
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nickel/photoredox-catalyzed carbonylative transformations of α-phosphorus-, α-sulfur-, and α-boron-substituted alkyl halides†
Le-Cheng
Wang
ab and
Xiao-Feng
Wu
 *ab
*ab
aDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 116023 Dalian, Liaoning, China. E-mail: xwu2020@dicp.ac.cn
bLeibniz-Institut für Katalyse e.V., 18059 Rostock, Germany. E-mail: Xiao-Feng.Wu@catalysis.de
First published on 26th February 2024
Abstract
Organophosphorus compounds are important motifs in living organisms, medicinal chemistry, agricultural chemistry, materials science, catalysts, ligands, etc. However, catalytic carbonylative transformation of α-phosphorus, α-sulfur or α-boron substituted alkyl halides remains a formidable challenge due to α-heteroatom effects. In this report, we describe a nickel/photoredox dual-catalytic strategy for the direct amino- and alkoxycarbonylation of α-phosphorus, α-sulfur, and α-boron substituted organohalides with an array of reaction partners under low CO gas pressure which furnished various high-value products in excellent yields. The utility of this process was also demonstrated by the development of a new α-phosphine amide ligand. Additionally, this synergistic protocol also facilitates a sequential four-component carbonylation in the presence of vinyl phosphonate.
Introduction
Phosphorus is one of the essential elements for life and is closely related to living organisms. In addition, organophosphorus compounds are not only important structural motifs of genes, but also widely used in medicinal chemistry, agricultural chemistry, materials science, organic synthesis, and other fields.1 In particular, β-phosphonyl acids and derivatives, an indispensable class of phosphorus skeletons, are widely utilized as ligands and key intermediates in organometallic species-mediated reactions due to their unique chemical properties.2 Thus, developing efficient strategies to access β-phosphonyl acids from readily available starting materials remains an important task. One of the most attractive approaches is the use of broadly available carbon monoxide as the C1/carbonyl source toward organophosphorus molecules.Carbonylation reactions have become indispensable tools for constructing carbonyl-containing compounds in organic and medicinal chemistry as they enable the efficient and robust union of molecular fragments and carbon monoxide.3 Over the last few decades, multiple generations of catalytic systems have been explored that have elevated the transition metal-catalyzed carbonylation of organohalides to an essential transformation.4 Compared with mature noble metal catalysts, cheap metal catalysts such as nickel have also been explored successfully by taking advantage of slow CO-releasing reagents and specialized ligand complexes to minimize the generation of highly toxic and low catalytic activity Ni(CO)n.5 Concerning the substrates applied, aryl,6 benzylic,7 and alkyl halides8 have been relatively well studied, even with nickel catalysts (Fig. 1(a)); however, carbonylative transformations of α-heteroatom substituted organohalides to construct high-value α-heteroatom substituted amides or esters remain less developed, with some examples of α-phosphorus- and α-sulfur-substituted alkyl halides.9
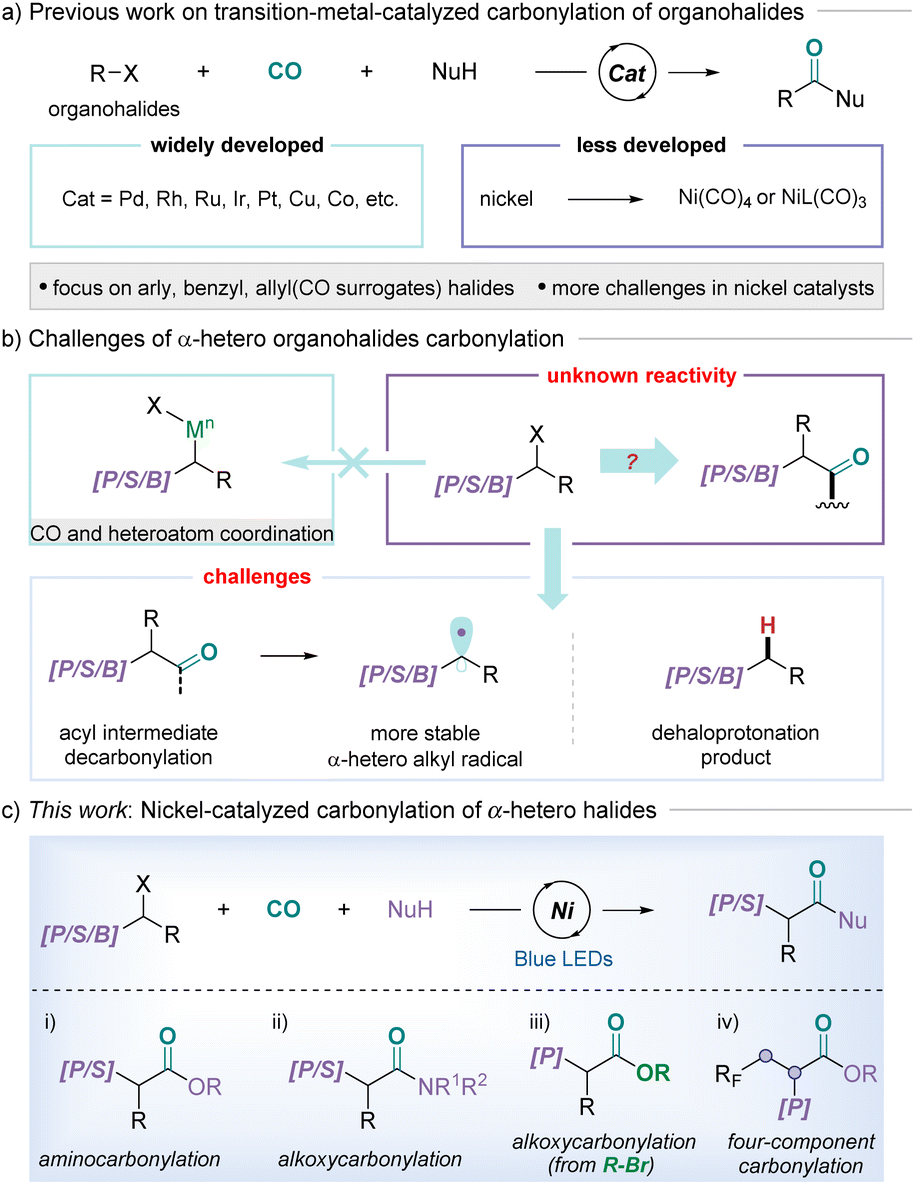 | ||
| Fig. 1 The background of α-heteroatom halide carbonylation. | ||
α-Heteroatom functionalization is a key and challenging strategy in organic synthesis.9 However, because of the unique properties (electron cloud density, bond energy, resonance, etc.) of heteroatoms, the substituents containing heteroatoms could change the properties and reaction characteristics of the molecules, especially in adjacent positions.9c,10 In addition, the coordination of π-acidic CO and heteroatoms with metal catalysts might decrease or even inhibit metal catalytic activity.11 On the other hand, the rate of decarbonylation depends strongly on the nature of substituents, and the α-heteroatoms can effectively stabilize the adjacent carbon radicals, resulting in acyl radicals that tend to decarbonylate to form a stable radical species, especially at lower CO pressures or higher temperatures.12,13 Additionally, owing to the relatively more polar carbon–halogen bonds, α-heteroatom substituted alkyl halides can readily undergo dehaloprotonation (Fig. 1(b)). To overcome these challenges, the development of novel and efficient strategies is highly in demand.
Considering the challenges discussed above, we questioned if photoredox catalysis may offer a unique pathway for the carbonylation of α-heteroatom substituted alkyl halides. Synergistic photoredox catalysis and metal catalysis offer a powerful catalytic platform for many challenging organic transformations. Several outstanding studies have shown that pairing a visible-light photocatalyst with a conventional nickel or palladium catalyst can accelerate challenging steps in reactions.14 Herein, we report the first example of nickel/photoredox dual-catalyzed carbonylation of α-heteroatom substituted alkyl halides with various nucleophiles under low CO pressure, furnishing a series of high-value compounds (Fig. 1(c)). Notably, this catalytic strategy also enables the multicomponent carbonylation of vinyl phosphonate and affords the target compounds in moderate yields and with high selectivity.
Results and discussion
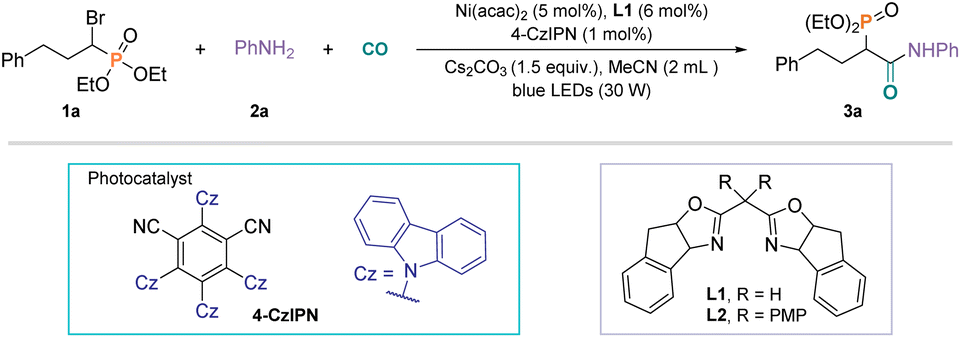
To establish this transformation and explore the optimal conditions, bromophosphate 1a and aniline 2a were selected as the model substrates. After a systematic evaluation of all the reaction parameters, the optimized reaction conditions were determined, as summarized in Table 1 (for more details, see the ESI†). Lower yields were obtained with other nickel catalysts than those with Ni(acac)2 (Table 1, entries 2 and 3). The nature of the ligand played an essential role in improving the yield of this reaction, and the bidentate nitrogen ligand L2 resulted in decreased yield (Table 1, entry 4). Next, Acr-Mes + ClO4− made the reaction almost impossible to occur (Table 1, entry 5). When fac-Ir(ppy)3 instead of 4-CzIPN was tested, a similar yet slightly diminished reactivity was observed (Table 1, entry 6). Diminished yield or no product was detected when using THF, DMAc, and DCE (the dehaloprotonation product was the main by-product), but PhCF3 could give a good yield (Table 1, entries 7 and 8). Screening of the bases highlighted the significant promotion of this carbonylation by Cs2CO3 (Table 1, entry 9). Notably, a good yield was also obtained with only 1 bar CO (Table 1, entries 10 and 14). Further assessment on the amount of bromophosphate indicated that 1.5 equivalents performed the best (Table 1, entry 11). In addition, the best yield of the desired product was observed in the presence of 10 mol% KI (Table 1, entry 12). We hypothesize that the reaction begins with the nucleophilic substitution of bromophosphate 1a with KI to form the corresponding iodophosphate. The desired product could not be detected under 60 bar CO in the presence of nickel or a photocatalyst (Table 1, entry 14). Experiments without a photocatalyst, nickel catalyst, ligand, Cs2CO3, or light failed to produce the desired carbonylated product, implying that all these components are necessary for the reaction to proceed (Table 1, entry 15). It is worthy of mention that easily accessible chiral dioxazole ligands were tested with the idea of introducing a chiral center, but low or no yield of the desired product was obtained without enantioselectivity.|
|
||
|---|---|---|
| Entry | Variation from standard conditions | 3a (%) |
| a Reaction conditions: 1a (1.2 equiv.), 2a (0.2 mmol), Ni(acac)2 (5 mol%), L1 (6 mol%), 4-CzIPN (1 mol%), Cs2CO3 (1.5 equiv.), CO (10 bar), MeCN (2 mL), 30 W blue LEDs, 18–25 °C, 24 h, isolated yields. acac = acetylacetone; TMHD = 2,2,6,6-tetramethyl-3,5-heptanedione. b 60 bar CO. | ||
| 1 | None | 68 |
| 2 | NiBr2(DME) instead of Ni(acac)2 | 17 |
| 3 | Ni(TMHD)2 instead of Ni(acac)2 | 36 |
| 4 | L2 instead of L1 | 20 |
| 5 | Acr-Mes+ClO4− instead of 4-CzIPN | 2 |
| 6 | fac-Ir(ppy)3 instead of 4-CzIPN | 59 |
| 7 | PhCF3 as solvent | 62 |
| 8 | THF, DMAc, DCE as solvent | 0–10 |
| 9 | DiPEA instead of Cs2CO3 | 0 |
| 10 | 1 bar CO instead of 10 bar CO | 60 |
| 11 | 1.5 equiv. of 1a was used | 75 |
| 12 | With 10 mol% KI | 93 |
| 13 | 1 bar CO, 1.5 equiv. of 1a, and 10 mol% KI | 82 |
| 14b | w/o Ni and PC | 0 |
| 15 | w/o PC or Ni or L1 or Cs2CO3 or light | 0 |
Encouraged by these results, we first investigated the scope of various nucleophiles under the optimized conditions (Fig. 2). Various aromatic amines substituted with an electron-donating group or an electron-withdrawing group were all suitable coupling partners. A variety of substituents including –F (3d), –Cl (3e), –Br (3f), and –CF3 (3g) survived, providing possibilities for further derivatization. The sterically bulky amine 3h also reacted equally well. In addition, a wide range of strongly nucleophilic alkylamines could be used, including benzylamine (3m), butylamine (3n), and amantadine (3o). Gratifyingly, heterocycle-containing substrates that readily coordinate with metals could successfully participate in this transformation to deliver the corresponding products in good yields (3p, 3q, 3r, and 3s). Next, some alcohols were also tested, such as long chain alcohols (4a, 4b, and 4c), cycloalcohols (4d and 4e), 3-methoxy-1-propanol (4f), chiral alcohol (4g) and benzyl alcohol (4h), giving the target products in moderate to excellent yields. Only a trace amount of the carbonylated product was detected when using sterically bulky tert-butanol as the substrate. In addition, phenols, which readily quench radicals, were also suitable substrates for this transformation (4j, 4k, 4l, and 4m). However, tert-butanol led to only a trace amount of the desired product (4i). Subsequently, we turned our attention to challenging nucleophiles. Several alcohols with various sensitive functional groups including trimethylsilyl (–TMS) and halogen atoms (–Cl and –I) were converted into the corresponding products in moderate to good yields (4n, 4o, and 4p). CD3OD was successfully converted into the corresponding D-containing product (4q) in 69% yield. Notably, the catalytic efficiency was unaffected when substrates with a carbon–carbon double bond (4r, 4s, 4t, and 4u) were used. Finally, we explored the reactivity of diols, and 1,6-hexanediol successfully delivered the diester product in good yield (4v). However, no target product could be obtained when thiophenol was used as the nucleophile.
 | ||
Fig. 2 Substrate scope. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reaction conditions: 1a (1.2 equiv.), 2 (0.2 mmol), Ni(acac)2 (5 mol%), L1 (6 mol%), 4-CzIPN (1 mol%), Cs2CO3 (1.5 equiv.), KI (10 mol%), CO (10 bar), MeCN (2 mL), 30 W blue LEDs, 18–25 °C, 24 h, isolated yields. bPhCF3 (2 mL). c1a (2.4 equiv.), 1,6-hexanediol (0.2 mmol), Ni(acac)2 (10 mol%), L1 (12 mol%), 4-CzIPN (2 mol%), Cs2CO3 (3 equiv.), KI (20 mol%), CO (10 bar), PhCF3 (3 mL). Reaction conditions: 1a (1.2 equiv.), 2 (0.2 mmol), Ni(acac)2 (5 mol%), L1 (6 mol%), 4-CzIPN (1 mol%), Cs2CO3 (1.5 equiv.), KI (10 mol%), CO (10 bar), MeCN (2 mL), 30 W blue LEDs, 18–25 °C, 24 h, isolated yields. bPhCF3 (2 mL). c1a (2.4 equiv.), 1,6-hexanediol (0.2 mmol), Ni(acac)2 (10 mol%), L1 (12 mol%), 4-CzIPN (2 mol%), Cs2CO3 (3 equiv.), KI (20 mol%), CO (10 bar), PhCF3 (3 mL). | ||
The conditions shown in Table 1 were then used for the transformation of various α-heteroatom substituted halides (Fig. 3). A series of phosphates with different steric hindrance, including diethyl, diisopropyl (5a), and dibutyl (5b) phosphonates, could be tolerated in this reaction. Other α-alkyl substituents of bromophosphates, such as long chain alkanes (5d, 5e, 5g, and 5j), cycloalkane (5f), heteroatom substituted alkane (6h), and halogen atom substituted alkane (5i), could be used in this reaction. However, both internal alkene (5k) and terminal alkene (5l) were all well tolerated in the transformation. Subsequently, α-bromoalkyldiarylphosphine oxides were successfully employed in this reaction, affording the target products in good to excellent yields (5m, 5n, 5o, 5p, 5q, and 5r). To demonstrate the practicality of this transformation, several natural products and bioactive molecules were also tested (Fig. 3). Aminoglutethimide (6a), sulfalen (6b), and amino acid derivatives (6c and 6d) were all suitable substrates. Likewise, geraniol (6e), phytol (6f), cholesterol (6g), menthol (6h) diacetonefructose (6i), lanosterol (6j), and epiandrosterone (6k) also reacted smoothly.
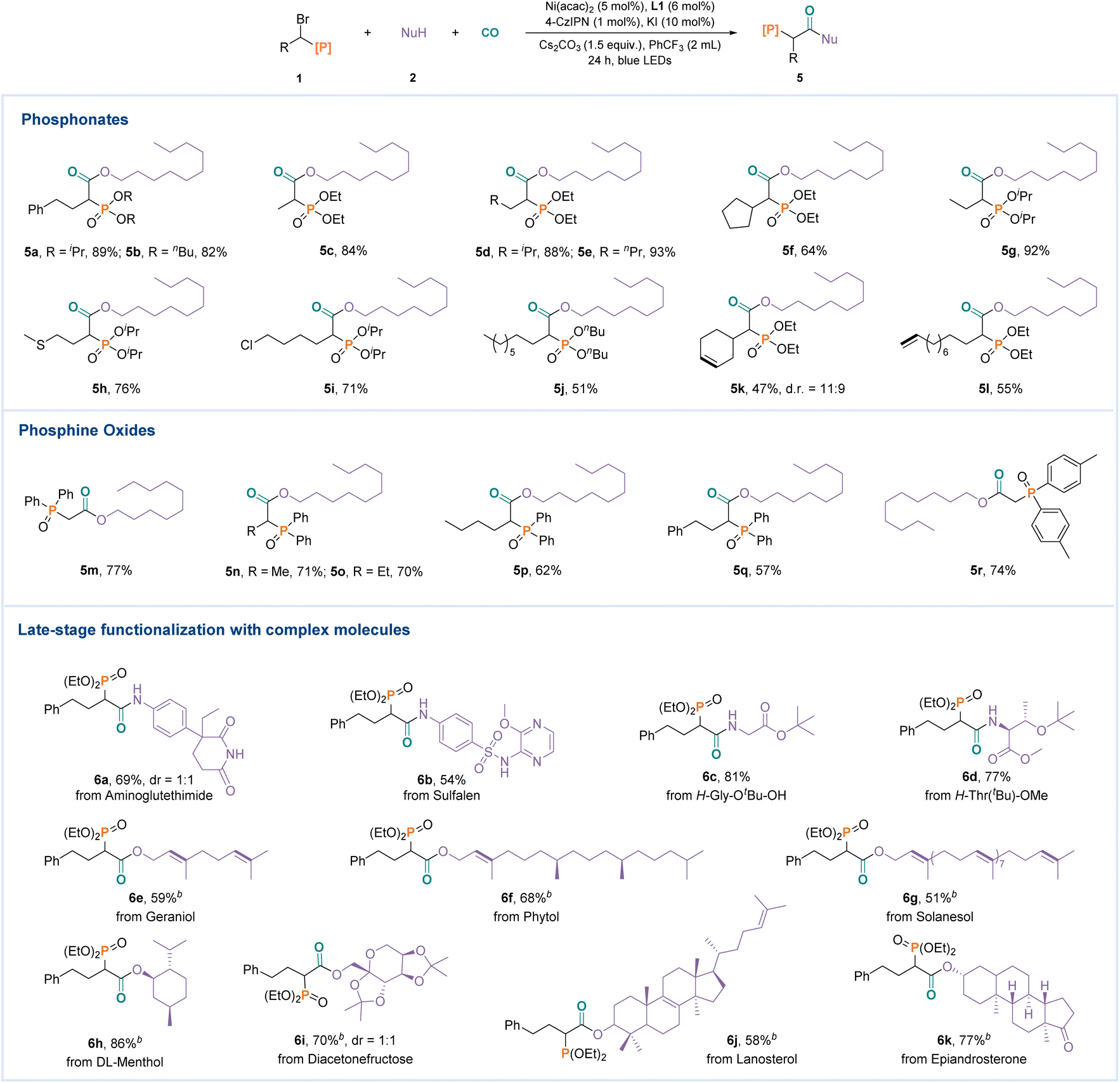 | ||
| Fig. 3 Substrate scope of α-phosphonates and complex molecules. Reaction conditions: 1a (1.2 equiv.), 2 (0.2 mmol), Ni(acac)2 (5 mol%), L1 (6 mol%), 4-CzIPN (1 mol%), Cs2CO3 (1.5 equiv.), KI (10 mol%), CO (10 bar), MeCN (2 mL), 30 W blue LEDs, 18–25 °C, 24 h, isolated yields. aPhCF3 (2 mL). | ||
Next, we found that various sulfur-containing compounds were also suitable carbonylated substrates (Fig. 4). Good yields were obtained with different functionalized α-bromo sulfone compounds (7a, 7b, 7c, 7d, and 7e). Finally, a carbonylative homologation of halomethylorganoborons, which are also important C1 reagents,15 was tested in this transformation. However, only acetylation products (7f and 7g) were obtained in good yields, which formed through a base-promoted protodeboronation of the originally produced carbonylative α-acylboron products.16
 | ||
| Fig. 4 Testing of reaction diversity. Reaction conditions: 1a (1.2 equiv.), 2 (0.2 mmol), Ni(acac)2 (5 mol%), L1 (6 mol%), 4-CzIPN (1 mol%), Cs2CO3 (1.5 equiv.), KI (10 mol%), CO (10 bar), MeCN (2 mL), 30 W blue LEDs, 18–25 °C, 24 h. aPhCF3 (2 mL). bYield reported post-deboronation. cR-Br (0.2 mmol), Cs2CO3 (3.5 equiv.), PhCF3 (2 mL). dVinyl phosphonate (1.5 equiv.), RF-I (2.5 equiv.), PhCF3 (2 mL). | ||
According to the literature, alkyl halides can be converted to the corresponding alcohols via oxyanions in the presence of bases.17 To our delight, we attempted to use alkyl halides instead of alcohols and the corresponding products were delivered in moderate yields (Fig. 4(c); 8a, 8b, 8c, and 8d). Catalytic carbonylative multicomponent reactions (CMCRs) represent a powerful and efficient strategy for the rapid construction of carbonyl-containing products in a single operation.18 Notably, a nickel/photoredox dual-catalyzed four-component α-heteroatom carbonylation reaction was also successfully achieved, and the target products were obtained in moderate yields (Fig. 4(d); 9a and 9b). In this way, the introduction of three useful fragments of phosphine, carbonyl, and fluorine into one molecule can be achieved.
To demonstrate the practicality and synthetic utility of this methodology, the carbonylation was performed on the 1 mmol scale and the target product 3a was delivered in 73% yield (Fig. 5(a)). Subsequently, we synthesized the Wittig–Horner reagent via a one-step reaction (Fig. 5(b)). Next, we successfully obtained the phosphine ligand 11 in 63% yield through the reduction reaction (Fig. 5(c)). The obtained phosphine ligand is a valuable and potential ligand in organic synthetic chemistry.19
 | ||
| Fig. 5 Synthetic applications. | ||
Subsequently, to better understand the pathway of this transformation, several control experiments were performed, as presented in Fig. 6. First, radical inhibition experiments were carried out by adding radical scavengers, and the results indicated that the reaction possibly proceeded via a radical pathway (Fig. 6(a)). The captured radical intermediate was detected by GC-MS and HRMS. Next, competition experiments between primary and secondary alkyl bromides with aniline were performed, and the primary substrate gave a slightly better yield (Fig. 6(b)). In order to test the reactivity of 3-iodopropanol, we carried out the reaction in the presence of 1.5 equivalents and 3.5 equivalents of Cs2CO3, respectively, and the corresponding product 8e was not detected in both cases (Fig. 6(c)). Finally, control experiments were performed to compare the current catalytic conditions with the several previously reported reactions. The developed strategy (Fig. 6(d); entry 1) afforded the desired product in high yields, whereas the previously reported carbonylation protocols (Fig. 6(d); entries 2–5) could not yield the target product (for more details, see the ESI†).
 | ||
| Fig. 6 Control experiments. | ||
Although elucidation of the detailed mechanism requires further studies, we proposed a possible catalytic pathway for this carbonylation reaction (Fig. 7), based on the above mechanistic studies and related literature.20 First, blue light irradiation of the photoredox catalyst would generate the excited-state PC*, which will oxidize the NiI species via a SET process to generate NiII species. Subsequently, the α-phosphonate alkyl radical A was delivered from α-bromophosphate through a SET reduction process. Then, the α-phosphonate alkyl radical A was trapped by CO to give the carbamoyl radical B, which was then quickly intercepted by the NiII species generated above to generate intermediate D. Alternatively, the α-phosphonate alkyl radical A was intercepted by the NiII species to afford the alkyl NiIII intermediate C, followed by migratory insertion with CO to deliver the same acyl species D. Next, the acyl complex D reacted with nucleophiles to afford the complex E. Finally, reductive elimination of the NiIII complex E provided the corresponding product and regenerated the nickel(I) complex for the next catalytic cycle.
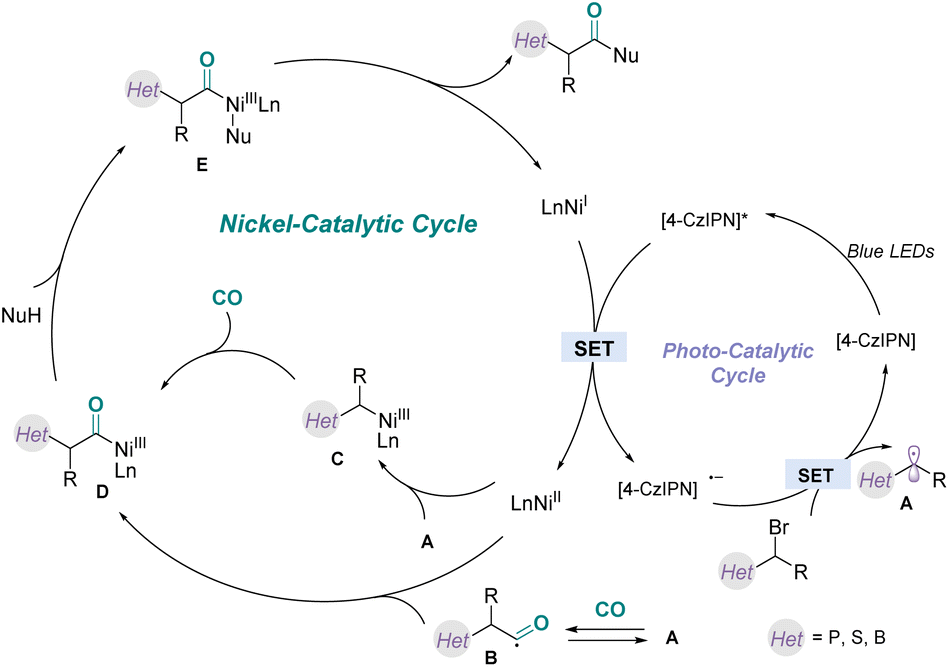 | ||
| Fig. 7 Proposed mechanism. | ||
Conclusions
In summary, we have identified a novel dual nickel/photoredox catalyzed direct amino- and alkoxycarbonylation of α-heteroatom substituted organohalides. The fundamental challenges posed by α-heteroatom effects, including decarbonylation, difficult oxidative addition, nucleophilic substitution, and reduction, can be circumvented by using the current nickel/photoredox catalyzed radical pathway. A variety of α-heteroatom substituted organohalides, including α-phosphorus, α-sulfur, and α-boron, reacted with amines, alcohols, phenols, and alkyl halides to deliver various α-heteroatom substituted amides and esters in excellent yields under mild conditions. In addition, a four-component carbonylation of vinyl phosphonate was also developed.Author contributions
X.-F. W. conceived and directed the project. L.-C. W. performed all the experiments. L.-C. W. and X.-F. W. wrote and revised the manuscript and the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2023YFA1507500) and the Dalian Institute of Chemical Physics DICP.References
- (a) R. Engel, Phosphonates as Analogues of Natural Phosphates, Chem. Rev., 1977, 77, 349–367 CrossRef CAS; (b) G. P. Horsman and D. L. Zechel, Phosphonate Biochemistry, Chem. Rev., 2017, 117, 5704–5783 CrossRef CAS PubMed; (c) K. M. Heidel and C. S. Dowd, Phosphonate Prodrugs: An Overview and Recent Advances, Future Med. Chem., 2019, 11, 1625–1643 CrossRef CAS PubMed; (d) S. J. Hecker and M. D. Erion, Prodrugs of Phosphates and Phosphonates, J. Med. Chem., 2008, 51, 2328–2345 CrossRef CAS PubMed; (e) W. Li and J. Zhang, Recent Developments in The Synthesis and Utilization of Chiral β-Aminophosphine Derivatives as Catalysts or Ligands, Chem. Soc. Rev., 2016, 45, 1657–1677 RSC; (f) W. S. Wadsworth and W. D. Emmons, The Utility of Phosphonate Carbanions in Olefin Synthesis, J. Am. Chem. Soc., 1961, 83, 1733–1738 CrossRef CAS; (g) B. E. Maryanoff and A. B. Reitz, The Wittig Olefination Reaction and Modifications Involving Phosphoryl-Stabilized Carbanions. Stereochemistry, Mechanism, and Selected Synthetic Aspects, Chem. Rev., 1989, 89, 863–927 CrossRef CAS.
- (a) M. Kitamura, M. Tokunaga and R. Noyori, Asymmetric Hydrogenation of .beta.-Keto Phosphonates: A Practical Way to Fosfomycin, J. Am. Chem. Soc., 1995, 117, 2931–2932 CrossRef CAS; (b) X. Li, H. Wang and T. Xu, Cross-Coupling of Aldehydes and α-Bromophosphonates to Modularly Access α-Substituted-β-Ketophosphonates under Dual Nickel/Photoredox Catalysis, Org. Chem. Front., 2023, 10, 3061–3066 RSC; (c) Q. Yang, C. Li, M.-X. Cheng and S.-D. Yang, Palladium-Catalyzed Migratory Insertion of Isocyanides for Synthesis of C-Phosphonoketenimines, ACS Catal., 2016, 6, 4715–4719 CrossRef CAS.
- (a) G. Kiss, Palladium-Catalyzed Reppe Carbonylation, Chem. Rev., 2001, 101, 3435–3456 CrossRef CAS PubMed; (b) H. Matsubara, T. Kawamoto, T. Fukuyama and I. Ryu, Applications of Radical Carbonylation and Amine Addition Chemistry: 1,4-Hydrogen Transfer of 1-Hydroxylallyl Radicals, Acc. Chem. Res., 2018, 51, 2023–2035 CrossRef CAS PubMed; (c) J.-B. Peng, F.-P. Wu and X.-F. Wu, First-Row Transition-Metal-Catalyzed Carbonylative Transformations of Carbon Electrophiles, Chem. Rev., 2019, 119, 2090–2127 CrossRef CAS PubMed; (d) J.-B. Peng, H.-Q. Geng and X.-F. Wu, The Chemistry of CO: Carbonylation, Chem, 2019, 5, 526–552 CrossRef CAS; (e) D.-M. Yan, C. M. Crudden, J.-R. Chen and W.-J. Xiao, A Career in Catalysis: Howard Alper, ACS Catal., 2019, 9, 6467–6483 CrossRef CAS; (f) L. Kollár, Modern Carbonylation Methods, Wiley-VCH, Weinheim, 2008 CrossRef; (g) R. Skoda-Földes and L. Kollár, Synthetic Applications of Palladium Catalysed Carbonylation of Organic Halides, Curr. Org. Chem., 2002, 6, 1097–1119 CrossRef.
- (a) M. Beller and X.-F. Wu, Transition Metal Catalyzed Carbonylation Reactions: Carbonylative Activation of C-X Bonds, Springer, Amsterdam, 1st edn, 2013 CrossRef; (b) B. T. Sargent and E. J. Alexanian, Palladium-Catalyzed Alkoxycarbonylation of Unactivated Secondary Alkyl Bromides at Low Pressure, J. Am. Chem. Soc., 2016, 138, 7520–7523 CrossRef CAS PubMed; (c) Y. Liu, C. Zhou, M. Jiang and B. A. Arndtsen, Versatile Palladium-Catalyzed Approach to Acyl Fluorides and Carbonylations by Combining Visible Light- and Ligand-Driven Operations, J. Am. Chem. Soc., 2022, 144, 9413–9420 CrossRef CAS PubMed; (d) H.-J. Ai, B. N. Leidecker, P. Dam, C. Kubis, J. Rabeah and X.-F. Wu, Iron-Catalyzed Alkoxycarbonylation of Alkyl Bromides via a Two-Electron Transfer Process, Angew. Chem., Int. Ed., 2022, 61, e202211939 CrossRef CAS PubMed; (e) P. Tung and N. P. Mankad, Light-Mediated Synthesis of Aliphatic Anhydrides by Cu-Catalyzed Carbonylation of Alkyl Halides, J. Am. Chem. Soc., 2023, 145, 9423–9427 CrossRef CAS PubMed; (f) H.-Y. Zhao, Z. Feng, Z. Luo and X. Zhang, Carbonylation of Difluoroalkyl Bromides Catalyzed by Palladium, Angew. Chem., Int. Ed., 2016, 55, 10401–10405 CrossRef CAS PubMed; (g) B. Lu, M. Xu, X. Qi, M. Jiang, W.-J. Xiao and J.-R. Chen, Switchable Radical Carbonylation by Philicity Regulation, J. Am. Chem. Soc., 2022, 144, 14923–14935 CrossRef CAS PubMed; (h) B. Lu, Y. Cheng, L.-Y. Chen, J.-R. Chen and W.-J. Xiao, Photoinduced Copper-Catalyzed Radical Aminocarbonylation of Cycloketone Oxime Esters, ACS Catal., 2019, 9, 8159–8164 CrossRef CAS; (i) B. Lu, Z. Zhang, M. Jiang, D. Liang, Z.-W. He, F.-S. Bao, W.-J. Xiao and J.-R. Chen, Photoinduced Five-Component Radical Relay Aminocarbonylation of Alkenes, Angew. Chem., Int. Ed., 2023, 62, e202309460 CrossRef CAS PubMed.
- (a) Y. Hoshimoto, T. Ohata, Y. Sasaoka, M. Ohashi and S. Ogoshi, Nickel(0)-Catalyzed [2 + 2 + 1] Carbonylative Cycloaddition of Imines and Alkynes or Norbornene Leading to γ-Lactams, J. Am. Chem. Soc., 2014, 136, 15877–15880 CrossRef CAS PubMed; (b) T. L. Andersen, A. S. Donslund, K. T. Nuemann and T. Skrydstrup, Carbonylative Coupling of Alkyl Zinc Reagents with Benzyl Bromides Catalyzed by a Nickel/NN2 Pincer Ligand Complex, Angew. Chem., Int. Ed., 2018, 57, 800–804 CrossRef CAS PubMed; (c) A. K. Ravn, M. B. T. Vilstrup, P. Noerby, D. U. Nielsen, K. Daasbjerg and T. Skrydstrup, Carbon Isotope Labeling Strategy for β-Amino Acid Derivatives via Carbonylation of Azanickellacycles, J. Am. Chem. Soc., 2019, 141, 11821–11826 CrossRef CAS PubMed; (d) J.-B. Peng, F.-P. Wu, C. Xu, X. Qi, J. Ying and X.-F. Wu, Nickel-Catalyzed Carbonylative Synthesis of Functionalized Alkyl Iodides, iScience, 2018, 8, 175–182 CrossRef CAS PubMed; (e) A. S. Donslund, S. S. Pedersen, C. Gaardbo, K. T. Neumann, L. Kingston, C. S. Elmore and T. Skrydstrup, Direct Access to Isotopically Labeled Aliphatic Ketones Mediated by Nickel(I) Activation, Angew. Chem., Int. Ed., 2020, 59, 8099–8103 CrossRef CAS PubMed.
- (a) N. Liu, X. Wu, C. Wang, J. Qu and Y. Chen, Nickel-Catalyzed Alkoxycarbonylation of Aryl Iodides with 1 atm CO, Chem. Commun., 2022, 58, 4643–4646 RSC; (b) J.-B. Peng, F.-P. Wu, D. Li, X. Qi, J. Ying and X.-F. Wu, Nickel-Catalyzed Molybdenum-Promoted Carbonylative Synthesis of Benzophenones, J. Org. Chem., 2018, 83, 6788–6792 CrossRef CAS PubMed.
- L. Hou, W. Huang, X. Wu, J. Qu and Y. Chen, Nickel-Catalyzed Carbonylation of Cyclopropanol with Benzyl Bromide for Multisubstituted Cyclopentenone Synthesis, Org. Lett., 2022, 24, 2699–2704 CrossRef CAS PubMed.
- (a) R. Shi and X. Hu, From Alkyl Halides to Ketones: Nickel-Catalyzed Reductive Carbonylation Utilizing Ethyl Chloroformate as the Carbonyl Source, Angew. Chem., Int. Ed., 2019, 58, 7454–7458 CrossRef CAS PubMed; (b) R. Yu, S.-Z. Cai, C. Li and X. Fang, Nickel-Catalyzed Asymmetric Hydroaryloxy- and Hydroalkoxycarbonylation of Cyclopropenes, Angew. Chem., Int. Ed., 2022, 61, e202200733 CrossRef CAS PubMed; (c) Z.-P. Bao and X.-F. Wu, Palladium-catalyzed carbonylation of activated alkyl halides via radical intermediates, Ind. Chem. Mater., 2024 10.1039/D3IM00078H.
- (a) S.-J. He, J.-W. Wang, Y. Li, Z.-Y. Xu, X.-X. Wang, X. Lu and Y. Fu, Nickel-Catalyzed Enantioconvergent Reductive Hydroalkylation of Olefins with α-Heteroatom Phosphorus or Sulfur Alkyl Electrophiles, J. Am. Chem. Soc., 2020, 142, 214–221 CrossRef CAS PubMed; (b) J. Choi, P. Martín-Gago and G. C. Fu, Stereoconvergent Arylations and Alkenylations of Unactivated Alkyl Electrophiles: Catalytic Enantioselective Synthesis of Secondary Sulfonamides and Sulfones, J. Am. Chem. Soc., 2014, 136, 12161–12165 CrossRef CAS PubMed; (c) L. Gong, H.-B. Sun, L.-F. Deng, X. Zhang, J. Liu, S. Yang and D. Niu, Ni-Catalyzed Suzuki–Miyaura Cross-Coupling of α-Oxo-vinylsulfones to Prepare C-Aryl Glycals and Acyclic Vinyl Ethers, J. Am. Chem. Soc., 2019, 141, 7680–7686 CrossRef CAS PubMed.
- (a) X. Li, M. Yuan, F. Chen, Z. Huang, F.-L. Qing, O. Gutierrez and L. Chu, Three-Component Enantioselective Alkenylation of Organophosphonates via Nickel Metallaphotoredox Catalysis, Chem, 2023, 9, 154–169 CrossRef CAS; (b) L. Qi, X. Pang, K. Yin, Q.-Q. Pan, X.-X. Wei and X.-Z. Shu, Mn-Mediated Reductive C(sp3)–Si Coupling of Activated Secondary Alkyl Bromides with Chlorosilanes, Chin. Chem. Lett., 2022, 33, 5061–5064 CrossRef CAS.
- (a) J. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, Sausalito, CA, 2010, pp. 301–320 Search PubMed; (b) B. T. Sargent and E. J. Alexanian, Palladium-Catalyzed Alkoxycarbonylation of Unactivated Secondary Alkyl Bromides at Low Pressure, J. Am. Chem. Soc., 2016, 138, 7520–7523 CrossRef CAS PubMed.
- C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chemistry of Acyl Radicals, Chem. Rev., 1999, 99, 1991–2070 CrossRef CAS PubMed.
- G. S. Lee, B. Park and S. H. Hong, Stereoretentive Cross-Coupling of Chiral Amino Acid Chlorides and Hydrocarbons through Mechanistically Controlled Ni/Ir Photoredox Catalysis, Nat. Commun., 2022, 13, 5200–5210 CrossRef CAS PubMed.
- (a) D. Kalyani, K. B. McMurtrey, S. R. Neufeldt and M. S. Sanford, Room-Temperature C–H Arylation: Merger of Pd-Catalyzed C–H Functionalization and Visible-Light Photocatalysis, J. Am. Chem. Soc., 2011, 133, 18566–18569 CrossRef CAS PubMed; (b) J. C. Tellis, D. N. Primer and G. A. Molander, Single-Electron Transmetalation in Organoboron Cross-Coupling by Photoredox/Nickel Dual Catalysis, Science, 2014, 345, 433–436 CrossRef CAS PubMed; (c) S. Z. Tasker and T. F. Jamison, Highly Regioselective Indoline Synthesis under Nickel/Photoredox Dual Catalysis, J. Am. Chem. Soc., 2015, 137, 9531–9534 CrossRef CAS PubMed; (d) K. L. Skubi, T. R. Blum and T. P. Yoon, Dual Catalysis Strategies in Photochemical Synthesis, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed; (e) G. M. Torres, Y. Liu and B. A. Arndtsen, A Dual Light-Driven Palladium Catalyst: Breaking the Barriers in Carbonylation Reactions, Science, 2020, 368, 318–323 CrossRef CAS PubMed; (f) K. Chami, E. Y. Liu, M. A. Belahouane, Y. Ma, P.-L. Lagueux-Tremblay and B. A. Arndtsen, A Visible Light Driven Nickel Carbonylation Catalyst: The Synthesis of Acid Chlorides from Alkyl Halides, Angew. Chem., Int. Ed., 2023, 62, e202213297 CrossRef PubMed; (g) A. M. Veatch, S. Liu and E. J. Alexanian, Cobalt-Catalyzed Deaminative Amino- and Alkoxycarbonylation of Aryl Trialkylammonium Salts Promoted by Visible Light, Angew. Chem., Int. Ed., 2022, 61, e202210772 CrossRef CAS PubMed.
- (a) K. A. C. Bastick and A. J. B. Watson, Pd-Catalyzed Organometallic-Free Homologation of Arylboronic Acids Enabled by Chemoselective Transmetalation, ACS Catal., 2023, 13, 7013–7018 CrossRef CAS PubMed; (b) B. Li, A. Bunescu and M. J. Gaunt, Multicomponent Synthesis of α-Chloro Alkylboronic Esters via Visible-Light-Mediated Dual Catalysis, Chem, 2023, 9, 1–11 CrossRef.
- (a) Z. Kuang, K. Yang, Y. Zhou and Q. Song, Base-Promoted Domino-Borylation-Protodeboronation Strategy, Chem. Commun., 2020, 56, 6469 RSC; (b) F.-P. Wu, Y. Yang, D. P. Fuentes and X.-F. Wu, Copper-Catalyzed Carbonylative Catenation of Olefins: Direct Synthesis of γ-Boryl Esters, Chem, 2022, 8, 1982–1992 CrossRef CAS.
- (a) H.-J. Ai, H. Wang, C.-L. Li and X.-F. Wu, Rhodium-Catalyzed Carbonylative Coupling of Alkyl Halides with Phenols under Low CO Pressure, ACS Catal., 2020, 10, 5147–5152 CrossRef CAS; (b) Y. Zhang, H.-Q. Geng and X.-F. Wu, Palladium-Catalyzed Perfluoroalkylative Carbonylation of Unactivated Alkenes: Access to β-Perfluoroalkyl Esters, Angew. Chem., Int. Ed., 2021, 60, 24292–24298 CrossRef CAS PubMed.
- (a) L. Albrecht, H. Jiang and K. A. Jorgensen, A Simple Recipe for Sophisticated Cocktails: Organocatalytic One-Pot Reactions—Concept, Nomenclature, and Future Perspectives, Angew. Chem., Int. Ed., 2011, 50, 8492–8509 CrossRef CAS PubMed; (b) A. Fusano, S. Sumino, S. Nishitani, T. Inouye, K. Morimoto, T. Fukuyama and I. Ryu, Pd/Light-Accelerated Atom-Transfer Carbonylation of Alkyl Iodides: Applications in Multicomponent Coupling Processes Leading to Functionalized Carboxylic Acid Derivatives, Chem. – Eur. J., 2012, 18, 9415–9422 CrossRef CAS PubMed; (c) J.-X. Xu, L.-C. Wang and X.-F. Wu, Non-Noble Metal-Catalyzed Carbonylative Multi-Component Reactions, Chem. – Asian J., 2022, 17, e202200928 CrossRef CAS PubMed.
- P. Braunstein, D. Matt, Y. Dusausoy, J. Fischer, A. Mitschler and L. Ricard, Complexes of Functional Phosphines. 4. Coordination Properties of (Diphenylphosphino)acetonitrile, Ethyl (Diphenylphosphino)acetate and Corresponding Carbanions. Characterization of a New Facile Reversible Carbon Dioxide Insertion into Palladium(II) Complexes, J. Am. Chem. Soc., 1981, 103, 5115–5125 CrossRef CAS.
- (a) H. Wang, P. Zheng, X. Wu, Y. Li and T. Xu, Modular and Facile Access to Chiral α-Aryl Phosphates via Dual Nickel- and Photoredox-Catalyzed Reductive Cross-Coupling, J. Am. Chem. Soc., 2022, 144, 3989–3997 CrossRef CAS PubMed; (b) H. Wang, X. Wu and T. Xu, Enantioconvergent Reductive C(sp)−C(sp3) Cross-Coupling to Access Chiral α-Alkynyl Phosphonates Under Dual Nickel/Photoredox Catalysis, Angew. Chem., Int. Ed., 2023, e202218299 CAS; (c) S. Zhao and N. P. Mankad, Metal-Catalysed Radical Carbonylation Reactions, Catal. Sci. Technol., 2019, 9, 3603–3613 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qo00167b |
| This journal is © the Partner Organisations 2024 |