DOI:
10.1039/D3RA06932J
(Paper)
RSC Adv., 2024,
14, 889-901
The effects of partially or fully linked boron with a cross-linking structure of an organic precursor on the purity and morphology of ZrB2 powder†
Received
11th October 2023
, Accepted 7th December 2023
First published on 2nd January 2024
Abstract
From a chemical infrastructure perspective, it is important to ensure that all ions constituting a target product, e.g., Zr and B ions for ZrB2, are fully linked with a cross-linking structure for synthesis via an organic precursor. In the present study, glycerol is used as a chelating ligand to prepare boron both partially and fully linked with the cross-linking structure of organic precursors by a sol–gel route. The results are far from expected, in that the more linked boron there is in the precursor, the purer the ZrB2 produced. In the case of a partially linked cross-linking structure, the carbothermic reduction reaction for ZrB2 is a multi-step process with an intermediary phase of ZrC, and then a high-purity prism-like ZrB2 powder with a larger size is obtained. A minimum of 0.26 wt% for the oxygen content of ZrB2 corresponds to a 0.67 molar ratio of glycerol to H3BO3. On the other hand, in the case with the boron fully linked, a single-phase of ZrB2 cannot be obtained, and instead a double-phase is obtained. Therefore, the amount of impurity is greater, even though the size is smaller. The carbothermic reduction reaction is direct, and has only one step.
1. Introduction
With the development of aerospace technology, the potential applications of ultra-high temperature ceramics (UHTCs) in the wing leading edges and nose cones of hypersonic aircraft, as well as engine components, have attracted considerable attention.1–6 As a member of the UHTCs, diboride, ZrB2, has become a potential candidate for thermal protection system materials of hypersonic aircraft because of its chemical inertness, low density (6.10 g cm−3), high thermal conductivity (56 W m−1 K−1), and high melting temperature (about 3200 °C).2,7,8
As a raw material, the characteristics of a ceramic powder, such as its purity and morphology, directly impact the performance of downstream products, for instance sintered parts and coatings.9 According to recent studies, reducing the oxygen content for ZrB2 powder could lead to a higher density of sintered ZrB2-based ceramics10 and enhance the mechanical properties of ZrB2–SiC composites.11–13 In addition, various morphologies of ZrB2 powders have been obtained, such as equiaxed,14–19 flake-like,20–24 and rod-like.9,25,26 Flake-like and rod-like ZrB2 particles can enhance the mechanical properties of ZrB2-based composite ceramics.27–29 Furthermore, equiaxed nano-ZrB2 particles are favourable for increasing the density of ceramic materials.30
Solid-state and solution-based synthesis methods are currently the two most effective processes for producing ZrB2 powders on an industrial scale. Generally, solid-state synthesis methods have higher efficiency. However, their disadvantages are inevitable, such as high energy consumption, lower powder purity, and difficulty in controlling the morphology.31–33 Solution-based synthesis methods, including solvothermal and sol–gel synergized with carbothermic reduction reaction methods, could improve the homogeneity of each component, significantly shorten the diffusion distance on the molecular or atomic scale, and reduce the energy barrier of reaction.26,34 Specifically, the sol–gel route synergized with a carbothermic reduction strategy is advantageous for obtaining high-purity powders and could further achieve a near-net-shape in one step, in contrast to the other routes. In general, boric acid (H3BO3) is commonly used as a boron source to synthesize ZrB2 using a sol–gel synergized with carbothermic reduction reaction method.14,35–37 Due to its poor solubility in organic solvents, H3BO3 has been substituted by an organic boron source to improve the solubility.15 However, if boron does not participate in the formation of a sol–gel network structure, just dissolving it may result in heterogeneous distribution of boron during the drying process and affect the properties of the ZrB2 powder.32 Walker et al. used B(C2H5O)3 to synthesize a precursor in which boron was involved in the sol–gel structure, which was used to obtain spherical and elongated rod-like or plate-like ZrB2 particles.33 However, organic boron sources are expensive and difficult to store. Subsequently, researchers used polyols or polysaccharides as ligands and carbon sources to chelate with H3BO3 promoting the uniformity of boron in precursors, such as D-fructose,38 sorbitol,32,39,40 glycerol,34,41 chitosan,42 and mannitol.26,43 The purities and morphologies of ZrB2 particles using different ligands were different.
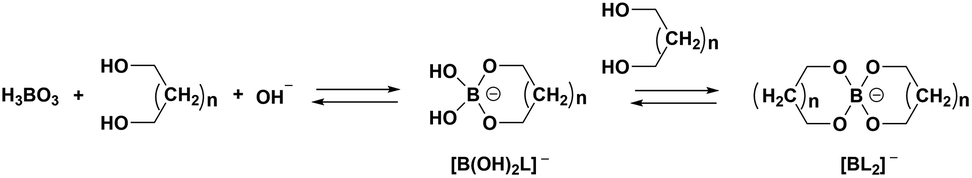
Under appropriate conditions, polyol or polysaccharide could be induced to form mono- or dicyclic structure complexes with H3BO3, which can enhance the reaction degree of H3BO3 and reduce the deficiencies of boron. The mechanism is illustrated employing polyol as an example (Fig. 1).44 As shown in Fig. 1, each polyol molecule can form a monocyclic structure complex with one H3BO3 molecule, and two polyol molecules can chelate with one H3BO3 molecule to obtain a complex with a dicyclic structure. Therefore, the B–OH groups are all involved in forming the complex when the molar ratio between polyol and H3BO3 is equal to or greater than 2. In this case, it is generally considered that the boron is fully linked with a cross-linking structure in the precursor. Alternatively, if the molar ratio is less than 2, the linked case of boron in the precursor is considered to be partial. It seems that the different linked cases of boron could affect the cross-linking structure of the precursor. From a chemical synthesis perspective, improving the homogeneity of ZrB2 precursor at the molecular or atomic scale requires the boron to be linked with a cross-linking structure in the precursor as much as possible. However, it is not clear whether it is necessary for all of the boron to be linked in the precursor to obtain high-quality ZrB2 powder. Additionally, there is limited literature exploring and discussing the effects of boron partially or fully linked with a cross-linking structure of an organic precursor on the properties of ZrB2 powder.
 |
| | Fig. 1 Schematic illustration of the chelation mechanism of polyol and H3BO3.44 | |
In this work, glycerol is employed as a chelating ligand to synthesize complexes with varying free B–OH contents, and then ZrB2 organic precursors with boron partially or fully linked with a cross-linking structure are prepared by a sol–gel method. The effects of the two cases on the purity and morphology of the ZrB2 powder are discussed. The gelation process of the precursor and the formation mechanism of ZrB2 are also investigated. Additionally, high-purity, prism-like ZrB2 powder is ultimately synthesized by a carbothermic reduction reaction.
2. Experimental
2.1 Raw materials
Polyzirconoxane (PNZ) and phenolic resin (char yield 62 wt%) were supplied by the Institute of Chemistry Chinese Academy of Sciences. Boric acid (H3BO3, Beijing Chemical Works, Beijing, China), glycerol (Lanyi Reagent Co. Ltd., Beijing, China), and ethanol (EtOH, Beijing Chemical Works, Beijing, China) are of analytical grade.
2.2 Synthesis of complexes
Given that the reaction temperature and time could potentially impact the complexation reaction between glycerol and H3BO3, independent experiments were designed to investigate the effect of different conditions on complexation. The molar ratio of functional groups between glycerol and H3BO3 (A), reaction temperature (B, °C), and reaction time (C, h) were selected as the factors for the complexation reaction of glycerol and H3BO3, and each factor was assigned to 4 levels. The percentage content of free B–OH (D, mol%) in the products served as the evaluation index and measured by acid–base titration. Details of the test procedures can be found in the ESI.† The test conditions and corresponding results are presented in Table S1.† As shown in Table S1,† the greatest influence on the complexation reaction between glycerol and H3BO3 is had by the molar ratio of functional groups, followed by reaction time and reaction temperature. In detail, for glycerol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H3BO3 (mol) = 0.67 (200 °C × 2.5 h), the complex, labelled as Complex 1, contains the highest content of free B–OH. Then, the content of free B–OH in the complex synthesized with glycerol:H3BO3 (mol) = 1 (180 °C × 1.5 h) is at an intermediate level. Finally, for glycerol:H3BO3 (mol) = 2 (160 °C × 1 h), there is almost no free B–OH detected in the complex, which is referred to as Complex 2. The impact of reaction time and temperature on the experimental results is negligible, and thus could be disregarded. Overall, an increase in the amount of glycerol leads to a decrease in the free B–OH content in the complexes when H3BO3 is kept constant (Table S1†). For convenience, the corresponding reaction temperature and time are omitted when the molar ratios of glycerol and H3BO3 are mentioned below. Specific details are shown in Table S1.†
:H3BO3 (mol) = 0.67 (200 °C × 2.5 h), the complex, labelled as Complex 1, contains the highest content of free B–OH. Then, the content of free B–OH in the complex synthesized with glycerol:H3BO3 (mol) = 1 (180 °C × 1.5 h) is at an intermediate level. Finally, for glycerol:H3BO3 (mol) = 2 (160 °C × 1 h), there is almost no free B–OH detected in the complex, which is referred to as Complex 2. The impact of reaction time and temperature on the experimental results is negligible, and thus could be disregarded. Overall, an increase in the amount of glycerol leads to a decrease in the free B–OH content in the complexes when H3BO3 is kept constant (Table S1†). For convenience, the corresponding reaction temperature and time are omitted when the molar ratios of glycerol and H3BO3 are mentioned below. Specific details are shown in Table S1.†
2.3 Synthesis of precursors
The synthesis process of ZrB2 powders is displayed in Fig. 2. Prior to the main experiment, the molar ratio of B and Zr was optimized through changing the reaction conditions and determined to be 4.75:1. In a typical synthesis method, an appropriate amount of EtOH was added to one of the complexes, and stirred at 70 °C until the complex was fully dissolved. After that, PNZ was added to the above solution with continuous stirring at room temperature for 1 h to form Sol 1. Afterwards, some of Sol 1 was subjected to aging at 70 °C for 24 h to form a wet gel, which was then heated (5 °C min−1) for the curing process in sequence at 120 °C (2 h), 160 °C (2 h), 200 °C (2 h), 250 °C (2 h) in an oven, then hard and brown dry gel was obtained. Phenolic resin solution (50 wt%, EtOH) was added to another portion of Sol 1 with continuous stirring for 1 h at room temperature to obtain Sol 2, and then the Sol 2 was aged and cured to prepare a dry gel. For comparison, a portion of Sol 1 that used Complex 1 was cured directly to form a dry gel without aging. Finally, the dry gels were all ground and passed through a 100-mesh sieve to obtain loosely packed brown precursor powders. The raw material compositions of all precursor samples are shown in Table 1.
 |
| | Fig. 2 Flow diagram for the synthesis of ZrB2 powders. | |
Table 1 Raw material compositions for ZrB2 precursor powders
| Precursor no. |
Raw materiala (g) |
Glycerol:H3BO3 (mol) |
Aged or not |
| Glycerol |
Phenolic resin |
| The mass of polyzirconoxane (PNZ) and H3BO3 as raw materials is fixed, with values of 7.7 g and 5 g, respectively. |
| 1 |
5 |
0 |
0.67 |
Aged |
| 2 |
15 |
0 |
2 |
Aged |
| 3 |
5 |
0 |
0.67 |
Non-aged |
| 4 |
5 |
1 |
0.67 |
Aged |
| 5 |
15 |
1 |
2 |
Aged |
| 6 |
7.4 |
1 |
1 |
Aged |
| 7 |
4.3 |
1 |
0.57 |
Aged |
| 8 |
6 |
1 |
0.8 |
Aged |
| 9 |
13 |
1 |
1.8 |
Aged |
| 10 |
16.7 |
1 |
2.3 |
Aged |
2.4 Synthesis of ZrB2 powders
The precursor samples listed in Table 1 were heated in a tube furnace in an argon atmosphere at 1150 °C for 2 h and 1450 °C for 1 h. Afterwards, they were cooled down to room temperature and ash-black ZrB2 powders were prepared. Eqn (1) shows the carbothermic reduction reaction that occurred during the calcination process of the precursors. To investigate the pyrolysis process, Precursor 4 and Precursor 5 were selected and heated in an argon atmosphere to the target temperature. The specimens were then held at this temperature for 1 h before being cooled down to room temperature. The heating and cooling rates were both set to 5 °C min−1.| | |
ZrO2 + 5C + B2O3 (l,g) → ZrB2 + 5CO (g)
| (1) |
2.5 Characterization
Electrospray ionization-mass spectrometry (ESI-MS, Bruker ultrafleXt) was used to analyse the chemical formula and structure. Fourier transform infrared spectroscopy (FTIR, Nicolet 6700) was utilized to identify complexes formed by glycerol and H3BO3, as well as ZrB2 precursors, within a range of 4000 to 650 cm−1. The thermal behaviour of the precursors was investigated by differential thermal analysis (DTA, Mettler Toledo TGA/DSC 1) in an argon atmosphere. The precursors were heated from 35 to 1500 °C at a heating rate of 5 °C min−1. The crystallographic data of the ceramic powders were collected by X-ray diffraction (XRD, Rigaku D/Max 2500 Diffractometer), and the relative content of each crystalline phase in the as-prepared powders was determined by the relative intensity ratio (RIR) method.45,46 The morphologies and chemical compositions of the ceramic powders were observed using a scanning electron microscope (SEM, JSM-7001F) fitted with an energy-dispersive X-ray spectroscopy (EDX) instrument. The oxygen content in the ceramic powders was measured by a nitrogen–oxygen analyser (LECO, ONH836).
3. Results and discussion
3.1 Characterization of complexes and precursors
Complex 1 (glycerol:H3BO3 (mol) = 0.67, 200 °C × 2.5 h) and Complex 2 (glycerol:H3BO3 (mol) = 2, 160 °C × 1 h) were selected for further study, with the highest content of free B–OH and almost no free B–OH, respectively. To better understand the chemical formulae and structures of the two complexes, as well as the chemical behaviour of the ZrB2 precursors synthesized using the two complexes, FTIR and ESI were used for characterization.
3.1.1 Characterization of complexes. FTIR spectra of glycerol, H3BO3, and the two complexes are shown in Fig. 3. The absorption peaks at 1032 cm−1 (Fig. 3a) and 1195 cm−1 (Fig. 3b) are assigned to the C–O stretching vibration of glycerol and B–O–H bending vibration of H3BO3, respectively.41 After the reaction between glycerol and H3BO3, the peaks at 1032 cm−1 for Complex 2 (Fig. 3c) and Complex 1 (Fig. 3d) are clearly decreased, especially in Fig. 3d. Meanwhile, a weak peak at 1195 cm−1 remains for Complex 1, as shown in the inserted image, while this peak disappears for Complex 2. Furthermore, the absorption peaks at 1172 cm−1 and 1277 cm−1 for both Complex 1 and Complex 2 are assigned to B–O–C,41,47 which is attributed to the reaction between B–OH and C–OH. Therefore, both Complex 1 and Complex 2 contain B–O–C, while Complex 1 retains a small amount of free B–OH. The peaks in the range of 3700 to 3000 cm−1 correspond to the stretching vibration of O–H. By comparing the absorption peaks of Complex 1 and Complex 2 within this range, it can be inferred that: (1) the peaks of Complex 1 at 3230 cm−1 and 3410 cm−1 indicate the presence of two types of O–H, namely (C)O–H and (B)O–H, which are connected to carbon and boron atoms, respectively;48,49 (2) Complex 2 only has (C)O–H at 3340 cm−1. Given the coordination polyhedron around the boron atom can assume either a triangle or a tetrahedral shape,50 the peak at 1085 cm−1 in both Complex 1 and Complex 2 is attributed to B(4)–O, indicating the existence of tetrahedral boron51 in the two complexes. The asymmetric stretching vibration of B(3)–O at 1378 cm−1 in Complex 1 demonstrates the existence of trigonal boron,52 implying different bonding states of boron in the complexes.
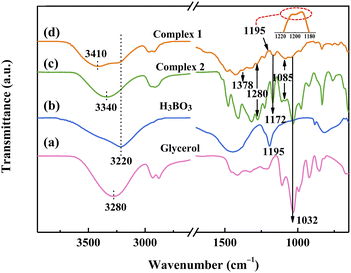 |
| | Fig. 3 FTIR spectra of (a) glycerol, (b) H3BO3, (c) Complex 2, and (d) Complex 1. | |
To further determine the molecular structures of the two complexes, mass spectrometry analysis was performed. According to ESI mass spectra in the negative mode of the liquid medium (Fig. 4), there is almost only one strong peak at m/z 191 (Fig. 4a) in Complex 2; however, Complex 1 exhibits not only a strong peak at m/z 191, but also two weak peaks at m/z 117 and 131 (Fig. 4b). The strong peak is attributed to the [B(C3H6O3)2]− anion with a double spirocyclic structure,53 indicating the presence of H[B(C3H6O3)2] with tetrahedral boron in both Complex 1 and Complex 2.54 The molecular structure of H[B(C3H6O3)2] is shown in Fig. 5a. The weak peak of Complex 1 at m/z = 117 could be associated with the HOB(C3H6O3).53 Additionally, the weak peak at m/z = 131 may be because of the formation of H3B3O6 by the dehydration of H3BO3. HOB(C3H6O3) and H3B3O6 both have trigonal boron atoms, as shown in Fig. 5b and c. In other words, Complex 2 almost exclusively contains the tetrahedral boron, whereas both tetrahedral and trigonal boron are present in Complex 1. Therefore, the difference in the amount of glycerol results in the formation of Complex 1 and Complex 2 with distinct structures, leading to different bonding states of boron. This finding further supports the FTIR analysis results (Fig. 3).
 |
| | Fig. 4 ESI mass spectra in the negative mode of (a) Complex 2 and (b) Complex 1. | |
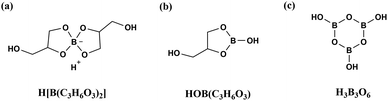 |
| | Fig. 5 Molecular structure formulae of (a) H[B(C3H6O3)2], (b) HOB(C3H6O3), and (c) H3B3O6. | |
3.1.2 Characterization of precursors. FTIR spectra of PNZ, Precursor 1, and Precursor 2 (Table 1) are shown in Fig. 6. PNZ is a zirconium-containing polymer produced by the condensation of Zr(OPr)4 followed by complexation with acetylacetone.36 The structure of PNZ is shown in Fig. S1.† The peaks at 1015 cm−1 and 1049 cm−1 for PNZ are assigned to Zr–O–C (Fig. 6a).36 After adding Complex 1 and Complex 2 to PNZ to obtain Precursor 1 and Precursor 2, respectively, the peaks shifting to 1098 cm−1 and 1041 cm−1 (Fig. 6b and c) may imply that the complexes and PNZ are bonded together to form Zr–O–C–B.25,34,40 Precursor 1 exhibits a peak at 3218 cm−1 associated with (B)–OH (Fig. 6b), which is similar to Complex 1 (Fig. 3d), suggesting that not all of the boron in Precursor 1 is coordinated to the gel network. In this case, it is considered that the boron is partially linked with a cross-linking structure. However, Precursor 2 displays no peak at this position, resembling Complex 2 (Fig. 3c), indicating that the boron in Precursor 2 is linked with a cross-linking structure almost without free B–OH, and boron in this case is designated as fully linked. It can be inferred that the linked boron in the organic precursor could be modulated by the bonding state of boron in the complex.
 |
| | Fig. 6 FTIR spectra of (a) polyzirconoxane (PNZ), (b) Precursor 1, and (c) Precursor 2. | |
3.2 The sol-to-gel transition
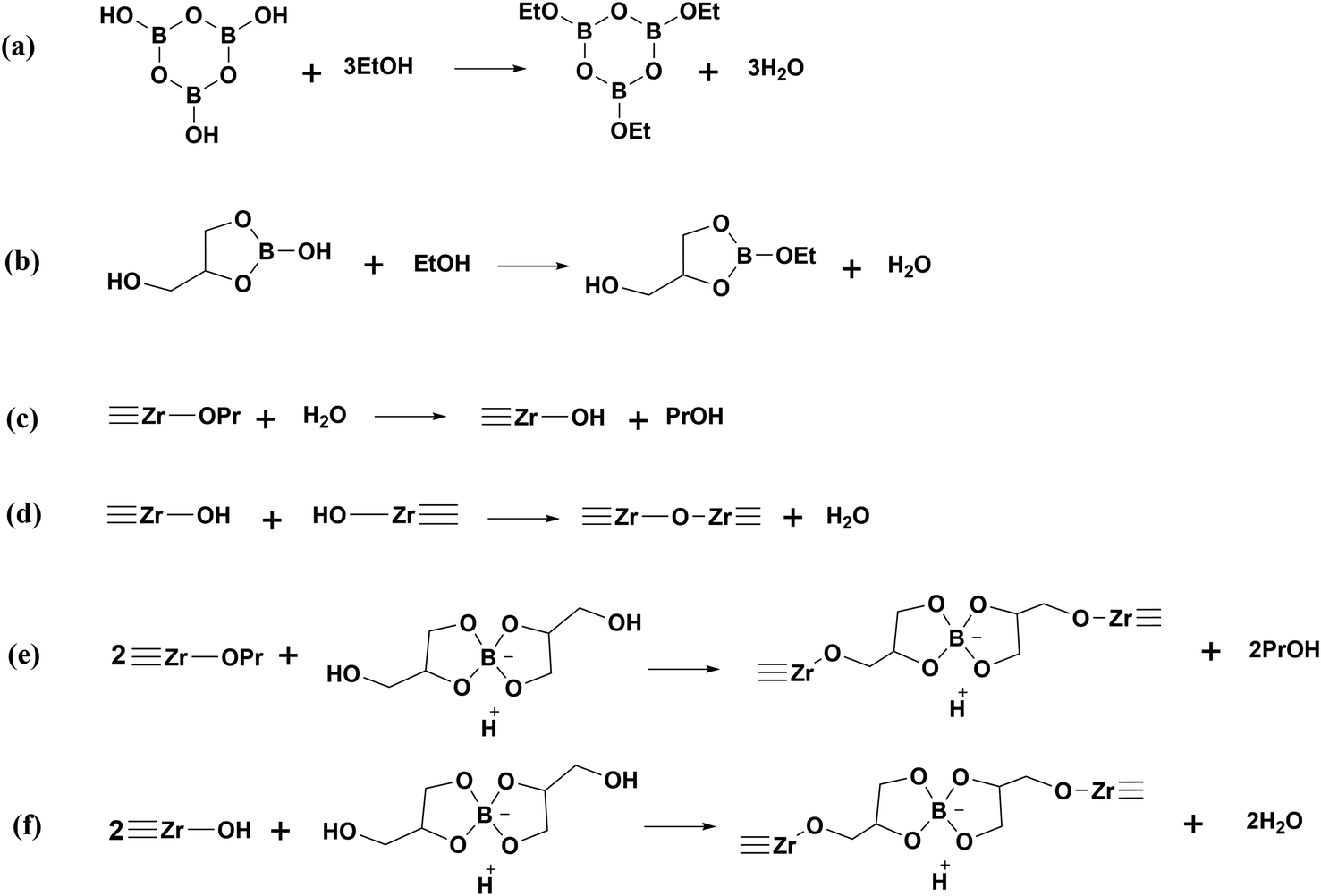
The different linking states of boron in Precursor 1 and Precursor 2 lead to distinct appearances of the gels formed after aging, as shown in Fig. 7. The sols of Precursors 1 and 2 both show a transparent deep orange colour (Fig. 7a and b). After aging, the gel of Precursor 1 becomes opaque and pale-yellow with phase separation (Fig. 7c), whereas the gel of Precursor 2 remains transparent and exhibits a typical Tyndall effect (Fig. 7d). Since boron is all tetrahedral in Precursor 2 and without free B–OH, a comparative experiment is performed by directly curing the sol of Precursor 1 without aging. The resulting precursor powder is labelled as Precursor 3 and the FTIR spectrum of Precursor 3 is shown in Fig. S2.† The peak of (B)–OH at 3218 cm−1 is stronger, while the peaks of B–O–C at 1280 cm−1 and 1163 cm−1 are weaker in Precursor 3, as can be seen by comparing Fig. 6b and S2.† This may be because of the esterification reaction between B–OH groups in HOB(C3H6O3) or H3B3O6 and C–OH groups in EtOH, leading to the formation of B–O–C during aging, as demonstrated in Scheme 1a and b.
 |
| | Fig. 7 Images of the sol of (a) Precursor 1 and (b) Precursor 2 and the gel of (c) Precursor 1 and (d) Precursor 2. (d) shows the Tyndall effect. | |
 |
| | Scheme 1 Esterification reaction of EtOH with (a) H3B3O6 and (b) HOB(C3H6O3); (c) hydrolysis of ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Zr–OPr groups in PNZ; (d) self-condensation of Zr–OH groups generated from (c); (e) alcoholysis between H[B(C3H6O3)2] and Zr–OPr groups; (f) condensation of H[B(C3H6O3)2] and Zr–OH groups. Zr–OPr groups in PNZ; (d) self-condensation of Zr–OH groups generated from (c); (e) alcoholysis between H[B(C3H6O3)2] and Zr–OPr groups; (f) condensation of H[B(C3H6O3)2] and Zr–OH groups. | |
The water generated from the reactions in Scheme 1a and b leads to a rapid hydrolysis of PNZ (Scheme 1c), followed by the self-condensation of Zr to form Zr–O–Zr chains (Scheme 1d). As a result, the increase in the size of gel particles renders the gel of Precursor 1 opaque (Fig. 7c). Moreover, H[B(C3H6O3)2] reacts with Zr–OPr groups in PNZ or Zr–OH groups generated from the reaction in Scheme 1c (as illustrated in Scheme 1e and f), which could drive the formation of Zr–O–C–B. This is consistent with the FTIR analysis results (Fig. 6). In the case of Precursor 1, the cross-linking structure is mainly composed of Zr–O–Zr and Zr–O–C–B.
Because of the absence of B–OH in Precursor 2 (Fig. 6), it could not produce water, which prevents the hydrolysis of PNZ. This inhibits the self-condensation of Zr to obtain the Zr–O–Zr structure, resulting in small gel particles. Therefore, the gel of Precursor 2 is transparent with a Tyndall effect (Fig. 7d). In addition, the formation of a Zr–O–C–B network skeleton in the Precursor 2 system during gelation and aging primarily relies on the alcoholysis reaction between H[B(C3H6O3)2] and Zr–OPr groups (Scheme 1e). When the system does not contain complexes formed by glycerol and H3BO3, the gel cannot be formed after aging (not shown). This confirms that the synthesized complexes act as a “bridge” for the formation of the Zr–O–C–B cross-linking structure.
3.3 Effects of the partially or fully linked case for the precursor on ZrB2 powder
In this study, glycerol serves as a chelating ligand as well as a carbon source. Although the linking states of boron in Precursors 1 and 2 are partial and full, respectively, the different amounts of glycerol in the two precursors may result in varying carbon yields during the pyrolysis process. Hence, based on Precursors 1 and 2, Precursors 4 and 5 (Table 1) are synthesized containing phenolic resin. A comparative experiment using ZrB2 powders prepared from these precursors is conducted to eliminate the interference caused by the different amounts of glycerol, and to investigate the influence of the two linking states on the purity and morphology of the ZrB2 powder.
3.3.1 Influence on the purity of ZrB2 powder. The XRD patterns of the powders obtained from Precursors 1 and 2 after calcination for 2 h at 1150 °C and 1 h for 1450 °C are displayed in Fig. 8, exhibiting a major phase of ZrB2 and also containing m-ZrO2 and t-ZrO2. Based on the RIR method, the relative contents of each phase in the powders were found to be very similar, as shown in Table S2.† This finding suggests that although glycerol serves as a chelating ligand and carbon source, pure ZrB2 cannot be produced. In addition, the significant difference in the amount of glycerol between Precursor 1 and Precursor 2 does not affect the purity of the ZrB2 powder, and pure ZrB2 powder could not be prepared solely using glycerol as a carbon source. According to the mechanism of carbothermic reduction, a sufficient carbon source is essential for obtaining pure ZrB2. Phenolic resin produced by Zhao et al.55 shows a high residual carbon yield, and can act as an effective carbon source with good solubility in organic systems. Therefore, equal amounts of phenolic resin are added to the sols of Precursors 1 and 2, and the resulting mixtures are used to synthesize ZrB2 precursors according to Fig. 2, labelled Precursor 4 and Precursor 5, respectively. By comparing the FTIR spectra and images of the sols and gels from the precursors (Fig. 5, 6 and S3†), it can be inferred that the addition of an equivalent amount of phenolic resin in Precursors 4 and 5 scarcely affects the linking state of the boron. Therefore, the linking states of boron in Precursors 4 and 5 are partial and full, respectively. Moreover, the cross-linking structures of Precursors 4 and 5 are consistent with those of Precursors 1 and 2, respectively.
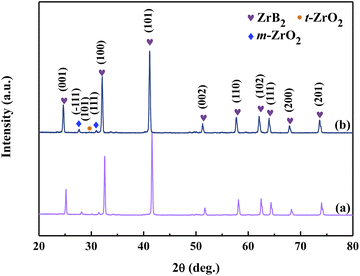 |
| | Fig. 8 XRD patterns of ZrB2 powders obtained from (a) Precursor 1 and (b) Precursor 2. | |
The XRD patterns of the powders prepared from Precursors 4 and 5 after annealing at 1150 °C for 2 h and 1450 °C for 1 h are shown in Fig. 9. ZrB2 is the only single crystalline phase as observed. It seems that phenolic resin improves the purity of the ZrB2 powders by providing enough carbon in both systems. The oxygen contents for the ZrB2 powders produced by Precursors 4 and 5 are 0.26 wt% and 0.38 wt%, respectively, and were investigated by nitrogen–oxygen analysis. This suggests that the boron in the partially linked case is more conducive to obtaining a low-oxygen ZrB2 powder. This may be related to the synthesis mechanism of the ZrB2 powder.
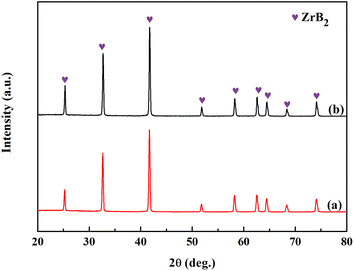 |
| | Fig. 9 XRD patterns of ZrB2 powders obtained from (a) Precursor 4 and (b) Precursor 5. | |
To investigate the formation mechanism of ZrB2 under conditions in which the linking states of the boron in the precursor are partial and full, DTA analysis was performed in an argon atmosphere with a heating rate of 5 °C min−1 (Fig. 10). The exothermic peaks of the two complexes and precursors in the low-temperature region may be attributed to the formation of amorphous ZrO2, carbon and B2O3.38,56,57 In the high-temperature region, both Complex 1 and Complex 2 exhibit broad endothermic peaks at 1220 °C and 1314 °C (Fig. 10a and c), respectively. These peaks could be ascribed to the vaporization of B2O3, due to the fact that B2O3 could begin to evaporate at 1200 °C and undergo significant evaporation at 1400 °C.33 A higher gasification temperature of B2O3 for Complex 2 indicates that it is more effective for holding boron. Therefore, this suggests that Precursor 5 using Complex 2 could inhibit the gasification of B2O3 better than Precursor 4 using Complex 1. The endothermic peaks observed at 1460 °C for Complex 1 and 1406 °C for Complex 2 may be attributed to the formation of B4C.58,59 For Precursors 4 and 5, the endothermic peaks observed in the high-temperature region are due to the carbothermic reduction reaction.41 The endothermic peaks for Precursor 4 are shown at 1356 °C, 1409 °C and 1460 °C (Fig. 10b), and those for Precursor 5 are exhibited at 1383 °C and 1406 °C (Fig. 10d). Notably, the peaks at 1460 °C for Precursor 4 and 1406 °C for Precursor 5 are consistent with the formation temperatures of B4C by the corresponding complexes. Based on the different linking states in Precursors 4 and 5, it is possible that the carbothermic reduction processes of the two precursors may involve different reaction pathways.34
 |
| | Fig. 10 DTA curves from 35 to 1500 °C for (a) Complex 1, (b) Precursor 4, (c) Complex 2, and (d) Precursor 5. | |
Due to the agglomeration and phase separation of Zr–O–Zr colloidal particles in the gelation of Precursor 4, the distribution of ZrO2, C, and B2O3 may be heterogeneous during the pyrolysis process. At the outset of the carbothermic reduction, the system may not provide enough energy for the diffusion of B2O3, resulting in ZrO2 which is located far from B2O3 reacting with the adjacent C and forming ZrC (eqn (2)).
| | |
ZrO2 + 3C → ZrC + 2CO (g)
| (2) |
The initial formation temperature of ZrC by the sol–gel method synergized with a carbothermic reduction strategy has been observed to be ∼1140 °C.33 This could provide the possibility for eqn (2), which is a low-energy reaction pathway for producing ZrB2. Therefore, the endothermic peak of Precursor 4 at 1356 °C is due to the formation of ZrC. Once ZrC is generated by eqn (2), it may serve as an intermediary phase to form ZrB2 by eqn (3) and (4).33,41
| | |
2.5ZrC + 1.5B2O3 (l,g) → 1.5ZrB2 + ZrO2 + CO (g)
| (3) |
| | |
ZrC + B2O3 (g) + 2C → ZrB2 + 3CO (g)
| (4) |
Based on the results of standard Gibbs free energy calculations for eqn (3) and (4), both reactions are spontaneous at ∼1150 °C, with eqn (3) being thermodynamically more favorable than eqn (4).33 As the temperature increases, the diffusion of B2O3 becomes extensive and sufficient energy could be provided by the Precursor 4 system to ensure full contact between the remaining ZrO2 and B2O3 and C, leading to the formation of ZrB2 (eqn (1)). Therefore, another endothermic peak appears at 1409 °C.
Because of the absence of free B–OH in Precursor 5, a lower degree of Zr self-condensation occurs and smaller gel particles are formed compared with Precursor 4 in gelation. Consequently, smaller ZrO2 particles may be obtained during the pyrolysis process, improving the reactivity of ZrO2. Owing to the fact that the cross-linking structure of Precursor 5 is mainly composed of Zr–O–C–B, the mixture of ZrO2, C, and B2O3 generated after calcination may be more homogeneous than that of Precursor 4. According to the results of DTA (Fig. 10), mass transfer through liquid B2O3 could occur more for Precursor 5 than Precursor 4 during the carbothermic reduction reaction. As a result, the energy provided by the Precursor 5 system is sufficient to overcome the reaction barrier of eqn (1), resulting in an endothermic peak at 1383 °C (Fig. 10d).
To further investigate the formation mechanism of ZrB2, Precursors 4 and 5 were calcined at different temperatures for 1 h and the obtained powders were analyzed by XRD, as shown in Fig. 11. The peaks of t-ZrO2 appear in the powder prepared from Precursor 4 at 600 °C, and are enhanced at 700 °C. The powder produced by Precursor 5 retains an amorphous structure at 600 °C, followed by the appearance of t-ZrO2 at 700 °C. Up to 900 °C, both Precursors 4 and 5 exhibit the peaks of m-ZrO2. With an increase in the temperature, the m-ZrO2 peaks strengthen gradually while the t-ZrO2 peaks diminish steadily. The ZrB2 phase is first detected at 1200 °C, then becomes the main phase with only a trace amount of t-ZrO2 at 1400 °C. At 1500 °C, no obvious change in the XRD patterns (Fig. 11b) could be observed for the powder generated from Precursor 5. However, the sample of Precursor 4 only contains the ZrB2 phase (Fig. 11a).
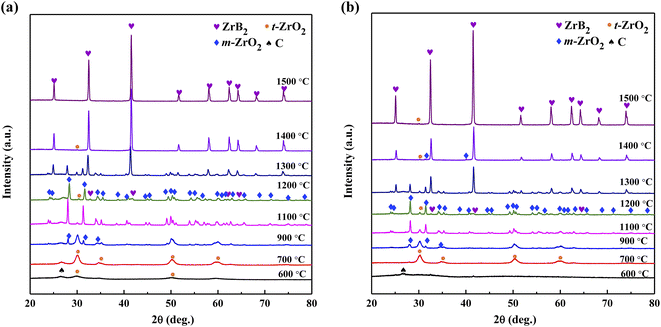 |
| | Fig. 11 XRD patterns of ceramic powders obtained from (a) Precursor 4 and (b) Precursor 5 after heating at different temperatures for 1 h. | |
The relative content of t-ZrO2 in the powders prepared from Precursors 4 and 5 from 1100 to 1500 °C was calculated using the RIR method, as shown in Fig. 12. Combined with Fig. 11, the content of t-ZrO2 in the Precursor 5 system gradually decreases with the generation of ZrB2, suggesting that t-ZrO2 is progressively transformed into ZrB2 through eqn (1); the content of t-ZrO2 first increases and then decreases in the Precursor 4 system, indicating a secondary generation of t-ZrO2 in the early stage of the carbothermic reduction reaction, which could result from eqn (3). This is consistent with the inference drawn from the DTA analysis. Due to the high-temperature stability of t-ZrO2 (>1200 °C), the ZrO2 formed in eqn (3) should be t-ZrO2. Nevertheless, pure ZrB2 cannot be obtained solely through eqn (3). Thus, this reaction pathway is only effective in the early stage of the carbothermic reduction reaction. When the temperature increases above 1300 °C, the system can obtain enough energy to facilitate the full contact of ZrO2, C, and B2O3 with each other, resulting in eqn (1), which leads to the decrease of the t-ZrO2 content. Therefore, the carbothermic reduction reaction for the case in which the boron is partially linked is a multi-step process. Moreover, the formation of the ZrC intermediary phase may reduce the initial generation temperature of ZrB2. This could potentially enable a more thorough carbothermic reduction, consequently reducing the oxygen content of the ZrB2 powder. This further supports the results of the nitrogen–oxygen analysis.
 |
| | Fig. 12 Relative contents of the t-ZrO2 phase. | |
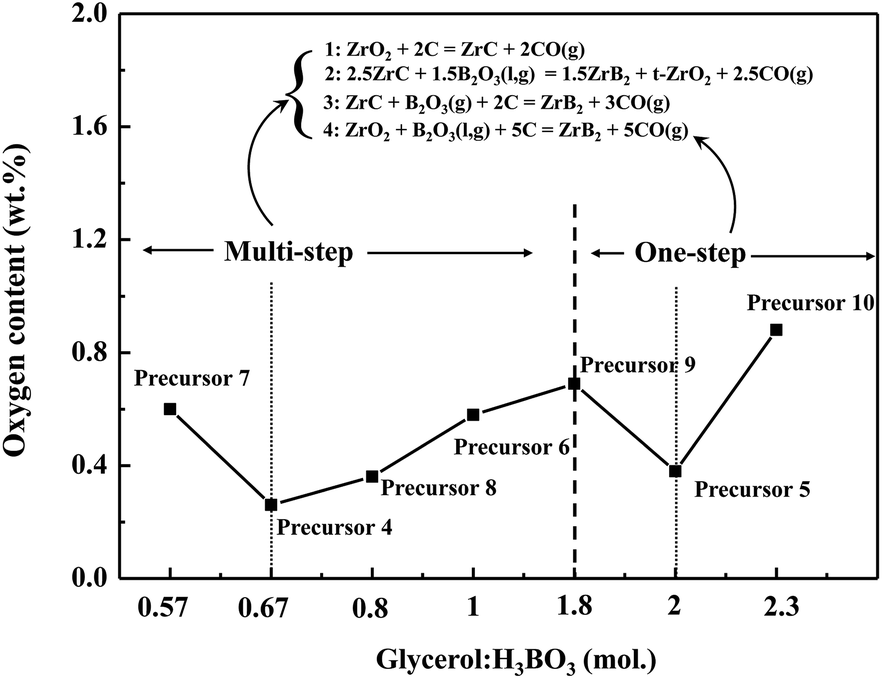
For further confirmation, some experiments are performed. A precursor labelled Precursor 6 (Table 1) is synthesized, using the complex with an intermediate level of free B–OH content. The gel of Precursor 6 is translucent (Fig. S4†) and the Tyndall effect is less pronounced than that of Precursor 5 (Fig. S3b†). It can be inferred that the extent of boron linked with a cross-linking structure of Precursor 6 and the size of the gel particles are between those of Precursors 4 and 5. The linking state of boron for Precursor 6 is also partial. Then, a single-phase ZrB2 powder is prepared after heat treatment at 1150 °C for 2 h and 1450 °C for 1 h (not shown), and its residual oxygen is 0.58 wt%. It seems that the oxygen content for ZrB2 powders first increases and then decreases as the extent of boron linked with organic precursors is increased. In other words, the partially linked case may not be always beneficial for reducing the oxygen content, which is contrary to the previous result. To further investigate this, four precursors are synthesized by changing the amount of glycerol, using molar ratios of glycerol to H3BO3 set at 0.67 and 2 as reference points. The four precursors are labelled Precursors 7 to 10 (Table 1), and the images of corresponding gels are shown in Fig. S5.† According to the Fig. S3b–S5,† it can be observed that the gels gradually become transparent as the molar ratio is increased, indicating a reduction in particle size of the gels. Since the free B–OH content decreases with an increasing amount of glycerol when H3BO3 is kept constant, it can be inferred that the extent of boron linked with a cross-linking structure of the precursors is enhanced with an increased amount of glycerol.
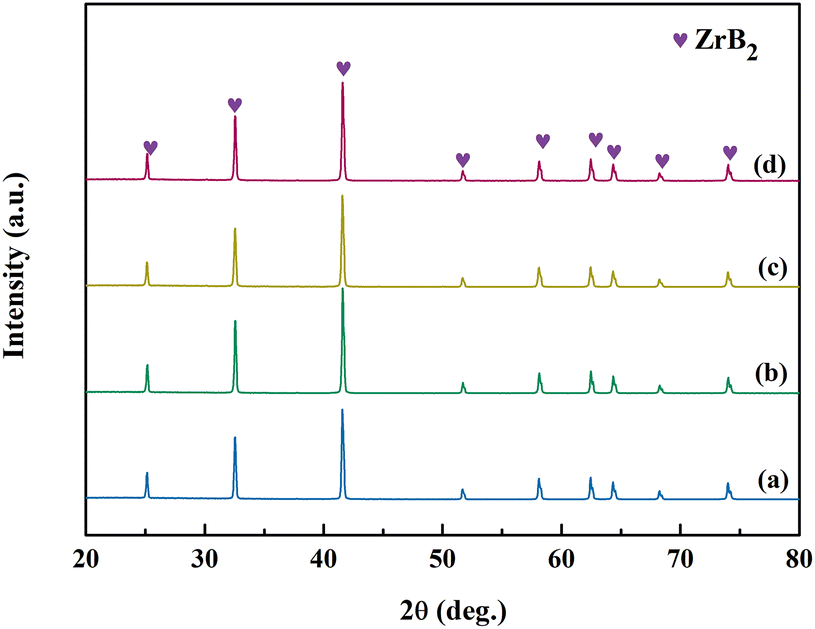
The XRD patterns of ZrB2 powders obtained from Precursors 7 to 10 after annealing at 1150 °C for 2 h and 1450 °C for 1 h are shown in Fig. 13. Single-phase ZrB2 powders are produced by Precursors 7 to 10, which are consistent with the results of Precursors 4 and 5 (Fig. 9). However, there is a significant difference in the oxygen contents for the as-prepared ZrB2 powders among Precursors 7 to 10, which are measured to be 0.60 wt%, 0.36 wt%, 0.69 wt%, and 0.88 wt%, respectively. Fig. 14 shows the oxygen content trend of the as-prepared ZrB2 powders for Precursors 4 to 10 using different glycerol:H3BO3 ratios (mol). An obvious “W”-shaped trend is observed with increasing amount of glycerol. In general, particle size, concentration, reaction medium, and other factors could affect the carbothermic reduction reaction. For glycerol:H3BO3 (mol) = 0.67 (Precursor 4), the system cannot successfully inhibit the vaporization of B2O3, resulting in relatively poor mass transfer. In this case, the generation of ZrC is not suppressed, which may decrease the initial generation temperature of ZrB2 and further enable a more thorough carbothermic reduction. Therefore, the oxygen content reaches its minimum value of 0.26 wt%. When the ratio is between 0.67 and 2, the extent of boron linked with a cross-linking structure in the precursor could be enhanced with an increased amount of glycerol, which may further potentially reduce the loss of B2O3. This facilitates mass transfer of liquid B2O3 during a carbothermic reduction, which may suppress the formation of ZrC. Therefore, the conversion of ZrC to ZrB2 is suppressed. However, at this time, the energy may not be sufficient to overcome the reaction barrier of eqn (1). Consequently, the carbothermic reduction gradually becomes insufficient, leading to an increase in oxygen content for the ZrB2 powders. The fully linked case, namely Precursor 5, is exactly formed when the molar ratio is 2. At this stage, mass transfer of B2O3(l) has probably reached its optimum level. Furthermore, the boron fully linked case may cause a lower self-condensation degree of Zr and smaller gel particles, potentially resulting in smaller and more active ZrO2 particles during the cracking process. Therefore, the energy provided by the Precursor 5 system could be enough to overcome the reaction barrier of eqn (1) at 1383 °C (Fig. 10d), whereas the Precursor 4 system may require a higher temperature, namely 1409 °C (Fig. 10b). ZrB2 powders obtained from precursors with the ratios at 0.57 (Precursor 7) and 2.3 (Precursor 10) show clearly higher oxygen contents than those of the adjacent points, which may be attributed to the lack or excessive amount of glycerol.
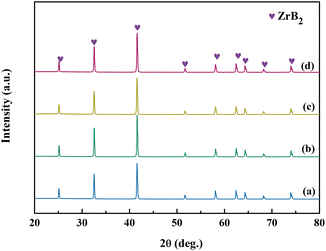 |
| | Fig. 13 XRD patterns of ZrB2 powders obtained from (a) Precursor 7, (b) Precursor 8, (c) Precursor 9, and (d) Precursor 10. | |
 |
| | Fig. 14 Oxygen content of ZrB2 powders prepared from Precursor 4 to Precursor 10 with different glycerol:H3BO3 ratios (mol). | |
3.3.2 Influence on the morphology of ZrB2 powder. SEM images of the ZrB2 powders obtained from Precursors 1 and 2 synthesized at 1150 °C for 2 h and 1450 °C for 1 h are shown in Fig. 15. The ZrB2 powders both exhibit a rod-like morphology with micron-scale sizes. The ZrB2 particles prepared from Precursor 1 have an average length of 2.69 μm and an average diameter of 1.34 μm, while the values for Precursor 2 are 2.67 μm and 1.40 μm, respectively. The morphology of the ZrB2 particles shows no significant difference between Precursors 1 and 2, indicating that the large difference in the amount of glycerol in these two precursors barely affects the morphology of ZrB2. It is noteworthy that Ji et al.34 and Yan et al.41 also used glycerol as a chelating ligand and carbon source. However, the generated ZrB2 powders were nano-sized spherical and submicron-sized nearly spherical particles, respectively, rather than micron-sized rod-like ZrB2 particles. This might be due to the fact that Ji et al. and Yan et al. also used other chelating ligands besides glycerol to stabilize the Zr source, which may cause different crystal growth mechanisms and further result in varying morphologies and sizes of the ZrB2 powders.
 |
| | Fig. 15 SEM images of ZrB2 powders obtained from (a) Precursor 1 and (b) Precursor 2. | |
Fig. 16 shows micrographs of ZrB2 powders obtained from Precursors 4 and 5 after annealing at 1150 °C for 2 h and 1450 °C for 1 h. The morphologies of the ZrB2 particles prepared from Precursors 4 and 5 are both prism-like, as shown in Fig. 16a and b, respectively. Additionally, few polyhedral-like particles could be observed in the square in Fig. 16b, which shows dark particles in the backscattered image (the inserted image). This indicates that they are composed of elements with low atomic numbers. According to the studies by Li et al.60 and Avcioğlu et al.,56 the polyhedral-like particles are B4C, and this is also confirmed by EDX elemental mapping (Fig. 16d). Because of the ability to hold liquid B2O3 better in the case in which boron is fully linked, excess B2O3 in the Precursor 5 system may consume carbon to form a byproduct, B4C. Thus, it can be inferred that the as-prepared ZrB2 powder is actually double-phase for the boron fully linked case and it may not effectively produce high-purity ZrB2 powder as compared to the partially linked case. This further supports the results shown in Fig. 14.
 |
| | Fig. 16 Micrographs: SEM images of ZrB2 powders prepared from (a) Precursor 4 and (b) Precursor 5 (the inset in (b) is the backscatter electron image); (c) enlarged image of the square in (a); (d) EDX elemental mappings of the polyhedral-like particles in the square of (b). | |
Notably, the particle size of ZrB2 in Fig. 16b is significantly smaller than that illustrated in Fig. 16a. Since the raw materials for both Precursors 4 and 5 contain the same amount of phenolic resin, there is a distinction between the morphologies of the ZrB2. This may be attributed to two main factors: (1) the amount of glycerol; and (2) the linked case of boron in the precursors. Due to the fact that the morphologies of ZrB2 particles generated from Precursors 1 and 2 are not significantly different despite the various amounts of glycerol (Fig. 15), the second factor is the determinant.
To explore the impacts of partially and fully linked boron cases on the morphology of ZrB2 powder, the morphology evolution of powders generated from Precursors 4 and 5 is analyzed. Fig. 17a–d and e–h show the SEM images of ceramic powders obtained from Precursor 4 and Precursor 5 at different temperatures, respectively. ZrO2 particles and rod-like ZrB2 particles (pointed to by the arrow) are generated at 1200 °C in both Precursors 4 and 5 (Fig. 17a and e). The content of rod-like ZrB2 particles increases, while it decreases for ZrO2 particles significantly at 1300 °C (Fig. 17b and f). The ZrO2 particles almost disappear and the crystallinity of ZrB2 particles is improved as the temperature is increased further (Fig. 17c and g). At 1500 °C, prism-like ZrB2 particles are obtained with some sintered excessively, which leads to adhesion among the particles (Fig. 17d and h). The average length, diameter, and aspect ratio of ZrB2 particles produced by Precursors 4 and 5 at different temperatures are shown in Fig. 18a–c. The ZrB2 particles prepared from Precursor 5 consistently present smaller lengths and diameters, and a larger aspect ratio than those generated from Precursor 4. The obvious growth steps on the top of a prism-like ZrB2 particle, pointed to by the arrow in Fig. 16c, suggest that ZrB2 is formed layer by layer from the surface to the core of ZrO2 particles.40 Due to the fact that the particle size could be tuned by controlling the size of starting powders,61 the size of ZrO2 particles generated during the calcination process is likely to affect the size of the resulting ZrB2 particles.
 |
| | Fig. 17 SEM images of ceramic powders prepared from (a–d) Precursor 4 and (e–h) Precursor 5 after calcination for 1 h from 1200 to 1500 °C. | |
 |
| | Fig. 18 The size of ZrB2 particles obtained from Precursors 4 and 5 at different temperatures: (a) average length; (b) average diameter; (c) average aspect ratio. (d) The size of ZrO2 particles generated from Precursors 4 and 5 at different temperatures. | |
Fig. S6† shows SEM images of the ZrO2 particles produced by Precursors 4 and 5 at different temperatures. Based on Fig. S6,† the average size of ZrO2 particles at different temperatures is shown in Fig. 18d. It is observed that the average size of ZrO2 particles obtained from Precursor 5 is smaller than that from Precursor 4 at all sintering temperatures. Due to the higher degree of Zr self-condensation in Precursor 4, the amorphous Zr–O–Zr structure may previously be formed, which could be more prone to transforming into an ordered crystalline ZrO2 structure during the ceramic conversion process. As shown in Fig. 11b, the powder obtained from Precursor 5 at 600 °C displays no crystalline phase, whereas a broad peak attributed to the t-ZrO2 phase is observed in the powder prepared from Precursor 4. As a consequence, smaller ZrO2 particles generated from Precursor 5 may lead to the generation of smaller ZrB2 particles, which are gradually formed layer by layer from the surface to the core of the ZrO2 particles. Therefore, the fully linked case could decrease the size of the ZrB2 particles. In general, increasing the liquid phase could promote particle growth, but in the fully linked case, the opposite occurs. Despite the ability to hold more liquid B2O3, the smaller ZrB2 particle size possibly results from the ineffective particle growth at 1450 °C.
Although the growth of anisotropic particles is related to the difference in solubility between large and small particles, it is generally believed that the growth of elongated crystals is mainly influenced by diffusion.62 This is due to the fact that the Precursor 5 system could reduce the loss of B2O3, which facilitates diffusion during the carbothermic reduction reaction and further results in ZrB2 particles with a larger aspect ratio.
4. Conclusions
In summary, ZrB2 organic precursors with boron partially and fully linked with a cross-linking structure are synthesized by a sol–gel route. The different linking cases of boron in the precursor affect the purity and morphology of the ZrB2 powder. In the case of partially linked boron, the free B–OH could drive the self-condensation of Zr and further influence the formation mechanism of ZrB2 with a multi-step process during carbothermic reduction, in which ZrC is an intermediary phase. The partially linked case could result in the oxygen content of the ZrB2 powder being decreased, with a minimum value of 0.26 wt%, and a high-purity prism-like ZrB2 powder with a larger size is obtained. On the other hand, the case of fully linked boron may lead to minimal self-condensation of Zr, which could reduce the particle size for ZrO2 and further result in a smaller size of the ZrB2 particles. In this case, the ZrB2 powder has a double-phase with a B4C impurity, and the carbothermic reduction reaction has one step.
Author contributions
Xi Yang contributed to conceptualization, formal analysis, investigation, and writing – original draft; Weijian Han contributed to writing – review & editing, supervision, and validation; Tong Zhao contributed to project administration, resources, and supervision; Ruixing Li contributed to conceptualization, writing – review & editing, supervision, and funding acquisition.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors appreciate the financial support from the National Natural Science Foundation of China (NSFC52072016).
References
- Y. D. Blum, J. Marschall, D. Hui and S. Young, J. Am. Ceram. Soc., 2008, 91, 1453–1460 CrossRef CAS.
- E. L. Corral and R. E. Loehman, J. Am. Ceram. Soc., 2008, 91, 1495–1502 CrossRef CAS.
- J. K. Sonber and A. K. Suri, Adv. Appl. Ceram., 2011, 110, 321–334 CrossRef CAS.
- F. Monteverde and R. Savino, J. Am. Ceram. Soc., 2012, 95, 2282–2289 CrossRef CAS.
- B. R. Golla, A. Mukhopadhyay, B. Basu and S. K. Thimmappa, Prog. Mater. Sci., 2020, 111, 100651 CrossRef CAS.
- D. W. Ni, Y. Cheng, J. P. Zhang, J. X. Liu, J. Zou, B. W. Chen, H. Y. Wu, H. J. Li, S. M. Dong, J. C. Han, X. H. Zhang, Q. G. Fu and G. J. Zhang, J. Adv. Ceram., 2022, 11, 1–56 CrossRef CAS.
- S. Q. Guo, J. Eur. Ceram. Soc., 2009, 29, 995–1011 CrossRef CAS.
- A. Nisar, R. Hassan, A. Agarwal and K. Balani, Ceram. Int., 2022, 48, 8852–8881 CrossRef CAS.
- Z. Ding, Q. Deng, D. Shi, X. Zhou, Y. Li, S. Ran and Q. Huang, J. Am. Ceram. Soc., 2014, 97, 3037–3040 CrossRef CAS.
- W. G. Fahrenholtz, G. E. Hilmas and R. X. Li, Sci. Sintering, 2020, 52, 1–14 CrossRef CAS.
- W. G. Fahrenholtz, G. E. Hilmas, S. C. Zhang and S. Zhu, J. Am. Ceram. Soc., 2008, 91, 1398–1404 CrossRef CAS.
- M. Thompson, W. G. Fahrenholtz and G. Hilmas, J. Am. Ceram. Soc., 2011, 94, 429–435 CrossRef CAS.
- L. Xu, Q. Y. Liu, Y. Z. Zhou, W. M. Guo, Y. Zhang, Y. You and H. T. Lin, Ceram. Int., 2022, 48, 31060–31064 CrossRef CAS.
- R. X. Li, Y. Zhang, H. J. Lou, J. P. Li and Z. H. Feng, J. Sol-Gel Sci. Technol., 2011, 58, 580–585 CrossRef CAS.
- C. M. Xie, M. W. Chen, X. Wei, M. Ge and W. G. Zhang, J. Am. Ceram. Soc., 2012, 95, 866–869 CrossRef CAS.
- Z. Amirsardari, R. M. Aghdam, M. Salavati-Niasari and S. Shakhesi, Mater. Manuf. Processes, 2016, 31, 134–140 CrossRef CAS.
- N. Patra, D. Jayaseelan and W. Lee, Adv. Appl. Ceram., 2016, 115, 36–42 CrossRef CAS.
- C. Q. Liu, X. J. Chang, Y. T. Wu, X. F. Wang and X. H. Hou, Vacuum, 2020, 177, 109430 CrossRef CAS.
- M. Rahmani-Azad, A. Najafi, N. Rahmani-Azad and G. Khalaj, J. Sol-Gel Sci. Technol., 2022, 103, 87–96 CrossRef CAS.
- C. F. Hu, J. Zou, Q. Huang, G. J. Zhang, S. Q. Guo and Y. Sakka, J. Am. Ceram. Soc., 2012, 95, 85–88 CrossRef CAS.
- R. Nowak, N. Sobczak, G. Bruzda, J. Wojewoda-Budka, L. Litynska-Dobrzynska, M. Homa, I. Kaban, L. Xi and L. Jaworska, J. Mater. Eng. Perform., 2016, 25, 3310–3316 CrossRef CAS.
- X. L. Fan, X. W. Huang, Q. Liu, H. M. Ding, H. Q. Wang and C. Hao, Results Phys., 2019, 14, 102494 CrossRef.
- J. F. Ma, S. Q. Cao, T. Q. Li, Q. Chen and D. H. Zhang, Mater. Sci. Eng., B, 2020, 261, 114698 CrossRef CAS.
- M. Velashjerdi, M. Soleymani, H. Sarpoolaky and A. Mirhabibi, Ceram. Int., 2020, 46, 28639–28651 CrossRef CAS.
- S. L. Song, R. Li, L. C. Gao, C. C. Sun, P. F. Hu and Q. Zhen, Ceram. Int., 2018, 44, 4640–4645 CrossRef CAS.
- T. Yin, B. Y. Jiang, Z. A. Su and Q. Z. Huang, J. Sol-Gel Sci. Technol., 2018, 85, 41–47 CrossRef CAS.
- L. Xu, C. Huang, H. Liu, B. Zou, H. Zhu, G. Zhao and J. Wang, Mater. Des., 2013, 49, 226–233 CrossRef CAS.
- J. J. Sha, Z. F. Zhang, S. X. Di, Z. Z. Lv, J. Li and J. X. Dai, Mater. Sci. Eng., A, 2017, 693, 145–150 CrossRef CAS.
- Y. H. Lin, J. H. Liu, S. L. Song, J. B. Liu, S. Bashir, Y. Guo and Q. Zhen, Ceram. Int., 2019, 45, 4016–4021 CrossRef CAS.
- J. B. He, Y. Gao, Y. G. Wang, J. Y. Fang and L. N. An, Ceram. Int., 2017, 43, 1602–1607 CrossRef CAS.
- Y.-N. Cao, S. Du, J.-K. Wang, H. Zhang, F.-L. Li, L.-L. Lu, S.-W. Zhang and X.-G. Deng, J. Sol-Gel Sci. Technol., 2014, 72, 130–136 CrossRef CAS.
- H. Ji, M. Yang, M. Li, G. Ji, H. Fan and X. Sun, Adv. Powder Technol., 2014, 25, 910–915 CrossRef CAS.
- L. S. Walker and E. L. Corral, J. Am. Ceram. Soc., 2014, 97, 399–406 CrossRef CAS.
- G. Ji, H. Ji, M. Li, X. Li and X. Sun, J. Sol-Gel Sci. Technol., 2014, 69, 114–119 CrossRef CAS.
- Y. Yan, Z. Huang, S. Dong and D. Jiang, J. Am. Ceram. Soc., 2006, 89, 3585–3588 CrossRef CAS.
- Y. Xie, T. H. Sanders Jr and R. F. Speyer, J. Am. Ceram. Soc., 2008, 91, 1469–1474 CrossRef CAS.
- Y. Zhang, R. Li, Y. Jiang, B. Zhao, H. Duan, J. Li and Z. Feng, J. Solid State Chem., 2011, 184, 2047–2052 CrossRef CAS.
- B. Yang, J. Li, B. Zhao, Y. Hu, T. Wang, D. Sun, R. Li, S. Yin, Z. Feng and Q. Tang, Powder Technol., 2014, 256, 522–528 CrossRef CAS.
- H. Shen, X. Y. Li, C. G. Hu, Z. Wang, X. Y. Hu, Y. Li and J. Yan, Surf. Interfaces, 2021, 25, 101162 CrossRef CAS.
- M. Hu, Z. Huang, X. Li, Y. Cheng, Z. Wang, K. Li, T. Wang, X. Hu, Y. Li and X. Zhang, ACS Appl. Eng. Mater., 2023, 1, 769–779 CrossRef CAS.
- C. Yan, R. Liu, C. Zhang, Y. Cao and X. Long, RSC Adv., 2015, 5, 78606–78613 RSC.
- C. L. Yan, R. J. Liu, C. R. Zhang and Y. B. Cao, J. Sol-Gel Sci. Technol., 2015, 76, 686–692 CrossRef CAS.
- Z. H. Ding, X. L. Huang, W. L. Liu, I. J. Kim and Y. H. Han, Adv. Appl. Ceram., 2021, 120, 222–226 CrossRef CAS.
- C. Chiappe, F. Signori, G. Valentini, L. Marchetti, C. S. Pomelli and F. Bellina, J. Phys. Chem. B, 2010, 114, 5082–5088 CrossRef CAS PubMed.
- F. H. Chung, J. Appl. Crystallogr., 1974, 7, 519–525 CrossRef.
- B. Jin, L. Wen, C.-z. LI, Z.-q. BAI and B.-q. LI, J. Fuel Chem. Technol., 2009, 37, 134–138 CrossRef.
- X. Chen, S. Dong, Y. Kan, H. Zhou, J. Hu and Y. Ding, RSC Adv., 2016, 6, 9338–9343 RSC.
- N. Shawgi, S. X. Li and S. Wang, Ceram. Int., 2017, 43, 10554–10558 CrossRef CAS.
- R. Ramzi, S. Wang, N. Shawgi and T. Tang, Mater. Res. Express, 2018, 6, 035007 CrossRef.
- R. L. Frost, Y. Xi, R. Scholz, F. M. Belotti and M. Cândido Filho, J. Mol. Struct., 2013, 1037, 23–28 CrossRef CAS.
- J. S. Li, L. F. Hao, X. H. Xu and T. H. Ren, Ind. Lubr. Tribol., 2012, 64, 217–223 CrossRef.
- M. N. Yang, S. L. Fan, H. Y. Huang, Y. J. Zhang, Z. Q. Huang, H. Y. Hu and J. Liang, Int. J. Biol. Macromol., 2020, 156, 280–288 CrossRef CAS PubMed.
- T. y. G. Khonina, N. V. Kungurov, N. y. V. Zilberberg, N. y. P. Evstigneeva, M. M. Kokhan, A. I. Polishchuk, E. V. Shadrina, E. Y. Nikitina, V. V. Permikin and O. N. Chupakhin, J. Sol-Gel Sci. Technol., 2020, 95, 682–692 CrossRef CAS.
- L. Xia, J. Liu, Z. Li, X. B. Wang, P. Wang, D. Wang and X. H. Hu, J. Macromol. Sci., Part A, 2020, 57, 560–568 CrossRef CAS.
- X. Tao, Z. Xiang, S. Zhou, Y. Zhu, W. Qiu and T. Zhao, J. Ceram. Sci. Technol., 2016, 7, 107–112 Search PubMed.
- S. Avcioğlu, M. Buldu, F. Kaya, C. B. Üstündağ, E. Kam, Y. Z. Menceloğlu, H. Y. Kaptan and C. Kaya, Ceram. Int., 2020, 46, 343–352 CrossRef.
- S. Avcioglu, F. Kaya and C. Kaya, Ceram. Int., 2020, 46, 17938–17950 CrossRef CAS.
- A. Najafi, F. Golestani-Fard and H. R. Rezaie, Ceram. Int., 2018, 44, 21386–21394 CrossRef CAS.
- M. GahremanDerakhshan, L. Nikzad, M. Farvizi and T. Ebadzadeh, Int. J. Appl. Ceram. Technol., 2021, 18, 32–39 CrossRef CAS.
- X. Li, M. Lei, S. Gao, D. Nie, K. Liu, P. Xing and S. Yan, Int. J. Appl. Ceram. Technol., 2020, 17, 1079–1087 CrossRef CAS.
- H. Y. Qiu, W. M. Guo, J. Zou and G. J. Zhang, Powder Technol., 2012, 217, 462–466 CrossRef CAS.
- Z. Shen, Z. Zhao, H. Peng and M. Nygren, Nature, 2002, 417, 266–269 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Weijian Han*b,
Tong Zhao
a,
Weijian Han*b,
Tong Zhao