
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of surface functionalization on DNA sequencing using MXene-based nanopores
You-Sheng Yu ab,
Rong-Ri Tan*c and
Hong-Ming Ding*d
ab,
Rong-Ri Tan*c and
Hong-Ming Ding*d
aSchool of Science, East China University of Technology, Nanchang 330013, China
bLaboratory of Solid State Microstructures, Nanjing University, Nanjing 210093, China
cDepartment of Physics, Jiangxi Science & Technology Normal University, Nanchang 330013, China. E-mail: rogertanr@hotmail.com
dCenter for Soft Condensed Matter Physics and Interdisciplinary Research, Soochow University, Suzhou 215006, China. E-mail: dinghm@suda.edu.cn
First published on 5th January 2024
Abstract
As one of the most promising types of label-free nanopores has great potential for DNA sequencing via fast detection of different DNA bases. As one of the most promising types of label-free nanopores, two-dimensional nanopore materials have been developed over the past two decades. However, how to detect different DNA bases efficiently and accurately is still a challenging problem. In the present work, the translocation of four homogeneous DNA strands (i.e., poly(A)20, poly(C)20, poly(G)20, and poly(T)20) through two-dimensional transition-metal carbide (MXene) membrane nanopores with different surface terminal groups is investigated via all-atom molecular dynamics simulations. Interestingly, it is found that the four types of bases can be distinguished by different ion currents and dwell times when they are transported through the Ti3C2(OH)2 nanopore. This is mainly attributed to the different orientation and position distributions of the bases, the hydrogen bonding inside the MXene nanopore, and the interaction of the ssDNA with the nanopore. The present study enhances the understanding of the interaction between DNA strands and MXene nanopores with different functional groups, which may provide useful guidelines for the design of MXene-based devices for DNA sequencing in the future.
1 Introduction
In the last two decades, nanopore-based DNA sequencing has become a rapidly growing research field, since it can provide fast, label-free and reliable single molecule detection, and ultimately may facilitate personalized medicine applied to individuals.1–12 It was first demonstrated in biological pores, such as α-hemolysin13 and MspA,14 but the applications of such pores share some disadvantages (e.g., low mechanical strength and susceptibility to pH, temperature and salt concentration).15–17 Synthetic solid-state nanopores (e.g., SiNx, SiO2, Al2O3 and others), as an alternative to biological nanopores, can overcome the above limitations of biological nanopores18–20 and do not need the excessive use of biochemical reagents.21–26 Nevertheless, the thickness of conventional solid-state pores is usually over 10 nm, thus it is likely that many DNA bases will be present inside the nanopore at the same time, which limits the single-read capability of the solid-state pores.27Graphene, with a sub-nanometer thickness (comparable to the spacing between neighboring DNA nucleotides), shows high-resolution DNA detection in theory and has been extensively investigated in simulations and experiments.28–33 However, a ssDNA molecule adsorbed on graphene cannot be energetically desorbed and traverse the pore, limiting its practical applications.34 Moreover, great attention has also been paid to nanopores formed by compound two-dimensional (2D) materials (e.g., molybdenum disulfide (MoS2), boron nitride (BN), tungsten disulfide (WS2), HfO2, etc.) owing to their higher hydrophilicity compared to graphene.35 For example, Aluru et al.36 demonstrated that a MoS2 nanopore shows four distinct ion current signals for single-base discrimination with low noise. Zhou et al.37 showed that it is possible to detect different dsDNAs from the recording of ion current during translocation of the DNA through BN nanopores. With more 2D materials being discovered and their physical properties elucidated, the natural progression is to appraise their performances in nanopore-based DNA sequencing.38
Recently, MXenes (a new family of 2D materials), consisting of transition-metal carbides and nitrides, have attracted great attention. Their general formula is Mn+1XnTx (n = 1, 2, or 3), where M and X represent a transition metal, and a carbon and/or nitrogen, respectively. Tx indicates different functional groups on the MXene surfaces, and it can be –O, –OH and –F. Generally, MXenes are produced by liquid exfoliation techniques and through selective etching of A-group elements.39 Due to their multi-element contents and tunable compositions, MXenes have shown great promise in various fields, such as gas sensing,40,41 water purification,42,43 energy storage, and so on. For example, Gogotsi et al.41 reported that lamellar stacked MXene membranes exhibit excellent gas separation performance with H2/CO2 selectivity >160. Wang et al.44 demonstrated that MXene membranes exhibit high oil rejection without compromising water permeance.
Very recently, MXenes have been proved to be a promising tool for molecular detection, due to their biocompatibility, hydrophilicity and near-infrared adsorption.41 For example, Wanunu et al.45 demonstrated the feasibility of MXene nanopores for DNA sequencing, and also found that MXene membranes exhibit low ion current leakage and noise characteristics comparable to other two-dimensional membranes. Pumera et al.46 probed the interaction of ssDNA and dsDNA with Ti3C2Tx via fluorescence spectroscopy and molecular dynamics (MD) simulations, and found that the MXene showed some potential for biosensing with unequivocal detection at picomole levels and single-base discrimination. Farimani et al.38 found that this nanopore material was able to detect different types of DNA bases, with base A having the longest residence time due to the strong physisorption and deflection. Notably, the surface terminal groups of the MXene can have great impacts on its physicochemical properties, which in turn may affect its DNA detection ability. However, to the best of our knowledge, there are no systematic studies that investigate the effect of surface decoration of the MXene nanopore on DNA sequencing.
In this work, we mainly focus on the effect of different surface terminal groups of MXenes (Ti3C2(OH)2, Ti3C2O2 and Ti3C2F2) on DNA sequencing by using all-atom molecular dynamics simulations, i.e., comparing the ionic current and dwell time produced by poly(A)20, poly(C)20, poly(G)20, and poly(T)20 in the case of three types of MXene nanopores. As we show below, the four types of DNA bases can be effectively identified by using Ti3C2(OH)2. Moreover, the underlying mechanism for why the ionic current and dwell time are different in different cases will be investigated in detail.
2 Methods
As shown in Fig. 1, our simulation system contains an MXene nanopore, a homogeneous single-stranded DNA, ions and water molecules. The structure of Ti3C2Tx MXene membranes was taken from the literature.47 The size of each Ti3C2Tx MXene membrane was set to about 7.6 × 7.6 nm2. The nanopore, with a radius of 1.0 nm, was constructed by deleting the atoms within 1.0 nm of the center of the MXene membrane. For the sake of simplicity, four types of homogeneous single-stranded DNA (ssDNA) with twenty identical bases generated using the make-na server (i.e., poly(A)20, poly(C)20, poly(G)20, and poly(T)20) were employed to model the DNA fragment. This length of single-stranded DNA fragment (two full turns of the helix48) has been successfully detected by graphene nanopores and biological nanopores in experiments49,50 and is commonly employed in DNA sequencing based on nanopores using molecular dynamics simulations.51–53 The size of the simulated box was about 7.8 × 7.8 × 9.0 nm3, where the beginning base of the ssDNA was placed at the pore center and perpendicular to the surface of the MXene membrane, and then was solvated with water molecules. Finally, a suitable number of Na+ ions were added to neutralize the net charge of the system, while NaCl salt with a concentration of 1.0 M was added into the system. In total, the simulation system contained about 51![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 atoms, including 349 Na+ ions, 330 Cl− ions, and appr. 15100 water molecules.
000 atoms, including 349 Na+ ions, 330 Cl− ions, and appr. 15100 water molecules.
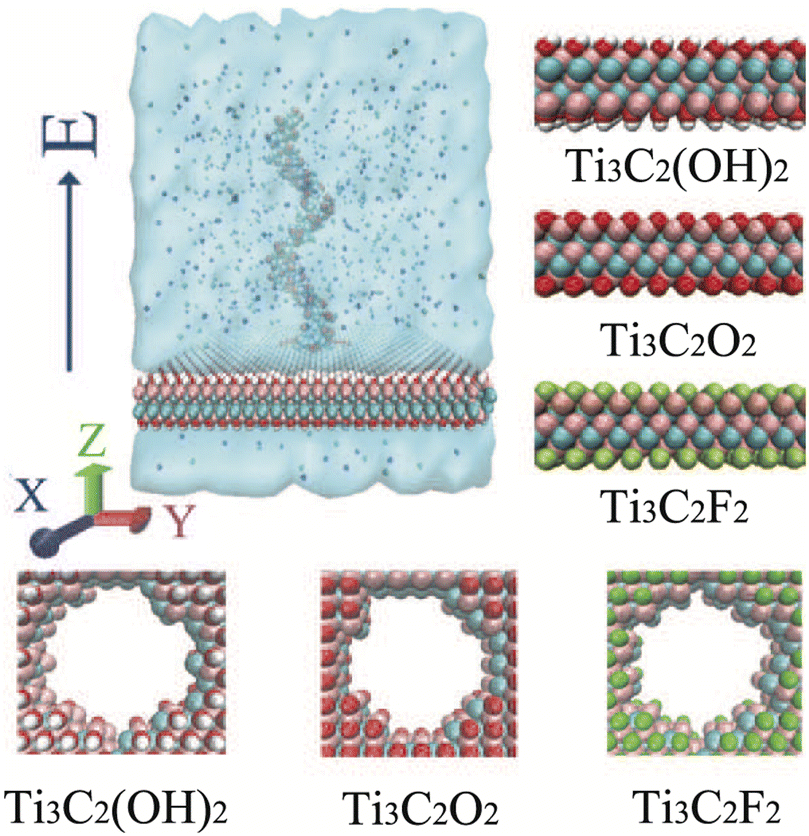 | ||
| Fig. 1 The simulation model for ssDNA translocation through an MXene nanopore. For clarity, water molecules filling the simulation cell are not displayed. The right column shows the atomic structures of the three types of MXene membrane (mixed atom termination at the nanopore). The Ti, C, F, O and H atoms are depicted as pink, cyan, green, red and white spheres, respectively. | ||
In this work, all-atom MD simulations were performed using the Gromacs 2020.4 package54 with the CHARMM27 force field, while the simulation trajectories were analyzed using Gromacs utilities and home-written codes. The TIP3P water model was chosen. The force-field parameters for the MXene membrane were taken from the work by Xu et al.46,55 The particle mesh Ewald method was used when calculating the electrostatic interactions, and the Lennard-Jones (LJ) interactions were cut off at a distance of 1.2 nm. Periodic boundary conditions were adopted in all three directions. The systems were firstly energy-minimized for 10000 steps. Then 1.0 ns NVT equilibrations were performed, where the DNA was harmonically restrained in the nanopore.56 Finally the translocation process of ssDNA through nanopores under an electric field of E = 0.2 V nm−1 was performed in the NVT ensemble, where the DNA was not restrained. The atoms of the MXene were frozen in all the simulations, since the nanosheets are rather rigid.41 The Nosé–Hoover method was used to keep the temperature at 300 K.57 There is no pore-surface functionalization and no special treatments are applied to the charges of the pore-surface atoms. Each system was repeated fifteen times by changing the random seed of the velocity.
The integration time step was 2 fs. Ionic current was calculated using the following equation from ref. 58:
 | (1) |
3 Results and discussion
We first investigated the ion currents (total, Na+ and Cl−) as a function of simulation time for the three types of MXene nanopores in the absence of DNA. As shown in Fig. 2a, the mean ion current for the Ti3C2(OH)2 nanopore was larger than those for the other two MXene nanopores. It indicated weaker resistance of ion movement through the Ti3C2(OH)2 nanopore compared to the other two MXene nanopores. Additionally, the Cl− ion current of each MXene nanopore was larger than the Na+ ion current (see the inset of Fig. 2a). This result was consistent with previous results,59 probably due to the fact that the Cl− ion has a larger mobility compared to the Na+ ion,60,61 and there is a large proportion of high electric potential in the vicinity of the nanopore (see Fig. 2b). | ||
| Fig. 2 (a) The mean ion currents for the three types of MXene nanopore with a radius of 1.0 nm, in the absence of DNA. The inset shows the ion currents as a function of simulation time. The black, red and green lines represent the total, Na+ and Cl− ion currents, respectively. The shadows (error bars) represent the standard deviation obtained from 7 independent runs. The electric field is 0.2 V nm−1. (b) The electrostatic potential map of the three types of MXene nanopore (obtained by using the PMEPot plugin of VMD). | ||
We then investigated the ion current and the dwell time for each homogeneous ssDNA through the three types of MXene nanopore (Fig. 3). Notably, the ion current and the dwell time are typical parameters for distinguishing different DNA types.38,62 For the Ti3C2(OH)2 nanopore, the difference in ion current with bases A, C and G was not significant (about 4.6 nA), but the ion current of base T was the smallest (about 4.2 nA) (see Fig. 3a). Additionally, the ion current of the Ti3C2(OH)2 nanopore for ssDNA translocation was the largest, which was in agreement with the case in the absence of DNA. The order of the dwell time for this nanopore was A > G > C ≈ T (see Fig. 3b). This indicates that the translocation of pyrimidines is faster than that of purines; moreover, base A exhibits remarkably slow translocation, probably due to the strong resistance force (see detailed discussion below).
 | ||
| Fig. 3 Ion currents (a, c and e) and dwell times (b, d and f) for each homogeneous ssDNA through the three types of MXene nanopore (Ti3C2(OH)2, Ti3C2O2 and Ti3C2F2). The dwell time is defined as the time for the ending base to leave the nanopore. In the left column, the black represents Na+ current and the red represents Cl− current. Error bars represent the standard deviation obtained from 15 independent runs. The radius of the nanopore is 1.0 nm and the electric field is 0.2 V nm−1. | ||
For the Ti3C2O2 nanopore, base G had the largest value for the ion current (about 3.1 nA) and base C had the smallest one (about 2.6 nA), while the ion currents of bases A and T showed little difference (see Fig. 3c). The order of the dwell time was not very obvious (see Fig. 3d). For the Ti3C2F2 nanopore, pyrimidines (bases C and T) had the same values for the ion current and dwell time, while those of purines (base A and G) were almost the same (see Fig. 3e and f). Additionally, the dwell time and ion current of pyrimidines was smaller than those of purines. This indicated that the transport velocity of pyrimidines was greater than that of purines.
We also computed the contribution of Na+ and Cl− ions to the total ion current (Fig. 3a, c and e). It was found that Cl− ion current was larger than Na+ ion current, no matter which base translocated into these three types of nanopore, indicating that the contribution of Cl− ions was larger than that of Na+ ions. Specifically, Na+ ion current was only about 18% of the total ion current for Ti3C2O2 and Ti3C2F2 nanopores, and about 23% for Ti3C2(OH)2. This was mainly attributed to the following reasons. First and as mentioned above, there was some repulsion between the MXene nanopore and the Na+ ion, which can decrease the Na+ ion current. Second, some salt bridges may exist between the ssDNA and Na+ ion, which results in a reverse Na+ ion current. In general, the Cl− ion played an important role in DNA sequencing using MXene nanopores.
To intuitively compare the ability of DNA sequencing among the three MXene nanopores, the ion current and the dwell time for the four types of DNA bases were plotted in Fig. 4a. It was found that the Ti3C2O2 nanopore can distinguish bases C and G, but it can not distinctly discriminate bases A and T. For the Ti3C2F2 nanopore, it can distinguish the purines and pyrimidines, but it can not further recognize bases C and T in the pyrimidines, or bases A and G in the purines. Importantly, the four types of bases can be distinguished by the Ti3C2(OH)2 nanopore. Additionally, only about one single base per frame was in the nanopore when the DNA was transported through the Ti3C2(OH)2 nanopore (see Table 1), which can further improve the ability for DNA detection. On the basis of the above discussion, the Ti3C2(OH)2 nanopore could be the most appropriate one for real applications. The presence of cations significantly affects hydrogen bonding in DNA structures, which stabilizes the double-helical structure and is specific to the DNA sequence.63 Also, the ions (especially divalent ions) interact strongly with single-stranded DNA, such that the double-helix stability decreases at very high salt concentrations (>1 M).64,65 Hence, we further investigated the effect of ion concentration on ion current and dwell time for the Ti3C2(OH)2 nanopore. It is found that the resolution of DNA-base detection increases as the ion concentration increases for the Ti3C2(OH)2 nanopore (see Fig. 4b).
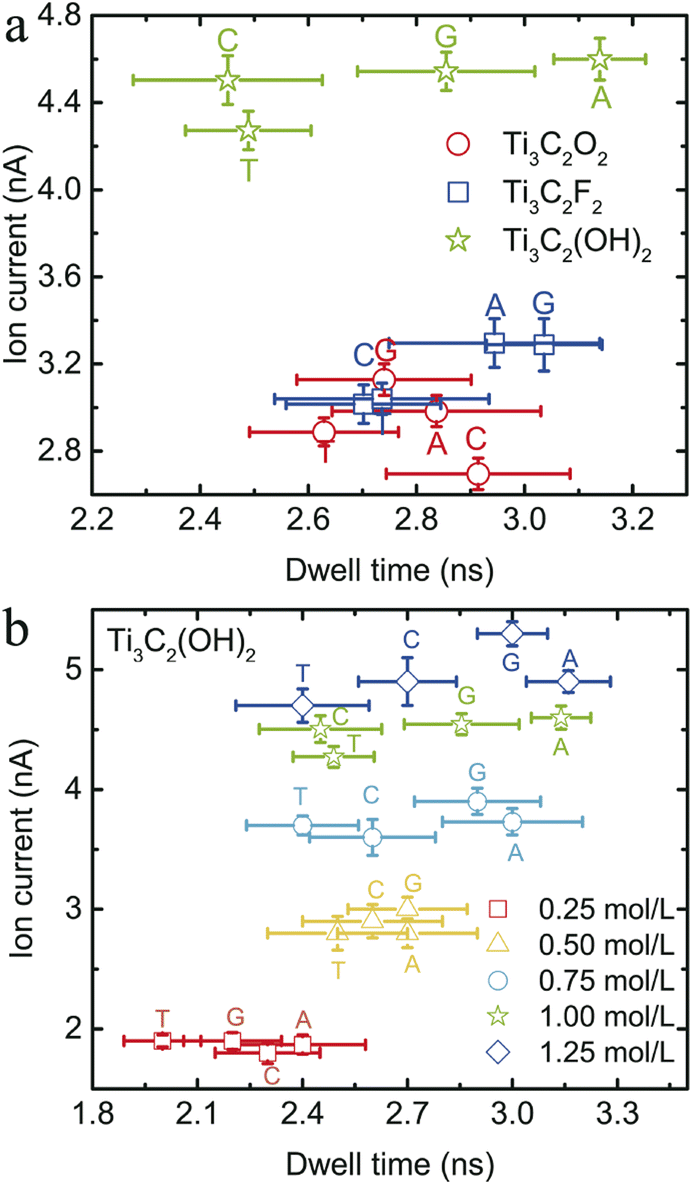 | ||
| Fig. 4 (a) Ion current vs. dwell time for the three types of MXene nanopore. The green stars, red circles and blue squares represent the nanopores of Ti3C2(OH)2, Ti3C2O2 and Ti3C2F2, respectively. (b) The ion currents and dwell times under different ion concentrations for the Ti3C2(OH)2 nanopore. | ||
| MXene nanopore | Base A | Base C | Base G | Base T |
|---|---|---|---|---|
| Ti3C2(OH)2 | 1.02 ± 0.17 | 1.31 ± 0.32 | 1.17 ± 0.18 | 0.97 ± 0.15 |
| Ti3C2O2 | 1.28 ± 0.17 | 1.63 ± 0.35 | 1.29 ± 0.19 | 1.03 ± 0.22 |
| Ti3C2F2 | 1.23 ± 0.19 | 1.65 ± 0.19 | 1.32 ± 0.12 | 1.00 ± 0.23 |
To provide some possible explanation for the above phenomena, we first calculated the probability distributions for the orientation and position of each nucleobase in the three types of MXene nanopore (Fig. 5). The orientation was characterized by the angle β (between the normal direction of the base and the normal direction of the MXene surface) and r, which denoted the distance between the central axis of the MXene nanopore and the mass center of the base inside the nanopore. All the distribution centers of β for the four types of bases through these three types of MXene nanopore were less than 90°, indicating that the four bases translocated through the MXene nanopore in a slanted manner.
 | ||
| Fig. 5 The distribution of orientation and position for the four types of bases through the Ti3C2(OH)2 (a), Ti3C2O2 (b) and Ti3C2F2 (c) nanopores. From top to bottom, the bases are A, C, G and T in turn. r denotes the distance between the central axis of the MXene nanopore and the mass center of the base inside the nanopore; the center of the nanopore is r = 0. β represents the angle between the normal direction of the base and the normal direction of the MXene surface; β = 0 means that the base plane and MXene surface are parallel to each other. | ||
For the Ti3C2(OH)2 nanopore, the distribution regions of bases A and G were smaller than those of the other two bases (Fig. 5a), meaning that these two bases had a stronger interaction with this MXene nanopore. These small regions led to an increment in ion movement through the nanopore due to a decrement in the blockage effect. Hence, bases A and G showed large ion currents when they were transported through the Ti3C2(OH)2 nanopore. Notably, base C had a large distribution region, but its probability appeared at about β < 30°, which was smaller than that for base T. Thus, base C had a small probability of the base plane being approximately parallel to the MXene surface and the ability of base C to block anions was reduced (because the anions are mainly distributed in the region of high electric potential). These phenomena led to an increment in the ion current for base C. As a result, the ion currents of bases A, C and G were nearly the same and much larger than that of base T.
Meanwhile, for the Ti3C2O2 nanopore, base C had the largest orientation and position distribution region, while base G had the smallest one (Fig. 5b). The larger the distribution region, the greater the fluctuation, which further shows that the blockage effect is stronger. Hence, base C had the smallest ion current, and base G had the largest one. Moreover, as shown in Fig. 5c, the distribution region for base G translocating through the Ti3C2F2 nanopore was small, meaning that base G inside the nanopore had a small blockage effect. Thus, its ion current was the largest. Although base C showed almost the same distribution region as base A, there were nearly two bases of C inside the nanopore (see Table 1), which could also lead to a decrement in the ion current. Thus, base C had a smaller ion current.
To analyze the mechanism for the different dwell times of ssDNA translocating through the MXene nanopore, we further calculated the number of hydrogen bonds in the hydrogen-bond “network” inside the MXene nanopore as ssDNA translocates through it and the interaction energy between the ssDNA and the MXene nanopore. These two parameters were used to describe the resistance of ssDNA translocation through the MXene nanopore (Fig. 6). The number of hydrogen bonds inside the nanopore mainly represents the hydration force around the ssDNA, where a greater number of hydrogen bonds corresponds to a stronger resistance for ssDNA to pass through the nanopore. The interaction energy describes the interaction between the ssDNA and the MXene nanopore, where a greater interaction energy indicates a stronger resistance. The hydrogen-bond network mainly consists of three parts: (1) the hydrogen bonding between the ssDNA and water molecules as well as the MXene nanopore (HBnmw), (2) the hydrogen bonding between the water molecules inside the nanopore (HBww), and (3) the hydrogen bonding between the water molecules and the MXene nanopore (HBwm). It is found that the number of hydrogen bonds as base A translocates through the Ti3C2(OH)2 nanopore had the largest value, which originates from the maximum hydrogen bonding between water molecules (HBww). It also indicates a decrease in the blockage effect. Meanwhile, base A had the largest interaction energy among these four bases. Hence, base A had the largest dwell time. For the pyrimidines (base C and T), the number of hydrogen bonds and interaction energy were both small compared with the case of the purines (base A and G), meaning that their resistances were small. Therefore, they had a small dwell time. Furthermore, the number of hydrogen bonds (HBnmw) for base C passing through the MXene nanopore was slightly larger than that for base T passing through it, mainly due to the fact that more C bases reside in the nanopore. Additionally, the values of HBnmw were large as the purines translocated through the MXene nanopore compared with those for the pyrimidines passing through the MXene nanopore.
 | ||
| Fig. 6 (a) The number of hydrogen bonds inside the three types of MXene nanopore, computed using the VMD plugin; the angle and distance cutoffs are 30° and 0.35 nm, respectively. (b) The interaction energy of ssDNA with the three types of MXene nanopore; this incorporates the Coulomb and Lennard-Jones energies. The error bars represent the standard deviation obtained from 15 independent runs. | ||
Meanwhile, for the Ti3C2O2 nanopore, the number of hydrogen bonds as the four types of bases translocated through the nanopore showed a tiny difference. However, base C had the largest interaction energy with the MXene nanopore among these four bases, attributed to there being more C bases inside the nanopore. Therefore, the dwell time of base C was the longest. Moreover, the numbers of hydrogen bonds as the purines translocated through the Ti3C2F2 nanopore were larger than those for the pyrimidines, while there was little difference in the interaction energies between the purines and pyrimidines. This meant that the purines translocating through the Ti3C2F2 nanopore exhibited a stronger resistance, so the dwell times of the purines were larger than those of the pyrimidines. Base C had a small hydrogen bonding value between water molecules (HBww), which is attributed to there being more bases inside the nanopore; this is also the reason for the large hydrogen bonding value between the ssDNA and water molecules (HBnmw). Additionally, the number of hydrogen bonds (HBwm) between the water molecules and the nanopore was larger for Ti3C2(OH)2 than for the other two nanopores, mainly attributed to the fact that the terminal groups (–OH) are both donors and acceptors.
4 Conclusions
In summary, we have systematically investigated the translocation of four DNA strands through three types of MXene nanopore (Ti3C2O2, Ti3C2F2 and Ti3C2(OH)2) by using all-atom molecular dynamics simulations. Importantly, it is found that the four bases can be discriminated by the Ti3C2(OH)2 nanopore due to their distinct ion currents and/or dwell time. Nevertheless, the four bases cannot be distinguished by the Ti3C2O2 and Ti3C2F2 nanopores since the Ti3C2O2 nanopore cannot distinguish bases A and T while the Ti3C2F2 nanopore can only distinguish the purines and pyrimidines. Moreover, the physical mechanism underlying the under different situations is revealed. It is found that the ion current is highly related to the orientation and position distributions of the bases within the nanopore, and the hydrogen bonds and interaction energy of the bases with the nanopore determines the dwell time. In general, the present work highlights the importance of the surface decoration of MXene materials in DNA detection, which could offer some physical insights into the experimental design of novel devices for efficient DNA sequencing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 12164002, 11947043 and 12222506), Jiangxi Provincial Natural Science Foundation (No. 20232BAB201034) and the PhD research startup foundation of East China University of Technology (No. DHBK2019203). We are grateful to the High Performance Computing Center (HPCC) of Nanjing University for carrying out the numerical calculations in this paper on its blade cluster system.References
- H. Qiu, A. Girdhar, K. Schulten and J. P. Leburton, ACS Nano, 2016, 10, 4482–4488 CrossRef CAS PubMed.
- C. Sathe, X. Zou, J. P. Leburton and K. Schulten, ACS Nano, 2011, 5, 8842–8851 CrossRef CAS PubMed.
- J. Prasongkit, G. T. Feliciano, A. R. Rocha, Y. He, T. Osotchan, R. Ahuja and R. H. Scheicher, Sci. Rep., 2015, 5, 17560 CrossRef CAS PubMed.
- E. Krueger, J. Shim, A. Fathizadeh, A. N. Chang, B. Subei, K. M. Yocham, P. H. Davis, E. Graugnard, F. Khalili-Araghi and R. Bashir, et al., ACS Nano, 2016, 10, 8910–8917 CrossRef CAS PubMed.
- D. P. Hoogerheide, B. Lu and J. A. Golovchenko, ACS Nano, 2014, 8, 7384–7391 CrossRef CAS PubMed.
- M. Shankla and A. Aksimentiev, Nat. Commun., 2014, 5, 5171 CrossRef CAS PubMed.
- Z. S. Siwy and M. Davenport, Nat. Nanotechnol., 2010, 5, 697–698 CrossRef CAS PubMed.
- Y. Cho, S. K. Min, W. Y. Kimb and K. S. Kim, Phys. Chem. Chem. Phys., 2011, 13, 14293–14296 RSC.
- J. Clarke, H. C. Wu, L. Jayasinghe, A. Patel, S. Reid and H. Bayley, Nat. Nanotechnol., 2009, 4, 265–270 CrossRef CAS PubMed.
- G. M. Cherf, K. R. Lieberman, H. Rashid, C. E. Lam, K. Karplus and M. Akeson, Nat. Biotechnol., 2012, 30, 344–348 CrossRef CAS PubMed.
- M. Belkin and A. Aksimentiev, ACS Appl. Mater. Interfaces, 2016, 8, 12599–12608 CrossRef CAS PubMed.
- Y. S. Yu, R. R. Tan and H. M. Ding, Phys. Chem. Chem. Phys., 2020, 22, 16855–16861 RSC.
- J. J. Kasianowicz, E. Brandin, D. Branton and D. W. Deamer, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13770–13773 CrossRef CAS PubMed.
- T. Z. Butler, M. Pavlenok, I. M. Derrington, M. Niederweis and J. H. Gundlach, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20647–20652 CrossRef CAS PubMed.
- D. B. Wells, M. Belkin, J. Comer and A. Aksimentiev, Nano Lett., 2012, 12, 4117–4123 CrossRef CAS PubMed.
- M. Kulkarni and A. Mukherjee, RSC Adv., 2016, 6, 46019–46029 RSC.
- H. S. Kim and Y. H. Kim, Biosens. Bioelectron., 2015, 69, 186–198 CrossRef CAS PubMed.
- H. Arjmandi-Tash, L. A. Belyaeva and G. F. Schneider, Chem. Soc. Rev., 2016, 45, 476–493 RSC.
- H. Qiu, A. Sarathy, J. P. Leburton and K. Schulten, Nano Lett., 2015, 15, 8322–8330 CrossRef CAS PubMed.
- M. Belkin, S. H. Chao, M. P. Jonsson, C. Dekker and A. Aksimentiev, ACS Nano, 2015, 9, 10598–10611 CrossRef CAS PubMed.
- S. Chang, S. Huang, J. He, F. Liang, P. Zhang, S. Li, X. Chen, O. Sankey and S. Lindsay, Nano Lett., 2010, 10, 1070–1075 CrossRef CAS PubMed.
- S. Huang, J. He, S. Chang, P. Zhang, F. Liang, S. Li, M. Tuchband, A. Fuhrmann, R. Ros and S. Lindsay, Nat. Nanotechnol., 2010, 5, 868–873 CrossRef CAS PubMed.
- A. P. Ivanov, E. Instuli, C. M. McGilvery, G. Baldwin, D. W. McComb, T. Albrecht and J. B. Edel, Nano Lett., 2011, 11, 279–285 CrossRef CAS PubMed.
- L. Xue, H. Yamazaki, R. Ren, M. Wanunu, A. P. Ivanov and J. B. Edel, Nat. Rev. Mater., 2020, 5, 931–951 CrossRef CAS.
- Y. He, M. Tsutsui, Y. Zhou and X.-S. Miao, NPG Asia Mater., 2021, 13, 48 CrossRef.
- S. Su, X. Wang and J. Xue, Mater. Horiz., 2021, 8, 1390–1408 RSC.
- B. M. Venkatesan and R. Bashir, Nat. Nanotechnol., 2011, 6, 615–624 CrossRef CAS PubMed.
- X. Lu, P. Xu, H. M. Ding, Y. S. Yu and Y. Q. Ma, Nat. Commun., 2019, 10, 4520 CrossRef PubMed.
- Y. Yu, X. Lu, H. Ding and Y. Ma, Phys. Chem. Chem. Phys., 2018, 20, 9063–9069 RSC.
- S. J. Heerema and C. Dekker, Nat. Nanotechnol., 2016, 11, 127–136 CrossRef CAS PubMed.
- S. W. Kowalczyk, D. B. Wells, A. Aksimentiev and C. Dekker, Nano Lett., 2012, 12, 1038–1044 CrossRef CAS PubMed.
- Z. Kong, W. Zheng, Q. Wang, H. Wang, F. Xi, L. Liang and J. W. Shen, J. Mater. Chem. B, 2015, 3, 4814–4820 RSC.
- A. A. Balandin, Nat. Nanotechnol., 2013, 8, 549–555 CrossRef CAS PubMed.
- A. Smolyanitsky and B. Luan, Phys. Rev. Lett., 2021, 127, 138103 CrossRef CAS PubMed.
- B. Luan and R. Zhou, ACS Nano, 2018, 12, 3886–3891 CrossRef CAS PubMed.
- A. B. Farimani, K. Min and N. R. Aluru, ACS Nano, 2014, 8, 7914–7922 CrossRef CAS PubMed.
- Z. Gu, Y. Zhang, B. Luan and R. Zhou, Soft Matter, 2016, 12, 817–823 RSC.
- P. Yadav, Z. Cao and A. B. Farimani, ACS Nano, 2021, 15, 4861–4869 CrossRef CAS PubMed.
- M. Naguib, V. N. Mochalin, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2013, 26, 992–1005 CrossRef PubMed.
- L. Li, T. Zhang, Y. Duan, Y. Wei, C. Dong, L. Ding, Z. Qiao and H. Wang, J. Mater. Chem. A, 2018, 6, 11734–11742 RSC.
- L. Ding, Y. Wei, L. Li, T. Zhang, H. Wang, J. Xue, L. X. Ding, S. Wang, J. Caro and Y. Gogotsi, Nat. Commun., 2018, 9, 155 CrossRef PubMed.
- E. Y. M. Yang, T. Y. Ng, J. Yeo, R. Lin, Z. Liu and K. R. Geethalakshmi, J. Membr. Sci., 2020, 598, 117653 CrossRef.
- S. Shen, T. Ke, K. Rajavel, K. Yang and D. Lin, Small, 2020, 2002433 CrossRef CAS PubMed.
- Z. K. Li, Y. Liu, L. Li, Y. Wei and H. Wang, J. Membr. Sci., 2019, 592, 117361 CrossRef.
- M. Mojtabavi, A. Vahidmohammadi, W. Liang, M. Beidaghi and M. Wanunu, ACS Nano, 2019, 13, 3042–3053 CrossRef CAS PubMed.
- C. L. Manzanares-Palenzuela, A. M. Pourrahimi, J. Gonzalez-Julian, Z. Sofer and M. Pumera, Chem. Sci., 2019, 10, 10010–10017 RSC.
- M. Khazaei, M. Arai, T. Sasaki, C. Y. Chung, N. S. Venkataramanan, M. Estili, Y. Sakka and Y. Kawazoe, Adv. Funct. Mater., 2013, 23, 2185–2192 CrossRef CAS.
- S. Kesselheim, W. Müller and C. Holm, Phys. Rev. Lett., 2014, 112, 018101 CrossRef PubMed.
- J. J. Nakane, M. Akeson and A. Marziali, J. Phys.: Condens. Matter, 2003, 15, R1365–R1393 CrossRef CAS.
- Y. Deng, Q. Huang, Y. Zhao, D. Zhou, C. Ying and D. Wang, Nanotechnology, 2016, 28, 045302 CrossRef PubMed.
- J. Li, Y. Zhang, J. Yang, K. Bi, Z. Ni, D. Li and Y. Chen, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2013, 87, 062707 CrossRef PubMed.
- W. Lv and M. Chen, et al., Soft Matter, 2013, 9, 960–966 RSC.
- D. Zhao, H. Chen, Y. Wang, B. Li, C. Duan, Z. Li and L. Li, Front. Chem. Sci. Eng., 2021, 15, 922–934 CrossRef CAS.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- K. Xu, Z. Lin, C. Merlet, P.-L. Taberna, L. Miao, J. Jiang and P. Simon, ChemSusChem, 2018, 11, 1892–1899 CrossRef CAS PubMed.
- M. Shankla and A. Aksimentiev, Nat. Commun., 2014, 5, 5171 CrossRef CAS PubMed.
- W. G. Hoover, Phys. Rev. A, 1985, 31, 1695–1697 CrossRef PubMed.
- W. Lv, M. Chen and R. Wu, Soft Matter, 2013, 9, 960–966 RSC.
- S. Go and M. Eun Suk, Electrochem. Commun., 2023, 147, 107434 CrossRef CAS.
- S. H. Lee and J. C. Rasaiah, J. Phys. Chem., 1996, 100, 1420–1425 CrossRef CAS.
- Q. Ying-Hua, L. Kun, C. Wei-Yu, S. Wei, T. Qi-Yan and C. Yun-Fei, Chin. Phys. B, 2015, 24, 108201 CrossRef.
- A. B. Farimani, P. Dibaeinia and N. R. Aluru, ACS Appl. Mater. Interfaces, 2017, 9, 92–100 CrossRef PubMed.
- A. Singh and N. Singh, Phys. A, 2015, 419, 328–334 CrossRef CAS.
- D. Sardana, P. Alam, K. Yadav, N. S. Clovis, P. Kumar and S. Sen, Phys. Chem. Chem. Phys., 2023, 25, 27744–27755 RSC.
- A. Maity, A. Singh and N. Singh, Eur. Biophys. J., 2017, 46, 33–40 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |