
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of annulated rosarins via iminium activation†
Duong D.
Nguyen
a,
Jorge
Labella
 *b,
Marta
Gómez-Gómez
b,
Tomás
Torres
*bcd and
Jonathan L.
Sessler
*a
*b,
Marta
Gómez-Gómez
b,
Tomás
Torres
*bcd and
Jonathan L.
Sessler
*a
aDepartment of Chemistry, The University of Texas at Austin, 105 E 24th Street A5300, Austin, TX 78712, USA. E-mail: sessler@cm.utexas.edu
bDepartamento de Química Orgánica, Universidad Autónoma de Madrid, Campus de Cantoblanco, C/Francisco Tomás y Valiente 7, 28049 Madrid, Spain. E-mail: jorge.labella@uam.es; tomas.torres@uam.es
cInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, 28049 Madrid, Spain
dIMDEA-Nanociencia, Campus de Cantoblanco, 28049 Madrid, Spain
First published on 14th September 2024
Abstract
Applications of expanded porphyrins, pyrrole-based macrocycles known for their unique optoelectronic properties, have been hampered by challenges associated with their synthesis. Typically, these compounds are prepared via condensation–macrocyclization reactions under acidic conditions, using pyrrolic nucleophiles and aldehydes as electrophiles. The yield of these transformations is heavily influenced by the electrophilicity of the aldehydes, thus limiting the scope of the reaction. In this study, we demonstrate that activation of aldehydes by pre-conversion to the corresponding iminium species can be exploited for the construction of expanded porphyrins. Specifically, we employ iminium-based activation to prepare a series of naphthorosarins (NRos) using various aldehydes as electrophiles, resulting in different meso substituents. This approach has proved to be more efficient than classical methodologies, paving the way for the synthesis of previously inaccessible systems. Based on a combination UV-vis spectroscopy, electrochemistry, and density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations, the meso-substituent affect slightly the optoelectronic properties of the NRos considered in this study. Theoretical calculations provide support for the conclusion that the iminium pathway makes the condensation reaction both kinetically and thermodynamically more favorable. The present approach may provide a general tool useful in the synthesis of expanded porphyrins and other pyrrolic macrocycles.
Introduction
Expanded porphyrins, macrocycles featuring more than four pyrrolic or related aromatic subunits,1,2 have garnered significant attention due to their versatile electronic states and ability to stabilize radical forms and various aromatic configurations, including Hückel, Möbius, and excited-state (anti-)aromaticity.3–7 Despite considerable progress in their chemistry, the synthesis of most expanded porphyrins remains a substantial challenge. The most common approach involves condensing a nucleophilic pyrrole derivative with an aldehyde electrophile, activated by either Brønsted or Lewis acids, followed by oxidation.8 Other methods involving oxidative couplings9,10 or cross-coupling reactions,11–13 have also been reported. The success of the condensation strategy is significantly influenced by the electrophilicity of the aldehyde; higher electrophilicity makes the key condensation reaction more favourable, thereby increasing the yield of the entire cyclization process. For this reason, fluorinated aldehydes are often employed, while less electrophilic aldehydes, on the contrary, frequently result in unacceptably poor yields or even failed macrocyclizations.14 This limitation significantly restricts the choice of meso-substituents, which play a crucial role in modulating the photophysical properties and potential applications of porphyrinoids.15–21 Therefore, there is an incentive to enhance the reactivity of the aldehyde. Doing so is expected to improve the synthesis of known expanded porphyrins and may allow the preparation of derivatives nominally derived from less-activated aldehydes.Iminium catalysis has emerged in recent decades as a powerful tool for the activation of aldehydes under simple, mild reaction conditions.22 This kind of catalysis has been exploited in many transformations, including Michael additions, Diels–Alder cycloadditions, Knoevenagel condensations, and, more recently, photochemical reactions. Remarkably, more than 15 years ago, the groups of Singh23,24 and Benaglia25 reported the iminium-catalysed synthesis of dipyrromethenes, key intermediates in the formation of expanded porphyrins. However, to our knowledge, the preparation of expanded porphyrins via iminium chemistry has never been reported.
Herein we report the synthesis of a representative class of expanded porphyrins, namely the naphthorosarins (NRos; Fig. 1), employing iminium-catalysed condensation conditions. As detailed below, we have found that the addition of secondary amines significantly enhances the macrocyclization yield, enabling the synthesis of NRos with various electron-donating meso-substituents. Density Functional Theory (DFT) calculations reveal that the origin of this enhancement lies in the higher concentration of “activated” aldehyde available to initiate the condensation and a reaction sequence that involves a more favourable rate-determining step. To provide a preliminary insight into the effect of meso-groups on the optoelectronic properties of the macrocycle, the present NRos were studied by a combination of UV-vis spectroscopic analyses and electrochemical measurements, as well as supporting DFT and time-dependent DFT (TD-DFT) calculations.
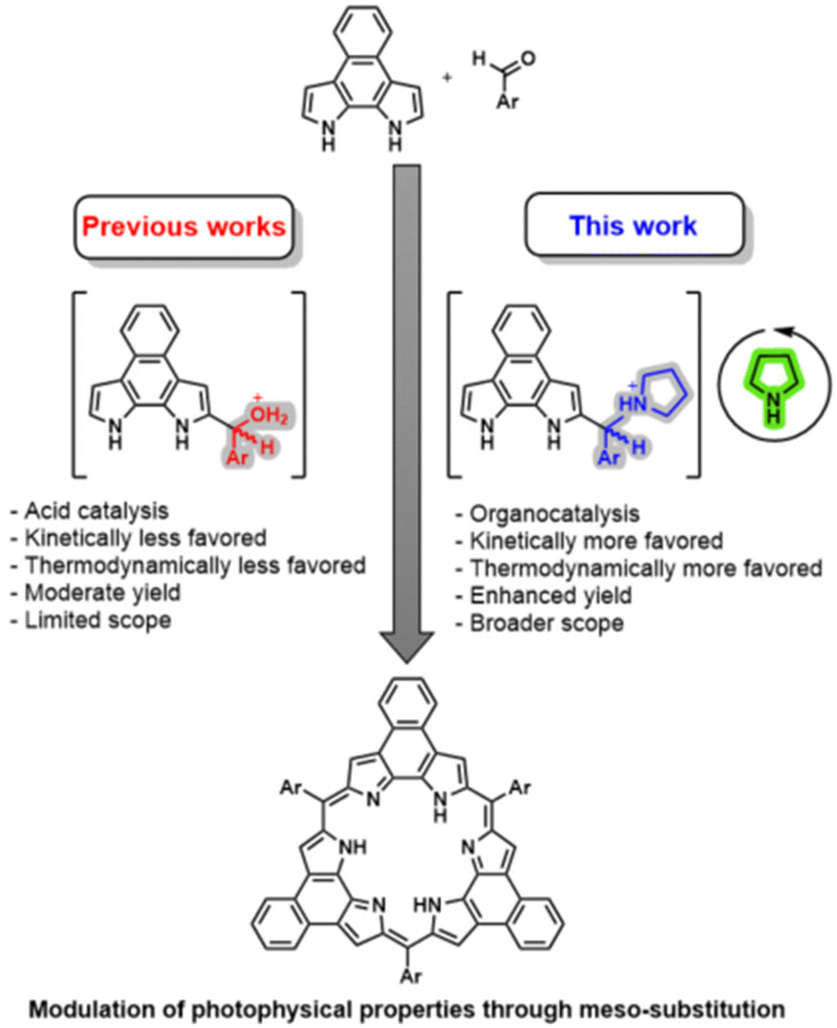 | ||
| Fig. 1 New strategy for the synthesis of expanded porphyrins. | ||
Results and discussion
Optimization, scope, and reaction mechanism of iminium-based macrocyclization
NRos are an intriguing class of expanded porphyrins characterized by an antiaromatic, planar π-skeleton.26–30 The synthesis of these derivatives relies on the acid-catalysed condensation of naphthobipyrrole (NBP) with a suitable aldehyde,31–33 followed by DDQ oxidation. The yield of this condensation is particularly dependent on the aldehyde reactivity. We thus considered NRos to be a near-ideal system with which to explore the iminium-catalysed synthesis of expanded porphyrins. Thus, NRos 1, featuring 2,6-dimethylphenyl groups as meso-substituents, was selected as a model substrate since 2,6-dimethylbenzaldehyde (a) did not show any reactivity under conditions effective for the preparation of NRos bearing electron-withdrawing substituents; even in such cases, the yields are often low (e.g., 5–15%; cf. Scheme where the yields using the previous standard protocol are shown in parentheses). The same conditions were then tested but in the presence of 50% mol morpholine. Again, no detectable NRos 1 was found in the reaction mixture. Adding 3 equiv. of trifluoroacetic acid (TFA) to promote the formation of a putative iminium intermediate, resulted in the formation of trace quantities of NRos 1. The addition of 6 equiv. further enhanced the macrocyclization process and allowed for the isolation of NRos 1 in 3% yield. Other secondary amines, namely piperidine and pyrrolidine, were then tested. Using the latter secondary amine, allowed a yield of 8% to be achieved. The use of 6 equiv. of TFA in the absence of pyrrolidine did not result in the formation of NRos 1, providing support for the notion that the reaction takes place via an iminium intermediate. Consistent with this conclusion was the finding that N-methyl-pyrrolidine in the presence of TFA was ineffective as a catalyst. Specifically, no NRos 1 was detected in the reaction medium under these conditions.Using the above conditions, we next explored reactions between NBP and various aldehydes. For these studies, several highly electron-rich aldehydes, such as 4-(tert-butyl)benzaldehyde, 4-(dimethylamino)benzaldehyde and 3,5-dimethoxybenzaldehyde, were studied. While evidence of reactivity was observed the resulting compounds suffered from low solubility and stability, precluding their full characterization. In order to quantitatively evaluate the effect of iminium catalysis, we decided to employ aldehydes known to afford stable and soluble NRos under conventional conditions (Scheme 1). The goal was to see if these yields could be increased. For this portion of the study, we selected several different aldehydes, including (i) b, since it is among the most common aldehydes employed for the synthesis of expanded porphyrins, (ii) c–e as they present functional groups capable of undergoing post-synthetic functionalization, and (iii) f–h, which bear methoxy groups at different positions. In all cases, the iminium-catalysed reaction led to higher yields, with increases of 33–200% being seen compared to the conventional method. The relative yield enhancement was greatest for aldehydes bearing fewer fluorine atoms, systems that give rise to the lowest macrocyclization yield using the conventional conditions. This highlights the positive effect of the present iminium-based approach.
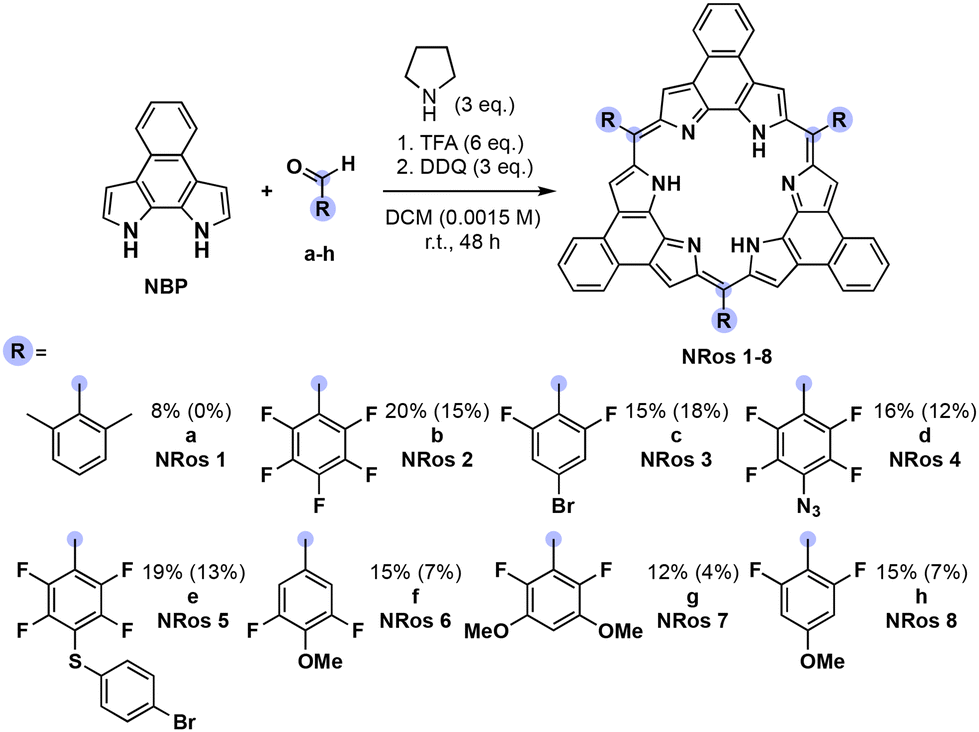 | ||
| Scheme 1 Iminium-based macrocyclization reaction of NBP with various aldehydes. Reagents and conditions: (1) aldehydes (1.2 equiv.), pyrrolidine (3 equiv.), TFA (6 equiv.), DCM, 25 °C, 48 h. (2) 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) (3 equiv.), DCM, 25 °C, 2.5 h. Yields in the absence of catalyst are given in parentheses. | ||
To shed light on the influence of iminium generation on the macrocyclization process, the reaction pathway was quantitatively compared to the non-iminium pathway by means of DFT calculations (see ESI† for computational details). Fig. 2 shows the calculated free energy profiles considering a as the aldehyde (i.e., NRos 1 formation). Our model assumes that the reaction proceeds from an activated carbonyl species, derived from either classic protonation (path α) or iminium formation (path β) of the carbonyl moiety in a (Reacα and β, respectively). The formation of these species proved to be energetically favoured in path β (Boltzmann distribution: F(a)/F(Reacα) = 4.7 × 1027 while F(a)/F(Reacβ) = 2.3 × 1012). The first C–C bond-forming step of the reaction then involves nucleophilic attack of NBP on either Reacα and β. This step is kinetically more favourable in the former case, which is consistent with the higher stability of Reacβ. This reaction, which leads to IN1α/β, seems to be highly reversible in the case of path β. The next step along the reaction coordination is a 1,3-H shift, an isomerization characterized by the highest activation energy in both pathways (37.3 kcal mol−1 and 30.3 kcal mol−1 for paths α and β, respectively). Crucially, this reaction step is kinetically and thermodynamically more favoured in the iminium-catalysed process. Indeed, the formation of IN2 is endergonic in the case of path α, while it is exergonic in the case of path β. Finally, IN3, which is common to both pathways, undergoes an SN1-type reaction, giving rise to the corresponding dipyrromethene (Prod), which by sequential reactions with activated carbonyl species would afford the reduced form of NRos 1. Based on these results, we suggest that the enhanced reactivity observed when using secondary amines is essentially due to (i) a higher concentration of activated carbonyl (Reacα/β) available to start the condensation and (ii) the lower activation barrier and greater exergonicity of the rate-determining 1,3-H shift step.
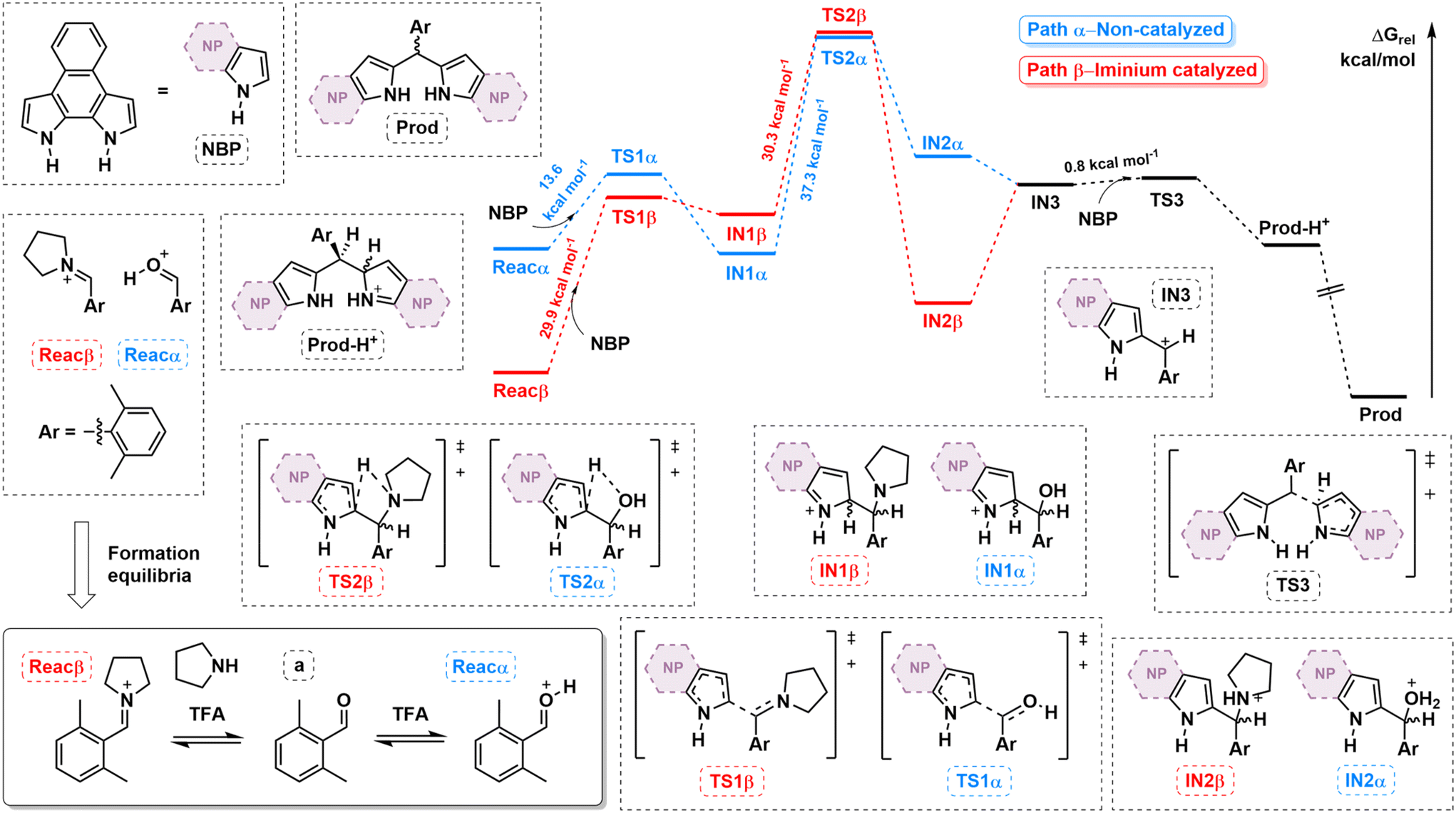 | ||
| Fig. 2 Relative energy profile for the DFT reaction mechanism of the macrocyclization process calculated at the B3LYP/6-31G(d,p) level, considering aldehyde a as model substrate. The influence of the iminium generation (red) is compared to the non-iminium pathway (blue). | ||
Role of meso-groups on the opto-electronic properties of NRos
To assess the influence of meso-substituents on the optical properties of the macrocycle, UV-vis spectra of NRos 2–8 were recorded in dichloromethane at a concentration of 5.0 × 10–6 (M). As depicted in Fig. 3, the absorption spectrum of NRos 2–8 is primarily characterized by a broad band that covers the 350 to 700 nm spectral region. Although the absorption maxima show minimal variations (467–481 nm), a discernible influence of the meso-group is observed. Specifically, the absorption maxima of NRos 6–8, all of which possess relatively electron-donating meso-aryl substituents are all above 470 nm, while the more electron-deficient NRos 2–5 display maxima below this value. As revealed by time-dependent density functional calculations (TD-DFT), the broad absorption seen for the NRos arises mainly from HOMO → LUMO+1 and HOMO → LUMO+2 transitions, corresponding to S0 → S5 and S0 → S6 excitations. Note: selected TD-DFT transition energies, oscillator strengths (f), and molecular orbital configurations are shown in Tables S7.1–7.5.† As illustrated in Fig. 3b, the methine bridge is minimally involved in these orbitals, explaining the slight effect of the meso-groups on the absorption maxima. In all cases, the absorption profiles exhibit a similar shape, suggesting that the meso-groups do not significantly perturb the electronic distribution of the macrocycle. Regarding the emission properties, these NRoss displayed no appreciable fluorescence, which is consistent with previous results.26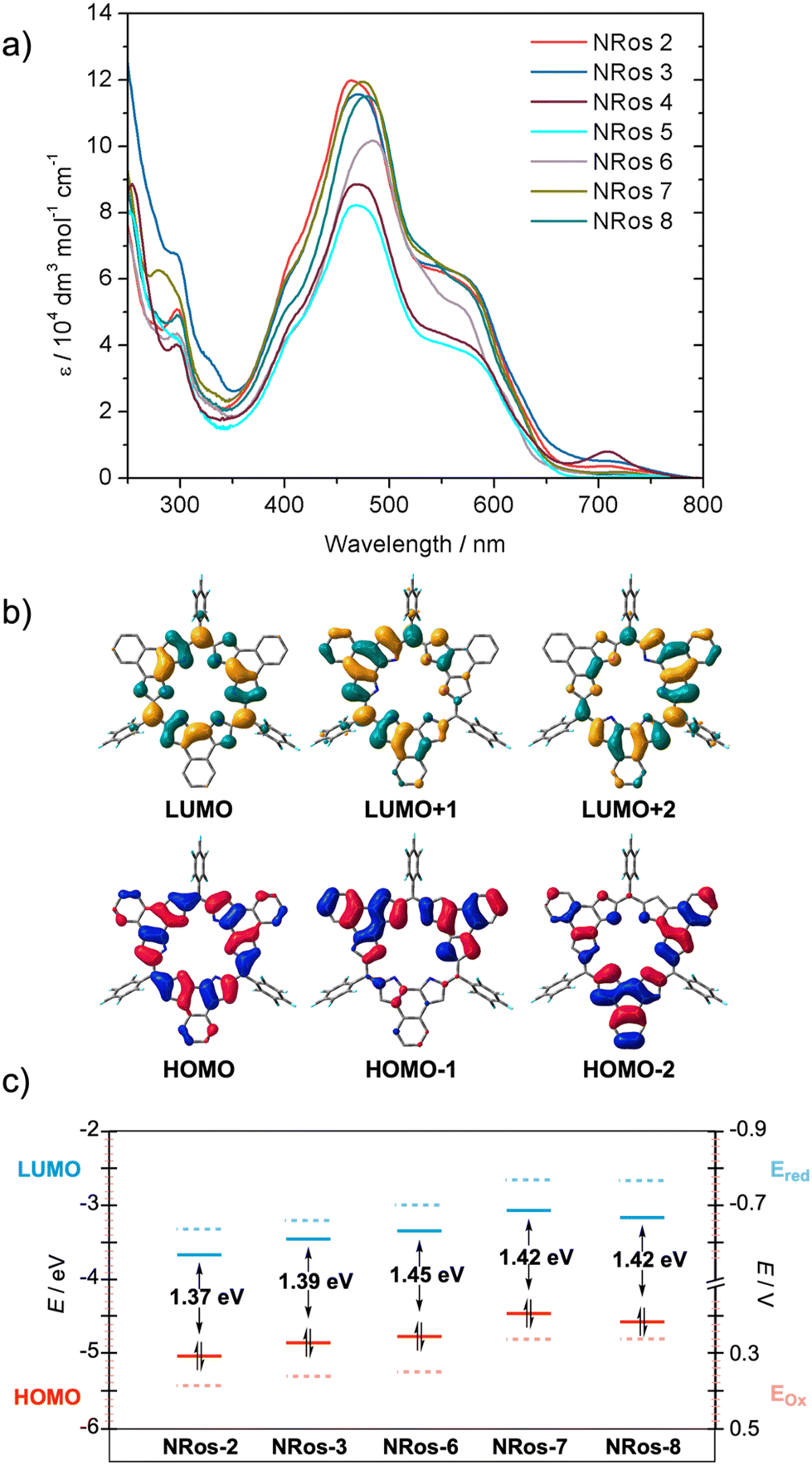 | ||
| Fig. 3 Optical and electrochemical properties of NRos 2–8 (a) UV-vis-NIR spectra in dichloromethane (5.0 × 10−6 M) at room temperature. (b) Selected molecular orbitals of NRos-2, which serves as example, calculated by DFT at the CAM-B3LYP/6-31G(d,p) level of theory. Isosurface value = 0.02. Hydrogen atoms are omitted for clarity. (c) Calculated HOMO–LUMO energies (solid) and electrochemical oxidation and reduction potentials obtained by cyclic voltammetry in CH2Cl2 (dashed lines) of NRos 2, 3, 6, 7 and 8. | ||
The effect of meso-groups on the oxidation/reduction potential was evaluated using cyclic and differential pulse voltammetry (see Fig. 3c and S6.1†) in CH2Cl2. To ensure a reliable comparison, we mainly focused on soluble NRos 2, 3, 6, 7 and 8. Fig. 3c shows the potentials for the first oxidation/reduction processes (see ESI† for second oxidation/reduction potentials and more details) and the calculated HOMO–LUMO levels thought to govern these redox processes. A strong correlation between the observed redox potential and the calculated HOMO–LUMO gap was observed. As expected, NRos 2 exhibits the deepest LUMO level and accordingly the lowest reduction potential. A clear influence of the group at the para-position of the meso-aryl is noticeable when comparing NRos 3 and NRos 8. Thus, a bromine atom makes meso-aryls less electron-rich compared to methoxy group, leading to higher oxidation and reduction potential. On the other hand, NRos 6 is easier to reduce than NRos 8, despite fluorine atoms that are farther from the meso-carbon. We hypothesize that this difference arises from the possible planarization of the electron-deficient meso-aryl in NRos 6, thus enabling better conjugation. On the other hand, the introduction of two methoxy groups (NRos 7) lowers the reduction/oxidation potentials. It should be noted that although the position of HOMO–LUMO levels in all these species varies slightly, the bandgap of the NRos is rather similar, consistent with the UV-vis absorption experiments.
Conclusions
In summary, we have shown that the generation of iminium species is a promising approach to increase the yield and broaden the preparative scope in the case of a test expanded porphyrin, naphthorosarin (NRos). Specifically, the presence of secondary amines, in conjunction with an acid catalyst, significantly improves the macrocyclization yield and provides access to previously inaccessible NRos. DFT calculations provide support for the suggestion that iminium generation lowers the activation energy of key steps during macrocyclization, including rate-determining proton transfers, while stabilizing most intermediates. This method has enabled the preparation of multiple NRos, which differ in their meso-substituents. The nature of the meso-groups, in turn, was found to affect slightly key properties of the products, such as the absorption maximum and the HOMO–LUMO levels.Ultimately, the iminium-based method described here is expected to provide a new tool that can be exploited for the synthesis and thus study of expanded porphyrins.
Data availability
The data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work in Austin was supported by the Robert A. Welch Foundation (F-0018 to J. L. S.) and by the National Science Foundation (CHE-2304731) subsequent to Nov. 1, 2023. T. T. acknowledges financial support from the Spanish MCIN/AEI/10.13039/501100011033 and European Union NextGenerationEU/PRTR (PID2020-116490GB-I00, TED2021-131255B-C43), MCIU/AEI/10.13039/501100011033/FEDER, UE (PID2023-151167NB-I00), the Comunidad de Madrid and the Spanish State through the Recovery, Transformation and Resilience Plan [“Materiales Disruptivos Bidimensionales (2D)” (MAD2D-CM) (UAM1)-MRR Materiales Avanzados], and the European Union through the Next Generation EU funds. IMDEA Nanociencia acknowledges support from the “Severo Ochoa” Programme for Centres of Excellence in R&D (MINECO, Grant SEV2016-0686). T. T. also acknowledges the Alexander von Humboldt Foundation (Germany) for the A. v. Humboldt – J. C. Mutis Research Award 2023 (Ref 3.3 - 1231125 - ESP-GSA). M. G-G. acknowledges MECD, Spain, for a F.P.U. fellowship. We acknowledge the generous allocation of computer time at the Centro de Computación Científica at the Universidad Autónoma de Madrid (CCC-UAM).References
- (a) V. V. Roznyatovskiy, C. Lee and J. L. Sessler, π-Extended isomeric and expanded porphyrins, Chem. Soc. Rev., 2013, 42, 1921–1933 Search PubMed; (b) S. Saito and A. Osuka, Expanded Porphyrins: Intriguing Structures, Electronic Properties, and Reactivities, Angew. Chem., Int. Ed., 2011, 50, 4342–4373 Search PubMed.
- (a) T. K. Chandrashekar and S. Venkatraman, Core-Modified Expanded Porphyrins: New Generation Organic Materials, Acc. Chem. Res., 2003, 36, 676–691 Search PubMed; (b) R. Misra and T. K. Chandrashekar, Structural Diversity in Expanded Porphyrins, Acc. Chem. Res., 2008, 41, 265–279 Search PubMed.
- J. Kim, J. Oh, A. Osuka and D. Kim, Porphyrinoids, a unique platform for exploring excited-state aromaticity, Chem. Soc. Rev., 2022, 51, 268–292 Search PubMed.
- M. Stępień, L. Latos-Grażyński, N. Sprutta, P. Chwalisz and L. Szterenberg, Expanded Porphyrin with a Split Personality: A Hückel–Möbius Aromaticity Switch, Angew. Chem., Int. Ed., 2007, 46, 7869–7873 Search PubMed.
- Z. S. Yoon, A. Osuka and D. Kim, Möbius aromaticity and antiaromaticity in expanded porphyrins, Nat. Chem., 2009, 1, 113–122 Search PubMed.
- G. I. Vargas-Zúñiga and J. L. Sessler, Advances in Inorganic Chemistry, ed. R. van Eldik and R. Puchta. 2018. pp. 327–377 Search PubMed.
- A. Alka, V. S. Shetti and M. Ravikanth, Coordination chemistry of expanded porphyrins, Coord. Chem. Rev., 2019, 401, 213063 Search PubMed.
- T. Tanaka and A. Osuka, Chemistry of meso-Aryl-Substituted Expanded Porphyrins: Aromaticity and Molecular Twist, Chem. Rev., 2017, 117, 2584–2640 Search PubMed.
- D. Seidel, V. Lynch and J. L. Sessler, Cyclo[8]pyrrole: A Simple-to-Make Expanded Porphyrin with No Meso Bridges, Angew. Chem., Int. Ed., 2002, 41, 1422–1425 Search PubMed.
- M. Takase, V. Enkelmann, D. Sebastiani, M. Baumgarten and K. Müllen, Annularly Fused Hexapyrrolohexaazacoronenes: An Extended π System with Multiple Interior Nitrogen Atoms Displays Stable Oxidation States, Angew. Chem., Int. Ed., 2007, 46, 5524–5527 Search PubMed.
- M. W. Harry, A. Bhaskar, G. Ramakrishna, T. Goodson III, M. Imamura, A. Mawatari, K. Nakao, H. Enozawa, T. Nishinaga and M. Iyoda, Giant Thienylene-Acetylene-Ethylene Macrocycles with Large Two-Photon Absorption Cross Section and Semishape-Persistence, J. Am. Chem. Soc., 2008, 130, 3252–3253 Search PubMed.
- J. Krömer, I. Rios-Carreras, G. Fuhrmann, C. Musch, M. Wunderlin, T. Debaerdemaeker, E. Mena-Osteritz and P. Bäuerle, Synthesis of the First Fully α-Conjugated Macrocyclic Oligothiophenes: Cyclo[n]thiophenes with Tunable Cavities in the Nanometer Regime, Angew. Chem., Int. Ed., 2000, 39, 3481–3486 Search PubMed.
- G. Fuhrmann, T. Debaerdemaeker and P. Bäuerle, C–C bond formation through oxidatively induced elimination of platinum complexes—A novel approach towards conjugated macrocycles, Chem. Commun., 2003, 948–949 Search PubMed.
- J. S. Lindsey, Synthetic Routes to meso-Patterned Porphyrins, Acc. Chem. Res., 2010, 43(2), 300–311 Search PubMed.
- H. Hata, H. Shinokubo and A. Osuka, Anthracene-Bridged Z-Shaped [26]Hexaphyrin(1.1.1.1.1.1.1) Dimer from the Regioselective Diels–Alder Reaction of a Hexaphyrin with Bis-o-xylylene Equivalents, Angew. Chem., Int. Ed., 2005, 44, 932–935 Search PubMed.
- J.-Y. Shin, T. Tanaka, A. Osuka, Q. Miao and D. Dolphin, BODIPY–Hexaphyrin Hybrids, Chem. – Eur. J., 2009, 15, 12955–12959 Search PubMed.
- A. Werner, M. Michels, L. Zander, J. Lex and E. Vogel, “Figure Eight” Cyclooctapyrroles: Enantiomeric Separation and Determination of the Absolute Configuration of a Binuclear Metal Complex, Angew. Chem., Int. Ed., 1999, 38, 3650–3653 Search PubMed.
- A. K. Burrell, G. Hemmi, V. Lynch and J. L. Sessler, Uranylpentaphyrin: an actinide complex of an expanded porphyrin, J. Am. Chem. Soc., 1991, 113, 4690–4692 Search PubMed.
- J. L. Sessler, D. Seidel, A. E. Vivian, V. Lynch, B. L. Scott and D. W. Keogh, Hexaphyrin(1.0.1.0.0.0): An Expanded Porphyrin Ligand for the Actinide Cations Uranyl (UO2(2+) and Neptunyl (NpO2+), Angew. Chem., Int. Ed., 2001, 40, 591–594 Search PubMed.
- X. J. Zhu, S. T. Fu, W. K. Wong, J. P. Guo and W. Y. Wong, A Near-Infrared-Fluorescent Chemodosimeter for Mercuric Ion Based on an Expanded Porphyrin, Angew. Chem., Int. Ed., 2006, 45, 3150–3154 Search PubMed.
- D. Wu, A. B. Descalzo, F. Weik, F. Emmerling, Z. Shen, X. Z. You and K. A. Rurack, A Core-Modified Rubyrin with meso-Aryl Substituents and Phenanthrene-Fused Pyrrole Rings: A Highly Conjugated Near-Infrared Dye and Hg2+ Probe, Angew. Chem., Int. Ed., 2008, 47, 193–197 Search PubMed.
- A. Erkkila, I. Majander and P. M. Pihko, Iminium Catalysis, Chem. Rev., 2007, 107, 5416–5470 Search PubMed.
- K. Singh, S. Behal and M. S. Hundal, Efficient and versatile single pot approach to dipyrromethanes and bis(heterocyclyl)methanes, Tetrahedron, 2005, 61, 6614–6622 Search PubMed.
- K. Singh, S. Behal and P. K. Deb, Efficient and Versatile Single–Pot Alternative Approach to Dipyrromethanes, Synth. Commun., 2005, 35, 929–934 Search PubMed.
- C. Biaggi, M. Benaglia, L. Raimondi and F. Cozzi, Organocatalytic synthesis of dipyrromethanes by the addition of N-methylpyrrole to aldehydes, Tetrahedron, 2006, 62, 12375–12379 Search PubMed.
- M. Ishida, S. J. Kim, C. Preihs, K. Ohkubo, J. M. Lim, B. S. Lee, J. S. Park, V. M. Lynch, V. V. Roznyatovskiy, T. Sarma, P. K. Panda, C. H. Lee, S. Fukuzumi, D. Kim and J. L. Sessler, Protonation-coupled redox reactions in planar antiaromatic meso-pentafluorophenyl-substituted o-phenylene-bridged annulated rosarins, Nat. Chem., 2013, 5, 15–20 Search PubMed.
- S. Samala, R. Dutta, Q. He, Y. Park, B. Chandra, V. M. Lynch, Y. M. Jung, J. Sessler and C. Lee, A robust bis-rhodium(I) complex of π-extended planar, anti-aromatic hexaphyrin[1.0.1.0.1.0], Chem. Commun., 2020, 56, 758–761 Search PubMed.
- S. Fukuzumi, K. Ohkubo, M. Ishida, C. Preihs, B. Chen, W. T. Borden, D. Kim and J. L. Sessler, Formation of Ground State Triplet Diradicals from Annulated Rosarin Derivatives by Triprotonation, J. Am. Chem. Soc., 2015, 137, 9780–9783 Search PubMed.
- G. Kim, R. Dutta, W. Y. Cha, S. J. Hong, J. Oh, D. Firmansyah, H. Jo, K. M. Ok, C. H. Lee and D. Kim, Noncovalent Intermolecular Interaction in Cofacially Stacked 24π Antiaromatic Hexaphyrin Dimer, Chem. – Eur. J., 2020, 26, 16434–16440 Search PubMed.
- J. Chen, A. C. Sedgwick, S. Sen, Y. Ren, Q. Sun, C. Chau, J. F. Arambula, T. Sarma, L. Song, J. L. Sessler and C. Liu, Expanded porphyrins: functional photoacoustic imaging agents that operate in the NIR-II region, Chem. Sci., 2021, 12, 9916–9921 Search PubMed.
- D. D. Nguyen, J. Labella, J. Laforga-Martin, C. L. Folcia, J. Ortega, T. Torres, T. Sierra and J. L. Sessler, Columnar liquid crystals based on antiaromatic expanded porphyrins, Chem. Commun., 2024, 60, 3401–3404 Search PubMed.
- D. Firmansyah, S. J. Hong, R. Dutta, Q. He, J. Bae, H. Jo, H. Kim, K. M. Ok, V. M. Lynch, H. R. Byon, J. L. Sessler and C. H. Lee, Trapping of Stable [4n+1] π-Electron Species from Peripherally Substituted, Conformationally Rigid, Antiaromatic Hexaphyrins, Chem. – Eur. J., 2019, 25, 3525–3531 Search PubMed.
- S. Lee, Y. Wang, R. Dutta, C. H. Lee, J. L. Sessler and D. Kim, Xanthene-Separated 24 π-Electron Antiaromatic Rosarin Dimer, Chem. – Eur. J., 2023, 29, e202301501 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qo01559b |
| This journal is © the Partner Organisations 2024 |