
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
1,2,3-Triarylazulenes as precursors of azulene-embedded polycyclic aromatic hydrocarbons†‡
Justyna
Biesaga
 ,
Sławomir
Szafert
and
Bartłomiej
Pigulski
*
,
Sławomir
Szafert
and
Bartłomiej
Pigulski
*
Faculty of Chemistry, University of Wrocław, 14 F. Joliot-Curie, 50-383 Wrocław, Poland. E-mail: bartlomej.pigulski@uwr.edu.pl
First published on 27th September 2024
Abstract
A series of 1,2,3-triarylazulenes was obtained using new synthetic methodology and subsequently subjected to Scholl-type oxidation aiming for conjugated azulene-embedded polycyclic aromatic hydrocarbons (PAHs). The oxidation yielded either unexpected azulen-1(8aH)-ones or desired purely hydrocarbon azulene-embedded PAHs, depending on the substitution pattern. Different reaction pathways were rationalized using DFT calculations, leading to the observation that 2-pyrenyl substituents facilitate formation of the desired conjugated molecules. The fully hydrocarbon azulene-embedded PAHs exhibit a relatively small electrochemical energy gap below 2 eV and optical absorption reaching the near-infrared (NIR) region. These properties are attributed to their non-alternant topology and retained azulene-like electronic structure.
Introduction
The discovery of fullerenes, carbon nanotubes, and graphene has initiated extensive research into new carbon allotropes and carbon-rich molecules.1 A diverse array of benzenoid polycyclic aromatic hydrocarbons (PAHs), or in other words, nanographenes, has been synthesized using a bottom-up approach.2,3 There is also a growing interest in the chemistry of non-alternant π-scaffolds containing five- or seven-membered rings due to their distinctive properties compared to fully benzenoid PAHs.4–6Azulene, in particular, is a highly sought-after non-alternant structural motif in PAHs due to its unique electronic properties, including a small highest occupied molecular orbital-lowest unoccupied molecular orbital (HOMO–LUMO) gap or emission from the S2 state, which disobeys Kasha's rule.7,8 These properties stem from azulene's electronic structure, which effectively functions as a fused tropylium ion and cyclopentadienyl anion with an inherent dipole moment 1.08 D and significant spatial separation between the HOMO and LUMO orbitals (Scheme 1a).9 PAHs containing azulene structural motifs exhibit unique properties such as warped structure,10 biradical characteristics,11,12 and near-infrared (NIR) absorption,13 which make them suitable for applications in organic electronics,14,15 solar cells,16–20 bioimaging,21 and single-molecule devices.22
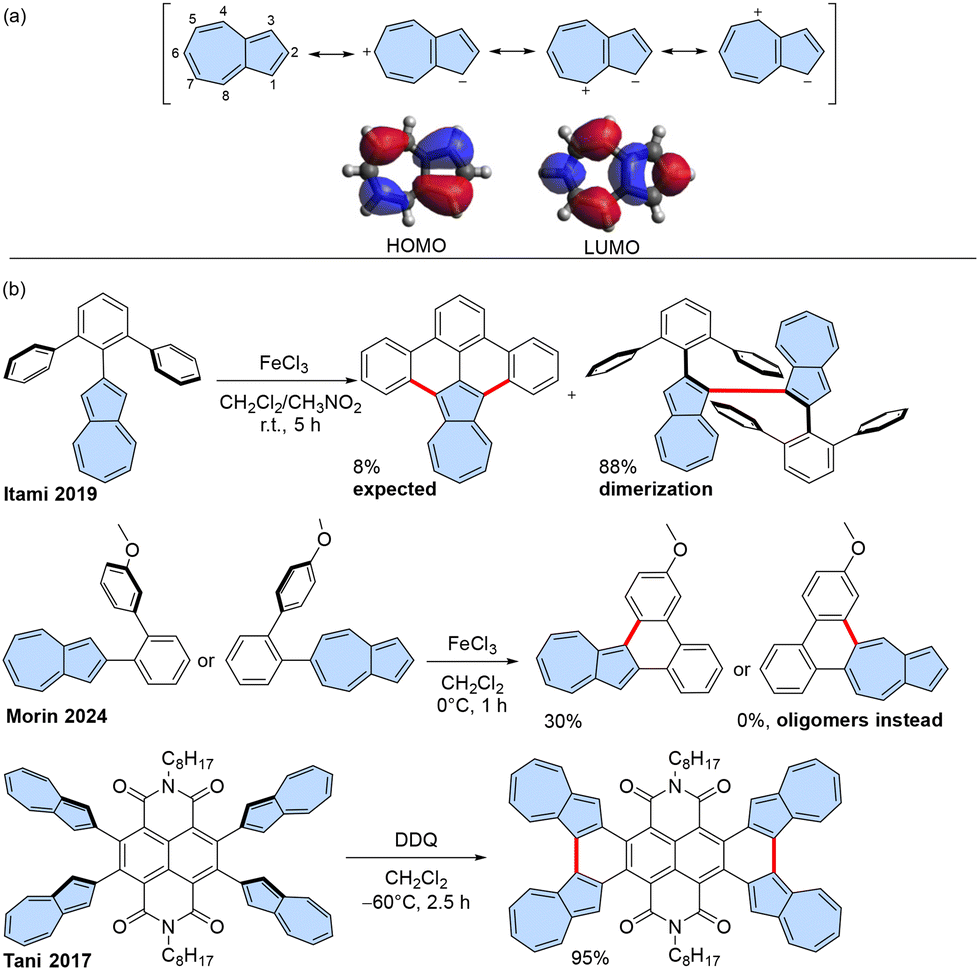 | ||
| Scheme 1 (a) Azulene: selected resonance structures, molecular frontier orbitals, and position numbering. (b) Synthesis of conjugated azulene-embedded PAHs using Scholl-type oxidation. | ||
The rapid development of synthetic methods for functionalized azulenes23–25 and azulene-embedded PAHs4,26 can be attributed to the diverse and intriguing properties of these compounds. In some cases, azulene or a ‘formal azulene’ moiety is formed in the last step of the synthetic pathway. However, the key step often involves the fusion of precursors that already contain azulene subunits, finally resulting in a fully conjugated π-system. Various chemical transformations have been employed to achieve conjugated π-systems, such as the annulation of alkenes27 or annulation of alkynes.28,29 Additionally, more complex approaches such as tandem Suzuki coupling followed by condensation might be realized in two steps30,31 or in one step.32
Several additional strategies have been developed for the synthesis of azulene-embedded PAHs, including palladium-catalysed Heck-type intramolecular cyclization,33 [3 + 3] palladium-catalysed tandem Suzuki coupling and C–H arylation,13 or cyclization of substituted 1-nitroazulenes.34 However, there are limitations to all these methods, and therefore, the need remains for more efficient synthetic methodologies for azulene-embedded PAHs.
Scholl oxidation is a very potent synthetic tool leading to various PAHs, but in many cases, the exact reactivity pattern is not easy to predict.35,36 Consequently, Scholl-type oxidation and related Mallory photo-oxidation have been employed for some azulene-embedded PAHs (Scheme 1b), although these reactions often yield suboptimal results in terms of yield and selectivity.
Because positions 1 and 3 of the azulene moiety are the most electron-rich, precursors of azulene-embedded PAHs are typically targeted for oxidation in the synthesis of azulene-embedded PAHs. However, the tendency of azulene to form 1,3-polyazulene upon oxidation37 might result in dimerization of azulene rather than the desired fused molecule. For instance, Itami and co-workers38 reported that the expected fully fused product was isolated only in 8% yield after oxidation with FeCl3, while the main product of 1,1′-biazulene was obtained in 88% yield. Recently, Morin and co-workers reported attempts to achieve fused products.39 However, the reaction yield was low (30%) when position 1 of azulene was involved in oxidation, and attempts to fuse position 5 resulted exclusively in oligomeric products.
Intramolecular fusion of azulene units is more efficient when performed in an electron-deficient system, as demonstrated by Tani and co-workers40 in their synthesis of azulene-fused tetracene diimide. Mallory photo-oxidation has also proven useful, as shown by Zhang and coworkers.41 All examples of known syntheses of azulene-embedded PAHs involve oxidation positions 1 and 3 of azulene, where the HOMO is primarily located, and which are the most electron-rich positions. However, having such positions unsubstituted can lead to competitive intermolecular oxidation and a lower yield of the desired intramolecular product, such as in the example reported by Itami.38
Herein, we report the synthesis of a series of 1,2,3-triarylazulenes, and the application of these triarylazulenes in Scholl-type reactions for the first time. Interestingly, depending on the substitution pattern, oxidation leads either to a 1,2-shift of a phenyl group or to partially and fully fused products.
Results and discussion
Synthesis of precursors
The primary rationale for using 1,2,3-triarylazulenes as precursors for fused azulene-embedded PAHs was the fact that the highly reactive positions 1 and 3 are pre-blocked before the oxidation step. It is expected that this approach will prevent the formation of oligomeric products and lead to the desired fully-fused azulene-embedded PAHs.Despite interesting photophysical properties,42 known examples of 1,2,3-triarylazulenes are quite limited. Existing syntheses typically start from substituted tolanes where the crucial step involves expansion of a phenyl ring, which significantly reduces the available substitution patterns. These reactions are usually catalysed using sulfenyl chloride/AlCl3,43,44 palladium catalyst,45 or gold catalyst.46
Hence, we developed a novel modular approach for 1,2,3-triarylazulenes (Scheme 2) that enables selective placement of substituents at positions 1, 2, and 3. We utilized two types of selective reactions: borylation (position 2) and iodination (positions 1 and 3). The synthesis began with the known 6-tert-butylazulene,47 which was selected for its increased solubility compared to unsubstituted azulene. Subsequently, 6-tert-butylazulene underwent borylation at position 2 via modified iridium(I)-catalysed C–H borylation,48 giving compound 5 in 66% isolated yield. Azulen-2-ylboronic acid pinacol ester 5 was later subjected to the Suzuki coupling with haloarenes. The lower yield of compound 6b was likely due to its poor solubility, which resulted in difficulties during its isolation. All compounds 6a–6c underwent double iodination using N-iodosuccinimide (NIS), yielding compounds 7a–7c in high yields from 84% to 98%.
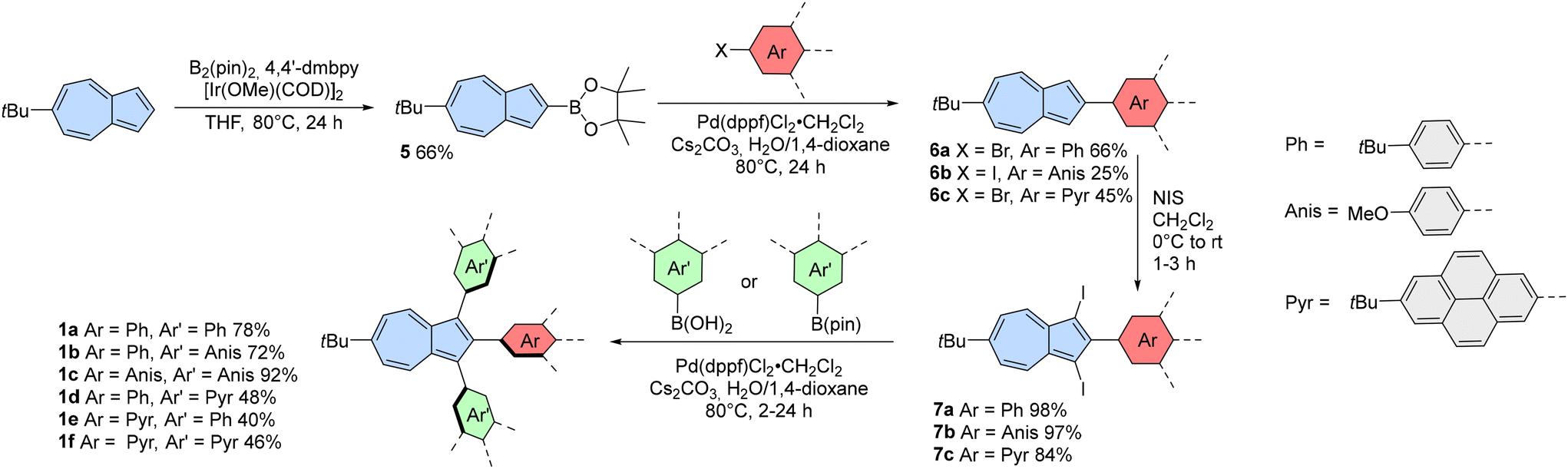 | ||
| Scheme 2 Synthesis of azulene-containing precursors 1. B2(pin)2 = bis(pinacolato)diboron, 4,4′-dmbpy = 4,4′-dimethyl-2,2-dipyridyl, COD = 1,5-cyclooctadiene, THF = tetrahydrofuran, dppf = 1,1′-bis(diphenylphosphino)ferrocene, NIS = N-iodosuccinimide. | ||
The final step in the synthesis of 1,2,3-triarylazulenes was the double Suzuki coupling with boronic acids (4-anisyl and 4-tert-butylphenyl substituents) or boronic acid pinacol ester (2-(7-tert-butylpyrenyl) substituent). The final coupling reactions were catalysed using Pd(dppf)Cl2·CH2Cl2, and the products were isolated in decent yields from 46% to 92%. Finally, a set of 1,2,3-triarylazulenes 1a–1f was synthesized using this modular approach and subsequently employed in Scholl-type oxidation.
Scholl-type oxidation
With a series of 1,2,3-arylazulenes available, we aimed to optimize the oxidation conditions to achieve fully fused products. Precursor 1a was selected as a convenient model substrate for the Scholl reaction, and various conditions were tested, aiming for fully fused product 2a (Table 1). Initially, we used 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) as an oxidant (entries 1 and 2). However, in the presence of MsOH, the substrate was recovered after the reaction, while in the absence of acid, the reaction resulted only in the decomposition of the starting material.|
|
|||
|---|---|---|---|
| Oxidant | Conditions | Result | |
| a Yields after isolation. | |||
| 1 | DDQ, MsOH | CH2Cl2, 0 °C, 24 h | 1a recovered |
| 2 | DDQ | CH2Cl2, rt, 17 h | Decomposition |
| 3 | AlCl3 | PhCl, 80 °C, 48 h | 1g (quant.) |
| 4 | MoCl5 | CH2Cl2, rt, 17 h | Decomposition |
| 5 | FeCl3 | CH2Cl2/MeNO2, −78 °C to 0 °C, 20 h | 1a recovered |
| 6 | FeCl3 | CH2Cl2/MeNO2, rt, 20 h | Decomposition |
| 7 | FeCl 3 , K 2 CO 3 | DCE/MeNO 2 , 80 °C, 19 h | 3a (93%)a |
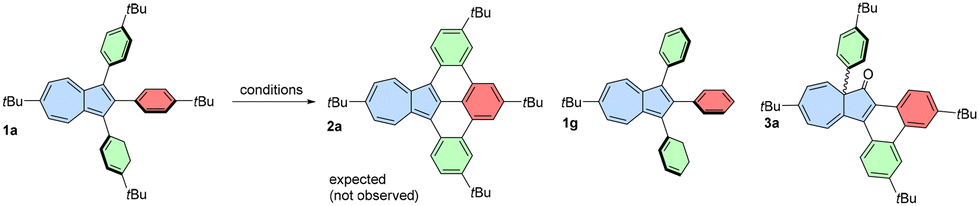
This fact suggested that the protonated form of azulene might be not oxidized under these conditions. Surprisingly, the reaction using AlCl3 in chlorobenzene (entry 3) led to almost quantitative removal of tert-butyl groups from phenyl rings and formation of 1g. Oxidation using MoCl5 led only to an unidentified mixture of products (entry 4), but after oxidation using FeCl3 at low temperature (entry 5), we recovered only the starting material. Additionally, when oxidation was conducted at room temperature, then only decomposition of the substrate was observed (entry 6).
We noted that the reaction mixture turned from green to very intense red after addition of FeCl3/MeNO2 to the solution of starting azulene. This suggested that in the presence of residual moisture, azulene can be protonated, and this hinders the desired reaction. Based on this observation, we hypothesized that adding a base to the reaction system could drive the reaction in the desired direction. Indeed, the addition of K2CO3 and switching to 1,2-dichloroethane (DCE) from CH2Cl2 along with a higher reaction temperature led to the product that was initially believed to be oxidized 1a with one new C–C bond formed (entry 7). However, the 13C NMR spectrum of the pale yellow product revealed a typical carbonyl signal above 200 ppm, indicating that azulen-1(8aH)-one 3a was the actual product of oxidation. This unprecedented oxidation involves a 1,2-aryl shift, oxidation of the most electron-rich position of azulene, and formation of a new C–C bond between adjacent phenyl groups.
We extended our investigation to the oxidation of a broader range of 1,2,3-triarylazulenes under designed conditions (Scheme 3). Precursor 1b gave similar pale-yellow azulen-1(8aH)-one 3b in 58% yield after extending the reaction time to 96 h. Substrate 1c bearing three methoxy groups also gave the product of 1,2 phenyl shift 3c, but no formation of C–C bond was observed even after two days. Additionally, a more prolonged reaction time led only to a gradual decomposition of 3c. To the best of our knowledge, this represents the first oxidation and 1,2 phenyl shift leading from azulene to azulen-1(8aH)-one. Previously, azulen-1(8aH)-ones have been synthesized through rhodium-49 or gold-catalysed50 cyclizations of diazo compounds or cycloadditions involving ketenes and alkynes.51,52
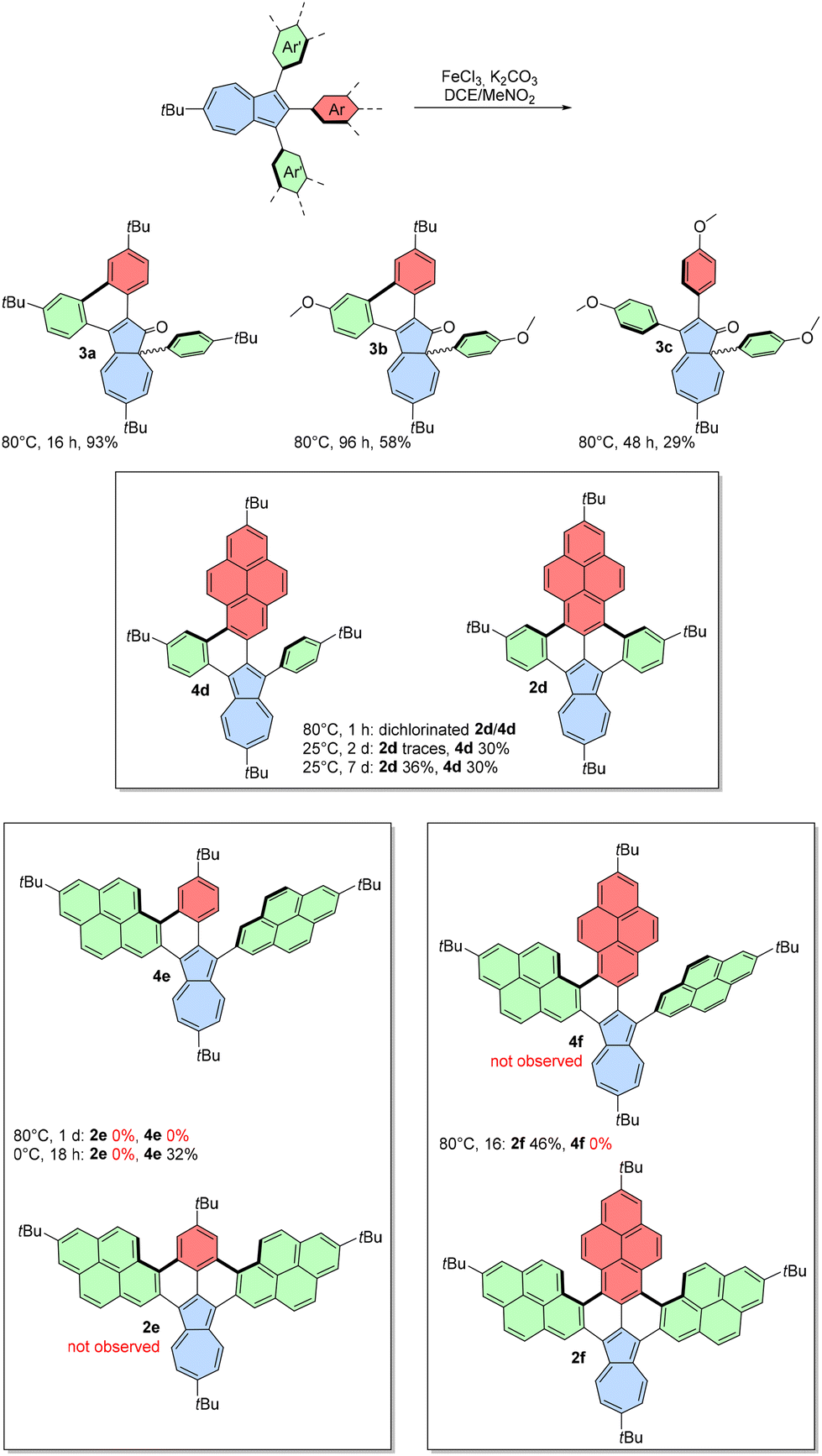 | ||
| Scheme 3 Scholl-type oxidation of azulenes 1a–1f (yields after isolation). | ||
We also attempted to verify the origin of the oxygen atom in products 3a–3c. It is highly unlikely that residual O2 was involved, as the reaction mixture was carefully degassed. We hypothesized that the oxygen might originate from residual moisture in K2CO3 and/or FeCl3, or possibly from the decomposition of K2CO3. To test this hypothesis, we conducted oxidation reactions of 1a and 1b with the addition of isotopically labelled H2O (97% 18O). Unfortunately, the added water quenched the reaction, which prevented us from drawing any definitive conclusions.
The formation of azulen-1(8aH)-ones was an intriguing observation, but it was not the initial goal of using 1,2,3-triarylazulenes as substrates in the Scholl oxidation. Inspired by literature precedents,53 we aimed to use azulenes 1d–1f bearing one, two, or three phenyl substituents replaced with 2-pyrenyl substituents to obtain fully fused azulene-embedded PAHs. Indeed, incorporating one or more 2-pyrenyl substituents shifted the reactivity pattern in the desired direction. The oxidation of 1d was initially carried out at 80 °C, which unfortunately led to significant decomposition and an undesired side reaction, as evidenced by atmospheric-pressure chemical ionization (APCI) mass spectra showing a mixture of oxidized products containing two chlorine atoms. However, reducing the reaction temperature provided greater control, and we isolated green compound 4d with one new C–C bond in 30% yield. We also observed a trace amount of double-oxidized 2d. Extension of the reaction time to 7 d increased the amount of 2d, which was isolated in 36% yield.
Oxidation of 1e at 80 °C led only to decomposition of the substrate and formation of a black reaction mixture with no detectable amount of desired products. Reducing the temperature to 0 °C resulted in the selective formation of single oxidized green product 4e in 32% isolated yield. Attempts to achieve the fully oxidized product were unsuccessful and resulted only in decomposition. Finally, tri(2-pyrenyl)azulene 1f was subjected to Scholl-type oxidation, resulting in the fully oxidized brown compound 2f in 46% isolated yield. The presence of K2CO3 was also important for pyrene-containing substrates, and without K2CO3, we observed mainly decomposition.
Compound 2f is a double [5]helicene, which may have a significant racemization barrier,54 leading to the potential formation of different isomers: the meso (P,M) form or a racemic mixture of (P,P) and (M,M) stereoisomers. Only one set of signals was observed in the 1H NMR spectrum, suggesting the presence of only one form in solution. Despite numerous attempts, we were unable to obtain crystals of 2f suitable for X-ray single crystal diffraction. Density functional theory (DFT) calculations indicated that there was 8.72 kJ mol−1 lower energy for the meso form than the chiral form (ESI, Fig. S16‡), and thus, the meso conformer was used for further analysis.
A particularly intriguing question is why different substitution patterns lead to different reactivity patterns. It is known that a 1,2-shift of a phenyl substituent can sometimes occur during Scholl oxidation, resulting in unexpected products.55,56 However, no examples of such a 1,2-shift involving an azulene precursor have been reported to date. Generally, two mechanisms are considered for Scholl oxidation: the arene cation mechanism and radical cation mechanism. In our case, the reaction occurs under basic conditions, leading us to assume that the radical cation mechanism occurred, where the initial step involves the abstraction of one electron from the substrate. Analysis of the spin density might be employed as a convenient tool to understand the observed reactivity53 because carbon atoms with positive spin density are expected to be involved in the formation of new C–C bonds. To gain further insights into the reactivity observed during Scholl oxidation, we conducted DFT calculations of possible intermediate radical cations at the UB3LYP/6-31G(d) level of theory.
The spin densities calculated for radical cations directly formed from compounds 1 showed significant difference regarding the substituent in position 2 of azulene (ESI, Fig. S15‡). When a 2-pyrenyl substituent is present in position 2 of azulene (1d)˙+ and (1f)˙+, significant spin density is observed on the relevant carbon atoms contrary to the compounds (1a)˙+ and (1e)˙+, which should facilitate the desired oxidation of the former. Comparing the DFT-calculated spin densities of radical cations after the formation of the first C–C bond (Fig. 1) highlights notable differences.
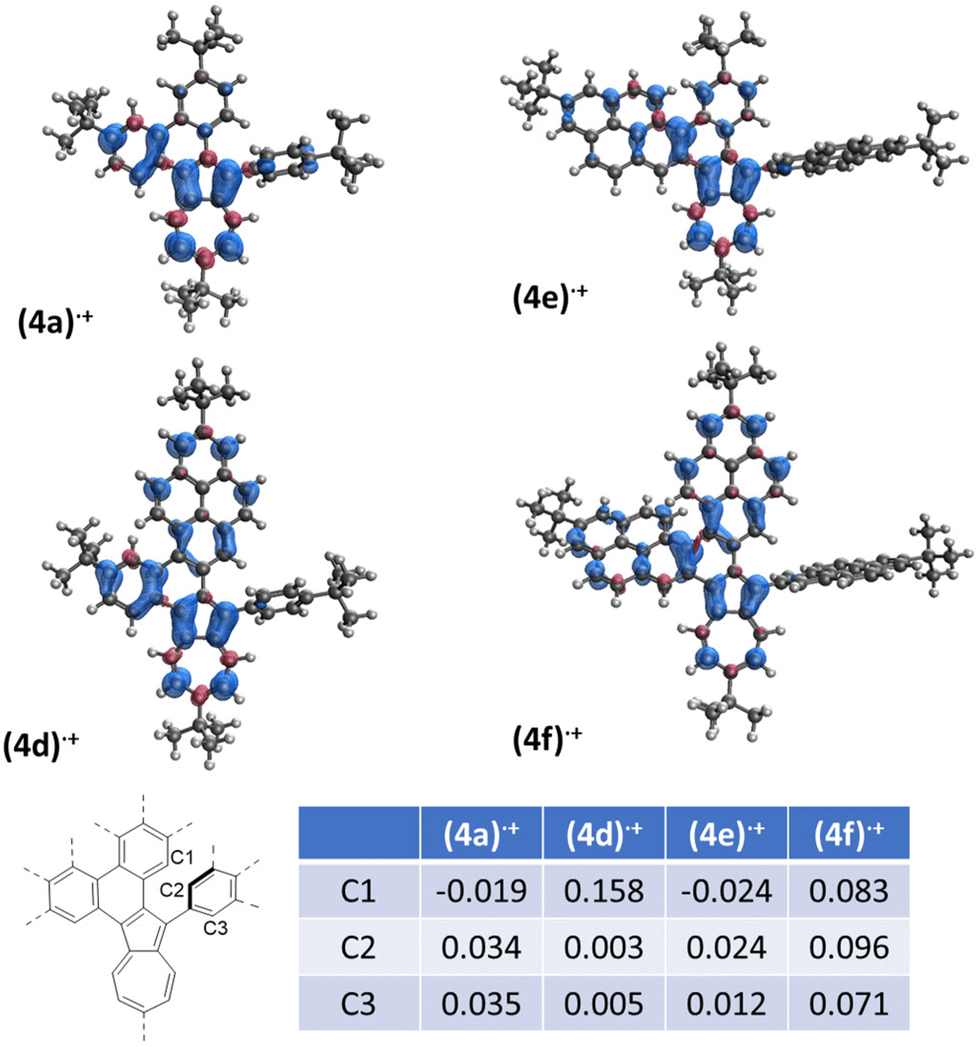 | ||
| Fig. 1 Spin density distribution of radical cations after formation of the first C–C bond, UB3LYP/6-31G(d). | ||
The carbon atoms involved in the formation of the second C–C bond are depicted in C1–C3. In the case of radical cations (4d)˙+ and (4f)˙+, significant positive spin density was observed at the C1 carbon atom. The spin density at the C2/C3 atoms was smaller in the case of (4d)˙+ as compared to (4f)˙+, which explains the easy double oxidation of 1f. There is no C1 positive spin for (4a)˙+ and (4e)˙+, which explains the alternative 1,2-phenyl shift pathway in the case of the first, and decomposition at higher temperature for the latter. Interestingly, a similar distribution of spin density over the pyrene moiety has been shown to play a key role in the selectivity of the Scholl reaction in fully benzenoid substrates.53
We acknowledge that fully elucidating the mechanism of the Scholl reaction is a great challenge, and many aspects of such reactions remain debatable in the literature. In our view, the key distinction between fully benzenoid and azulene-containing precursors in the Scholl reaction is the high-lying HOMO of azulene. Consequently, the intermediate radical cation has its positive spin density primarily localized on the azulene moiety, facilitating alternative reaction pathways. This contrast is evident when compared to the reactivity of 1,2,3,4-tetraphenylnaphthalenes, which exclusively yield the typical products of the Scholl reaction under various oxidation conditions.57–59 Nonetheless, these results bring us closer to understanding the elusive reaction mechanism in azulene-containing substrates and provide insights into predicting the outcomes of Scholl-type oxidations.
X-ray single crystal diffraction
X-ray single crystal experiments were conducted to confirm the molecular structures of the oxidation products. Single crystals of compounds 1a and 3a were obtained (Fig. 2a and b), unequivocally confirming the occurrence of a 1,2-phenyl shift, followed by oxidation at position 1 of azulene and the formation of a new C–C bond. Compound 3a crystallizes with three molecules in the asymmetric unit in the chiral point group P212121, existing as a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of two enantiomers.
:1 mixture of two enantiomers.
 | ||
| Fig. 2 The molecular structures and selected bond lengths (Å) of (a) compound 1a; (b) compound 3a, one of three molecules in the asymmetric unit, where bond lengths are the mean of the bond lengths of the molecules in the asymmetric unit; (c) compound 4e, with ellipsoids given at 50% probability. | ||
Analysis of the experimental bond lengths reveals a significant change from the initial geometry and electronic structure of the azulene unit compared to substrate 1a. The bridging carbon atom adopts an sp3 hybridization, disrupting conjugation and resulting in an alternating pattern of double and single C–C bonds. Disappearance of azulene-like geometry and electronic structure explains the yellow colour observed in compounds 3a–3c.
Single crystals of compound 4e suitable for X-ray experiments were obtained by a slow diffusion of MeOH vapours into its solution in ortho-dichlorobenzene. X-ray crystallography unambiguously confirmed the identity of 4e (Fig. 2c). Notably, compound 4e retained the azulene unit's geometry, suggesting that the azulene-like electronic structure remained present. The carbon–carbon bond lengths of the azulene moiety in 4e are similar to the geometry of unsubstituted azulene.60 The entire π-scaffold is slightly warped due to the presence of the [4]helicene moiety.
Electronic and optical properties
The optical properties of all oxidation products were studied in CH2Cl2 solution. The absorption of compounds 3a–3c occurs mainly in the UV region due to the broken conjugation in the former azulene moiety, resulting in a pale yellow colour (ESI, Fig. S1–S3‡). In contrast, the absorption spectra of the fused azulenes 2d, 4d, 4e, and 2f are far more complex (Fig. 3a). Extended azulenes 4d and 4e exhibit the lowest energy transition above 680 nm (Table 2), which, according to the time-dependent (TD)-DFT calculations, corresponds to an almost forbidden S0 → S1 transition (f = 0.024 for 4d and f = 0.037 for 4e) primarily involving the HOMO to LUMO transition component (ESI, Tables S5–S8‡). This transition is responsible for the distinctive green colour of 4d and 4e (Fig. 3b). | ||
| Fig. 3 (a) UV/Vis/NIR spectra of compounds 4d, 2d, 4e, and 2f (CH2Cl2, c approximately 10−6 M, 20 °C). (b) Photographs of CH2Cl2 solutions of 4d, 2d, 4e, and 2f. | ||
| Compound | E ox1/2 [V] | E red1/2 [V] | ΔE [V] |
E
HOMOb [eV] |
E
LUMOb [eV] |
λ
maxc (exp.) [nm] |
λ
maxc (exp.) [eV] |
λ
maxd (DFT) [nm] |
λ
maxd (DFT) [eV] |
|---|---|---|---|---|---|---|---|---|---|
| a Working/counter electrodes: Pt disc/wire, respectively; reference electrode: Ag/AgCl; scan rate: 50 mV s−1; concentration: approximately 10−5 M in CH2Cl2 containing 0.1 M [NBu4][PF6] as the supporting electrolyte. All redox potentials were calibrated against the Fc/Fc+ standard. b Calculated according to the known procedure using the experimentally determined redox potentials (ELUMO = −(Ered + 4.8 eV) and EHOMO = −(Eox + 4.8 eV)) and the energy level of Fc+/Fc with respect to the vacuum level (−4.8 eV).61 c Measured for CH2Cl2 solutions, c approximately 10−5 M. d Calculated at B3LYP/6-31G(d) level of theory, S0 → S1 transitions. | |||||||||
| 4d | 0.30 | −1.92 | 2.22 | −5.10 | −2.88 | 687 | 1.80 | 617 | 2.01 |
| 2d | 0.10 | −1.81 | 1.91 | −4.90 | −2.99 | 764 | 1.62 | 692 | 1.79 |
| 4e | 0.35 | −1.90 | 2.25 | −5.15 | −2.90 | 689 | 1.80 | 605 | 2.05 |
| 2f | 0.29 | −1.60 | 1.89 | −5.09 | −3.20 | 733 | 1.69 | 677 | 1.83 |
Similar low-energy transitions are present in the spectra of the fully fused azulenes 2d and 2f, but these are more redshifted, reaching up to 764 nm in the case of compound 2d. This trend is in accordance with the outcome of the TD-DFT calculations (ESI, Tables S5–S8‡). Such a low energy transition is the result of the retained azulene-like electronic structure in fully and partially fused π-scaffolds. The spatial separation of the HOMO and LUMO orbitals resembles those of pristine azulene, as clearly visible in Fig. 4. Therefore, these compounds can be regarded as ‘true’ extended azulenes, unlike many azulene-embedded PAHs reported in the literature, which are merely benzenoid PAHs with seven- and five-membered rings serving as linkages between adjacent benzenoid moieties.
 | ||
| Fig. 4 DFT-calculated (B3LYP/6-31G(d), isovalue 0.05 A−3) and experimental energies of HOMO and LUMO orbitals of 4d, 2d, 4e, and 2f. | ||
In the case of fully fused 2d and 2f, a distinctive HOMO to LUMO+1 S0 → S2 transition above 500 nm was primarily responsible for their red colour, as it is much stronger than the almost forbidden S0 → S1 transition (oscillator strength f = 0.680 for 2d and f = 0.380 for 2f). Although some of the known extended azulenes exhibited fluorescence, we did not observe Kasha nor anti-Kasha emission for the compounds reported here.
Electrochemical measurements of the reported azulene-embedded PAHs were carried out to experimentally estimate the HOMO and LUMO energy levels. Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) measurements were implemented for all extended azulenes (ESI, Fig. S8–S14‡). All compounds exhibited relatively low, reversible first oxidation potentials, ranging from 0.10 V vs. Fc/Fc+ for 2d to 0.35 V vs. Fc/Fc+ for 4e. This corresponds to the relatively high-lying HOMO orbitals (Fig. 4), and is consistent with the highest DFT-calculated HOMO energy for 2d.
The difference between fully fused π-scaffolds (2d, 2f) and partially fused molecules (4d, 4e) was also evident in their first reduction potentials. Compounds 2d and 2f are easier to reduce, with Ered1/2 = −1.81 V for the former and Ered1/2 = −1.60 V for the latter compared with Ered1/2 = −1.90 V for 4e and Ered1/2 = −1.92 V for 4d. The electrochemical gap is smaller for fully fused molecules 2d and 2f (approximately 1.9 V), compared to partially fused azulenes, which have an electrochemical gap above 2.2 V. This trend is in alignment with the DFT calculations (Fig. 4) and is due to both the lower energies of the LUMO orbitals and the higher energies of the HOMO orbitals. Notably, the electrochemical and optical gaps of the fully fused-azulenes reported here are significantly narrower than those of benz[a]azulenes28 (Eelectrg ≈ 3.0 eV; Eoptg ≈ 2.6 eV) or azuleno[1,2,3-fg]benzo[op]tetracene38 (Eelectrg = 2.18 eV; Eoptg = 1.78 eV).
Finally, we calculated the NICS(0) values62 of azulene-embedded PAHs 4d, 2d, 4e, and 2f (ESI, Fig. S24‡). The NICS(0) values confirmed the retained aromatic characteristics of the azulene moiety, which are in agreement with the 1H NMR chemical shifts and all the optical and electrochemical properties. This serves as additional confirmation that the reported PAHs are truly extended azulenes, not simply benzenoid structures with bridging five- and seven-membered rings.
Conclusions
Our investigations revealed that Scholl oxidation of relatively simple 1,2,3-triarylazulenes can result in unexpected reactivity patterns, including a 1,2-shift of a phenyl group and the formation of azulen-1(8aH)-ones. However, incorporating 2-pyrenyl substituents facilitated the formation of fused azulene structures, which was likely due to the favourable spin density distribution in the intermediate radical cations. This synthetic route enabled us to obtain a series of ‘true’ π-extended azulenes with optical absorption reaching the NIR region and a relatively low electrochemical gap below 2 eV. A synthetic strategy involving 2-pyrenyl substituents is promising for the construction of larger, fully hydrocarbon nanographenes incorporating multiple azulene subunits. This approach is currently being further developed in our laboratory.Author contributions
All authors have given approval to the final version of the manuscript. CRediT: Justyna Biesaga: conceptualization, investigation, writing – review and editing; Sławomir Szafert: supervision; Bartłomiej Pigulski: conceptualization, funding acquisition, investigation, supervision, writing – original draft, writing – review and editing.Data availability
The data supporting this article have been included as part of the ESI.‡ The crystallographic data for 1a, 3a, and 4e have been deposited at the CCDC under 2371233–2371235.‡Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Science Centre Poland (Grant UMO-2022/47/D/ST4/03312) for support of this research. Calculations were carried out at the Wrocław Centre for Networking and Supercomputing (https://www.wcss.pl), Grant No. 523.References
- A. Hirsch, The era of carbon allotropes, Nat. Mater., 2010, 9, 868–871 CrossRef CAS PubMed.
- Z. Liu, S. Fu, X. Liu, A. Narita, P. Samorì, M. Bonn and H. I. Wang, Small Size, Big Impact: Recent Progress in Bottom-Up Synthesized Nanographenes for Optoelectronic and Energy Applications, Adv. Sci., 2022, 9, 2106055 CrossRef PubMed.
- A. Narita, X. Y. Wang, X. Feng and K. Müllen, New advances in nanographene chemistry, Chem. Soc. Rev., 2015, 44, 6616–6643 RSC.
- A. Konishi and M. Yasuda, Breathing new life into nonalternant hydrocarbon chemistry: Syntheses and properties of polycyclic hydrocarbons containing azulene, pentalene, and heptalene frameworks, Chem. Lett., 2021, 50, 195–212 CrossRef CAS.
- Chaolumen, I. A. Stepek, K. E. Yamada, H. Ito and K. Itami, Construction of Heptagon-Containing Molecular Nanocarbons, Angew. Chem., Int. Ed., 2021, 60, 23508–23532 CrossRef CAS PubMed.
- K. Kawasumi, Q. Zhang, Y. Segawa, L. T. Scott and K. Itami, A grossly warped nanographene and the consequences of multiple odd-membered-ring defects, Nat. Chem., 2013, 5, 739–744 CrossRef CAS PubMed.
- M. Beer and H. C. Longuet-Higgins, Anomalous Light Emission of Azulene, J. Chem. Phys., 1955, 23, 1390–1391 CrossRef CAS.
- D. Dunlop, L. Ludvíková, A. Banerjee, H. Ottosson and T. Slanina, Excited-State (Anti)Aromaticity Explains Why Azulene Disobeys Kasha's Rule, J. Am. Chem. Soc., 2023, 145, 21569–21575 CrossRef CAS PubMed.
- A. G. Anderson and B. M. Steckler, Azulene. VIII. Spectra and Dipole Moments of Substituted Azulenes A Study of the Visible Absorption Spectra and Dipole Moments of Some 1- and 1,3-Substituted Azulenes, J. Am. Chem. Soc., 1959, 81, 4941–4946 CrossRef CAS.
- C. Zhu, K. Shoyama and F. Würthner, Conformation and Aromaticity Switching in a Curved Non-Alternant sp2 Carbon Scaffold, Angew. Chem., Int. Ed., 2020, 59, 21505–21509 CrossRef CAS PubMed.
- J. Liu, S. Mishra, C. A. Pignedoli, D. Passerone, J. I. Urgel, A. Fabrizio, T. G. Lohr, J. Ma, H. Komber, M. Baumgarten, C. Corminboeuf, R. Berger, P. Ruffieux, K. Müllen, R. Fasel and X. Feng, Open-Shell Nonbenzenoid Nanographenes Containing Two Pairs of Pentagonal and Heptagonal Rings, J. Am. Chem. Soc., 2019, 141, 12011–12020 CrossRef CAS PubMed.
- A. Konishi, K. Horii, D. Shiomi, K. Sato, T. Takui and M. Yasuda, Open-shell and antiaromatic character induced by the highly symmetric geometry of the planar heptalene structure: Synthesis and characterization of a nonalternant isomer of bisanthene, J. Am. Chem. Soc., 2019, 141, 10165–10170 CrossRef CAS PubMed.
- B. Pigulski, K. Shoyama and F. Würthner, NIR-Absorbing π-Extended Azulene: Non-Alternant Isomer of Terrylene Bisimide, Angew. Chem., Int. Ed., 2020, 59, 15908–15912 CrossRef CAS PubMed.
- H. Xin, B. Hou and X. Gao, Azulene-Based π-Functional Materials: Design, Synthesis, and Applications, Acc. Chem. Res., 2021, 54, 1737–1753 CrossRef CAS PubMed.
- H. Xin and X. Gao, Application of Azulene in Constructing Organic Optoelectronic Materials: New Tricks for an Old Dog, ChemPlusChem, 2017, 82, 945–956 CrossRef CAS PubMed.
- H. Xin, C. Ge, X. Yang, H. Gao, X. Yang and X. Gao, Biazulene diimides: A new building block for organic electronic materials, Chem. Sci., 2016, 7, 6701–6705 RSC.
- E. Puodziukynaite, H. W. Wang, J. Lawrence, A. J. Wise, T. P. Russell, M. D. Barnes and T. Emrick, Azulene methacrylate polymers: Synthesis, electronic properties, and solar cell fabrication, J. Am. Chem. Soc., 2014, 136, 11043–11049 CrossRef CAS PubMed.
- Y. Yamaguchi, M. Takubo, K. Ogawa, K. I. Nakayama, T. Koganezawa and H. Katagiri, Terazulene Isomers: Polarity Change of OFETs through Molecular Orbital Distribution Contrast, J. Am. Chem. Soc., 2016, 138, 11335–11343 CrossRef CAS PubMed.
- T. Shoji, K. Miura, T. Araki, A. Maruyama, A. Ohta, R. Sekiguchi, S. Ito and T. Okujima, Synthesis of 2-Methyl-1-azulenyl Tetracyanobutadienes and Dicyanoquinodimethanes: Substituent Effect of 2-Methyl Moiety on the Azulene Ring toward the Optical and Electrochemical Properties, J. Org. Chem., 2018, 83, 6690–6705 CrossRef CAS PubMed.
- H. Xin, C. Ge, X. Jiao, X. Yang, K. Rundel, C. R. McNeill and X. Gao, Incorporation of 2,6−Connected Azulene Units into the Backbone of Conjugated Polymers: Towards High–Performance Organic Optoelectronic Materials, Angew. Chem., Int. Ed., 2018, 57, 1322–1326 CrossRef CAS PubMed.
- L. C. Murfin, M. Weber, S. J. Park, W. T. Kim, C. M. Lopez-Alled, C. L. McMullin, F. Pradaux-Caggiano, C. L. Lyall, G. Kociok-Köhn, J. Wenk, S. D. Bull, J. Yoon, H. M. Kim, T. D. James and S. E. Lewis, Azulene-Derived Fluorescent Probe for Bioimaging: Detection of Reactive Oxygen and Nitrogen Species by Two-Photon Microscopy, J. Am. Chem. Soc., 2019, 141, 19389–19396 CrossRef CAS PubMed.
- S. Cai, W. Deng, F. Huang, L. Chen, C. Tang, W. He, S. Long, R. Li, Z. Tan, J. Liu, J. Shi, Z. Liu, Z. Xiao, D. Zhang and W. Hong, Light–Driven Reversible Intermolecular Proton Transfer at Single–Molecule Junctions, Angew. Chem., Int. Ed., 2019, 58, 3829–3833 CrossRef CAS PubMed.
- A. Jarszak-Tyl, B. Pigulski and S. Szafert, Solvent-free C-H alkynylation of azulenes, Org. Chem. Front., 2021, 8, 5674–5680 RSC.
- A. H. M. Elwahy, I. A. Abdelhamid and M. R. Shaaban, Recent Advances in the Functionalization of Azulene Through Pd-Catalyzed Cross-Coupling Reactions, ChemistrySelect, 2021, 6, 13664–13723 CrossRef CAS.
- A. H. M. Elwahy, M. R. Shaaban and I. A. Abdelhamid, Recent advances in the functionalization of azulene through Rh-, Ir-, Ru-, Au-, Fe-, Ni-, and Cu-catalyzed reactions, Appl. Organomet. Chem., 2022, 36, e6772 CrossRef CAS.
- Y. Fei and J. Liu, Synthesis of Defective Nanographenes Containing Joined Pentagons and Heptagons, Adv. Sci., 2022, 9, 2201000 CrossRef CAS PubMed.
- M. Murai, S. Iba, H. Ota and K. Takai, Azulene-Fused Linear Polycyclic Aromatic Hydrocarbons with Small Bandgap, High Stability, and Reversible Stimuli Responsiveness, Org. Lett., 2017, 19, 5585–5588 CrossRef CAS PubMed.
- A. Vardanyan, A. Villinger, P. Ehlers and P. Langer, Synthesis and Properties of Carbo- and Heterocyclic Benz[a]azulenes, J. Org. Chem., 2023, 88, 11411–11423 CrossRef CAS PubMed.
- C. Duan, J. Zhang, J. Xiang, X. Yang and X. Gao, Azulene-Embedded [n]Helicenes (n=5, 6 and 7), Angew. Chem., Int. Ed., 2022, 61, e202201494 CrossRef CAS PubMed.
- R. Liu, Y. Fu, F. Wu, F. Liu, J. J. Zhang, L. Yang, A. A. Popov, J. Ma and X. Feng, Modular Synthesis of Structurally Diverse Azulene-Embedded Polycyclic Aromatic Hydrocarbons by Knoevenagel-Type Condensation, Angew. Chem., Int. Ed., 2023, 62, e202219091 CrossRef CAS PubMed.
- Q. Jiang, T. Tao, H. Phan, Y. Han, T. Y. Gopalakrishna, T. S. Herng, G. Li, L. Yuan, J. Ding and C. Chi, Diazuleno–s–indacene Diradicaloids: Syntheses, Properties, and Local (anti)Aromaticity Shift from Neutral to Dicationic State, Angew. Chem., Int. Ed., 2018, 57, 169737–116741 Search PubMed.
- Y. Liang, S. Wang, M. Tang, L. Wu, L. Bian, L. Jiang, Z.-B. Tang, J. Liu, A. Guan and Z. Liu, Cascade Synthesis of Benzotriazulene with Three Embedded Azulene Units and Large Stokes Shifts, Angew. Chem., Int. Ed., 2023, 62, e202218839 CrossRef CAS PubMed.
- J. Wang, F. G. Gámez, J. Marín-Beloqui, A. Diaz-Andres, X. Miao, D. Casanova, J. Casado and J. Liu, Synthesis of a Dicyclohepta[a,g]heptalene-Containing Polycyclic Conjugated Hydrocarbon and the Impact of Non-Alternant Topologies, Angew. Chem., Int. Ed., 2023, 62, e202217124 CrossRef CAS PubMed.
- H. Xin, J. Li, R. Q. Lu, X. Gao and T. M. Swager, Azulene-Pyridine-Fused Heteroaromatics, J. Am. Chem. Soc., 2020, 142, 13598–13605 CrossRef CAS PubMed.
- M. Grzybowski, K. Skonieczny, H. Butenschön and D. T. Gryko, Comparison of Oxidative Aromatic Coupling and the Scholl Reaction, Angew. Chem., Int. Ed., 2013, 52, 9900–9930 CrossRef CAS PubMed.
- M. Grzybowski, B. Sadowski, H. Butenschön and D. T. Gryko, Synthetic Applications of Oxidative Aromatic Coupling—From Biphenols to Nanographenes, Angew. Chem., Int. Ed., 2020, 59, 2998–3027 CrossRef CAS PubMed.
- H. N. Zeng, Z. M. Png and J. Xu, Azulene in Polymers and Their Properties, Chem. – Asian J., 2020, 15, 1904–1915 CrossRef CAS PubMed.
- Chaolumen, H. Ito and K. Itami, An axially chiral 1,1′-biazulene and its π-extended derivative: Synthesis, structures and properties, Chem. Commun., 2019, 55, 9606–9609 RSC.
- P. Mathey, I. Fernández and J. F. Morin, Exploring C-C bond formation reactions for expanding azulene derivatives linked at the 2- and/or 6-positions, New J. Chem., 2024, 48, 4801–4809 RSC.
- T. Koide, M. Takesue, T. Murafuji, K. Satomi, Y. Suzuki, J. Kawamata, K. Terai, M. Suzuki, H. Yamada, Y. Shiota, K. Yoshizawa and F. Tani, An Azulene-Fused Tetracene Diimide with a Small HOMO–LUMO Gap, ChemPlusChem, 2017, 82, 1010–1014 CrossRef CAS PubMed.
- L. Chen, B. Wu, L. Qin, Y. Y. Huang, W. Meng, R. Kong, X. Yu, K. ChenChai, C. Li, G. Zhang, X. S. Zhang and D. Zhang, A perylene five-membered ring diimide for organic semiconductors and π-expanded conjugated molecules, Chem. Commun., 2022, 58, 5100–5103 RSC.
- D. Eppel, N. Oberhof, M. C. Dietl, P. Cieslik, M. Rudolph, L. Eberle, P. Krämer, F. Stuck, F. Rominger, A. Dreuw and A. S. K. Hashmi, Gold(III) Meets Azulene: A Class of [(tBuC^N^C)AuIII(azulenyl)] Pincer Complexes, Organometallics, 2021, 40, 3865–3870 CrossRef CAS.
- P. de Wit, H. J. A. Lambrechts and H. Cerfontain, On the bromination of 1,2,3-triphenylazulene, Recl. Trav. Chim. Pays-Bas, 1985, 104, 68–69 CrossRef CAS.
- S. J. Assony and N. Kharasch, Derivatives of Sulfenic Acids. XXXII. The Synthesis of Azulenes via the Interactions of Arylacetylenes with Sulfenyl Halides. Part 1. 1,2,3-Triphenylazulene, J. Am. Chem. Soc., 1958, 80, 5978–5982 CrossRef CAS.
- C. Lambert, G. Nöll, M. Zabel, F. Hampel, E. Schmälzlin, C. Bräuchle and K. Meerholz, Highly substituted azulene dyes as multifunctional NLO and electron-transfer compounds, Chem. – Eur. J., 2003, 9, 4232–4239 CrossRef CAS PubMed.
- V. Claus, M. Schukin, S. Harrer, M. Rudolph, F. Rominger, A. M. Asiri, J. Xie and A. S. K. Hashmi, Gold-Catalyzed Dimerization of Diarylalkynes: Direct Access to Azulenes, Angew. Chem., Int. Ed., 2018, 57, 12966–12970 CrossRef CAS PubMed.
- T. D. Lash, J. A. El-Beck and G. M. Ferrence, Syntheses and reactivity of meso-unsubstituted azuliporphyrins derived from 6-tert-butyl- and 6-phenylazulene, J. Org. Chem., 2007, 72, 8402–8415 CrossRef CAS PubMed.
- M. Narita, T. Murafuji, S. Yamashita, M. Fujinaga, K. Hiyama, Y. Oka, F. Tani, S. Kamijo and K. Ishiguro, Synthesis of 2-Iodoazulenes by the Iododeboronation of Azulen-2-ylboronic Acid Pinacol Esters with Copper(I) Iodide, J. Org. Chem., 2018, 83, 1298–1303 CrossRef CAS PubMed.
- K. H. Chen, Y. J. Chiang and J. L. Zhu, Rhodium-catalyzed cyclization of acceptor-substituted biphenyl α-diazoketones: A study of the substitution effect on chemoselectivity, Org. Biomol. Chem., 2018, 16, 6975–6986 CAS.
- S. A. More, V. A. Sadaphal, T. C. Kuo, M. J. Cheng and R. S. Liu, Reactions of thioalkynes with diarylketenes via [3+2]-annulation versus benzannulation using Au and P(C6F5)3 catalysts, Chem. Commun., 2022, 58, 10064–10067 RSC.
- D. G. Brown, T. R. Hoye and R. G. Brisbois, Synthesis of Azulenone Skeletons by Reaction of 2-Phenyl-2-acylketenes [RCO(Ph)C=C=O] with Alkynyl Ethers: Mechanistic Aspects and Further Transformations, J. Org. Chem., 1998, 63, 1630–1636 CrossRef CAS.
- X. D. Yue, L. Chen and W. D. Z. Li, Synthesis and applications of angular acyl-substituted azulenones: Formal synthesis of (±)-acorenol and facile access to the hydroazulene core of pseudolaric acid, Tetrahedron, 2014, 70, 5505–5512 CrossRef CAS.
- S. H. Pun, E. C. H. Wen, Z. Xia, H. Chen, F. R. Fischer and Q. Miao, Reactivity, Regioselectivity, and Synthetic Application of 2-Pyrenyl Units in Scholl Reactions, CCS Chem., 2023, 5, 2069–2077 CrossRef CAS.
- P. Ravat, Carbo[n]helicenes Restricted to Enantiomerize: An Insight into the Design Process of Configurationally Stable Functional Chiral PAHs, Chem. – Eur. J., 2021, 27, 3957–3967 CrossRef CAS PubMed.
- M. Krzeszewski, K. Sahara, Y. M. Poronik, T. Kubo and D. T. Gryko, Unforeseen 1,2-Aryl Shift in Tetraarylpyrrolo[3,2-b] pyrroles Triggered by Oxidative Aromatic Coupling, Org. Lett., 2018, 20, 1517–1520 CrossRef CAS PubMed.
- J. Liu, A. Narita, S. Osella, W. Zhang, D. Schollmeyer, D. Beljonne, X. Feng and K. Müllen, Unexpected Scholl Reaction of 6,7,13,14-Tetraarylbenzo[k]tetraphene: Selective Formation of Five-Membered Rings in Polycyclic Aromatic Hydrocarbons, J. Am. Chem. Soc., 2016, 138, 2602–2608 CrossRef CAS PubMed.
- W. Kong, L. H. Finger, J. C. A. Oliveira and L. Ackermann, Rhodaelectrocatalysis for Annulative C−H Activation: Polycyclic Aromatic Hydrocarbons through Versatile Double Electrocatalysis, Angew. Chem., Int. Ed., 2019, 58, 6342–6346 CrossRef CAS PubMed.
- Z. Guo, J. Zhang, J. Zhang and M. Xie, Electrochemical Rhodium-Catalyzed C-H Cyclodimerization of Alkynes to Access Diverse Functionalized Naphthalenes: Involvement of RhIV/V and RhI Dual Catalysis, Org. Lett., 2022, 24, 7784–7789 CrossRef CAS PubMed.
- Y. Honjo, Y. Shibata, E. Kudo, T. Namba, K. Masutomi and K. Tanaka, Room Temperature Decarboxylative and Oxidative [2+2+2] Annulation of Benzoic Acids with Alkynes Catalyzed by an Electron-Deficient Rhodium(III) Complex, Chem. – Eur. J., 2018, 24, 317–321 CrossRef CAS PubMed.
- B. Dittrich, F. P. A. Fabbiani, J. Henn, M. U. Schmidt, P. Macchi, K. Meindl and M. A. Spackman, Azulene revisited: solid-state structure, invariom modeling and lattice-energy minimization of a classical example of disorder, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2018, 74, 416–426 CrossRef CAS PubMed.
- I. Seguy, P. Jolinat, P. Destruel, R. Mamy, H. Allouchi, C. Courseille, M. Cotrait and H. Bock, Crystal and Electronic Structure of a Fluorescent Columnar Liquid Crystalline Electron Transport Material, ChemPhysChem, 2001, 2, 448–452 CrossRef CAS PubMed.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta, P. Von and R. Schleyer, Nucleus-Independent Chemical Shifts (NICS) as an Aromaticity Criterion, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Prof. Frank Würthner on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures, details of X-ray single crystal diffraction experiments, copies of UV/Vis/NIR spectra, copies of NMR spectra, copies of electrochemical measurements, and Cartesian coordinates of DFT-optimized structures. CCDC 2371233–2371235. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qo01459f |
| This journal is © the Partner Organisations 2024 |