
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
NBN/BNB-doped phenalenyl homo- and heterodyads: structural uniformity but optoelectronic diversity†
Alexander S.
Scholz
a,
Thomas
Froitzheim
 *b,
Michael
Bolte
a,
Hans-Wolfram
Lerner
a,
Jan-M.
Mewes
b and
Matthias
Wagner
*a
*b,
Michael
Bolte
a,
Hans-Wolfram
Lerner
a,
Jan-M.
Mewes
b and
Matthias
Wagner
*a
aInstitut für Anorganische und Analytische Chemie, Goethe-Universität Frankfurt am Main, Max-von-Laue-Straße 7, 60438 Frankfurt am Main, Germany. E-mail: matthias.wagner@chemie.uni-frankfurt.de
bMulliken Center for Theoretical Chemistry, Institut für Physikalische und Theoretische Chemie, Rheinische Friedrich Wilhelms-Universität Bonn, Beringstr. 4, 53115 Bonn, Germany. E-mail: th.froitzheim@thch.uni-bonn.de
First published on 9th April 2024
Abstract
Phenalenyl anions and cations are fundamentally interesting but highly reactive complementary species. One way to tame this reactivity and prevent comproportionation as soon as the two species are allowed to interact is by switching to the isosteric NBN- or BNB-containing heterocycles. Herein, we show that the heteroatom-doped pair preserves to a certain extent the desirable complementarity of the original carbonaceous molecules. To this end, we synthesized a series of 1,4-phenylene-bridged (NBN)2- or (BNB)2-homodyads and (NBN,BNB)-heterodyads. The homodyads are accessible via treatment of a 1,4-diborylated benzene with a 1,8-diaminonaphthalene or of 1,4-diaminobenzene with 1,8-naphthalenediyl-bridged diborane(6), respectively. The heterodyads were prepared from the same diborane(6) and an NBN-phenalenyl with B-bonded p-NH2-aryl substituent. All products were characterized by NMR spectroscopy, X-ray crystallography, cyclic voltammetry, UV/vis absorption and emission spectroscopy, as well as state-of-the-art quantum-chemical calculations. The heterodyads undergo both oxidation and reduction and thus qualify as ambipolar compounds. Any intramolecular NBN-to-BNB charge-transfer (CT) emission is negligible on conformational grounds. In contrast, an appreciable aggregation-induced green emission is achieved by adding H2O to THF solutions of the heterodyads and is presumably caused by intermolecular CT between head-to-tail aligned molecules upon aggregation.
Introduction
Polycyclic aromatic hydrocarbons (PAHs) are an important compound class for the development of novel organic optoelectronic materials.1 One reason is that PAHs can be derivatized in various ways to generate a wide range of properties. A particularly powerful derivatization mode is the selective replacement of individual carbon atoms within the π-electron system by other p-block elements, especially boron and nitrogen (“B,N-doping” to obtain “B,N-PAHs”).2 Depending on the number and positioning of the dopant heteroatoms, the electronic structures of the resulting B,N-PAHs are influenced in different ways, while their molecular structures remain largely unchanged compared to those of the carbonaceous parent compounds.3 With this in mind, we recently set out to prepare largely isostructural B,N-PAHs with complementary electron-donor and -acceptor characteristics. As parent scaffold, we selected the triangular (D3h), odd-alternant hydrocarbon phenalenyl, which consists of three mutually annulated benzene rings and has a fully conjugated system of 13 π-electrons.4 The neutral radical has a nonbonding molecular orbital and can therefore, in principle, undergo redox transitions to form an antagonistic anion/cation couple (Fig. 1, I/II).5–7 Since an N or B atom has one electron more or fewer, respectively, than a C atom, the NBN-doped phenalenyl III8,9 can be considered a neutral equivalent of the phenalenyl anion, while the BNB-doped species IV10,11 is an isoster of the phenalenyl cation (Fig. 1). In a previous publication, we have already shown that equimolar co-precipitates of III- and IV-type compounds can exhibit photoluminescence properties not seen in the individual components; a thorough quantum-chemical study revealed an intermolecular III-to-IV charge transfer (CT) as the origin of the observed aggregation-induced emission.12 Herein, we now report the synthesis of III- and IV-based, 1,4-phenylene-bridged donor–donor (III,III), acceptor–acceptor (IV,IV), and donor–acceptor dyads (III,IV). We will show a truly ambipolar character of the donor–acceptor species and remarkably different optical properties of the three different kinds of dyads.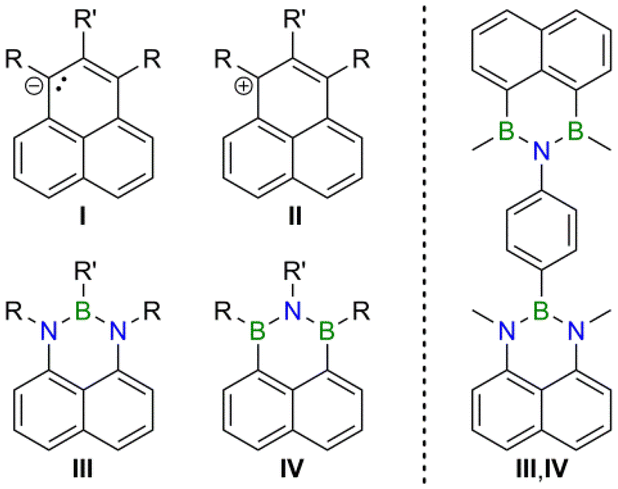 | ||
| Fig. 1 Carbonaceous phenalenyl anions (I) and cations (II), their respective NBN- (III) and BNB-doped (IV) analogs, and the corresponding 1,4-phenylene-bridged donor–acceptor dyad (III,IV). | ||
Results and discussion
To prove the claim that B,N-doping indeed provides access to structurally uniform but electronically diverse compounds, we prepared the series of (NBN)2- (1Me), (BNB)2- (2Me), and (NBN,BNB)-phenalenyls (3Me,Me, Scheme 1). Compound 1Me is accessible from 1,8-diaminonaphthalene A and the 1,4-diborylated benzene Bvia established coordination chemistry.13 Aminolysis of 2 equiv. of the 1,8-naphthalenediyl-bridged diborane(6) C with 1 equiv. of 1,4-diaminobenzene D furnishes the parent (BNB)2-phenalenyl 2H, which can be converted to its derivatives 2Me and 2Mes by treatment with MeMgBr and MesMgBr (Mes = mesityl), respectively. This fourfold nucleophilic substitution reaction is remarkably straightforward considering that H− ions act as the leaving groups. LiH elimination is key to the introduction of the 4-aminophenyl substituent into the NBN-phenalenyl E to give 5 or 6 (R′ = H, Me). After deprotection of the NH2 group, the BNB functionality is again assembled via an aminolysis step. Reaction of the resulting BH intermediate with MeMgBr or MesMgBr affords the donor–acceptor dyads 3Me,Me and 3Me,Mes, respectively. Compound 4Me,Mes, which contains a sterically more demanding doubly methylated phenylene linker, was prepared in an analogous manner.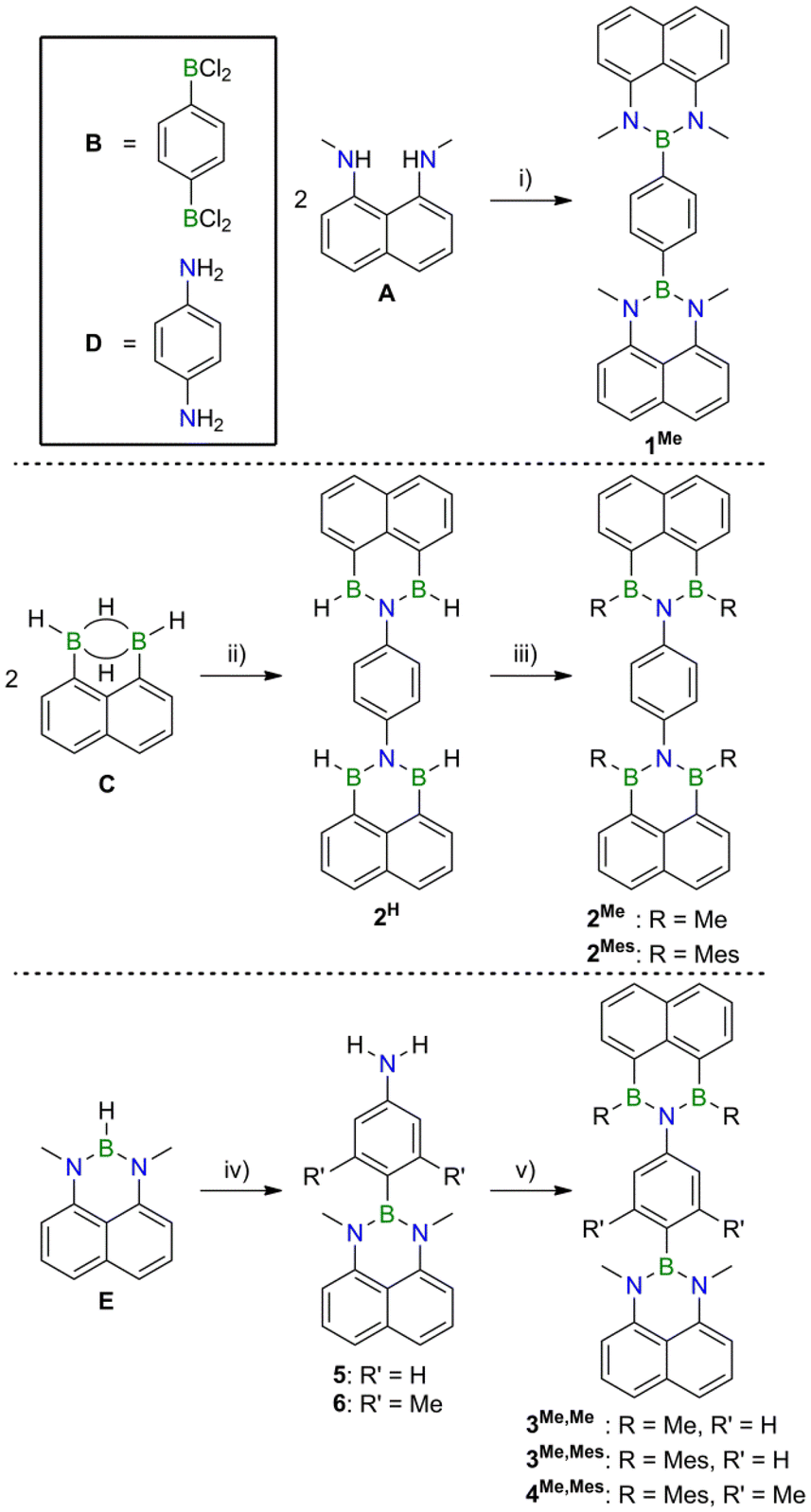 | ||
| Scheme 1 Syntheses of compounds 1Me–6. Reagents and conditions: (i) 1 equiv. of B and 4 equiv. of i-Pr2NEt, toluene, −78 °C to room temperature; yield: 80%. (ii) 1 equiv. of D, CH2Cl2, heated under reflux; yield: 78%. (iii) R = Me: 4 equiv. of MeMgBr, Et2O, 0 °C to room temperature; yield: 76%. R = Mes: 4.3 equiv. of MesMgBr, THF, −78 °C to room temperature; yield: 55%. (iv) R′ = H: 1. 1 equiv. of 4-(Me3Si)2N-C6H4-Li, THF, −78 °C to room temperature; 2. 1 vol% conc. aqueous HCl in MeOH, THF, room temperature; yield: 65%. R′ = Me: 1. 1 equiv. of 2,6-Me2-4-(Me3Si)2N-C6H2-Li, THF, −78 °C to room temperature; 2. 1 vol% conc. aqueous HCl in MeOH, THF, room temperature; yield: 47%. (v) R = Me, R′ = H: 1. 1 equiv. of C, C6H6, room temperature; 2. 2.2 equiv. of MeMgBr, THF, 0 °C to room temperature; yield: 68%. R = Mes, R′ = H: 1 equiv. of C, C6H6, room temperature; 2. 2 equiv. of MesMgBr, THF, room temperature; yield: 75%. R = Mes, R′ = Me: 1 equiv. of C, C6H6, room temperature; 2. 2.3 equiv. of MesMgBr, THF, 0 °C to room temperature; yield: 53%. | ||
NMR-spectroscopic and crystallographic characterization
The 1H NMR spectrum of the (NBN)2-dyad 1Me shows one singlet for the NMe groups, two doublets as well as one virtual triplet for the 1,8-naphthalenediyl moieties, and one singlet for the four equivalent protons of the 1,4-phenylene bridge. A qualitatively similar spectrum is obtained for the (BNB)2 compound 2Me. The dyad 3Me,Me is characterized by the proton-signal patterns of the NBN- and BNB-phenalenyl moieties with equal integral values; the 1,4-phenylene protons give rise to an AA′BB′ spin system. The NMR spectra of all other products were also fully assigned and are consistent with the proposed molecular structures. With respect to electronic structure elucidation, the following feature from the 13C{1H} NMR spectra is particularly revealing: in all cases, the NBN-phenalenyl C-nuclei ortho and para to the N atoms are significantly better shielded than the corresponding C-nuclei within the BNB-phenalenyl fragments (cf.1Me: o-C: 103.8/120.7 ppm, p-C: 118.4 ppm vs.2Me: 138.2/136.9 ppm, 133.0 ppm). Given that the magnetic shielding of a C(sp2) atom within a delocalized π-electron system increases with increasing electron density at this position,14 NMR spectroscopy provides experimental support for the classification of NBN- and BNB-phenalenyls as electron donors and acceptors, respectively.The molecular structures of all products 1Me·CH2Cl2, 2H, 2Me·CH2Cl2, 2Mes·C6H12, 3Me,Me·THF, 3Me,Mes·C6H14, and 4Me,Mes were confirmed by X-ray crystallography (all solid-state structures, including B, 5, and 6, are shown in Fig. S56–S67†). We refrain from a detailed discussion of bond lengths and bond angles, as these are very similar to those of corresponding published heterocycles.12 One result, however, is noteworthy: in the crystal lattice of 3Me,Me·THF, the ambipolar mixed (NBN,BNB)-dyad is located on a crystallographic mirror plane, rendering the NBN and BNB halves of the molecules indistinguishable. We take this as confirmation of our basic claim that NBN- and BNB-phenalenyls have essentially identical molecular scaffolds despite their different electron-donor and -acceptor character. For an assessment of possible intramolecular charge-transfer (CT) interactions between the NBN- and BNB-phenalenyl residues (see below), one should also consider the dihedral angles between these heteroatomic units and the respective 1,4-phenylene bridges (C6) in the ambipolar species 3Me,Me (NBN//C6 = BNB//C6 = 77.60(6)°), 3Me,Mes (NBN//C6 = 62.82(11)°, BNB//C6 = 62.58(10)°), and 4Me,Mes (NBN//C6 = 79.44(9)°, BNB//C6 = 74.94(11)°). Although the molecular conformations in solution are likely different from those in the solid state (see the ESI† for a comparison to the calculated structures), it is safe to conclude that (i) dihedral angles in the range of 60°–80° are accessible and (ii) dihedral angles of about 80° are assumed both with and without bulky Me substituents at the 1,4-phenylene bridge (but will certainly be favored in the former case).
Optoelectronic properties
To gain further information about their electronic structures, the NBN- and BNB-phenalenyls were investigated by cyclic voltammetry (CV; [n-Bu4N][PF6], vs. FcH/FcH+; Table 1). As expected, the NBN-containing species undergo electrochemical oxidation with peak potentials in the range of Epa = 0.08 to 0.39 V, but in most cases the corresponding transitions are not (fully) reversible (Fig. 2a and S53–S54†). The BNB-containing congeners are electrochemically better behaved and undergo reduction at half-wave potentials of E1/2 = −2.33 to −2.43 V. Of the (BNB)2-dyads, 2Me shows one reversible reduction event at E1/2 = −2.43 V (Fig. S51†), while the more sterically protected 2Mes gives rise to two barely resolved redox waves at approx. E1/2 = −2.33 V (Fig. 2b). In any case, this points towards an only weak electronic communication between the two BNB-phenalenyl halves in 2Mes. In terms of electrochemistry, our best-performing compound is 4Me,Mes, as it undergoes both reversible oxidation (E1/2 = 0.22 V) and reversible reduction (E1/2 = −2.35 V, Fig. 2c).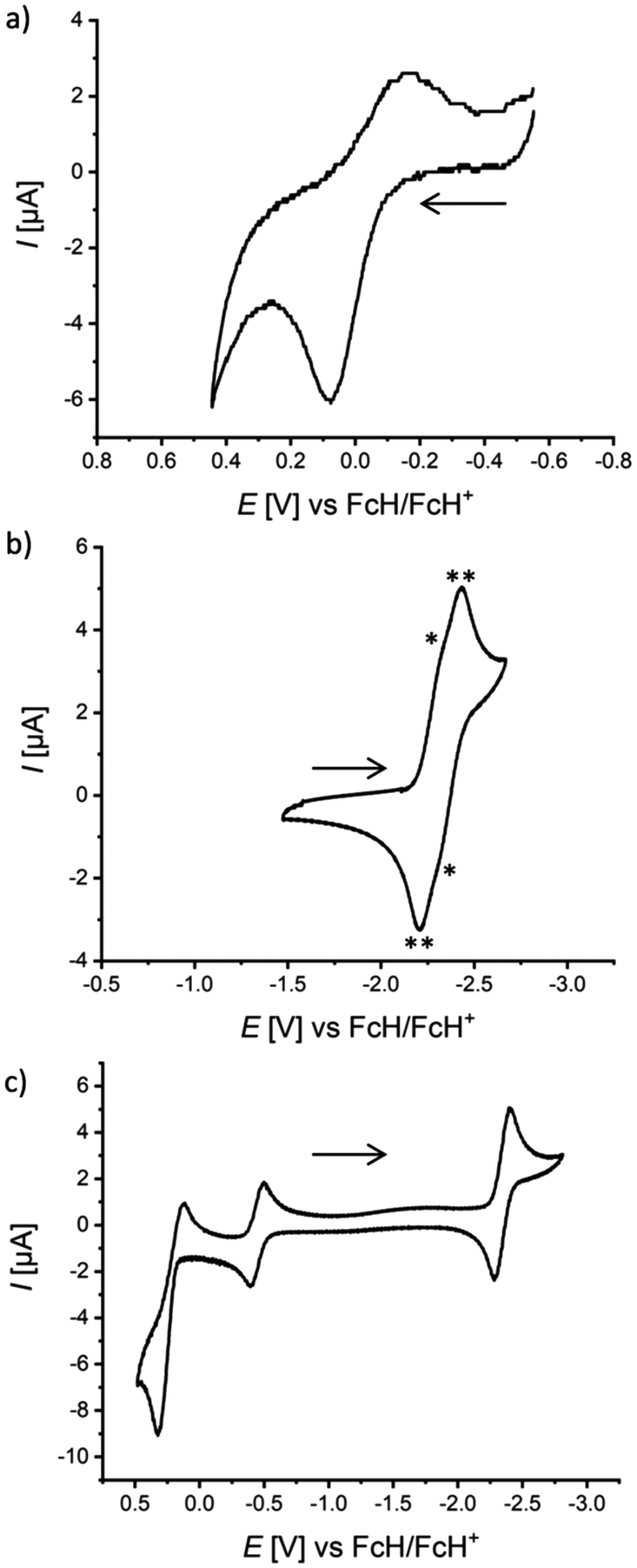 | ||
| Fig. 2 Cyclic voltammograms of (a) 1Me, (b) 2Mes, and (c) 4Me,Mes in THF (against the FcH/FcH+ redox couple, room temperature, [n-Bu4N][PF6] (0.1 M), scan rate 200 mV s−1). The cyclic voltammogram of 2Mes in (b) shows two partly resolved reduction waves with very similar E1/2 values (marked with “*” and “**”). The redox wave at −0.446 V in (c) corresponds to the Fc*H/Fc*H+ redox couple (Fc*H: decamethylferrocene; internal standard). | ||
| E 1/2 [V] | E pa [V] | E pc [V] |
λ
abs![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b [nm] b [nm] |
λ
emd [nm] |
Φ
PLe [%] |
|
|---|---|---|---|---|---|---|
| a The cyclic voltammogram of 2Mes shows two partly resolved reduction waves with very similar E1/2 values (cf. “*” and “**” in Fig. 2b). b Most intense maximum of each absorption spectrum in C6H12. c For solubility and/or comparability reasons, this measurement was performed in CH2Cl2. d In case of vibrationally structured emission bands (C6H12), the most intense maximum is given. λex [nm] = 330 (2Me), 335 (2Mes), 327 (3Me,Mes), and 328 (4Me,Mes). e Quantum yields were determined by using a calibrated integrating sphere. | ||||||
| 1Me | — | 0.08 | — | 333c | — | — |
| 2Me | −2.43 | — | −2.52 | 316c | 371c | 2c |
| 2Mes | ca. −2.33a | — | −2.43 | 342 | 434c | 22c |
| 3Me,Me | −2.43 | 0.39 | −2.55 | 332c | — | — |
| 3Me,Mes | −2.35 | 0.33 | −2.47 | 327 | 395 | 6 |
| 4Me,Mes | 0.22, −2.35 | 0.32 | −2.40 | 328 | 392 | 3 |
The electrochemical measurements align well with the computed adiabatic redox potentials at the ωB97X-D3/ma-def2-QZVPP/SMD[THF]15–22 level of theory for fully relaxed r2SCAN-3c/SMD[THF]23 structures of the neutral, anionic, and cationic species (against the FcH/FcH+ redox couple, see the ESI† for computational details):47,48 for compounds containing the NBN-motif, theory predicts first oxidation potentials between 0.26 V and 0.37 V, while the first reduction potentials of the BNB-containing congeners range between −2.49 V and −2.69 V (see Table S15†). Deviations between the experimental and theoretical redox potentials therefore remain within 0.3 V, as is expected from the limitations of DFT and implicit solvation for radical and charged species.20,24–26 The inspection of the highest occupied (HOMO) and lowest unoccupied molecular orbitals (LUMO) (see Fig. S88–S93†) confirms that electrochemical activity is primarily localized on the NBN- and BNB-phenalenyl units and to a much lesser extent on the 1,4-phenylene bridges. Although the heterocyclic moieties belonging to the same molecule appear largely independent from each other (due to the orthogonal conformation of the bridge), the predicted reduction or oxidation of the second subunit within a given homodyad is less favorable (min. ± 0.75 V). The only notable exception is the reduction of the second BNB-subunit of compound 2Mes (see Fig. S90†), which appears only −0.13 V more cathodically shifted than the first (E2.red = −2.68 V vs. E1.red = −2.55 V). This theoretical result again agrees well with the observation of the two partly resolved reduction waves at approx. −2.33 V in the CV of 2Mes.
The propensity of mixed (NBN,BNB)-dyads to experience charge-transfer (CT) transitions between the different heterocycles in the excited state was investigated by UV/vis absorption and emission spectroscopy. We first consider the three structurally closely related species 1Me, 2Me, and 3Me,Me in CH2Cl2 solutions (Fig. 3a and Table 1). As to be anticipated, no broad, bathochromic CT bands are visible in the absorption spectra of the two homodyads 1Me and 2Me. Moreover, either no (1Me) or only very weak luminescence is detectable (2Me; λem = 371 nm, Fig. 3b), suggesting that any local emission from the individual heterocyclic moieties can be neglected. Importantly, a switch from 2Me to 2Mes shifts the emission bathochromically (λem = 434 nm, Fig. 3b) and increases the fluorescence quantum yield by one order of magnitude (from ΦPL = 2 to 22%; Table 1). According to quantum-chemical calculations (see below), the origin of this emission is mainly a CT from the mesityl substituents to the BNB-phenalenyl moiety.12,27 The heterodyad 3Me,Me gives rise to a low-intensity absorption band between 400 and 550 nm that is most likely attributable to a CT transition from the NBN- to the BNB-phenalenyl fragment; 3Me,Me is non-emissive (see below for computed oscillator strengths and electron–hole plots).
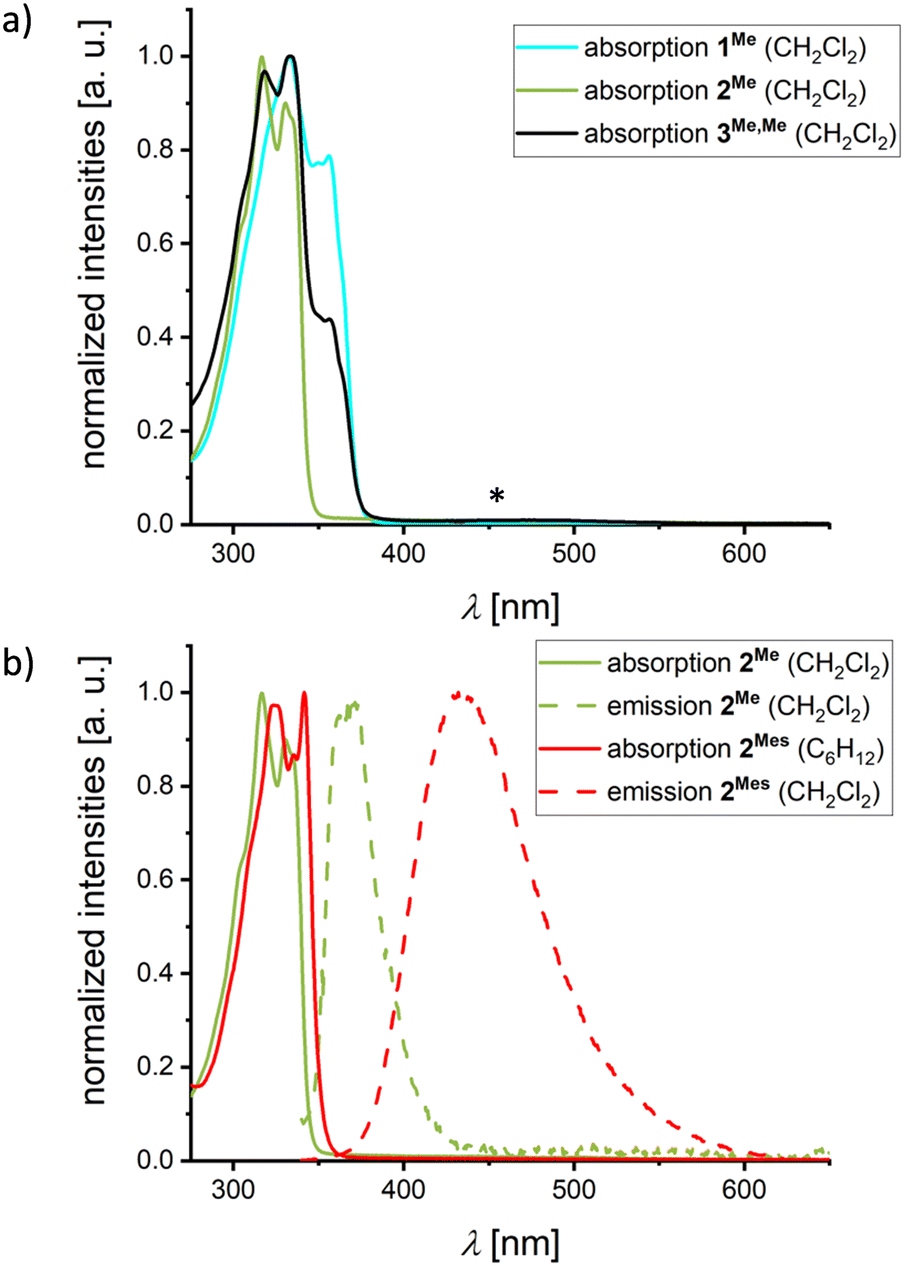 | ||
| Fig. 3 (a) Normalized UV/vis absorption spectra of 1Me, 2Me, and 3Me,Me in CH2Cl2. The “*” symbol marks a low-intensity absorption maximum of 3Me,Me (an enlarged plot of this maximum is shown in Fig. S40†). (b) Normalized UV/vis absorption and emission spectra of 2Me (λex = 330 nm) and 2Mes (λex = 335 nm) in the specified solvents. | ||
To gain deeper insight into the low-lying excited states involved in absorption and emission, we performed time-dependent DFT (TD-DFT)28 calculations in the Tamm-Dancoff approximation (TDA)29 with the dispersion corrected,16,30,31 optimally-tuned,32–34 range-separated hybrid functional OT-LRC-ωPBEh-D4.35,47,48 For vertical absorption energies (Ecabs), we combined TD-DFT with non-equilibrium perturbative ptSS-PCM36,37 solvation (CH2Cl2) at the r2SCAN-3c/SMD optimized ground state geometries. Conversely, for vertical emission energies (Ecem), we used iterative SS-PCM equilibrium solvation38–40 at ΔSCF/UKS/PCM41 optimized excited state geometries. This specific combination of ΔSCF/UKS and TD-DFT with continuum solvation mitigates some inherent shortcomings of TD-DFT for polar CT states in solution (see the ESI† for computational details).12,42–44
Due to their large oscillator strengths (fosc), local excitations (LE) on one or both of the NBN/BNB-moieties are responsible for absorption. Moreover, the orthogonal arrangement of the NBN/BNB-fragments relative to the respective 1,4-phenylene bridge causes all intramolecular CT states to be dark (see the ESI† for a detailed discussion, including difference density plots and absorption energies calculated in different solvents). In the largely isostructural compounds 1Me, 2Me, and 3Me,Me, two states dominate the low-energy absorption: (i) a lower LE state on the NBN-fragments (1Me/3Me,Me; Ecabs = 3.99 eV, fosc = 0.052–0.104), and (ii) a strongly absorbing mixed LE state simultaneously on both NBN- and/or BNB-fragments (1Me/2Me/3Me,Me; Ecabs = 4.19 eV, fosc = 0.672–0.705). The more hypsochromic onset of absorption observed for 2Me compared to 1Me/3Me,Me is therefore likely attributable to the lack of an NBN moiety in 2Me (see Fig. 3a).
Regarding the emission of 1Me, 2Me, and 3Me,Me, structural and solvent relaxation give rise to the following low-lying polar CT states (see Fig. 4): (i) a dark CT from the NBN-phenalenyl to the 1,4-phenylene bridge in 1Me (Ecem = 2.51 eV, fosc = 0.008), (ii) a weakly allowed CT from the 1,4-phenylene bridge to the BNB unit in 2Me (Ecem = 2.85 eV, fosc = 0.010), and (iii) a dipole-forbidden CT with vanishing transition density from the NBN to the BNB unit in 3Me,Me (Ecem = 1.68 eV, fosc = 0.000). While the CT states of both type (i) and, in particular, type (iii) lie substantially below the respective LE (ΔE12 = 0.63 eV and 1.31 eV),45 the gap for 2Me is significantly smaller (0.38 eV). Considering the known artificial destabilization of LE states in NBN/BNB-phenalenyls by TD-DFT,12 the state ordering of 2Me falls within the error margins of our computational protocol. Consequently, 2Me might retain some population for emission from the bright local S2 state (cf.Fig. 3b and 4), while 1Me and 3Me,Me decay purely non-radiatively through the dark CT states.
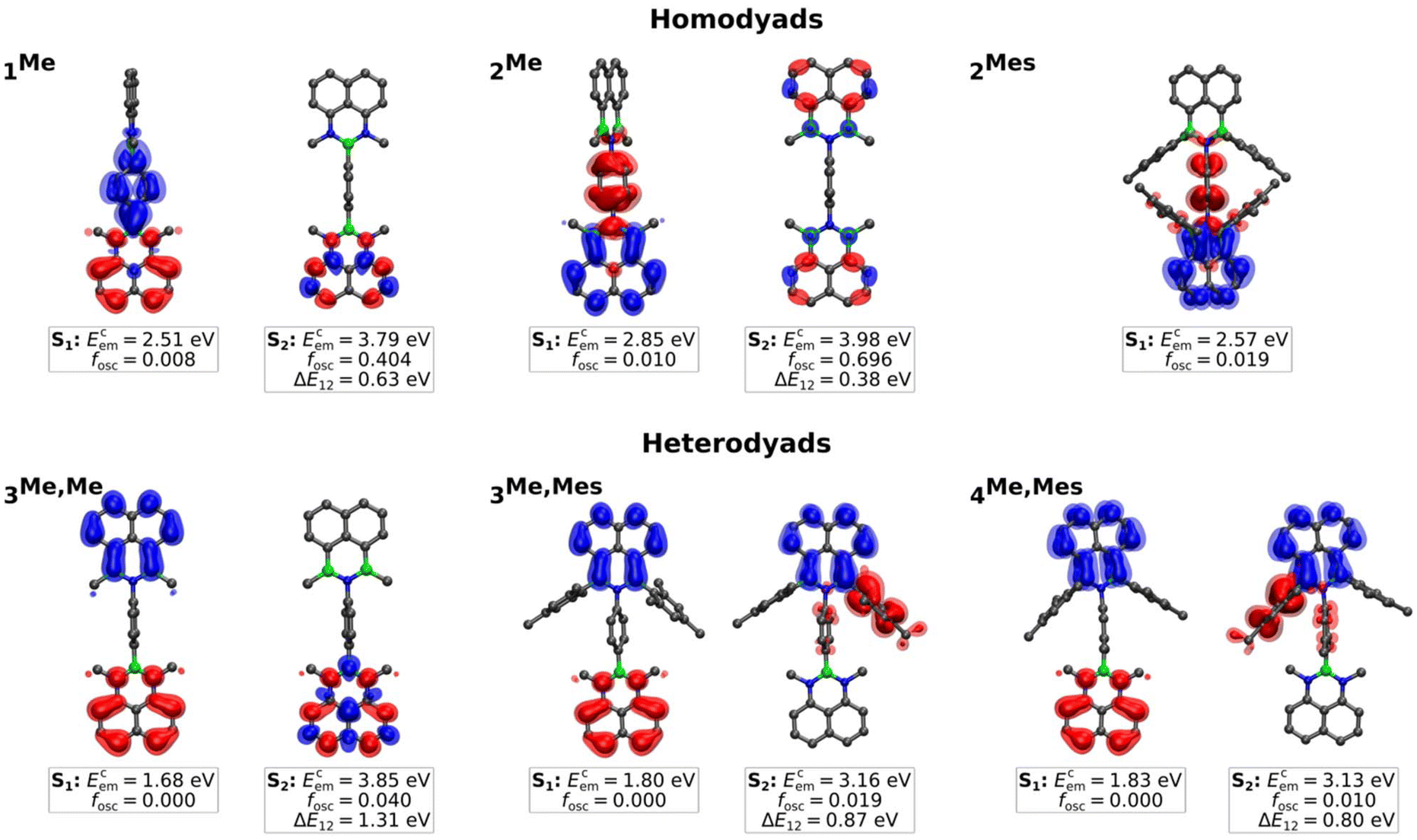 | ||
| Fig. 4 Isosurface plots of the unrelaxed difference density (hole [red] and electron [blue]), calculated emission energies (Ecem), and oscillator strengths (fosc) in CH2Cl2 for the first two excited states of the homo- (1Me, 2Me, 2Mes) and heterodyads (3Me,Me, 3Me,Mes, 4Me,Mes) at the TDA-OT-LRC-ωPBEh-D4/def2-TZVPP/SS-PCM[CH2Cl2]//ΔSCF/UKS/PCM level of theory. 0.997 [opaque] and 0.999 [translucent] were used as isovalues.45 | ||
The introduction of Mes substituents in 2Mes lowers the CT state and increases its oscillator strength by mixing donor contributions from the 1,4-phenylene bridge and the Mes groups. Although the oscillator strength remains too low to significantly affect the absorption spectrum of 2Mes (Ecabs = 3.78 eV, fosc = 0.010), relaxation of both the molecular structure in the excited state and the solvent shell leads the CT to be the relevant state for emission (Ecem = 2.57 eV, fosc = 0.019), which explains the broad, solvatochromically shifted band observed for 2Mes (Fig. 3b and S36†).
Having studied the extent of intramolecular optoelectronic communication between the peripheral heterocycles in 1Me, 2Me, and 3Me,Me, the next step was to investigate possible intermolecular interactions. A viable method for producing the required intimately mixed aggregates is precipitation of the individual component molecules from THF by adding H2O.12 Since 3Me,Me lacks long-term stability against hydrolysis, it was necessary to use its Mes-shielded derivative 3Me,Mes instead. As a reference system, in which the 1,4-phenylene bridge is locked in a largely orthogonal conformation, 4Me,Mes was also included in this study. In C6H12 solution, 3Me,Mes and 4Me,Mes have emission maxima at 395 and 392 nm with ΦPL values of 6 and 3%, respectively (Fig. 5a and S46;†Tables 1 and S1†); in THF, the quantum yields drop to 1 and 0%. Calculations confirm a weakly allowed Mes-to-BNB CT emission in C6H12 (Ecem = 3.60–3.64 eV, fosc = 0.014–0.030). In polar solvents, a dipole-forbidden NBN-to-BNB CT state is dominant and effectively quenches any luminescence (Ecem = 1.87–1.91 eV, fosc = 0.000; in THF). Upon addition of H2O to their THF solutions, the emission behavior of 3Me,Mes and 4Me,Mes changes dramatically: 3Me,Mes shows a continuous decrease of the original blue emission46 and a concomitant increase of a bathochromically shifted band at λem = 531 nm with ΦPL = 6% (95% H2O fraction, a further increase of the H2O content lowers the emission intensity again; Fig. 5b). For 4Me,Mes the aggregation-induced emission is even more pronounced with ΦPL = 12% (λem = 509 nm, 95% H2O fraction; Fig. S47†). An in-depth quantum chemical investigation of intermolecular interactions in aggregates of 3Me,Mes or 4Me,Mes is not feasible due to prohibitively high computational costs. Instead, we have to refer to previous studies on intermolecular CT in mixtures of individual NBN- with BNB-phenalenyls, where the heteromolecular aggregates emit about 0.7 eV bathochromically shifted compared to the individual constituents. For 3Me,Mes, the energy difference between its emission in THF solution (λem = 370 nm) and in the aggregated state (λem = 531 nm) amounts to a similar value of 1.0 eV and can likely be traced back to head-to-tail aggregates of 3Me,Mes resulting in intermolecular NBN-to-BNB CT.
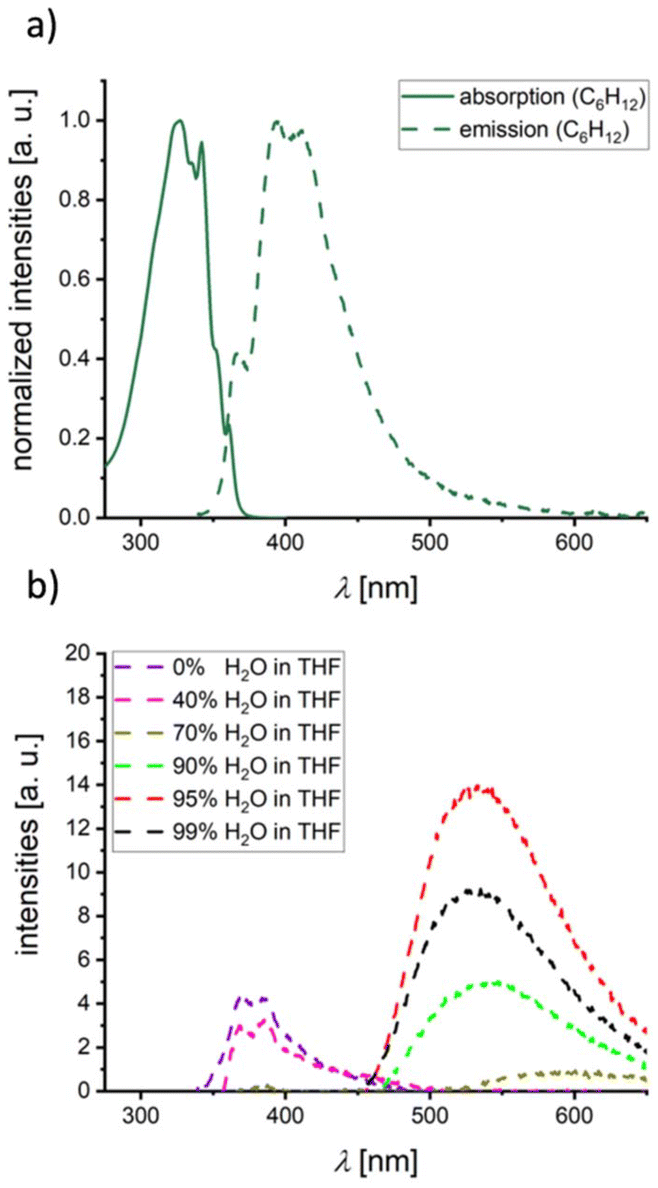 | ||
| Fig. 5 (a) Normalized UV/vis absorption and emission spectra of 3Me,Mes (C6H12, λex = 327 nm). (b) Emission spectra of 3Me,Mes (λex = 327 nm) with differing H2O fractions from 0% to 99% H2O in THF. For the complete picture including all measured H2O fractions, see Fig. S43 in the ESI.† | ||
Conclusions
NBN- and BNB-doped phenalenyls are isosters of phenalenyl anions and cations, respectively. Cyclic voltammetry on 1,4-phenylene-bridged (NBN)2- or (BNB)2-homodyads showed that this formal consideration is also relevant in practice, since the former are only oxidizable and the latter are only reducible within the THF solvent window. Remarkably, the NBN/BNB-heterodyad 4Me,Mes undergoes both oxidation and reduction and can therefore be considered an ambipolar material. We have thus shown that the tool of heteroatom doping is suitable for generating (largely) isostructural species that still differ markedly in their electronic structures. Initially, we had hoped that the heterodyads would also show a bright intramolecular NBN-to-BNB charge-transfer emission. Yet, it turned out that the orthogonal conformation of the 1,4-phenylene linker prevents a pronounced interaction between the heterocyclic moieties and leads to a vanishing transition dipole. Aggregates of the heterodyads, however, show significant intermolecular CT emission, presumably due to head-to-tail alignment in the solid state. In order to also promote an intramolecular CT in the future, we are currently aiming to replace the 1,4-phenylene linker in the heterodyads with an 1,2-ethynediyl bridge.Author contributions
A. S. synthesized and characterized all compounds. T. F. performed all quantum-chemical calculations. M. B. is responsible for the X-ray crystal structure analyses. H.-W. L., J.-M. M., and M. W. supervised the project. The manuscript was written by M. W., A. S., and T. F. and edited by all co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. F. thanks the Fonds der Chemischen Industrie (FCI) for funding through a Kekulé scholarship.References
- Q. Li, Y. Zhang, Z. Xie, Y. Zhen, W. Hu and H. Dong, Polycyclic aromatic hydrocarbon-based organic semiconductors: ring-closing synthesis and optoelectronic properties, J. Mater. Chem. C, 2022, 10, 2411–2430 RSC.
- For reviews regarding heteroatom doping, see: (a) Z. Liu and T. B. Marder, B–N versus C–C: How Similar Are They?, Angew. Chem., Int. Ed., 2007, 47, 242–244 CrossRef; (b) P. G. Campbell, A. J. V. Marwitz and S.-Y. Liu, Recent Advances in Azaborine Chemistry, Angew. Chem., Int. Ed., 2012, 51, 6074–6092 CrossRef CAS; (c) G. Bélanger-Chabot, H. Braunschweig and D. K. Roy, Recent Developments in Azaborinine Chemistry, Eur. J. Inorg. Chem., 2017, 2017, 4353–4368 CrossRef; (d) E. von Grotthuss, A. John, T. Kaese and M. Wagner, Doping Polycyclic Aromatics with Boron for Superior Performance in Materials Science and Catalysis, Asian J. Org. Chem., 2018, 7, 37–53 CrossRef CAS; (e) Z. X. Giustra and S.-Y. Liu, The State of the Art in Azaborine Chemistry: New Synthetic Methods and Applications, J. Am. Chem. Soc., 2018, 140, 1184–1194 CrossRef CAS; (f) H. Helten, Doping the Backbone of π-Conjugated Polymers with Tricoordinate Boron: Synthetic Strategies and Emerging Applications, Chem. – Asian J., 2019, 14, 919–935 CrossRef CAS PubMed; (g) X.-Y. Wang, X. Yao, A. Narita and K. Müllen, Heteroatom-Doped Nanographenes with Structural Precision, Acc. Chem. Res., 2019, 52, 2491–2505 CrossRef CAS; (h) X. Chen, D. Tan and D.-T. Yang, Multiple-boron-nitrogen (multi-BN) doped π-conjugated systems for optoelectronics, J. Mater. Chem. C, 2022, 10, 13499–13532 RSC.
- For selected papers on B,N-PAHs, see: (a) A. J. V. Marwitz, M. H. Matus, L. N. Zakharov, D. A. Dixon and S.-Y. Liu, A Hybrid Organic/Inorganic Benzene, Angew. Chem., Int. Ed., 2009, 48, 973–977 CrossRef CAS; (b) M. Krieg, F. Reicherter, P. Haiss, M. Ströbele, K. Eichele, M.-J. Treanor, R. Schaub and H. F. Bettinger, Construction of an Internally B3N3-Doped Nanographene Molecule, Angew. Chem., Int. Ed., 2015, 54, 8284–8286 CrossRef CAS PubMed; (c) F.-D. Zhuang, J.-M. Han, S. Tang, J.-H. Yang, Q.-R. Chen, J.-Y. Wang and J. Pei, Efficient Modular Synthesis of Substituted Borazaronaphthalene, Organometallics, 2017, 36, 2479–2482 CrossRef CAS; (d) T. Kaehler, M. Bolte, H.-W. Lerner and M. Wagner, Introducing Perylene as a New Member to the Azaborine Family, Angew. Chem., Int. Ed., 2019, 58, 11379–11384 CrossRef CAS; (e) M. Fingerle, S. Stocker and H. F. Bettinger, New Synthesis of a Dibenzoperylene Motif Featuring a Doubly Boron–Nitrogen-Doped Bay Region, Synthesis, 2019, 51, 4147–4152 CrossRef CAS; (f) K. Boknevitz, C. Darrigan, A. Chrostowska and S.-Y. Liu, Cation-π binding ability of BN indole, Chem. Commun., 2020, 56, 3749–3752 RSC; (g) M. Chen, K. S. Unikela, R. Ramalakshmi, B. Li, C. Darrigan, A. Chrostowska and S.-Y. Liu, A BN-Doped Cycloparaphenylene Debuts, Angew. Chem., Int. Ed., 2021, 60, 1556–1560 CrossRef CAS; (h) O. Ouadoudi, T. Kaehler, M. Bolte, H.-W. Lerner and M. Wagner, One tool to bring them all: Au-catalyzed synthesis of B,O- and B,N-doped PAHs from boronic and borinic acids, Chem. Sci., 2021, 12, 5898–5909 RSC; (i) W. Li, C.-Z. Du, X.-Y. Chen, L. Fu, R.-R. Gao, Z.-F. Yao, J.-Y. Wang, W. Hu, J. Pei and X.-Y. Wang, BN–Anthracene for High–Mobility Organic Optoelectronic Materials through Periphery Engineering, Angew. Chem., Int. Ed., 2022, 61, e202201464 CrossRef CAS PubMed.
- J. Ahmed and S. K. Mandal, Phenalenyl Radical: Smallest Polycyclic Odd Alternant Hydrocarbon Present in the Graphene Sheet, Chem. Rev., 2022, 122, 11369–11431 CrossRef CAS PubMed.
- For papers regarding phenanyl radicals, see: (a) S. H. Glarum and J. H. Marshall, ESR of the Perinaphthenyl Radical in a Liquid Crystal, J. Chem. Phys., 1966, 44, 2884–2890 CrossRef CAS; (b) K. Goto, T. Kubo, K. Yamamoto, K. Nakasuji, K. Sato, D. Shiomi, T. Takui, M. Kubota, T. Kobayashi, K. Yakusi and J. Ouyang, A Stable Neutral Hydrocarbon Radical: Synthesis, Crystal Structure, and Physical Properties of 2,5,8-Tri-tert-butyl-phenalenyl, J. Am. Chem. Soc., 1999, 121, 1619–1620 CrossRef CAS; (c) Y. Morita, T. Ohba, N. Haneda, S. Maki, J. Kawai, K. Hatanaka, K. Sato, D. Shiomi, T. Takui and K. Nakasuji, New Persistent Radicals: Synthesis and Electronic Spin Structure of 2,5-Di-tert-butyl-6-Oxophenalenoxyl Derivatives, J. Am. Chem. Soc., 2000, 122, 4825–4826 CrossRef CAS; (d) P. A. Koutentis, Y. Chen, Y. Cao, T. P. Best, M. E. Itkis, L. Beer, R. T. Oakley, A. W. Cordes, C. P. Brock and R. C. Haddon, Perchlorophenalenyl Radical, J. Am. Chem. Soc., 2001, 123, 3864–3871 CrossRef CAS; (e) E. Turco, A. Bernhardt, N. Krane, L. Valenta, R. Fasel, M. Juríček and P. Ruffieux, Observation of the Magnetic Ground State of the Two Smallest Triangular Nanographenes, JACS Au, 2023, 3, 1358–1364 CrossRef CAS; (f) T. Kodama, Y. Hirao and T. Kubo, Synthesis and Properties of a Through-Space Interacting Diradicaloid, Precis. Chem., 2023, 1, 183–191 CrossRef CAS.
- For papers regarding phenalenyl anions, see: (a) V. Boekelheide and C. E. Larrabee, An Investigation of the Preparation and Some Properties of Perinaphthene, J. Am. Chem. Soc., 1950, 72, 1245–1249 CrossRef CAS; (b) R. L. Shannon and R. H. Cox, On the alkali metal reduction of phenalene, Tetrahedron Lett., 1973, 14, 1603–1605 CrossRef.
- For papers regarding phenalenyl cations, see: (a) R. Pettit, The Synthesis and Properties of the Perinaphthenylium Cation, J. Am. Chem. Soc., 1960, 82, 1972–1975 CrossRef CAS; (b) A. Mukherjee, S. C. Sau and S. K. Mandal, Exploring Closed-Shell Cationic Phenalenyl: From Catalysis to Spin Electronics, Acc. Chem. Res., 2017, 50, 1679–1691 CrossRef CAS PubMed; (c) T. Murata, K. Yoshida, S. Suzuki, A. Ueda, S. Nishida, J. Kawai, K. Fukui, K. Nakasuji and Y. Morita, Design and Synthesis of a C3 Symmetrical Phenalenyl Derivative with Three Oxo Groups by Regioselective Deoxygenation/Oxygenation, Org. Lett., 2022, 24, 1033–1037 CrossRef CAS PubMed.
- For studies regarding NBN-phenalenyls, see: (a) Y. Lu, A. Bolag, J.-i. Nishida and Y. Yamashita, Synthesis and characterization of 1,3,2-diazaboroine derivatives for organic thin-film transistor applications, Synth. Met., 2010, 160, 1884–1891 CrossRef CAS; (b) W.-M. Wan, D. Tian, Y.-N. Jing, X.-Y. Zhang, W. Wu, H. Ren and H.-L. Bao, NBN-Doped Conjugated Polycyclic Aromatic Hydrocarbons as an AIEgen Class for Extremely Sensitive Detection of Explosives, Angew. Chem., Int. Ed., 2018, 57, 15510–15516 CrossRef CAS PubMed; (c) H. E. Hackney, M. Paladino, H. Fu and D. G. Hall, Diazaboryl-naphthyl-ketone: A New Scaffold with Bright Fluorescence, Aggregation-Induced Emission, and Application in the Quantitation of Trace Boronic Acids in Drug Intermediates, Chem. – Eur. J., 2020, 26, 14324–14329 CrossRef CAS; (d) Y. Zeng, J. Yang and X. Zheng, Aggregation effects on photophysical properties of NBN-doped polycyclic aromatic hydrocarbons: a theoretical study, Phys. Chem. Chem. Phys., 2021, 23, 23986–23997 RSC.
- For examples of π-extended NBN-aromatics, see: (a) M. Numano, N. Nagami, S. Nakatsuka, T. Katayama, K. Nakajima, S. Tatsumi, N. Yasuda and T. Hatakeyama, Synthesis of Boronate-Based Benzo[fg]tetracene and Benzo[hi]hexacene via Demethylative Direct Borylation, Chem. – Eur. J., 2016, 22, 11574–11577 CrossRef CAS; (b) X. Wang, F. Zhang, K. S. Schellhammer, P. Machata, F. Ortmann, G. Cuniberti, Y. Fu, J. Hunger, R. Tang, A. A. Popov, R. Berger, K. Müllen and X. Feng, Synthesis of NBN-Type Zigzag-Edged Polycyclic Aromatic Hydrocarbons: 1,9-Diaza-9a-boraphenalene as a Structural Motif, J. Am. Chem. Soc., 2016, 138, 11606–11615 CrossRef CAS; (c) D.-T. Yang, T. Nakamura, Z. He, X. Wang, A. Wakamiya, T. Peng and S. Wang, Doping Polycyclic Arenes with Nitrogen–Boron–Nitrogen (NBN) Units, Org. Lett., 2018, 20, 6741–6745 CrossRef CAS; (d) Y. Fu, K. Zhang, E. Dmitrieva, F. Liu, J. Ma, J. J. Weigand, A. A. Popov, R. Berger, W. Pisula, J. Liu and X. Feng, NBN-embedded Polycyclic Aromatic Hydrocarbons Containing Pentagonal and Heptagonal Rings, Org. Lett., 2019, 21, 1354–1358 CrossRef CAS PubMed; (e) P. Qiang, Z. Sun, M. Wan, X. Wang, P. Thiruvengadam, C. Bingi, W. Wei, W. Zhu, D. Wu and F. Zhang, Successive Annulation to Fully Zigzag-Edged Polycyclic Heteroaromatic Hydrocarbons with Strong Blue–Green Electroluminescence, Org. Lett., 2019, 21, 4575–4579 CrossRef CAS PubMed; (f) C.-W. Ju, B. Li, L. Li, W. Yan, C. Cui, X. Ma and D. Zhao, Modular Synthesis of Pentagonal and Hexagonal Ring-Fused NBN-Phenalenes Leading to an Excited-State Aromatization-Induced Structural Planarization Molecular Library, J. Am. Chem. Soc., 2021, 143, 5903–5916 CrossRef CAS PubMed; (g) M. Zhao and Q. Miao, Design, Synthesis and Hydrogen Bonding of B3N6-[4]Triangulene, Angew. Chem., Int. Ed., 2021, 60, 21289–21294 CrossRef CAS; (h) M. Franceschini, M. Crosta, R. R. Ferreira, D. Poletto, N. Demitri, J. P. Zobel, L. González and D. Bonifazi, Peri-Acenoacene Ribbons with Zigzag BN-Doped Peripheries, J. Am. Chem. Soc., 2022, 144, 21470–21484 CrossRef CAS.
- A. S. Scholz, J. G. Massoth, M. Bursch, J.-M. Mewes, T. Hetzke, B. Wolf, M. Bolte, H.-W. Lerner, S. Grimme and M. Wagner, BNB-Doped Phenalenyls: Modular Synthesis, Optoelectronic Properties, and One-Electron Reduction, J. Am. Chem. Soc., 2020, 142, 11072–11083 CrossRef CAS PubMed.
- Examples of BNB-phenalenyls: (a) H. Wei, Y. Liu, T. Y. Gopalakrishna, H. Phan, X. Huang, L. Bao, J. Guo, J. Zhou, S. Luo, J. Wu and Z. Zeng, B–N–B Bond Embedded Phenalenyl and Its Anions, J. Am. Chem. Soc., 2017, 139, 15760–15767 CrossRef CAS PubMed; (b) M. Crumbach, O. Ayhan, L. Fritze, J. A. P. Sprenger, L. Zapf, M. Finze and H. Helten, BNB-doped phenalenyls - aromaticity switch upon one-electron reduction, Chem. Commun., 2021, 57, 2408–2411 RSC. For references regarding π-extended or related systems, see: (c) M. Fingerle, C. Maichle-Mössmer, S. Schundelmeier, B. Speiser and H. F. Bettinger, Synthesis and Characterization of a Boron–Nitrogen–Boron Zigzag-Edged Benzo[fg]tetracene Motif, Org. Lett., 2017, 19, 4428–4431 CrossRef CAS PubMed; (d) M. Crumbach, J. Bachmann, L. Fritze, A. Helbig, I. Krummenacher, H. Braunschweig and H. Helten, Dithiophene-Fused Oxadiborepins and Azadiborepins: A New Class of Highly Fluorescent Heteroaromatics, Angew. Chem., Int. Ed., 2021, 60, 9290–9295 CrossRef CAS; (e) J. Bachmann, A. Helbig, M. Crumbach, I. Krummenacher, H. Braunschweig and H. Helten, Fusion of Aza- and Oxadiborepins with Furans in a Reversible Ring-Opening Process Furnishes Versatile Building Blocks for Extended π-Conjugated Materials, Chem. – Eur. J., 2022, 28, e202202455 CrossRef CAS PubMed; (f) T. Bischof, L. Beßler, I. Krummenacher, L. Erhard, H. Braunschweig and M. Finze, Construction of a Diverse Range of Boron Heterocycles via Ring Expansion of a Carboranyl–Substituted 9-Borafluorene, Chem. – Eur. J., 2023, 29, e202300210 CrossRef CAS; (g) S. Zhou, Y. Liu, W. Jin, T. Qin, X. Liu, C. Zhao, Z. Liu and X. Yu, Synthesis, Structures, and Photophysical Properties of Zigzag BNBNB-Embedded Anthracene-Fused Fluoranthene, Org. Lett., 2023, 25, 1573–1577 CrossRef CAS.
- A. S. Scholz, J. G. Massoth, L. Stoess, M. Bolte, M. Braun, H.-W. Lerner, J.-M. Mewes, M. Wagner and T. Froitzheim, NBN- and BNB-Phenalenyls: the Yin and Yang of Heteroatom-doped π Systems, Chem. – Eur. J., 2024, e202400320 CrossRef PubMed.
- (a) F. F. Caserio Jr., J. J. Cavallo and R. I. Wagner, Preparation of 8-Bora-7,9-diazaro-peri-naphthene and Derivatives, J. Org. Chem., 1961, 26, 2157–2158 CrossRef; (b) V. M. Jiménez-Pérez, B. M. Muñoz-Flores, H. W. Roesky, T. Schulz, A. Pal, T. Beck, Z. Yang, D. Stalke, R. Santillan and M. Witt, Monomeric Boron and Tin(II) Heterocyclic Derivatives of 1,8-Diaminonaphthalenes: Synthesis, Characterization and X-ray Structures, Eur. J. Inorg. Chem., 2008, 2008, 2238–2243 CrossRef.
- M. Karplus and J. A. Pople, Theory of Carbon NMR Chemical Shifts in Conjugated Molecules, J. Chem. Phys., 1963, 38, 2803–2807 CrossRef CAS.
- Y.-S. Lin, G.-D. Li, S.-P. Mao and J.-D. Chai, Long-Range Corrected Hybrid Density Functionals with Improved Dispersion Corrections, J. Chem. Theory Comput., 2013, 9, 263–272 CrossRef CAS PubMed.
- S. Grimme, A. Hansen, J. G. Brandenburg and C. Bannwarth, Dispersion-Corrected Mean-Field Electronic Structure Methods, Chem. Rev., 2016, 116, 5105–5154 CrossRef CAS PubMed.
- J. Zheng, X. Xu and D. G. Truhlar, Minimally augmented Karlsruhe basis sets, Theor. Chem. Acc., 2011, 128, 295–305 Search PubMed.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Accurate Coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS.
- P. Winget, D. M. Dolney, D. J. Giesen, C. J. Cramer and D. G. Truhlar, Minnesota Solvent Descriptor Database, https://comp.chem.umn.edu/solvation/, (accessed 4 March 2024).
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, A. Hansen, S. Ehlert and J.-M. Mewes, r2SCAN-3c: A “Swiss army knife” composite electronic-structure method, J. Chem. Phys., 2021, 154, 064103 CrossRef CAS.
- A. V. Marenich, C. P. Kelly, J. D. Thompson, G. D. Hawkins, C. C. Chambers, D. J. Giesen, P. Winget, C. J. Cramer and D. G. Truhlar, Minnesota Solvation Database (MNSOL) version 2012, https://doi.org/10.13020/3eks-j059, (accessed 5 March 2024).
- D. L. Mobley and J. P. Guthrie, FreeSolv: a database of experimental and calculated hydration free energies, with input files, J. Comput.-Aided Mol. Des, 2014, 28, 711–720 CrossRef CAS PubMed.
- C. Plett, M. Stahn, M. Bursch, J.-M. Mewes and S. Grimme, Improving Quantum Chemical Solvation Models by Dynamic Radii Adjustment for Continuum Solvation (DRACO), J. Phys. Chem. Lett., 2024, 15, 2462–2469 CrossRef CAS PubMed.
- This interpretation is in line with a continuous bathochromic shift of the emission band upon increasing the solvent polarity from C6H12 (λem = 399 nm) to C6H6 (408 nm), THF (421 nm), and CH2Cl2 (434 nm, Fig. S36†).
- E. Runge and E. K. U. Gross, Density-Functional Theory for Time-Dependent Systems, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- S. Hirata and M. Head-Gordon, Time-dependent density functional theory within the Tamm–Dancoff approximation, Chem. Phys. Lett., 1999, 314, 291–299 CrossRef CAS.
- E. Caldeweyher, C. Bannwarth and S. Grimme, Extension of the D3 dispersion coefficient model, J. Chem. Phys., 2017, 147, 034112 CrossRef PubMed.
- E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth and S. Grimme, A generally applicable atomic-charge dependent London dispersion correction, J. Chem. Phys., 2019, 150, 154122 CrossRef PubMed.
- T. Stein, L. Kronik and R. Baer, Prediction of charge-transfer excitations in coumarin-based dyes using a range-separated functional tuned from first principles, J. Chem. Phys., 2009, 131, 244119 CrossRef PubMed.
- R. Baer, E. Livshits and U. Salzner, Tuned Range-Separated Hybrids in Density Functional Theory, Annu. Rev. Phys. Chem., 2010, 61, 85–109 CrossRef CAS PubMed.
- J. Shee and M. Head-Gordon, Predicting Excitation Energies of Twisted Intramolecular Charge-Transfer States with the Time-Dependent Density Functional Theory: Comparison with Experimental Measurements in the Gas Phase and Solvents Ranging from Hexanes to Acetonitrile, J. Chem. Theory Comput., 2020, 16, 6244–6255 CrossRef CAS PubMed.
- M. A. Rohrdanz, K. M. Martins and J. M. Herbert, A long-range-corrected density functional that performs well for both ground-state properties and time-dependent density functional theory excitation energies, including charge-transfer excited states, J. Chem. Phys., 2009, 130, 054112 CrossRef PubMed.
- J.-M. Mewes, Z.-Q. You, M. Wormit, T. Kriesche, J. M. Herbert and A. Dreuw, Experimental Benchmark Data and Systematic Evaluation of Two a Posteriori, Polarizable-Continuum Corrections for Vertical Excitation Energies in Solution, J. Phys. Chem. A, 2015, 119, 5446–5464 CrossRef CAS.
- S. Chibani, Š. Budzák, M. Medved’, B. Mennucci and D. Jacquemin, Full cLR-PCM calculations of the solvatochromic effects on emission energies, Phys. Chem. Chem. Phys., 2014, 16, 26024–26029 RSC.
- J. M. Herbert, Dielectric continuum methods for quantum chemistry, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, 11, e1519 CAS.
- R. Improta, V. Barone, G. Scalmani and M. J. Frisch, A state-specific polarizable continuum model time dependent density functional theory method for excited state calculations in solution, J. Chem. Phys., 2006, 125, 054103 CrossRef.
- J.-M. Mewes, J. M. Herbert and A. Dreuw, On the accuracy of the general, state-specific polarizable-continuum model for the description of correlated ground- and excited states in solution, Phys. Chem. Chem. Phys., 2017, 19, 1644–1654 RSC.
- A. T. B. Gilbert, N. A. Besley and P. M. W. Gill, Self-Consistent Field Calculations of Excited States Using the Maximum Overlap Method (MOM), J. Phys. Chem. A, 2008, 112, 13164–13171 CrossRef CAS PubMed.
- T. Froitzheim, S. Grimme and J.-M. Mewes, Either Accurate Singlet–Triplet Gaps or Excited-State Structures: Testing and Understanding the Performance of TD-DFT for TADF Emitters, J. Chem. Theory Comput., 2022, 18, 7702–7713 CrossRef CAS PubMed.
- L. Kunze, A. Hansen, S. Grimme and J.-M. Mewes, PCM-ROKS for the Description of Charge-Transfer States in Solution: Singlet–Triplet Gaps with Chemical Accuracy from Open-Shell Kohn–Sham Reaction-Field Calculations, J. Phys. Chem. Lett., 2021, 12, 8470–8480 CrossRef CAS PubMed.
- J.-M. Mewes, Modeling TADF in organic emitters requires a careful consideration of the environment and going beyond the Franck–Condon approximation, Phys. Chem. Chem. Phys., 2018, 20, 12454–12469 RSC.
- ΔE12 is the energy difference between the first and second excited states with fully relaxed equilibrium solvation at their respective ΔSCF/UKS/PCM geometries. Due to the differences in structure and solvent environment, ΔE12 deviates substantially from the difference of either emission or absorption energies.
- The emission maximum of 3Me,Mes in pure THF is λem = 389 nm at a concentration of 1.5 × 10−5 mol L−1; the mixtures used for the H2O-precipitation studies had a higher concentration of 1.5 × 10−4 mol L−1 and gave a λem of 370 nm. The origin of this slight hypsochromic shift is unknown.
- E. Epifanovsky, A. T. B. Gilbert, X. Feng, J. Lee, Y. Mao, N. Mardirossian, P. Pokhilko, A. F. White, M. P. Coons, A. L. Dempwolff, Z. Gan, D. Hait, P. R. Horn, L. D. Jacobson, I. Kaliman, J. Kussmann, A. W. Lange, K. U. Lao, D. S. Levine, J. Liu, S. C. McKenzie, A. F. Morrison, K. D. Nanda, F. Plasser, D. R. Rehn, M. L. Vidal, Z.-Q. You, Y. Zhu, B. Alam, B. J. Albrecht, A. Aldossary, E. Alguire, J. H. Andersen, V. Athavale, D. Barton, K. Begam, A. Behn, N. Bellonzi, Y. A. Bernard, E. J. Berquist, H. G. A. Burton, A. Carreras, K. Carter-Fenk, R. Chakraborty, A. D. Chien, K. D. Closser, V. Cofer-Shabica, S. Dasgupta, M. de Wergifosse, J. Deng, M. Diedenhofen, H. Do, S. Ehlert, P.-T. Fang, S. Fatehi, Q. Feng, T. Friedhoff, J. Gayvert, Q. Ge, G. Gidofalvi, M. Goldey, J. Gomes, C. E. González-Espinoza, S. Gulania, A. O. Gunina, M. W. D. Hanson-Heine, P. H. P. Harbach, A. Hauser, M. F. Herbst, M. Hernández Vera, M. Hodecker, Z. C. Holden, S. Houck, X. Huang, K. Hui, B. C. Huynh, M. Ivanov, Á. Jász, H. Ji, H. Jiang, B. Kaduk, S. Kähler, K. Khistyaev, J. Kim, G. Kis, P. Klunzinger, Z. Koczor-Benda, J. H. Koh, D. Kosenkov, L. Koulias, T. Kowalczyk, C. M. Krauter, K. Kue, A. Kunitsa, T. Kus, I. Ladjánszki, A. Landau, K. V. Lawler, D. Lefrancois, S. Lehtola, R. R. Li, Y.-P. Li, J. Liang, M. Liebenthal, H.-H. Lin, Y.-S. Lin, F. Liu, K.-Y. Liu, M. Loipersberger, A. Luenser, A. Manjanath, P. Manohar, E. Mansoor, S. F. Manzer, S.-P. Mao, A. V. Marenich, T. Markovich, S. Mason, S. A. Maurer, P. F. McLaughlin, M. F. S. J. Menger, J.-M. Mewes, S. A. Mewes, P. Morgante, J. W. Mullinax, K. J. Oosterbaan, G. Paran, A. C. Paul, S. K. Paul, F. Pavošević, Z. Pei, S. Prager, E. I. Proynov, Á. Rák, E. Ramos-Cordoba, B. Rana, A. E. Rask, A. Rettig, R. M. Richard, F. Rob, E. Rossomme, T. Scheele, M. Scheurer, M. Schneider, N. Sergueev, S. M. Sharada, W. Skomorowski, D. W. Small, C. J. Stein, Y.-C. Su, E. J. Sundstrom, Z. Tao, J. Thirman, G. J. Tornai, T. Tsuchimochi, N. M. Tubman, S. P. Veccham, O. Vydrov, J. Wenzel, J. Witte, A. Yamada, K. Yao, S. Yeganeh, S. R. Yost, A. Zech, I. Y. Zhang, X. Zhang, Y. Zhang, D. Zuev, A. Aspuru-Guzik, A. T. Bell, N. A. Besley, K. B. Bravaya, B. R. Brooks, D. Casanova, J.-D. Chai, S. Coriani, C. J. Cramer, G. Cserey, A. E. DePrince III, R. A. DiStasio Jr., A. Dreuw, B. D. Dunietz, T. R. Furlani, W. A. Goddard III, S. Hammes-Schiffer, T. Head-Gordon, W. J. Hehre, C.-P. Hsu, T.-C. Jagau, Y. Jung, A. Klamt, J. Kong, D. S. Lambrecht, W. Liang, N. J. Mayhall, C. W. McCurdy, J. B. Neaton, C. Ochsenfeld, J. A. Parkhill, R. Peverati, V. A. Rassolov, Y. Shao, L. V. Slipchenko, T. Stauch, R. P. Steele, J. E. Subotnik, A. J. W. Thom, A. Tkatchenko, D. G. Truhlar, T. Van Voorhis, T. A. Wesolowski, K. B. Whaley, H. L. Woodcock III, P. M. Zimmerman, S. Faraji, P. M. W. Gill, M. Head-Gordon, J. M. Herbert and A. I. Krylov, Software for the frontiers of quantum chemistry: An overview of developments in the Q-Chem 5 package, J. Chem. Phys., 2021, 155, 084801 CrossRef CAS PubMed.
- F. Neese, The ORCA program system, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details, analytical data. CCDC 2338779–2338788. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qo00468j |
| This journal is © the Partner Organisations 2024 |