
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvent dependent hierarchical dual helicity in a fused [4]carbohelicene/quinoline oligoamide foldamer†
Rita
Borges-Anastácio
 ab,
Kevin
Moreno
a,
Eric
Merlet
b,
Thierry
Buffeteau
a,
Brice
Kauffmann
c,
Nathan
McClenaghan
a,
Yann
Ferrand
*b and
Céline
Olivier
*a
ab,
Kevin
Moreno
a,
Eric
Merlet
b,
Thierry
Buffeteau
a,
Brice
Kauffmann
c,
Nathan
McClenaghan
a,
Yann
Ferrand
*b and
Céline
Olivier
*a
aInstitut des Sciences Moléculaires, UMR 5255 CNRS, Université de Bordeaux, 351 Cours de la Libération, 33405 Talence, France. E-mail: celine.olivier@u-bordeaux.fr
bUniv. Bordeaux, CNRS, Bordeaux Institut National Polytechnique, CBMN UMR 5248, 2 rue Escarpit, 33600 Pessac, France. E-mail: yann.ferrand@u-bordeaux.fr
cUniv. Bordeaux, CNRS, INSERM, Institut Européen de Chimie Biologie (UMS3033/US001), 2 rue Escarpit, 33600 Pessac, France
First published on 18th March 2024
Abstract
Two classes of helical motif, namely a [4]carbohelicene and an aromatic oligoamide foldamer, were fused to form a hybrid molecular construct in which their helical axes are parallel. Depending on the solvent, the [4]carbohelicene can bias the dynamic screw sense of the aromatic oligoamide with a reverse handedness.
Helices are ubiquitous in nature at various length scales, from macroscopic to nanoscopic.1 This specific geometry imparts intrinsic chirality to these systems, also called helicity, due to a left (M) or a right (P) helix screw sense. At the molecular level, helical self-organization is found in biological macromolecules such as peptides, proteins, DNA, and polysaccharides;2 equally, many examples of artificial helically folded polymers have been designed,3 but smaller molecules can also adopt helical patterns. Among them, helicenes are ortho-fused polyaromatic systems, in which a well-defined nonplanar screw shape is imposed by strong steric hindrance.4 The chirality of these helical structures is typically evidenced by electronic circular dichroism (ECD) based on enantiopure helicene samples, yielding characteristic strong Cotton effects, and also by circularly polarized luminescence (CPL) spectroscopy.5 In contrast to these rigid tridimensional structures, helical small molecules may also be flexible and thereby self-organize through dynamic folding. Thus, in the absence of any chiral induction, these systems exist as dynamic mixtures of interconverting racemic right- and left-handed helices in solution. This type of molecule can be exemplified by foldamers, which are flexible oligomers able to fold into a helical conformation due to non-covalent intramolecular interactions, such as hydrogen bonding, electronic repulsion, and, in the case of aromatic monomers, aromatic stacking interactions.6 However, for chiroptical7 or chiral recognition applications,8 the prime importance of foldamer handedness control was highlighted. The helical bias of aromatic oligoamides, in particular quinoline-derived oligomers, has been accomplished by tethering a chiral inducer at the helix N-terminus such as camphanyl;9 at C-terminus with phenyl- or naphthylethyl amino groups,10 β-pinene-derived pyridyl11 or oxazolylaniline12 moieties; or by the insertion of a chiral aromatic δ-amino acid building block directly within the oligomeric sequence.13
In this study, we surmised that embedding an enantiomerically pure [4]carbohelicene within the sequence of a helical aromatic oligoamide would allow biasing of its handedness (Fig. 1a). Thus, we took advantage of the complementary curvature fit between the naphthyl core of the dimethyl[4]helicene dicarboxylic acid and the 8-amino-2-quinolinecarboxylic acid unit to implement them in the same sequence. We demonstrated the possibility of segregating each chiral contribution in this dual-helical construct using various spectroscopic techniques. We also evidenced the superior chiroptical activity of the hybrid oligomeric system over the more classical sterically constrained small helicene.
 | ||
| Fig. 1 (a) Cartoon representation of the dual helicity in the fused helicene-foldamer architecture and the equilibrium between the PP and PM diastereoisomers. Note that the small diameter helix has a fixed handedness whereas the large diameter helix handedness is dynamic. Left (M) and right (P) handedness are colored in red and blue, respectively. (b) Molecular formulas of the fused helicene/foldamer, the [4]carbohelicenes and the quinoline oligomers. | ||
The helicene motif employed for this study, 1,12-dimethyl[4]helicene-5,8-dicarboxylic acid 2, was prepared according to the procedure reported by Yamaguchi et al. (Fig. 1b).14 Optical resolution of the two enantiomers of 2 was performed by forming the diastereoisomeric carboxylate salts with two equivalents of quinine, followed by their selective recrystallization from chloroform–methanol and acidification to recover optically pure P-2 or M-2. The dicarboxylic acid moiety (either P or M) was subsequently coupled with two quinoline oligoamide tetramers 4,15 thereby yielding the new hybrid construct 1, which combines the two different types of molecular helices described in the introduction (Fig. 1a).
The structure of P-1 was first evidenced in the solid state.16 Single crystals were grown by slow liquid–liquid diffusion of hexane into a chloroform solution of P-1. The X-ray crystal structure was solved in the monoclinic P2(1) space group. The crystalline structure confirmed the canonical folding of the two helical quinoline segments that span 1.6 turns each and exhibit the same handedness. Additionally, a dual helicity is observed in the structure as the quinoline segments adopt a left-handed helicity (M) while the helicene motif shows a right-handed helicity (P) (Fig. 2a–c). Single crystals could also be obtained from DMSO solution of P-1. To our surprise, the new crystalline structure, also solved in the P2(1) space group, revealed the existence of a pair of diastereoisomers with the aromatic oligoamide segments forming either a left-handed or a right-handed helix, P-1-M or P-1-P, respectively (Fig. 2d–f). This quasi-racemic structure10,17 hints at a partial foldamer handedness induction in solution, the latter may be solvent dependent.
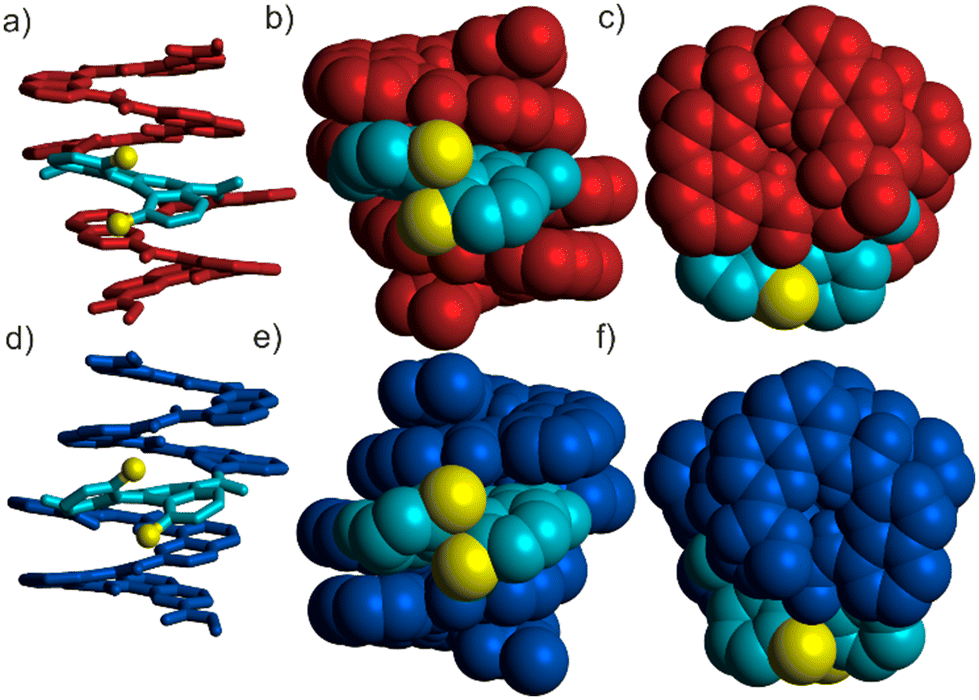 | ||
| Fig. 2 Side views (a and b) and top view (c) of the X-ray structure of P-1-M obtained from a chloroform/hexane solution.16 Side views (d and e) and top view (e) of the X-ray structure of P-1-P obtained from a DMSO solution. In that case, the P-1-M diastereoisomer present in the lattice is not shown, yet it is similar to the one in a–c. In (a and d) 1 is shown in tube representation whereas it is shown in CPK representation in (b, c, e and f). In (a–f) the helicene methyl groups are colored in yellow. The P-[4]helicene is colored in light blue, whereas the M and P quinoline oligoamide segments are colored in red and blue, respectively. Hydrogen atoms and isobutoxy chains were omitted for clarity. | ||
The solvent dependent chiral induction in P-1 was then demonstrated by electronic circular dichroism spectroscopy. Chiral induction was tested in two polar solvents, DMSO and acetone, as well as in three chlorinated solvents, dichloromethane (CH2Cl2), chloroform (CHCl3) and tetrachloroethane (C2H2Cl4). In each solvent a negative CD band around 400 nm was observed which is usually indicative of left handedness for the oligoamide segments (Fig. 3a).9 Although P-1 proved to be CD active in all these media, the intensity of the chiroptical signature varied dramatically with the solvent nature. Whereas the maxima at 386 nm was found to be quasi similar in the two polar solvents and in dichloromethane (∼65 L mol−1 cm−1), the CD response was almost twice as intense in CHCl3 (∼113 L mol−1 cm−1). The more intense band was found in C2H2Cl4 (∼150 L mol−1 cm−1).
 | ||
| Fig. 3 (a) ECD spectra of P-1 (150 μM) recorded in DMSO (green), acetone (pink), dichloromethane (black), chloroform (purple) and tetrachloroethane (orange) at 298 K. Parts of the 1H NMR spectra (700 MHz) showing the amide signals of P-1 (1.0 mM) at 298 K in (b) acetone-d6; (c) DMSO-d6; (d) dichloromethane-d2; (e) chloroform-d; and (f) tetrachloroethane-d2. The amide signals of the major diastereoisomer are denoted with full black circles (●), while those of the minor species are marked with black circles (○). | ||
The extent of chiral induction was quantitatively assessed by 1H NMR spectroscopy. For example in DMSO, the two diastereoisomers P-1-M and P-1-P were shown to be in equilibrium. At 298K, the two diastereoisomeric helices interconvert slowly on the NMR time scale leading to two sets of signals, one for the major diastereoisomer and the second one for the minor diastereoisomer (Fig. 3b). The diastereomeric excess (de) in each solvent could be determined by direct integration of the major and minor species (Fig. 3b–f) and are reported in Table 1. The de values obtained by 1H NMR correlate well the CD results as a minimum de of 30% was measured in DMSO-d6 whereas a maximum de of 90% was obtained in C2D2Cl4.
| Solvent | proportionsa | de (NMR) (%) | Δεb at 386 nm |
|---|---|---|---|
| a Calculated from integrating 1H NMR signals of the major and minor diastereoisomers under slow exchange on NMR timescale. b Δε are expressed in L mol−1 cm−1. | |||
| DMSO | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.54 :0.54 |
30 | −63 |
| Acetone | 1:0.51 |
32 | −65 |
| Dichloromethane | 1:0.48 |
35 | −68 |
| Chloroform | 1:0.22 |
64 | −113 |
| Tetrachloroethane | 1:0.06 |
90 | −150 |
The optical and chiroptical properties of the new supramolecular architecture were further studied in solution in order to assess the dual helicity through chiroptical techniques. The optoelectronic properties of 1 were evaluated by UV-visible absorption and fluorescence emission spectroscopies in chloroform (Fig. S1†), along with those of the parent helicene 2 for comparison. The photophysical properties of quinoline oligomers 5 and 6 were reported previously.18 Helicene 2 presents a maximum absorption band at λ = 310 nm (ε = 25400 L mol−1 cm−1) with a shoulder at 360 nm (ε = 5200 L mol−1 cm−1). The absorption spectrum of 1 shows a similar profile, although with significantly higher molar absorption coefficients, λmax = 320 nm (ε = 62200 L mol−1 cm−1) and 362 nm (ε = 40700 L mol−1 cm−1). These features are in line with those reported for a quinoline tetramer 5 and a hexamer 6 in the same solvent. Upon excitation, the helicene precursor 2 shows weak fluorescence emission with a maximum at λ = 445 nm and a fluorescence quantum yield Φem = 0.001. By tethering two quinoline tetramers, the hybrid construct 1 shows a slightly red-shifted emission spectrum with a maximum at λ = 460 nm and a fluorescence quantum yield Φem = 0.09. This value is higher than those reported for quinoline tetramer 5 (Φem = 0.014) or even for hexamer 6 (Φem = 0.06)18b evidencing the cumulative effect of the quinoline monomers on the optoelectronic properties of the system described herein, comprising eight monomers. As described by Jiang et al. the fluorescence quantum yield of oligoquinoline foldamers is increasing with the number of monomers presumably because of a more-rigid structure in the excited state that originates from the strong aromatic stacking existing in longer oligomers.12a
The chiroptical emission properties of the hybrid system 1 were investigated by circularly polarized luminescence (CPL). The CPL spectrum of the polyhelical compound P-1 recorded in tetrachloroethane is shown in Fig. S2,† along with that of the helicene precursor P-2. The CPL properties of quinoline oligomers 5 and 6 were reported previously.12a,18 Helicene P-2 shows a maximum CPL signal at λ = 430 nm with corresponding dissymmetry factor glum = −4 × 10−3, while the CPL intensity is higher for the supramolecular system P-1, which shows polarized emission at λ = 430 nm with corresponding dissymmetry factor glum = −1 × 10−2. This intense chiroptical signal originates from the oligoquinoline and is consistent with previous reports.18
One striking feature of the chiroptical spectra is the sign of the signals recorded for the dual-helical architecture 1. Indeed when the P-[4]carbohelicene precursor was employed, the CPL and lowest-energy ECD signals of the resulting assembly are negative (Fig. 3, S2 and S3†) which corresponds to M-helicity of the quinoline oligomers.9,18 Thus, the sign conservation of the chiroptical signals from P-2 to P-1 (Fig. S2 and S3†) indicates the opposite helicity of the two helical systems coexisting in the dual architecture. However, the chiroptical contribution of the quinoline segments overwhelms those of the helicene moiety in 1. This gave us the impetus to employ another chiroptical technique, namely Vibrational Circular Dichroism (VCD), to detect simultaneously the chiral signature of each helical system. The polyhelical system 1, as well as the helicene and quinoline oligoamide tetrameric precursors, were thus analyzed by infrared (IR) and VCD spectroscopies. In order to allow higher solubility in chlorinated solvents, the dicarboxylic acid helicene precursor 2 was converted into the corresponding diamide derivative 3 (see ESI†). To allow a direct comparison of the VCD properties, optically pure quinoline tetramers 5 were also prepared.18a
The IR absorption spectra of 1, 3, and 5 recorded in CDCl3 are represented in Fig. S4.† The helicene derivative 3 shows two main bands band at 1515 cm−1 (ε = 685 L mol−1 cm−1) for the νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching mode of the polyaromatic system and at 1655 cm−1 (ε = 800 L mol−1 cm−1) for the νCO mode of the amide groups. Tetramer 5 shows multiple bands from 1800 cm−1 to 1000 cm−1, that were attributed in previous reports.18b,19 The main features are the strong absorption bands at 1540 cm−1 (ε = 3800 L mol−1 cm−1) and at 1680 cm−1 (ε = 1900 L mol−1 cm−1) corresponding to the amide 2 (δNH + νCN) and amide 1 (νCO) modes of the amide groups, respectively, and a small band at 1795 cm−1 (ε = 600 L mol−1 cm−1) corresponding to the νCO stretching vibration of the ketone group of camphanyl chiral inducer. Oligomer 1 combines the features of both precursors however with a superposition of the amide 1 modes around 1680 cm−1 (ε = 2200 L mol−1 cm−1) and the disappearance of the νCO band of the ketone group of the camphanyl chiral inducer of 5 which is not present anymore in the supramolecular system.
C stretching mode of the polyaromatic system and at 1655 cm−1 (ε = 800 L mol−1 cm−1) for the νCO mode of the amide groups. Tetramer 5 shows multiple bands from 1800 cm−1 to 1000 cm−1, that were attributed in previous reports.18b,19 The main features are the strong absorption bands at 1540 cm−1 (ε = 3800 L mol−1 cm−1) and at 1680 cm−1 (ε = 1900 L mol−1 cm−1) corresponding to the amide 2 (δNH + νCN) and amide 1 (νCO) modes of the amide groups, respectively, and a small band at 1795 cm−1 (ε = 600 L mol−1 cm−1) corresponding to the νCO stretching vibration of the ketone group of camphanyl chiral inducer. Oligomer 1 combines the features of both precursors however with a superposition of the amide 1 modes around 1680 cm−1 (ε = 2200 L mol−1 cm−1) and the disappearance of the νCO band of the ketone group of the camphanyl chiral inducer of 5 which is not present anymore in the supramolecular system.
VCD analyses of the two enantiomers of each compound were also performed and the raw VCD spectra are reported in Fig. S5–S7.† Selected baseline-corrected VCD spectra are shown in Fig. 4. First considering the VCD response of the helicene 3 (Fig. 4, cyan), the main chiral signatures consist of a small band at 1600 cm−1 and a slightly more intense signal at 1650 cm−1, both of which are negative for the P-enantiomer. By contrast, the VCD spectrum of tetramer 5 shows much more intense signals, notably (i) an intense band at 1487 cm−1, that is negative for the M-enantiomer (Fig. 4, red), (ii) a very strong bisignate band (−,+) with exciton-coupling shape, around 1540 cm−1, and (iii) a medium-intensity bisignate band (+,−) around 1677 cm−1. Those features are in line with previous reports on quinoline-based tetramers and hexamers.18b,19 Finally, the VCD spectrum of P-1 (Fig. 4, brown) combines the chiral signatures of the P-2 precursor and of a quinoline foldamer with M-helicity. Notably, a strong negative band is observed at 1485 cm−1 in line with M-5 spectrum, and a negative band is observed at 1665 cm−1 corresponding to amide 1 (νCO) mode of the two amide groups allowing the linkage between the helicene and the two quinoline oligoamide foldamers, in line with P-3 spectrum. This spectroscopically proves the occurrence of two helicities in the same molecular architecture, i.e. right-handed helicene and left-handed foldamers.
 | ||
| Fig. 4 VCD spectra of selected enantiomers of 1 and precursors, recorded in CDCl3. Cyan: P-3; red: M-5; brown: P-1. | ||
Consistent with the ECD and CPL results, the VCD study underlines the intensity difference of the chiral signatures of each class of helical motifs that are the self-organized oligomer vs. the small helicene. The intensity of the VCD response of 5 is one order of magnitude higher than that of 3. Thus the helicene response is masked by those of the quinoline tetramers in 1, except in the region where the two motifs absorb differently (1600–1700 cm−1); allowing to assess spectroscopically the occurrence of dual helicity in the helicene-foldamer intertwined system.
In conclusion, a rigid small helicene can induce the reversed helicity of foldamer segments. The direct observation of such dual helicity in a polyhelical system was possible directly in the solid state by X-ray crystallography but also in solution by VCD. ECD and CPL spectroscopies only provided indirect evidence of this structural arrangement. New designs incorporating helicene/foldamer hybrids that can self-assemble are currently under investigation in our labs.20
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the IdEx University of Bordeaux, and the Grand Research Program “GPR LIGHT S&T” for financial support. RBA thanks ANR for PhD grant (ANR NLOChiraMat grant no. ANR-21-CE06-0018).References
- G. H. Wagnière, in “On Chirality and the Universal Asymmetry. Reflections on Image and Mirror Image”, Wiley-VCH, 2007 Search PubMed.
- V. Percec and Qi Xiao, Helical Self-Organizations and Emerging Functions in Architectures, Biological and Synthetic Macromolecules, Bull. Chem. Soc. Jpn., 2021, 94, 900–928 CrossRef CAS.
- (a) E. Yashima, K. Maeda, H. Iida, Y. Furusho and K. Nagai, Helical Polymers: Synthesis, Structures, and Functions, Chem. Rev., 2009, 109, 6102–6211 CrossRef CAS PubMed; (b) J. H. K. K. Hirschberg, L. Brunsveld, A. Ramzi, J. A. J. M. Vekemans, R. P. Sijbesma and E. W. Meijer, Helical Self-Assembled Polymers from Cooperative Stacking of Hydrogen-Bonded Pairs, Nature, 2000, 407, 167–170 CrossRef CAS PubMed; (c) M. Shigeno, Y. Kushida and M. Yamaguchi, Heating/Cooling Stimulus Induces Three-State Molecular Switching of Pseudoenantiomeric Aminomethylenehelicene Oligomers: Reversible Nonequilibrium Thermodynamic Processes, J. Am. Chem. Soc., 2014, 136(22), 7972–7980 CrossRef CAS PubMed; (d) T. Yamamoto, R. Murakami and M. Suginome, Single-Handed Helical Poly(quinoxaline-2,3-diyl)s Bearing Achiral 4-Aminopyrid-3-yl Pendants as Highly Enantioselective, Reusable Chiral Nucleophilic Organocatalysts in the Steglich Reaction, J. Am. Chem. Soc., 2017, 139, 2557–2560 CrossRef CAS PubMed; (e) Y. Li, L. Bouteiller and M. Raynal, Catalysts Supported by Homochiral Molecular Helices: A New Concept to Implement Asymmetric Amplification in Catalytic Science, ChemCatChem, 2019, 11, 5212–5226 CrossRef CAS.
- (a) Y. Shen and C.-F. Chen, Helicenes: Synthesis and Applications, Chem. Rev., 2012, 112, 1463–1535 CrossRef CAS PubMed; (b) M. Gingras, One Hundred Years of Helicene Chemistry, Chem. Soc. Rev., 2013, 42, 968–1006 RSC.
- (a) W.-L. Zhao, M. Li, H.-Y. Lu and C.-F. Chen, Advances in Helicene Derivatives with Circularly Polarized Luminescence, Chem. Commun., 2019, 55, 13793–13803 RSC; (b) T. Mori, Chiroptical Properties of Symmetric Double, Triple, and Multiple Helicenes, Chem. Rev., 2021, 121, 2373–2412 CrossRef CAS PubMed; (c) M. Cei, L. Di Bari and F. Zinna, Circularly Polarized Luminescence of Helicenes: A Data-Informed Insight, Chirality, 2023, 35, 192–210 CrossRef CAS PubMed.
- (a) I. Huc, Aromatic Oligoamide Foldamers, Eur. J. Org. Chem., 2004, 17–29 CrossRef CAS; (b) X. Hu, A. Schulz, J. O. Lindner, M. Grüne, D. Bialas and F. Würthner, Folding and Fluorescence Enhancement with Strong Odd–Even Effect for a Series of Merocyanine Dye Oligomers, Chem. Sci., 2021, 12, 8342–8352 RSC; (c) J. C. Nelson, J. G. Saven, J. S. Moore and P. G. Wolynes, Solvophobically Driven Folding of Nonbiological Oligomers, Science, 1997, 277, 1793–1796 CrossRef CAS PubMed; (d) H. Goto, H. Katagiri, Y. Furusho and E. Yashima, Oligoresorcinols Fold into Double Helices in Water, J. Am. Chem. Soc., 2006, 128, 7176–7178 CrossRef CAS PubMed; (e) Y. Hua, Y. Liu, C.-H. Chen and A. H. Flood, Hydrophobic Collapse of Foldamer Capsules Drives Picomolar-Level Chloride Binding in Aqueous Acetonitrile Solutions, J. Am. Chem. Soc., 2013, 135, 14401–14412 CrossRef CAS PubMed; (f) K.-J. Chang, B.-N. Kang, M.-H. Lee and K.-S. Jeong, Oligoindole-Based Foldamers with a Helical Conformation Induced by Chloride, J. Am. Chem. Soc., 2005, 127, 12214–12215 CrossRef CAS PubMed; (g) C. J. Massena, D. A. Decato and O. B. Berryman, A Long-Lived Halogen-Bonding Anion Triple Helicate Accommodates Rapid Guest Exchange, Angew. Chem., Int. Ed., 2018, 57, 16109–16113 CrossRef CAS PubMed; (h) S. Peddi, M. C. Bookout, G. N. Vemuri and C. S. Hartley, Guest-Driven Control of Folding in a Crown-Ether-Functionalized ortho-Phenylene, J. Org. Chem., 2022, 87, 3686–3690 CrossRef CAS PubMed; (i) H. Jiang, J.-M. Léger and I. Huc, Aromatic δ-Peptides, J. Am. Chem. Soc., 2003, 125, 3448–3449 CrossRef CAS PubMed; (j) L. Yuan, H. Zeng, K. Yamato, A. R. Sanford, W. Feng, H. S. Atreya, D. K. Sukumaran, T. Szyperski and B. Gong, Helical Aromatic Oligoamides: Reliable, Readily Predictable Folding from the Combination of Rigidified Structural Motifs, J. Am. Chem. Soc., 2004, 126, 16528–16537 CrossRef CAS PubMed.
- D. Verreault, K. Moreno, E. Merlet, F. Adamietz, B. Kauffmann, Y. Ferrand, C. Olivier and V. Rodriguez, Hyper-Rayleigh Scattering as a New Chiroptical Method: Uncovering the Nonlinear Optical Activity of Aromatic Oligoamide Foldamers, J. Am. Chem. Soc., 2020, 142, 257–263 CrossRef CAS PubMed.
- G. Lautrette, B. Wicher, B. Kauffmann, Y. Ferrand and I. Huc, Iterative Evolution of an Abiotic Foldamer Sequence for the Recognition of Guest Molecules with Atomic Precision, J. Am. Chem. Soc., 2016, 138, 10314–10322 CrossRef CAS PubMed.
- A. M. Kendhale, L. Poniman, Z. Dong, K. Laxmi-Reddy, B. Kauffmann, Y. Ferrand and I. Huc, Absolute Control of Helical Handedness in Quinoline Oligoamides, J. Org. Chem., 2011, 76, 195–200 CrossRef CAS PubMed.
- C. Dolain, H. Jiang, J.-M. Léger, P. Guionneau and I. Huc, Chiral Induction in Quinoline-Derived Oligoamide Foldamers: Assignment of Helical Handedness and Role of Steric Effects, J. Am. Chem. Soc., 2005, 127, 12943–12951 CrossRef CAS PubMed.
- L. Zheng, Y. Zhan, C. Yu, F. Huang, Y. Wang and H. Jiang, Controlling Helix Sense at N- and C-Termini in Quinoline Oligoamide Foldamers by β-Pinene-Derived Pyridyl Moieties, Org. Lett., 2017, 19, 1482–1485 CrossRef CAS PubMed.
- (a) D. Zheng, L. Zheng, C. Yu, Y. Zhan, Y. Wang and H. Jiang, Significant Enhancement of Circularly Polarized Luminescence Dissymmetry Factors in Quinoline Oligoamide Foldamers with Absolute Helicity, Org. Lett., 2019, 21, 2555–2559 CrossRef CAS PubMed; (b) L. Yang, M. Chunmiao, B. Kauffmann, L. Dongyao and Q. Gan, Absolute Handedness Control of Oligoamide Double Helices by Chiral Oxazolylaniline Induction, Org. Biomol. Chem., 2020, 18, 6643–6650 RSC.
- D. Bindl, E. Heinemann, P. K. Mandal and I. Huc, Quantitative Helix Handedness Bias Through a Single H vs. CH3 Stereochemical Differentiation, Chem. Commun., 2021, 57, 5662–5665 RSC.
- H. Okubo, M. Yamaguchi and C. Kabuto, Macrocyclic Amides Consisting of Helical Chiral 1,12-Dimethylbenzo[c]phenanthrene-5,8-dicarboxylate, J. Org. Chem., 1998, 63, 9500–9509 CrossRef CAS.
- T. Qi, T. Deschrijver and I. Huc, Large-Scale and Chromatography-Free Synthesis of an Octameric Quinoline-Based Aromatic Amide Helical Foldamer, Nat. Protoc., 2013, 8, 693–708 CrossRef PubMed.
- Note: P-1 stands for the enantiomer of 1 obtained from the [4]carbohelicene P-2. P-1-M corresponds to the diastereoisomer of 1 where the quinoline oligomers adopt an M helicity, and P-1-P corresponds to the diastereoisomer of 1 where the quinoline oligomers adopt a P helicity.
- G. Lautrette, B. Kauffmann, Y. Ferrand, C. Aube, N. Chandramouli, D. Dubreuil and I. Huc, Structure Elucidation of Host–Guest Complexes of Tartaric and Malic Acids by Quasi-Racemic Crystallography, Angew. Chem., Int. Ed., 2013, 52, 11517 CrossRef CAS PubMed.
- (a) E. Merlet, K. Moreno, A. Tron, N. McClenaghan, B. Kauffmann, Y. Ferrand and C. Olivier, Aromatic Oligoamide Foldamers as Versatile Scaffolds for Induced Circularly Polarized Luminescence at Adjustable Wavelengths, Chem. Commun., 2019, 55, 9825–9828 RSC; (b) K. Moreno, E. Merlet, N. McClenaghan, T. Buffeteau, Y. Ferrand and C. Olivier, Influence of Positional Isomerism on the Chiroptical Properties of Functional Aromatic Oligoamide Foldamers, ChemPlusChem, 2021, 86, 496–503 CrossRef CAS PubMed; (c) V. Laffilé, K. Moreno, E. Merlet, N. McClenaghan, Y. Ferrand and C. Olivier, CPL-Active Water-Soluble Aromatic Oligoamide Foldamers, Org. Biomol. Chem., 2023, 21, 3644–3649 RSC.
- T. Buffeteau, L. Ducasse, L. Poniman, N. Delsuc and I. Huc, Vibrational Circular Dichroism and Ab Initio Structure Elucidation of an Aromatic Foldamer, Chem. Commun., 2006, 2714–2716 RSC.
- V. Koehler, G. Bruschera, E. Merlet, P. K. Mandal, E. Morvan, F. Rosu, C. Douat, L. Fischer, I. Huc and Y. Ferrand, High-Affinity Hybridization of Complementary Aromatic Oligoamide Strands in Water, Angew. Chem., Int. Ed., 2023, e202311639 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic protocols, crystallographic studies and characterisation of the new compounds. CCDC 2304953 and 2333345. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qo00320a |
| This journal is © the Partner Organisations 2024 |