
Iridium/nickel dual catalyzed hydroacylation of hetero-bicyclic alkenes under visible-light irradiation†
Li
Meng
a,
Chunhui
Yang
a,
Jun
Dong
a,
Wanliu
Wen
a,
Jingchao
Chen
 *a and
Baomin
Fan
*ab
*a and
Baomin
Fan
*ab
aKey Laboratory of Chemistry in Ethnic Medicinal Resources, Yunnan Minzu University, Yuehua Street, Kunming, 650500, China. E-mail: chenjingchao84@163.com; adams.bmf@hotmail.com
bYunnan Key Laboratory of Chiral Functional Substance Research and Application, Yunnan Minzu University, Yuehua Street, Kunming 650504, China
First published on 14th November 2023
Abstract
Hetero-bicyclic alkenes are versatile synthetic motifs for the synthesis of highly substituted ring systems. The transition metal-catalyzed reactions of hetero-bicyclic alkenes with various kinds of nucleophiles have been well developed. We report herein an iridium/nickel dual catalyst umpolung hydroacylation of hetero-bicyclic alkenes under visible-light irradiation. By using easily accessible and stable carboxylic acids as acyl radical sources, a wide range of hetero-bicyclic alkenes, including both oxabenzonorbornadienes and azabenzonorbornadienes, were transformed into the corresponding hydroacylation products at room temperature. This protocol is distinguished by its mild conditions, wide scope, and easily accessible acyl radical sources, thus providing a photoredox/nickel synergistic catalysis platform for hydroacylation reactions.
Ketones are versatile building blocks that are frequently derivatized for the synthesis of structurally diverse compounds through various transformations. The hydroacylation of unsaturated hydrocarbons both intermolecularly and intramolecularly using abundant and readily available aldehydes represents the most valuable synthetic approach to create carbonyl compounds with complete atom economy. The conventional ionic transformation processes for this important transformation are C–H bond insertion initiated by the chelation-assisted activation of aldehydes using a transition-metal catalyst1 and the umpolung of carbonyl groups utilizing N-heterocyclic carbene catalysts.2 Unfortunately, most of the traditional strategies for generating acyl radicals require harsh conditions or using highly reactive acid derivatives. Therefore, methods that generate acyl radicals under mild conditions are highly desired. In recent years, a surge of interest from the synthetic community has put the use of photoredox manifolds at the forefront of catalysis, and has provided enormous opportunities for alternative efficient strategies for the hydroacylation of hydrocarbons via an acyl radical process. In this context, aldehydes,3 acyl halides,4 anhydrides,5 α-keto acids6 and some other pre-generated precursors7 can be transformed into acyl radicals to enable their coupling with alkenes under photoirradiation (Fig. 1a). Carboxylic acids are versatile synthons for the preparation of numerous commodity chemicals with several advantages in organic synthetic chemistry: (i) carboxylic acids are abundant, inexpensive, and structurally diverse feedstocks; and (ii) carboxylic acids are much more stable and easier to handle than other acyl sources such as aldehydes, acyl halides, and anhydrides. Therefore, the photochemical deoxygenation of carboxylic acids has been developed for the acyl radical coupling reactions.8 Notably, the Xie and Zhu group has pioneered the visible-light mediated deoxygenation of carboxylic acids with triphenylphosphine (Fig. 1b). For instance, they developed the synthesis of aromatic ketones from aromatic carboxylic acids using ethylene, other alkenes or organohalides. This reaction proceeds via the intramolecular 1,5-aryl migration of α-aminobenzoic acids.9 In addition, an iridium-catalyzed deoxygenative allylation reaction of carboxylic acids with alkenes and electron-deficient 1,3-dienes was also reported.10 By taking advantage of the visible-light mediated deoxygenation strategy, the Studer group has developed a photocatalytic phosphine-mediated water activation for radical hydrogenation.11 During this period of flourishing identification and development of photochemical methods, new catalytic methods are in great demand to expand the scope and synthetic utility of these valuable hydroacylation reactions.
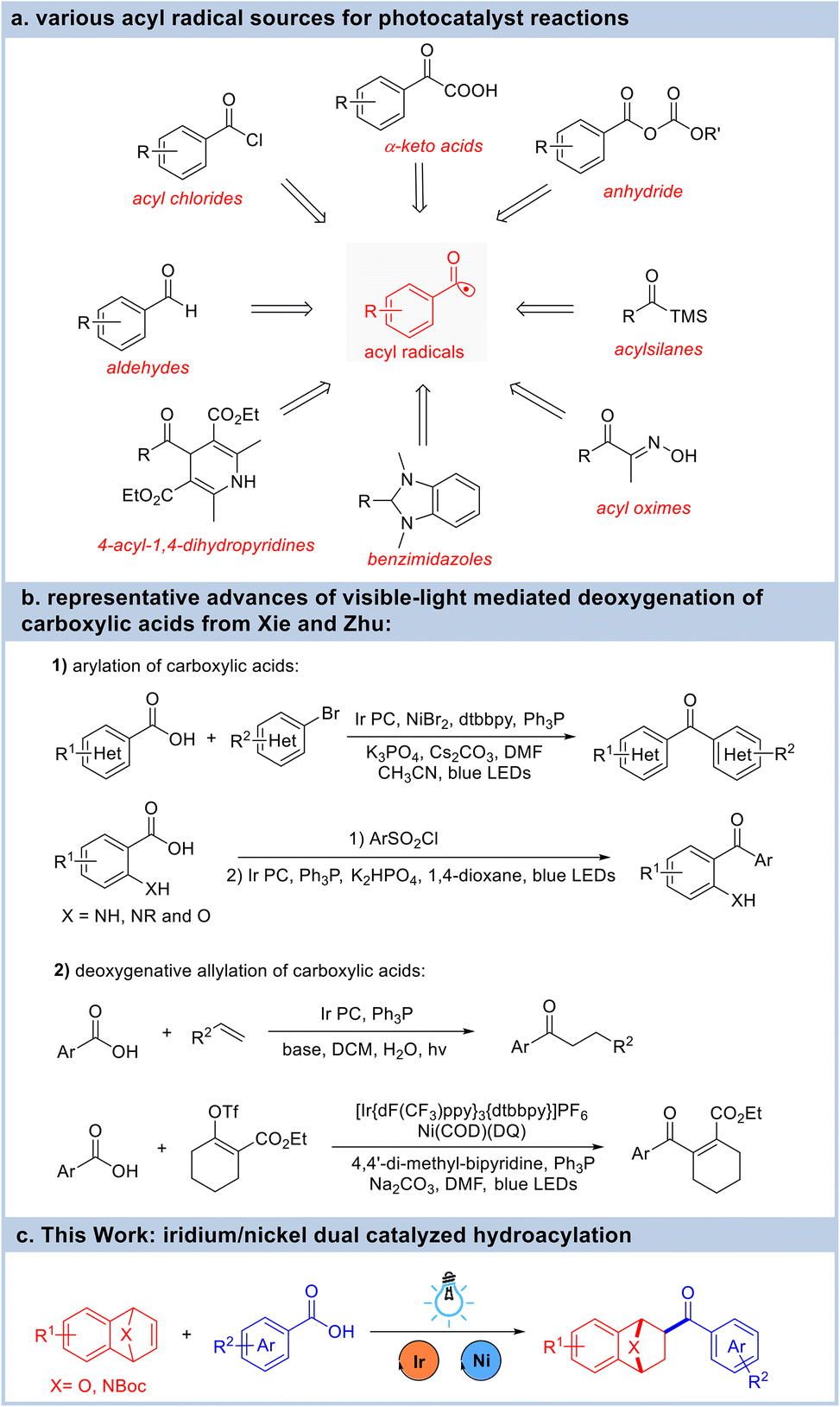 | ||
| Fig. 1 Summary of the generation of acyl radicals and their use in hydroacylation reactions. | ||
Hetero-bicyclic alkenes are multipurpose foundational synthetic motifs for the synthesis of highly substituted ring systems. Benefiting from the seminal works of Lautens, Cheng, and many others, the catalytic reactions of hetero-bicyclic alkenes and their coupling partners have been developed rapidly in the past two decades.12 In general, the ring-opening reactions of hetero-bicyclic alkenes with nucleophiles using transition metal catalysts such as rhodium, iridium, palladium, cobalt, and nickel were extensively investigated. However, reports regarding the addition reactions of hetero-bicyclic alkenes have mainly utilized aromatic compounds,13 and, although these have provided straightforward access to exo-substituted oxa/azabenzonorbornadienes, reports of reactions with other kinds of coupling partners have been scarce.14 Our group has systematically studied the asymmetric ring-opening reactions of hetero-bicyclic alkenes with alcohols, amines, acids, alkynes, and aromatic nucleophiles.15 Inspired by the quick development of photo-catalyzed reactions, which were quite effective for the generation of acyl radicals, we wish to develop a free radical addition reaction of hetero-bicyclic alkenes (Fig. 1c). Herein, we report the hydroacylation of hetero-bicyclic alkenes under visible-light irradiation by merging a photoredox approach with the use of a transition metal catalyst.
Initially, the model hydroacylation of oxabenzonorbornadiene (1a) with benzoic acid (2a) was explored with various metallaphotoredox catalysts (Table 1). We were delighted to isolate the desired hydroacylation product 3aa in an 82% yield when the reaction was carried out in the presence of 2 mol% of [Ir{dF(CF3)ppy}3{dtbbpy}]PF6 as a photosensitizer and a complex of Ni(cod)2 (3 mol%) and bpy (6 mol%) as the transition metal catalyst with PPh3 as the reductant in acetone under blue LED irradiation (Table 1, entry 1). Control experiments showed that no reaction proceeded in the absence of [Ir{dF(CF3)ppy}3{dtbbpy}]PF6 or Ni(cod)2, indicating that both photosensitizer and transition metal catalyst are essential for the present transformation (Table 1, entries 2 and 3). Triphenylphosphine was shown to be an effective reductant for the deoxygenative C–C coupling, as no reaction took place in its absence (Table 1, entry 4), due to the strong P–O affinity that exists between the Ph3P radical cation and the carboxylate anion. This affinity facilitates homolytic C–O bond cleavage of the carboxylic acids. When DMF, MeCN, DMAc, or PhCF3 were used as the solvent instead of acetone, the yield declined significantly (Table 1, entry 5). Essentially no hydroacylation product was observed when using fac-Ir(ppy)3, 4CzIPN, or Ru(bpy)3 as the photosensitizer (Table 1, entry 6). The reaction with NiCl2 or CoCl2 as the metal precursor gave the product in a reduced yield (Table 1, entry 7). The use of 1,10-phenanthroline as a ligand was also effective, but no desired product was detected when using 2,2′:6′,2′′-terpyridine (Table 1, entry 8). The screening of other light sources indicated the use of a blue LED (450 nm) is optimal (Table 1, entry 9). It should be noted that triphenyl phosphorous oxide was isolated in an 85% yield (0.085 mmol) under the standard reaction conditions (Table 1, entry 1), indicating that the oxidation of PPh3 occurred during the transformation.
|
|
||
|---|---|---|
| Entry | Variation from the above conditions | Yieldb (%) |
| a Reaction conditions: unless otherwise specified, reactions were performed with 1a (0.15 mmol), 2a (0.1 mmol), PPh3 (0.15 mmol) using (Ir[dF(CF3)ppy]2(dtbpy))PF6 (2 mol%), Ni(cod)2 (3 mol%), and 2,2′-bipyridyl (6 mol%) in acetone (1 mL) with 20 W blue LED irradiation at room temperature under an argon atmosphere for 18 hours. b Yield of the isolated product after column chromatography. | ||
| 1 | None | 82 |
| 2 | Without [Ir{dF(CF3)ppy}3{dtbbpy}]PF6 | NR |
| 3 | Without Ni(cod)2 | NR |
| 4 | Without PPh3 | NR |
| 5 | DMF/MeCN/DMAc/PhCF3 instead of acetone | 30/33/18/22 |
| 6 | fac-Ir(ppy)3/4CzIPN/Ru(bpy)3 instead of PC | NR/trace/NR |
| 7 | NiCl2/CoCl2 instead of Ni(cod)2 | 53/37 |
| 8 | 1,10-Phenanthroline/2,2′:6′,2′′-terpyridine instead of bpy | 54/trace |
| 9 | Purple LED (365 nm)/purple LED (390 nm)/green LED (530 nm) instead of blue LED (450 nm) | 60/61/NR |

With the optimized reaction conditions (Table 1, entry 1) in hand, the scope of the carboxylic acid substrates was investigated (Scheme 1). In general, all of the aromatic carboxylic acids gave hydroacylation products, the reaction yields were slightly affected by the electronic properties of the substituents on the aromatic rings. Benzoic acids bearing electron-donating groups such as methyl, methoxyl, OBn, iPr, and tBu at the para, meta, and ortho positions on the phenyl rings reacted smoothly to afford the deoxygenative C–C coupling hydroacylation products 3aa–ak. In contrast, electron-withdrawing groups including halogens and phenyl-substituted benzoic acids delivered the desired coupling products 3al–as in slightly reduced yields. Notably, the halogen atoms remained intact under the present reaction conditions, allowing further potential functionalization. 2-Naphthoic acid, furan-3-carboxylic acid, and thiophene-3-carboxylic acid also furnished hydroacylation products 3at–av. However, no desired product was detected by using 4-methylpentanoic acid and cyclopentanecarboxylic acid, indicating that alkyl carboxylic acids are not applicable in the present reaction. The use of nicotinic acid, phenyl carboxylic acids containing strong electron-withdrawing groups, and alkenyl carboxylic acids failed to give the desired products.
 | ||
| Scheme 1 Scope of carboxylic acids in the hydroacylation reaction. Reaction conditions: unless otherwise specified, reactions were performed with 1a (0.15 mmol), 2 (0.1 mmol), PPh3 (0.15 mmol) using [Ir{dF(CF3)ppy}3{dtbbpy}]PF6 (2 mol%), Ni(cod)2 (3 mol%), and 2,2′-bipyridyl (6 mol%) in acetone (1 mL) with 20 W blue LED irradiation at room temperature under an argon atmosphere for 18 hours. Yields were calculated from the isolated product after column chromatography. | ||
Subsequently, the scope of this transformation for different hetero-bicyclic alkenes was studied under the optimal reaction conditions (Scheme 2). The present deoxygenative hydroacylation reaction proceeded well with dimethyl-substituted oxabenzonorbornadiene 1b and oxabenzonorbornadiene 1c with relatively bulky groups. However, the use of dimethyl-substituted oxabenzonorbornadiene gave 3da in an abnormally low yield. Sterically hindered oxabenzonorbornadiene 1e afforded 3ea in a 45% yield. Unsymmetrical oxabenzonorbornadiene 1f, that possesses a bridging methyl group, delivered 3fa in a 33% yield together with its regioisomer which was isolated in a 17% yield. The dibromo-substituted oxabenzonorbornadiene 3ga and naphthalene-derived oxabenzonorbornadiene 3ha reacted well. Notably, the azabenzonorbornadienes 1i–l with electron-donating and electron-withdrawing groups successfully transformed into the desired deoxygenative hydroacylation products 3ia–la in good yields. These results indicated that both oxabenzonorbornadienes and azabenzonorbornadienes are all suitable substrates in this dual catalytic system.
 | ||
| Scheme 2 Scope of hetero-bicyclic alkenes in the hydroacylation. Reaction conditions: unless otherwise specified, reactions were performed with 1a (0.15 mmol), 2a (0.1 mmol), PPh3 (0.15 mmol) using [Ir{dF(CF3)ppy}3{dtbbpy}]PF6 (2 mol%), Ni(cod)2 (3 mol%), and 2,2′-bipyridyl (6 mol%) in acetone (1 mL) with 20 W blue LED irradiation at room temperature under an argon atmosphere for 18 hours. Yields were calculated from the isolated product after column chromatography. | ||
The present photocatalyzed deoxygenative hydroacylation reaction between hetero-bicyclic alkenes with carboxylic acids provides straightforward access to exo-acylated oxa/azabenzonorbornadienes. The catalytic system was sufficiently robust to convert 1 gram of benzoic acid to 1.35 g of the desired hydroacylation product in a slightly reduced yield (Scheme 3a). To illustrate the potential synthetic applications of the current transformation, 3aa was converted to the terminal olefine 4aa in an 81% yield via the classical Witting reaction (Scheme 3b). In order to verify the reaction pathway, the hydroacylation reaction of azabenzonorbornadiene 1a with benzoic acid 2a under the standard reaction conditions was completely quenched in the presence of 1,1-diphenylethylene, and the trapped product 6a was analyzed using GC-MS (Scheme 3c). The results indicated that an active benzoyl radical was generated and participated in the hydroacylation. In addition, we observed a quantum yield of Φ = 0.23, which ruled out the possibility of a radical chain process in the present reaction (see ESI† for details).
 | ||
| Scheme 3 Reaction application and radical trapping reactions. Reaction conditions: (a) 1a (12.3 mmol), 2a (8.2 mmol), PPh3 (12.3 mmol) using [Ir{dF(CF3)ppy}3{dtbbpy}]PF6 (2 mol%), Ni(cod)2 (3 mol%), and 2,2′-bipyridyl (6 mol%) in acetone (30 mL) were irradiated with a 20 W blue LED at room temperature under an argon atmosphere for 24 hours; (b) 3aa (25 mg, 0.1 mmol), MePPh3Br (107 mg, 0.3 mmol), and tBuOK (34 mg, 0.3 mmol) in THF (1.0 mL) were stirred at room temperature for 6 h; (c) 1a (0.15 mmol), 2a (0.1 mmol), PPh3 (0.15 mmol), (Ir[dF(CF3)ppy]2(dtbpy))PF6 (mol%), Ni(cod)2 (3 mol%), 2,2′-bipyridyl (6 mol%), and 1,1-diphenylethylene (0.2 mmol) in acetone (1 mL) were irradiated with a 20 W blue LED at room temperature under an argon atmosphere for 18 hours. | ||
Based on the reports regarding the photochemical deoxygenation of carboxylic acids and the radical trapping experimental results, a possible reaction mechanism is proposed (Scheme 4). Under irradiation with a blue LED, the photocatalyst [Ir{dF(CF3)ppy}3{dtbbpy}]PF6 is excited to provide its excited species IrIII*, which then undergoes a single-electron transfer (SET) process with PPh3 to generate the reactive radical cation intermediate A and IrII. Next, the radical deoxygenation of carboxylate anion with the triphenylphosphine radical cation affords the active benzoyl radical B, which enables the generation of an acyl–nickel complex C. Then, subsequent coordination and a migratory insertion coupling of complex C to oxabenzonorbornadiene 1a affords the alkyl–nickel complex D. Further protonation of D produces the hydroacylation product 3aa with regeneration of NiIL, which is reduced by IrII and gives Ni0L, simultaneously completing both the photocatalytic and transition metal catalytic cycles.
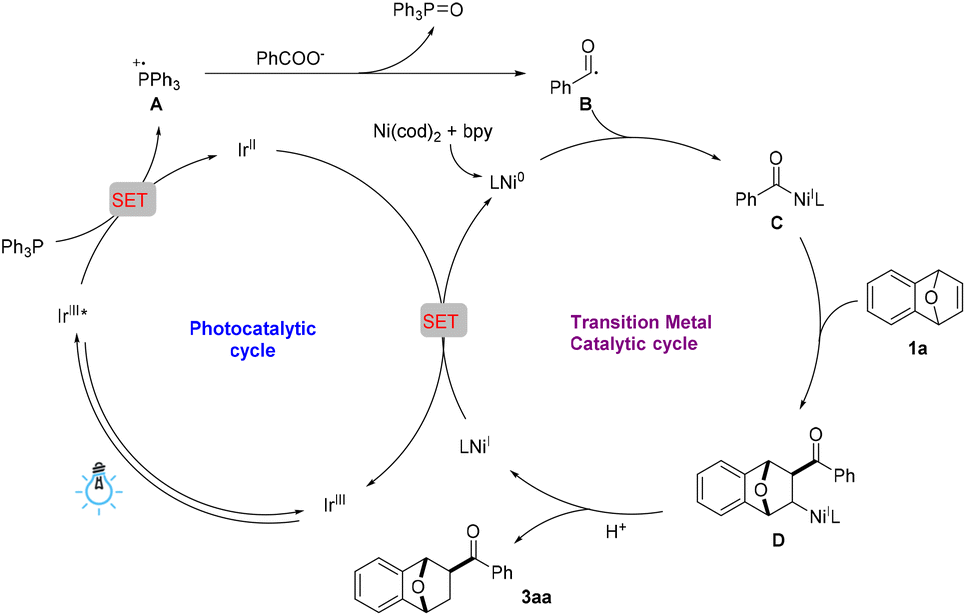 | ||
| Scheme 4 Proposed mechanism. | ||
Conclusions
In summary, the umpolung hydroacylation of hetero-bicyclic alkenes is accomplished by the merger of a photoredox process with transition metal catalysis. The iridium/nickel dual catalyst hydroacylation reaction tolerates a wide range of aryl carboxylic acids and oxa/azabenzonorbornadienes, affording exo-acylated oxa/azabenzonorbornadienes in good yields under mild reaction conditions. Radical trapping reactions verified the generation of acyl radicals and a possible reaction mechanism is proposed.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Natural Science Foundation of China (21961045, 22361052, 21572198), the Applied Basic Research Project of Yunnan Province (2017FA004, 2018FB021), the Department of Education of Yunnan Province (2019Y0198), and Yunnan Provincial Key Laboratory Construction Plan Funding of Universities for their financial support.References
- (a) C.-H. Jun, E.-A. Jo and J.-W. Park, Intermolecular Hydroacylation by Transition–Metal Complexes, Eur. J. Org. Chem., 2007, 1869–1881 CrossRef CAS; (b) M. C. Willis, Transition metal catalyzed alkene and alkyne hydroacylation, Chem. Rev., 2010, 110, 725–748 CrossRef CAS PubMed; (c) J. C. Leung and M. J. Krische, Catalytic intermolecular hydroacylation of C–C π-bonds in the absence of chelation assistance, Chem. Sci., 2012, 3, 2202–2209 RSC; (d) Y.-Y. Jiang, H.-Z. Yu and J. Shi, Mechanistic study on the regioselectivity of Co-catalyzed hydroacylation of 1, 3-dienes, Chin. Chem. Lett., 2015, 26, 58–62 CrossRef CAS; (e) A. Ghosh, K. F. Johnson, K. L. Vickerman, J. A. Walker and L. M. Stanley, Recent advances in transition metal-catalysed hydroacylation of alkenes and alkynes, Org. Chem. Front., 2016, 3, 639–644 RSC; (f) H.-R. Wang and M.-C. Ye, Research Advance on Enantioselective Transition Metal-Catalyzed Hydroacylation Reactions, Chin. J. Org. Chem., 2022, 42, 3152–3166 CrossRef CAS.
- (a) A. T. Biju, N. Kuhl and F. Glorius, Extending NHC-catalysis: coupling aldehydes with unconventional reaction partners, Acc. Chem. Res., 2011, 44, 1182–1195 CrossRef CAS PubMed; (b) M. Schedler, D.-S. Wang and F. Glorius, NHC–Catalyzed Hydroacylation of Styrenes, Angew. Chem., Int. Ed., 2013, 52, 2585–2589 CrossRef CAS PubMed; (c) D. Janssen-Müller, M. Fleige, D. Schlüns, M. Wollenburg, C. G. Daniliuc, J. Neugebauer and F. Glorius, NHC-catalyzed enantioselective dearomatizing hydroacylation of benzofurans and benzothiophenes for the synthesis of spirocycles, ACS Catal., 2016, 6, 5735–5739 CrossRef; (d) X. Bugaut, F. Liu and F. Glorius, N-heterocyclic carbene (NHC)-catalyzed intermolecular hydroacylation of cyclopropenes, J. Am. Chem. Soc., 2011, 133, 8130–8133 CrossRef CAS PubMed; (e) I. Piel, M. Steinmetz, K. Hirano, R. Fröhlich, S. Grimme and F. Glorius, Highly Asymmetric NHC–Catalyzed Hydroacylation of Unactivated Alkenes, Angew. Chem., Int. Ed., 2011, 50, 4983–4987 CrossRef CAS PubMed.
- (a) K. Kawaai, T. Yamaguchi, E. Yamaguchi, S. Endo, N. Tada, A. Ikari and A. Itoh, Photoinduced generation of acyl radicals from simple aldehydes, access to 3-acyl-4-arylcoumarin derivatives, and evaluation of their antiandrogenic activities, J. Org. Chem., 2018, 83, 1988–1996 CrossRef CAS PubMed; (b) A. Chinchole, M. A. Henriquez, D. Cortes-Arriagada, A. R. Cabrera and O. Reiser, Iron(III)-light-induced homolysis: A dual photocatalytic approach for the hydroacylation of alkenes using acyl radicals via direct HAT from aldehydes, ACS Catal., 2022, 12, 13549–13554 CrossRef CAS; (c) Y. Luo, Q. Wei, L.-K. Yang, Y.-Q. Zhou, W.-D. Cao, Z.-S. Su, X.-H. Liu and X.-M. Feng, Enantioselective Radical Hydroacylation of α, β-Unsaturated Carbonyl Compounds with Aldehydes by Triplet Excited Anthraquinone, ACS Catal., 2022, 12, 12984–12992 CrossRef CAS; (d) Y. L. Kuang, K. Wang, X. C. Shi, X. Q. Huang, E. Meggers and J. Wu, Asymmetric Synthesis of 1, 4-Dicarbonyl Compounds from Aldehydes by Hydrogen Atom Transfer Photocatalysis and Chiral Lewis Acid Catalysis, Angew. Chem., Int. Ed., 2019, 58, 16859–16863 CrossRef CAS PubMed.
- (a) C.-M. Wang, D. Song, P.-J. Xia, J. Wang, H.-Y. Xiang and H. Yang, Visible–Light–Promoted Synthesis of 1, 4-Dicarbonyl Compounds via Conjugate Addition of Aroyl Chlorides, Chem. – Asian J., 2018, 13, 271–274 CrossRef CAS PubMed; (b) Y. Ueda, T. Iwai and M. Sawamura, Nickel–Copper–Catalyzed Hydroacylation of Vinylarenes with Acyl Fluorides and Hydrosilanes, Chem. – Eur. J., 2019, 25, 9410–9414 CrossRef CAS PubMed; (c) Q.-S. Zhao, G.-Q. Xu, H. Liang, Z.-Y. Wang and P.-F. Xu, Aroylchlorination of 1, 6-dienes via a photoredox catalytic atom-transfer radical cyclization process, Org. Lett., 2019, 21, 8615–8619 CrossRef CAS PubMed.
- (a) Y.-T. Hong, A. Barchuk and M. J. Krische, Branch–Selective Intermolecular Hydroacylation: Hydrogen–Mediated Coupling of Anhydrides to Styrenes and Activated Olefins, Angew. Chem., Int. Ed., 2006, 45, 6885–6888 CrossRef CAS PubMed; (b) S. P. Dong, G. B. Wu, X. Q. Yuan, C. C. Zou and J. X. Ye, Visible-light photoredox catalyzed hydroacylation of electron-deficient alkenes: carboxylic anhydride as an acyl radical source, Org. Chem. Front., 2017, 4, 2230–2234 RSC.
- (a) J.-J. Zhao, H.-H. Zhang, X. Shen and S.-Y. Yu, Enantioselective radical hydroacylation of enals with α-ketoacids enabled by photoredox/amine cocatalysis, Org. Lett., 2019, 21, 913–916 CrossRef CAS PubMed; (b) G. N. Papadopoulos, E. Voutyritsa, N. Kaplaneris and C. G. Kokotos, Green Photo–Organocatalytic C–H Activation of Aldehydes: Selective Hydroacylation of Electron–Deficient Alkenes, Chem. – Eur. J., 2018, 24, 1726–1731 CrossRef CAS PubMed; (c) J.-Y. Yang, M.-H. Song, H.-Y. Zhou, G.-G. Wang, B. Ma, Y.-Y. Qi and C.-D. Huo, Visible-light-mediated hydroacylation of azobenzenes with α-keto acids, Org. Lett., 2020, 22, 8407–8412 CrossRef CAS PubMed.
- (a) L. Zheng, P.-J. Xia, Q.-L. Zhao, Y.-E. Qian, W.-N. Jiang, H.-Y. Xiang and H. Yang, Photocatalytic Hydroacylation of Alkenes by Directly Using Acyl Oximes, J. Org. Chem., 2020, 85, 11989–11996 CrossRef CAS PubMed; (b) Y. Saga, Y. Nakayama, T. Watanabe, M. Kondo and S. Masaoka, Visible-Light-Driven Hydroacylation of Unactivated Alkenes Using Readily Available Acyl Donors, Org. Lett., 2023, 25, 1136–1141 CrossRef CAS PubMed; (c) L. Capaldo, R. Riccardi, D. Ravelli and M. Fagnoni, Acyl radicals from acylsilanes: photoredox-catalyzed synthesis of unsymmetrical ketones, ACS Catal., 2018, 8, 304–309 CrossRef CAS; (d) X.-X. Zhao, B. Li and W.-J. Xia, Visible-Light-Promoted Photocatalyst-Free Hydroacylation and Diacylation of Alkenes Tuned by NiCl2·DME, Org. Lett., 2020, 22, 1056–1061 CrossRef CAS PubMed.
- (a) L.-L. Zhang, Y.-H. Lia, Z.-Y. Guo, Y.-T. Li, N. Li, W.-P. Li, C.-J. Zhu and J. Xie, Photoredox deoxygenative allylation of carboxylic acids via selective 1, 6-addition of acyl radicals to electron-deficient 1, 3-dienes, Chin. J. Catal., 2023, 50, 215–221 CrossRef CAS; (b) E. E. Stache, A. B. Ertel, T. Rovis and A. G. Doyle, Generation of phosphoranyl radicals via photoredox catalysis enables voltage–independent activation of strong C–O bonds, ACS Catal., 2018, 8, 11134–11139 CrossRef CAS PubMed; (c) R. Mao, S. Bera, A. Cheseaux and X. Hu, Deoxygenative trifluoromethylthiolation of carboxylic acids, Chem. Sci., 2019, 10, 9555–9559 RSC; (d) G. Bergonzini, C. Cassani and C.-J. Wallentin, Acyl Radicals from Aromatic Carboxylic Acids by Means of Visible–Light Photoredox Catalysis, Angew. Chem., Int. Ed., 2015, 54, 14066–14069 CrossRef CAS PubMed; (e) Y.-Y. Ning, S.-S. Wang, M.-Z. Li, J. Han, C.-J. Zhu and J. Xie, Site-specific Umpolung amidation of carboxylic acids via triplet synergistic catalysis, Nat. Commun., 2021, 12, 4637 CrossRef CAS PubMed; (f) M.-L. Zhang, X.-A. Yuan, C.-J. Zhu and J. Xie, Deoxygenative Deuteration of Carboxylic Acids with D2O, Angew. Chem., Int. Ed., 2019, 58, 312–322 CrossRef CAS PubMed.
- (a) M.-L. Zhang, J. Xie and C.-J. Zhu, A general deoxygenation approach for synthesis of ketones from aromatic carboxylic acids and alkenes, Nat. Commun., 2018, 9, 3517–3526 CrossRef PubMed; (b) R. Ruzi, J.-Y. Ma, X.-A. Yuan, W.-L. Wang, S.-S. Wang, M.-L. Zhang, J. Dai, J. Xie and C.-J. Zhu, Deoxygenative Arylation of Carboxylic Acids by Aryl Migration, Chem. – Eur. J., 2019, 25, 12724–12729 CrossRef CAS PubMed; (c) L.-L. Zhang, S. Chen, H.-C. He, W.-P. Li, C.-J. Zhu and J. Xie, Chem. Commun., 2021, 57, 9064–9067 RSC.
- (a) R. Ruzi, K. Liu, C.-J. Zhu and J. Xie, Upgrading ketone synthesis direct from carboxylic acids and organohalides, Nat. Commun., 2020, 11, 3312–3320 CrossRef CAS PubMed; (b) L.-L. Zhang, Y.-H. Li, Z.-Y. Guo, Y.-T. Li, N. Li, W.-P. Li, C.-J. Zhu and J. Xie, Photoredox deoxygenative allylation of carboxylic acids via selective 1,6-addition of acyl radicals to electron-deficient 1,3-dienes, Chin. J. Catal., 2023, 50, 215–221 CrossRef CAS; (c) Y.-T. Li, Q.-Z. Shao, H.-C. He, C.-J. Zhu, X.-S. Xue and J. Xie, Highly selective synthesis of all-carbon tetrasubstituted alkenes by deoxygenative alkenylation of carboxylic acids, Nat. Commun., 2022, 13, 10 CrossRef CAS PubMed.
- J.-J. Zhang, C. Mück-Lichtenfeld and A. Studer, Photocatalytic phosphine-mediated water activation for radical hydrogenation, Nature, 2023, 619, 506–513 CrossRef CAS PubMed.
- (a) M. Lautens, K. Fagnou and S. Hiebert, Transition metal-catalyzed enantioselective ring-opening reactions of oxabicyclic alkenes, Acc. Chem. Res., 2003, 36, 48–58 CrossRef CAS PubMed; (b) D. K. Rayabarapun and C.-H. Cheng, New catalytic reactions of oxa-and azabicyclic alkenes, Acc. Chem. Res., 2007, 40, 971–983 CrossRef PubMed; (c) R. Khan, J.-C. Chen and B.-M. Fan, Versatile Catalytic Reactions of Norbornadiene Derivatives with Alkynes, Adv. Synth. Catal., 2020, 362, 1564–1601 CrossRef CAS; (d) H.-C. Cheng, X.-L. Liang, X.-L. Li, Y.-H. Long and D.-Q. Yang, Progress in Transition Metal-Catalyzed Asymmetric Ring-Opening Reactions of Azabicyclic Alkenes, Chin. J. Org. Chem., 2012, 3, 433–443 CrossRef; (e) D.-Q. Yang and H.-P. Zeng, Regio-and diasteroselectivity of rhodium-catalyzed ring opening reaction of oxabenzonorbornadienes with heteroatom nucleophiles, Chin. Chem. Lett., 2003, 7, 697–699 Search PubMed; (f) Y.-F. Han, D.-Q. Yang, E.-C. Liu and J.-X. Dong, Rhodium-catalyzed asymmetric ring opening reaction of oxabenzonorbornadiene with substituted phenolic nucleophiles, Chin. Chem. Lett., 2006, 17, 296–298 CAS; (g) E.-C. Liu, D.-Q. Yang, Y.-F. Han and J.-X. Dong, The Studies of Rhodium-catalyzed Ring Opening Reaction of N-Boc azabenzonorbornadiene with Heteroatom Nucleophiles, Chin. Chem. Lett., 2006, 17, 717–719 CAS; (h) L. Xie, D.-Q. Yang, S.-Q. Zhao, H. Wang, L.-H. Liang and R.-S. Luo, Rhodium-catalyzed asymmetric ring opening of azabenzonorbornadiene with substituted piperazine nucleophiles, Chin. Chem. Lett., 2007, 18, 127–129 CrossRef CAS.
- (a) H.-C. Cheng, W.-R. Dong, C. A. Dannenberg, S.-X. Dong, Q.-Q. Guo and C. Bolm, Ruthenium-catalyzed hydroarylations of oxa-and azabicyclic alkenes, ACS Catal., 2015, 5, 2770–2773 CrossRef CAS; (b) S. Prakash, Y.-C. Chang and C.-H. Cheng, Rhenium(I)–Catalyzed ortho–C–H Addition to Bicyclic Alkenes, Chem. – Asian J., 2018, 13, 1664–1668 CrossRef CAS PubMed.
- M. Nagamoto and T. Nishimura, Stereoselective hydroacylation of bicyclic alkenes with 2-hydroxybenzaldehydes catalyzed by hydroxoiridium/diene complexes, Chem. Commun., 2015, 51, 13791–13794 RSC.
- (a) B.-M. Fan, S.-F. Li, H.-L. Chen, Z.-W. Lu, S.-S. Liu, Q.-J. Yang, L. Yu, J.-B. Xu, Y.-Y. Zhou and J. Wang, Palladium/Copper Complexes Co–Catalyzed Highly Enantioselective Ring Opening Reaction of Azabenzonorbornadienes with Terminal Alkynes, Adv. Synth. Catal., 2013, 355, 2827–2832 CrossRef CAS; (b) J.-C. Chen, L.-L. Zou, C.-Y. Zeng, Y.-Y. Zhou and B.-M. Fan, Rhodium-Catalyzed Asymmetric Arylative Ring-Opening Reactions of Heterobicyclic Alkenes with Anilines, Org. Lett., 2018, 20, 1283–1286 CrossRef CAS PubMed; (c) F. Yang, J.-C. Chen, J.-B. Xu, F.-J. Ma, Y.-Y. Zhou, M. V. Shinde and B.-M. Fan, Palladium/Lewis Acid Co-catalyzed Divergent Asymmetric Ring-Opening Reactions of Azabenzonorbornadienes with Alcohols, Org. Lett., 2016, 18, 4832–4835 CrossRef CAS PubMed; (d) X. Xu, J.-C. Chen, Z.-X. He, Y.-Y. Zhou and B.-M. Fan, Rhodium-catalyzed asymmetric ring opening reaction of oxabenzonorbornadienes with amines using ZnI2 as the activator, Org. Biomol. Chem., 2016, 14, 2480–2486 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, product characterization, copies of 1H and 13C NMR spectra. See DOI: https://doi.org/10.1039/d3qo01377d |
| This journal is © the Partner Organisations 2024 |