
Interception of RCONCl2: late-stage hydrolysis and esterification of primary amides†
Xiao
Wan
,
Jiaxin
Lin
,
Junjie
Bai
,
Jingjing
Li
,
Kaifeng
Wang
,
Zhiyu
Cao
,
Minggang
Li
,
Xiaoguang
Bao
 * and
Xiaobing
Wan
*
* and
Xiaobing
Wan
*
Key Laboratory of Organic Synthesis of Jiangsu Province, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, China. E-mail: xgbao@suda.edu.cn; wanxb@suda.edu.cn
First published on 21st November 2023
Abstract
Current strategies for activating amides primarily focus on secondary or tertiary amides. However, the activation of primary amides, the simplest form of amides, remains a significant challenge. In this study, we propose a novel approach that involves generating and intercepting RCONCl2in situ to activate primary amides under mild conditions. This method allows for hydrolysis and esterification of primary amides without the use of strong Lewis acid or transition-metal catalysts typically used in amide activation. We demonstrate the synthetic potential of this approach through late-stage modifications on complex molecules, tolerance toward over 20 heterocycles, and scalability. Mechanistic experiments and DFT calculations suggest that the formation of RCONCl2 and subsequent nucleophilic attack with hydrogen bonding assistance play crucial roles in this transformation.
Introduction
Amide bonds are crucial structures commonly found in bioactive molecules, natural products, drugs, polymers, and functional materials.1 However, despite their significance and prevalence, the chemical community often overlooks them as synthetic modules. This is because amides have lower reactivity compared to other carboxylic acid derivatives like acyl halides, anhydrides, and acids due to amidic resonance (RE = 15–20 kcal mol−1 in planar amides), which is explained in basic organic chemistry textbooks. Consequently, there has been a historical perception that amides are too inert to be used as acylation reagents.Despite the formidable challenges, recent years have seen significant success in C(O)–N bond activation. This has greatly expanded the toolkit available to chemists, allowing them to utilize the synthetic potential of typically inert amides. Since Ghosez's pioneering work,2 the use of trifluoromethanesulfonic anhydride for amide electrophilic activation has become a powerful tool in synthetic chemistry (Fig. 1a).3 This is thanks to significant contributions from Huang,4 Maulide,5 and other research groups.6 Furthermore, there have been recent conceptual advancements in amide C(O)–N bond activation from the groups of Garg,7 Szostak,8 and Zou.9 Through a ground-state destabilization strategy, twisted amides were used as electrophiles (Fig. 1b) to obtain acylation or decarbonylation products under transition-metal or transition-metal-free reaction conditions.10 Both electrophilic activation and ground-state destabilization of amides have now become mainstream techniques in synthetic chemistry. These methods enable the transformation of inert amide bonds into functional groups that can be modified, facilitating a wide range of previously challenging reactions.
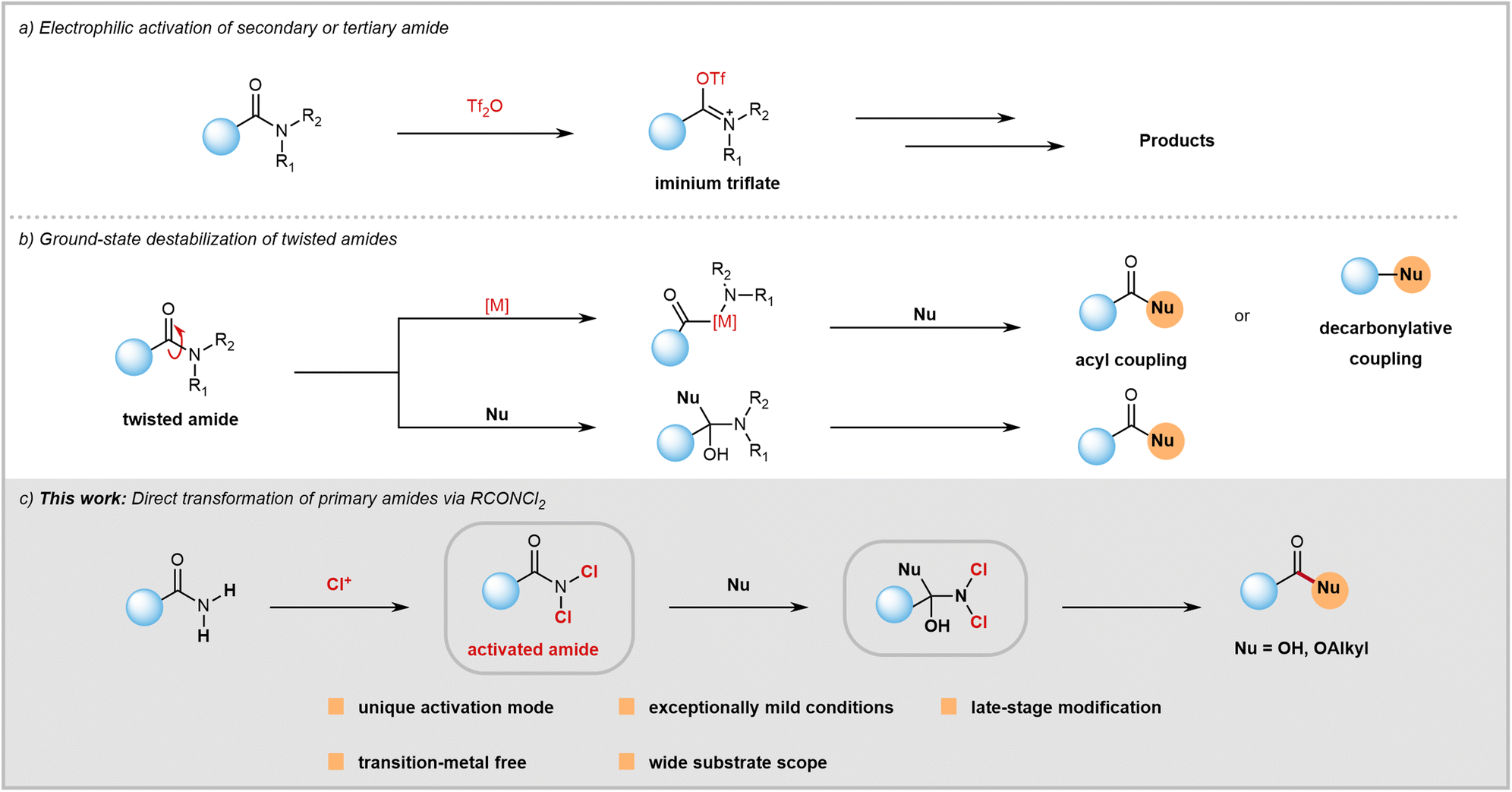 | ||
| Fig. 1 Activation strategies for amides. (a) Electrophilic activation of secondary or tertiary amide. (b) Ground-state destabilization of twisted amides. (c) This work: Direct transformation of primary amides via RCONCl2. | ||
Despite progress in the field, research on amide activation lags behind that of other carboxylic acid derivatives. The chemical community is actively seeking novel activation strategies to overcome existing challenges. Compared to secondary and tertiary amides, the activation of primary amides has barely been explored and remains a huge challenge. Primary amides, unlike secondary and tertiary amides, cannot react with trifluoromethanesulfonic anhydride to form the required iminium intermediate due to the weak Lewis basicity of their oxygen atoms. Therefore, electrophilic activation is not suitable for primary amides. While ground-state destabilization has proven successful with sterically twisted tertiary amides, achieving geometric alterations in primary amides is difficult. Twisted tertiary amides are not commercially available and often require pre-synthesis from primary or secondary amides, which can be time-consuming and inefficient in terms of atom usage. In contrast, primary amides are readily available as commercial feedstock chemicals (with nearly 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 options) or can be easily prepared from various carboxylic acids with diverse structures. Therefore, directly transforming primary amides bearing diverse structures not only provides a powerful method for acyl transfer but also holds significant potential to impact amide chemistry and contribute to drug discovery efforts.
000 options) or can be easily prepared from various carboxylic acids with diverse structures. Therefore, directly transforming primary amides bearing diverse structures not only provides a powerful method for acyl transfer but also holds significant potential to impact amide chemistry and contribute to drug discovery efforts.
In order to enhance the versatility of primary amides as a synthetic module, we propose a unique method to weaken their planar resonance. Our hypothesis is that the electron-withdrawing nature of chlorine atoms allows for predictable activation of amides by reducing planar resonance (Fig. 1c). Furthermore, the small size of chlorine can facilitate attack on RCONCl2 by nucleophiles. By using this strategy, a mild approach for hydrolysis and esterification reactions of both aromatic and aliphatic primary amides has been successfully developed. This innovative approach offers several distinct advantages such as late-stage modification, transition-metal-free conditions, resistance to air and moisture, broad substrate compatibility, and operational simplicity.
Results and discussion
Initially, we performed preliminary theoretical calculations to test our hypothesis. We used the COSNAR method developed by Greenberg et al.11 to calculate the resonance energy of the amide bond in PhCONH2 and PhCONCl2. The calculated values were 19.7 kcal mol−1 for PhCONH2 and 4.5 kcal mol−1 for PhCONCl2 (Fig. 2a). These results indicate a significant weakening of the C(O)–N bond in PhCONCl2, which could enhance nucleophilic attack on the carbonyl group. A similar outcome can be achieved through an isodesmic reaction as shown in Fig. 2b. | ||
| Fig. 2 DFT calculations on the effect of chlorine atoms. | ||
Our hypothesis was that RCONCl2, with weakened planar resonance, is more reactive than RCONH2 in nucleophilic substitution reactions. This is the key to the success of our designed scenario, which we intend to verify with hydrolysis reactions. Hydrolysis of primary amides is typically unfavorable both thermodynamically and kinetically, requiring harsh conditions such as strong acids/bases and high temperatures.12 In 2020, Garg and coworkers developed a one-pot, two-step hydrolysis strategy, in which 2-(trimethylsilyl)ethanol was used for nickel-catalyzed esterification followed by fluoride-promoted deprotection to generate the corresponding carboxylic acid.13 We conducted hydrolysis experiments on RCONCl2 and RCONH2.14 As anticipated, the experimental findings showed that RCONCl2 exhibited higher reactivity compared to RCONH2, albeit with low yields (Fig. 3). These results effectively demonstrate the viability of our concept: by introducing chlorine atoms, we can greatly reduce the planar resonance of amides and transform them into acylating agents with significant synthetic potential.
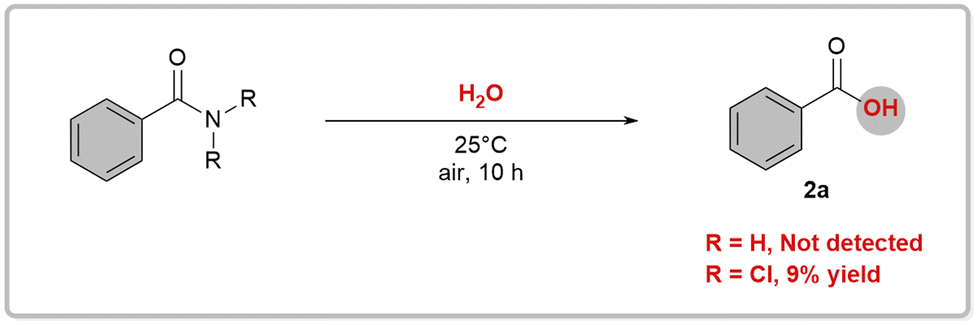 | ||
| Fig. 3 Initial hydrolysis test. | ||
Although the initial experimental results mentioned above show great promise, there were still significant challenges to overcome in activating primary amides. Chlorinating reagents often lead to chlorination or oxidation of nucleophiles (water or alcohol).15 Hence, it is crucial to identify a suitable chlorinating reagent that reacts preferably with primary amides to obtain RCONCl2, rather than causing undesired side reactions with nucleophiles. Additionally, the identified chlorinating reagents should react quickly and cleanly with primary amides, eliminating the need for further purification. The resulting RCONCl2 can then undergo smooth nucleophilic substitution to produce the desired products.
After testing various chlorinating reagents, we found tert-butyl hypochlorite to be the most effective option for hydrolysis of primary amides. By mixing p-chlorobenzamide and tert-butyl hypochlorite in acetone/H2O at room temperature for 24 hours, we achieved a high yield of 99% for the desired acid 2b (Table S1, entry 1 in ESI†). Hydrolysis reactions of amides are known to be extremely slow, with a half-life of several hundred years.16 However, by generating and intercepting RCONCl2in situ, we successfully established a mild hydrolysis method for primary amides under neutral conditions. It is important to note that other commercially available chlorinating reagents yielded inferior results (Table S1, entries 2–4†), and that changing the solvent type also resulted in poorer product formation outcomes (Table S1, entries 5–15†). One notable advantage of this reaction setup is its simplicity; it does not require air removal or the use of metal catalysts.
After developing a practical protocol, we assessed its generality for primary amides. Scheme 1 demonstrates that the optimized reaction conditions successfully tolerated a wide range of common functional groups, including halide (2b, 2c, 2e, 2k, 2o–2q), alkyl (2f and 2g), carboxyl (2l), sulfone (2h), CF3 (2i, 2j, 2n, 2p) and nitro (2d and 2o). Notably, a tertiary amide (2x) was also compatible with this system. Oxidizable functional groups such as free hydroxyl groups (2aa) and easily hydrolyzable groups, such as ester (2w), BocO (2m), and TsO (2t), could be preserved during the reaction. Encouragingly, positional changes in ortho-, meta-, or para-substituents on aromatic amides did not impact the reaction outcomes. Desired carboxylic acids were obtained in good to nearly quantitative yields regardless of these substitutions. Compared to aromatic amides containing electron-withdrawing groups, those with electron-donating groups had lower activity and required heating conditions for efficient hydrolysis (2f and 2g). This indicates that the rate-determining step for these hydrolysis reactions is nucleophilic attack by water on RCONCl2. We then focused on the hydrolysis of aliphatic amides, which are typically considered unreactive in twisted amide activation reactions due to their resistance to oxidative addition with transition-metal catalysts.10y This transition-metal-free approach was expected to work for both aromatic and aliphatic amides. Various structurally diverse aliphatic amides (2m, 2t, 2v–2x) were treated with standard reaction conditions, resulting in high yields of the desired carboxylic acids.
 | ||
| Scheme 1 Scope of amides for hydrolysis. Reaction conditions: amide (0.5 mmol), tert-butyl hypochlorite (4.0 equiv.), acetone/H2O (2.0 mL/2.0 mL), under air at 25 °C for 24 h. Isolated yield. a40 °C. btert-Butyl hypochlorite (6.0 equiv.). | ||
Heterocycles are commonly found in biologically active molecules, pharmaceuticals, agrochemicals, and advanced materials. A limitation of transition metal-catalyzed amide activations is their reduced feasibility for substrates bearing heterocycles due to the poisoning of transition metal catalyst. Fortunately, our hydrolysis system was found to be compatible with various medicinally relevant heterocycles such as thiazole (2u), pyridine (2ac), pyrrolidin-2-one (2x), phthalimide (2v), 1,2,3-triazole (2z), and 1,2,4-triazole (2aa). Amide hydrolysis is a crucial step in the synthesis of drug molecules and complex products.17 However, it typically requires strong acids or bases. Our methodology offers excellent functional group compatibility and mild reaction conditions, making it suitable for late-stage modification of complex molecules. As expected, dinitolmide, an approved anticoccidial agent, yielded the desired product 2y almost quantitatively. Highly functionalized drug molecules like rufinamide (2z) with a 1,2,3-triazole moiety and ribavirin (2aa) with a 1,2,4-triazole moiety were both converted into the corresponding acids with excellent yields. Probenecid, an antipodagrics compound bearing a tertiary sulfonamide group, also smoothly reacted to produce the desired product 2ab in a yield of 93%. Considering the ease of further transforming carboxylic acids, we believe this hydrolysis methodology will enhance the toolkit for drug discovery programs exploring these pharmacophores. To demonstrate the robustness of this methodology further, we scaled up the amide hydrolysis using 4-chlorobenzamide as the raw material. Without modifying the original protocol, we successfully transformed 10 mmol of 4-chlorobenzamide into product 2b without any significant decrease in yield.
Both secondary amide (N-methylbenzamide) and tertiary amide (N,N-dimethylbenzamide) were found to be unsuitable substrates for the hydrolysis process. The Winkler–Dunitz parameters (τ + χN) are often used to describe the geometric distortion of amides, which can predict the reactivity of twisted amides. Calculations show that PhCONCl2 (Fig. 4) has less distortion compared to classical twisted amides like N,N-Boc2-benzamide18 and N-benzoyl-glutarimide.19 However, despite this higher distortion, the expected hydrolysis reaction did not occur in the case of twisted amides. We also tested Weinreb amide,20 a commonly used acylation reagent. Surprisingly, it did not undergo hydrolysis; instead, most starting materials were recovered. These results clearly demonstrate our approach offers a complementary and fundamentally distinct strategy for activating amides, which may be due to the significant pyramidalization of nitrogen atom and small size of the chlorine atom.
 | ||
| Fig. 4 The geometric distortion of PhCONCl2. | ||
The activity of carboxylic esters is generally higher than that of amides. Therefore, the esterification of amides typically requires harsh reaction conditions such as strong acidity or basicity, toxic reagents, high temperatures, and/or large amounts of alcohol solvents.21 Since the pioneering work from the group of Garg,7 a series of esterification methods for twisted amides10y,22 have been successfully developed under transition-metal or metal-free conditions. However, these methods are mainly applicable to aromatic tertiary amides. Building on the success achieved in hydrolysis reactions, we aimed to extend this strategy to other nucleophilic substitution reactions of amides. Interestingly, although hydrolysis and esterification share similar elementary steps, there is currently no general method that can be applied to both processes. We were excited to find that this innovative activation strategy can be used not only for amide hydrolysis, but also for achieving amide esterification under mild conditions. By intercepting RCONCl2 with alcohols, both aromatic and aliphatic amides can effectively function as acylating agents, resulting in high yields of the desired ester products.
At the outset, a systematic survey of esterification reaction conditions was carried out using amide 3a and alcohol 4a as model substrates (for more information on reaction optimization, see Table S2 in the ESI†). The reaction was performed in cyclohexane with TCCA (trichloroisocyanuric acid) as the chlorinating reagent and PhCOOLi as the base. This resulted in the successful synthesis of desired ester 5a, with an impressive yield of 87%. With the optimal conditions in hand, the scope of amides in this process was investigated (Scheme 2). Remarkably, the esterification reaction exhibited a wide range of functional group tolerance; various groups such as halide (5a, 5c, 5d, 5n, 5s–5u), NO2 (5e and 5n), CF3 (5m), carbonyl (5i), ester (5j and 5u), ether (5k), and benzyl chloride (5h), remained unaffected under these mild reaction conditions. Pentafluorobenzamide (5l) proved to be a suitable substrate for this protocol. Both trichloroacetamide (5s) and dichloroacetamide (5t) yielded desired esters with satisfactory yields. It is worth noting that this esterification system is compatible with both biologically relevant carbon rings such as cyclopentane 5q and naphthalene 5r, as well as heterocycles like isoxazole 5o and benzofuran 5p. As expected, when ethyl oxamate was used as a reactant, the desired ester 5u was obtained with a yield of 79%. Not surprisingly, secondary amide, tertiary amide, twisted amides, and the Weinreb amide were found to be unreactive under these conditions.
 | ||
| Scheme 2 Scope of amides for esterification. Reaction conditions: amides 3 (0.2 mmol), alcohol 4a (2.0 equiv.), TCCA (2.0 equiv.), PhCOOLi (2.5 equiv.), cyclohexane (2.0 mL), under air at 25 °C for 1 h, then 40 °C for 48 h. Isolated yield. a25 °C for 1 h, then 60 °C for 48 h. | ||
After defining the scope of primary amides, we evaluated the generality of the alcohols (Scheme 3). Methanol (5v) and butanol (5w) smoothly produced esters with high yields. A major limitation of current amide alcoholysis is its compatibility only with simple alcohols.21,22 In this study, a wide range of structurally diverse alcohols were successfully converted to valuable functionalized esters by intercepting RCONCl2. This demonstrated an extensive tolerance for different functional groups, including alkyl halides (5x, 5an, 5as, and 5ax), aryl halides (5aa–5ac), CF3 (5ad and 5bd), carbonate (5bb), Boc (5am, 5ap, 5az and 5bc), ketal (5ag), phosphate (5af), silyl (5ah and 5ar), ketone (5ak), ester (5be), ether (5y), tertiary alcohol (5aq), and epoxide (5ai and 5aj) groups which remained unaffected in the reaction. Primary amides are often used as nucleophiles to form C–N bonds by replacing the hydroxy group of benzyl alcohols.23 However, in this study, benzyl alcohols were unexpectedly found to be suitable nucleophiles instead of electrophiles. As a result, desired benzyl esters (5z, 5aa, 5ac–5ae) were obtained with high yields. It is worth mentioning that when secondary alcohols were used as reactants, moderate yields of the corresponding esters (5bc–5bg) were also obtained. Additionally, under these mild esterification conditions, optically pure substrates (5am, 5az, and 5be) were converted into corresponding esters without changing their configuration. This advantage is expected to benefit the application of this method in chiral drug discovery.
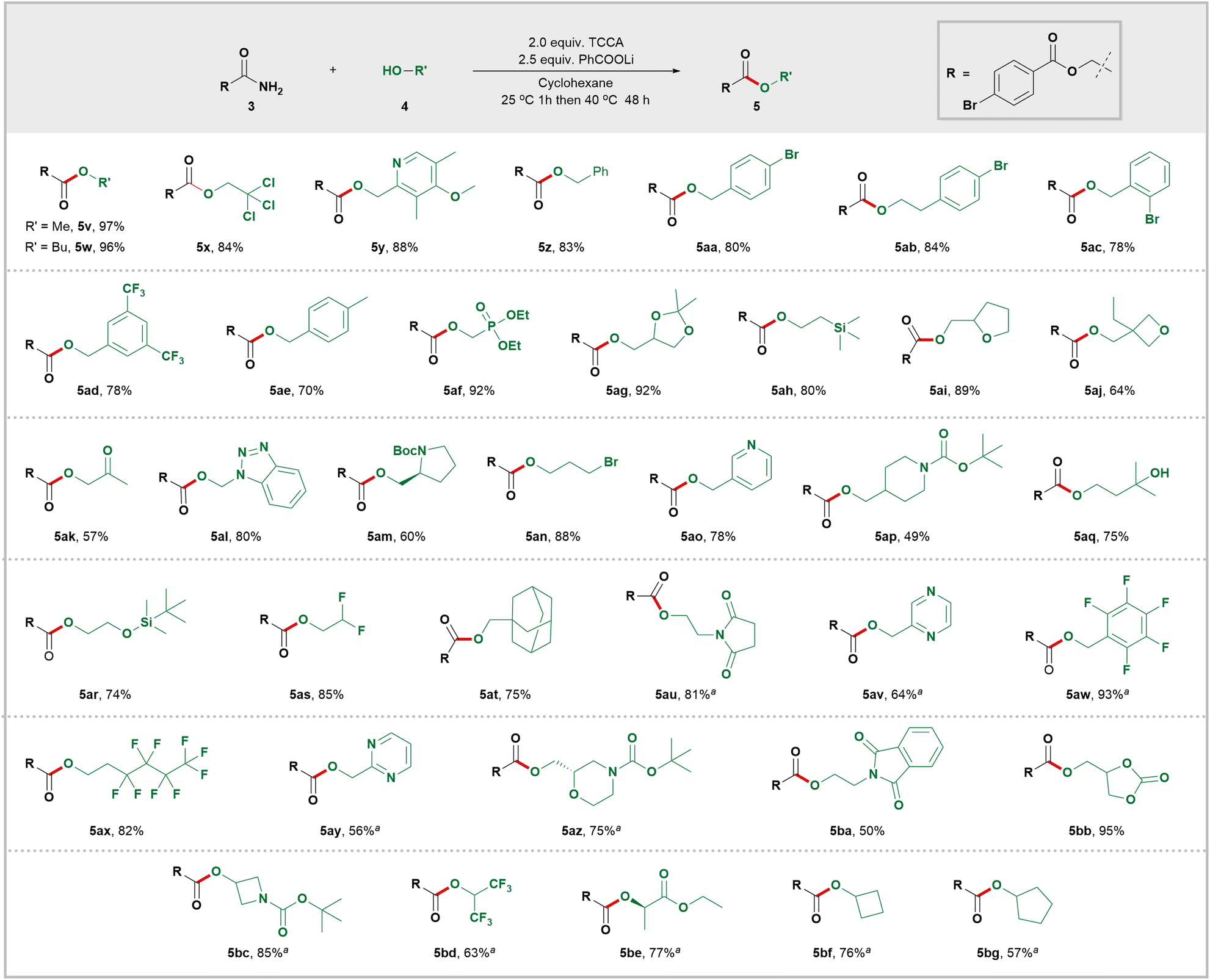 | ||
| Scheme 3 Scope of alcohols for esterification. Amides (0.2 mmol), alcohols (2.0 equiv.), TCCA (2.0 equiv.), PhCOOLi (2.5 equiv.), cyclohexane (2.0 mL), under air at 25 °C for 1 h, then 40 °C for 48 h. Isolated yield. a25 °C for 1 h, then 60 °C for 48 h. | ||
Given the prevalence of cyclic scaffolds in drugs and agrochemicals, we conducted experiments to assess the efficacy of various alcohols in synthesizing esters with different carbo- and heterocyclic scaffolds. This transformation readily accommodated carbocyclic compounds like cyclobutane (5bf), cyclopentane (5bg), and adamantane (5at). Heterocyclic compounds containing oxygen atoms such as oxetane (5aj), tetrahydrofuran (5ai), 1,3-dioxolane (5ag), and 1,3-dioxolan-2-one (5bb)—common motifs in drug scaffolds—also underwent efficient esterification. Medicinally relevant nitrogen heterocycles including pyridine (5y and 5ao), pyrrolidine (5am), piperidine (5ap), azetidine (5bc), and succinimide (5au and 5ba) successfully delivered the desired esters with good to excellent efficiency. Multiple-nitrogen-heterocycles such as pyrimidine (5ay), pyrazine (5av), and benzotriazole (5al) were also converted into corresponding esters with high yields.
In order to further demonstrate the effectiveness of our novel strategy, we next conducted late-stage modifications of complex molecules or drug scaffolds. As shown in Scheme 4, primary amides progenitors of nicotinamid (5bh), aspirin (5bi), rufinamide (5bj), temozolomide (5bk), fenofibric acid (5bl), UV absorber THUV-328 (5bm), dinitolmide (5bn), and probenecid (5bo) were subjected to our mild conditions, generating the corresponding esters in moderate to excellent yields. We also tested a variety of structurally diverse alcohols for this transformation. For instance, diacetonefructose (5bp) and diacetonegalactose (5bt and 5bw) both delivered the desired esters with satisfactory yields. Cyclandelate (5bq), an α-hydroxy ester, was smoothly converted into its corresponding ester with excellent yield. Esterification of the primary alcohols in menthone (5br and 5bu) delivered the corresponding esters in high yields. When using icaridine as the alcohol source, high yields of the desired ester 5bs and 5bv were achieved. These findings demonstrate both the practicality and versatility of our innovative amide activation strategy, which can accommodate a wide range of pharmaceutically relevant functional groups including heterocycles, sulfonamides, aryl bromides, and more.
 | ||
| Scheme 4 Late-stage modification on complex molecules. Reaction conditions: amides (0.2 mmol), alcohols (2.0 equiv.), TCCA (2.0 equiv.), PhCOOLi (2.5 equiv.), cyclohexane (2.0 mL), under air at 25 °C for 1 h, then 40 °C for 48 h. Isolated yield. a25 °C for 1 h, then 60 °C for 48 h. | ||
During the hydrolysis of amides, the presence of RCONCl2 was detected. Under standard conditions, RCONCl2 can be efficiently converted into a carboxylic acid in nearly quantitative yield (Fig. 5a). When RCONCl2 and RCONH2 were separately reacted under standard conditions for one hour (Fig. 5b), it was observed that the conversion rate of RCONCl2 was significantly higher than that of RCONH2. Based on these results, we conclude that RCONCl2 serves as an intermediate product in the hydrolysis of amides. The competition experiment demonstrates that electron-withdrawing groups are more favorable for the hydrolysis of amides (Fig. 5c). This indicates that the nucleophilic attack of water molecules on RCONCl2 is the rate-determining step of this process.
 | ||
| Fig. 5 Preliminary mechanistic study. | ||
Computational studies were performed to shed light on the hydrolysis mechanism of PhCONCl2 (Fig. 6). When a single water molecule attacks the carbonyl carbon of PhCONCl2 to undergo hydrolysis, the predicted activation barrier, however, is very high (for details, see Fig. S1 in ESI†). The presence of more water molecules might stabilize the leaving group (–NCl2) via a H-bonding interaction, and hence could assist the hydrolysis of PhCONCl2. Computational results suggest that the activation energy would be lowered significantly (24.2 kcal mol−1) when five water molecules are involved in the hydrolysis of PhCONCl2 (for hydrolysis involving two-four water molecules, see Fig. S2–S4 in ESI†).
 | ||
| Fig. 6 Energy profile (in kcal mol−1) for the hydrolysis of primary amides. | ||
Conclusions
Through the interception of RCONCl2, we have developed a mechanistically distinct and versatile method for activating primary amides. This protocol allows the conversion of various types of primary amides, including aromatic and aliphatic ones, into acids or esters under metal-free conditions. It uses commercially available primary amides as the source of acyl, greatly simplifying operations, shortening the synthetic sequence, and enhancing overall efficiency in atom utilization. It can tolerate over 20 heterocycles and nearly all common functional groups. Furthermore, we demonstrate the synthetic potential of this approach by performing late-stage modifications of complex products and drug-like molecules. Mechanistic investigation and DFT calculations suggest that this transformation involves the generation of RCONCl2 followed by nucleophilic attack facilitated by hydrogen bonding.Data availability
Full experimental results are available in the ESI.†Author contributions
X. Wan and X.B. conceived and supervised the project. X.W., J.B., J.L., Z.C. and M.L. performed the experiments and developed the reactions. J.L. and K.W. performed the DFT calculations for the mechanistic studies. X. Wan wrote the manuscript with feedback from all authors. All authors analyzed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), NSFC (grant numbers 21973068, 21971175).References
- (a) A. Greenberg, C. M. Breneman and J. F. Liebman, The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science, Wiley-Interscience, New York, NY, USA, 2000 Search PubMed; (b) J. M. Humphrey and A. R. Chamberlin, Chemical Synthesis of Natural Product Peptides: Coupling Methods for the Incorporation of Noncoded Amino Acids into Peptides, Chem. Rev., 1997, 97, 2243–2266 CrossRef CAS.
- J.-B. Falmagne, J. Escudero, S. Taleb-Sahraoui and L. Ghosez, Cyclobutanone and Cyclobutenone Derivatives by Reaction of Tertiary Amides with Alkenes or Alkynes, Angew. Chem., Int. Ed. Engl., 1981, 20, 879–880 CrossRef.
- (a) M. Feng, H. Zhang and N. Maulide, Challenges and Breakthroughs in Selective Amide Activation, Angew. Chem., Int. Ed., 2022, 61, e202212213 CrossRef CAS; (b) D. Kaiser, A. Bauer, M. Lemmerer and N. Maulide, Amide Activation: an Emerging Tool for Chemoselective Synthesis, Chem. Soc. Rev., 2018, 47, 7899–7925 RSC; (c) P. Q. Huang, Direct Transformations of Amides: Tactics and Recent Progress, Acta Chim. Sin., 2018, 76, 357–365 CrossRef CAS; (d) D. Kaiser and N. Maulide, Making the Least Reactive Electrophile the First in Class: Domino Electrophilic Activation of Amides, J. Org. Chem., 2016, 81, 4421–4428 CrossRef CAS.
- (a) K. L. Ji, S. F. He, D. D. Xu, W. X. He, J. F. Zheng and P. Q. Huang, Concise Total Synthesis of (–)-Quinocarcin Enabled by Catalytic Enantioselective Reductive 1,3-Dipolar Cycloaddition of Secondary Amides, Angew. Chem., Int. Ed., 2023, 62, e202302832 CrossRef CAS; (b) X. Z. Huang, L. H. Gao and P. Q. Huang, Enantioselective Total Syntheses of (+)-Stemofoline and Three Congeners Based on A Biogenetic Hypothesis, Nat. Commun., 2020, 11, 5314–5322 CrossRef CAS; (c) Z. Xu, X. G. Wang, Y. H. Wei, K. L. Ji, J. F. Zheng, J. L. Ye and P. Q. Huang, Organocatalytic, Enantioselective Reductive Bis-functionalization of Secondary Amides: One-Pot Construction of Chiral 2,2-Disubstituted 3-Iminoindoline, Org. Lett., 2019, 21, 7587–7591 CrossRef CAS; (d) Y. H. Huang, S. R. Wang, D. P. Wu and P. Q. Huang, Intermolecular Dehydrative [4 + 2] Aza-Annulation of N-Arylamides with Alkenes: a Direct and Divergent Entrance to Aza-Heterocycles, Org. Lett., 2019, 21, 1681–1685 CrossRef CAS; (e) T. Fan, A. Wang, J. Q. Li, J. L. Ye, X. Zheng and P. Q. Huang, Versatile One-Pot Synthesis of Polysubstituted Cyclopent-2-enimines from α, β-Unsaturated Amides: Imino-Nazarov Reaction, Angew. Chem., Int. Ed., 2018, 57, 10352–10356 CrossRef CAS PubMed; (f) P. Q. Huang, W. Ou, K. J. Xiao and A. E. Wang, Tertiary Amide-Based Knoevenagel-Type Teactions: A Direct, General, and Chemoselective Approach to Enaminones, Chem. Commun., 2014, 50, 8761–8763 RSC; (g) K. J. Xiao, Y. Wang, Y. H. Huang, X. G. Wang and P. Q. Huang, A Direct and General Method for the Reductive Alkylation of Tertiary Lactams/Amides: Application to the Step Economical Synthesis of Alkaloid (–)-Morusimic Acid D, J. Org. Chem., 2013, 78, 8305–8311 CrossRef CAS; (h) K. J. Xiao, A. E. Wang and P. Q. Huang, Direct Transformation of Secondary Amides into Secondary Amines: Triflic Anhydride Activated Reductive Alkylation, Angew. Chem., Int. Ed., 2012, 51, 8314–8317 CrossRef CAS; (i) K. J. Xiao, Y. Wang, K. Y. Ye and P. Q. Huang, Versatile One-Pot Reductive Alkylation of Lactams/Amides via Amide Activation: Application to the Concise Syntheses of Bioactive Alkaloids (±)-Bgugaine, (±)-Coniine, (+)-Preussin, and (–)-Cassine, Chem. – Eur. J., 2010, 16, 12792–12796 CrossRef CAS PubMed; (j) K. J. Xiao, J. M. Luo, K. Y. Ye, Y. Wang and P. Q. Huang, Direct, One-pot Sequential Reductive Alkylation of Lactams/Amides with Grignard and Organolithium Reagents through Lactam/Amide activation, Angew. Chem., Int. Ed., 2010, 49, 3037–3040 CrossRef CAS.
- (a) M. Feng, A. J. Fernandes, R. Meyrelles and N. Maulide, Direct Enantioselective α-Amination of Amides Guided by DFT Prediction of E/Z Selectivity in A Sulfonium Intermediate, Chem, 2023, 9, 1538–1548 CrossRef CAS; (b) S. Heindl, M. Riomet, J. Matyasovsky, M. Lemmerer, N. Malzer and N. Maulide, Chemoselective γ-Oxidation of β,γ-Unsaturated Amides with TEMPO, Angew. Chem., Int. Ed., 2021, 60, 19123–19127 CrossRef CAS PubMed; (c) V. Porte, G. Di Mauro, M. Schupp, D. Kaiser and N. Maulide, Chemoselective α-Deuteration of Amides via Retro-Ene Reaction, Chem. – Eur. J., 2020, 26, 15509–15512 CrossRef CAS; (d) C. J. Teskey, P. Adler, C. R. Goncalves and N. Maulide, Chemoselective α,β-Dehydrogenation of Saturated Amides, Angew. Chem., Int. Ed., 2019, 58, 447–451 CrossRef CAS PubMed; (e) J. Li, M. Berger, W. Zawodny, M. Simaan and N. Maulide, A Chemoselective α-Oxytriflation Enables the Direct Asymmetric Arylation of Amides, Chem, 2019, 5, 1883–1891 CrossRef CAS; (f) C. R. Goncalves, M. Lemmerer, C. J. Teskey, P. Adler, D. Kaiser, B. Maryasin, L. Gonzalez and N. Maulide, Unified Approach to the Chemoselective α-Functionalization of Amides with Heteroatom Nucleophiles, J. Am. Chem. Soc., 2019, 141, 18437–18443 CrossRef CAS; (g) P. Adler, C. J. Teskey, D. Kaiser, M. Holy, H. H. Sitte and N. Maulide, α-Fluorination of Carbonyls with Nucleophilic Fluorine, Nat. Chem., 2019, 11, 329–334 CrossRef CAS PubMed; (h) D. Kaiser, C. J. Teskey, P. Adler and N. Maulide, Chemoselective Intermolecular Cross-Enolate-Type Coupling of Amides, J. Am. Chem. Soc., 2017, 139, 16040–16043 CrossRef CAS PubMed; (i) D. Kaiser, A. de la Torre, S. Shaaban and N. Maulide, Metal-Free Formal Oxidative C−C Coupling by In Situ Generation of an Enolonium Species, Angew. Chem., Int. Ed., 2017, 56, 5921–5925 CrossRef CAS.
- (a) Z. J. Niu, L. H. Li, X. S. Li, H. C. Liu, W. Y. Shi and Y. M. Liang, Formation of o-Allyl- and Allenyl-Modified Amides via Intermolecular Claisen Rearrangement, Org. Lett., 2021, 23, 1315–1320 CrossRef CAS; (b) D. Matheau-Raven and D. J. Dixon, General α-Amino 1,3,4-Oxadiazole Synthesis via Late-Stage Reductive Functionalization of Tertiary Amides and Lactams, Angew. Chem., Int. Ed., 2021, 60, 19725–19729 CrossRef CAS; (c) J. W. Jiao and X. M. Wang, Merging Electron Transfer with 1,2-Metalate Rearrangement: Deoxygenative Arylation of Aromatic Amides with Arylboronic Esters, Angew. Chem., Int. Ed., 2021, 60, 17088–17093 CrossRef CAS; (d) G. He, B. List and M. Christmann, Unified Synthesis of Polycyclic Alkaloids by Complementary Carbonyl Activation, Angew. Chem., Int. Ed., 2021, 60, 13591–13596 CrossRef CAS; (e) A. Dubart and G. Evano, Divergent Synthesis of α-Fluorinated Carbonyl and Carboxyl Derivatives by Double Electrophilic Activation of Amides, Org. Lett., 2021, 23, 8931–8936 CrossRef CAS; (f) M. Leypold, K. A. D'Angelo and M. Movassaghi, Chemoselective α-Sulfidation of Amides Using Sulfoxide Reagents, Org. Lett., 2020, 22, 8802–8807 CrossRef CAS PubMed; (g) M. Bhunia, S. R. Sahoo, A. Das, J. Ahmed, P. Sreejyothi and S. K. Mandal, Transition Metal-Free Catalytic Reduction of Primary Amides Using an Abnormal NHC Based Potassium Complex: Integrating Nucleophilicity with Lewis Acidic Activation, Chem. Sci., 2020, 11, 1848–1854 RSC; (h) S. Katahara, S. Kobayashi, K. Fujita, T. Matsumoto, T. Sato and N. Chida, An Iridium-Catalyzed Reductive Approach to Nitrones from N-Hydroxyamides, J. Am. Chem. Soc., 2016, 138, 5246–5249 CrossRef CAS PubMed; (i) J. W. Medley and M. Movassaghi, Direct Dehydrative N-Pyridinylation of Amides, J. Org. Chem., 2009, 74, 1341–1344 CrossRef CAS.
- L. Hie, N. F. F. Nathel, T. K. Shah, E. L. Baker, X. Hong, Y.-F. Yang, P. Liu, K. N. Houk and N. K. Garg, Conversion of Amides to Esters by the Nickel-Catalysed Activation of Amide C−N Bonds, Nature, 2015, 524, 79–83 CrossRef CAS PubMed.
- G. Meng and M. Szostak, Sterically Controlled Pd-Catalyzed Chemoselective Ketone Synthesis via N−C Cleavage in Twisted Amides, Org. Lett., 2015, 17, 4364–4367 CrossRef CAS PubMed.
- X. Li and G. Zou, Acylative Suzuki Coupling of Amides: Acyl-Nitrogen Activation via Synergy of Independently Modifiable Activating Groups, Chem. Commun., 2015, 51, 5089–5092 RSC.
- (a) D. Zuo, Q. Wang, L. Liu, T. Huang, M. Szostak and T. Chen, Highly Chemoselective Transamidation of Unactivated Tertiary Amides by Electrophilic N−C(O) Activation by Amide-to-Acyl Iodide Re-routing, Angew. Chem., Int. Ed., 2022, 61, e202202794 CrossRef CAS PubMed; (b) J. Zhang, P. Zhang, L. Shao, R. Wang, Y. Ma and M. Szostak, Mechanochemical Solvent-Free Suzuki-Miyaura Cross-Coupling of Amides via Highly Chemoselective N−C Cleavage, Angew. Chem., Int. Ed., 2022, 61, e202114146 CrossRef CAS; (c) Y. Y. Ping, X. Li, Q. Pan and W. Q. Kong, Ni-Catalyzed Divergent Synthesis of 2-Benzazepine Derivatives via Tunable Cyclization and 1,4-Acyl Transfer Triggered by Amide N−C Bond Cleavage, Angew. Chem., Int. Ed., 2022, 61, e202201574 CrossRef CAS; (d) T. B. Boit, M. M. Mehta, J. Kim, E. L. Baker and N. K. Garg, Reductive Arylation of Amides via a Nickel-Catalyzed Suzuki-Miyaura-Coupling and Transfer-Hydrogenation Cascade, Angew. Chem., Int. Ed., 2021, 60, 2472–2477 CrossRef CAS; (e) Z. Wang, A. Matsumoto and K. Maruoka, Efficient Cleavage of Tertiary Amide Bonds via Radical-Polar Crossover Using a Copper(II) Bromide/Selectfluor Hybrid System, Chem. Sci., 2020, 11, 12323–12328 RSC; (f) G. J. Wang, Q. Q. Shi, W. Y. Hu, T. Chen, Y. Y. Guo, Z. L. Hu, M. H. Gong, J. C. Guo, D. H. Wei, Z. Q. Fu and W. Huang, Organocatalytic Asymmetric N-Sulfonyl Amide C−N Bond Activation to Access Axially Chiral Biaryl Amino Acids, Nat. Commun., 2020, 11, 946–955 CrossRef CAS PubMed; (g) W. Sun, L. Wang, Y. Hu, X. D. Wu, C. G. Xia and C. Liu, Chemodivergent Transformations of Amides Using gem-Diborylalkanes as Pro-Nucleophiles, Nat. Commun., 2020, 11, 3113–3123 CrossRef CAS; (h) D. Y. Ong, D. Fan, D. J. Dixon and S. Chiba, Transition-Metal-Free Reductive Functionalization of Tertiary Carboxamides and Lactams for α-Branched Amine Synthesis, Angew. Chem., Int. Ed., 2020, 59, 11903–11907 CrossRef CAS; (i) G. C. Li, S. Y. Ma and M. Szostak, Amide Bond Activation: The Power of Resonance, Trends Chem., 2020, 2, 914–928 CrossRef CAS; (j) T. B. Boit, A. S. Bulger, J. E. Dander and N. K. Garg, Activation of C−O and C−N Bonds Using Non-Precious-Metal Catalysis, ACS Catal., 2020, 10, 12109–12126 CrossRef CAS PubMed; (k) D. Y. Ong, Z. Yen, A. Yoshii, J. Revillo Imbernon, R. Takita and S. Chiba, Controlled Reduction of Carboxamides to Alcohols or Amines by Zinc Hydrides, Angew. Chem., Int. Ed., 2019, 58, 4992–4997 CrossRef CAS; (l) G. Li, C.-L. Ji, X. Hong and M. Szostak, Highly Chemoselective, Transition-Metal-Free Transamidation of Unactivated Amides and Direct Amidation of Alkyl Esters by N−C/O−C Cleavage, J. Am. Chem. Soc., 2019, 141, 11161–11172 CrossRef CAS; (m) C. Liu and M. Szostak, Decarbonylative Cross-Coupling of Amides, Org. Biomol. Chem., 2018, 16, 7998–8010 RSC; (n) G. C. Li and M. Szostak, Highly Selective Transition-Metal-Free Transamidation of Amides And Amidation of Esters at Room Temperature, Nat. Commun., 2018, 9, 4165–4172 CrossRef PubMed; (o) H. Yue, L. Guo, S. C. Lee, X. Liu and M. Rueping, Selective Reductive Removal of Ester and Amide Groups from Arenes and Heteroarenes through Nickel-Catalyzed C−O and C−N Bond Activation, Angew. Chem., Int. Ed., 2017, 56, 3972–3976 CrossRef CAS PubMed; (p) J. A. Walker, Jr., K. L. Vickerman, J. N. Humke and L. M. Stanley, Ni-Catalyzed Alkene Carboacylation via Amide C−N Bond Activation, J. Am. Chem. Soc., 2017, 139, 10228–10231 CrossRef; (q) J. M. Medina, J. Moreno, S. Racine, S. J. Du and N. K. Garg, Mizoroki-Heck Cyclizations of Amide Derivatives for the Introduction of Quaternary Centers, Angew. Chem., Int. Ed., 2017, 56, 6567–6571 CrossRef CAS; (r) C. W. Liu and M. Szostak, Decarbonylative Phosphorylation of Amides by Palladium and Nickel Catalysis: The Hirao Cross-Coupling of Amide Derivatives, Angew. Chem., Int. Ed., 2017, 56, 12718–12722 CrossRef CAS; (s) C. L. Ji and X. Hong, Factors Controlling the Reactivity and Chemoselectivity of Resonance Destabilized Amides in Ni-Catalyzed Decarbonylative and Nondecarbonylative Suzuki-Miyaura Coupling, J. Am. Chem. Soc., 2017, 139, 15522–15529 CrossRef CAS; (t) H. Wu, Y. Li, M. Cui, J. Jian and Z. Zeng, Suzuki Coupling of Amides via Palladium-Catalyzed C−N Cleavage of N-Acylsaccharins, Adv. Synth. Catal., 2016, 358, 3876–3880 CrossRef CAS; (u) N. A. Weires, E. L. Baker and N. K. Garg, Nickel-Catalysed Suzuki-Miyaura Coupling of Amides, Nat. Chem., 2016, 8, 75–79 CrossRef CAS PubMed; (v) S. Shi, G. Meng and M. Szostak, Synthesis of Biaryls through Nickel-Catalyzed Suzuki-Miyaura Coupling of Amides by Carbon-Nitrogen Bond Cleavage, Angew. Chem., Int. Ed., 2016, 55, 6959–6963 CrossRef CAS; (w) G. Meng and M. Szostak, Rhodium-Catalyzed C−H Bond Functionalization with Amides by Double C−H/C−N Bond Activation, Org. Lett., 2016, 18, 796–799 CrossRef CAS PubMed; (x) J. Hu, Y. Zhao, J. Liu, Y. Zhang and Z. Shi, Nickel-Catalyzed Decarbonylative Borylation of Amides: Evidence for Acyl C−N Bond Activation, Angew. Chem., Int. Ed., 2016, 55, 8718–8722 CrossRef CAS PubMed; (y) L. Hie, E. L. Baker, S. M. Anthony, J. N. Desrosiers, C. Senanayake and N. K. Garg, Nickel-Catalyzed Esterification of Aliphatic Amides, Angew. Chem., Int. Ed., 2016, 55, 15129–15132 CrossRef CAS.
- A. Greenberg, D. T. Moore and T. D. DuBois, Small and Medium-Sized Bridgehead Bicyclic Lactams: A Systematic ab Initio Molecular Orbital Study, J. Am. Chem. Soc., 1996, 118, 8658–8668 CrossRef CAS.
- (a) S. M. A. H. Siddiki, M. N. Rashed, A. S. Touchy, M. A. R. Jamil, Y. Jing, T. Toyao, Z. Maeno and K.-i. Shimizu, Hydrolysis of amides to carboxylic acids catalyzed by Nb2O5, Catal. Sci. Technol., 2021, 11, 1949–1960 RSC; (b) X. Lopez, J. I. Mujika, G. M. Blackburn and M. Karplus, Alkaline Hydrolysis of Amide Bonds: Effect of Bond Twist and Nitrogen Pyramidalization, J. Phys. Chem. A, 2003, 107, 2304–2315 CrossRef CAS; (c) R. S. Brown, A. J. Bennet and H. Slebocka-Tilk, Recent perspectives concerning the mechanism of H3O+- and OH−-promoted amide hydrolysis, Acc. Chem. Res., 1992, 25, 481–488 CrossRef CAS.
- R. R. Knapp, A. S. Bulger and N. K. Garg, Nickel-Catalyzed Conversion of Amides to Carboxylic Acids, Org. Lett., 2020, 22, 2833–2837 CrossRef CAS.
- L. D. Luca, G. Giacomelli and G. Nieddu, Efficient N-Chlorination of Amides and Carbamates, Synlett, 2005, 223–226 Search PubMed.
- (a) S. Gaspa, M. Carraro, L. Pisano, A. Porcheddu and L. De Luca, Trichloroisocyanuric Acid: a Versatile and Efficient Chlorinating and Oxidizing Reagent, Eur. J. Org. Chem., 2019, 3544–3552 CrossRef CAS; (b) F. Munyemana, I. George, A. Devos, A. Colens, E. Badarau, A.-M. Frisque-Hesbain, A. Loudet, E. Differding, J.-M. Damien, J. Rémion, J. Van Uytbergen and L. Ghosez, A Mild Method for the Replacement of A Hydroxyl Group by Halogen, Tetrahedron, 2016, 72, 420–430 CrossRef CAS; (c) Z. Li, S. Crosignani and B. Linclau, A Mild, Phosphine-free Method for the Conversion of Alcohols into Halides (Cl, Br, I) via the Corresponding O-Alkyl Isoureas, Tetrahedron Lett., 2003, 44, 8143–8147 CrossRef CAS; (d) N. E. Leadbeater, H. M. Torenius and H. Tye, Ionic Liquids as Reagents and Solvents in Conjunction with Microwave Heating: Rapid Synthesis of Alkyl Halides from Alcohols and Nitriles from Aryl Halides, Tetrahedron, 2003, 59, 2253–2258 CrossRef CAS; (e) G. A. Hiegel and M. Rubino, Conversion of Alcohols into Alkyl Chlorides Using Trichloroisocyanuric Acid with Triphenylphosphine, Synth. Commun., 2002, 32, 2691–2694 CrossRef CAS; (f) L. D. Luca, G. Giacomelli and A. Porcheddu, An Efficient Route to Alkyl Chlorides from Alcohols Using the Complex TCT/DMF, Org. Lett., 2001, 4, 553–555 CrossRef PubMed.
- A. Radzicka and R. Wolfenden, Rates of Uncatalyzed Peptide Bond Hydrolysis in Neutral Solution and the Transition State Affinities of Proteases, J. Am. Chem. Soc., 1996, 118, 6105–6109 CrossRef CAS.
- (a) M. J. R. Richter, M. Schneider, M. Brandstatter, S. Krautwald and E. M. Carreira, Total Synthesis of (–)-Mitrephorone A, J. Am. Chem. Soc., 2018, 140, 16704–16710 CrossRef CAS PubMed; (b) K. Mitachi, B. A. Aleiwi, C. M. Schneider, S. Siricilla and M. Kurosu, Stereocontrolled Total Synthesis of Muraymycin D1 Having a Dual Mode of Action against Mycobacterium tuberculosis, J. Am. Chem. Soc., 2016, 138, 12975–12980 CrossRef CAS PubMed.
- G. R. Meng, S. C. Shi and M. Szostak, Palladium-Catalyzed Suzuki-Miyaura Cross-Coupling of Amides via Site-Selective N−C Bond Cleavage by Cooperative Catalysis, ACS Catal., 2016, 6, 7335–7339 CrossRef CAS.
- G. Meng and M. Szostak, General Olefin Synthesis by the Palladium-Catalyzed Heck Reaction of Amides: Sterically Controlled Chemoselective N−C Activation, Angew. Chem., Int. Ed., 2015, 54, 14518–14522 CrossRef CAS PubMed.
- R. Senatore, L. Ielo, S. Monticelli, L. Castoldi and V. Pace, Weinreb Amides as Privileged Acylating Agents for Accessing α-Substituted Ketones, Synthesis, 2019, 51, 2792–2808 CrossRef CAS.
- (a) B. Villoria-del-Alamo, S. Rojas-Buzo, P. Garcia-Garcia and A. Corma, Zr-MOF-808 as Catalyst for Amide Esterification, Chem. – Eur. J., 2021, 27, 4588–4598 CrossRef CAS PubMed; (b) M. N. Rashed, S. Siddiki, A. S. Touchy, M. A. R. Jamil, S. S. Poly, T. Toyao, Z. Maeno and K. Shimizu, Direct Phenolysis Reactions of Unactivated Amides into Phenolic Esters Promoted by a Heterogeneous CeO2 Catalyst, Chem. – Eur. J., 2019, 25, 10594–10605 CrossRef CAS PubMed; (c) X. L. Chen, S. Y. Hu, R. X. Chen, J. Wang, M. H. Wu, H. B. Guo and S. F. Sun, Fe-Catalyzed Esterification of Amides via C−N Bond Activation, RSC Adv., 2018, 8, 4571–4576 RSC; (d) Y. Kita, Y. Nishii, A. Onoue and K. Mashima, Combined Catalytic System of Scandium Triflate and Boronic Ester for Amide Bond Cleavage, Adv. Synth. Catal., 2013, 355, 3391–3395 CrossRef CAS; (e) Y. Kita, Y. Nishii, T. Higuchi and K. Mashima, Zinc-Catalyzed Amide Cleavage and Esterification of β-Hydroxyethylamides, Angew. Chem., Int. Ed., 2012, 51, 5723–5726 CrossRef CAS.
- (a) C. Huang, J. Li, J. Wang, Q. Zheng, Z. Li and T. Tu, Hydrogen-Bond-Assisted Transition-Metal-Free Catalytic Transformation of Amides to Esters, Sci. China: Chem., 2021, 64, 66–71 CrossRef CAS; (b) H. Chen, D. H. Chen and P. Q. Huang, Ni-Catalyzed Direct Alcoholysis of N-Acylpyrrole-Type Tertiary Amides under Mild Conditions, Sci. China: Chem., 2020, 63, 370–376 CrossRef CAS; (c) H. Nagae, T. Hirai, D. Kato, S. Soma, S. Y. Akebi and K. Mashima, Dinuclear Manganese Alkoxide Complexes as Catalysts for C−N Bond Cleavage of Simple Tertiary N, N-Dialkylamides to Give Esters, Chem. Sci., 2019, 10, 2860–2868 RSC; (d) H. X. Wu, W. J. Guo, S. Daniel, Y. Li, C. Liu and Z. Zeng, Fluoride-Catalyzed Esterification of Amides, Chem. – Eur. J., 2018, 24, 3444–3447 CrossRef CAS PubMed; (e) K. Subramanian, S. L. Yedage and B. M. Bhanage, Electrodimerization of N-Alkoxyamides for Zinc(II) Catalyzed Phenolic Ester Synthesis under Mild Reaction Conditions, Adv. Synth. Catal., 2018, 360, 2511–2521 CrossRef CAS; (f) P. Q. Huang and H. Geng, Ni-Catalyzed Chemoselective Alcoholysis of N-Acyloxazolidinones, Green Chem., 2018, 20, 593–599 RSC; (g) N. A. Weires, D. D. Caspi and N. K. Garg, Kinetic Modeling of the Nickel-Catalyzed Esterification of Amides, ACS Catal., 2017, 7, 4381–4385 CrossRef CAS PubMed; (h) T. Deguchi, H.-L. Xin, H. Morimoto and T. Ohshima, Direct Catalytic Alcoholysis of Unactivated 8-Aminoquinoline Amides, ACS Catal., 2017, 7, 3157–3161 CrossRef CAS; (i) Y. Bourne-Branchu, C. Gosmini and G. Danoun, Cobalt-Catalyzed Esterification of Amides, Chem. – Eur. J., 2017, 23, 10043–10047 CrossRef CAS PubMed.
- (a) A. Charvieux, L. Le Moigne, L. G. Borrego, N. Duguet and E. Metay, Solvent-Free N-Alkylation of Amides with Alcohols Catalyzed by Nickel on Silica−Alumina, Eur. J. Org. Chem., 2019, 6842–6846 CrossRef CAS; (b) J. Das and D. Banerjee, Nickel-Catalyzed Phosphine Free Direct N-Alkylation of Amides with Alcohols, J. Org. Chem., 2018, 83, 3378–3384 CrossRef CAS PubMed; (c) S. Kerdphon, X. Quan, V. S. Parihar and P. G. Andersson, C−N Coupling of Amides with Alcohols Catalyzed by N-Heterocyclic Carbene−Phosphine Iridium Complexes, J. Org. Chem., 2015, 80, 11529–11537 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data, and NMR spectra. See DOI: https://doi.org/10.1039/d3qo01730c |
| This journal is © the Partner Organisations 2024 |