
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-pot multistep synthesis of 1-fluoroalkylisoquinolines and fused fluoroalkylpyridines from N-fluoroalkyl-1,2,3-triazoles†
Anna
Kubíčková
 ab,
Svatava
Voltrová
a,
Adam
Kleman
a,
Blanka
Klepetářová
a and
Petr
Beier
*a
ab,
Svatava
Voltrová
a,
Adam
Kleman
a,
Blanka
Klepetářová
a and
Petr
Beier
*a
aInstitute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo nám. 2, 16610 Prague 6, Czechia. E-mail: beier@uochb.cas.cz
bUniversity of Chemistry and Technology, Technická 5, 166 28 Prague 6, Czechia
First published on 18th June 2024
Abstract
An efficient one-pot microwave-assisted potassium fluoride-mediated synthesis of 1-fluoroalkyl-3-fluoroisoquinolines and fused fluoroalkylpyridines from N-fluoroalkylated 1,2,3-triazoles was developed. The reaction has a wide scope and allows the preparation of structurally diverse 3-fluoroisoquinolines with a fluoroalkyl group in position 1, a substituent in position 4 and a substituent on the fused benzene (or heteroaromatic) ring. N-Fluoroalkylated ketenimines, which undergo stereoselective formal 1,3-fluorine shift to difluoroazadienes, were identified as intermediates in the reaction sequence. The presence of fluorine in position 3 and a halogen in position 4 of the resulting isoquinolines allowed for further modification by nucleophilic aromatic substitution and cross-coupling reactions, respectively. The developed methodologies were utilized for the synthesis of derivatives of drug candidates.
Introduction
The isoquinoline core is present in a variety of drugs and a large number of naturally occurring alkaloids, which in many cases possess compelling biological activities (Fig. 1).1–4 Numerous synthetic approaches exist leading to these benzopyridines, such as multistep sequences of reactions including the well-known Bischler–Napieralski,5 Pomeranz–Fritsch6 and Pictet–Spengler7 reactions, as well as processes involving transition metal catalysis.8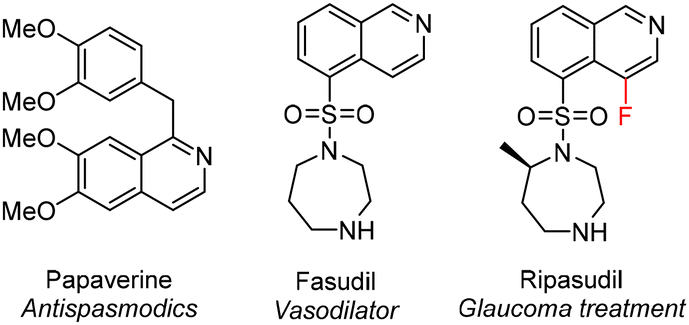 | ||
| Fig. 1 Examples of an isoquinoline containing alkaloid (papaverine) and synthetic drugs (fasudil and ripasudil). | ||
The introduction of fluorine atoms or fluoroalkyl groups into a lead molecule is a widely used strategy to enhance the pharmacologically relevant properties9 and several 1-trifluoromethylisoquinolines exploit this trend (Fig. 2), for example valiglurax10 – a positive allosteric modulator of mGlu4 receptors and a candidate for the treatment of Parkinson's disease. Yet, the procedures for their preparation remain underdeveloped, substrate-specific, low-yielding, or require expensive, non-selective and atom non-economical fluoroalkylation methods or transition metal catalysts.13,14
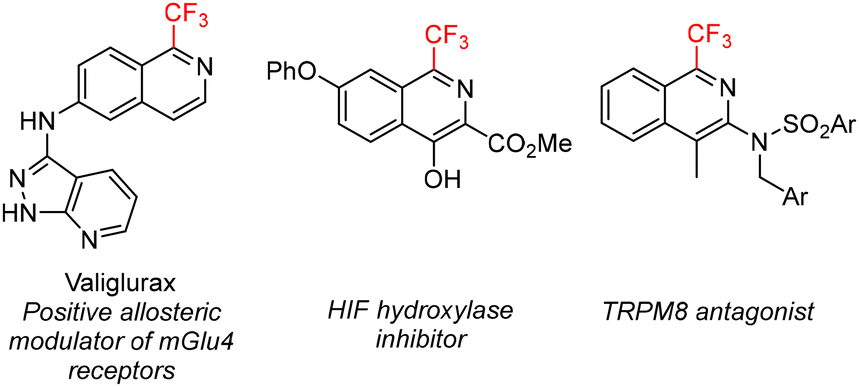 | ||
| Fig. 2 Examples of bioactive 1-trifluoromethylisoquinolines.10–12 | ||
The first multi-step approach leading to 1-perfluoroalkylisoquinolines with the Bischler–Napieralski type cyclization was demonstrated by Pastor15 in 1979. A similar approach was also used in 2019 by Lindsley (Scheme 1A) for the preparation of valiglurax, which allowed the construction of the isoquinoline core in an overall 21% yield.10 Another possible approach involves direct C–H bond perfluoroalkylation of isoquinolines16 (Scheme 1B) or isoquinoline-N-oxides.17,18 Trifluoromethylation via a coupling reaction of iodoisoquinolines with copper19–22 or palladium23 catalysts was also reported. However, the most common strategy towards 1-fluoroalkylated isoquinolines involves the insertion of fluoroalkyl radicals into isonitriles, followed by radical cyclization (Scheme 1C).13,24–29
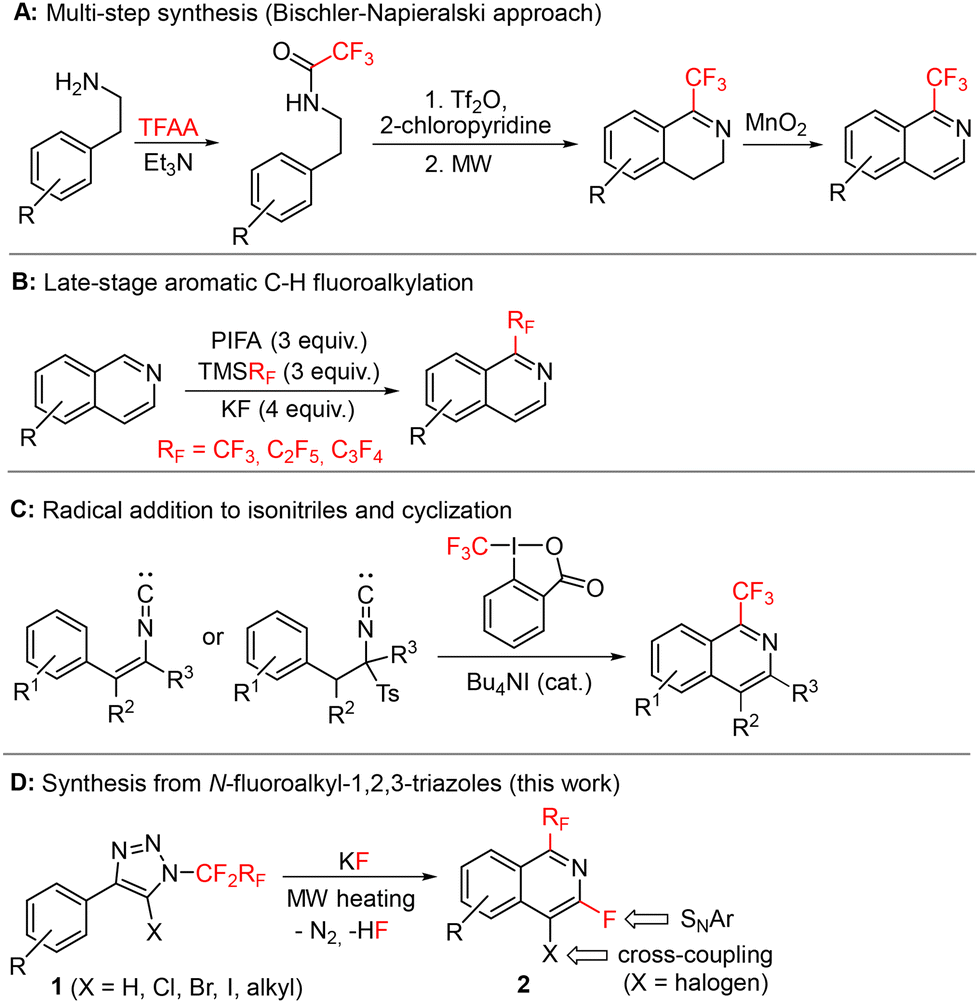 | ||
| Scheme 1 Selected literature syntheses of 1-fluoroalkylated isoquinolines (A–C) and our new approach from triazoles (D). | ||
Herein, we report a high-yielding and novel one-pot synthetic strategy to prepare substituted 1-fluoroalkyl-3-fluoroisoquinolines 2 based on thermal decomposition of N-fluoroalkyl-1,2,3-triazoles 1, formal 1,3-fluorine shift, and cyclization (Scheme 1D). The presence of fluorine in position 3 and a halogen in position 4 of the isoquinoline ring enabled further modifications by nucleophilic aromatic substitution and/or cross-coupling reactions, respectively. The procedure is applicable also to heteroaryl substituted N-fluoroalkyl-1,2,3-triazoles affording heteroarenes with fused fluoroalkylated pyridine rings. The methodology thus allows the expansion of known chemical space to new selectively substituted fluoroalkylated isoquinoline-type structures with potential applications in life sciences.
Results and discussion
Recently, we reported thermal rearrangement of N-fluoroalkyl-1,2,3-triazoles30–331 leading to N-fluoroalkylated ketenimines 3.34 We noticed that a prolonged heating of 3 led to new products identified by HRMS and NMR as isoquinolines 2 and enamides 5. Their formation can be explained by a thermally induced 1,3-fluorine shift of ketenimines 3 to two geometric isomers of azadienes 4. Although four isomers of 4 can be theoretically formed by the fluorine shift, only the formation of two isomers was observed. The isomer (Z,E)-4 cyclized to isoquinolines 2 while the isomer (Z,Z)-4 only hydrolysed to enamides 5 (Scheme 2) (see the ESI† for three examples of isolated enamides 5). A high-temperature NMR kinetic study revealed the time course of intermediate and product formation (Fig. 3).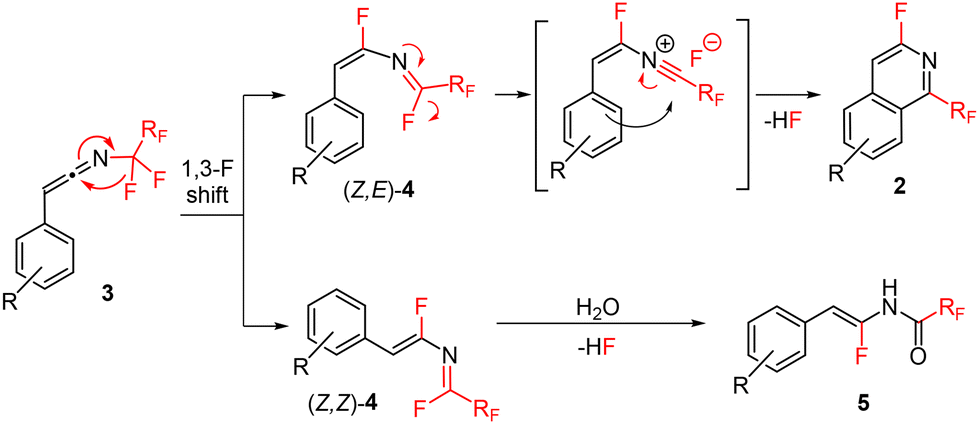 | ||
| Scheme 2 Proposed reaction mechanism of thermal additive-free decomposition of ketenimines 3 to isoquinolines 2 and side-products enamides 5. | ||
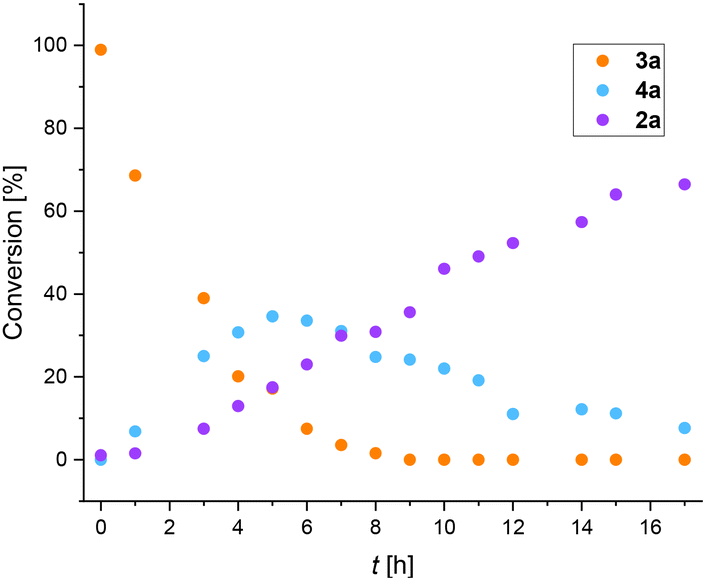 | ||
| Fig. 3 Conversion of 3a (R = H, RF = CF3) vs. reaction time for the formation of intermediates 4a and product 2a determined by 1H NMR (C2D4Cl2; 140 °C). | ||
A related transformation was briefly reported by Lermontov in 2002.35 Thermal Huisgen cycloaddition of diphenylacetylene with ethyl 3-azido-2,2,3,3-tetrafluoropropanoate afforded isoquinolines and enamides in low yields. The authors wrongly assumed antiaromatic 1H-azirines to be the reactive intermediates (Scheme 3A), which we disproved with ab initio calculations in our previous study.34 In another report, Molina showed the formation of an isoquinoline by ring closure of an N-styryl-substituted ketenimine (Scheme 3B).36–38
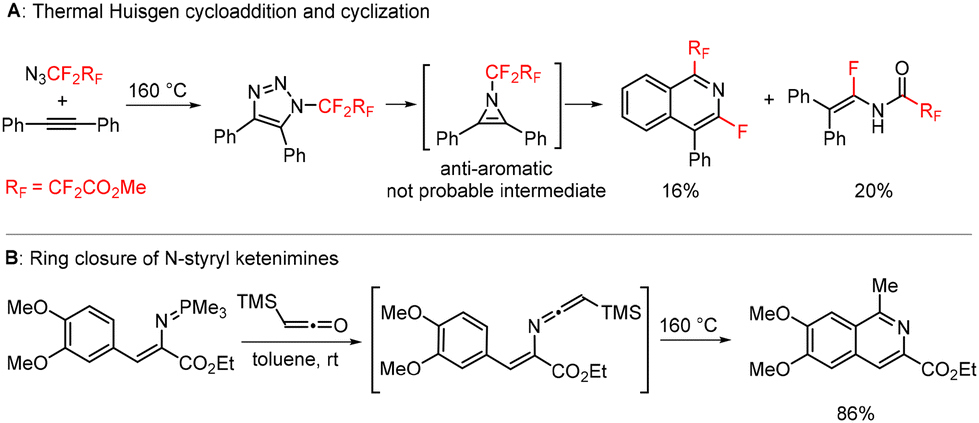 | ||
| Scheme 3 Published preparations of isoquinolines by thermal Huisgen cyclization (A) and from N-styryl-substituted ketenimines (B). | ||
In order to develop a general synthesis of 1-fluoroalkylated-2-fluoroisoquinolines 2 from triazoles 1 or ketenimines 3 we studied the effect of additives on the formation of 4. Ideally, the formation of 4 should proceed stereoselectively to the Z,E-isomer. Therefore, the influence of additives on the stereoselectivity of the formal 1,3-fluorine shift of 3a at room temperature was studied. While the addition of Et3N, DBU, or BF3·OEt2 did not lead to efficient formation of 4a, the addition of other basic additives or fluoride salts proved beneficial (Table 1). Carbonates induced the stereoselective transformation to the required (Z,E)-isomer of 4a with Cs2CO3 reacting much faster than K2CO3 (entries 2 and 3) and Na2CO3 being unreactive (presumably due to its low solubility). However, decomposition of 4a was observed in the basic conditions over time. A similar trend was observed in the case of inorganic fluorides with NaF being unreactive and CsF inducing the formation much faster than KF, but product decomposition and isomerization precluded its use in preparative experiments (entries 4–6; see the ESI† for the isomerization study of 4a with CsF). Therefore, mildly basic KF was used as the additive of choice, accelerating the formal 1,3-fluorine shift of ketenimines 3 and providing a high selectivity to the required isomer of 4 for further cyclization. The origin of the stereoselectivity of the thermal or heterogeneous additive-mediated 1,3-fluorine shift is unknown; however, we propose the steric factor to be dominant with fluoride addition to the central sp carbon atom of the ketenimine proceeding trans to the large aryl group followed by fluoride elimination from the CF2 group (Scheme 4).
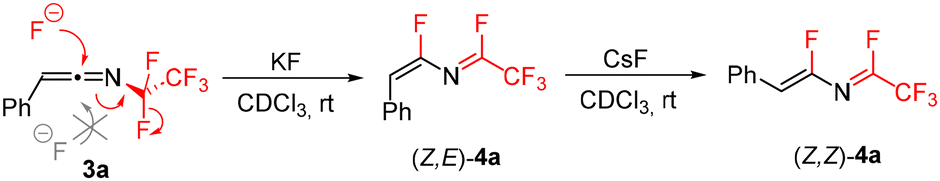 | ||
| Scheme 4 Fluoride-mediated formation of azadienes 4a from ketenimine 3a. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Additive | 19F NMR yield 3a/(Z,E)-4a/(Z,Z)-4a (%) | ||
| 1 h | 5 h | 24 h | ||
| 1 | KHCO3 | 78![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :22:0 :22:0 |
49:45:0 |
23:62:0 |
| 2 | K2CO3 | 83:17:0 |
57:38:0 |
0:83:0 |
| 3 | Cs2CO3 | 0:86:0 |
0:43:0 |
0:0:0 |
| 4 | NaF | 98:0:0 |
98:0:0 |
98:0:0 |
| 5 | KF | 83:17:0 |
50:50:0 |
13:80:0 |
| 6 | CsF | 0:89:0 |
0:79:0 |
0:50:12 |
Difluoroazadiene (Z,E)-4a was prepared using CsF (Scheme 5). The structure of its derivative 4m was confirmed by X-ray crystallography. Furthermore, addition of sodium acetate to 3a efficiently afforded acetate 6a, confirming that indeed a suitable nucleophile can add to the sp carbon of ketenimine 3a, followed by fluoride elimination. Attempts to use chloride or iodide salts were unsuccessful.
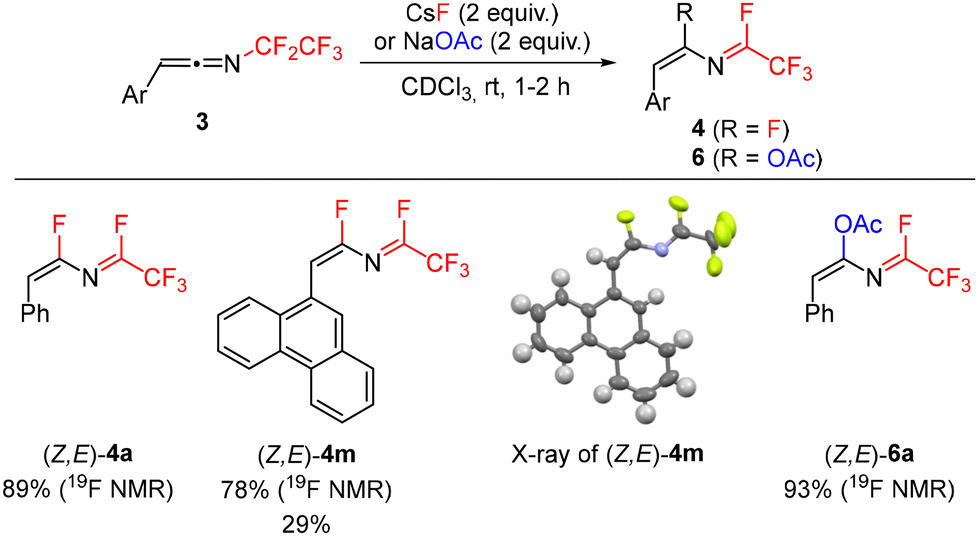 | ||
| Scheme 5 Characterized azadienes 4 and 6. | ||
An optimization study revealed that under microwave heating conditions a slight excess of KF afforded the formation of isoquinoline 2a directly from triazole 1a without the need to isolate the intermediates ketenimine 3a or difluoroazadiene 4a (Table 2). As for the solvent effect, we previously reported that the formation of ketenimines 3 from triazoles 1 works best in DCE but other solvents (chloroform, THF, toluene, cyclohexane, acetone) can also be used.34 In this study we chose DCE as the optimal solvent.
|
|
||
|---|---|---|
| Entry | KF (equiv.) | 19F NMR ratio 3a/(Z,E)-4a/2a |
| 1 | 0.05 | 70:20:10 |
| 2 | 0.2 | 48:28:24 |
| 4 | 0.7 | 7:5:88 |
| 5 | 1.0 | 3:3:94 |
| 6 | 1.1 | 0:0:99 |
| 7 | 2.0 | 0:0:99 |
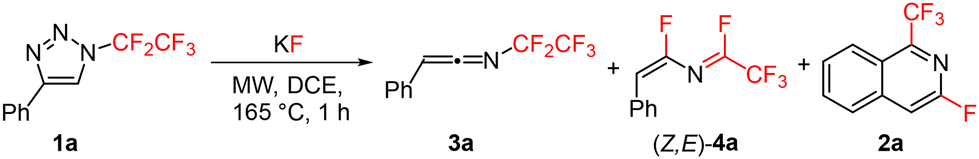
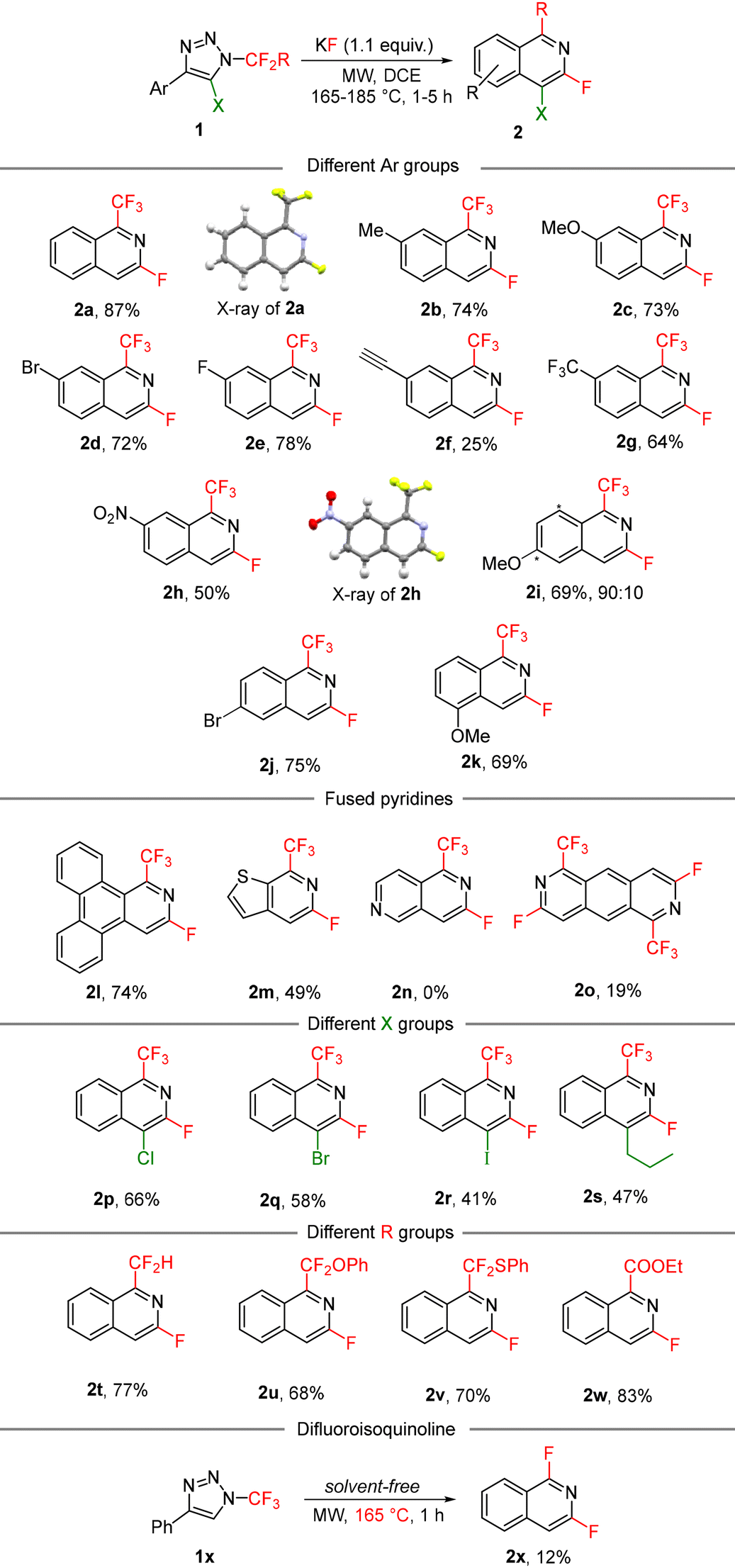
With the optimized set of conditions, we expanded the multistep one-pot process to diversely substituted N-fluoroalkylated 1,2,3-triazoles 1 (Table 3). The method tolerated various functional groups on the aryl moiety, including electron-neutral, electron-rich and electron-poor substituents on the phenyl group with slightly decreased yields in the last case. Different substitution positions on the aryl group were also well tolerated with differently substituted isoquinolines being produced from o-, m- or p-substituted aryls. In the case of m-substituted aryls, two isomers of the products were formed (2i) with good regioselectivity. In another case, the reaction was regiospecific (2j). Substrates with large (1l) or heteroaromatic (1m) groups also underwent the reaction to afford unique isoquinolines or fused pyridines; however, the pyridyl-substituted triazole (1n) was found to be unreactive and only decomposition to a complex mixture of products was observed.
|
Position 5 of the starting triazole ring can be substituted with a halogen or alkyl group, which introduced these functions into position 4 of the final isoquinoline with various degrees of efficiency. The observed trend can be explained by steric factors where bulky substituents on the ketenimine sp2 carbon atom hindered the attack of the fluoride ion to form the productive isomer of azadienes 4.
The methodology was found to display an excellent robustness with regards to the fluoroalkyl substituent in position 1 of the products. Not only the trifluoromethyl group, but also difluoromethyl, substituted difluoromethylene and ethoxycarbonyl substituents can be introduced efficiently. Under solvent free conditions difluoroisoquinoline 2x was prepared in low yield due to its high volatility and some side reactions.
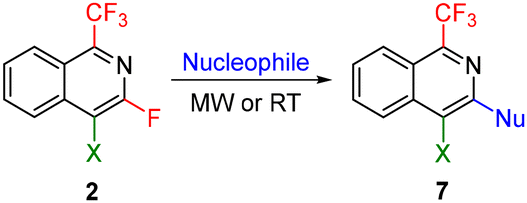
The presence of a fluorine substituent in isoquinolines 2 in the activated position called for the investigation of substitution with various nucleophiles by SNAr which expanded the diversity of accessible 1-fluoroalkylated isoquinolines. Thus, the fluorine atom of isoquinolines 2 was readily substituted with various oxygen, sulfur, and nitrogen nucleophiles in polar solvents to obtain heteroatom-substituted 1-trifluoromethyl isoquinolines 7 (Table 4).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Nucleophile (equiv.) | X | Solvent | Temp. (°C) | 7, yield (%) |
| 1 | NaOH (15) | H | H2O | 155 | 7a, 88 |
| 2 | EtONa (12) | H | EtOH | 80 | 7b, 96 |
| 3 | EtONa (12) | Cl | EtOH | 155 | 7c, 99 |
| 4 | t-BuOK (1.2) | H | t-BuOH | 80 | 7d, 80 |
| 5 | PhONa (1.5) | H | DMA | 80 | 7e, 89 |
| 6 | MeSNa (5) | H | DMA | 20 | 7f, 85 |
| 7 | MeSNa (2) | Ph | DMF | 20 | 7g, 91 |
| 8 | p-Tol-SNa (1) | H | DMA | 80 | 7h, 91 |
| 9 | p-Tol-SO2Li (2.5) | H | DMSO | 155 | 7i, 58 |
| 10 | NH2NH2 (20) | H | i-PrOH | 100 | 7j, 95 |
| 11 | p-Tol-CH2NH2 (2) | H | DMSO | 155 | 7k, 42 |
Furthermore, isoquinolines 2 were used for the preparation of a small library of nine analogues of the TRPM8 antagonist shown in Fig. 2. Compounds 8 were easily accessed by nucleophilic sulfonamidation of 2 (Scheme 6), demonstrating the value of our approach in the synthesis of fluorinated and fluoroalkylated isoquinolines and their structurally diverse derivatives in drug development.
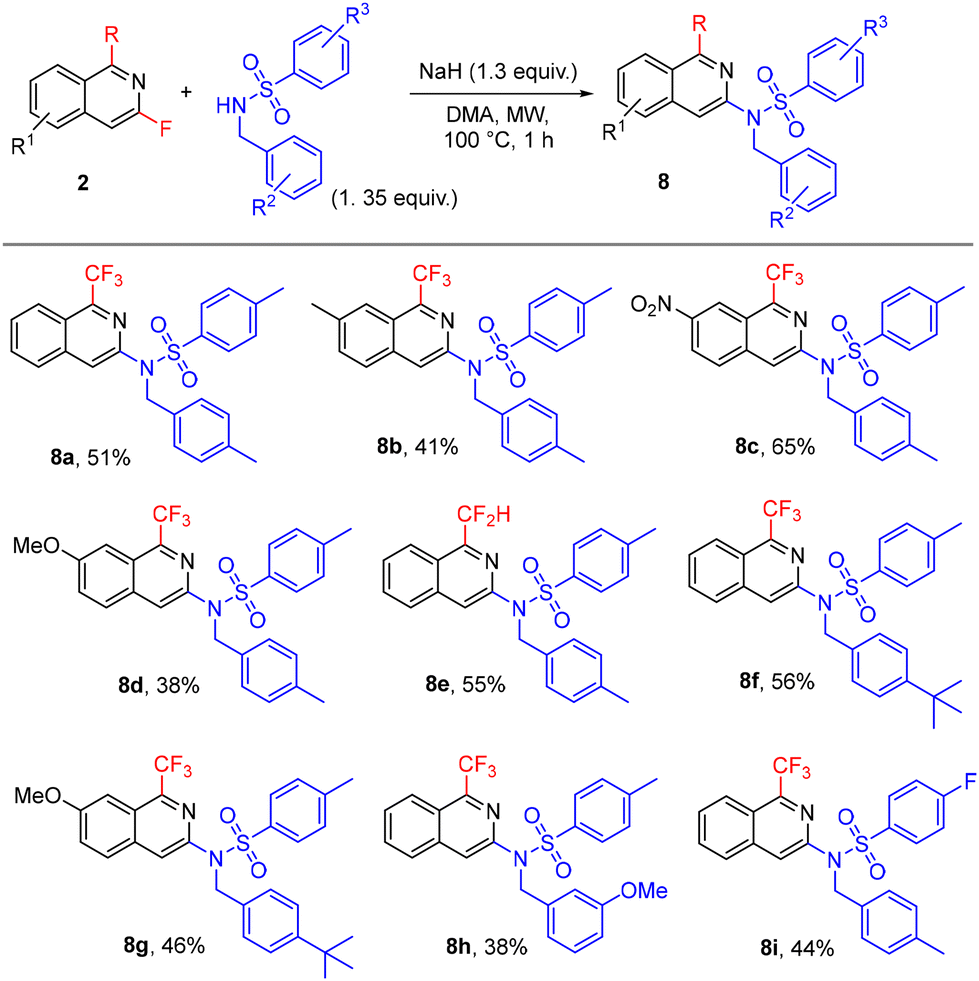 | ||
| Scheme 6 Analogues of the TRPM8 antagonist. | ||
Other investigated follow-up derivatizations of compounds 2 were the cross-coupling reactions. Suzuki–Miyaura coupling of arylboronic acids with chloroisoquinoline 2p afforded coupling products 9a–c in high yields (Scheme 7). Heck, Sonogashira and Buchwald–Hartwig reactions of bromoisoquinoline 2q also worked well giving the coupling products 10a–c (Scheme 8).
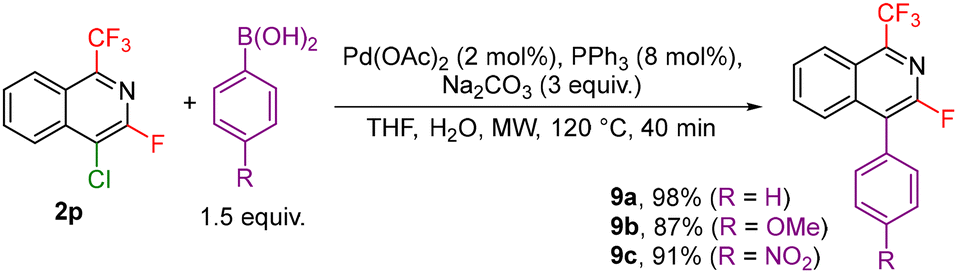 | ||
| Scheme 7 Suzuki–Miyarura coupling reactions with 2p. | ||
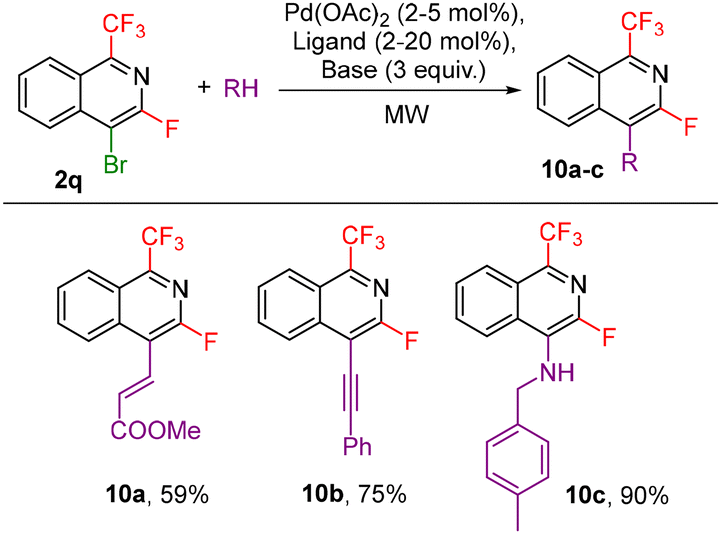 | ||
| Scheme 8 Heck, Sonogashira and Buchwald–Hartwig coupling reactions of 2r (see the ESI† for detailed conditions). | ||
The developed method for the synthesis of fluorinated isoquinolines was used for the preparation of 3-fluoro analogue 12 of the drug candidate valiglurax. The brominated isoquinoline 2j was used for Pd-catalyzed amination, followed by protecting group removal to give analogue 12 in high yields (Scheme 9).
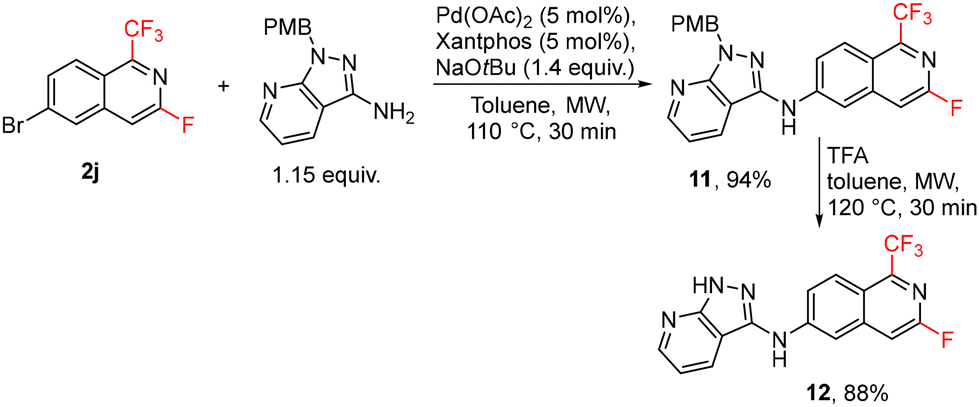 | ||
| Scheme 9 Synthesis of valiglurax analogue 12. | ||
Conclusions
In conclusion, microwave heating of N-fluoroalkyl-1,2,3-triazoles in the presence of potassium fluoride led to a series of events involving triazole ring opening, nitrogen molecule elimination, rearrangement, stereoselective formal 1,3-fluorine shift, and finally cyclization to produce diverse 1-fluoroalkylated 3-fluoroisoquinolines in good yields and with excellent substrate scope. Nucleophilic aromatic substitution of the fluorine atom in position 3 with heteroatom nucleophiles afforded 1-fluoroalkylated 3-substituted isoquinolines. Cross-coupling reactions of halogen atoms in position 4 of the isoquinolines gave derivatives with aryl, alkenyl, alkynyl or alkylamino groups. This synthetic approach to novel selectively fluorinated isoquinolines was applied in the synthesis of analogues of two families of drug candidates.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Czech Academy of Sciences (research plan RVO: 61388963) and the Czech Science Foundation (project 23-04659S). This work was supported by the grant of Specific University Research – grant no. A2_FCHT_2024_001.References
- E. Plazas, M. M. C. Avila, D. R. Muñoz and S. L. E. Cuca, Natural Isoquinoline Alkaloids: Pharmacological Features and Multi-Target Potential for Complex Diseases, Pharmacol. Res., 2022, 177, 106126, DOI:10.1016/j.phrs.2022.106126.
- X.-F. Shang, C.-J. Yang, S. L. Morris-Natschke, J.-C. Li, X.-D. Yin, Y.-Q. Liu, X. Guo, J.-W. Peng, M. Goto, J.-Y. Zhang and K.-H. Lee, Biologically Active Isoquinoline Alkaloids Covering 2014–2018, Med. Res. Rev., 2020, 40(6), 2212–2289, DOI:10.1002/med.21703.
- G. Diaz, I. L. Miranda and M. A. N. Diaz, Quinolines, Isoquinolines, Angustureine, and Congeneric Alkaloids—Occurrence, Chemistry, and Biological Activity, in Phytochemicals - Isolation, Characterisation and Role in Human Health, IntechOpen, 2015. DOI:10.5772/59819.
- A. Vijayakumar, M. Manod, R. B. Krishna, A. Mathew and C. Mohan, Diversely Functionalized Isoquinolines and Their Core-Embedded Heterocyclic Frameworks: A Privileged Scaffold for Medicinal Chemistry, RSC Med. Chem., 2023, 14, 2509–2534, 10.1039/D3MD00248A.
- A. Bischler and B. Napieralski, Zur Kenntniss Einer Neuen Isochinolinsynthese, Ber. Dtsch. Chem. Ges., 1893, 26(2), 1903–1908, DOI:10.1002/cber.189302602143.
- C. Pomeranz, Über eine neue Isochinolinsynthese, Monatsh. Chem., 1893, 14(1), 116–119, DOI:10.1007/BF01517862.
- A. Pictet and T. Spengler, Über Die Bildung von Isochinolin-Derivaten Durch Einwirkung von Methylal Auf Phenyl-Äthylamin, Phenyl-Alanin Und Tyrosin, Ber. Dtsch. Chem. Ges., 1911, 44(3), 2030–2036, DOI:10.1002/cber.19110440309.
- R. Gujjarappa, N. Vodnala and C. C. Malakar, Comprehensive Strategies for the Synthesis of Isoquinolines: Progress Since 2008, Adv. Synth. Catal., 2020, 362(22), 4896–4990, DOI:10.1002/adsc.202000658.
- C. Zhang, K. Yan, C. Fu, H. Peng, C. J. Hawker and A. K. Whittaker, Biological Utility of Fluorinated Compounds: From Materials Design to Molecular Imaging, Therapeutics and Environmental Remediation, Chem. Rev., 2022, 122(1), 167–208, DOI:10.1021/acs.chemrev.1c00632.
- J. D. Panarese, D. W. Engers, Y.-J. Wu, J. J. Bronson, J. E. Macor, A. Chun, A. L. Rodriguez, A. S. Felts, J. L. Engers, M. T. Loch, K. A. Emmitte, A. L. Castelhano, M. J. Kates, M. A. Nader, C. K. Jones, A. L. Blobaum, P. J. Conn, C. M. Niswender, C. R. Hopkins and C. W. Lindsley, Discovery of VU2957 (Valiglurax): An mGlu4 Positive Allosteric Modulator Evaluated as a Preclinical Candidate for the Treatment of Parkinson's Disease, ACS Med. Chem. Lett., 2019, 10(3), 255–260, DOI:10.1021/acsmedchemlett.8b00426.
- Y. Tsuzuki, D. Sawamoto, T. Sakamoto, T. Kato, Y. Niwa and N. Awai, Sulfonamide Compounds Having TRPM8 Antagonistic Activity. US9540360B2, 2017.
- W.-B. Ho, H. Zhao, S. Deng, D. Ng, L. R. Wright, M. Wu, X. Zhou, M. P. Arend and L. A. Flippin, 4-Hydroxy-Isoquinoline Compounds as Hif Hydroxylase, Inhibitors, 2866556, 2020 Search PubMed.
- K. Aradi and L. Kiss, Recent Advances in the Trifluoromethylation Reactions of Isonitriles To Construct CF3-Substituted N-Heterocycles, Synthesis, 2023, 55(12), 1834–1843, DOI:10.1055/a-2020-9090.
- J. C. Sloop, Advances in the Preparation of Fluorinated Isoquinolines: A Decade of Progress, J. Chem., 2017, 2017, e2860123, DOI:10.1155/2017/2860123.
- P. R. Pastor and A. Cambon, Synthese d'isoquinoleines F-Alkyl Substituees, J. Fluor. Chem., 1979, 13(4), 279–296, DOI:10.1016/S0022-1139(00)82079-5.
- Y. Pan, J. Li, Z. Li, F. Huang, X. Ma, W. Jiao and H. Shao, An Efficient and Facile Strategy for Trifluoromethylation and Perfluoroalkylation of Isoquinolines and Heteroarenes, Chem. Commun., 2020, 56(56), 7813–7816, 10.1039/D0CC00963F.
- C. Fan, J. Song, G. Qiu, G. Liu and J. Wu, Generation of 1-(Trifluoromethyl)Isoquinolines via a Copper-Catalyzed Reaction of Isoquinoline-N-Oxide with Togni Reagent, Org. Chem. Front., 2014, 1(8), 924–928, 10.1039/C4QO00173G.
- D. E. Stephens, G. Chavez, M. Valdes, M. Dovalina, H. D. Arman and O. V. Larionov, Synthetic and Mechanistic Aspects of the Regioselective Base-Mediated Reaction of Perfluoroalkyl- and Perfluoroarylsilanes with Heterocyclic N-Oxides, Org. Biomol. Chem., 2014, 12(32), 6190–6199, 10.1039/C4OB01088D.
- M. Chen and S. L. Buchwald, Rapid and Efficient Trifluoromethylation of Aromatic and Heteroaromatic Compounds Using Potassium Trifluoroacetate Enabled by a Flow System, Angew. Chem., Int. Ed., 2013, 52(44), 11628–11631, DOI:10.1002/anie.201306094.
- Z. Gonda, S. Kovács, C. Wéber, T. Gáti, A. Mészáros, A. Kotschy and Z. Novák, Efficient Copper-Catalyzed Trifluoromethylation of Aromatic and Heteroaromatic Iodides: The Beneficial Anchoring Effect of Borates, Org. Lett., 2014, 16(16), 4268–4271, DOI:10.1021/ol501967c.
- T. Uchikura, N. Kamiyama, T. Ishikawa and T. Akiyama, Catalytic Trifluoromethylation of Iodoarenes by Use of 2-Trifluoromethylated Benzimidazoline as Trifluoromethylating Reagent, Beilstein J. Org. Chem., 2020, 16(1), 2442–2447, DOI:10.3762/bjoc.16.198.
- Y. Liu, X. Shao, P. Zhang, L. Lu and Q. Shen, Trifluoromethyl-Substituted Sulfonium Ylide: Rh-Catalyzed Carbenoid Addition to Trifluoromethylthioether, Org. Lett., 2015, 17(11), 2752–2755, DOI:10.1021/acs.orglett.5b01170.
- K. Aikawa, H. Serizawa, K. Ishii and K. Mikami, Palladium-Catalyzed Negishi Cross-Coupling Reaction of Aryl Halides with (Difluoromethyl)Zinc Reagent, Org. Lett., 2016, 18(15), 3690–3693, DOI:10.1021/acs.orglett.6b01734.
- B. Zhang and A. Studer, 1-Trifluoromethylated Isoquinolines via Radical Trifluoromethylation of Isonitriles, Org. Biomol. Chem., 2014, 12(48), 9895–9898, 10.1039/C4OB02145B.
- L. Wang and A. Studer, 1-Trifluoromethylisoquinolines from α-Benzylated Tosylmethyl Isocyanide Derivatives in a Modular Approach, Org. Lett., 2017, 19(20), 5701–5704, DOI:10.1021/acs.orglett.7b02882.
- Y. Liu, K. Zhang, W. Jiang, Y. Yang, Y. Jiang, X. Liu, Y. Xie, J. Wu, J. Cai and X.-H. Xu, Synthesis of 1-Difluoroalkylated Isoquinolines via Palladium-Catalyzed Radical Cascade Difluoroalkylation–Cyclization of Vinyl Isocyanides with Bromodifluoroacetic Derivatives, Chem. – Asian J., 2017, 12(5), 568–576, DOI:10.1002/asia.201601645.
- F. Ding, Y. Jiang, K. Lin and L. Shi, Tandem Radical Cyclization for the Construction of 1-Difluoroalkylated Isoquinolines via Cu Catalyzed and Visible Light-Promoted Pathways, Org. Biomol. Chem., 2018, 16(11), 1812–1815, 10.1039/C8OB00232K.
- Y. Wang, Z. Cao, Q. He, X. Huang, J. Liu, H. Neumann, G. Chen and M. Beller, Activation of Perfluoroalkyl Iodides by Anions: Extending the Scope of Halogen Bond Activation to C(Sp3)–H Amidation, C(Sp2)–H Iodination, and Perfluoroalkylation Reactions, Chem. Sci., 2023, 14(7), 1732–1741, 10.1039/D2SC06145G.
- R. Ruzziconi and F. Buonerba, Recent Advances in the Synthesis of Regioselectively Fluorinated Homo- and Heterocyclic Compounds by Complementary Cyclization Methods, J. Fluor. Chem., 2013, 152, 12–28, DOI:10.1016/j.jfluchem.2013.05.001.
- Z. E. Blastik, S. Voltrová, V. Matoušek, B. Jurásek, D. W. Manley, B. Klepetářová and P. Beier, Azidoperfluoroalkanes: Synthesis and Application in Copper(I)-Catalyzed Azide-Alkyne Cycloaddition, Angew. Chem., Int. Ed., 2017, 56, 346–349, DOI:10.1002/anie.201609715.
- S. Voltrová, I. Putovný, V. Matoušek, B. Klepetářová and P. Beier, Reintroducing Azidodifluoromethane: Synthesis, Stability and [3 + 2] Cycloadditions, Eur. J. Org. Chem., 2018, 5087–5090, DOI:10.1002/ejoc.201800650.
- E. Shaitanova, V. Matoušek, T. Herentin, M. Adamec, R. Matyáš, B. Klepetářová and P. Beier, Synthesis and Cycloaddition Reactions of 1-Azido-1,1,2,2-tetrafluoroethane, J. Org. Chem., 2023, 88, 14969–14977, DOI:10.1021/acs.joc.3c01346.
- M. Ziabko, B. Klepetářová and P. Beier, Synthesis of Azidodifluoromethyl Phenyl Sulfone and Its Use as a Synthetic Equivalent of the Azidodifluoromethyl Anion, J. Org. Chem., 2023, 88, 6939–6946, DOI:10.1021/acs.joc.3c00256.
- A. Kubíčková, A. Markos, S. Voltrová, A. Marková, J. Filgas, B. Klepetářová, P. Slavíček and P. Beier, Aza-Wolff Rearrangement of N-Fluoroalkyl Triazoles to Ketenimines, Org. Chem. Front., 2023, 10(13), 3201–3206, 10.1039/D3QO00618B.
- S. A. Lermontov, S. V. Shkavrov, A. N. Pushin and V. V. Tkachev, Unusual Fluorination of Diphenylacetylene with Methyl 3-Azidotetrafluoropropionate, Russ. J. Gen. Chem., 2002, 72(8), 1289–1290, DOI:10.1023/A:1020804502390.
- P. Molina, M. Alajarín, A. Vidal, J. Feneau-Dupont and J. P. Declercq, One-Pot Preparation of Derivatives of the Unknown Fluoreno[2,3,4-i,j]Isoquinoline Ring from Conjugated Ketenimines by a Consecutive Electrocyclic Ring-Closure/Claisen Rearrangement/Intramolecular Diels–Alder Cycloaddition/Double Aromatization Process, J. Chem. Soc., Chem. Commun., 1990,(no. 11), 829–831, 10.1039/C39900000829.
- P. Molina, M. Alajarin, A. Vidal, J. Fenau-Dupont and J. P. Declerq, Domino Reactions. One-Pot Preparation of Fluoreno[2,3,4-ij]Isoquinoline Derivatives from Conjugated Ketene Imines, J. Org. Chem., 1991, 56(12), 4008–4016, DOI:10.1021/jo00012a039.
- P. Molina, A. Vidal and I. Barquero, Regiospecific Electrocyclization of β-Arylvinyl Ketenimines. Formal Syntheses of the Alkaloid from Marine Origin Aaptamine, Synthesis, 1996,(10), 1199–1202, DOI:10.1055/s-1996-4367.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2336080–2336082. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qo00713a |
| This journal is © the Partner Organisations 2024 |