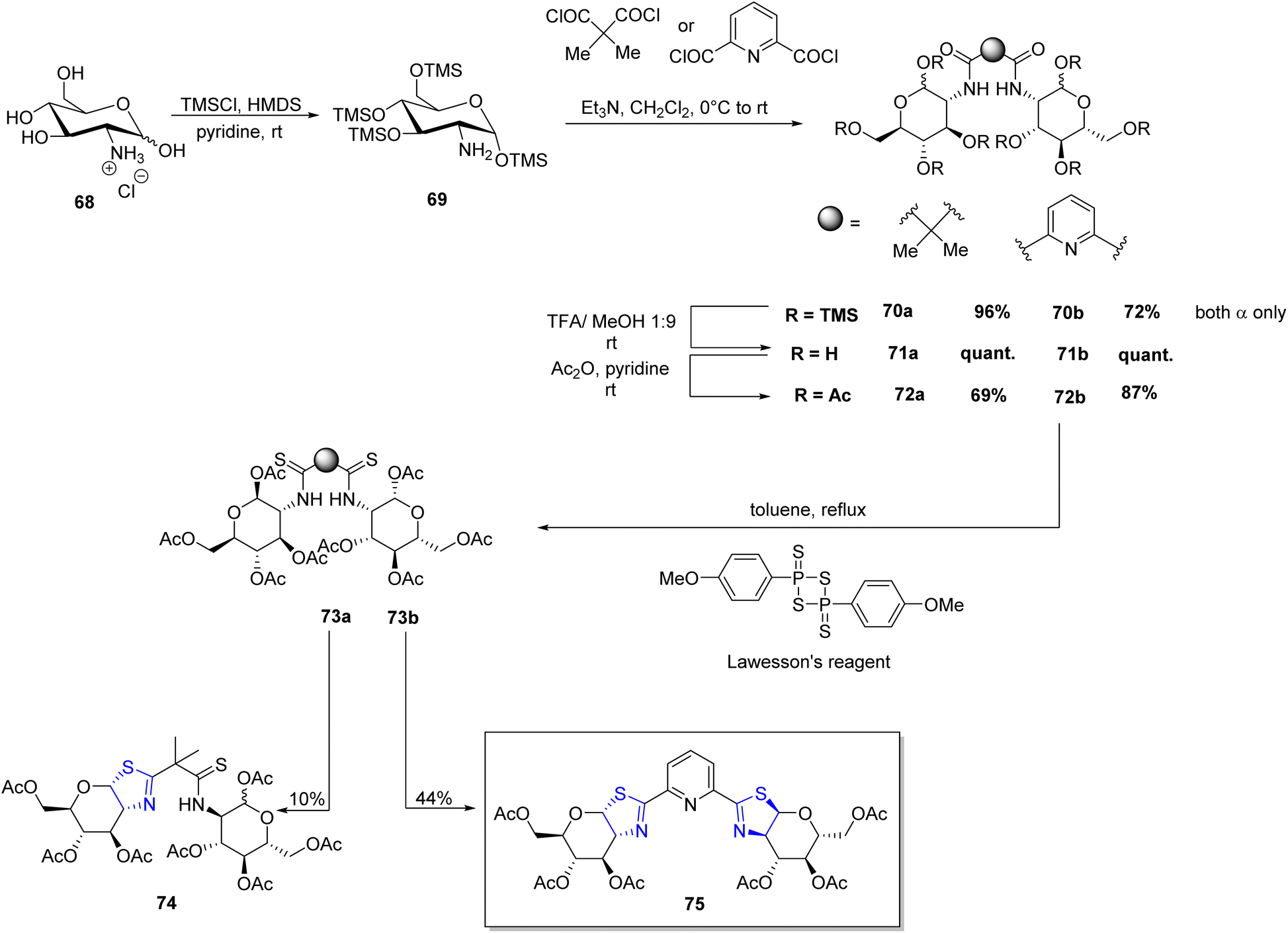 | ||
| Scheme 18 Synthesis of a ligand 75. | ||
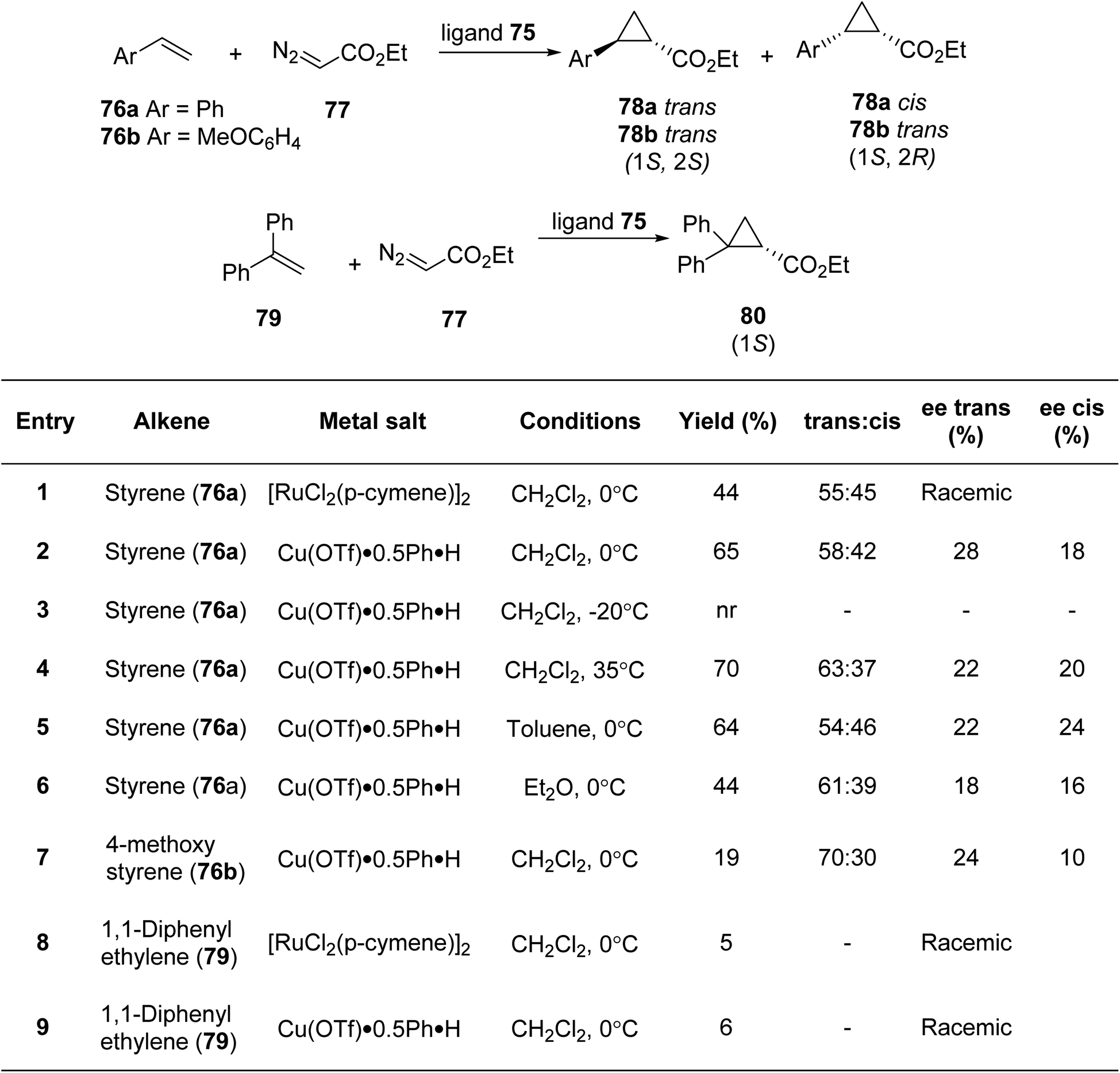 | ||
| Scheme 19 Asymmetric cyclopropanation processes. | ||
Sudharsan et al.87 conducted an investigation into the formation of thiazoline–ligand complexes with Pd(II), yielding both homo- and heteroleptic complexes, as depicted in Scheme 20. The synthesized compounds were subsequently assessed for their catalytic capabilities. These compounds served as catalysts in reactions involving the generation of Csp2–Csp2 bonds. Characterization of Pd(II) complexes 82, 83, and 84a–b, as well as ligand 60, was achieved through spectroscopic analysis, elucidating their crystal and molecular structures. Under optimized reaction conditions, Pd(II) complex 83 exhibited exceptional catalytic activity in the synthesis of biaryls 87 from phenylhydrazine 86 and aryl halides 85, achieving a remarkable turnover frequency of 49.5 h−1 (Scheme 21).
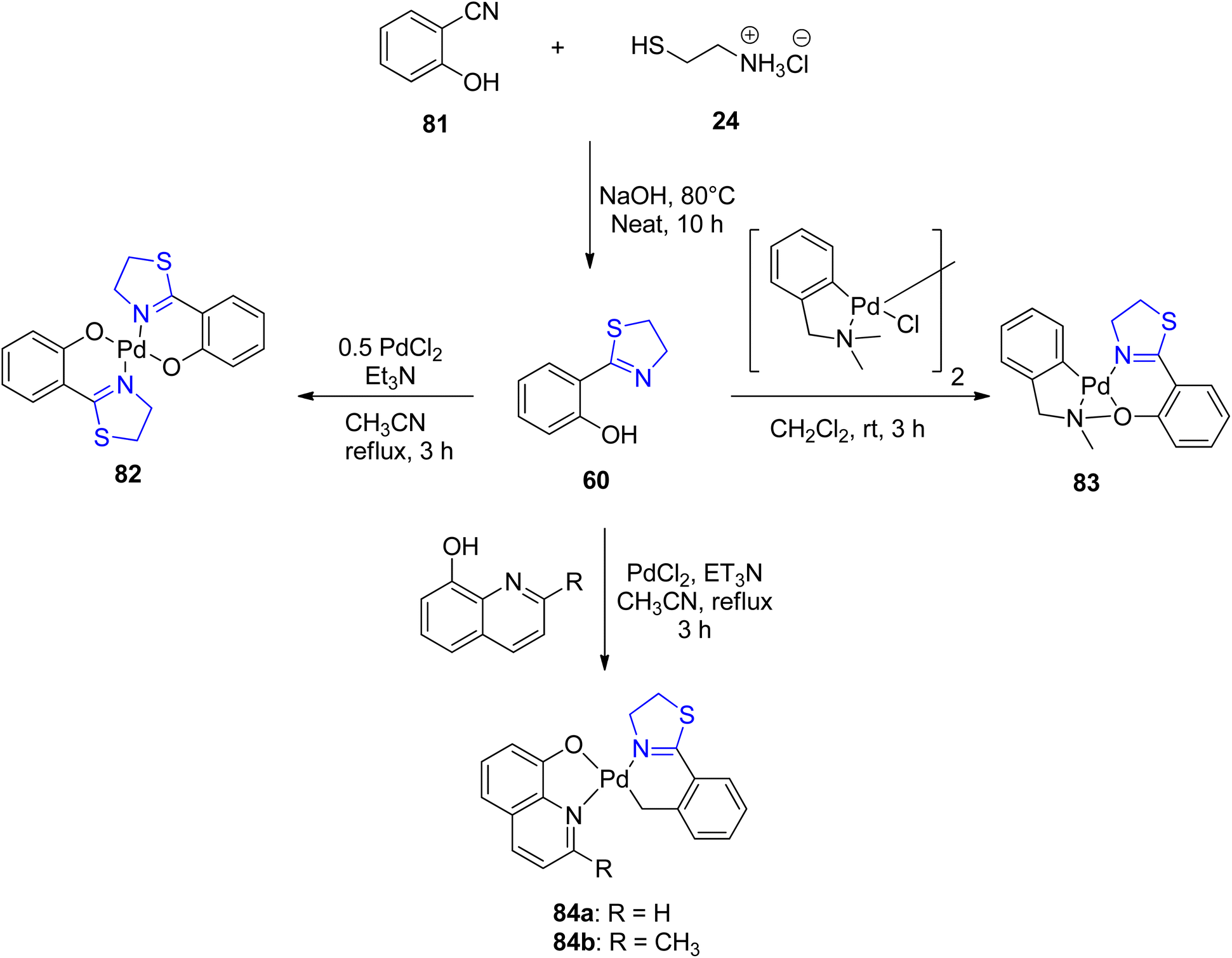 | ||
| Scheme 20 Synthetic scheme for ligand 60 and its corresponding complexes 82, 83 and 84a–b with Pd. | ||
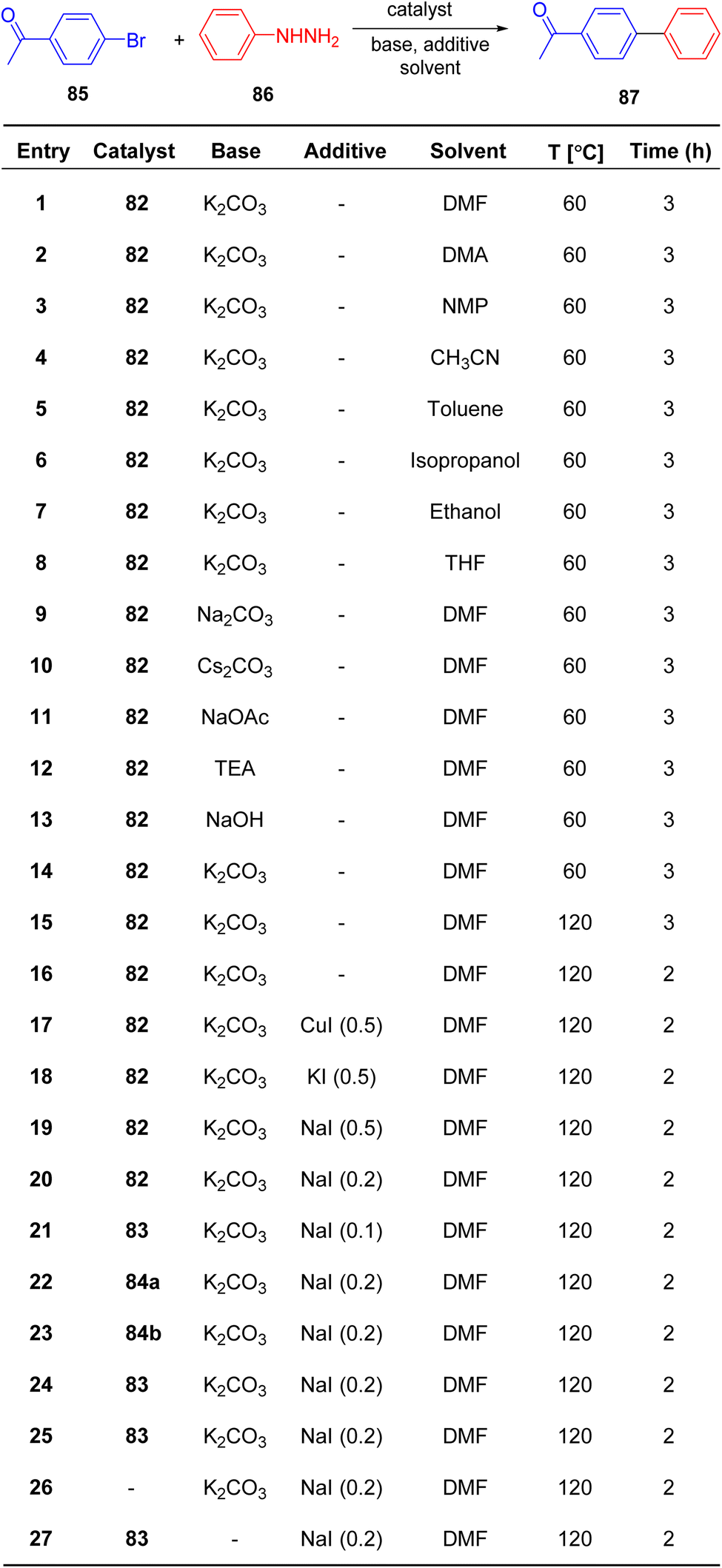 | ||
| Scheme 21 Synthesis of bi-aryls 87 from phenylhydrazine 86 and aryl halides 85. | ||
Mckeon et al.88 reported the convergent preparation of thiazoline & oxazoline containing six non-C2-symmetric ligands 90a–f by microwave irradiation. The amination reaction was catalysed by Pd(II) (Scheme 22). In a zinc catalysed Friedel–Crafts alkylation reaction involving trans-β-nitrostyrene 92 & indole 91, the ligands 90a–f gave enantioselectivity as high as 76% (Scheme 23).
 | ||
| Scheme 22 Synthetic scheme for non-C2-symmetric ligands 90a–f. | ||
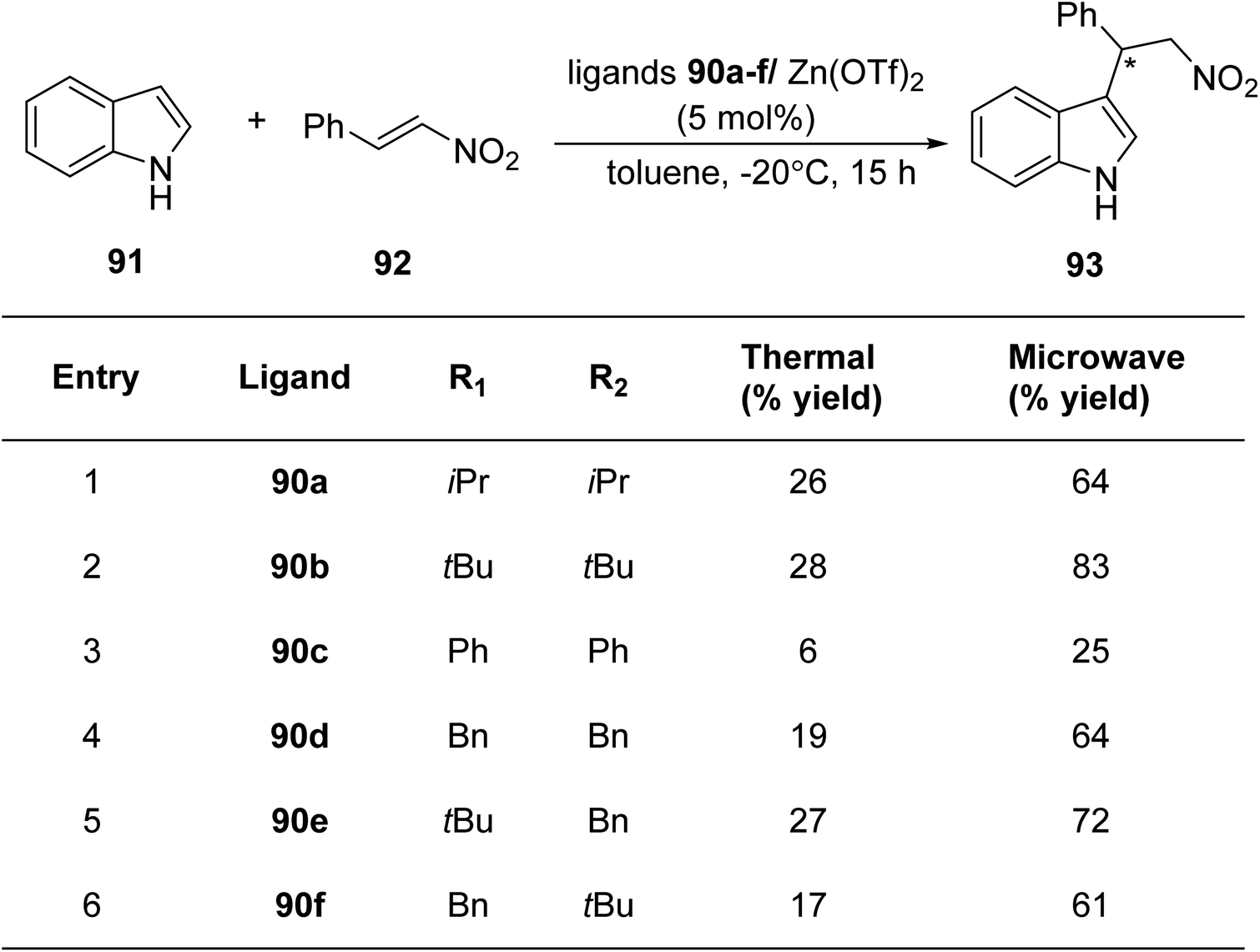 | ||
| Scheme 23 Zinc catalyzed Friedel–Crafts alkylation reaction. | ||
Mckeon et al.89 reported the synthesis of ten novel ligand analogues denoted as 90g–p (Scheme 24), aiming to enhance enantioselectivity in the Friedel–Crafts reaction, as outlined in Scheme 25. Furthermore, these ligands were employed in the NHK allylation reaction with benzaldehyde 94, catalyzed by chromium (Scheme 26). The X-ray analysis of the Fe(II) complex of the ligand revealed its tridentate ligating behavior. Based on this crystallographic evidence, the remarkable enantioselectivity observed with ligand 90e (R1 = t-Bu, R2 = Bn) in the allylation reaction of benzaldehyde [85% (R)] was elucidated through the proposal of a transition state.
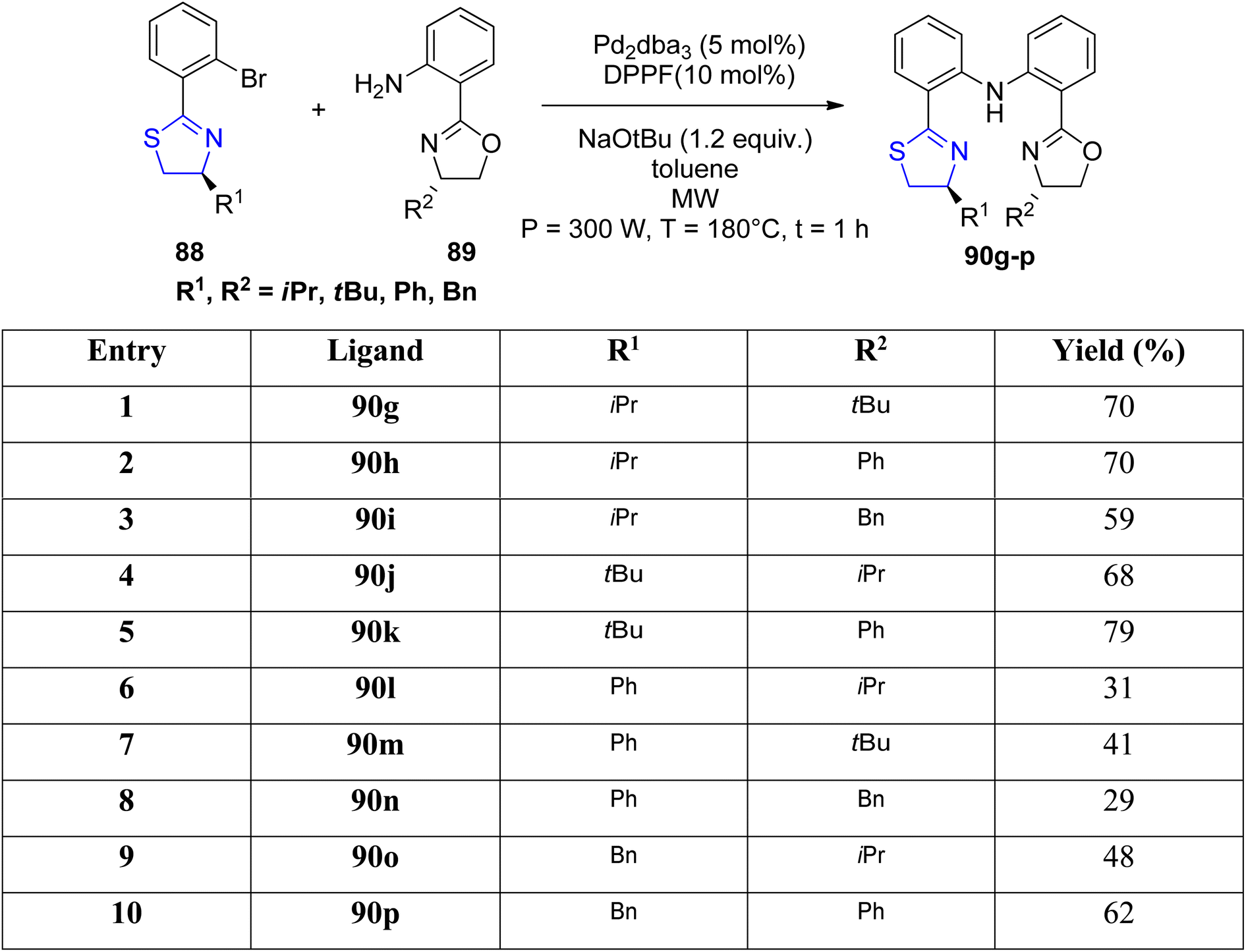 | ||
| Scheme 24 Synthesis of ligand class 90g–p. | ||
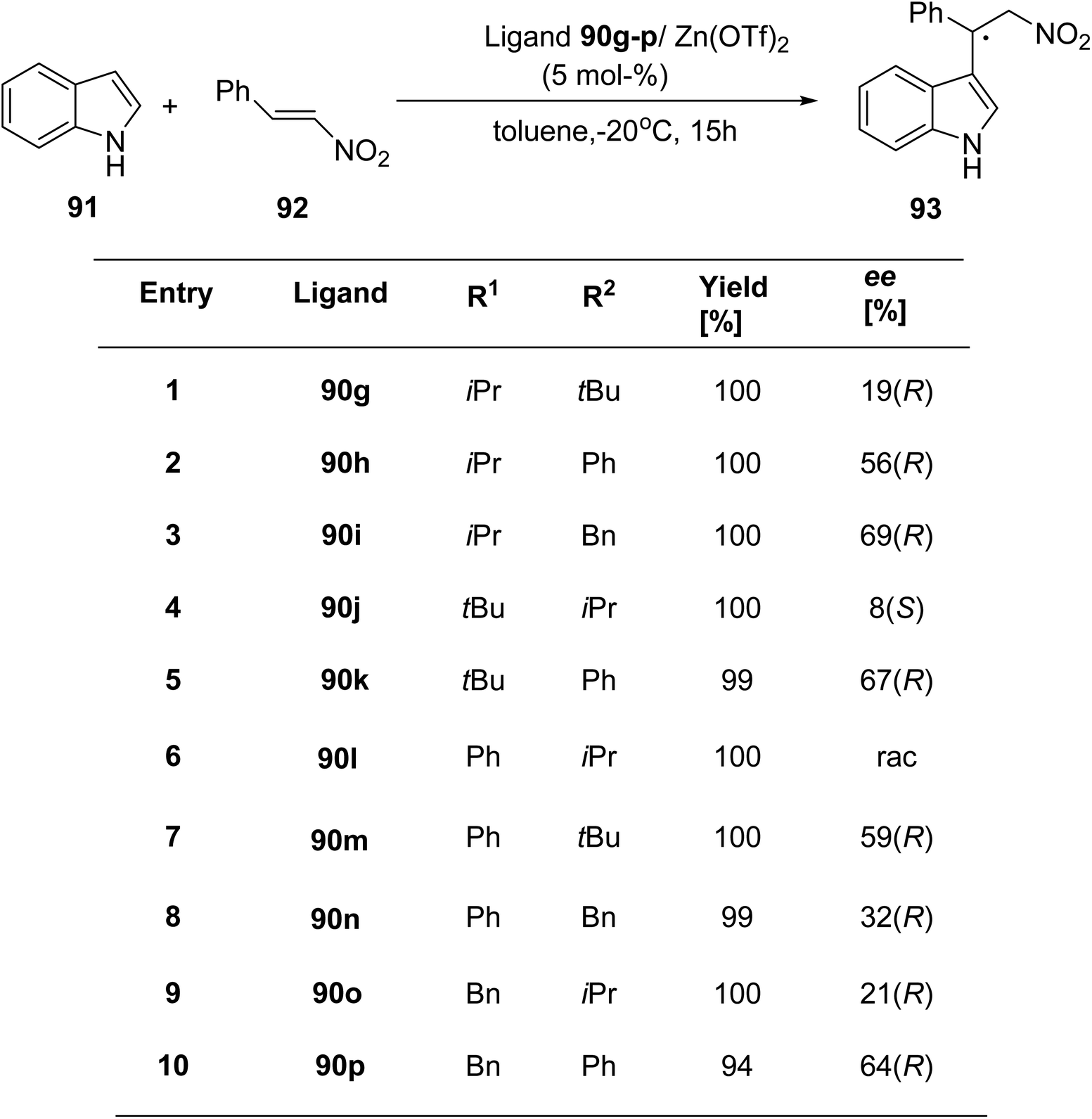 | ||
| Scheme 25 Zinc catalyzed Friedel–Crafts alkylation reaction. | ||
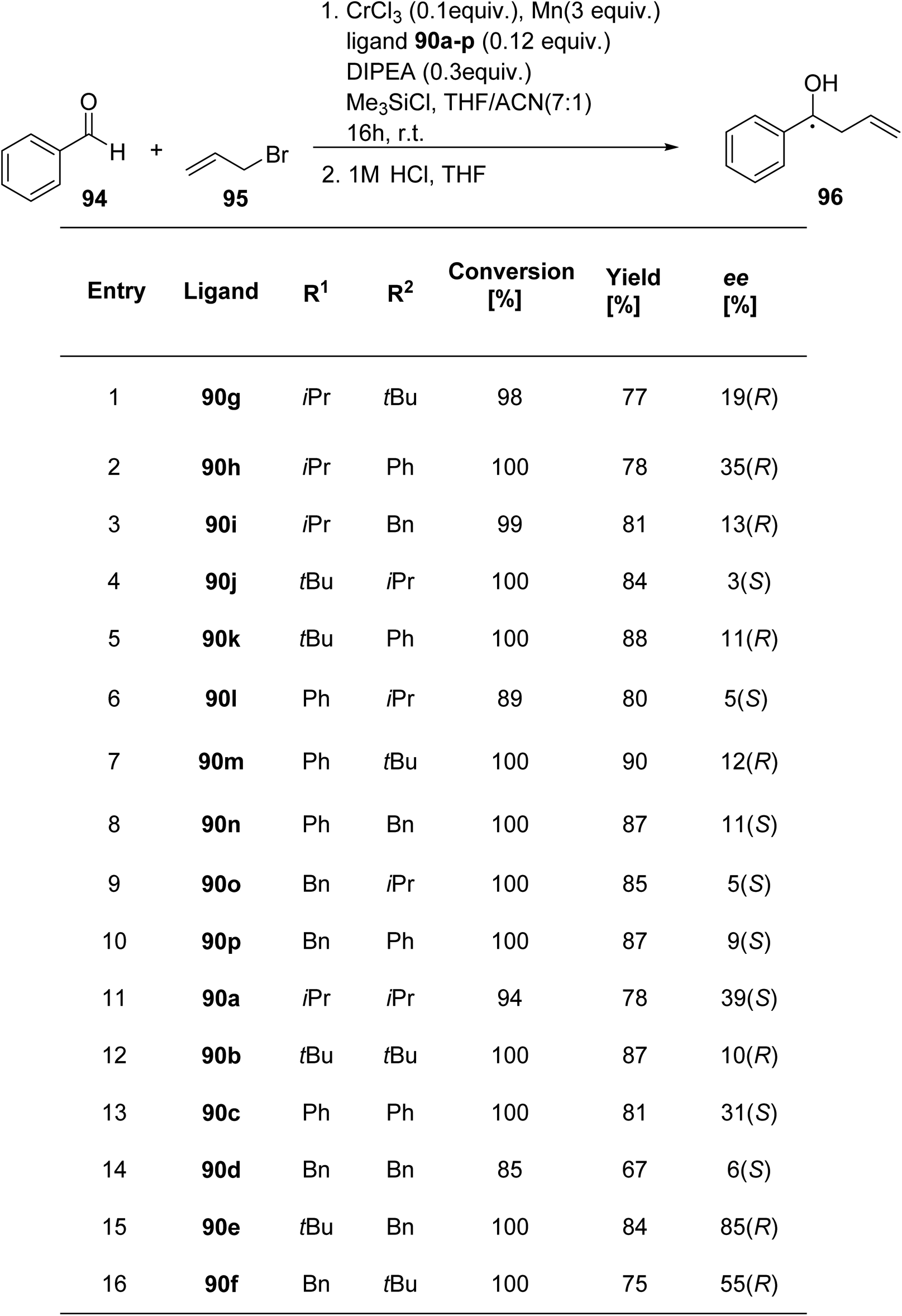 | ||
| Scheme 26 NHK allylation reaction with benzaldehyde 94. | ||
Liu et al.90 reported the synthesis of four new ligand analogues 99a–d as part of an effort to improve the enantioselectivity in the Friedel–Crafts reaction, as illustrated in Scheme 27. The primary focus of the research was to investigate the asymmetric Friedel–Crafts alkylation of indole derivatives 91 and pyrrole 100 with trans-β-nitrostyrene 92, as shown in Scheme 28a and b. The reason behind the observed enantioselectivity was attributed to the NH–π interaction between the catalyst and the incoming aromatic system in the transition state. This interaction was confirmed by comparing the enantioselectivity and the absolute configuration of the products in reactions catalysed by the specially designed ligands.
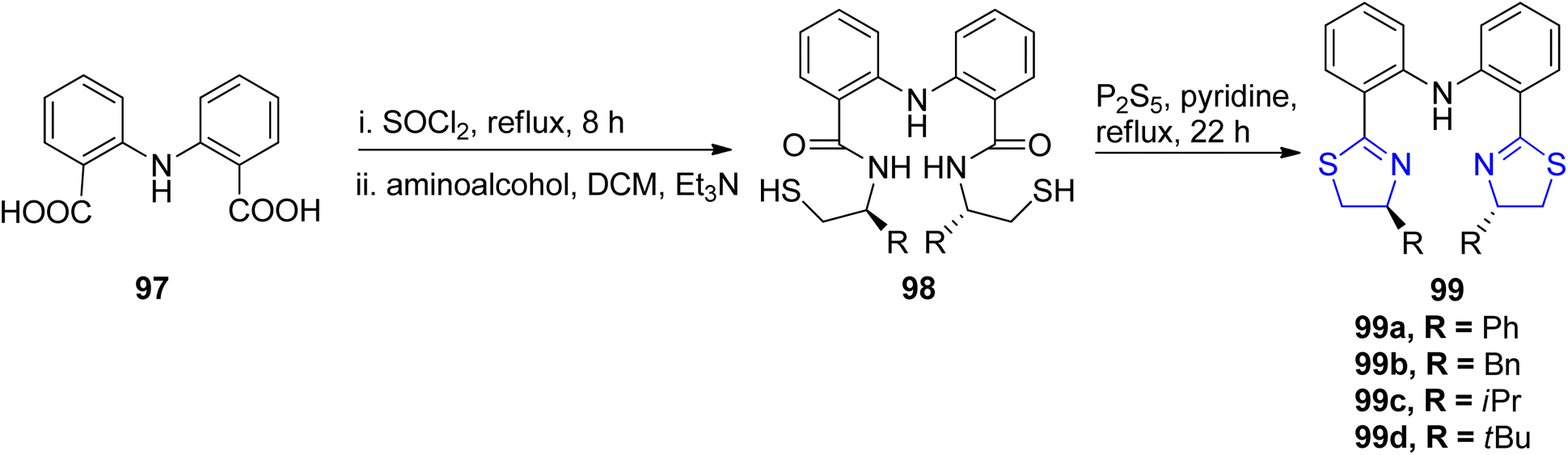 | ||
| Scheme 27 Diphenylamine-tethered bis(thiazoline) ligands 99a–d. | ||
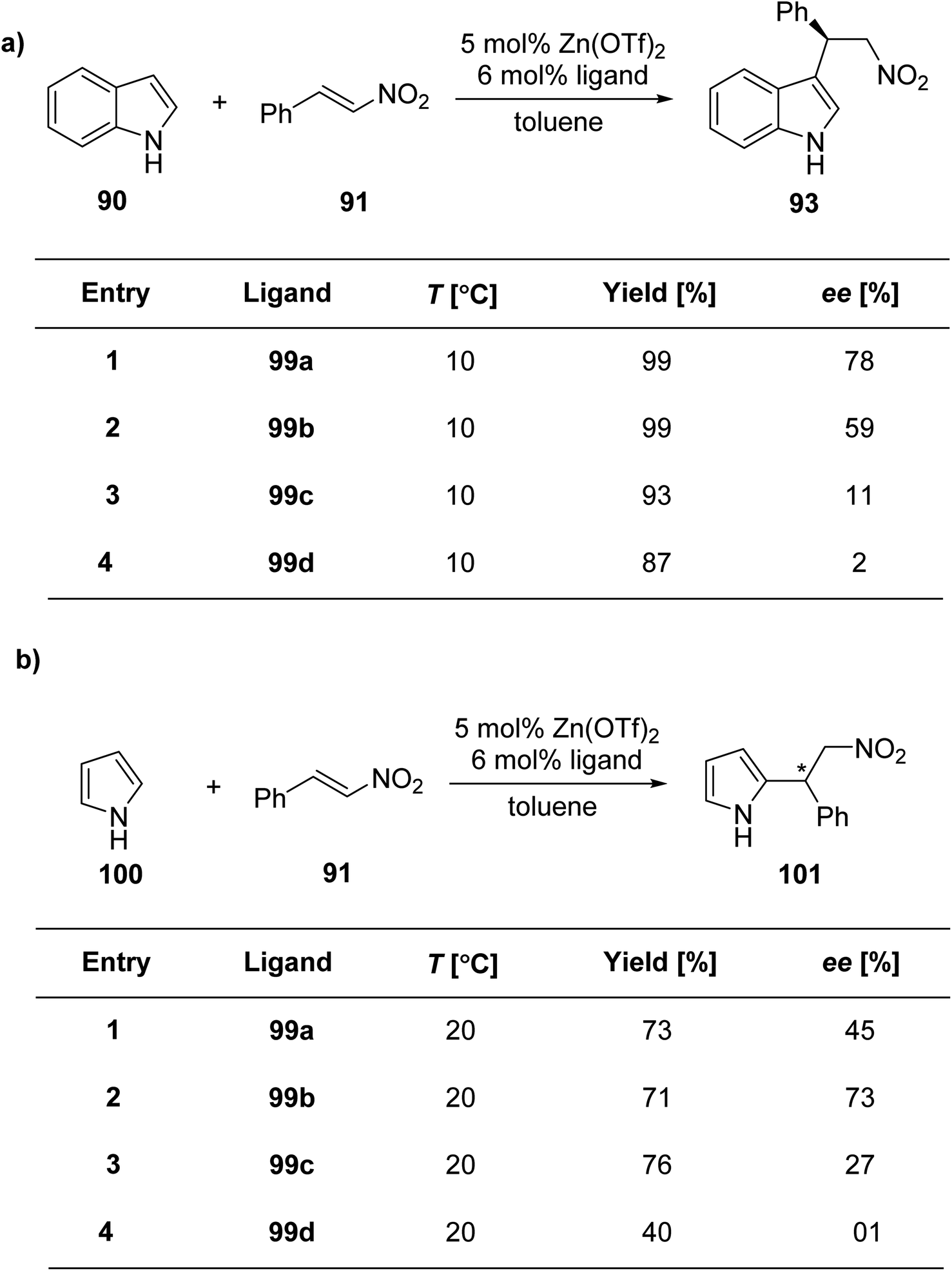 | ||
| Scheme 28 (a and b) Asymmetric Friedel–Crafts alkylation. | ||
Abrunhosa-Thomas et al.91 reported a series of thiazoline ligands substituted with a sulfinyl or sulfanyl moiety. The ligands were assessed for their catalytic properties in an allylic substitution reaction catalysed by palladium. Using t-BuLi, deprotonation was performed at the α-position followed by addition of diphenyl disulfide. Thus, the enantiopure 2-isopropyl-thiazolines 102a–c,92 were transformed into their corresponding sulfanyl-thiazolines 103a–c (Scheme 29a). Similar to this, thiazoline (R)-102a as well as the (R)/(S) tert-butyl thiosulfinate were used to obtain α-sulfinyl-thiazolines (R, Rs)-104a as well as (R, Ss)-104b as a diastereomeric mixture (Scheme 29a).93 α,β-unsaturated thiazolines 105 when treated with thiol, gave β-sulfanyl-thiazolines 106 (Scheme 29b). Starting with commercially available (S)-methioninol 108 and phenyldithioic methyl ester 107,94 compound 110 was synthesized in 2 steps; thioacylation followed by intramolecular cyclization (Scheme 29c). Sulfanyl-thiazoline polymer 114a–b was synthesized as the first immobilized version of the ligand from vinyl benzene 111 (Scheme 29d). When analyzed for their ability to asymmetrically induce the allylic substitution reaction catalysed by palladium, none of the newly synthesized ligands produced a significant excess of the desired enantiomer. Following 24 hours, the thiazoline polymer P-114a produced complete conversion (similar to the monomeric homologue 110), but with reduced ee (36% as compared to 66% ee) (Scheme 30).
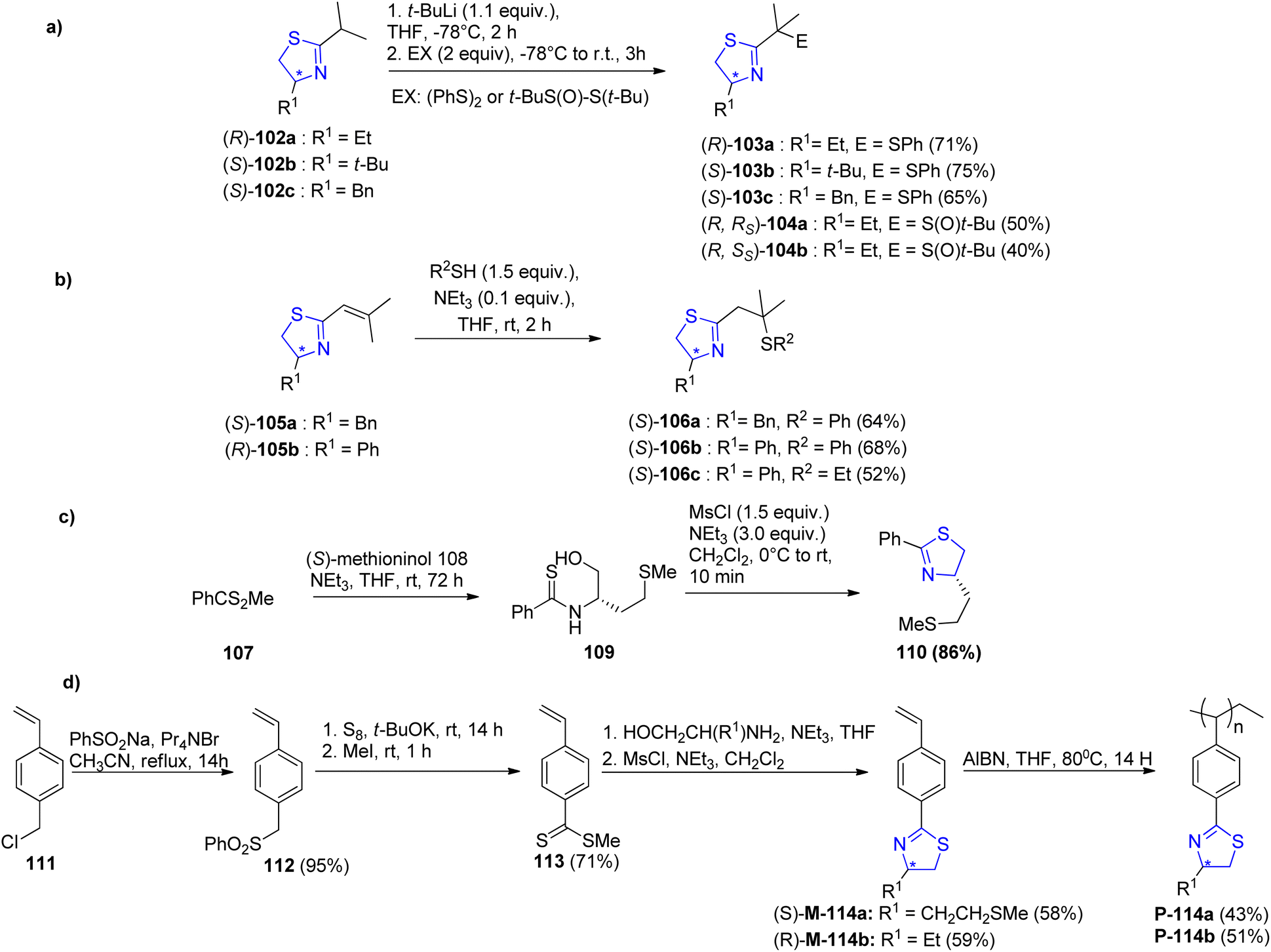 | ||
| Scheme 29 (a–d) Synthetic scheme for chiral thiazoline ligands. | ||
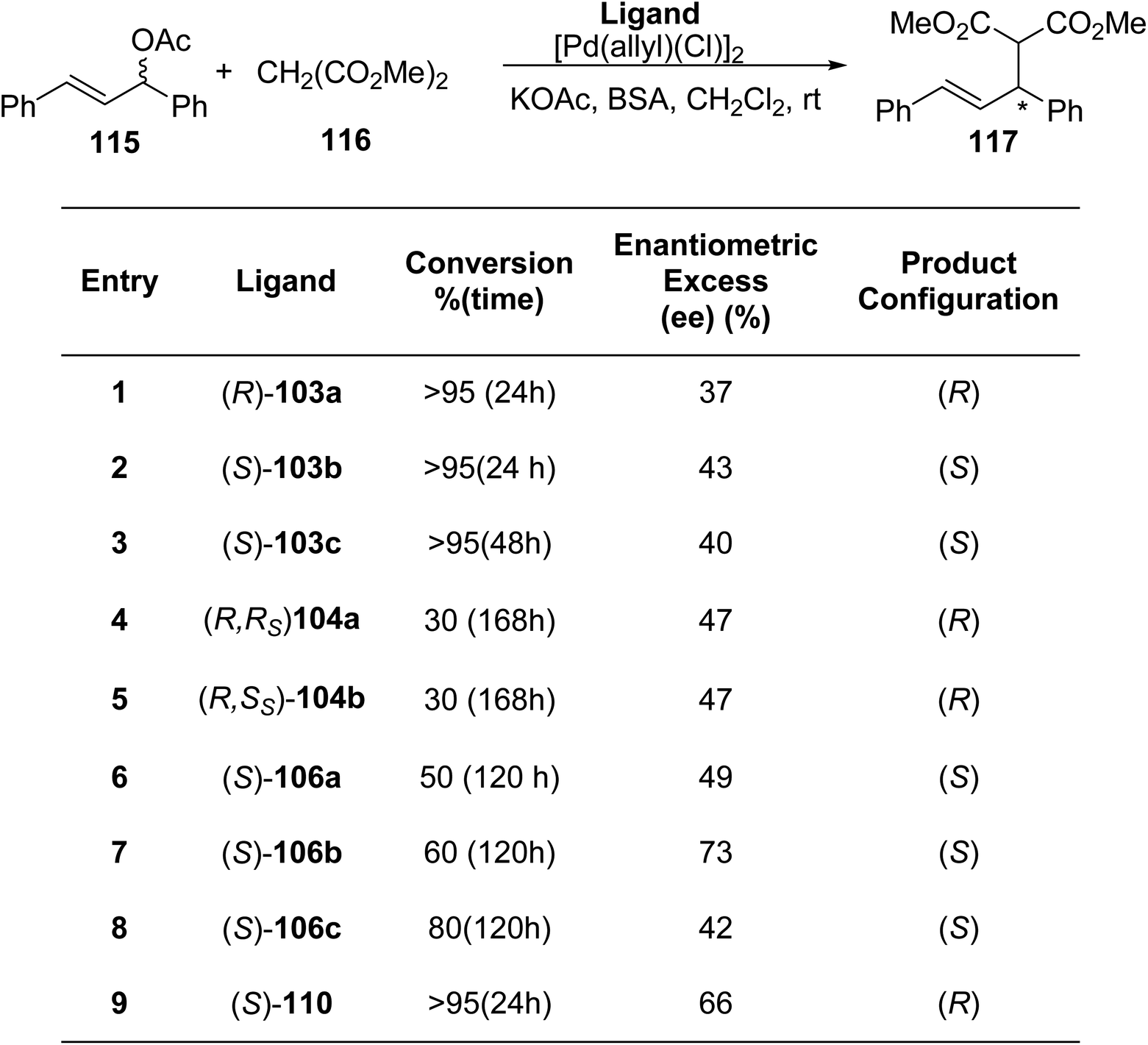 | ||
| Scheme 30 Asymmetrically induced allylic substitution reaction catalysed by palladium. | ||
3.3. Pharmacological
Numerous thiazoline derivatives exhibit anticancer properties, encompassing a range of molecular characteristics and biological diversity. Oligothiazoline compounds, sourced from nature, include marine compounds like tantazole B, mirabazole, and thiangazole, which demonstrate selective toxicity against murine solid tumors.98,99 Oligomers based on the 2-thiazoline moiety are cytotoxic to the cell lines HCT-116 (colon cancer), HPAC (pancreatic cancer) and PC-3 (prostate cancer).100 Additionally, artificial thiazoline derivatives with antiproliferative action have been thoroughly researched and described in literature.101–104 Ability to induce apoptosis and impede cell division105 have been identified as anticancer action mechanisms.
The structurally distinct diastereomers, ulbactin F 118 as well as ulbactin G 119, with a tricyclic ring structure including nitrogen and sulfur, were identified by Igarashi et al.106 from sponge-derived Brevibacillus sp. With the use of X-ray crystallography and NMR measurements, compounds 118 and 119 were structurally characterized (Scheme 31). These substances in micromolar quantities prevent tumor cells from metastasizing. Compounds 118 (IC50 = 6.4 μM) and 119 (IC50 = 6.1 μM) display non-cytotoxic inhibition of the metastasis of the A431 cancer cells. Isomer 118 also prevents cell invasion of 26-L5 cells (IC50 = 1.7 μM) as well as the metastasis of the EC109 cancer cells (IC50 = 2.1 μM). Thus, it displays anti-metastatic properties.
 | ||
| Scheme 31 Structures of ulbactin F 118 and ulbactin G 119. | ||
Wang et al.107 employed dibromides 122 in the synthesis of a series of novel multi-thioether derivatives of thiazoline 123, as illustrated in Scheme 32. Structural characterization was conducted through spectroscopic analysis, elemental analysis, and infrared (IR) measurements. Furthermore, the synthesized compounds underwent evaluation for their anti-cancer efficacy. The biological assessment revealed that compound 123g exhibited notably enhanced anti-tumor effects, with IC50 values of 22.58 μg mL−1 for A-549 and 19.41 μg mL−1 for Bcap-37, respectively.
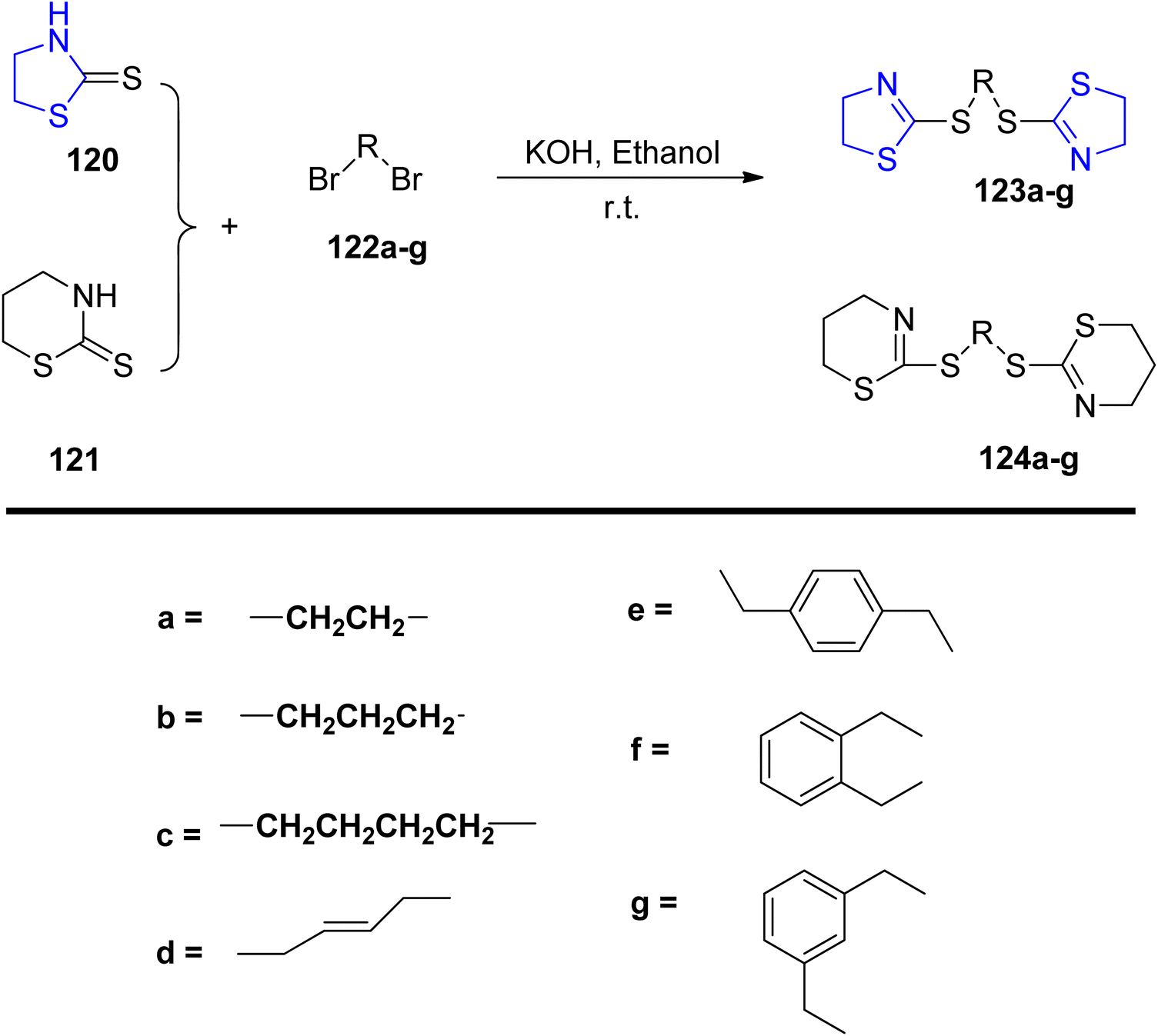 | ||
| Scheme 32 Synthesis of thiazoline and thiazine multi-thioether. | ||
El-Helw et al.108 reported the synthesis of two newly identified active N-heterocycles: the thiazoline derivative 128 and 2-cyano acetohydrazide 127. The N-condensation product, equivalent to 127, was generated through refluxing an ethanolic solution containing carboxaldehyde 125 and hydrazide 126 for a duration of 2 hours, as described in Scheme 33. Subsequently, compound 127 was subjected to an initial treatment with Et3N in dioxane as the solvent, followed by a reaction with phenyl isothiocyanate in the presence of elemental sulfur, yielding compounds 128 and 129 as the final products. The in vitro anticancer properties of the synthesized compounds were assessed in two distinct cancer cell lines, MCF7 and HepG2. Remarkably, compound 127 demonstrated the highest efficacy against both cell lines.
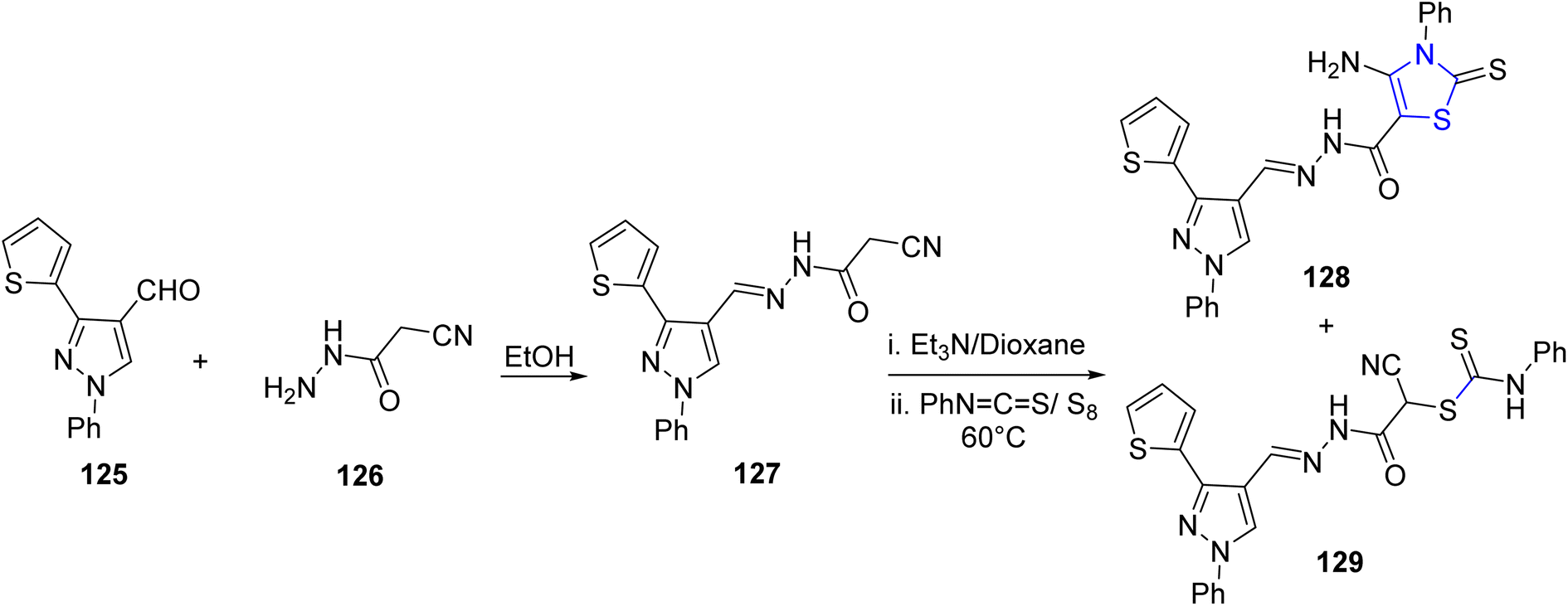 | ||
| Scheme 33 Synthesis of thiazoline derivative 128. | ||
Turan-Zitouni et al.109 established a hydrazine bridge between the thiazoline and the tetralin rings. Tetrahydro naphthol 130 on treatment with 2-chloro acetate gave intermediate 131. Further, acetohydrazide 132 was synthesized from this intermediate.110 The acetohydrazide 132 thus obtained was treated with ethanolic solution of cyclohexyl as well as phenyl isothiocyanate to give carbothioamide 133 according to the literature report.111 The final acetohydrazides 134a–k were synthesized by reacting carbothioamide 133 with phenacyl bromdie (Scheme 34). The anticancer potency of 134a–k derivatives was assessed on the MCF-7, NIH/3T3 & the A549 cancer cell lines by following the MTT method, studying the inhibition of DNA synthesis as well as analysis of flow cytometry. Compound 134e consisting of a 4-methoxyphenyl moiety displayed excellent anti-cancer efficacy against the MCF-7 cell line with enhanced apoptotic cell percentage as well as improved inhibition of DNA synthesis. Compounds 134f (4-bromo), 134g (4-chloro) and 134h (4-fluorophenyl) exhibited significant apoptotic levels in A549 cancer cell line with concentrations lower than that of cisplatin. While testing the compound's anti-cholinesterase activity, it was revealed that compound 134h inhibited acetylcholinesterase (AChE) by 49.92%.
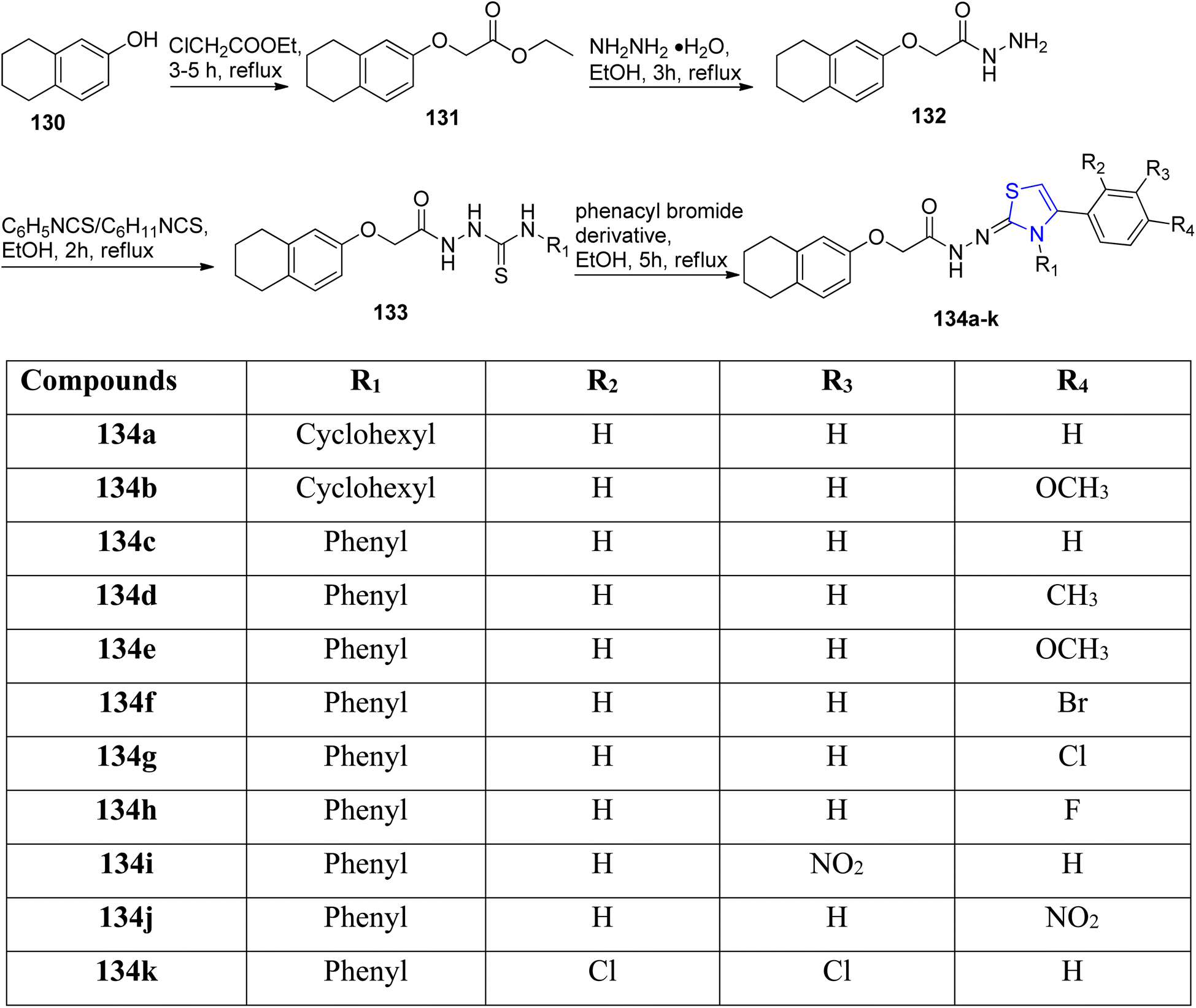 | ||
| Scheme 34 Synthesis of the compounds 134a–k. | ||
Kelly's synthetic method for thiazoline preparation112 was utilized in the process of total synthesis of the new anticancer natural product cyclic depsipeptide largazole, which was discovered in 2008.113 A good yield of thiazoline esters 136a–b were produced by treating amides 135a–b with triphenylphosphine oxide and trifluoroanhydride (Scheme 35). In refluxing toluene, the double dehydrative cyclization of the tripeptide 137 resulted in the formation of (bis)thiazoline 138, which was easily oxidized to 135a (Scheme 36a). Using this methodology, Numajiri et al.114 achieved the total synthesis of largazole 139 and its derivative 140–142 (Scheme 36b). The depsipeptide largazole 139, obtained from cyanobacterium of Symploca genus, exhibits excellent anti-cancer efficacy.115 Strong inhibitory effect against histone deacetylases was observed in biological testing of the synthetic largazole (HDAC) as well as the S-modified analogs.
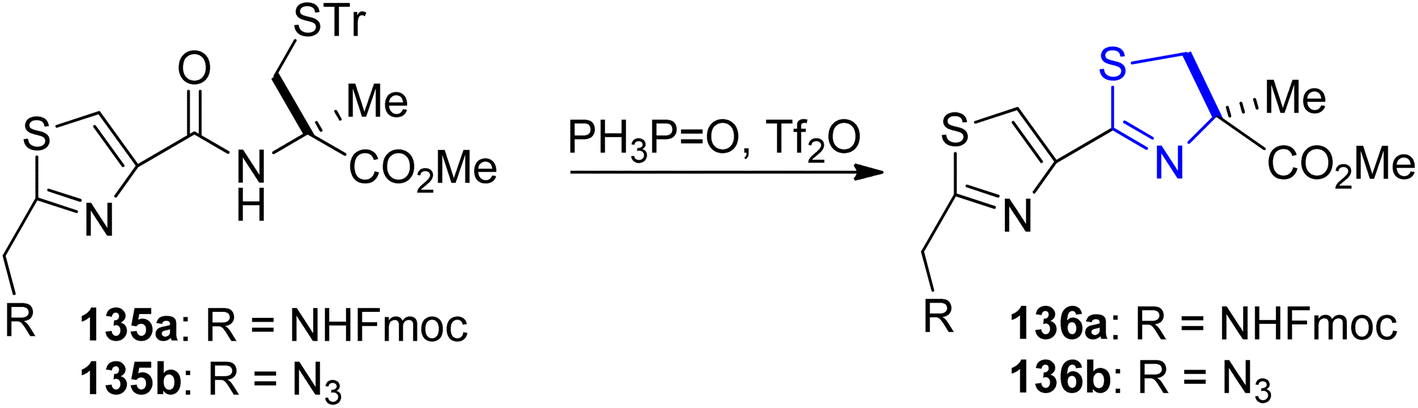 | ||
| Scheme 35 Synthesis of thiazoline esters 136a–b. | ||
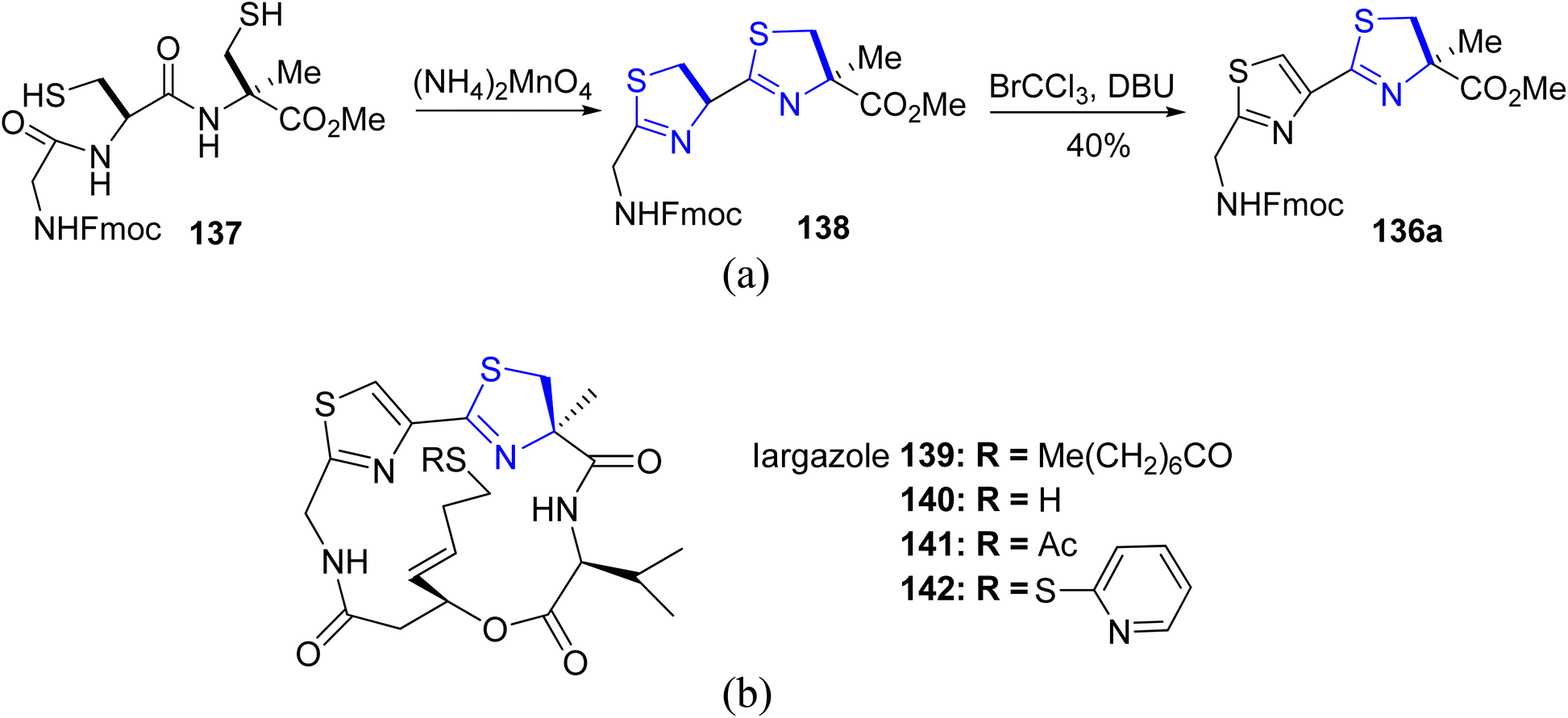 | ||
| Scheme 36 (a) Synthesis of thiazoline esters 136a. (b) Synthetic largazole 139 and its derivatives 140–142. | ||
As a potential alternative for cancer therapeutics, largazole, which was first discovered by Luesch.33,115 Guerra-Bubb et al.116 reported the synthesis of an analogue of largazole 139. Beginning with the well-known oxazole 143, the thiazoline–oxazole fragment 144 was obtained (Scheme 37a).117 Acrolein 145 was used to formulate the heptenoic acid fragment 148 (Scheme 37a). The first step in the macrocycle construction involved synthesis of peptide 149 by treatment of acid 148 with N-Fmoc-Val-OH in presence of EDCI (Scheme 37a).118 The required substrate 150 was obtained in two phases of deprotection, coupling with PyBOP and Hunigs base with a yield of 91%. T3P and Hunigs base were used to produce the required macrocycle 151 in 30% yield after conversion of compound 150 to amino acid in a one-pot reaction. Using TFA and iPr3SiH8 in degassed dichloromethane, the macrocycle 151 was detritylated to give disulfide 152 instead of the anticipated thiol (Scheme 37b). Interestingly, it was found that the required thiol 153 was prepared when the trityl residue of 151 was removed with Et3SiH rather than iPr3SiH in degassed dichloromethane. This thiol was then instantly acylated to produce the desired octanoyl-thioester 154. Using a previously established optimized homogeneous assay carried out on a 384-well plate, compounds 152–154 were examined for their inhibitory efficacy against HDACs 1–9.119 According to the findings of these studies, compound 153 was found to be more active and potent in comparison to the largazole. However, it is less potent than largazole thiol. 153 was substantially less active than in the biochemical model with an IC50 value of 6.2 μM, which is probably because the free thiol gets degraded in the cellular environment. Similar to largazole, the prodrugs 152 and 154 showed IC50 values of 0.91 & 0.12 μM, respectively.
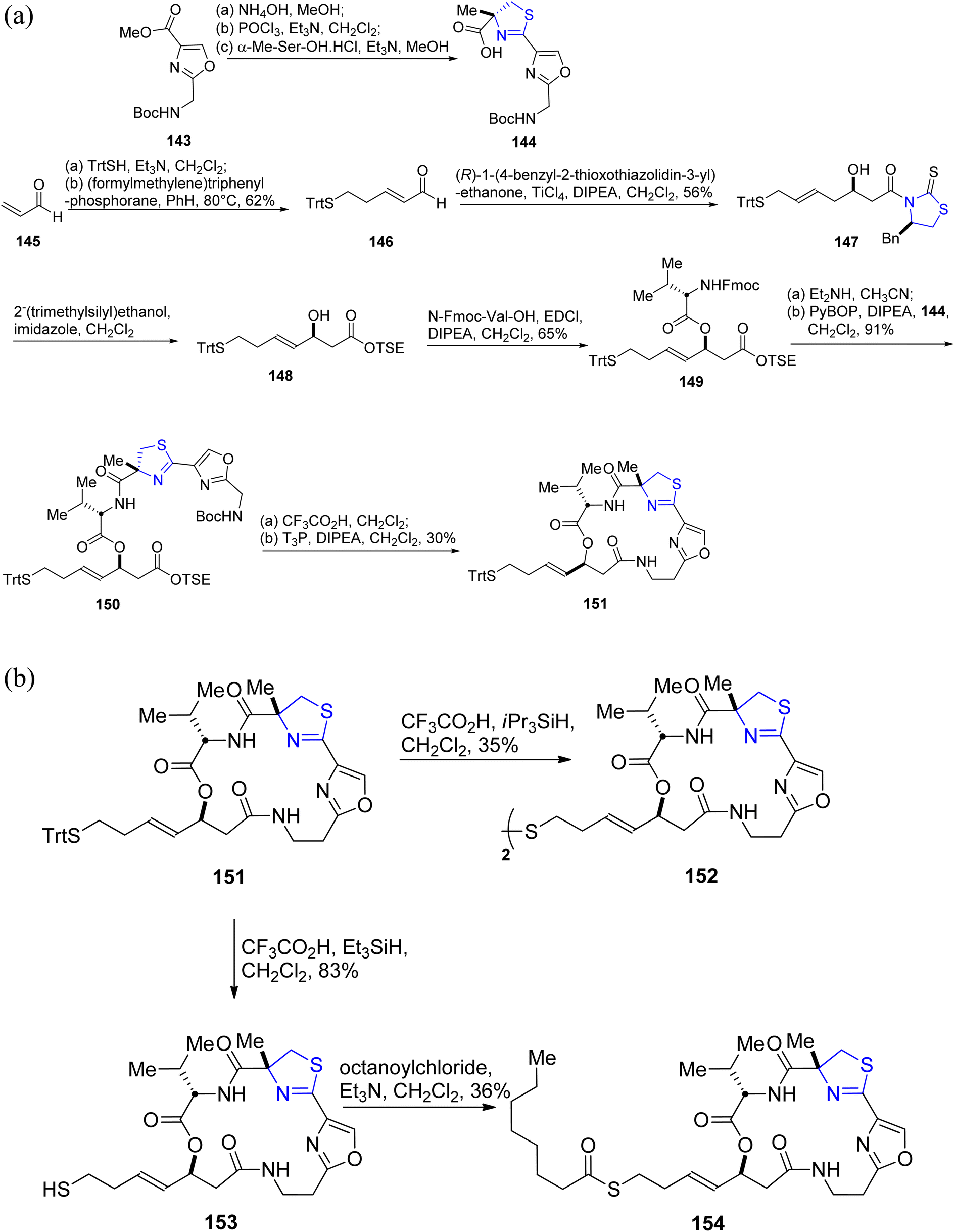 | ||
| Scheme 37 (a) Synthetic scheme for largazole analogue 151. (b) Synthesis of largazole analogues 152–154. | ||
Taher et al.120 conducted a condensation reaction involving Mannich bases 156a–h and thiazoline 157, resulting in the synthesis of a series of novel isatin-thiazoline derivatives denoted as 158a–h, as outlined in Scheme 38. These newly synthesized compounds underwent comprehensive characterization through spectroscopic analysis. Notably, all the prepared compounds exhibited efficacy against MCF-7.
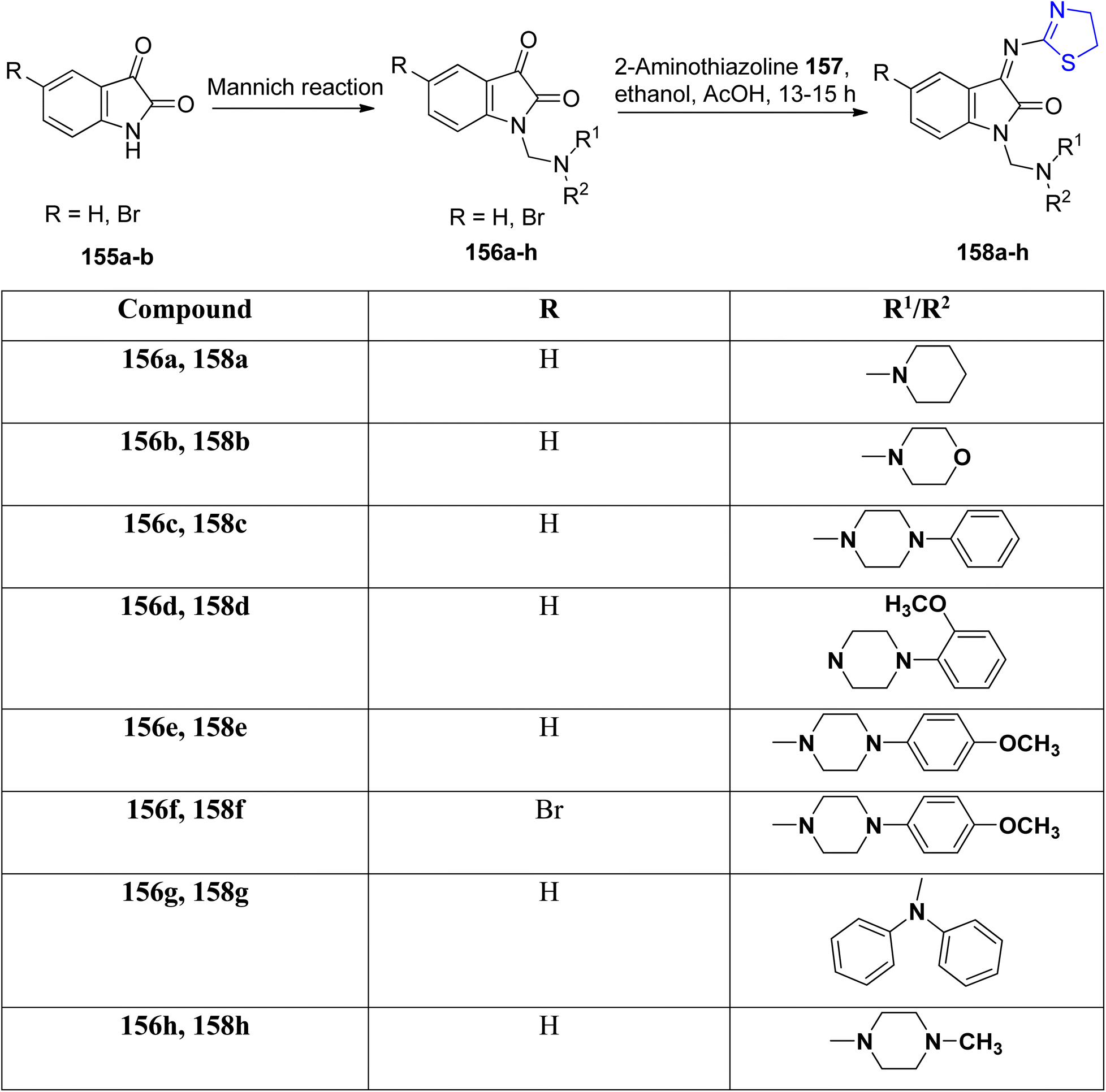 | ||
| Scheme 38 Synthesis of novel isatin–thiazoline 158a–h. | ||
One of the most commonly used chemotherapy drugs in the treatment of various malignancies is cisplatin,121 but it is associated with a high level of toxicity and adverse effects.122 New chemotherapeutic drugs must therefore be developed in order to obtain less harmful and efficient drugs for cancer treatment. A Pd(II) complex, PdPyTT 162 was synthesized and described by Espino et al.123 using the ligand PyTT 161 (Scheme 39). The ligand was prepared according to the reported methodology,124 with a few minor adjustments as per published literature.125 Additionally, its cytotoxicity and pro-apoptotic potential were examined in the HL-60 cancer cell line. The Pd complex, PdPyTT 162 also reduced cell viability in a time- & dose-dependent manner, similar to cisplatin. In addition, the palladium compound boosted caspase-3 and caspase-9 activation and the percentage of cells exhibiting apoptotic morphology. Also, PdPyTT enhanced DNA oxidative damage and intracellular ROS generation, identical with that of cisplatin.
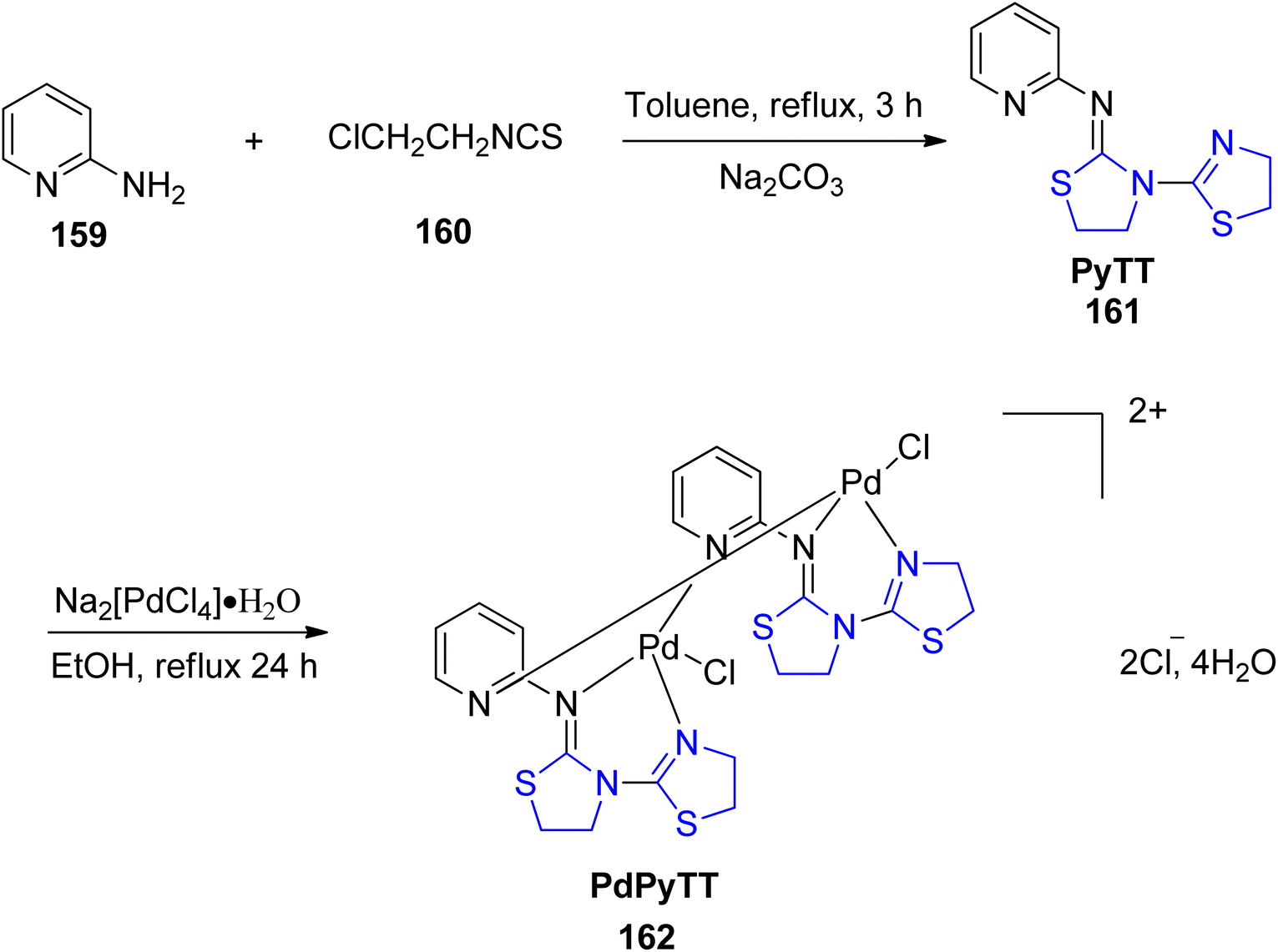 | ||
| Scheme 39 Synthesis of thiazoline-based palladium(II) complex 162. | ||
Kwan et al.126 identified a series of cyclic depsipeptides, specifically compounds 163–165, featuring bis-thiazoline moieties as constituents, within the Lyngbya confervoides extract, as depicted in Scheme 40. Compound 165 exhibited enhanced metal affinity, potentially contributing to its increased potency. Structure–activity relationships among these analogues were evident in their in vitro cytotoxic effects. While the activity experienced only a modest decrease (3–4 fold) upon replacing the ethyl group in compound 163 with a methyl group in compound 164, a notable enhancement in potency (16–23 fold) was observed when the phenyl moiety flanking the thiazoline molecule was inverted. Both compounds 163 and 165 demonstrated binding to Cu2+ and Zn2+ metal ions, with the capacity to arrest the cell cycle at the G1 phase at lower doses and at the G2/M phases at higher concentrations.
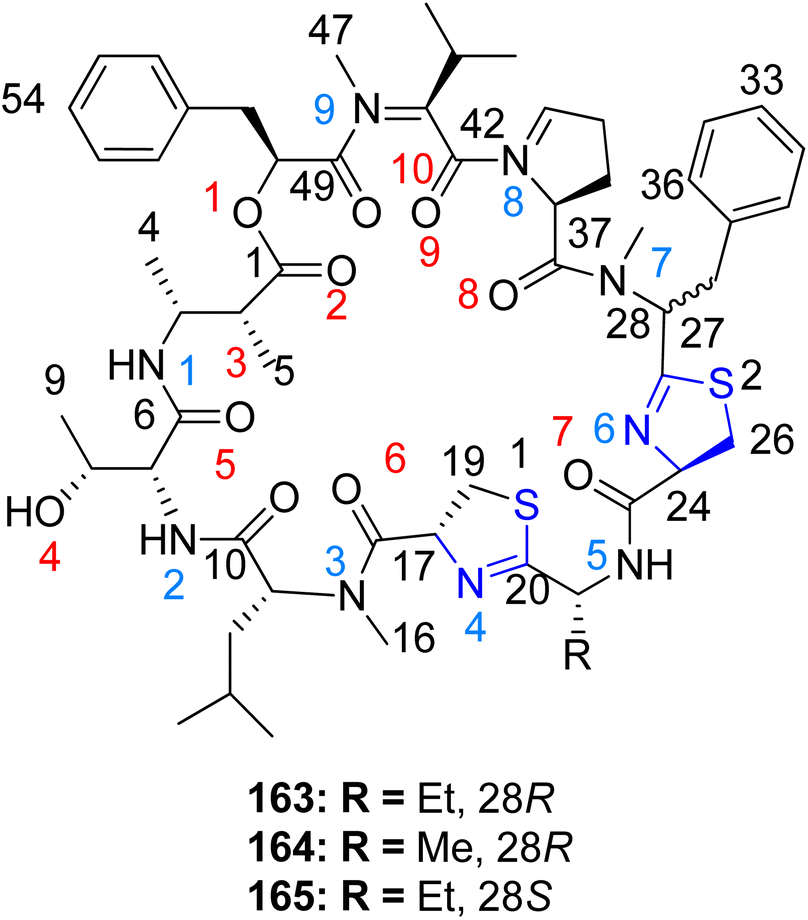 | ||
| Scheme 40 Grassypeptolides 163–165, the cyclic depsipeptides. | ||
Altıntop et al.127 synthesized acetohydrazide compounds 179–218, which were subsequently evaluated for their antibacterial properties and cytotoxic effects against NIH/3T3 cells. Thiol compounds 166a–e were combined with potassium carbonate to obtain thioacetate derivatives 167a–e. The 2-[(aryl)thio]acetohydrazides were then synthesized by treating ester derivatives 167a–e with hydrazine hydrate 168a–e. These hydrazides 168a–e were further transformed into 1-(arylthioacetyl)-4-phenyl thiosemicarbazides 170a–e by reacting them with phenyl isothiocyanate 169. The final target compounds 179–218 were produced by ring-closure through treatment of thiosemicarbazides 170a–e with various analogues of 2-bromoacetophenones 171–178 (Scheme 41). Among the compounds tested, compound 200 exhibited the highest antibacterial activity against Pseudomonas aeruginosa, while compound 201 displayed the most potent antifungal activity against Candida albicans. In terms of cytotoxicity against C6 glioma cells, compound 195 emerged as the most effective with an IC50 value of 8.3 ± 2.6 μg mL−1, surpassing cisplatin (IC50 range: 13.7 ± 1.2 μg mL−1). Compound 195 also demonstrated DNA synthesis inhibition on C6 cells and exhibited lower toxicity to NIH/3T3 cells with an IC50 value in the range of 416.7 ± 28.9 μg mL−1.
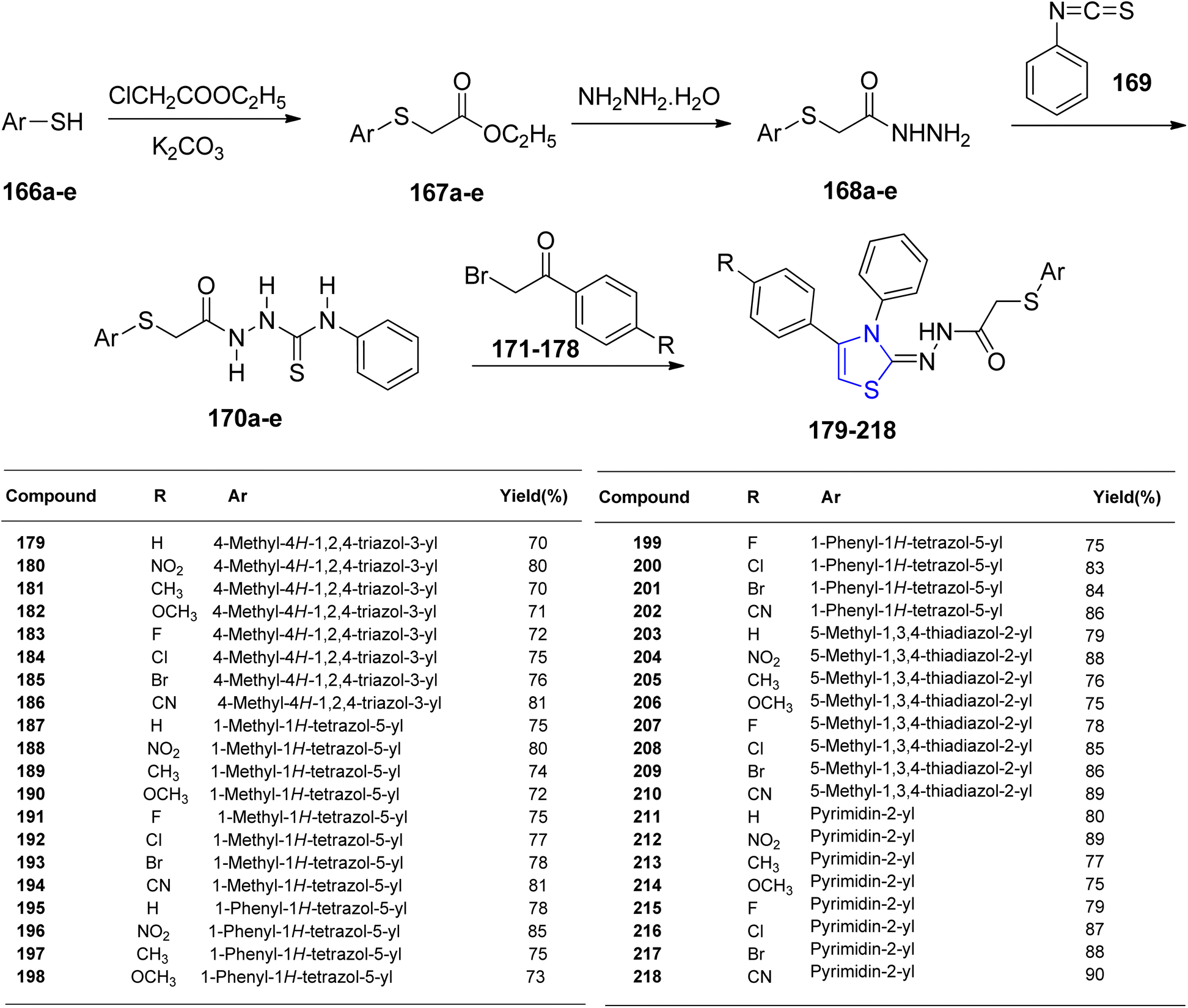 | ||
| Scheme 41 Synthetic scheme for thiazoline analogues 179–218. | ||
Mabkhot et al.128 successfully synthesized a series of novel thiazoline compounds. The reaction sequence for the synthesis of thiazolines 221a–e involved treatment of dione 219a/b with 1° amines 220a–c at room temperature in ethanolic media. Compound 221a resulted in the formation of thiazolines 223a–e when refluxed with the suitable aniline derivatives (Scheme 42). Next, the reaction of the thiazoline-2-thione derivative 221a with 2-oxo-N′-phenylpropane hydrazonoyl chloride 224 produced the spiro-compound 225 (Scheme 42). The compounds were assessed for their anti-tumor properties against HepG2 as well as HCT-116 cancer cell lines. The outcomes showed that the thiazoline compounds, 223b and 221c, had a considerable effect on the two cell lines. The inhibitory activity of thiazolines 221c, 223b, and 223d against Salmonella sp. was found to be promising as determined by anti-microbial screening. Additionally, the reference compound gentamycin and the thiazolines 221e and 223b were found to have equivalent inhibitory activity against Escherichia coli.
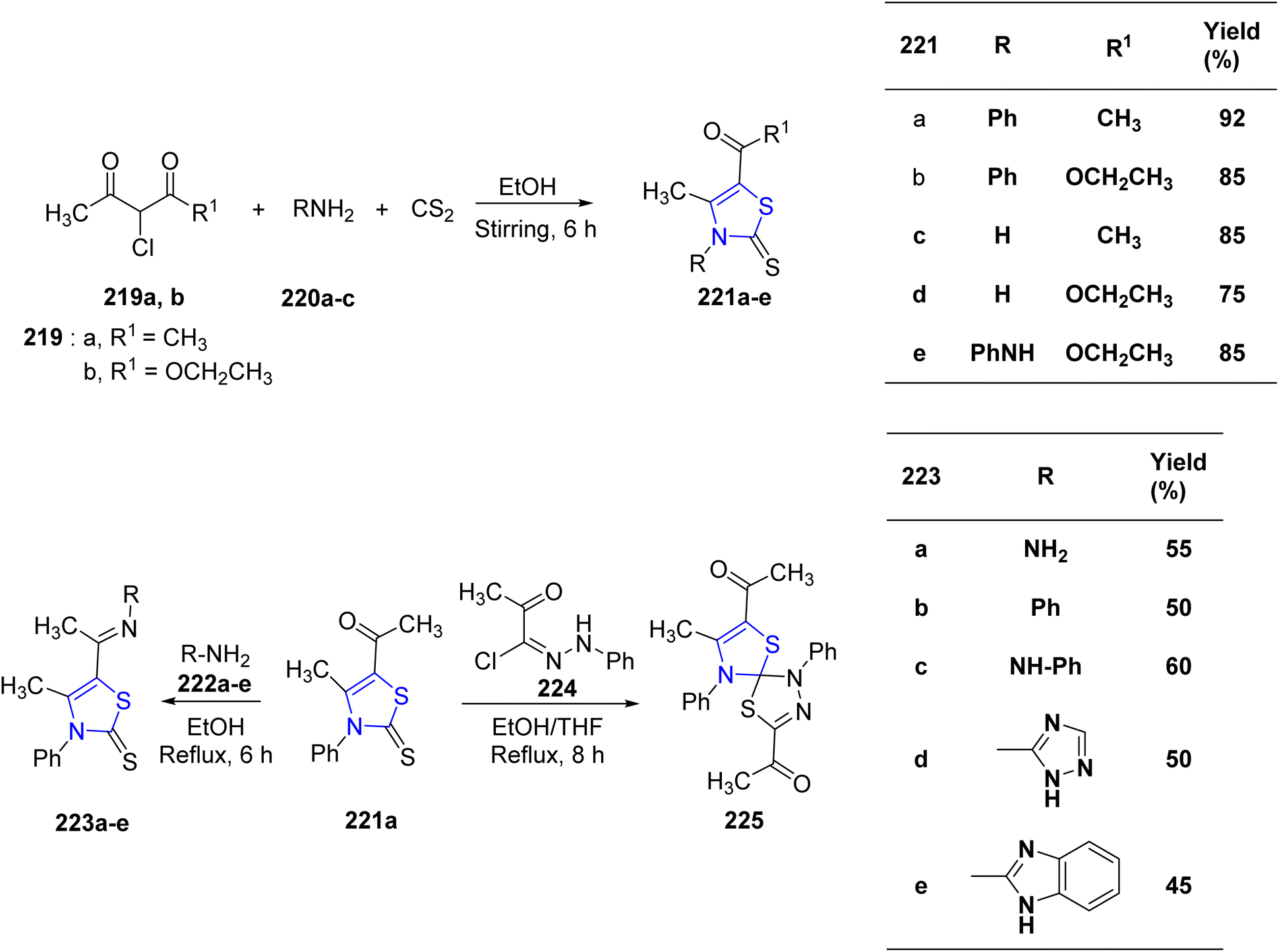 | ||
| Scheme 42 Synthesis of new thiazoline derivatives. | ||
Recent research has shown that multitargeting kinase inhibitors is an efficient strategy for restricting cancer growth. Thiazoline-based derivatives 229a/b were developed and synthesized by Alamshany et al.129 Initially, the intermediate 227 was prepared by treatment of p-toluidine 226 in ethanolic media with phenacyl bromide 177.130 Further, the target thiazolines 229a/b were synthesized by a condensation reaction between intermediate 227 and isothiocyanates 228a/b in a refluxing ethanolic solution containing Et3N as the catalyst (Scheme 43). Compound 227 was the only effective one against a broad range of bacteria (Gram-positive as well as Gram-negative) & fungi. Additionally, in vitro analysis was also carried out against HepG-2, HCT-116 & MCF-7 cancer cell lines.
 | ||
| Scheme 43 Synthesis of thiazoline-based derivatives 229a–b. | ||
000 people.131 Due to the capacity of microorganisms to develop resistance to treatments, bacterial and fungal infections have emerged as significant contributors to global morbidity and mortality.132 Microbe's resilience in harsh environments pose challenges for finding effective treatments. The demand for potent antimicrobial drugs that can inhibit or eradicate pathogens without harming host cells has increased due to the sharp rise in life-threatening bacterial and fungal infections.133,134 In light of the rapid emergence of drug resistance, the development of novel antimicrobial agents necessitates distinctive chemical characteristics compared to currently employed drugs. Heterocyclic compounds featuring nitrogen and sulfur represent well-established chemical moieties present in various natural products and essential medications. A paramount concern in contemporary medicine revolves around the proliferation of antimicrobial resistance, particularly in multi-drug resistant microorganisms. Thiazolines have gained prominence due to their significant synthetic and biological relevance, serving as vital scaffolds. Compounds incorporating the thiazoline moiety have been associated with diverse biological effects, including antimicrobial activity, underscoring their biomedical importance.135–137Asiri et al.138 developed a synthetic methodology to obtain twelve 2-thione analogues of thiazoline (Scheme 44). The compounds thus synthesized were characterized by spectroscopic experiments, IR measurements, X-ray and elemental analysis. Additionally, the biological activities of the compounds were also investigated against various microorganisms and human cancer cell lines. The thiazoline derivatives 233a, 233b, 235a, 235c and 236 demonstrated significant anti-fungal properties when tested against Aspergillus fumigatus, in comparison to the conventional medicines. Additionally, all the thiazoline derivatives were found to be effective against Candida albicans, with the exception of compound 233b. Moreover, the compounds also exhibited anti-bacterial activity against both Gram-positive as well as Gram-negative bacteria. On evaluation of the cytotoxic effects of compounds 232, 235b and 236 against HCT-116 as well as the HepG-2 cancer cell lines, compound 235b was found to be the most efficient with IC50 values of 79 μg mL−1 and 49 μg mL−1, respectively.
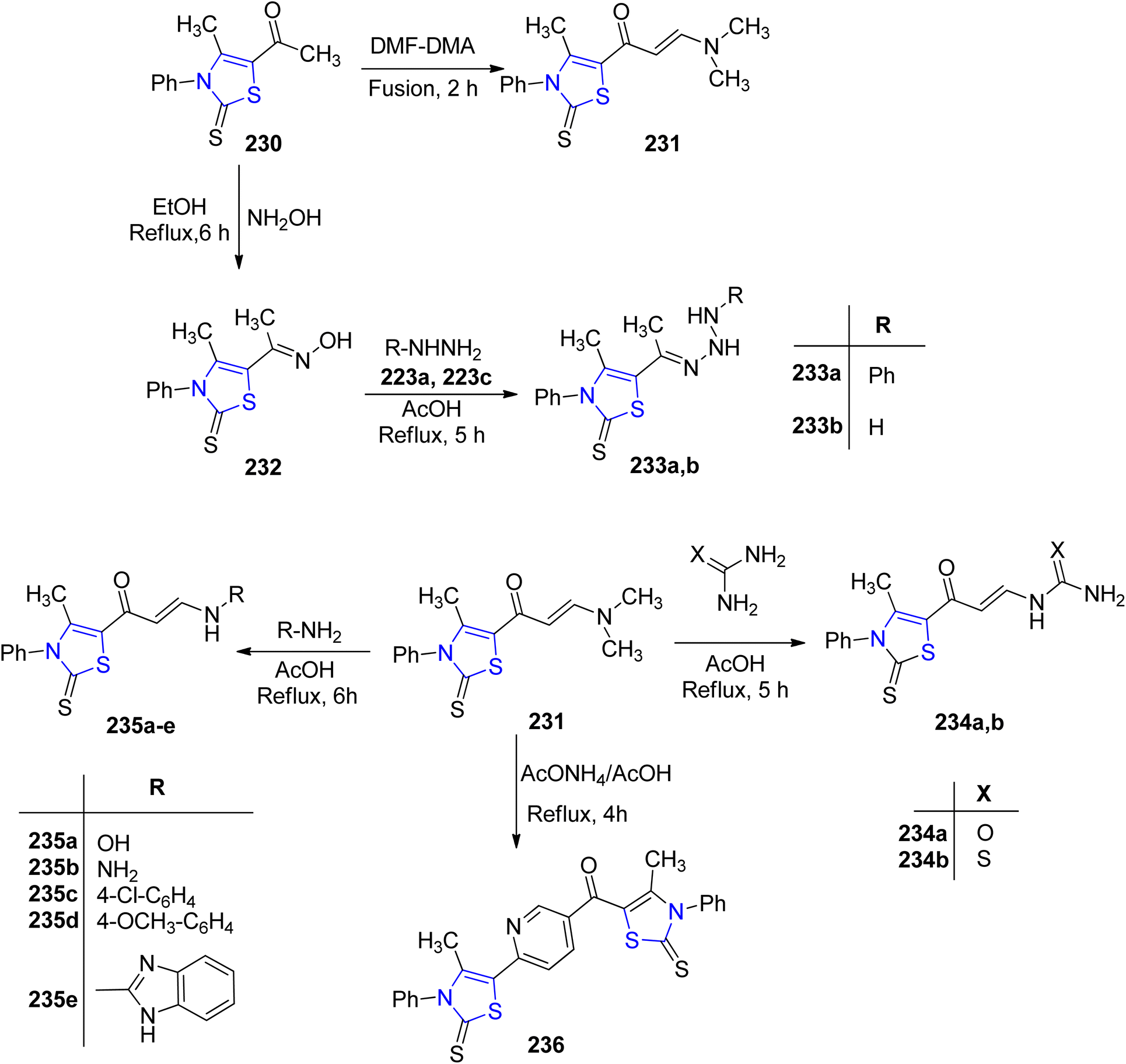 | ||
| Scheme 44 Synthesis of novel thiazoline-2-thione derivatives. | ||
Bondock et al.139 outlined a practical synthetic approach to generate novel thiazoline derivatives intended for antimicrobial evaluation. This synthetic methodology involved the reaction of cyanoacetic acid hydrazide 238 with α-halocarbonyl compound 237, as depicted in Scheme 45. The interaction of the aldehyde functionality in compound 237 with cyanoacetohydrazide led to the formation of compound 239. Subsequently, the key intermediate 240 was derived through reaction with phenyl isothiocyanate 228b. The final cyclized pyrimidinone analogue 241, was synthesized by treating intermediate 240 with a mixture of triethylorthoformate and acetic anhydride. The compounds were further assessed for their antimicrobial properties.
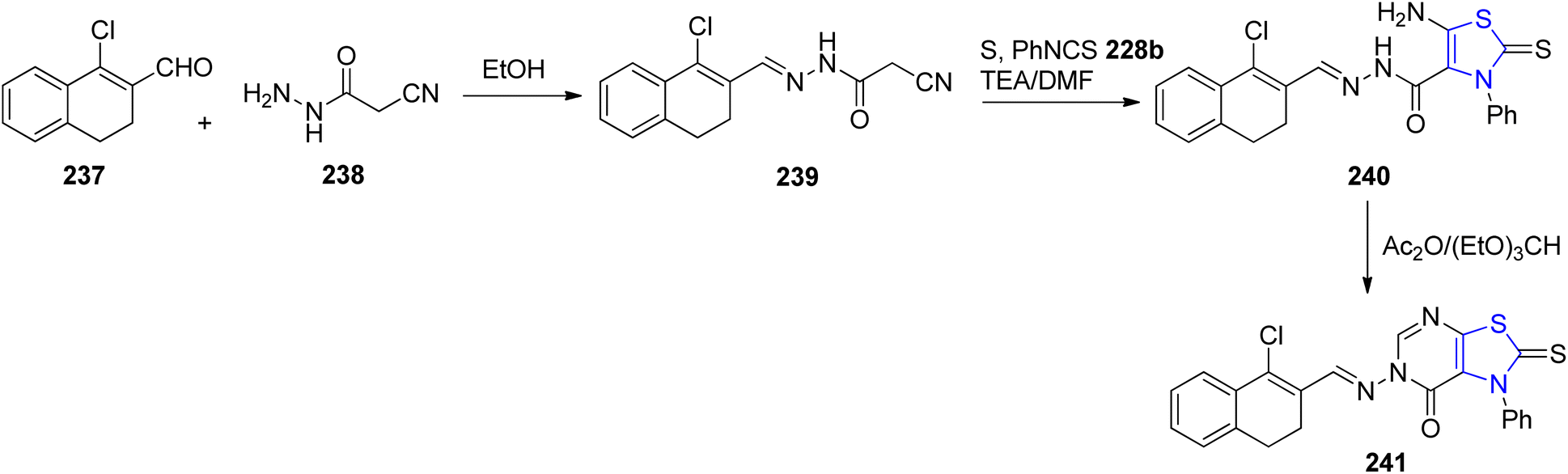 | ||
| Scheme 45 Synthesis of thiazoline 240 and thiazolo[5,4-d]pyrimidinone derivative 241. | ||
Viñuelas-Zahínos et al.140 reported the synthesis of Schiff base ligand 244 via condensation reaction of thiazoline 242 with thiosemicarbazide 243. The ligand thus obtained was complexed with different metal ions (Co, Ni, Zn and Cd) to yield complexes 245, 246, 247, 248, 249 and 250 (Scheme 46). The precursor 242 was synthesized by using the procedure outlined by Doornbos and Peer.141 The anti-bacterial efficacy of the ligand 244 and the complexes 245–250 was also investigated against B. subtilis, S. epidermidis, E. faecalis, E. coli and S. aureus. Antimicrobial studies have demonstrated that cadmium complexes exhibit the most potent antimicrobial activity against various microorganisms. The MIC values for cadmium(II) complexes 249 and 250 against E. faecalis, E. coli, S. epidermidis, and S. aureus were found to be 50, 25, 12.5, and 25 mg mL−1, respectively. In these instances, the antibacterial activity is enhanced compared to both the free HATtsc ligand and cadmium(II) salts. Concerning B. subtilis, the activity of complexes 249 and 250 matches that of the salts and surpasses that of HATtsc. However, it's important to note that these complexes exhibit no activity against P. aeruginosa. The observed low minimum inhibitory concentration (MIC) values for B. subtilis can be attributed to the interference of Cd(II) compounds with the process of cell separation.
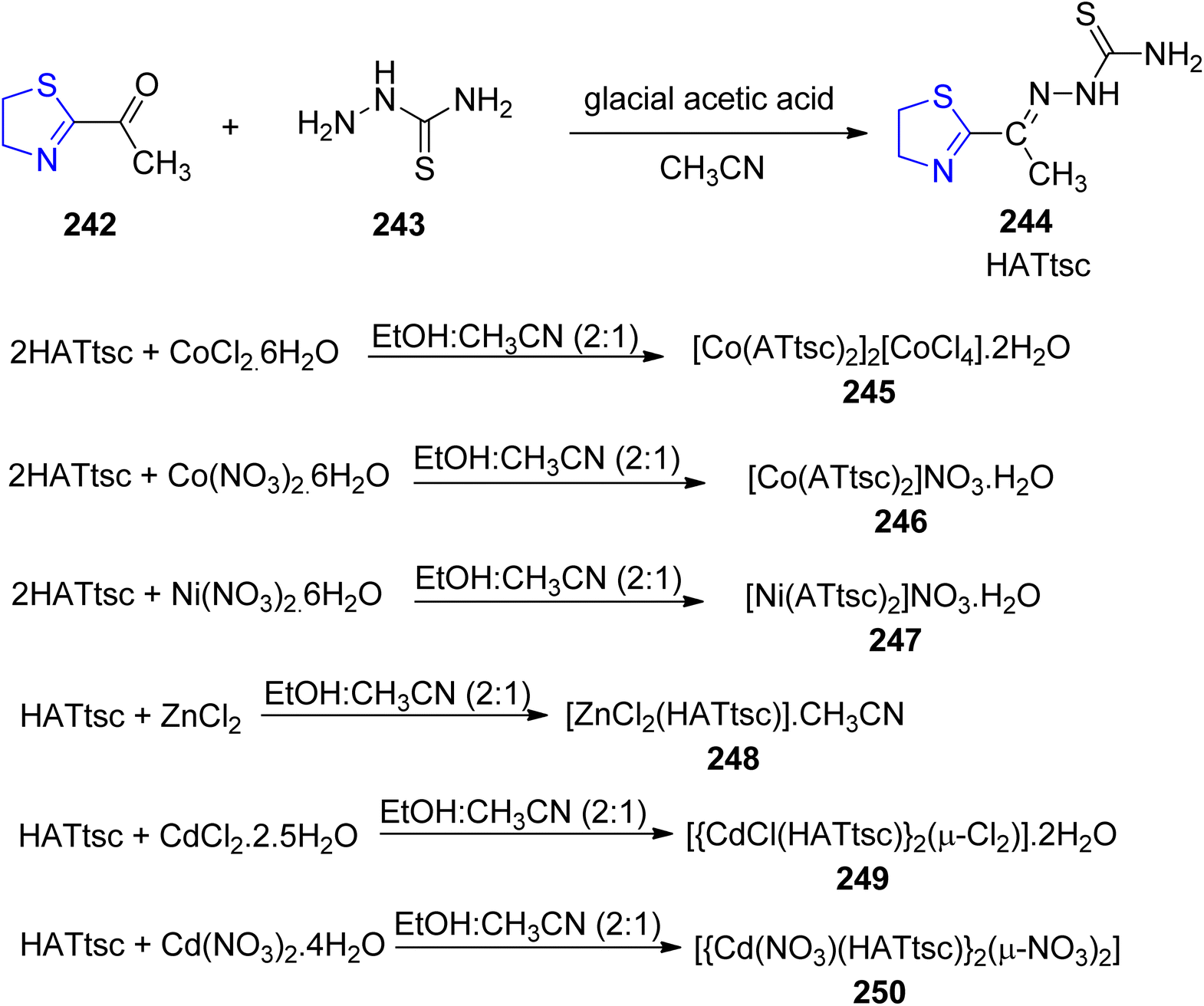 | ||
| Scheme 46 Synthesis of novel ligand, HATtsc 244 and its complexes 245–250. | ||
Ahmad et al.142 designed and synthesized a diverse library of thiazoline analogues, incorporating long-chain esters of fatty acids. These analogues were developed to inhibit CYP51 in Candida albicans and PDF in Escherichia coli. The corresponding thiazoline derivatives, denoted as 252a–d, were obtained by subjecting the dibromo derivative to thiourea treatment, as depicted in Scheme 47. Comprehensive investigations into the antibacterial and antifungal activities of these compounds were conducted. Characterization involved spectroscopic analyses, infrared (IR) and mass analysis. Notably, these molecules exhibited remarkable antibacterial efficacy against the tested microbes, comparable to commonly used drugs like fluconazole and ciprofloxacin. Among all the synthesized compounds, compound 252a (MIC: 25 μg mL−1) showed highest antibacterial activity against E. coli. These results are almost equivalent to approved drug ciprofloxacin.
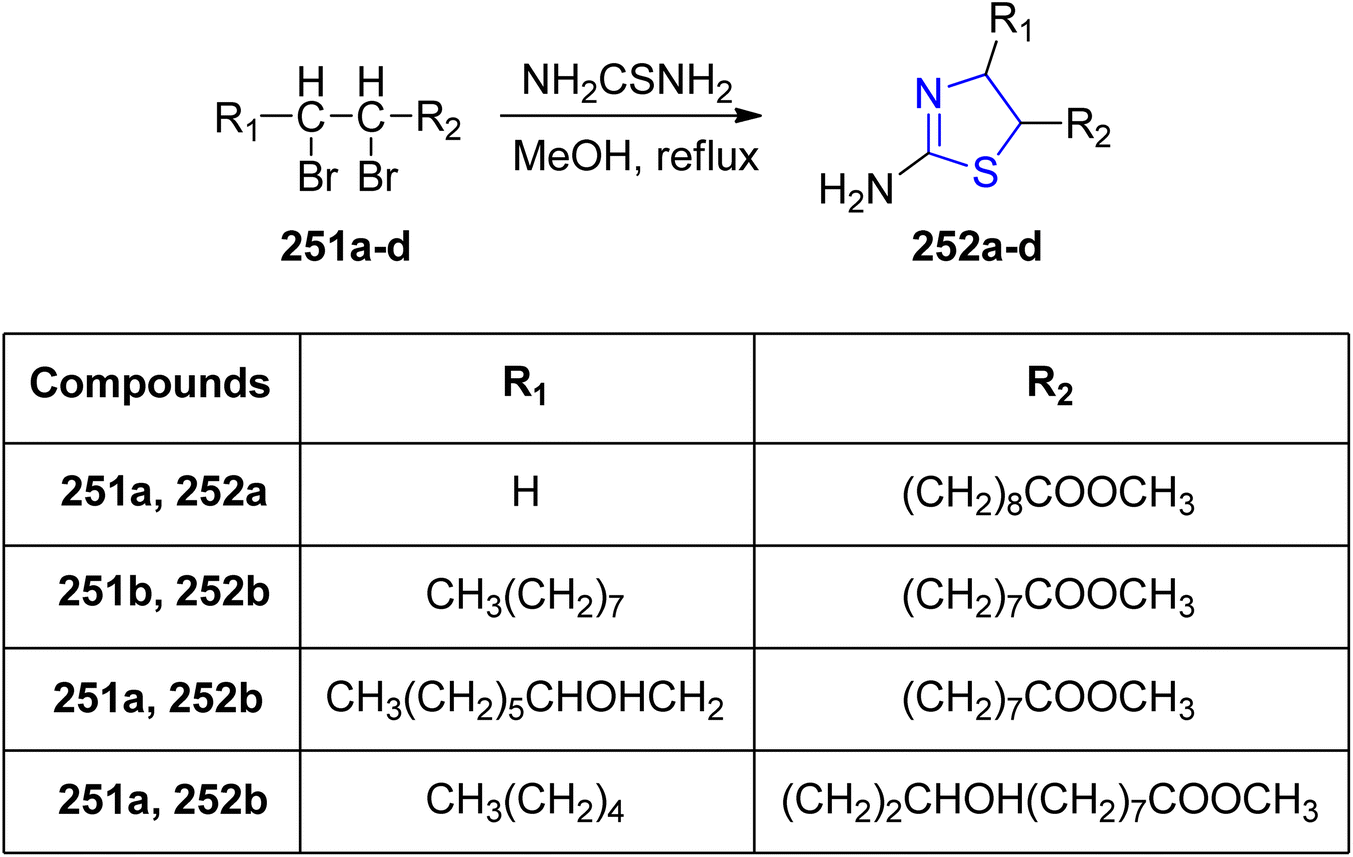 | ||
| Scheme 47 Synthesis of thiazoline derivative 252a–d. | ||
Furthermore, compounds 252a and 252d displayed antifungal properties when tested against clinical isolates of Candida that had developed resistance to itraconazole and fluconazole.
A facile and practical methodology for developing novel heterocyclic compounds based on anthraquinone for testing their anti-bacterial properties was disclosed by Gouda et al.143 The thiazoline analogue 254 was synthesized by reaction of compound 253 with PhNCS 228b (Scheme 48). The precursor acetamide 253 was obtained by following the previously described scheme.144
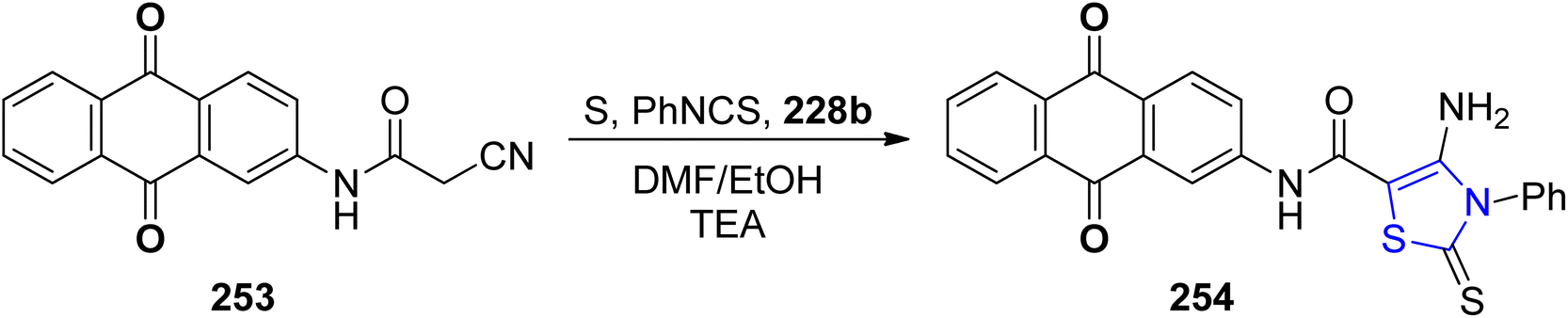 | ||
| Scheme 48 Synthesis of thiazoline derivative 254. | ||
Ke et al.145 investigated steroidal analogues of thiazoline as potential antiviral agents, leveraging the diverse biological properties inherent in naturally derived heterocyclic molecules. The steroid-based thiazoline heterocycles were synthesized by following a simple condensation reaction (Scheme 49). The literature reported procedure was modified appropriately while preparing the intermediates 257a–c.146,147 The target thiazolines 260a–k, 261a–i, 262a–i were obtained by treating the intermediates 257a–c with the required amount of acetophenone 259. Dihydrothiazole 263 was synthesized to explore the impact of introducing various fragments on its activity. The synthesized compounds were assessed for their potential antiviral effectiveness against CVB3 and EV71 viruses. Compounds 260b, 260g, and 260i demonstrated efficiency against EV71 having EC50 values of 0.61 μmol L−1, 0.95 μmol L−1, and 2.31 μmol L−1, respectively; while compounds 260b, 260e, 261c, and 261g with EC50 values of 1.83 μmol L−1, 6.79 μmol L−1, 7.09 μmol L−1 and 2.49 μmol L−1, respectively, exhibited enhanced antiviral activity against CVB3 compared to control such as ribavirin or pirodavir, as determined through in vitro analysis.
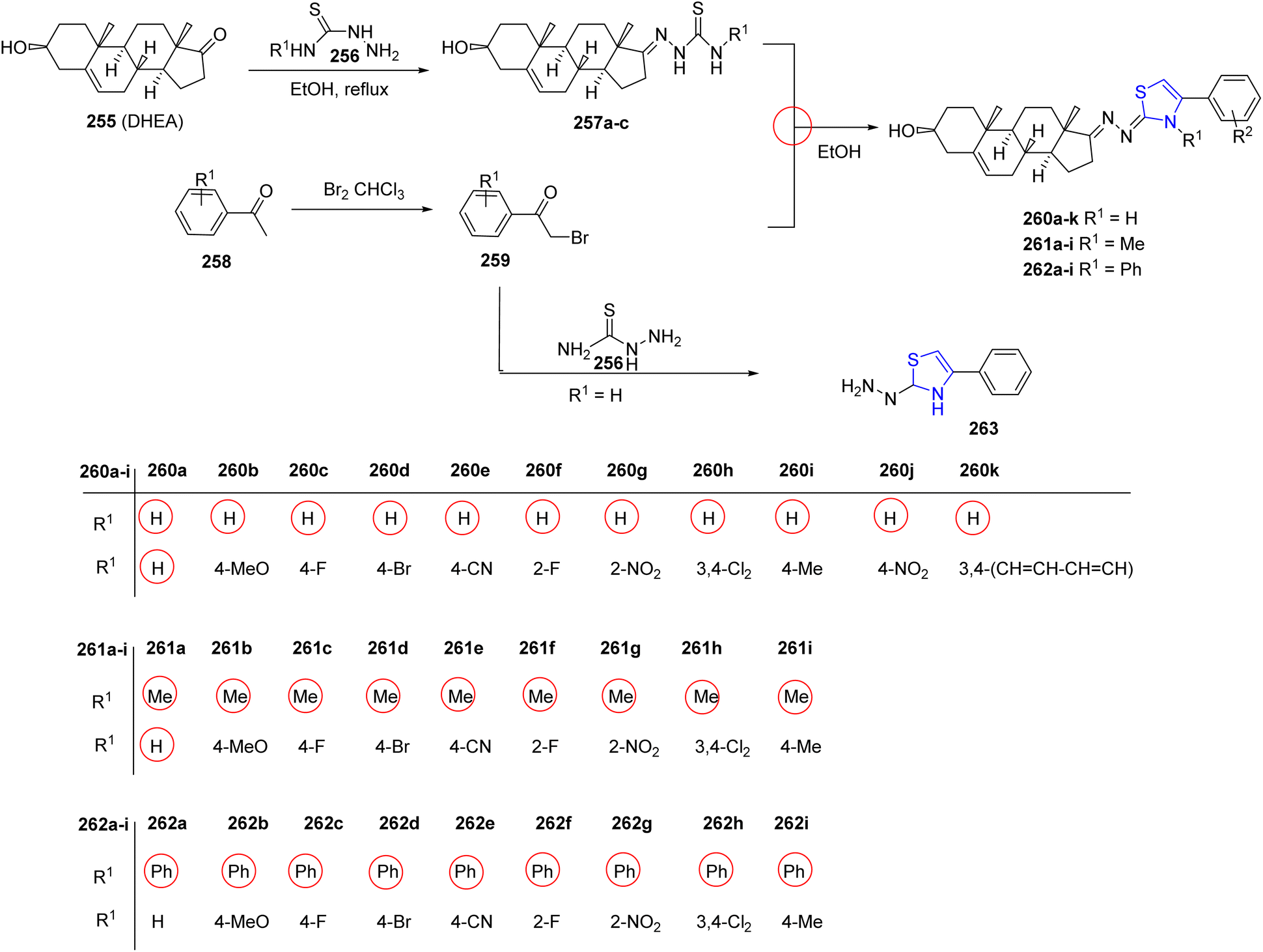 | ||
| Scheme 49 Synthesis of a number of steroid derivatives with thiazoline heterocycles. | ||
Meleddu et al.148 detailed the synthesis and development of a series of indol-2-one analogues, which were subsequently investigated for their impact on HIV-1 reverse transcriptase (RT). Initially, thiosemicarbazones 266a–b were synthesized by refluxing an ethanolic solution containing substituted isatins 265a–b and compound 264. Subsequently, through treatment of compound 266a–b with variously substituted bromo- or chloro-acetophenones 267a–m in isopropanol, the resulting compounds 268a–m and 269a–l were obtained in high yields, as outlined in Scheme 50. These compounds demonstrated micromolar-level activity against ribonuclease H and DNA polymerase. IC50 values for 268a–m were in the range 15–29 μM, whereas 10–27 μM for compounds 269a–l.
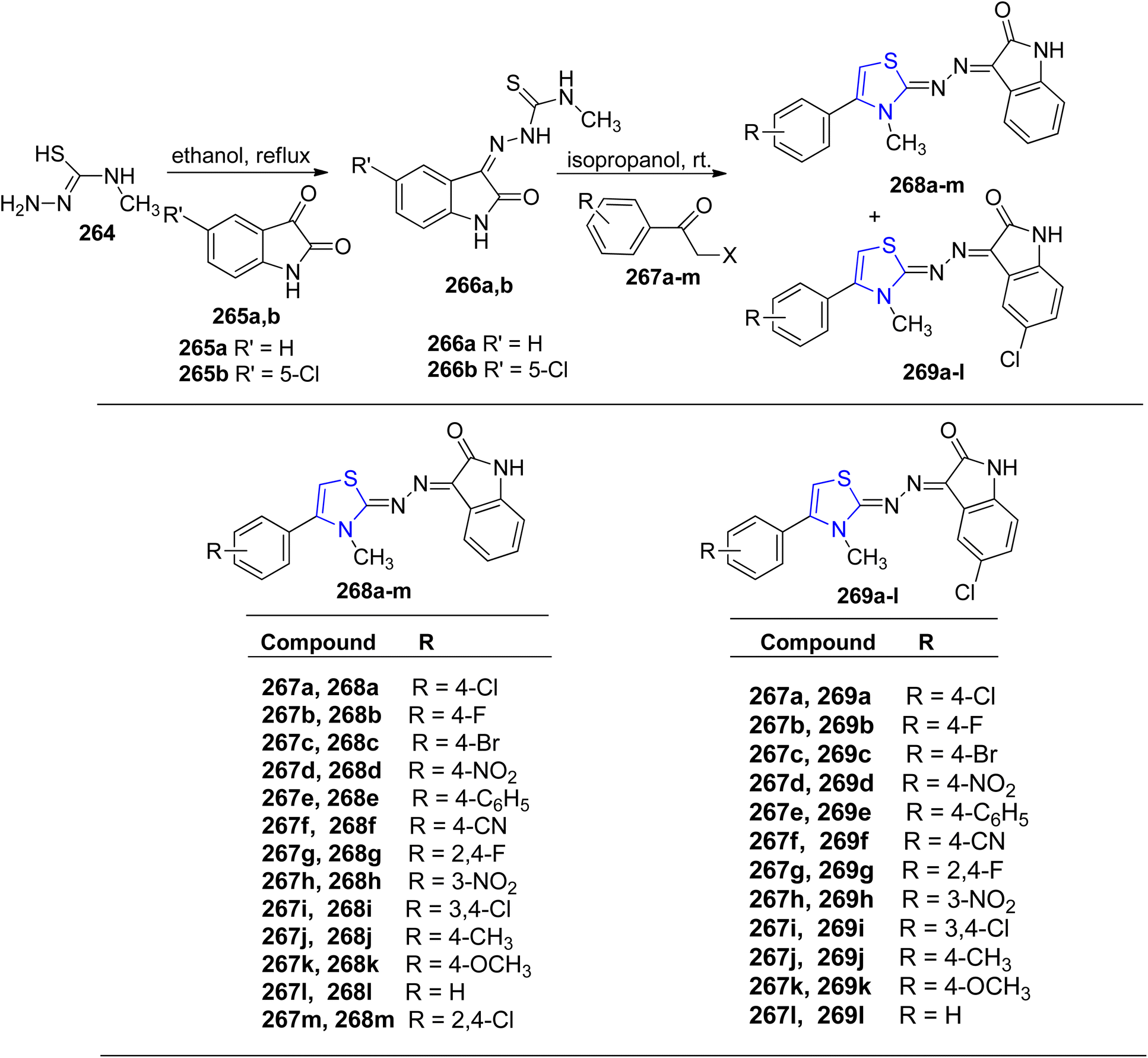 | ||
| Scheme 50 Synthesis of isatin thiazoline hybrids 268a–m and 269a–l. | ||
Hussein et al.149 designed and synthesized a novel series of thiazoline quinoline derivatives through the cyclization of quinoline thiosemicarbazone. The preparation involved the synthesis of hydrazones 276a–e and 281a–c by treating ethanolic solutions of thiosemicarbazones 274a–e with phenacyl bromides 275a–e, as depicted in Scheme 51. Characterization of the prepared compounds was conducted through spectroscopic measurements and elemental analysis. A total of 28 novel compounds were evaluated for their potential antibacterial properties. In comparison to the reference drug gatifloxacin, most of the investigated compounds displayed moderate efficacy against various bacterial strains. Similar results were observed when assessing the compounds for their antifungal properties, using ketoconazole as the reference drug. Notably, these compounds demonstrated significant anti-inflammatory activity, with indomethacin serving as the reference. Furthermore, when the most potent compounds, 277b and 280e, were tested on mice, they were found to be non-toxic even at high doses of 400 mg kg−1.
 | ||
| Scheme 51 Synthetic scheme for quinoline–thiazoline analogues 276a–e to 281a–c. | ||
The two incretin hormones, GLP-1 and GIP, are deactivated by the enzyme dipeptidyl peptidase IV (DPP-4). Therefore, DPP-4 inhibitors play a vital role by slowing down the activity of GIP and GLP-1 hormones, thereby maintaining glucose homeostasis. The therapeutic potential of the DPP-4 enzyme makes it a preferred target in pharmacology.155
Ali et al.156 synthesized a distinct class of thiazoline derivatives linked to quinazoline. Initially, three different derivatives of anthranilic acid 282a–c were reacted with acetyl chloride 283 to give the cyclized benzoxazin-4-ones 284a–c. Further, a condensation reaction of these benzoxain-4-ones 284a–c with substituted thiazoles 285a/b gave benzoxain-4-ones 286a–f.157 In the final stage, quinazolin-4-ones 287a–f were prepared by refluxing alcoholic solution of 286a–f with hydrazine hydrate for 4 hours. Additionally, the quinazoli-4-ones 287a–f thus obtained were condensed with aldehydes 288a–d to yield a sequence of Schiff bases 289a–x (Scheme 52).158 The compounds thus obtained were also assessed for their inhibitory action against dipeptidyl peptidase IV (DPP-4) invitro. Utilizing linagliptin as a benchmark, compounds that displayed good to moderate activity were contrasted. The results for compound 289x (IC50 of 1.12 nM) were the most encouraging. Compound 289x possessed unique chemical features which provided DPP-4 with better inhibitory selectivity in comparison to DPP-8 or DPP-9.
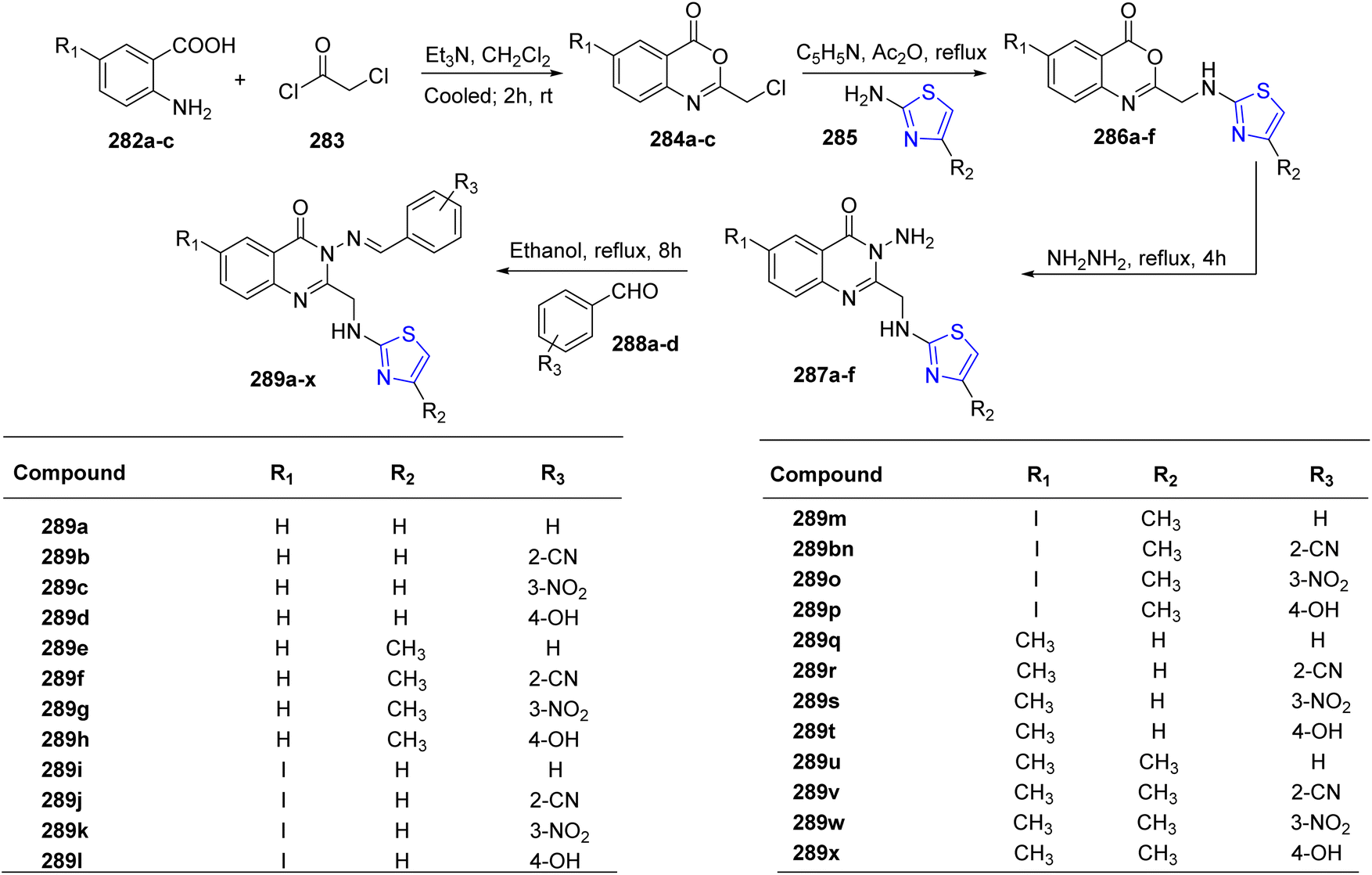 | ||
| Scheme 52 Synthetic scheme for quinoline linked thiazoline derivatives. | ||
Utilizing small heterocyclic compounds to inhibit aldose reductase (ALR2) is a viable approach for developing innovative anti-diabetic medications. To identify a lead as a potential novel anti-diabetic drug, Shehzad et al.159 synthesized thiazoline analogues 294a–k, 295a–f, 296a–l and 297a–j. In order to obtain the thiazoline derivatives 294–297, four different types of carbonyl group-bearing compounds were used, with yields ranging from 76 to 92% (Scheme 53). Compounds 296b (IC50 = 1.39 ± 2.21 μM) and 297e (IC50 = 1.52 ± 0.78 μM) were identified to be the most efficient ones in comparison to the reference drug, sorbinil (IC50 = 3.14 ± 0.02 μM). Compound 296b demonstrated good selectivity for the intended ALR2 with just 23.4% inhibition for ALR1.
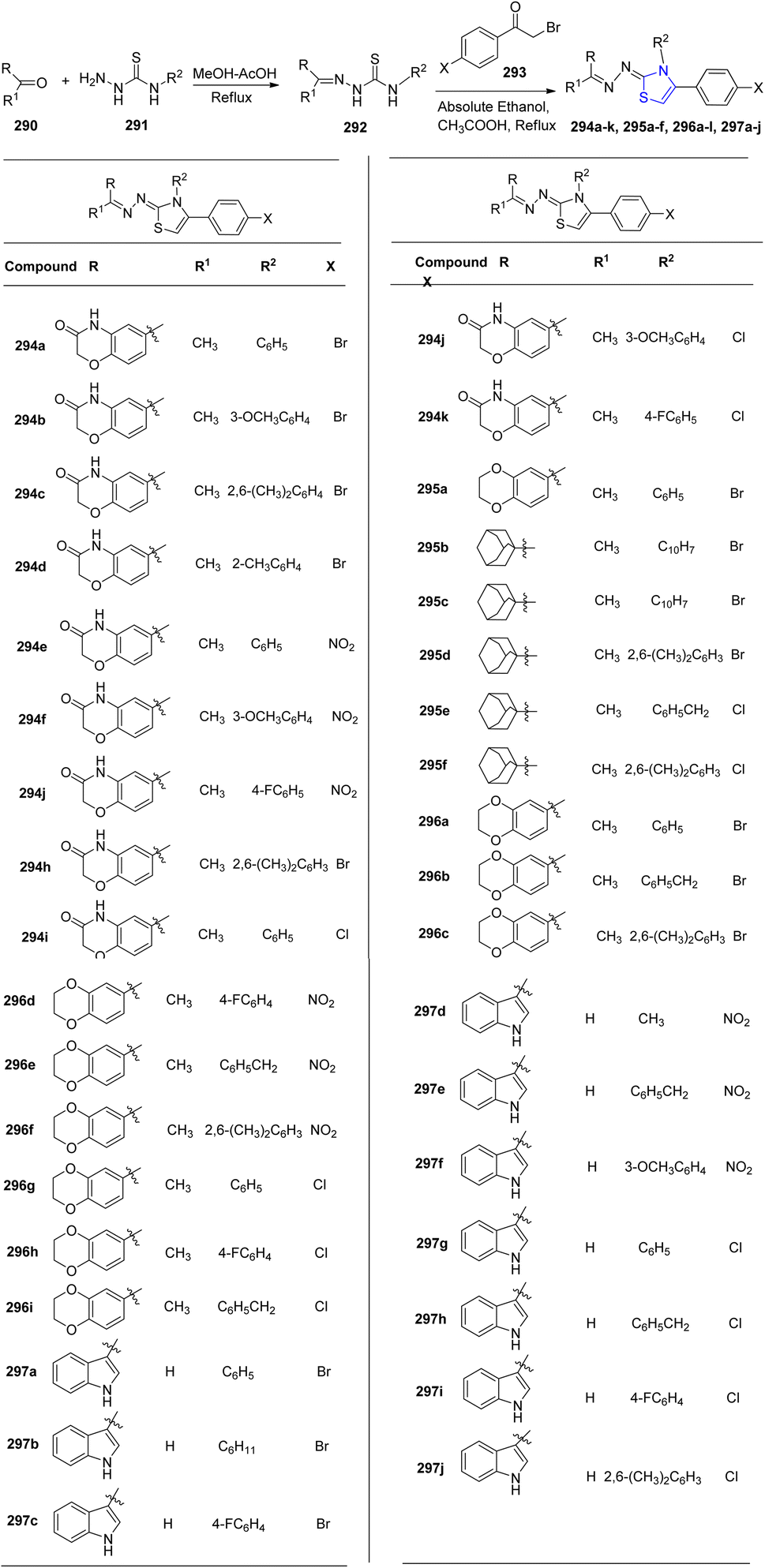 | ||
| Scheme 53 Synthesis of thiazoline derivatives 294a–k, 295a–f, 296a–l and 297a–j. | ||
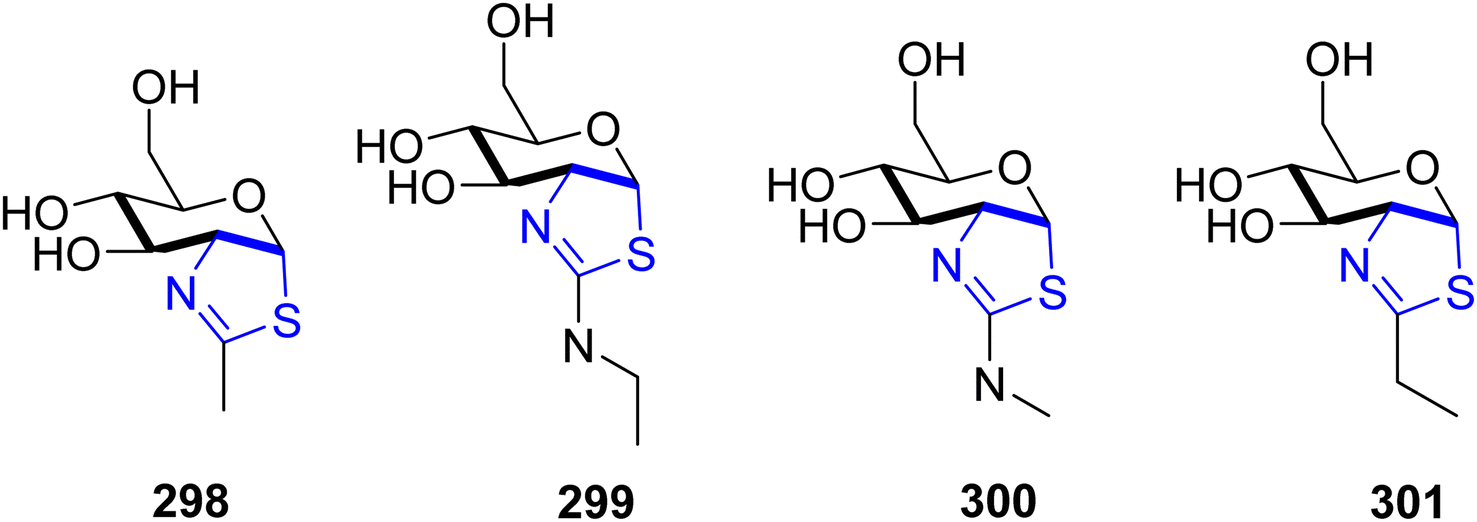 | ||
| Scheme 54 Structures of NGT and its derivatives. | ||
Human skin and hair contain the biological pigment melanin. Pigmentation development in animals depends on α-MSH.161 A novel thiazoline analogue, KHG22394 302 was synthesized by Kim et al.162 as a skin whitener (Scheme 55). Although, KHG22394 does not inhibit the tyrosinase enzyme directly, but according to the research data it has been shown that it greatly reduces melanin synthesis in a dose-dependent way. ERK activity has been shown to inhibit the transcription factor linked to microphthalmia, which in turn decreases melanin formation (Mitf). In B16 melanoma cells, KHG22394 upregulates the ERK pathway while downregulating the protein levels of Mitf and tyrosinase. Despite not directly inhibiting tyrosinase activity, KHG22394's hypopigmentary impact is due to the downregulation of Mitf and tyrosinase as a result.
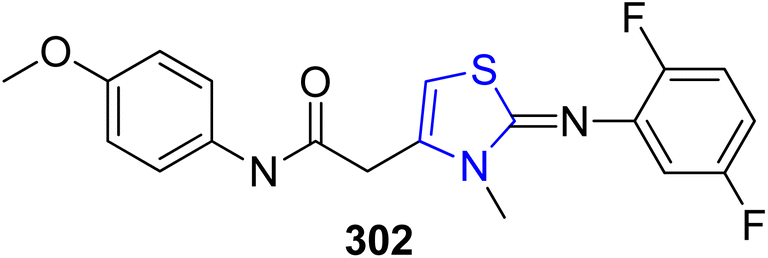 | ||
| Scheme 55 Structure of KHG22394. | ||
Hosamani et al.163 employed microwave irradiation to establish an efficient methodology for synthesizing coumarin–thiazolines 307a–j, as illustrated in Scheme 56. Characterization of these compounds 307a–j was accomplished through spectroscopic and elemental analyses. Notably, compound 307b exhibited remarkable efficacy (MIC = 0.09 μg mL−1) with minimal toxicity to normal cells, as determined via in vitro anti-tubercular screening on Vero cells. Furthermore, compounds 307b and 307i demonstrated complete DNA cleavage, establishing them as the most effective in vitro agents against the MtbH37 strain.
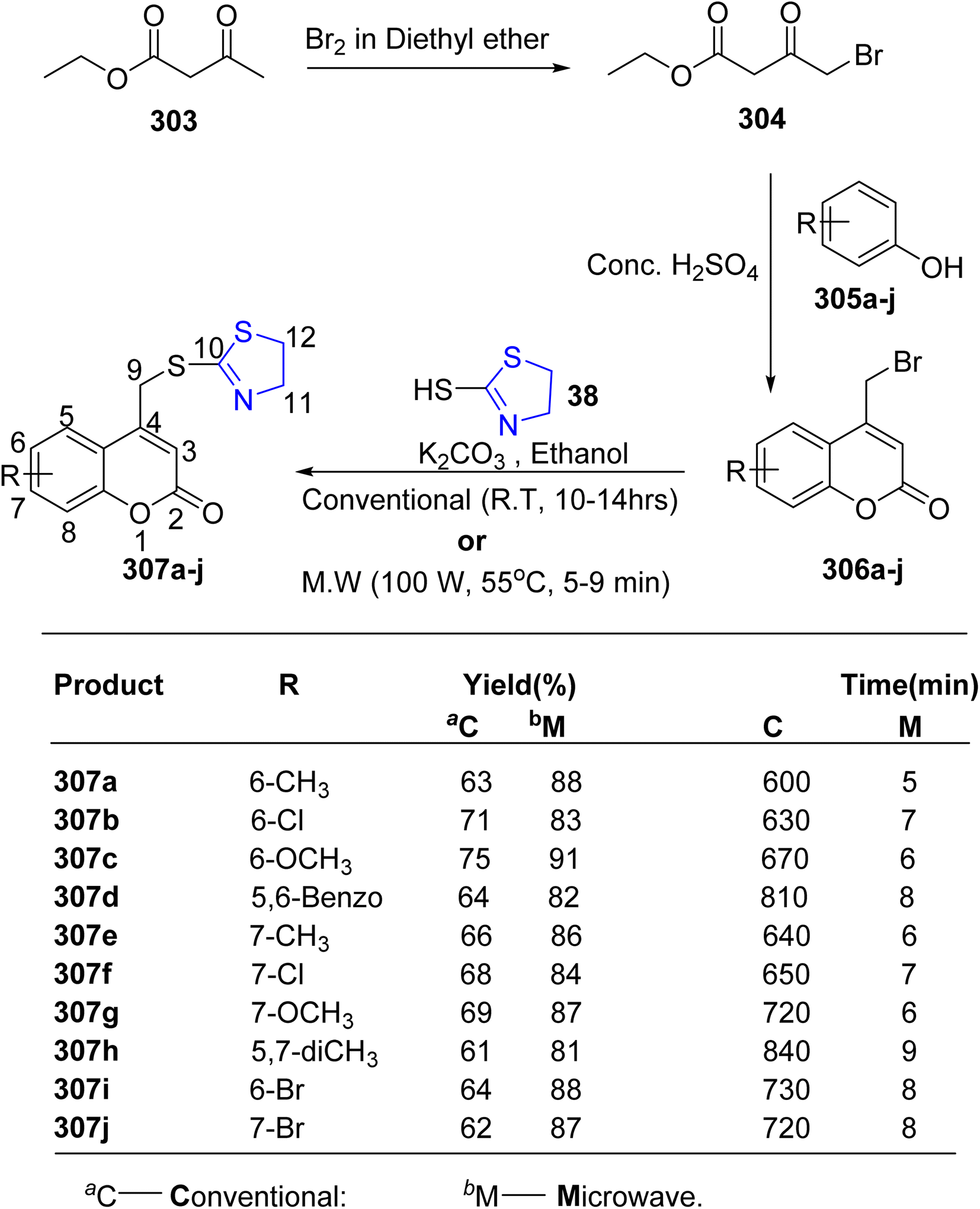 | ||
| Scheme 56 Synthesis of coumarin–thiazoline hybrids 307a–j. | ||
4. Miscellaneous
Nonlinear optical (NLO) materials are cost-effective and easily tunable in terms of absorption wavelength, making them versatile for applications in information technology, telecommunications, and healthcare sector. Researchers, both experimental and computational, are drawn to these NLO compounds for their diverse applications in optical computing, biophysics, solid physics, dynamic image processing, nuclear science, and biophysics.164,165 Thiazoline derivatives are fascinating molecules because of their potential usage in nonlinear optics (NLO). Haroon et al.166 described four new thiazoline derivatives with the chemical formulae C27H24ClN3O2S, C27H23ClN4O2S, C26H21N5O2S, and C28H30ClN3S. Characterization of the synthesized compounds was done by using spectroscopic experiments, elemental analysis and FTIR measurements. The two-step process used to obtain the new thiazoline derivatives 312a–d with varied substitutions is detailed in Scheme 57. The first step of the synthetic procedure began by condensing varied carbonyl molecules 318a–d with thiosemicarbazides 319 in methanolic solution to yield thiosemicarbazone intermediates 310. These intermediates were then subjected to a cyclization reaction in ethanolic media with two different phenacyl bromides 311a/b to yield the thiazoline analogues. The energy gap of the compounds was in the order: 312a–d >312c > 312b > 312d > 312a. The least HOMO–LUMO energy gap in 312a makes it the sensitive and reactive molecule, while 312c has the highest energy gap, and thus is the most stable of all the examined compounds. Using a variety of various functionals, including HF, B3LYP, LC-BLYP, CAM-B3LYP, M06 & M062X in coupling with 6-311+G(d,p) basis set, the NLO characteristics of the examined compounds 312a–d were estimated. Furthermore, at 0.02389 and 0 nm, the signals γ(−ω,ω,0,0) and γ(−2ω,ω,ω,0) indicating the electro-optic Kerr effect and second harmonic production, respectively, were also studied. The NLO results clearly showed that compounds 312a–d have attractive NLO traits and are suitable NLO aspirants for next-generation optoelectronic devices.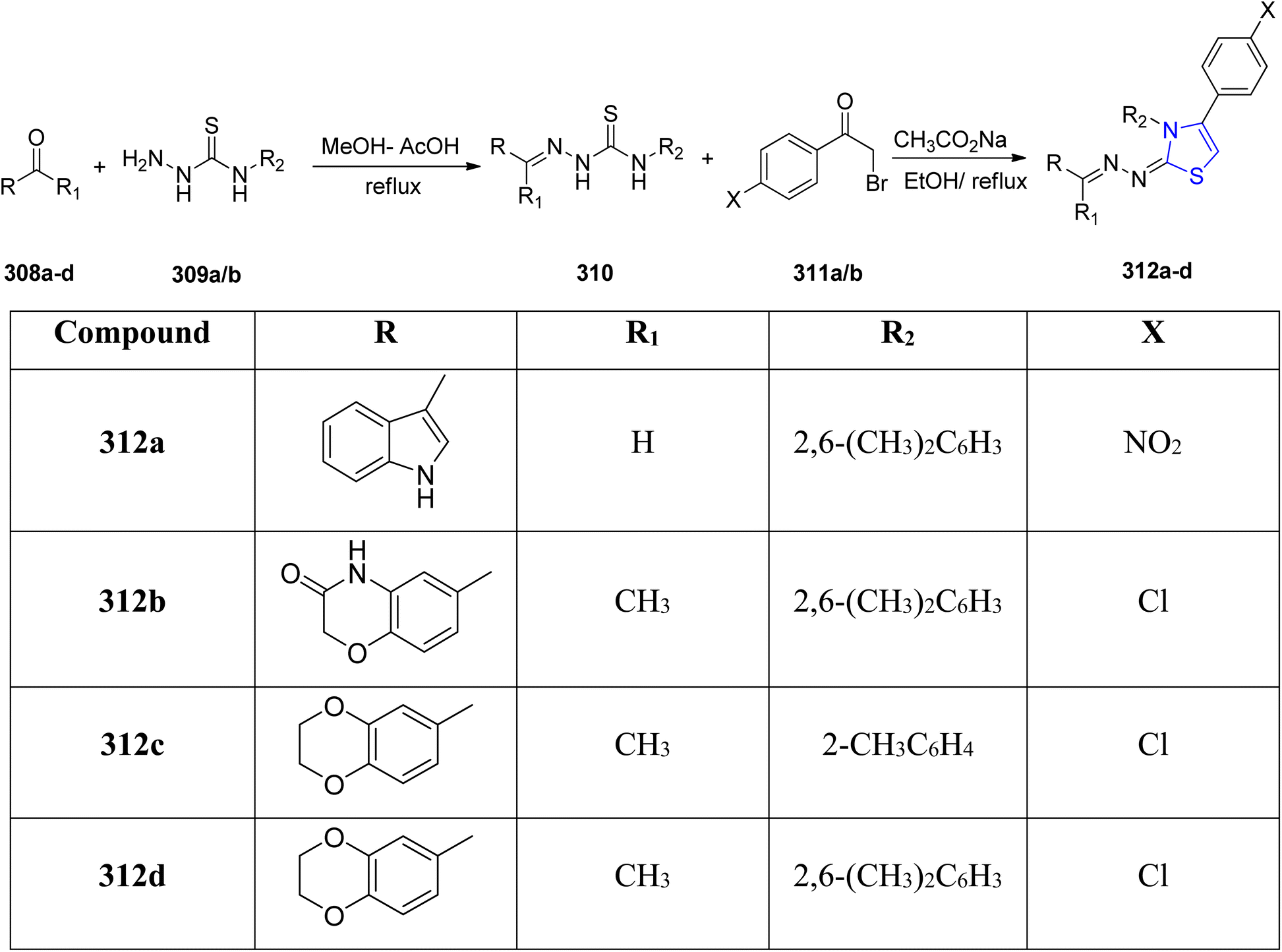 | ||
| Scheme 57 Synthesis of thiazoline derivatives 312a–d. | ||
The thiazoline moiety can also be synthesized through cyclodehydration of compounds containing the β-hydroxy thioamide functionality. The synthetic pathways employed for the production of cyclopeptide YM-216391 317 and thiopeptide-based antibiotics such as GE2270C1, GE2270T, and GE2270A exemplify the utility of this approach.167,168 When β-hydroxy thioamides 313 and 315 were exposed to DAST, they underwent intramolecular cyclization, leading to the formation of thiazolines 314 and 316 (Scheme 58). Compound 316 underwent oxidation to ultimately yield the cyclopeptide YM-216391 317 in the presence of MnO2. YM-216391 dose-dependently inhibited the growth of human cervical cancer HeLa S3 cells with an IC50 value of 14 nM. YM-216391 also showed potent cytotoxic activity against a human cancer cell line panel.
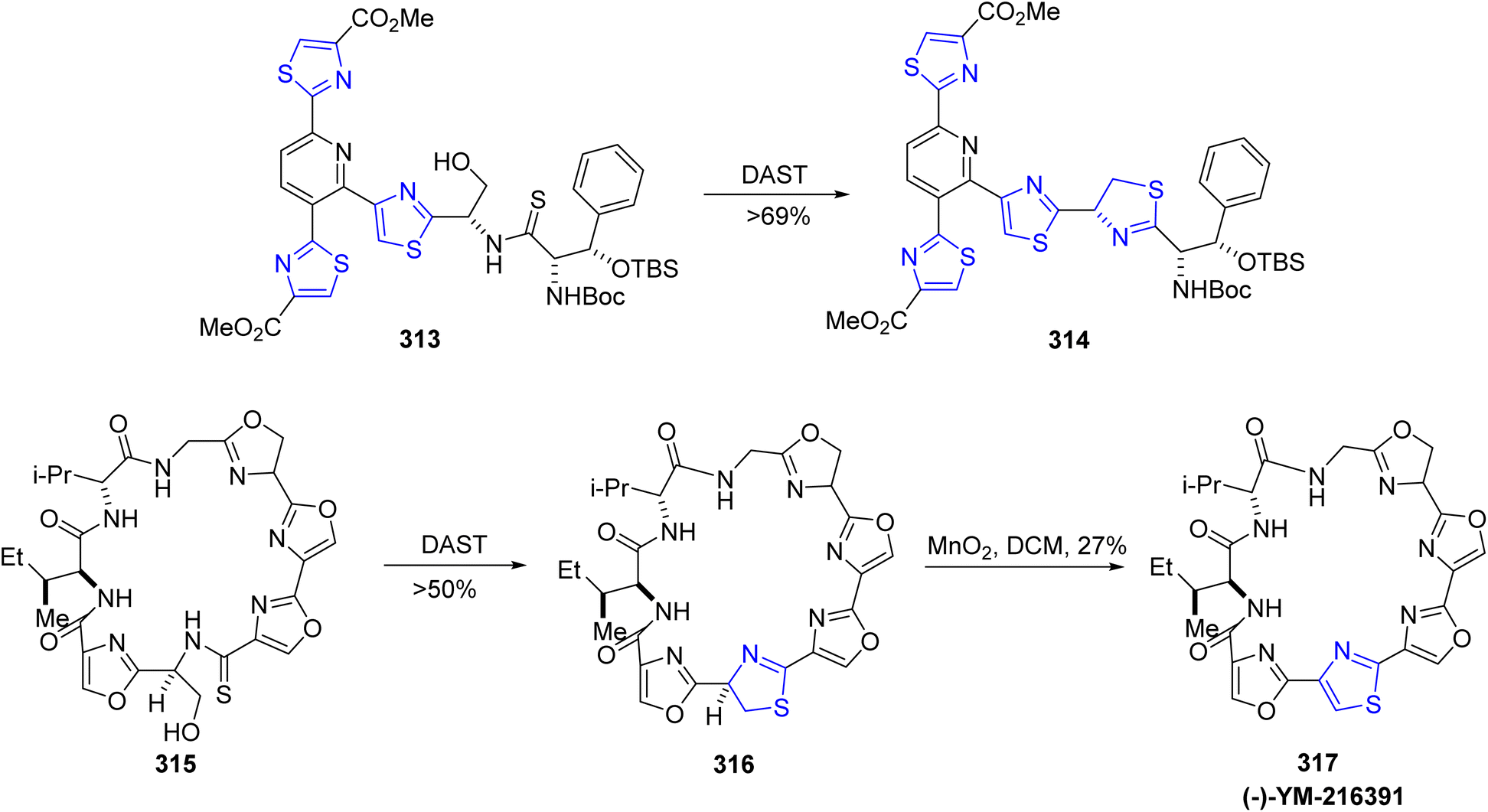 | ||
| Scheme 58 Synthesis of thiazoline 314 and (−)-YM-216391 317. | ||
Scientific literature has documented the existence of over 3000 compounds containing thiazole-4-carbonitrile. These derivatives, including 4-carbonitrile and 4-carbimidate variations of thiazole, can be readily synthesized under mild conditions, yielding satisfactory yields, from thiazole–oxazoline and thiazole–thiazolines, respectively.
Diness et al.169 synthesized one such fragment of the natural product, largazole, in five steps. The synthesis began by transforming Fmoc-Gly-NH2 318 with Lawesson's reagent to yield the Fmoc-protected product, which was further condensed with bromopyruvate to give thiazole acid 319 by following literature procedure.170 The resulting amide 320 was then transformed to the appropriate nitrile derivative 321 by dehydrating the amide with trifluoroacetic anhydride (TFAA). The Fmoc group was retained by rapid condensation with cysteine 322.171 The Fmoc-protected glycine amide 318 was transformed into the building block 323 with an overall yield of 47% (Scheme 59a). The other building blocks were protected by Boc-group since there was a chance that the Fmoc-protecting group might be lost during the final condensation. A modified Hantzsch process was utilized to condense the Boc-protected amino acids with 2-bromo-pyruvate ethyl ester to yield the corresponding thioamides 324a–c.172 The direct condensation reaction between the unprotected thioamide 324c and bromopyruvate resulted in a partially racemized product, thus making this approach crucial. The thus obtained ethyl esters 325a–c then underwent ammonolysis on treatment with aqueous ammonia to give compounds 326a–c. This was followed by dehydration to yield the compounds 327a–c.173 In order to obtain the desired compounds 328a–c, a condensation reaction was performed between cysteine 322 and carbonitriles 327a–c with an overall yield of 43–72% (Scheme 59b). However, Scheme 59c describes a more effective method for creating a library of these building blocks. Fortunately, substituting the pyruvate derivative with nitrile 330 did result in good yields of the appropriate carbonitriles 331a–c.174 A final condensation reaction with the cysteine derivative 322 resulted in the formation of the required thiazole-thiazoline building blocks 332a–c (Scheme 59c).
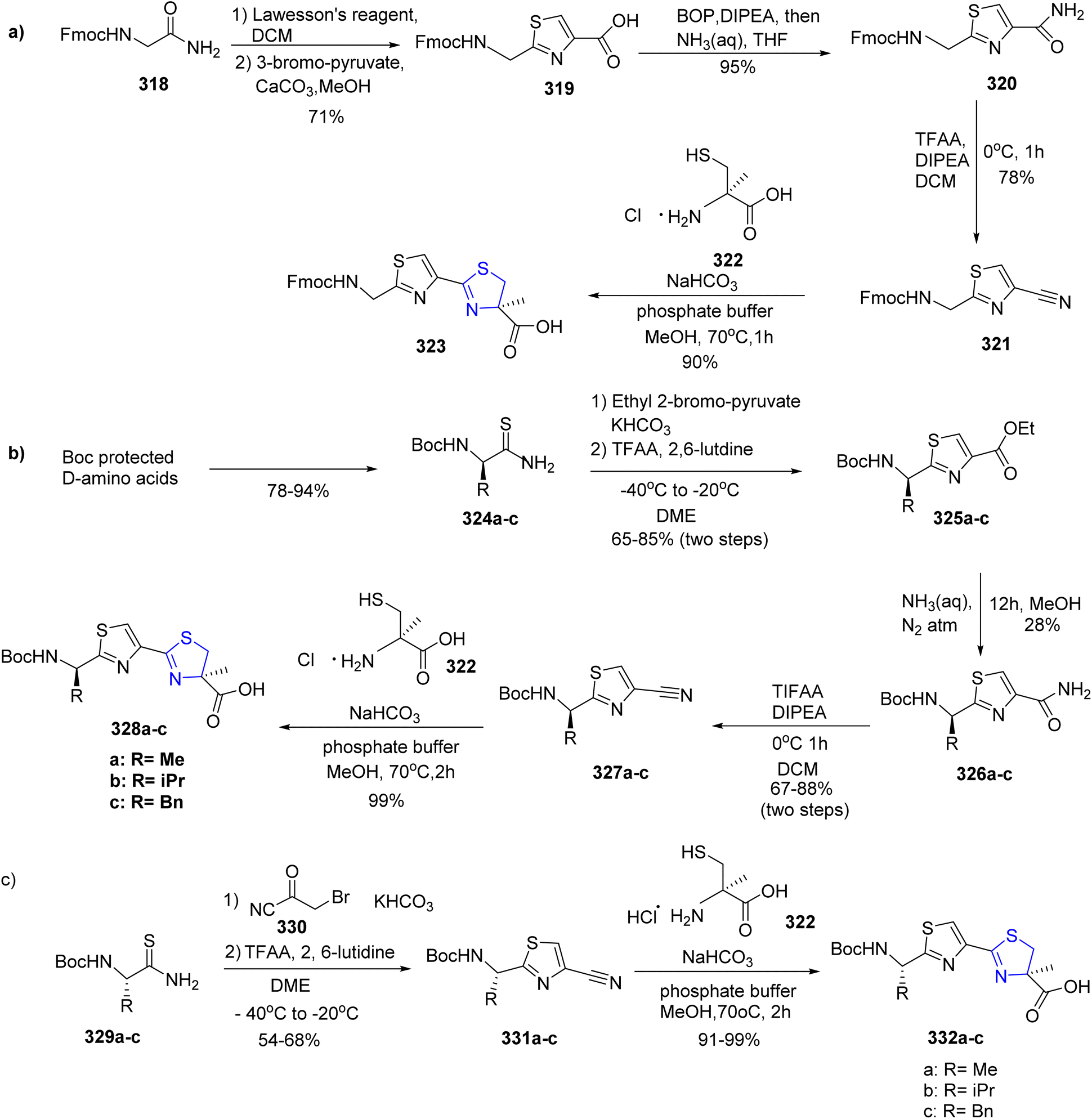 | ||
| Scheme 59 (a–c) Synthetic scheme for thiazoline fragment of the largazole derivatives. | ||
Cone snails have been associated with various actinomycetes and other bacteria, some of which possess neurologically active properties upon extraction.175 Actinomycetes and other bacteria have a specialized environment provided by the Philippine cone snail Conus pulicarius. Lin et al.176 conducted research on Streptomyces sp. CP32, one of the active isolates from C. pulicarius. Five known analogues 333–337 and five novel analogues, 338–342 known as pulicatins A–E, were isolated using the assay-guided fractionation (Scheme 60). These molecules attach themselves to the human receptors, specifically the human 5-HT2B serotonin receptor. Additionally, 338 was discovered to be an important constituent of the Streptomyces sp. strain CT8, obtained from the hepatopancreas of the cone snail C. tribblei.
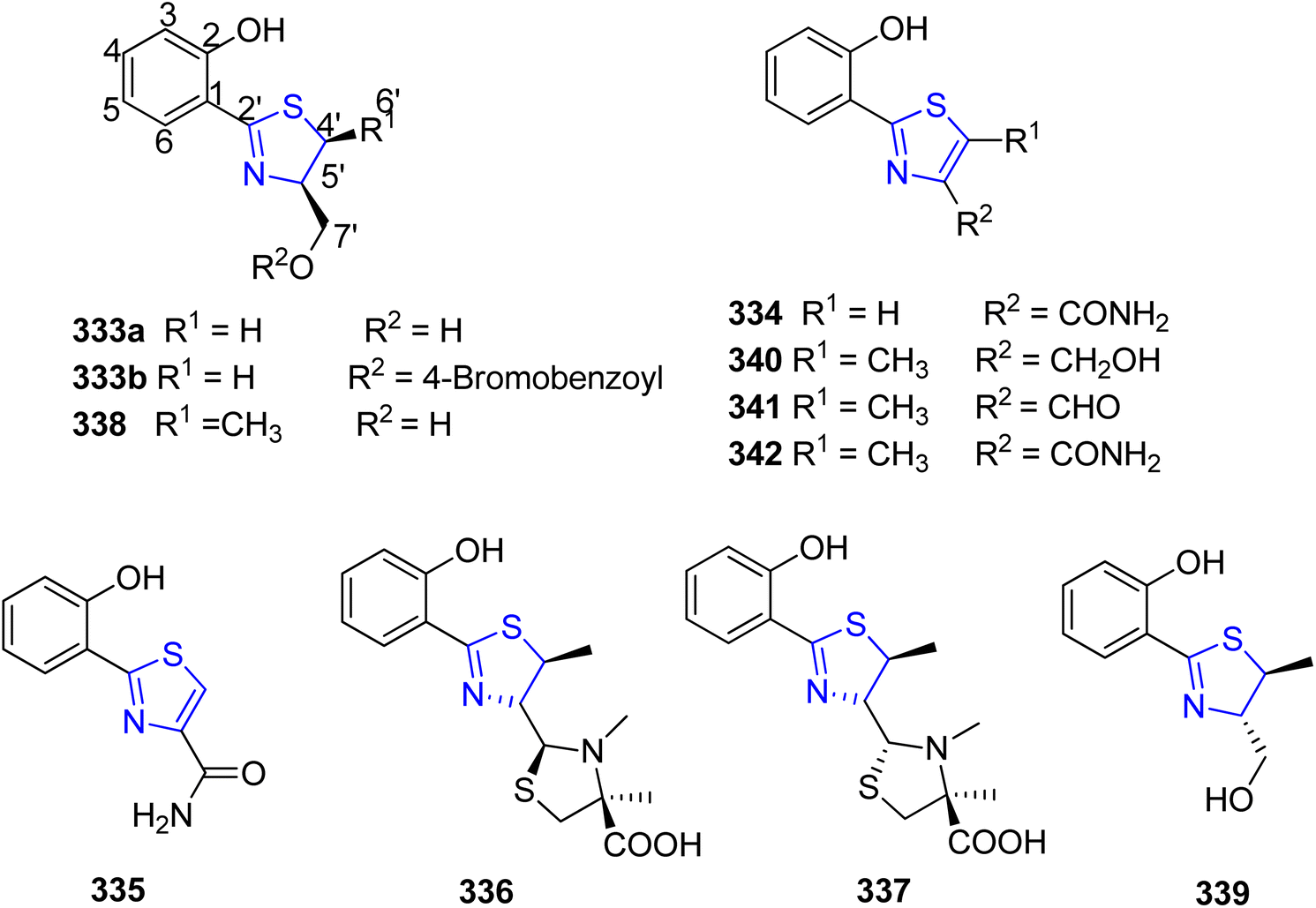 | ||
| Scheme 60 Neuroactive thiazoline metabolites. | ||
5. Future prospectus & conclusions
Thiazolines, also known as dihydrothiazoles, represent a class of 5-membered heterocyclic compounds characterized by the presence of both sulfur and nitrogen atoms within the ring structure. This isomeric heterocyclic family has garnered significant attention in the realm of chemistry due to its multifaceted role as efficient ligands in asymmetric catalysis and as crucial intermediates in synthetic organic chemistry. Thiazolines exhibit notable chelating capabilities with transition metal ions. Despite their relative novelty among chiral ligands compared to their oxygen counterparts (oxazolines), thiazolines exhibit distinct behavior in a variety of metal-catalysed reactions.The versatility of these novel heterocyclic molecules has led to their wide-ranging applications, spanning organic synthesis, pharmaceuticals, agrochemistry, and catalysis. In recent years, a plethora of thiazoline analogues have been synthesized and explored for their intriguing therapeutic potential, encompassing anti-cancer properties, anti-oxidant effects, anti-inflammatory, anti-viral, and anti-microbial characteristics. Extensive research has also delved into their capacity to inhibit various enzymes, including urease, butyrylcholinesterase, and carboxylesterase.
Natural sources have yielded substances with key structural motifs known as thiazolines, exemplified by compounds such as thiangazole, luciferin, kalkitoxin, curacin A, and mirabazole B and C. Many of these compounds exhibit remarkable biological attributes, including neurological effects, anti-HIV activity, anti-cancer potential, and bioluminescence.
The burgeoning significance of thiazolines is indisputable, given their prevalence in chemical synthesis, pharmaceuticals, and natural products. However, it is noteworthy that the development of their synthetic strategies is still evolving, with advancements trailing behind their oxygen analogues. Over the past 15 years, the research landscape in this domain, encompassing the synthesis and applications of thiazoline derivatives, has expanded significantly. To fully harness the myriad unique properties of these compounds, the establishment of comprehensive structure–activity relationships is imperative.
In our assessment, thiazolines represent a captivating class of compounds within the catalysis field. It is crucial to acknowledge that their chemistry is still evolving, and their complete potential remains unrealized. It is plausible that further advancements will transpire as the distinctive attributes of the sulfur atom are more fully exploited.
6. Abbreviations
| HIV | Human Immunodeficiency Virus |
| MCRs | Multicomponent Reactions |
| DFBP | N-(α,α-Difluorobenzyl)pyrrolidine |
| DFMPP | N-(1,1-Difluoro-2,2-dimethylpropyl)-pyrrolidine |
| NMR | Nuclear Magnetic Resonance |
| DNA | Deoxyribonucleic acid |
| ATHTd | N-(2-Acetyl-2-thiazoline)-N′-(2-thiazolidin-2-one)azine |
| HFIP | Hexafluoroisopropanol |
| QDs | Quantum Dots |
| ICT | Intramolecular Charge Transfer |
| TFA | Trifluoroacetic acid |
| HCl | Hydrochloric acid |
| TEA | Triethylamine |
| RNA | Ribonucleic acid |
| Ppb | Parts per billion |
| Ppm | Parts per million |
| DFT | Density Functional Theory |
| HOMO | Highest Occupied Molecular Orbital |
| Mcf-7 | Human breast cancer cell line |
| LOD | Limit of detection |
| OLED | Organic light-emitting diode |
| Dipp | N-2,6-Diisopropylphenyl |
| Cbz | Carbazole |
| A549 | Human lung carcinoma cell line |
| LUMO | Lowest Occupied Molecular Orbital |
| WOLED | White organic light-emitting diode |
| MeCbz | 1,8-Dimethylcarbazole |
| HBr | Hydrobromic acid |
| EtOH | Ethanol |
| NIR | Near infra-red |
| EL | Electroluminescence |
| CRI | Colour Rendering Index |
| Htzol | 2-(2′-Hydroxyphenyl)-2-thiazoline |
| NHK | Nozaki–Hiyama–Kishi |
| TMS | Tetramethylsilane |
| WHO | World Health Organization |
| HPAC | Human Pancreatic Cancer |
| PC-3 | Classical prostate cancer cell line |
| HCT-116 | Human colorectal carcinoma cell line |
| IC50 | Half-maximal Inhibitory Concentration |
| MS | Mass spectrometry |
| HepG2 | Liver hepatocellular carcinoma cell line |
| NIH/3T3 | Mouse embryoblast cell line |
| AChE | Acetylcholinesterase |
| HDAC | Histone deacetylase |
| EDCI | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| ROS | Reactive oxygen species |
| mL | Millilitre |
| μg | Microgram |
| μM | Micromole |
| mM | Millimole |
| HATtsc | 2-Acetyl-2-thiazoline thiosemicarbazone |
| CVB3 | Coxsackie type B3 |
| EV71 | Enterovirus 71 |
| kg | Kilogram |
| RT | Reverse transcriptase |
| LD50 | Lethal dose |
| DM | Diabetes mellitus |
| CVD | Cardiovascular disease |
| GLP-1 | Glucagon-like peptide 1 |
| Mitf | Microphthalmia-associated transcription factor |
| DPP-4 | Dipeptidyl peptidase IV |
| GIP | Glucose-dependent insulin tropic polypeptide |
| nM | Nano-mole |
| ALR | Aldose reductase |
| β-GlcNAcases | β-Acetylglucosaminidases |
| NGT | NAG-thiazoline |
| α-MSH | α-Melanocyte-stimulating hormone |
| ERK | Extracellular signal-regulated kinase |
| MIC | Minimum inhibitory concentration |
| NLO | Nonlinear optical |
| EIMS | Electron ionization mass spectral |
| TFAA | Trifluoroacetic anhydride |
| Fmoc | Fluorenylmethoxycarbonyl |
| Boc | tert-Butyloxycarbonyl |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate the University of Delhi's Institute of Imminence for providing financial support to research and development. Sumit Kumar (File No. 09/045(1798)/2020-EMR-I) and Aditi Arora (File No. 09/0045(11270)/2021-EMR-I) thank CSIR for awarding SRF & JRF Awards, respectively.References
- A. I. Meyers, Heterocycles in Organic Synthesis, Wiley, 1974 Search PubMed
.
- P. M. Abreu and P. S. Branco, Natural Product-Like Combinatorial Libraries, J. Braz. Chem. Soc., 2003, 14(5), 675–712, DOI:10.1590/S0103-50532003000500002
- A. C. Gaumont, M. Gulea and J. Levillain, Overview of the chemistry of 2-thiazolines, Chem. Rev., 2009, 109(3), 1371–1401 CrossRef CAS PubMed
- C. T. Walsh and E. M. Nolan, Morphing Peptide Backbones into Heterocycles, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(15), 5655–5656, DOI:10.1073/pnas.0802300105
- G. M. Leod and J. M. Ames, Effect of Water on the Production of Cooked Beef Aroma Compounds, J. Food Sci., 1987, 52(1), 42–45, DOI:10.1111/j.1365-2621.1987.tb13968.x
- G. MacLeod and J. M. Ames, The Effect of Heat on Beef Aroma: Comparisons of Chemical Composition and Sensory Properties, Flavour Fragrance J., 1986, 1(3), 91–104, DOI:10.1002/ffj.2730010302
- J. S. Elmore, D. S. Mottram, M. Enser and J. D. Wood, Novel Thiazoles and 3-Thiazolines in Cooked Beef Aroma, J. Agric. Food Chem., 1997, 45(9), 3603–3607, DOI:10.1021/jf970066m
- P. K. C. Ong and T. E. Acree, Gas Chromatography/Olfactory Analysis of Lychee (Litchi Chinesis Sonn.), J. Agric. Food Chem., 1998, 46(6), 2282–2286, DOI:10.1021/jf9801318
- S. Carmeli, R. E. Moore, G. M. L. Patterson, T. H. Corbett and F. A. T. Valeriote, Unusual Cytotoxic Alkaloids from the Blue-Green Alga Scytonema Mirabile, J. Am. Chem. Soc., 1990, 112(22), 8195–8197, DOI:10.1021/ja00178a070
- P. Wipf, Synthetic Studies of Biologically Active Marine Cyclopeptides, Chem. Rev., 1995, 95(6), 2115–2134, DOI:10.1021/cr00038a013
- H. Luesch, W. Y. Yoshida, R. E. Moore, V. J. Paul and T. H. Corbett, New Apratoxins of Marine Cyanobacterial Origin from Guam and Palau, Bioorg. Med. Chem., 2002, 10(6), 1973–1978, DOI:10.1016/s0968-0896(02)00014-7
- R. S. Roy, A. M. Gehring, J. C. Milne, P. J. Belshaw and C. T. Walsh, Thiazole and Oxazole Peptides: Biosynthesis and Molecular Machinery, Nat. Prod. Rep., 1999, 16(2), 249–263, 10.1039/a806930a
- B. McKeever and G. Pattenden, Total Synthesis of the Prenylated Cyclopeptide Trunkamide A, a Cytotoxic Metabolite from Lissoclinum sp, Tetrahedron Lett., 2001, 42(13), 2573–2577, DOI:10.1016/S0040-4039(01)00194-0
- J. S. Elmore and D. S. Mottram, Investigation of the Reaction Between Ammonium Sulfide, Aldehydes, and α-Hydroxyketones or α-Dicarbonyls to Form Some Lipid− Maillard Interaction Products Found in Cooked Beef, J. Agric. Food Chem., 1997, 45(9), 3595–3602, DOI:10.1021/jf970065u
- R. J. Boyce, G. C. Mulqueen and G. Pattenden, Total Synthesis of Thiangazole, a Novel Inhibitor of HIV-1 from Polyangium Sp, Tetrahedron Lett., 1994, 35(31), 5705–5708, DOI:10.1016/S0040-4039(00)77284-4
- R. W. Boyle, L. Y. Xie and D. Dolphin, Meso-Phenyl Substituted Porphocyanines: A New Class of Functionalized Expanded Porphyrins, Tetrahedron Lett., 1994, 35(30), 5377–5380, DOI:10.1016/S0040-4039(00)73504-0
- J.-H. Lai, J. Yu, B. Mekonnen and J. R. Falck, Synthesis of Curacin A, an Antimitotic Cyclopropane-Thiazoline from the Marine Cyanobacterium Lyngbya Majuscula, Tetrahedron Lett., 1996, 37(40), 7167–7170, DOI:10.1016/0040-4039(96)01616-4
- R. Willstätter and T. Wirth, Ü. Thioformamid, Ber. Dtsch. Chem. Ges., 1909, 42(2), 1908–1922, DOI:10.1002/cber.19090420267
- K. Drauz, I. Grayson, A. Kleemann, H. P. Krimmer, W. Leuchtenberger and C. Weckbecker, Amino Acids, Ullmann's Encyclopedia of Industrial Chemistry, 2007 Search PubMed
- A. Ino and A. Murabayashi, Synthetic Studies of Thiazoline and Thiazolidine-Containing Natural
Products—1. Phosphorus Pentachloride-Mediated Thiazoline Construction Reaction, Tetrahedron, 1999, 55(34), 10271–10282, DOI:10.1016/S0040-4020(99)00582-7
- A. Ino, Y. Hasegawa and A. Murabayashi, Synthetic Studies of Thiazoline and Thiazolidine-Containing Natural Products — 2. Total Synthesis of the Antimycoplasma Antibiotic Micacocidin, Tetrahedron, 1999, 55(34), 10283–10294, DOI:10.1016/S0040-4020(99)00583-9
- W. D. McElroy, The Energy Source for Bioluminescence in an Isolated System, Proc. Natl. Acad. Sci. U. S. A., 1947, 33(11), 342–345, DOI:10.1073/pnas.33.11.342
- J. D. White, T.-S. Kim and M. Nambu, Synthesis of Curacin A: A Powerful Antimitotic from the Cyanobacterium Lyngbya Majuscula, J. Am. Chem. Soc., 1995, 117(20), 5612–5613, DOI:10.1021/ja00125a034
- T. Fukuyama and L. Xu, Total Synthesis of (-)-Tantazole B, J. Am. Chem. Soc., 1993, 115(18), 8449–8450, DOI:10.1021/ja00071a065
- M. Mellah, A. Voituriez and E. Schulz, Chiral Sulfur Ligands for Asymmetric Catalysis, Chem. Rev., 2007, 107(11), 5133–5209, DOI:10.1021/cr068440h
- S. Zhang, R. Pattacini and P. Braunstein in Advances in Organometallic Chemistry and Catalysis, John Wiley & Sons Inc., 2013, pp. 185–198 Search PubMed
- A. C. Gaumont, M. Gulea and J. Levillain, Chem. Rev., 2009, 109, 1371–1401 CrossRef CAS PubMed
- J. I. Badillo-Gómez, M. Gouygou, M. C. Ortega-Alfaro and J. G. López-Cortés, Org. Biomol. Chem., 2021, 19(35), 7497–7517 RSC
- H. Jeon, D. Kim, J. H. Lee, J. Song, W. S. Lee, D. W. Kang, S. Kang, S. B. Lee, S. Choi and K. B. Hong, Hypervalent Iodine-Mediated Alkene Functionalization: Oxazoline and Thiazoline Synthesis via Inter-/Intramolecular Aminohydroxylation and Thioamination, Adv. Synth. Catal., 2018, 360(4), 779–783, DOI:10.1002/adsc.201701087
- A. Dömling, W. Wang and K. Wang, Chemistry and Biology of Multicomponent Reactions, Chem. Rev., 2012, 112(6), 3083–3135, DOI:10.1021/cr100233r
- K. Appalanaidu, T. Dadmal, N. Jagadeesh Babu and R. M. Kumbhare, An Improved One-Pot Multicomponent Strategy for the Preparation of Thiazoline, Thiazolidinone and Thiazolidinol Scaffolds, RSC Adv., 2015, 5(107), 88063–88069, 10.1039/C5RA17278K
- G. C. Mulqueen, G. Pattenden and D. A. Whiting, Synthesis of the Thiazoline-Based Siderophore (S)-Desferrithiocin, Tetrahedron, 1993, 49(24), 5359–5364, DOI:10.1016/S0040-4020(01)82385-1
- Y. Ying, K. Taori, H. Kim, J. Hong and H. Luesch, Total Synthesis and Molecular Target of Largazole, a Histone Deacetylase Inhibitor, J. Am. Chem. Soc., 2008, 130(26), 8455–8459, DOI:10.1021/ja8013727
- C. G. Nasveschuk, D. Ungermannova, X. Liu and A. J. Phillips, A Concise Total Synthesis of Largazole, Solution Structure, and Some Preliminary Structure Activity Relationships, Org. Lett., 2008, 10(16), 3595–3598, DOI:10.1021/ol8013478
- T. Seiser, F. Kamena and N. Cramer, Synthesis and Biological Activity of Largazole and Derivatives, Angew Chem. Int. Ed. Engl., 2008, 47(34), 6483–6485, DOI:10.1002/anie.200802043
- Q. Ren, L. Dai, H. Zhang, W. Tan, Z. Xu and T. Ye, Synlett, 2008, 2379 CAS
- V. Padmavathi, B. C. O. Reddy, A. V. N. Mohan and A. Padmaja, Synthesis of a New Class of Sulfone Linked Bisheterocycles, J. Heterocycl. Chem., 2007, 44(2), 459–462, DOI:10.1002/jhet.5570440229
- N. E. Alom, F. Wu and W. Li, One-Pot Strategy for Thiazoline Synthesis from Alkenes and Thioamides, Org. Lett., 2017, 19(4), 930–933, DOI:10.1021/acs.orglett.7b00079
- T. Fukuhara, C. Hasegawa and S. Hara, A Facile Synthesis of Oxazolines, Thiazolines, and Imidazolines Using α, α-Difluoroalkylamines, ChemInform, 2007, 38(40), 1528–1534, DOI:10.1002/chin.200740127
- Z. A. Alsharif and M. A. Alam, Modular Synthesis of Thiazoline and Thiazole Derivatives by Using a Cascade Protocol, RSC Adv., 2017, 7(52), 32647–32651, 10.1039/C7RA05993K
- D. Bonnet-Delpon, J.-P. B′egúe and B. Crousse, Synlett, 2004, 18–29 CrossRef
- C. Shen, L. Wang, M. Wen, H. Shen, J. Jin and P. Zhang, Synthesis of Benzimidazo[1,2- c]Quinazolines via Metal-Free Intramolecular C–H Amination Reaction, Ind. Eng. Chem. Res., 2016, 55(11), 3177–3181, DOI:10.1021/acs.iecr.5b04452
- S. Kamila and E. R. Biehl, Synthesis of Thiazolines by the Reaction of Aryl Ketonitriles with Cysteamine via Microwave Irradiation, J. Heterocycl. Chem., 2007, 44(2), 407–409, DOI:10.1002/jhet.5570440220
- A. Sakakura, R. Kondo, S. Umemura and K. Ishihara, Catalytic Synthesis of Peptide-Derived Thiazolines and Oxazolines Using Bis(Quinolinolato)Dioxomolybdenum(VI) Complexes, Adv. Synth. Catal., 2007, 349(10), 1641–1646, DOI:10.1002/adsc.200700068
- E. Viñuelas-Zahínos, F. Luna-Giles, P. Torres-García and A. Bernalte-García, Synthesis, Characterization and Crystal Structure of a Novel Thiazoline/Thiazolidine Azine Ligand and Its Nickel, Copper and Zinc Complexes, Polyhedron, 2009, 28(18), 4056–4064, DOI:10.1016/j.poly.2009.09.032
- O. A. Attanasi, S. Berretta, L. D. De Crescentini, G. Favi, P. Filippone, G. Giorgi, S. Lillini and F. Mantellini, Unexpected Regioselectivity in the Reaction between Cycloalkenyl-1-Diazenes and Thioamides: Useful Entry to Fused Cycloalkyl-Thiazoline and Cycloalkyl-Thiazoline–Pyrazole Systems, Tetrahedron Lett., 2007, 48(14), 2449–2451, DOI:10.1016/j.tetlet.2007.02.041
- A. I. Meyers, J. L. Durandetta and R. Munavu, J. Org. Chem., 1975, 40, 2025 CrossRef CAS
- J. Laduranty, F. Barbot and L. Miginiac, Bull. Soc. Chim. Fr., 1989, 6, 850 Search PubMed
- M. W. Nötzel, T. Labahn, M. Es-Sayed and A. de Meijere, Eur. J. Org Chem., 2001, 3025 CrossRef
- R. Brossmer and H. Mack, Tetrahedron Lett., 1981, 22, 933 CrossRef CAS
- D. R. Williams, P. D. Lowder, Y.-G. Gu and D. A. Brooks, Tetrahedron Lett., 1997, 38, 331 CrossRef CAS
- R. S. Brown, J. Dowden, C. Moreau and B. V. L. Potter, Tetrahedron Lett., 2002, 43, 6561 CrossRef CAS
- R. S. Brown, J. Dowden, C. Moreau and B. V. L. Potter, Tetrahedron Lett., 2002, 43, 6561 CrossRef CAS
- R. Caujolle, G. Baziard-Mouysset, J. D. Favrot, M. Payard, P. R. Loiseau, H. Amarouch, M. D. Linas, J. P. Seguela, P. M. Loiseau, C. Bories and P. Gayral, Eur. J. Med. Chem., 1993, 28, 29 CrossRef CAS
- M. C. Elliot, A. E. Monk, E. Kruiswijk, D. E. Hibbs, R. L. Jenkins and D. V. Jones, Synlett, 1999, 1379 CrossRef
- K. Inami and T. Shiba, Tetrahedron Lett., 1984, 25, 2009 CrossRef CAS
- K. C. Nicolaou, M. Nevalainen, M. Zak, S. Bulat, S. Bella and B. S. Safina, Angew. Chem., Int. Ed., 2003, 42, 3418 CrossRef CAS PubMed
- S. F. Lu, D. M. Du and J. Xu, Org. Lett., 2006, 8, 2115 CrossRef CAS PubMed
- T. Nishio, Y. Kodama and Y. Tsurumi, Phosphorus, Sulfur Silicon Relat. Elem., 2005, 180, 1449 CrossRef CAS
- S. Muthusamy, K. Rajalakshmi, D. Zhu, W. Zhu, S. Wang, K.-B. Lee, H. Xu and L. Zhao, Dual Detection of Mercury (II) and Lead (II) Ions Using a Facile Coumarin-Based Fluorescent Probe via Excited State Intramolecular Proton Transfer and Photo-Induced Electron Transfer Processes, Sens. Actuators, B, 2021, 346, 130534, DOI:10.1016/j.snb.2021.130534
- M. Colon, J. L. Todolí, M. Hidalgo and M. Iglesias, Development of Novel and Sensitive Methods for the Determination of Sulfide in Aqueous Samples by Hydrogen Sulfide Generation-Inductively Coupled Plasma-Atomic Emission Spectroscopy, Anal. Chim. Acta, 2008, 609(2), 160–168, DOI:10.1016/j.aca.2008.01.001
- P. Wu and X. P. Yan, A Simple Chemical Etching Strategy to Generate “Ion-Imprinted” Sites on the Surface of Quantum Dots for Selective Fluorescence Turn-On Detecting of Metal Ions, Chem. Commun., 2010, 46(37), 7046–7048, 10.1039/c0cc01762k
- L. Zou, Z. Gu and M. Sun, Review of the Application of Quantum Dots in the Heavy-Metal Detection, Toxicol. Environ. Chem., 2015, 97(3–4), 477–490, DOI:10.1080/02772248.2015.1050201
- P. Xue, R. Lu, P. Zhang, J. Jia, Q. Xu, T. Zhang, M. Takafuji and H. Ihara, Amplifying Emission Enhancement and Proton Response in Two-Component Gel, Langmuir, 2013, 29(1), 417–425, DOI:10.1021/la3037617
- C. M. Sousa, J. Berthet, S. Delbaere and P. J. Coelho, A Closer Look at the Photochromism of Vinylidene-Naphthofurans, Dyes Pigm., 2017, 137, 593–600, DOI:10.1016/j.dyepig.2016.11.001
- S. Erdemir, M. Oguz and S. Malkondu, A NIR Fluorescent Sensor Based on Thiazoline-Isophorone with Low Cytotoxicity in Living Cells for Hg2+ Detection through ICT Associated Hydrogen Bonding Effect, Anal. Chim. Acta, 2022, 1192, 339353, DOI:10.1016/j.aca.2021.339353
- S. Erdemir and S. Malkondu, Detection of Water Content in Alcohol Solvents over Al3+ Induced Colorimetric and NIR-Fluorescent Sensor Based on Isophorone-Phenylamine, Microchem. J., 2021, 160, 105677, DOI:10.1016/j.microc.2020.105677
- R. Tang, X. Wang, W. Zhang, X. Zhuang, S. Bi, W. Zhang and F. Zhang, Aromatic Azaheterocycle-Cored Luminogens with Tunable Physical Properties via Nitrogen Atoms for Sensing Strong Acids, J. Mater. Chem. C, 2016, 4(32), 7640–7648, 10.1039/C6TC02591A
- K. R. Solomon, G. J. M. Velders, S. R. Wilson, S. Madronich, J. Longstreth, P. J. Aucamp and J. F. Bornman, Sources, Fates, Toxicity, and Risks of Trifluoroacetic Acid and Its Salts: Relevance to Substances Regulated Under the Montreal and Kyoto Protocols, J. Toxicol. Environ. Health, Part B, 2016, 19(7), 289–304, DOI:10.1080/10937404.2016.1175981
- S. Chaudhary, M. Mukherjee, T. K. Paul, S. Taraphder and M. D. Milton, Novel Thiazoline-Phenothiazine Based ‘Push-Pull’ Molecules as Fluorescent Probes for Volatile Acids Detection, J. Photochem. Photobiol., A, 2020, 397, 112509, DOI:10.1016/j.jphotochem.2020.112509
- G. Sivaraman, M. Iniya, T. Anand, N. G. Kotla, O. Sunnapu, S. Singaravadivel, A. Gulyani and D. Chellappa, Coord. Chem. Rev., 2018, 357, 50–104 CrossRef CAS
- D. Wu, A. C. Sedgwick, T. Gunnlaugsson, E. U. Akkaya, J. Yoon and T. D. James, Fluorescent Chemosensors: The Past, Present and Future, Chem. Soc. Rev., 2017, 46(23), 7105–7123, 10.1039/c7cs00240h
- J. Wang, J. Liang, X. Liu, H. Xiao, F. Dong, Y. Wang, X. Shu, F. Huang and H. B. Liu, Thiazoline−Pyrene Selective and Sensitive Fluorescence ‘Turn-On’ Sensor for Detection of Cu2+, Spectrochim. Acta, Part A, 2019, 215, 260–265, DOI:10.1016/j.saa.2019.02.066
- A. Ruduss, B. Turovska, S. Belyakov, K. A. Stucere, A. Vembris, G. Baryshnikov, H. Ågren, J. C. Lu, W. H. Lin, C. H. Chang and K. Traskovskis, Thiazoline Carbene-Cu(I)-Amide Complexes: Efficient White Electroluminescence from Combined Monomer and Excimer Emission, ACS Appl. Mater. Interfaces, 2022, 14(13), 15478–15493, DOI:10.1021/acsami.2c00847
- I. Piel, M. D. Pawelczyk, K. Hirano, R. Fröhlich and F. Glorius, A Family of Thiazolium Salt Derived N-Heterocyclic Carbenes (NHCs) for Organocatalysis: Synthesis, Investigation and Application in Cross-Benzoin Condensation, Eur. J. Org Chem., 2011, 2011(28), 5475–5484, DOI:10.1002/ejoc.201100870
- J. S. Casas, M. S. García-Tasende and J. Sordo, Coord. Chem. Rev., 2000, 209, 197 CrossRef CAS
- E. Viñuelas-Zahínos, F. Luna-Giles, P. Torres-García, A. B. Rodríguez and A. Bernalte-García, Effects of a Derivative Thiazoline/Thiazolidine Azine Ligand and Its Cadmium Complexes on Phagocytic Activity by Human Neutrophils, Inorg. Chim. Acta, 2011, 366(1), 373–379, DOI:10.1016/j.ica.2010.11.037
- M. Mellah, A. Voituriez and E. Schulz, Chiral Sulfur Ligands for Asymmetric Catalysis, Chem. Rev., 2007, 107(11), 5133–5209, DOI:10.1021/cr068440h
- I. Abrunhosa, L. Delain-Bioton, A.-C. Gaumont, M. Gulea and S. Masson, Chiral Thiazoline Ligands: Application in Pd-Catalysed Allylic Substitution, Tetrahedron, 2004, 60(41), 9263–9272, DOI:10.1016/j.tet.2004.07.048
- G. Helmchen, A. Krotz, K. T. Ganz and D. Hansen, C2-Symmetric Bioxazolines and Bithiazolines as New Chiral Ligands for Metal Ion Catalyzed Asymmetric Syntheses: Asymmetric Hydrosilylation, Synlett, 1991, 04, 257–259 CrossRef
- R. Corona-Sánchez, R. A. Toscano, M. C. Ortega-Alfaro, C. Sandoval-Chávez and J. G. López-Cortés, 2-Ferrocenyl-2-Thiazoline as a Building Block of Novel Phosphine-Free Ligands, Dalton Trans., 2013, 42(33), 11992–12004, 10.1039/c3dt50451d
- I. Abrunhosa, M. Gulea, J. Levillain and S. Masson, Synthesis of New Chiral Thiazoline-Containing Ligands, Tetrahedron: Asymmetry, 2001, 12(20), 2851–2859, DOI:10.1016/S0957-4166(01)00481-5
- A.-C. Gaumont, M. Gulea and J. Levillain, Overview of the Chemistry of 2-Thiazolines, Chem. Rev., 2009, 109(3), 1371–1401, DOI:10.1021/cr800189z
- M. Amini, A. Bayrami, M. N. Marashi, A. Arab, A. Ellern and L. Keith Woo, Synthesis, Structure, and Catalytic Properties of Copper, Palladium and Cobalt Complexes Containing an N,O-Type BidentateThiazoline Ligand, Inorg. Chim. Acta, 2015, 443(2016), 22–27 Search PubMed
- M. Irmak, T. Lehnert and M. M. K. Boysen, First Synthesis of a Carbohydrate-Derived Pyridyl Bis(Thiazoline) Ligand, Tetrahedron Lett., 2007, 48(44), 7890–7893, DOI:10.1016/j.tetlet.2007.08.101
- S. Knapp, D. Vocadlo, Z. Gao, B. Kirk, J. Lou and S. G. Withers, NAG-Thiazoline, an N -Acetyl-Hexosaminidase Inhibitor That Implicates Acetamido Participation, J. Am. Chem. Soc., 1996, 118(28), 6804–6805, DOI:10.1021/ja960826u
- M. Sudharsan, K. Thirumoorthy, M. Nethaji and D. Suresh, Synthesis, Characterization and Theoretical Investigation on Thiazoline-Derived Palladium-Complexes-Catalyzed Denitrogenative Cross-Coupling of Aryl Halides with Arylhydrazines, ChemistrySelect, 2019, 4(32), 9253–9261, DOI:10.1002/slct.201902137
- S. C. McKeon, H. Müller-Bunz and P. J. Guiry, New Thiazoline–Oxazoline Ligands Application in Asymmetric Friedel–Crafts Reaction, Eur. J. Org. Chem., 2009, 28, 4833–4841 CrossRef
- S. C. McKeon, H. Müller-Bunz and P. J. Guiry, Synthesis of thiazoline–oxazoline Ligands and their application in asymmetric Catalysis, Eur. J. Org. Chem., 2011, 35, 7107–7115, DOI:10.1002/ejoc.201101335
- H. Liu, S. F. Lu, J. Xu and D.-M. Du, Asymmetric Friedel–Crafts Alkylation of Electron-Rich N-Heterocycles with Nitroalkenes Catalyzed by Diphenylamine-Tethered Bis(Oxazoline) and Bis(Thiazoline) ZnII Complexes, Chem.–Asian J., 2008, 3(7), 1111–1121, DOI:10.1002/asia.200800071
- I. Abrunhosa-Thomas, A. Betz, M. Denancé, I. Dez, A.-C. Gaumont and M. Gulea, Synthesis of Chiral Thiazoline Ligands Tethered to s Sulfur Function and First Immobilization of a Thiazoline-Ligand, Heteroat. Chem., 2010, 21(4), 242–249, DOI:10.1002/hc.20603
- I. Abrunhosa, M. Gulea, J. Levillain and S. Masson, Synthesis of New Chiral Thiazoline-Containing Ligands, Tetrahedron: Asymmetry, 2001, 12(20), 2851–2859, DOI:10.1016/S0957-4166(01)00481-5
- D. J. Weix and J. A. Ellman, Improved Synthesis of Tert-Butanesulfinamide Suitable for Large-Scale Production, Org. Lett., 2003, 5(8), 1317–1320, DOI:10.1021/ol034254b
- J. Meijer, P. Vermeer and L. Brandsma, A Simple Preparative Method for Dithioesters, Recl. Trav. Chim. Pays-Bas, 1973, 92(6), 601–604, DOI:10.1002/recl.19730920605
- A. Jemal, F. Bray, M. M. Center, J. Ferlay, E. Ward and D. Forman, Global Cancer Statistics, Ca-Cancer J. Clin., 2011, 61(2), 69–90, DOI:10.3322/caac.20107
- S.-K. Kim, Handbook of Anticancer Drugs from Marine Origin, Springer International Publishing, 2015 Search PubMed
- F. Rojo, J. Albanell, A. Rovira, J. M. Corominas and F. Manzarbeitia, Targeted Therapies in Breast Cancer, Semin. Diagn. Pathol., 2008, 25(4), 245–261, DOI:10.1053/j.semdp.2008.08.001
- F. S. Han, H. Osajima, M. Cheung, H. Tokuyama and T. Fukuyama, Novel Structural Motifs Consisting of Chiral Thiazolines: Synthesis, Molecular Recognition, and Anticancer Activity, Chemistry, 2007, 13(11), 3026–3038, DOI:10.1002/chem.200601446
- S. Carmeli, R. E. Moore and G. L. Patterson, Mirabazoles, Minor Tantazole-Related Cytotoxins from the Terrestrial Blue-Green Alga Scytonema Mirabile, Tetrahedron Lett., 1991, 32(23), 2593–2596, DOI:10.1016/S0040-4039(00)78793-4
- F. S. Han, H. Osajima, M. Cheung, H. Tokuyama and T. Fukuyama, Novel Structural Motifs Consisting of Chiral Thiazolines: Synthesis, Molecular Recognition, and Anticancer Activity, Chemistry, 2007, 13(11), 3026–3038, DOI:10.1002/chem.200601446
- S. M. Sondhi, M. Johar, N. Singh and S. G. Dastidar, Synthesis of Biscoupled Hemin-Thiazoline Derivatives and Their Anticancer Activity Evaluation, Indian J. Chem., 2004, 43, 162–167 Search PubMed
- D. D. Miller, J. T. Dalton, V. Gududuru and E. Hurh, Thiazoline Analogs as Cell Proliferation Inhibitors, Chem, 2005, 143, 326352 Search PubMed
- R. A. Ng and Z. Sui, Preparation of Thiazoline Derivatives as Selective Androgen Receptor Modulators (SARMs), Chem, 2005, 142, 74557 Search PubMed
- M. J. Pine, E. A. Mirand, J. L. Ambrus and F. G. Bock, Antitumor Studies of 2- Amino-2-Thiazoline and Other Tumor-Modifying Agents, J. Med., 1983, 14(5–6), 433–449, DOI:10.1016/S0022-5347(17)49541-3
- M. H. Shih and F. Y. Ke, Syntheses and Evaluation of Antioxidant Activity of Sydnonyl Substituted Thiazolidinone and Thiazoline Derivatives, Bioorg. Med. Chem., 2004, 12(17), 4633–4643, DOI:10.1016/j.bmc.2004.06.033
- Y. Igarashi, D. Asano, M. Sawamura, Y. In, T. Ishida and M. Imoto, Ulbactins F. and G, Polycyclic Thiazoline Derivatives with Tumor Cell Migration Inhibitory Activity from Brevibacillus sp, Org. Lett., 2016, 18(7), 1658–1661 CrossRef CAS PubMed
- W. Wang, B. Zhao, C. Xu and W. Wu, Synthesis and Atitumor Activity of the Thiazoline and Thiazine Multithioether, Int. J. Org. Chem., 2012, 02(2), 117–120, DOI:10.4236/ijoc.2012.22018
- E. A. E. El-Helw and A. A. El-Badawy, Synthesis of Chromenone, Pyrimidinone, Thiazoline, and Quinolone Derivatives as Prospective Antitumor Agents, J. Heterocycl. Chem., 2020, 57(6), 2354–2364, DOI:10.1002/jhet.3948
- G. Turan-Zitouni, L. Yurttaş, A. Tabbi, G. Akalın Çiftçi, H. E. Temel and Z. A. Kaplancıklı, New Thiazoline-Tetralin Derivatives and Biological Activity Evaluation, Molecules, 2018, 23(1), 135, DOI:10.3390/molecules23010135
- G. Turan-Zitouni, A. Ozdemir, Z. A. Kaplancikli, G. Revial and F. Demirci, Synthesis and Antimicrobial Activity Evaluation of New Hydrazide Derivatives, Turk. J. Pharm. Sci., 2011, 8, 199–206 Search PubMed
- G. Turan-Zitouni, Z. A. Kaplancıklı, K. Erol and F. S. Kılıç, Synthesis and Analgesic Activity of Some Triazoles and Triazolothiazines, Farmaco, 1999, 54(4), 218–223, DOI:10.1016/S0014-827X(99)00016-6
- S. L. You, H. Razavi and J. W. Kelly, A Biomimetic Synthesis of Thiazolines Using Hexaphenyloxodiphosphonium Trifluoromethanesulfonate, Angew. Chem., Int. Ed., 2003, 42(1), 83–85, DOI:10.1002/anie.200390059
- A. K. Ghosh and S. Kulkarni, Enantioselective Total Synthesis of (+)-Largazole, a Potent Inhibitor of Histone Deacetylase, Org. Lett., 2008, 10(17), 3907–3909, DOI:10.1021/ol8014623
- Y. Numajiri, T. Takahashi, M. Takagi, K. Shin-Ya and T. Doi, Synlett, 2008, 2483 CAS
- K. Taori, V. J. Paul and H. Luesch, Structure and Activity of Largazole, a Potent Antiproliferative Agent from the Floridian Marine Cyanobacterium Symploca Sp, J. Am. Chem. Soc., 2008, 130(6), 1806–1807, DOI:10.1021/ja7110064
- J. M. Guerra-Bubb, A. A. Bowers, W. B. Smith, R. Paranal, G. Estiu, O. Wiest, J. E. Bradner and R. M. Williams, Synthesis and HDAC Inhibitory Activity of Isosteric Thiazoline-Oxazole Largazole Analogs, Bioorg. Med. Chem. Lett., 2013, 23(21), 6025–6028, DOI:10.1016/j.bmcl.2013.06.012
- A. A. Bowers, N. West, T. L. Newkirk, A. E. Troutman-Youngman, S. L. Schreiber, O. Wiest, J. E. Bradner and R. M. Williams, Synthesis and Histone Deacetylase Inhibitory Activity of Largazole Analogs: Alteration of the Zinc-Binding Domain and Macrocyclic Scaffold, Org. Lett., 2009, 11(6), 1301–1304, DOI:10.1021/ol900078k
- A. Bowers, N. West, J. Taunton, S. L. Schreiber, J. E. Bradner and R. M. Williams, Total Synthesis and Biological Mode of Action of Largazole: A Potent Class I Histone Deacetylase Inhibitor, J. Am. Chem. Soc., 2008, 130(33), 11219–11222, DOI:10.1021/ja8033763
- A. A. Bowers, T. J. Greshock, N. West, G. Estiu, S. L. Schreiber, O. Wiest, R. M. Williams and J. E. Bradner, Synthesis and Conformation-Activity Relationships of the Peptide Isosteres of FK228 and Largazole, J. Am. Chem. Soc., 2009, 131(8), 2900–2905, DOI:10.1021/ja807772w
- A. T. Taher, N. A. Khalil and E. M. Ahmed, Synthesis of Novel Isatin-Thiazoline and Isatin-Benzimidazole Conjugates as Anti-breast Cancer Agents, Arch. Pharmacal Res., 2011, 34(10), 1615–1621, DOI:10.1007/s12272-011-1005-3
- A. M. Thayer, Platinum Drugs Take Their Toll, Chem. Eng. News Arch., 2010, 88(26), 24–28, DOI:10.1021/cen-v088n026.p024
- W. H. Ang, A. Casini, G. Sava and P. J. Dyson, Organometallic Ruthenium-Based Antitumor Compounds with Novel Modes of Action, J. Organomet. Chem., 2011, 696(5), 989–998, DOI:10.1016/j.jorganchem.2010.11.009
- J. Espino, E. Fernández-Delgado, S. Estirado, F. de la Cruz-Martinez, S. Villa-Carballar, E. Viñuelas-Zahínos, F. Luna-Giles and J. A. Pariente, Synthesis and Structure of a New Thiazoline-Based Palladium(II) Complex That Promotes Cytotoxicity and Apoptosis of Human Promyelocytic Leukemia HL-60 Cells, Sci. Rep., 2020, 10(1), 16745, DOI:10.1038/s41598-020-73488-0
- R. J. Outcalt, On the Reaction of 2-Chloroethylisothiocyanate with Aromatic Amines, J. Heterocycl. Chem., 1987, 24(5), 1425–1428, DOI:10.1002/jhet.5570240540
- P. Cintas, M. Avalos, R. Babiano, J. L. Jiménez, J. C. Palacios and C. Valencia, Structure of Adducts of 2-Arylaminothiazolines with Isocyanates and Isothiocyanates, Heterocycles, 1993, 35(2), 1237–1246, DOI:10.3987/COM-92-S(T)121
- J. C. Kwan, R. Ratnayake, K. A. Abboud, V. J. Paul and H. Luesch, Grassypeptolides A−C, Cytotoxic Bis-Thiazoline Containing Marine Cyclodepsipeptides, J. Org. Chem., 2010, 75(23), 8012–8023, DOI:10.1021/jo1013564
- M. D. Altıntop, Z. A. Kaplancıklı, G. A. Çiftçi and R. Demirel, Synthesis and Biological Evaluation of Thiazoline Derivatives as New Antimicrobial and Anticancer Agents, Eur. J. Med. Chem., 2014, 74, 264–277, DOI:10.1016/j.ejmech.2013.12.060
- Y. N. Mabkhot, H. Algarni, A. Alsayari, A. Bin Muhsinah, N. A. Kheder, Z. M. Almarhoon and F. A. Al-Aizari, Synthesis, X-ray analysis, biological evaluation and molecular docking study of new thiazoline derivatives, Molecules, 2019, 1654(9), 24 Search PubMed
- Z. M. Alamshany, N. Y. Tashkandi, I. M. M. Othman, M. M. Anwar and E. S. Nossier, New Thiophene, Thienopyridine and Thiazoline-Based Derivatives: Design, Synthesis and Biological Evaluation as Antiproliferative Agents and Multitargeting Kinase Inhibitors, Bioorg. Chem., 2022, 127, 105964, DOI:10.1016/j.bioorg.2022.105964
- H. Z. Shams, R. M. Mohareb, M. H. Helal and A. E. Mahmoud, Novel Synthesis and Antitumor Evaluation of Polyfunctionally Substituted Heterocyclic Compounds Derived from 2-Cyano-N-(3-Cyano-4, 5, 6, 7-Tetrahydrobenzo [b] Thiophen-2-yl)-Acetamide, Molecules, 2010, 16(1), 52–73, DOI:10.3390/molecules16010052
- https://www.cdc.gov/drugresistance/pdf/ar-threats-2013-508.
- R. S. Shapiro, N. Robbins and L. E. Cowen, Regulatory Circuitry Governing Fungal Development, Drug Resistance, and Disease, Microbiol. Mol. Biol. Rev., 2011, 75(2), 213–267, DOI:10.1128/MMBR.00045-10
- M. A. Pfaller and D. J. Diekema, Epidemiology of Invasive Candidiasis: A Persistent Public Health Problem, Clin. Microbiol. Rev., 2007, 20(1), 133–163, DOI:10.1128/CMR.00029-06
- M. K. Kathiravan, A. B. Salake, A. S. Chothe, P. B. Dudhe, R. P. Watode, M. S. Mukta and S. Gadhwe, The Biology and Chemistry of Antifungal Agents: A Review, Bioorg. Med. Chem., 2012, 20(19), 5678–5698, DOI:10.1016/j.bmc.2012.04.045
- S. M. El-Khawass, M. A. Khalil and G. G. Tawil, Synthesis of Some Novel Thiazoline and 4-Thiazolidone Derivatives of Potential Antimicrobial Activity, Sci. Pharm., 1980, 48, 219–224 CAS
- S. A. M. El-Hawash and A. E. A. Wahab, Arch. Pharm. Chem. Life Sci., 2006, 339, 437–447 CrossRef CAS PubMed
- C. G. Bonde and N. J. Gaikwad, Synthesis and Preliminary Evaluation of Some Pyrazine Containing Thiazolines and Thiazolidinones as Antimicrobial Agents, Bioorg. Med. Chem., 2004, 12(9), 2151–2161, DOI:10.1016/j.bmc.2004.02.024
- Y. I. Asiri, A. B. Muhsinah, A. Alsayari, H. A. Ghabbour, Z. M. Almarhoon, F. A. Al-aizari, K. Venkatesan, S. Tasqeeruddin, S. S. Sulthana and Y. N. Mabkhot, Design, Synthesis, X-Ray Analysis, and Biological Screening of New Oxime and Enaminone Thiazoline-2-Thione Derivatives, J. Mol. Struct., 2021, 1223, 128977, DOI:10.1016/j.molstruc.2020.128977
- S. Bondock, W. Khalifa and A. A. Fadda, Synthesis and Antimicrobial Evaluation of Some New Thiazole, Thiazolidinone and Thiazoline Derivatives Starting from 1-Chloro-3,4-Dihydronaphthalene-2-Carboxaldehyde, Eur. J. Med. Chem., 2007, 42(7), 948–954, DOI:10.1016/j.ejmech.2006.12.025
- E. Viñuelas-Zahínos, F. Luna-Giles, P. Torres-García and M. C. Fernández-Calderón, Co(III), Ni(II), Zn(II) and Cd(II) Complexes with 2-Acetyl-2-Thiazoline Thiosemicarbazone: Synthesis, Characterization, X-ray Structures and Antibacterial Activity, Eur. J. Med. Chem., 2011, 46(1), 150–159, DOI:10.1016/j.ejmech.2010.10.030
- T. Doornbos and H. G. Peer, Synthesis of 2-acyl-2-Thiazolines, Recl. Trav. Chim. Pays-Bas, 1972, 91(6), 711–728, DOI:10.1002/recl.19720910612
- A. Ahmad, A. Ahmad, R. Sudhakar, H. Varshney, N. Subbarao, S. Ansari, A. Rauf and A. U. Khan, Designing, Synthesis, and Antimicrobial Action of Oxazoline and Thiazoline Derivatives of Fatty Acid Esters, J. Biomol. Struct. Dyn., 2017, 35(15), 3412–3431, DOI:10.1080/07391102.2016.1255260
- M. A. Gouda, M. A. Berghot, A. I. Shoeib and A. M. Khalil, Synthesis and Antimicrobial of Certain New Thiazolidinone, Thiazoline, and Thiophene Derivatives, Phosphorus, Sulfur Silicon Relat. Elem., 2010, 185(7), 1455–1462, DOI:10.1080/10426500903074858
- D. R. Tatke and S. Seshadri, Indian J. Chem., 1983, 22(12), 1197 Search PubMed
- N. Li, T. Ke, L. Shi, Z. Zhang, W. Fang, Y. N. Zhang, K. Wang, R. Zhou, Z. Wan, Z. Yang, G. Zhang and Y. Wei, Synthesis and Evaluation of Steroidal Thiazoline Conjugates as Potential Antiviral Agents, Future Med. Chem., 2018, 10(22), 2589–2605, DOI:10.4155/fmc-2018-0075
- M. I. Merlani, L. S. Amiranashvili, M. G. Davitishvili, E. P. Kemertelidze, K. Papadopoulos and E. Yannakopoulou, Synthesis of Novel Steroidal Isonicotinylhydrazones and Thiosemicarbazones from Tigogenin, Chem. Nat. Compd., 2006, 42(2), 194–197, DOI:10.1007/s10600-006-0076-8
- S. Ke, L. Shi and Z. Yang, Discovery of Novel Isatin-Dehydroepiandrosterone Conjugates as Potential Anticancer Agents, Bioorg. Med. Chem. Lett., 2015, 25(20), 4628–4631, DOI:10.1016/j.bmcl.2015.08.041
- R. Meleddu, S. Distinto, A. Corona, E. Tramontano, G. Bianco, C. Melis, F. Cottiglia and E. Maccioni, Isatin Thiazoline Hybrids as Dual Inhibitors of HIV-1 Reverse Transcriptase, J. Enzyme Inhib. Med. Chem., 2017, 32(1), 130–136, DOI:10.1080/14756366.2016.1238366
- M. A. Hussein, A. H. Kafafy, S. G. Abdel-Moty and O. M. Abou-Ghadir, Synthesis and Biological Activities of New Substituted Thiazoline-Quinoline Derivatives, Acta Pharm., 2009, 59(4), 365–382, DOI:10.2478/v10007-009-0033-8
- R. Khursheed, S. K. Singh, S. Wadhwa, B. Kapoor, M. Gulati, R. Kumar, A. K. Ramanunny, A. Awasthi and K. Dua, Treatment Strategies Against Diabetes: Success so Far and Challenges Ahead, Eur. J. Pharmacol., 2019, 862, 172625, DOI:10.1016/j.ejphar.2019.172625
- American Diabetes Association, 12. Older Adults: Standards of Medical Care in Diabetes—2019, Diabetes Care, 2019, 42(1), S139–S147, DOI:10.2337/dc19-S012
- M. Blair, Diabetes Mellitus Review, Urol. Nurs., 2016, 36(1), 27–36, DOI:10.7257/1053-816X.2016.36.1.27
- Y. Shi and F. B. Hu, The Global Implications of Diabetes and Cancer, Lancet, 2014, 383, 1947 CrossRef PubMed
- American Diabetes Association, Standards of Medical Care in Diabetes—2014, Diabetes Care, 2014, 37(1), S14–S80, DOI:10.2337/dc14-S014
- H. U. Demuth, C. H. S. McIntosh and R. A. Pederson, Type 2 Diabetes–Therapy with Dipeptidyl Peptidase IV Inhibitors, Biochim. Biophys. Acta, 2005, 1751(1), 33–44, DOI:10.1016/j.bbapap.2005.05.010
- Z. Ali, M. J. Akhtar, A. A. Siddiqui, A. A. Khan, M. R. Haider and M. S. Yar, Design, Synthesis, and Biological Evaluation of Novel Quinazoline Clubbed Thiazoline Derivatives, Arch. Pharm., 2017, 350(2), 1600298, DOI:10.1002/ardp.201600298
- J. M. Pattanaik, M. Pattanaik and D. Bhatta, Indian J. Chem. Sect. B, 1998, 37, 1304–1306 Search PubMed
- A. K. Nanda, S. Ganguli and R. Chakraborty, Antibacterial Activity of Some 3-(Arylideneamino)-2-Phenylquinazoline-4(3H)-Ones: Synthesis and Preliminary QSAR Studies, Molecules, 2007, 12(10), 2413–2426, DOI:10.3390/12102413
- M. T. Shehzad, A. Imran, A. Hameed, M. A. Rashida al, M. Bibi, M. Uroos, A. Asari, S. Iftikhar, H. Mohamad, M. N. Tahir, Z. Shafiq and J. Iqbal, Exploring Synthetic and Therapeutic Prospects of New Thiazoline Derivatives as Aldose Reductase (ALR2) Inhibitors, RSC Adv., 2021, 11(28), 17259–17282, 10.1039/d1ra01716k
- T. Liu, M. Xia, H. Zhang, H. Zhou, J. Wang, X. Shen and Q. Yang, Exploring NAG-Thiazoline and Its Derivatives as Inhibitors of Chitinolytic-Acetylglucosaminidases, FEBS Lett., 2015, 589(1), 110–116, DOI:10.1016/j.febslet.2014.11.032
- G. Hunt, C. Todd, J. E. Cresswell and A. J. Thody, Alpha-Melanocyte Stimulating Hormone and Its Analogue Nle4DPhe7 Alpha-MSH Affect Morphology, Tyrosinase Activity and Melanogenesis in Cultured Human Melanocytes, J. Cell Sci., 1994, 107(1), 205–211, DOI:10.1242/jcs.107.1.205
- D. S. Kim, Y. M. Jeong, I. K. Park, H. G. Hahn, H. K. Lee, S. B. Kwon, J. H. Jeong, S. J. Yang, U. D. Sohn and K. C. Park, A New 2-Imino-1,3-Thiazoline Derivative, KHG22394, Inhibits Melanin Synthesis in Mouse B16 Melanoma Cells, Biol. Pharm. Bull., 2007, 30(1), 180–183, DOI:10.1248/bpb.30.180
- D. S. Reddy, K. M. Hosamani, H. C. Devarajegowda and M. M. Kurjogi, A Facile Synthesis and Evaluation of New Biomolecule-Based Coumarin–Thiazoline Hybrids as Potent Anti-tubercular Agents with Cytotoxicity, DNA Cleavage and X-ray Studies, RSC Adv., 2015, 5(79), 64566–64581, 10.1039/C5RA09508E
- M. Shahid, M. Salim, M. Khalid, M. N. Tahir, M. U. Khan and A. A. C. Braga, Synthetic, XRD, Non-covalent Interactions and Solvent Dependent Nonlinear Optical Studies of Sulfadiazine-ortho-Vanillin Schiff Base:(E)-4-((2-hydroxy-3-methoxy-benzylidene) Amino)-N-(Pyrimidin-2-yl) Benzene-Sulfonamide, J. Mol. Struct., 2018, 1161, 66–75, DOI:10.1016/j.molstruc.2018.02.043
- M. Ghiasuddin, M. Akram, M. Adeel, M. N. Khalid, M. N. Tahir, M. U. Khan, M. A. Asghar, M. A. Ullah and M. Iqbal, A Combined Experimental and Computational Study of 3-Bromo-5-(2,5-Difluorophenyl) Pyridine and 3, 5-bis (Naphthalen-1-yl) Pyridine: Insight into the Synthesis, Spectroscopic, Single Crystal XRD, Electronic, Nonlinear Optical and Biological Properties, J. Mol. Struct., 2018, 1160, 129–141, DOI:10.1016/j.molstruc.2018.01.100
- M. Haroon, M. Khalid, Z. Shafiq, M. U. Khan and M. R. S. A. Janjua, High-Throughput Calculations and Experimental Insights Towards the Development of Potent Thiazoline Based Functional Materials, Mater. Today Commun., 2021, 27, 102485, DOI:10.1016/j.mtcomm.2021.102485
- J. Deeley, A. Bertram and G. Pattenden, Novel Polyoxazole-Based Cyclopeptides from Streptomyces Sp. Total Synthesis of the Cyclopeptide YM-216391 and Synthetic Studies towards Telomestatin, Org. Biomol. Chem., 2008, 6(11), 1994–2010, 10.1039/b802477d
- K. C. Nicolaou, D. H. Dethe, G. Y. C. Leung, B. Zou and D. Y.-K. Chen, Total Synthesis of Thiopeptide Antibiotics GE2270A, GE2270T, and GE2270C1, Chem.–Asian J., 2008, 3(2), 413–429, DOI:10.1002/asia.200700361
- F. Diness, D. S. Nielsen and D. P. Fairlie, Synthesis of the Thiazole–Thiazoline Fragment of Largazole Analogues, J. Org. Chem., 2011, 76(23), 9845–9851, DOI:10.1021/jo201675r
- G. Videnov, D. Kaiser, C. Kempter and G. Jung, Synthesis of Naturally Occurring, Conformationally Restricted Oxazole- and Thiazole-Containing Di- and Tripeptide Mimetics, Angew Chem. Int. Ed. Engl., 1996, 35(1314), 1503–1506, DOI:10.1002/anie.199615031
- R. J. Bergeron, J. Wiegand, J. S. McManis, B. H. McCosar, W. R. Weimar, G. M. Brittenham and R. E. Smith, Effects of C-4 Stereochemistry and C-4‘ Hydroxylation on the Iron Clearing Efficiency and Toxicity of Desferrithiocin Analogues, J. Med. Chem., 1999, 42(13), 2432–2440, DOI:10.1021/jm990058s
- M. W. Bredenkamp, C. W. Holzapfel and W. J. van Zyl, The Chiral Synthesis of Thiazole Amino Acid Enantiomers, Synth. Commun., 1990, 20(15), 2235–2249, DOI:10.1080/00397919008053164
- T. Seiser, F. Kamena and N. Cramer, Synthesis and Biological Activity of Largazole and Derivatives, Angew Chem. Int. Ed. Engl., 2008, 47(34), 6483–6485, DOI:10.1002/anie.200802043
- Q. Zeng, J. G. Allen, M. P. Bourbeau, C. Dominguez, C. H. Fotsch, N. Han, F.-T. Hong, X. Huang, M. R. Lee, A. Li, Q. Liu, J. T. Rider, S. Tadesse, A. S. Tasker, V. N. Viswanadhan, X. Wang, K. E. Weiler, G. E. Wohlhieter, G. Yao and C. C. Yuan, WO2007084391, 2007
- O. Peraud, J. S. Biggs, R. W. Hughen, A. R. Light, G. P. Concepcion, B. M. Olivera and E. W. Schmidt, Microhabitats within Venomous Cone Snails Contain Diverse Actinobacteria, Appl. Environ. Microbiol., 2009, 75(21), 6820–6826, DOI:10.1128/AEM.01238-09
- Z. Lin, R. R. Antemano, R. W. Hughen, M. D. B. Tianero, O. Peraud, M. G. Haygood, G. P. Concepcion, B. M. Olivera, A. Light and E. W. Schmidt, Pulicatins A−E, Neuroactive Thiazoline Metabolites from Cone Snail-Associated Bacteria, J. Nat. Prod., 2010, 73(11), 1922–1926, DOI:10.1021/np100588c
Footnote |
| † Authors have equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |