
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Emergence of Ruddlesden–Popper phases and other pitfalls for moderate temperature solution deposited chalcogenide perovskites†
Apurva A.
Pradhan‡
 a,
Shubhanshu
Agarwal‡
a,
Kiruba Catherine
Vincent
a,
Daniel C.
Hayes
a,
Jonas M.
Peterson
b,
Jonathan W.
Turnley
a,
Robert M.
Spilker
a,
Madeleine C.
Uible
b,
Suzanne C.
Bart
b,
Libai
Huang
b,
Kim
Kisslinger
c and
Rakesh
Agrawal
*a
a,
Shubhanshu
Agarwal‡
a,
Kiruba Catherine
Vincent
a,
Daniel C.
Hayes
a,
Jonas M.
Peterson
b,
Jonathan W.
Turnley
a,
Robert M.
Spilker
a,
Madeleine C.
Uible
b,
Suzanne C.
Bart
b,
Libai
Huang
b,
Kim
Kisslinger
c and
Rakesh
Agrawal
*a
aDavidson School of Chemical Engineering, Purdue University, West Lafayette, IN 47907, USA. E-mail: agrawalr@purdue.edu
bH.C. Brown Laboratory, Department of Chemistry, Purdue University, West Lafayette, IN 47907, USA
cCenter for Functional Nanomaterials, Brookhaven National Laboratory, Upton, NY 11973, USA
First published on 22nd August 2024
Abstract
Chalcogenide perovskites have recently attracted significant attention for renewable energy applications due to their predicted combination of air, moisture, and thermal stability, which has been experimentally validated, along with their excellent optoelectronic properties, which are still under experimental investigation. While historically requiring high synthesis temperatures, some solution-processed routes have recently emerged for synthesizing chalcogenide perovskites, such as BaZrS3 and BaHfS3, at temperatures below 600 °C. This study discusses several experimental challenges associated with the moderate-temperature synthesis of solution-deposited chalcogenide perovskites. Firstly, we identify Ruddlesden–Popper (RP) phases as thermodynamically stable competing secondary phases in perovskite synthesis. High sulfur pressures favor the formation of BaZrS3 or BaHfS3, whereas lower sulfur pressures result in a mixture of perovskite and RP phases. Additionally, we briefly discuss the mechanism of moderate-temperature synthesis of chalcogenide perovskites, including some of the morphological and optoelectronic challenges it presents, such as grain overgrowth, secondary phase contamination entrapment, and the presence of mid-band gap emissions. Finally, we address the importance of substrate selection and the potential presence of Ca- and Na-based impurities originating from cation out-diffusion from glass substrates. Addressing these challenges will be crucial as these unique materials continue to be investigated for applications in optoelectronic devices.
Introduction
Chalcogenide perovskites are a promising, non-toxic, and earth-abundant alternative to metal halide perovskites for applications in photovoltaics and many other optoelectronic devices. Similar to metal halide perovskites, II–IV–VI chalcogenide perovskites have an ABX3 composition where A is an alkaline earth metal, B is a Group 4 transition metal, and X is a chalcogen. BaZrS3 is the most widely studied of these materials due to its lower synthesis temperatures, thermodynamic favorability, and favorable bandgap for the top layer in a tandem device.1–17 Computational work has suggested that this material and BaHfS3 have high near-band edge absorption coefficients, good carrier transport, and band gap tunability.1–4,18–30 Moreover, chalcogenide perovskites have exhibited superior stability in air, moisture, and thermal conditions compared to their lead halide counterparts.31–35 However, most synthesis approaches in the literature for BaZrS3 and BaHfS3 utilize solid state or vacuum-deposited approaches, which involve high-temperature sulfurization or annealing steps exceeding 600 °C, making them incompatible with the glass substrates and conductive back contacts required for the synthesis of photovoltaic devices. Therefore, low-temperature synthesis methods are highly desirable for chalcogenide perovskites. One such method is solution processing.Solution processing of thin film materials is an appealing method to synthesize optoelectronic devices, including photovoltaics, due to its potential for reducing manufacturing costs by allowing for continuous, high throughput, roll-to-roll processing over large areas with high materials utilization. Literature is abundant on utilizing solution-processed methods to synthesize thin film photovoltaic materials like the halide perovskites36 and chalcogenide materials including, but not limited to Cu(In,Ga)Se2 (CIGSe), Cu2ZnSnSe4 (CZTSe), Ag2ZnSnSe4 (AZTSe).37–41 Several robust solvent systems, including the amine-thiol and alkylammonium-polyselenide chemistries, have been shown to dissolve a variety of different late and post-transition metals and their various compounds including Cu, Ag, In, Ga, Sn, Zn, As, Sb, and Bi metals and metal chalcogenides, and the resulting solutions have been used to synthesize various thin-film semiconductor materials including CIGSe, CZTSe, AZTSe, Cu3AsS4, and Cd(Se,Te).42–49 Hydrazine50,51 and DMSO/DMF-Thiourea52,53 based synthesis approaches for thin film photovoltaics have also been reported. However, none of the above approaches have yet been observed to successfully dissolve early transition metals, such as Zr, Hf, or their metal-containing species of interest, that would result in a successful synthesis of chalcogenide perovskites.
Recently, a few new routes for lower temperature synthesis of BaZrS3 have been reported, including a synthesis route by Zilevu et al. where metal amide precursors in oleylamine solution were used to create BaZrS3 nanoparticles at 365 °C,54 a route by Yang et al. where luminescent nanoparticles were synthesized at 330 °C utilizing barium dibutyldithiocarbamate and zirconium diethyldithiocarbamate,55 a mixed phase route reported by Turnley et al. where ZrH2 particles were suspended in a precursor slurry containing barium thiolates which were deposited on a glass substrate and created crystalline BaZrS3 after a 575 °C sulfurization step,56 and a fully solution-deposited route reported by Pradhan et al. where barium dithiocarboxylates and zirconium dithiocarbamates were fully dissolved and deposited on a glass substrate before crystalline material was grown after a 575 °C sulfurization step.57 Vincent et al. also recently demonstrated solution-processing methods that utilize metal sulfides and metal chlorides to synthesize BaZrS3 at temperatures below 600 °C.58 Explaining the low-temperature growth mechanism, Comparotto et al., Yang et al., and Vincent et al. have suggested that a liquid barium polysulfide (BaSx) flux may be playing a pivotal role in synthesizing crystalline BaZrS3 by overcoming kinetic- or diffusion-related barriers via the liquid flux to allow for lower-temperature synthesis methods.8,59,60 With the ability to synthesize BaZrS3, and related chalcogenide perovskites such as BaHfS3, at lower temperatures, there is reason to be optimistic about the potential of this class of materials. However, many challenges remain, including secondary phase contaminants, oxides, poor or broad photoluminescence (PL), and non-continuous growth of BaZrS3 films.8,55–57 Our experience utilizing molecular precursor inks for synthesizing BaZrS3 also highlights several challenges in working with chalcogenide perovskites that have sparsely been mentioned in the literature.
In this paper, we outline several experimental challenges, offer insights on addressing some of these challenges, and identify areas that necessitate further attention from the broader research community. Initially, we investigate the influence of the partial pressure of elemental sulfur on the formation of BaZrS3 and related secondary phases during the moderate-temperature sulfurization heat treatment step. Subsequently, we examine the challenges associated with achieving rapid, low-temperature growth of large BaZrS3 grains. Following this, we explore challenges related to photoluminescence (PL) and secondary phases exhibiting PL emissions near the band gap of chalcogenide perovskites. Finally, we demonstrate how sulfurization conditions affect substrates utilized in synthesizing chalcogenide perovskites and provide recommendations for advancing research in this field. Our findings can serve as a roadmap for researchers investigating chalcogenide perovskites.
Results and discussion
Impact of sulfur partial pressure during sulfurization heat treatment
The solution-processing of BaZrS3 films from molecular precursor inks, thus far, has shown promise yet presents several challenges. Unlike the solution-processed synthesis of other chalcogenides, such as Cu(In,Ga)(S,Se)2, BaZrS3 does not nucleate after annealing the as-coated molecular precursor film on the hotplate in an inert glovebox atmosphere. Instead, it forms an amorphous matrix of Ba–Zr–S, requiring subsequent moderate-temperature annealing of the film in the presence of sulfur. The precise cause of this challenge in direct-to-film nucleation of BaZrS3 is not entirely clear, but several factors may contribute to it. One possibility is the absence of a liquid medium on the film at the nucleation temperature, which could restrict the mobility of elements and impede nucleation. Alternatively, a sulfur-deficient environment during annealing on the hotplate may affect the kinetics of BaZrS3 nucleation. Additionally, zirconium's strong affinity for oxygen may result in small amounts of oxygen impurities in the inert atmosphere inhibiting nucleation. Another factor could be excessive residual carbon in the film, which is known to impact nucleation and grain growth.61 Some challenges discussed in this study stem from the inability to nucleate the ternary compound after hotplate annealing and the reliance on a liquid medium during subsequent heat treatment. Hence, developing soluble precursors capable of decomposition followed by subsequent nucleation under suitable annealing conditions without the need for an external sulfur atmosphere while maintaining a constant Ba/Zr ratio is crucial.As reported in previous work, BaZrS3 and BaHfS3 can be synthesized using barium dithiocarboxylates and group IV metal dithiocarbamate precursors dissolved in a pyridine-based ink.57 These precursors are synthesized through well-established CS2 insertion chemistry routes62–64 utilizing bis(pentamethylcyclopentadienyl)barium(II) (Cp*2Ba) and tetrakis(ethylmethylamino)zirconium(IV) (TEMAZ) or tetrakis(ethylmethylamino)hafnium(IV) (TEMAH). The resulting inks were blade coated onto low-alkali Eagle XG (EXG) glass or quartz substrates with hotplate annealing at 200 °C between each coated layer in an inert nitrogen atmosphere to create a film. However, the film was amorphous at this stage, showing no distinct peaks in Raman spectroscopy or powder X-ray diffraction (XRD).57 It should be noted that these measurements were performed in air where the precursor film may have degraded into related oxide-containing phases, obscuring accurate representation of the film before removal from the inert atmosphere. Nonetheless, to nucleate crystalline chalcogenide perovskites at temperatures of 575 °C, we found that an external sulfur source was required, and sulfurization heat treatment was accomplished by sealing the film in an evacuated ampule alongside sulfur and HfH2 powder. Due to the high oxophilicity of transition metals such as Zr and Hf, even trace quantities of oxygen in the system can limit the formation of crystalline chalcogenide perovskites and even form metal oxides such as ZrO2 and BaZrO3. This demonstrates the need for a system that preferentially traps O2 from the reaction environment and the film. For this case, the HfH2 acts as an oxygen sink for any residual oxygen present in the film or the ampule, forming HfO2, due to the thermodynamic driving force caused by hafnium's higher oxophilicty than zircoinum. Later work studying the mechanism of the HfH2 oxygen sink showed that in the presence of a sulfur atmosphere, HfH2 reacts with sulfur to form HfS3 and H2S.65 The H2S can then react with oxides in the ampule or the film, forming H2O. This H2O is then shuttled back to the HfS3, forming HfO2 and regenerating H2S. Changing the ratio of sulfur to HfH2 in the ampules during sulfurization makes it possible to control how much sulfur is entrapped as HfS3 and control the partial pressure of sulfur and H2S in the ampule. However, the coexistence of H2S and sulfur within the ampule environment complicates the independent control of each and the delineation of their individual effects on ternary perovskite synthesis.
The examples in the literature show that sulfur partial pressure can significantly affect the formation rate of BaZrS3 and whether secondary phases form.7,16,59 Wang et al. found that the presence of intermediate amounts of excess sulfur allowed for the formation of BaZrS3 at lower temperatures.7 Based on this, Sopiha et al. suggested that an ideal sulfur pressure exists under which BaZrS3 can form quickly, with excess sulfur leading to the formation of ZrS3 and BaS3 and too little sulfur resulting in the formation of ZrS2 and BaS2.16 Vincent et al. further explored this mechanism and found that while increasing sulfur leads to increasingly more crystalline BaZrS3 up to the highest sulfur (S6) partial pressure tested (0.74 bar), no formation of binary sulfides was reported.53 Kayastha et al. additionally computed that BaZrS3 cannot decompose into binaries at any temperature ranging from 230 °C to 1000 °C under sulfur pressures up to 1 × 107 Pa.66 Therefore, there is a considerably high sulfur pressure threshold preventing the breakdown of BaZrS3. However, no information has been documented regarding the lower limit of sulfur pressure required for the synthesis of BaZrS3.
Using a different synthesis route for the annealed film utilizing BaS and TEMAZ precursors, as described for Method 1 in the experimental section, along with increase in crystallinity, the partial pressure of sulfur plays a large role in the species and secondary phases that form after sulfurization heat treatment. Table 1 shows the initial amount of sulfur and HfH2 powder loaded into each 5 mL ampule along with the expected H2S and sulfur partial pressures, assuming that all free sulfur in the ampule is in the S6 allotrope. While we expect sulfur to be in a mixture of S8, S7, and S6 allotropes,67 S6 was used to determine an upper bound in pressure. Table S1 (ESI†) shows calculated mmol of H2S, HfS3, sulfur, H2, HfS2, and HfH2 in the ampules. We assumed that in the presence of excess sulfur, HfH2 will react to form HfS3 and H2S. If sulfur is not in excess, we expect the sulfur to preferentially react with Hf to form HfS3 or HfS2 at equilibrium before any excess reacts with H2 to form H2S as is described for ZrS2 and ZrS3 in previous work.65 Note that these calculations do not account for the sulfur consumed or evolved from the film.
| Sample | Initial sulfur in ampule (mmol) | Initial HfH2 in ampule (mmol) | Theoretical sulfur (S6) partial pressure at 575 °C (atm) | Theoretical H2S partial pressure at 575 °C (atm) |
|---|---|---|---|---|
| 1 | 0.45 | 0.03 | 0.8 | 0.42 |
| 2 | 0.30 | 0.03 | 0.4 | 0.42 |
| 3 | 0.15 | 0.03 | 0.1 | 0.42 |
| 4 | 0.06 | 0.03 | 0.0 | 0.00 |
| 5 | 0.03 | 0.03 | 0.0 | 0.00 |
It is important to note that while the inks were synthesized with stoichiometric amounts of barium and zirconium, there was a relative loss of zirconium during the hot plate annealing and/or sulfurization step as indicated by X-ray fluorescence (XRF) measurements in Table S2 (ESI†), resulting in Ba-rich films. This phenomenon is the opposite of what is observed when Cp*2Ba is used as the Ba source, as described in previous work where the relative loss of Ba was more significant, and the Ba/Zr ratio was found to decrease during the annealing and sulfurization steps.57 This presents yet another challenge in controlling the Ba/Zr ratio in the film while using dithiocarboxylate and dithiocarbamate precursors.
Fig. 1 shows the different species that form upon a one-hour sulfurization heat treatment at 575 °C for varying levels of sulfur in the ampule. Fig. 1a shows that for S6 partial pressures of 0.8 atm (Sample 1) and 0.4 atm (Sample 2), we form crystalline BaZrS3 with no prominent crystalline secondary phases observed. This result is noteworthy as there is a significant excess of Ba and S in Samples 1 and 2, as observed by XRF (Table S2, ESI†). This excess Ba and S is likely in the form of amorphous barium polysulfide. Fig. 1b shows that further reducing the S6 partial pressure to 0.1 atm (Sample 3) results in the formation of a mixture of BaZrS3 and Ruddlesden–Popper (RP) phases, Ba4Zr3S10 and Ba3Zr2S7, with small amounts of BaS. RP phases in the Ba–Zr–S system consist of a layer of BaS separated by n layers of BaZrS3 perovskite, resulting in a Ban+1ZrnS3n+1 composition where the n = 2 and n = 3 phases are believed to be the most stable.68 However, Kayastha et al. also reported the presence of impurities of Ba4Zr3S10 in their solid-state attempts to synthesize BaZrS3.69 This underscores the importance of reevaluating the relative thermodynamic stabilities of Ba–Zr–S RP phases. Remarkably, RP phases in chalcogenide perovskites exhibit narrower bandgaps than their parent distorted perovskite phase, rendering them suitable as absorber materials for single-junction solar cells.16 However, their presence as impurity phases alongside BaZrS3 could pose a challenge.
 | ||
| Fig. 1 XRD patterns of Ba–Zr–S films synthesized as described in Method 1 followed by 1 h sulfurization heat treatments at 575 °C in evacuated 5 mL ampules containing 0.03 mmol HfH2 and (a) 0.45 mmol sulfur in Sample 1 and 0.30 mmol in Sample 2 or (b) 0.15 mmol sulfur in Sample 3. Fig. S2 (ESI†) shows Rietveld refinement for Samples 1 and 3. | ||
There is a lack of concrete previous studies that report the effect of low partial pressure of sulfur during the synthesis. However, it plays a decisive role in determining whether RP phases form or crystalline BaZrS3 forms without detectable crystalline secondary phases. We hypothesize that a liquid barium polysulfide flux forms under sulfur-rich conditions, reducing the kinetic barriers and facilitating the nucleation and growth of ternary Ba–Zr–S material. Upon cooling at roughly 14 °C min−1, this liquid flux is quenched into an amorphous barium polysulfide, which is not detectable in XRD or Raman analyses. Vincent et al. have presented scanning electron microscopy (SEM) evidence of the quenching of this liquid barium polysulfide phase under similar cooling conditions.59 Under large excesses of sulfur, excessive amounts of barium polysulfide may form, reducing the kinetic barrier and allowing the thermodynamically favored phase of BaZrS3 to form under these temperature-pressure conditions, along with amorphous barium polysulfide. However, as the partial pressure of sulfur is reduced, the driving force for the formation of barium polysulfide decreases, and the mass transfer barrier leads to a mixture of the thermodynamically favored phase of BaZrS3 and RP phases, as evidenced by Sample 3 through XRD patterns (Fig. 1b) and Raman spectra (Fig. S1, ESI†). Rietveld refinement for Samples 1 and 3 is depicted in Fig. S2 (ESI†). The XRD patterns of Sample 1 were fully accounted for by BaZrS3, with any other ternary or binary phases tested contributing less than 0.5% to the data. As expected, Sample 3 consisted of a mixture of BaZrS3 and RP phases, including Ba3Zr2S7 and Ba4Zr3S10. It has been reported that the formation energies of BaZrS3 and its RP phase counterparts are similar at high temperatures. Kayastha et al. mentioned that mixtures of BaZrS3 and RP phases can form when dwelling at 900 °C in for multiple days.69 It is also plausible that RP phases are thermodynamically more stable than BaZrS3 under sulfur-poor conditions, and localized variations in the films may have rendered conditions suitable for BaZrS3 despite an otherwise preferred RP phase.
To ensure that these observations are not specific to a particular method, similar experiments were conducted under varying sulfur pressures and with different Ba![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zr ratios using the hybrid precursor ink (Method 2) previously reported by our group (see Table 2 for description).51 Please note that, unlike Method 1, this method does not employ an HfH2 oxygen trap. Instead, the in-situ H2S produced in this case originates from the zirconium precursor used, ZrH2. As depicted in Fig. 2 and Fig. S3 (ESI†), stoichiometric films synthesized at low sulfur pressures exhibited mixed phases of BaZrS3 and Ba3Zr2S7, along with some impurities of BaS3 and ZrS3—formation of these binaries appearing to contradict previous statements regarding the trisulfide formation only at higher pressures. The Rietveld refinement in Fig. S4 and S5 (ESI†) shows a significant fraction of Ba3Zr2S7 in the films. Conversely, phase-pure BaZrS3 was attained at high sulfur pressures for films synthesized from stoichiometric Ba:Zr inks, as shown in Fig. 2 and Fig. S6 (ESI†). In another significant experiment, we sulfurized the as-deposited film at 575 °C for 24 h under sulfur-poor conditions (Sample 9) and observed a significantly higher percentage of Ba3Zr2S7 in the film relative to BaZrS3, as determined by the Rietveld refinement (see Fig. S7–S9, ESI†). Additionally, we observed that using a solution with a Ba:Zr ratio of 1.5:1 also resulted in a mixture of BaZrS3, Ba3Zr2S7, and BaS3 when synthesized under sulfur-poor conditions, illustrated in Fig. S10–S12 (ESI†) for Sample 10.
:Zr ratios using the hybrid precursor ink (Method 2) previously reported by our group (see Table 2 for description).51 Please note that, unlike Method 1, this method does not employ an HfH2 oxygen trap. Instead, the in-situ H2S produced in this case originates from the zirconium precursor used, ZrH2. As depicted in Fig. 2 and Fig. S3 (ESI†), stoichiometric films synthesized at low sulfur pressures exhibited mixed phases of BaZrS3 and Ba3Zr2S7, along with some impurities of BaS3 and ZrS3—formation of these binaries appearing to contradict previous statements regarding the trisulfide formation only at higher pressures. The Rietveld refinement in Fig. S4 and S5 (ESI†) shows a significant fraction of Ba3Zr2S7 in the films. Conversely, phase-pure BaZrS3 was attained at high sulfur pressures for films synthesized from stoichiometric Ba:Zr inks, as shown in Fig. 2 and Fig. S6 (ESI†). In another significant experiment, we sulfurized the as-deposited film at 575 °C for 24 h under sulfur-poor conditions (Sample 9) and observed a significantly higher percentage of Ba3Zr2S7 in the film relative to BaZrS3, as determined by the Rietveld refinement (see Fig. S7–S9, ESI†). Additionally, we observed that using a solution with a Ba:Zr ratio of 1.5:1 also resulted in a mixture of BaZrS3, Ba3Zr2S7, and BaS3 when synthesized under sulfur-poor conditions, illustrated in Fig. S10–S12 (ESI†) for Sample 10.
| Sample | Ba:Zr ratio in the ink |
Initial sulfur in ampule (mmol) | Theoretical sulfur (S6) partial pressure at 575 °C (atm) | Theoretical H2S partial pressure at 575 °C (atm) | Sulfurization time |
|---|---|---|---|---|---|
| 6 | 1:1 |
0.31 | 0.61 | 0.22 | 15 min |
| 7 | 1:1 |
0.08 | 0.07 | 0.22 | 15 min |
| 8 | 1:1 |
0.05 | 0.03 | 0.00 | 15 min |
| 9 | 1:1 |
0.08 | 0.07 | 0.22 | 24 h |
| 10 | 1.5:1 |
0.08 | 0.07 | 0.22 | 15 min |
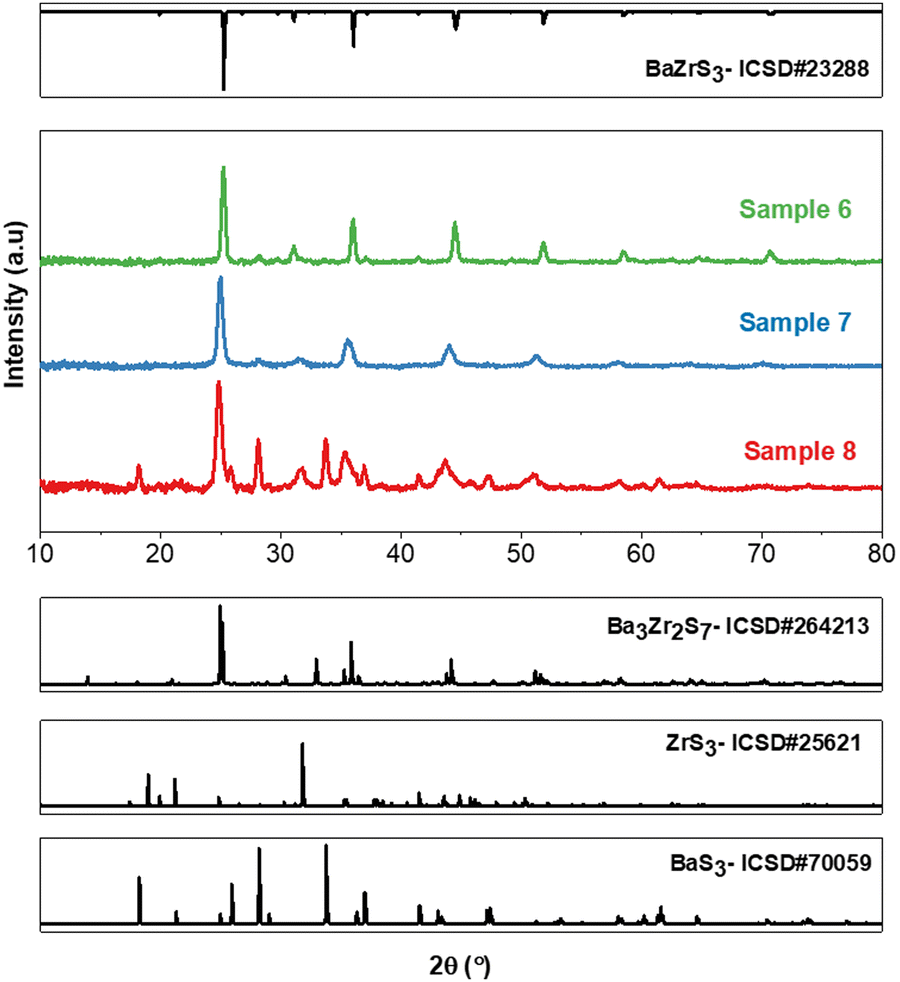 | ||
| Fig. 2 XRD patterns of Ba–Zr–S films synthesized as described in Method 2 followed by 15 min sulfurization heat treatments at 575 °C in evacuated 5 mL ampules containing varying amounts of sulfur 0.31 mmol sulfur in Sample 6, 0.08 mmol in Sample 7 and 0.05 mmol in Sample 8. Fig. S3–S5 (ESI†) show Rietveld refinement for Samples 6, 7 and 8. | ||
This underscores that the bulk Ba:Zr ratio and sulfurization time do not significantly influence the synthesis of competing RP phases, with sulfur pressure playing a more decisive role. Furthermore, the synthesis of RP phases does not appear to be specific to the synthesis method. Nevertheless, the potential to synthesize RP phases at 575 °C in as little as 15 minutes could also be seen as an exciting step towards producing phase-pure RP phases for applications in single-junction solar cells. However, further investigation is necessary to determine the conditions for their phase-pure synthesis at low-to-moderate temperatures. It is also crucial to determine whether small, localized impurities of RP phases consistently exist in bulk BaZrS3 films produced under high sulfur conditions. Until now, it has been assumed that BaZrS3 is the thermodynamically favored phase at low temperatures, but competing RP phases now challenge this belief. Similar observations were made for the Ba–Hf–S system using Ba:Hf ratios of 1.5:1 in the ink, as evidenced in Fig. S13 and S14 (ESI†), and should also be a focus of future studies.
Further decreasing the amount of sulfur in the ampules, as shown in Samples 4 and 5 (synthesized using Method 1), results in all the sulfur being consumed by the HfH2 and forming HfS2, and consequently, no sulfur vapor is expected to be present at equilibrium in both samples (Table S1, ESI†). It is also possible that HfH2 may be extracting sulfur from the annealed films themselves. Without sulfur vapor, a liquid barium polysulfide flux cannot form, and consequently, ternary BaZrS3 or RP phases cannot form in a short duration of a few minutes at 575 °C (see Fig. S15, ESI†). Instead, a mixture of BaS and BaS3 is observed. It is important to note that while a crystalline zirconium-containing phase is not observed in XRD, XRF measurements (Table S2, ESI†) do show the presence of zirconium in the film, indicating that the zirconium is in an amorphous phase. The strength of the XRD signal is a qualitative indicator of the abundance of each type of crystalline material. As indicated by the lower signal-to-noise ratio for Sample 5 in Fig. S15 (ESI†), the signal from Sample 5 is weaker than that of the other films, meaning there is a lower abundance of crystalline BaS and BaS3 present. The lower amount of BaS and BaS3 in Sample 5 compared with Sample 4 demonstrates that the small amount of sulfur present from the coated barium and zirconium dithiocarbamates complexes is not enough to convert the amorphous material into crystalline binary barium sulfides fully and the small amount of sulfur vapor present in the initial stages of sulfurization in Sample 4, before the HfH2 consumes it, aids in the crystallization of BaS and BaS3.
We have demonstrated that the partial pressure of sulfur in the ampule plays an essential role in which Ba–Zr–S phase forms after heat treatment. Sulfur present in the dithiocarbamate and dithiocarboxylate precursors alone does not result in the formation of crystalline ternary phase Ba–Zr–S material at temperatures below 600 °C without introducing an external sulfur source. Using sulfur partial pressures of 0.1 atm or lower for Ba–Zr–S films leads to Ruddlesden–Popper phases (Ba3Zr2S7) and binary sulfides such as BaS and BaS3. However, greater sulfur partial pressures approaching 0.4 atm drive Ba-Zr-S films toward the distorted perovskite phase, even in excess barium.
Impacts of rapid growth on the properties of chalcogenide perovskite grains
While the formation of chalcogenide perovskites, such as BaZrS3 and BaHfS3, at low temperatures with the assistance of a barium polysulfide flux, as demonstrated by Vincent et al. and Yang et al.,59,60 has allowed for the creation of large crystalline grains of perovskite material at moderate temperatures, the perceived fast growth of these grains has led to some significant challenges. Prior work in our group has demonstrated that large micron-sized grains of BaZrS3 and BaHfS3 can be synthesized at 575 °C in the presence of a sulfur atmosphere in heat treatment times as short as 10 minutes (excluding a ∼10-minute heat up).56,57 While rapid grain growth is typically advantageous for the synthesis of thin films, rapid grain growth in our work has led to defective and impure materials with regions of entrapped oxides and secondary phases. Furthermore, this uncontrolled grain growth has also resulted in forming isolated islands of material instead of a continuous film layer after sulfurization heat treatments, making these films unviable for use in renewable energy devices such as photovoltaics. Fig. S16 (ESI†) shows that within 10 minutes at 575 °C, cubic BaZrS3 grains on the order of 10 μm by 10 μm form from the suspension precursor solution of Method 3. Longer sulfurization times result in cubic BaZrS3 grains as large as 40 μm per edge, likely due to Ostwald ripening. As BaZrS3 thin film absorber materials would ideally be less than 1 μm thick, due to its anticipated high absorption coefficient, 10 μm grains are already far too large. The primary challenge with this synthesis approach is that BaZrS3 does not form in a phase pure manner below ∼550 °C due to diffusion limitations aided by the barium polysulfide liquid flux,59 but once these crystals of ternary phase chalcogenide perovskites do nucleate, they appear to grow rapidly, absorbing material around the crystals and leading to island growth and thus, non-continuous films. So far, attempts to uniformly nucleate BaZrS3 or BaHfS3 and grow dense, ≤1 μm-thick films utilizing a solution-deposited approach have been unsuccessful.In addition to the problems regarding film homogeneity, the rapid grain growth resulting from the barium polysulfide flux may also lead to a defective material with poor optoelectronic properties and regions of entrapped secondary phases. Fig. 3a shows a bright field transmission electron microscope (TEM) image of a thin lamella of a BaZrS3 grain prepared using a gallium-focused ion beam (FIB). This sample was synthesized as described in Method 3 and sulfurized for 16 h at 575 °C in a 5 mL borosilicate ampule with 0.1 mmol sulfur without HfH2. This would result in an estimated S6 partial pressure of 0.26 atm in the ampule at 575 °C. The TEM image shows many planar defects, including what appears to be many edge dislocations and internal grain boundaries. Furthermore, oxide impurities in the crystal can be identified as bright spots in the image. Fig. 3b is a high-angle annular dark field (HAADF)-scanning transmission electron microscopy (STEM) image of one of these bright spots from the bright field TEM image. Energy dispersive X-ray (EDX) analysis was carried out on this spot and is shown in Fig. 3c–f. We found that these dark regions in the HAADF image are oxygen-rich and poor in sulfur and barium, indicating the presence of a zirconium oxide secondary phase. Comparable Zr–O-rich regions were identified in the films synthesized using Method 4, reported earlier by our group,59 followed by sulfurization at 575 °C with sulfur in the presence of HfH2 (see Fig. S17, ESI†). The slower kinetics of oxygen transport to Hf compared to Zr, coupled with the faster kinetics of BaZrS3 growth in the presence of BaSx liquid flux, could possibly be resulting in the entrapment of oxide impurities within the crystal. Thus, it is necessary to further explore and optimize the sulfurization step.
 | ||
| Fig. 3 (a) TEM bright field image of a thin lamella prepared by FIB shows many planar defects and impurities in the grain. The red arrows point out examples of bright spots-indicating oxide impurities- in the image, one of which is analyzed using HAADF-STEM. (b) a HAADF-STEM image of a bright spot seen in the TEM bright field image. (c)–(f) EDX intensity maps of the HAADF image indicate the presence of zirconium oxide embedded in the BaZrS3 grain. | ||
In addition to the poor morphological properties of the synthesized BaZrS3 grains, the optoelectronic properties of these materials are also poor. BaZrS3 is expected to have a band gap of 1.7 to 1.9 eV,16 but as shown in Fig. 4a, we observed photoemission at 1.05 eV with no emission near the bandgap. This result was surprising given that multiple computational studies have predicted BaZrS3 to have excellent optoelectronic properties.70,71 Raman analysis ensured the samples measured were BaZrS3 and did not contain RP secondary phases. The mid-bandgap emission at 1.05 eV is consistently observed in BaZrS3 synthesized through all solution-based methods documented in this paper. As depicted in Fig. S18 (ESI†), it was also detected in solid-state BaZrS3 samples synthesized using liquid BaSx flux and I2 transport agents. This underscores that the mid-gap emission seem to be a common problem across a range of synthesis techniques. Additionally, the peak persists even with prolonged sulfurization times. Márquez et al. also observed mid-gap photoluminescence (PL) peaks at 1.14 and 1.35 eV for BaZrS3 samples sulfurized at temperatures above 1000 °C and attributed them to deep defect states.12 The 1.05 eV photoemission we observe is likely similar due to the material's deep transition state. Wu et al. have recently published computational work on defect tolerance in BaZrS3, which suggests that under moderate sulfur conditions, the shallow VS donor defect is the dominant defect in BaZrS3.72 The only deep-level defects in BaZrS3 are BaZr, VZr, Si, SBa, and SZr.72 BaZr and SZr are deep acceptors positioned 0.46 eV and 0.40 eV above the valence band maximum (VBM) respectively while VZr, Si, and SBa are deep acceptors positioned 1.28, 1.34, and 1.40 eV below the conduction band minimum (CBM) respectively.72 While all deep-level defects have been estimated to have high formation energies under moderate sulfur conditions, Si, SBa, and VZr become the dominant defects under sulfur-rich conditions. In this paper and previous work,59 we have demonstrated that low-temperature synthesis of crystalline BaZrS3 requires excess sulfur, but this approach may increase concentrations of harmful defects such as Si, SBa, and VZr. Additionally, RP phases are known to have band gaps in the region of ∼1.2 eV, and the presence of local nano-domains of RP phases in the final films cannot be completely ruled out at this stage. It is noteworthy that samples 7–10, which contain a significant amount of RP phases, also exhibit these emission peaks, albeit at slightly shifted energies. Sample 9, which includes the highest proportion of RP phases relative to BaZrS3, also displays a similar peak at 1.15 eV, noticeably shifted from the reported bandgaps of the RP phases in the Ba–Zr–S system (illustrated in Fig. S19, ESI†). This emission may originate from near-bandgap states of RP phases trapped within the bulk BaZrS3 crystals. These harmful defects, or nano-domains of RP phases, may explain the weak band gap photoluminescence in BaZrS3 and the defect-related photoemission at 1.05 eV.
 | ||
| Fig. 4 (a) Photoluminescence (PL) spectrum of a Ba–Zr–S sample synthesized as described in Method 3 on a quartz substrate and sulfurized for 24 h in a 5 mL ampule containing 0.3 mmol sulfur and 0.03 mmol HfH2 and measured using a 632 nm excitation source. A strong photoemission is observed at 1.05 eV but no band-to-band emission was observed. (b) Time-resolved photoluminescence (TRPL) measurements of the grains in the BaZrS3 film show a biexponential decay curve with fast decay resulting in carrier lifetimes of 1.1 ns and a long decay resulting in carrier lifetimes of 309 ns. TRPL data show a statistical average minority carrier lifetime of 9.06 ns (τstat = (A1τ1 + A2τ2)/(A1 + A2)). | ||
Owing to the high light absorption coefficient of BaZrS3, photons across various wavelengths with energies greater than the bandgap are absorbed thoroughly within the first 200 nm of the film. The near-surface region of the micron-thick film may be suboptimal, resulting in weak band-to-band photoemission and necessitating substantial enhancement. Some of the impurities could potentially include ZrOx and BaSO4, as highlighted by Mukherjee et al. using X-ray photoelectron spectroscopy (XPS) measurements.73 The defective grains observed in Fig. 3a and the poor optoelectronic properties of the synthesized materials suggest that significant enhancements in sulfurization conditions are necessary. Additionally, exploring the possibility of etching away the few nanometers from near the top surface of the film could address some of these issues.
Time-resolved photoluminescence spectroscopy (TRPL) shown in Fig. 4b indicates that photogenerated minority carriers in BaZrS3 have relatively long lifetimes calculated via a biexponential decay. A fast decay with a lifetime of 1.1 ns and a slower decay with a lifetime of 309 ns were determined via curve fitting, resulting in a statistical average minority carrier lifetime of 9.06 ns. BaZrS3 synthesized using other approaches and on different substrates, such as low-alkali EXG, also showed these long carrier lifetimes. This lifetime is unexpectedly large for a defective material, which may suggest that minority carrier trapping/de-trapping is occurring. In this case, some photogenerated minority carriers may be trapped in localized shallow states with capture times similar to the fast decay lifetime of 1.1 ns we observe. The trap emission time could govern the longer decay time of 309 ns.74 Voltage-dependent and intensity-dependent TRPL would need to be carried out to confirm this hypothesis as described in Hages et al.74
Optoelectronic properties of secondary phases and substrate selection
Investigations of chalcogenide perovskites' photoluminescence properties are further complicated by secondary phases that can have PL emissions at the same energy as the related perovskites. These secondary phases likely contributed to emissions observed in photoluminescent grains in our previous studies.56,57Fig. 5a shows a PL image of a grain that showed luminescence centered at 1.72 eV in a Ba–Zr–S film synthesized as described in Method 3 on EXG glass and sulfurized for 16 h at 575 °C in a 5 mL borosilicate ampule with 0.1 mmol sulfur and no HfH2. The white dotted box is the region on which FIB was used to create a thin lamella of material for STEM-EDX analysis. It is important to note that this region contains bright, luminescent areas and darker, lower luminescence areas. Fig. 5b is a HAADF-STEM image of the thin lamella that shows gray areas and brighter white areas due to differences in the atomic number of the elements present. STEM-EDX in Fig. 5c–f reveals that the dark area in Fig. 5a, with less luminescence, contains BaZrS3, while the areas in Fig. 5a that show strong luminescence in the visible range (1.72 eV) are calcium sulfide. Calcium sulfide is usually a high, indirect band gap material, but studies have shown that when doped with certain elements, it can show strong luminescence at lower energies.75–77 We believe this calcium is diffusing from the EXG glass substrate and nucleating around particles of BaZrS3, forming luminescent CaS crystals at temperatures of 575 °C after long sulfurization times. As shown in Fig. 5g, these CaS grains show a strong PL emission at 1.72 eV, which is near the expected band gap of BaZrS3. Initially, these luminescent grains were believed to be BaZrS3 through Raman analysis. As CaS is not strongly Raman active due to the symmetry of its rock salt structure,78 collecting Raman on the CaS grains, as shown in Fig. 5h, resulted in a collection of only the Raman signal from the embedded BaZrS3. CaS was also not detectable in Raman using a higher energy 532 nm excitation laser. As shown in Fig. S20 (ESI†), grains in Ba–Hf–S films synthesized from Method 5 that showed luminescence near the expected band gap of BaHfS3 were also found to be CaS with Ba-Hf-S embedded within the grain. Similar to Fig. 5a, Fig. S20 (ESI†) demonstrates that the luminescent areas in the grain correspond to CaS, while the darker regions contained Ba–Hf–S. | ||
| Fig. 5 (a) PL image of a CaS grain with embedded BaZrS3. The white dotted box indicates where FIB was performed to create a thin lamella of material for STEM-EDX analysis. (b) A HAADF-STEM image of the lamella where the light grey regions correspond to the darker area in the PL image and the dark grey region corresponds to the luminescent area in the PL image. (c)–(f) EDX maps of the HAADF image indicate that the light grey areas are BaZrS3 while the dark grey areas are CaS. (g) PL spectrum of the luminescent CaS particle showing an emission centered at 1.72 eV. (h) Raman spectra of the CaS grain with embedded BaZrS3 compared with that of the BaZrS3 grain. | ||
Cationic diffusion from glass substrates is a well-established phenomenon in the photovoltaic industry. Specifically, soda-lime glass (SLG) is often used as substrates for Cu(In,Ga)Se2 (CIGSe) solar cells to incorporate Na intentionally. At temperatures between 500 °C and 600 °C, which is in the same range as SLG's strain point (514 °C), annealing point (550 °C), and glass transition temperature (570 °C), Na ions become more mobile and can diffuse from the glass substrate into the absorber layer, resulting in improved electronic properties in CIGSe.79 We also observed that under our sulfurization heat treatment conditions, Na ions diffuse from SLG into the absorber material and form a Na2ZrS3 secondary phase within 3 h, as shown in Fig. S21 (ESI†). For this reason, we investigated alternative substrates. While the strain and annealing point of the EXG glass (669 °C and 722 °C respectively) are significantly higher than that of SLG, at temperatures of 575 °C in a sulfur atmosphere, Ca may be diffusing from the substrate and nucleating around crystalline chalcogenide perovskite after several hours. Fig. S22 (ESI†) provides some evidence for Ca diffusion from the glass as SEM-EDX demonstrates that the ratio of Ca to Si in the EXG glass drops by 56% after exposing the glass substrate to a sulfur atmosphere at 575 °C for 10 days.
Although the band gap of CaS is known to be indirect at 4.43 eV,80 we found that CaS was responsible for photoemission at 1.72 eV in Ba–Zr–S films and at 2.05 eV in Ba–Hf–S films. Previous literature demonstrates that CaS can act as a host material for phosphors that can be activated by doping from transition metals such as Zr.75–77 For CaS:Zr phosphors, the phosphorescence emission band is reported to peak near 605 nm (2.05 eV).77 This emission can shift depending on doping concentration, with higher doping concentrations resulting in lower energy emissions.77 While CaS:Hf phosphors have not been reported, we believe they are the cause of the emissions we observe at 2.05 eV in the Ba–Hf–S films. BaS has also been shown to act as a host material for phosphors, so caution must also be taken to ensure PL in barium-based chalcogenide perovskites do not originate from BaS phosphors.75 Considering the preceding discussion, it is probable that the photoluminescence documented by our group in earlier publications also emanated from CaS with Ba–Zr–S integrated within the grains. This revelation points to a need to reevaluate previous reports of PL emission in low-temperature chalcogenide perovskites.50–52
Quartz substrates were initially considered an excellent alternative to EXG glass as diffusion of cations, such as Ca, from the glass would not be an issue. However, as depicted in Fig. S23 (ESI†), we found that barium can diffuse into the quartz through lattice diffusion under our heat treatment conditions, changing Ba/Zr ratios in the absorber layer. This film, which was synthesized as described in Method 4, does involve a 12 h heat treatment step at 700 °C to form crystalline BaZrO3 before this material is sulfurized at 575 °C for 24 h to form BaZrS3. This barium diffusion may be occurring during either (or both) of these heat treatment steps.
A sapphire substrate could be used instead of low-alkali EXG or quartz to minimize diffusion into and out of the substrate. A cheaper alternative would be to deposit an impermeable layer of graphite or a stable oxide material that will not react with sulfur at elevated temperatures, such as alumina, on the quartz substrate before layering with the chalcogenide perovskite to prevent Ba diffusion into the substrate. Furthermore, as BaS and CaS tend not to be strongly Raman active, they can be challenging to detect in small quantities. As both materials can act as phosphors in the presence of transition metals such as Zr or Hf, it is essential to ensure that any photoemission in experimentally synthesized chalcogenide perovskites is coming from the perovskite material instead of secondary phases.
Conclusion
Recent research has demonstrated the successful synthesis of chalcogenide perovskites through solution-based methods at temperatures below 600 °C. Despite significant advancements, several challenges require attention to sustain rapid progress. These challenges include the need for soluble molecular precursors capable of reacting to form chalcogenide perovskites without relying on an external sulfur atmosphere while maintaining a consistent Ba/Zr ratio. Additionally, addressing the elimination of trace amounts of oxygen from both the ampule and the film and carefully managing various sulfur source partial pressures throughout the heat treatment process is crucial. Inadequate sulfur atmospheres can result in the formation of binary BaS or Ruddlesden–Popper phases. Attempts to utilize this phenomenon to produce phase-pure RP phases at low temperatures have proved unsuccessful due to the comparable formation energies of the distorted perovskite and RP phase materials, leading to mixed-phase Ba–Zr–S films.Utilizing a barium polysulfide flux for moderate-temperature synthesis holds promise for advancing research on chalcogenide perovskites. However, the rapid growth of BaZrS3 and BaHfS3 presents several challenges. Firstly, this rapid growth leads to the entrapment of oxide contaminants within large multi-micron-sized grains. Under sulfurization heat treatment conditions, sulfur/H2S cannot diffuse and react with these oxide contaminants. Additionally, the rapid grain growth results in numerous planar defects within the grains, which persist even after extended annealing at 575 °C. Furthermore, this substantial grain growth produces large cubic grains on the order of tens of microns per side, hindering the formation of continuous, single-micron absorber layers necessary for synthesizing photovoltaic devices. The electronic defects in this material, as indicated by mid-band gap photoemissions, may also stem from this rapid grain growth or possibly from nano-domains of Ruddlesden–Popper phases present in the film. Enhancing the nucleation rate followed by controlled grain growth could enable the formation of a continuous film of chalcogenide perovskite with grains on the order of hundreds of nanometers, fewer entrapped secondary phases, and improved optoelectronic properties.
Furthermore, given the temperatures employed for sulfurization heat treatment, careful substrate selection is imperative during the solution-deposited synthesis of chalcogenide perovskites. To mitigate sodium diffusion from SLG substrates, we opted for low-alkali Corning EXG glass. However, we observed calcium diffusion from the EXG substrate with extended sulfurization heat treatment durations, leading to nucleation around chalcogenide perovskite crystals. This resulted in the formation of Zr and Hf-doped CaS phosphors, exhibiting photoemissions near the anticipated band gaps of BaZrS3 and BaHfS3 in Ba–Zr–S and Ba–Hf–S films. Moreover, as CaS lacks Raman activity, only the underlying chalcogenide perovskites were detectable during Raman analysis, and the abundance of CaS in the films was insufficient to be detected via XRD. Hence, meticulous characterization is essential to ensure the absence of difficult-to-detect secondary phases in synthesized films. Although quartz was initially considered a viable alternative to EXG as a substrate, we discovered that Ba diffusion into the quartz occurred under our heat treatment conditions, altering Ba/Zr ratios at the glass interface. Consequently, we advocate using quartz coated with graphite or an inert oxide layer, such as Al2O3 to prevent material diffusion into or out of the substrate.
Finally, many of these experiments were only feasible with the meticulous attention to ensuring oxygen- and water-free syntheses. Group IV transition metals, including Zr and Hf, rank among the most oxyphilic metals on the periodic table, rendering their conversion to chalcogenides exceptionally challenging and laborious. While we took the utmost care to conduct all methods described herein in an oxygen- and water-free environment, several steps were carried out using conventional equipment in our laboratory, such as our ampule sealing, which, until now, has been accomplished using a standard glass Schlenk line. This setup features numerous O-ring connections secured by stainless-steel pinch clamps, each representing a potential site for leaks. Given the delicate nature of this material system, these factors and their potential to influence experimental outcomes must be taken into account.
For the chalcogenide perovskite research community to advance BaZrS3 synthesis for optoelectronic applications, it is crucial to further investigate the following summarized challenges:
1. Solubility of metal precursors (for those taking a solution-based approach).
2. Nucleation of ternary phases.
3. Loss of Ba or Zr during annealing.
4. Sensitivity to sulfur partial pressure.
5. Sensitivity to oxygen.
6. Potential co-formation of the Ruddlesden–Popper (RP) phase impacting optoelectronic properties.
7. Formation of binary compounds at lower sulfur pressures.
8. Use of excess Ba to form phase-pure BaZrS3, potentially contaminating the material with trace quantities of BaSx.
9. Island growth leading to non-contiguous films and rapid growth in BaSx liquid flux.
10. Lack of photoluminescence near the band-to-band region and presence of midgap photoluminescence at 1.05 eV.
11. Photoluminescence from secondary phases with energy emissions near the band gap of the perovskite, with elements such as Ca diffusing into the film and showing photoluminescence via CaS phosphors.
Chalcogenide perovskites remain a compelling alternative to halide perovskites for optoelectronic applications, with their synthesis at temperatures compatible with device fabrication offering additional prospects. However, further refinement of growth conditions and enhancement of optoelectronic properties are prerequisites before integrating these materials into optoelectronic devices.
Experimental section
Chemicals and materials
Bis(pentamethylcyclopentadienyl)barium(II) (Cp*2Ba, 98%), barium acetate (99%), and zirconium acetylacetonate (97%) were purchased from Strem and used as received. Tetrakis(ethylmethylamino)zirconium(IV) (TEMAZ), >99.99%, Tetrakis(dimethylamino)zirconium(IV) (TDMAZ), >99.99%, tetrakis(ethylmethylamino)hafnium(IV) (TEMAH), >99.99%, barium sulfide (BaS, 99.9%), polyvinyl butyral, propionic acid (ACS Reagent, >99.5%), and carbon disulfide (CS2, anhydrous, >99%) were purchased from Sigma Aldrich and used as received. Butylamine (BA, >99.5%), Propylamine (PA, >99.0%), and 2-methyl-2-propanethiol (>99%) were purchased from Sigma Aldrich and stored over molecular sieves before use. Pyridine (Pyd, anhydrous, 99.8%) was purchased from Sigma-Aldrich and was further dried by stirring over calcium hydride for 36 h, followed by distillation. The pyridine was stored over molecular sieves and calcium hydride chunks in a nitrogen atmosphere. Sulfur flakes (S, 99.99%) were purchased from Sigma Aldrich, ground into a powder in an inert atmosphere, and dried under vacuum overnight at ambient temperature. Hafnium hydride nanopowder (HfH2, 99.9%) and Zirconium Hydride (ZrH2, 99.9%) were purchased from Nano Research Elements and used as received. All chemicals and solvents were handled inside inert nitrogen-filled gloveboxes. Carbon disulfide is a highly flammable and volatile species that is known to cause acute organ and nervous system damage. Only use CS2 in well-ventilated hoods or glove boxes and keep it away from open flames and hot surfaces.Low-alkali Eagle XG (EXG) glass (1.1 mm thick) was purchased from Corning and quartz glass (1 mm thick) was purchased from Quartz Scientific, cleaned with IPA, hexane, Alconox, ultrapure water, and acetone, and dried on a hotplate at annealing temperatures for 5 minutes immediately before use. 5 mL borosilicate glass ampules were purchased from Chemglass, cleaned with IPA, hexane, and acetone, and dried at 150 °C for at least 30 minutes immediately before use. The SEM-EDX confirmed the following elements in the EXG glass: Si, O, Al, Ca, and Mg.
Next, 120 μL of the ink was dropcast onto an EXG substrate and allowed to dry at 50 °C. The glass substrate was then transferred to a hotplate at 300 °C and allowed to anneal for 5 min to decompose the metal complexes and leave behind an amorphous Ba–Zr–S material.
Sulfurization heat treatment
Without any exposure to air, Ba–Zr–S, Ba–Hf–S, or BaZrO3 films were cut into 25 mm by 7 mm strips in a glovebox (except for the films from Method 2 which were already precut) and placed in 5 mL borosilicate glass ampules. A borosilicate glass NMR tube was cut to roughly two inches, and various amounts of sulfur and HfH2 were carefully weighed into it, as described in the main text (no HfH2 was used for samples from Method 2). Quartz wool was then placed into the tubes so that the solid powders could not escape from the NMR tube, but vapor phase sulfur and H2S could escape. This NMR tube was placed in the ampule alongside the film. The ampule was sealed using a butane torch at a pressure below 200 mtorr.The sealed ampule was placed in a refractory tube furnace and heated to 575 °C at a ramp rate of roughly 1 °C s−1. Once the setpoint temperature was reached, the ampule was held at the setpoint temperature. The furnace was cooled through forced convection at roughly 14 °C min−1. Samples were kept in sealed ampules until they were ready for analysis.
Characterization
X-ray diffraction (XRD) was performed with a Rigaku SmartLab diffractometer utilizing a Cu-Kα (λ = 1.5406 Å) source operated at 40 kV/44 mA in parallel beam mode at an incident angle of 0.5°. Raman spectroscopy was performed using a Horiba/Jobin-Yvon LabRAM HR800 confocal microscope setup through a 50× objective lens using a He:Ne laser (632.8 nm excitation wavelength). A Fischerscope X-Ray XAN 250 was used to collect X-ray fluorescence (XRF) elemental composition data.Photoluminescence (PL) measurements in the range of 1.38 to 3.10 eV and PL imaging were captured using an X-Cite Series 120 Q lamp as an excitation source. An Olympus BX53 microscope with a 330–385 nm bandpass filter for excitation and a dichroic mirror with a 400 nm cutoff wavelength was used for PL spectra measurement, and these PL spectra used a SpectraPro HRS-300 spectrometer using a silicon detector. For PL measurements in the range of 0.9 to 1.8 eV, a Horiba/Jobin-Yvon LabRAM HR800 confocal microscope set up through a 50× objective lens using a He:Ne laser (632.8 nm excitation wavelength) using an InGaAs detector. Transfer functions for both PL systems were acquired by utilizing a calibrated halogen light source to correct for optical losses due to the internal optics of the microscopes and account for changes in detector quantum efficiencies. PL measurements with 447 nm, 520 nm, 550 nm, and 600 nm lasers were performed using a home-built confocal micro-photoluminescence setup. Time-resolved photoluminescence measurements were carried out using a 637 nm diode laser (100 kHz to 1 MHz) and an InGaAs photomultiplier tube (Hamamatsu H10330A-45) under low-level injection conditions.
Scanning electron microscopy (SEM) and energy dispersive X-ray (EDX) analyses were conducted using a Quanta 3D FEG with an accelerating voltage of 10 kV, a spot size of 1.0, and a working distance of 10 mm. Transmission electron microscope (TEM) samples were prepared using a Ga-focused ion beam (FIB) on an FEI HELIOS 600 Dual Beam FIB-SEM, and TEM, HAADF, and EDX imaging was performed on an FEI Talos F200X scanning/transmission electron microscope (STEM).
Author contributions
Apurva A. Pradhan and Shubhanshu Agarwal: conceptualization, investigation, data curation, validation, formal analysis, visualization, writing – original draft, writing – review & editing. Kiruba Catherine Vincent: data curation, validation, formal analysis, writing – review & editing, visualization. Daniel C. Hayes: Data curation, formal analysis, writing – review & editing. Jonas M. Peterson: data curation, formal analysis. Jonathan W. Turnley: conceptualization, writing – review & editing. Robert M. Spilker: data curation. Madeleine C. Uible: data curation. Suzanne C. Bart: resources. Libai Huang: resources. Kim Kisslinger: data curation. Rakesh Agrawal: writing – review & editing, resources, supervision, project administration, funding acquisition.Data availability
The data supporting this article have been included in the ESI.†Conflicts of interest
The authors do not have any conflict of interest to declare regarding the work published in this article.Acknowledgements
This work was supported by the Research Traineeship Program (No. 1735282-NRT (SFEWS) and 10001536 (INFEWS)). This research used Electron Microscopy facilities of the Center for Functional Nanomaterials (CFN), a U.S. Department of Energy Office of Science User Facility, at Brookhaven National Laboratory under Contract No. DE-SC0012704.References
- X. Wei, H. Hui, S. Perera, A. Sheng, D. F. Watson, Y.-Y. Sun, Q. Jia, S. Zhang and H. Zeng, Ti-Alloying of BaZrS3 Chalcogenide Perovskite for Photovoltaics, ACS Omega, 2020, 5(30), 18579–18583, DOI:10.1021/acsomega.0c00740.
- Y. Nishigaki, T. Nagai, M. Nishiwaki, T. Aizawa, M. Kozawa, K. Hanzawa, Y. Kato, H. Sai, H. Hiramatsu, H. Hosono and H. Fujiwara, Extraordinary Strong Band-Edge Absorption in Distorted Chalcogenide Perovskites, Solar RRL, 2020, 4(5), 1900555, DOI:10.1002/solr.201900555.
- J. Xu, Y. Fan, W. Tian, L. Ye, Y. Zhang, Y. Tian, Y. Han and Z. Shi, Enhancing the Optical Absorption of Chalcogenide Perovskite BaZrS3 by Optimizing the Synthesis and Post-Processing Conditions, J. Solid State Chem., 2022, 307, 122872, DOI:10.1016/j.jssc.2021.122872.
- T. Gupta, D. Ghoshal, A. Yoshimura, S. Basu, P. K. Chow, A. S. Lakhnot, J. Pandey, J. M. Warrender, H. Efstathiadis, A. Soni, E. Osei-Agyemang, G. Balasubramanian, S. Zhang, S. Shi, T. Lu, V. Meunier and N. Koratkar, An Environmentally Stable and Lead-Free Chalcogenide Perovskite, Adv. Funct. Mater., 2020, 30(23), 2001387, DOI:10.1002/adfm.202001387.
- S. Perera, H. Hui, C. Zhao, H. Xue, F. Sun, C. Deng, N. Gross, C. Milleville, X. Xu, D. F. Watson, B. Weinstein, Y. Y. Sun, S. Zhang and H. Zeng, Chalcogenide Perovskites - an Emerging Class of Ionic Semiconductors, Nano Energy, 2016, 22, 129–135, DOI:10.1016/j.nanoen.2016.02.020.
- R. Lelieveld and D. J. W. IJdo, Sulphides with the GdFeO3 Structure, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1980, 36(10), 2223–2226, DOI:10.1107/S056774088000845X.
- Y. Wang, N. Sato and T. Fujino, Synthesis of BaZrS3 by Short Time Reaction at Lower Temperatures, J. Alloys Compd., 2001, 327(1–2), 104–112, DOI:10.1016/S0925-8388(01)01553-5.
- C. Comparotto, P. Ström, O. Donzel-Gargand, T. Kubart and J. J. S. Scragg, Synthesis of BaZrS3 Perovskite Thin Films at a Moderate Temperature on Conductive Substrates, ACS Appl. Energy Mater., 2022, 5(5), 6335–6343, DOI:10.1021/acsaem.2c00704.
- X. Wei, H. Hui, C. Zhao, C. Deng, M. Han, Z. Yu, A. Sheng, P. Roy, A. Chen, J. Lin, D. F. Watson, Y. Y. Sun, T. Thomay, S. Yang, Q. Jia, S. Zhang and H. Zeng, Realization of BaZrS3 Chalcogenide Perovskite Thin Films for Optoelectronics, Nano Energy, 2020, 68, 104317, DOI:10.1016/j.nanoen.2019.104317.
- I. Sadeghi, K. Ye, M. Xu, Y. Li, J. M. LeBeau and R. Jaramillo, Making BaZrS3 Chalcogenide Perovskite Thin Films by Molecular Beam Epitaxy, Adv. Funct. Mater., 2021, 31(45), 2105563, DOI:10.1002/adfm.202105563.
- V. K. Ravi, S. H. Yu, P. K. Rajput, C. Nayak, D. Bhattacharyya, D. S. Chung and A. Nag, Colloidal BaZrS3 Chalcogenide Perovskite Nanocrystals for Thin Film Device Fabrication, Nanoscale, 2021, 13(3), 1616–1623, 10.1039/D0NR08078K.
- J. A. Márquez, M. Rusu, H. Hempel, I. Y. Ahmet, M. Kölbach, I. Simsek, L. Choubrac, G. Gurieva, R. Gunder, S. Schorr and T. Unold, BaZrS3 Chalcogenide Perovskite Thin Films by H2S Sulfurization of Oxide Precursors, J. Phys. Chem. Lett., 2021, 12(8), 2148–2153, DOI:10.1021/acs.jpclett.1c00177.
- A. Clearfield, The Synthesis and Crystal Structures of Some Alkaline Earth Titanium and Zirconium Sulfides, Acta Crystallogr., 1963, 16(2), 135–142, DOI:10.1107/S0365110X6300030X.
- S. Niu, H. Huyan, Y. Liu, M. Yeung, K. Ye, L. Blankemeier, T. Orvis, D. Sarkar, D. J. Singh, R. Kapadia and J. Ravichandran, Bandgap Control via Structural and Chemical Tuning of Transition Metal Perovskite Chalcogenides, Adv. Mater., 2017, 29(9), 1604733, DOI:10.1002/adma.201604733.
- S. Sharma, Z. Ward, K. Bhimani, K. Li, A. Lakhnot, R. Jain, S.-F. Shi, H. Terrones and N. Koratkar, Bandgap Tuning in BaZrS3 Perovskite Thin Films, ACS Appl. Electron Mater., 2021, 3(8), 3306–3312, DOI:10.1021/acsaelm.1c00575.
- K. V. Sopiha, C. Comparotto, J. A. Márquez and J. J. S. Scragg, Chalcogenide Perovskites: Tantalizing Prospects, Challenging Materials, Adv. Opt. Mater., 2022, 2101704, DOI:10.1002/adom.202101704.
- A. Jess, R. Yang and C. J. Hages, On the Phase Stability of Chalcogenide Perovskites, Chem. Mater., 2022, 30(3), 679–684, DOI:10.1021/acs.chemmater.2c01289.
- W. Meng, B. Saparov, F. Hong, J. Wang, D. B. Mitzi and Y. Yan, Alloying and Defect Control within Chalcogenide Perovskites for Optimized Photovoltaic Application, Chem. Mater., 2016, 28(3), 821–829, DOI:10.1021/acs.chemmater.5b04213.
- Y.-Y. Sun, M. L. Agiorgousis, P. Zhang and S. Zhang, Chalcogenide Perovskites for Photovoltaics, Nano Lett., 2015, 15(1), 581–585, DOI:10.1021/nl504046x.
- D. Tiwari, O. S. Hutter and G. Longo, Chalcogenide Perovskites for Photovoltaics: Current Status and Prospects, J. Phys.: Energy, 2021, 3(3), 034010, DOI:10.1088/2515-7655/abf41c.
- S. Niu, J. Milam-Guerrero, Y. Zhou, K. Ye, B. Zhao, B. C. Melot and J. Ravichandran, Thermal Stability Study of Transition Metal Perovskite Sulfides, J. Mater. Res., 2018, 33(24), 4135–4143, DOI:10.1557/jmr.2018.419.
- I. Sadeghi, J. Van Sambeek, T. Simonian, M. Xu, K. Ye, T. Cai, V. Nicolosi, J. M. LeBeau and R. Jaramillo, Expanding the Perovskite Periodic Table to Include Chalcogenide Alloys with Tunable Band Gap Spanning 1.5–1.9 eV, Adv. Funct. Mater., 2023, 2304575, 1–7, DOI:10.1002/adfm.202304575.
- S. Mukherjee, S. Riva, C. Comparotto, F. O. L. Johansson, G. J. Man, D. Phuyal, K. Simonov, J. Just, K. Klementiev, S. M. Butorin, J. Scragg and H. Rensmo, Interplay between Growth Mechanism, Materials Chemistry, and Band Gap Characteristics in Sputtered Thin Films of Chalcogenide Perovskite BaZrS3, ACS Appl. Energy Mater., 2023, 6, 11642–11653, DOI:10.1021/acsaem.3c02075.
- N. Jaykhedkar, R. Bystrický, M. Sýkora and T. Bučko, How the Temperature and Composition Govern the Structure and Band Gap of Zr-Based Chalcogenide Perovskites: Insights from ML Accelerated AIMD, Inorg. Chem., 2023, 62(31), 12480–12492, DOI:10.1021/acs.inorgchem.3c01696.
- H. I. Eya and N. Y. Dzade, Density Functional Theory Insights into the Structural, Electronic, Optical, Surface, and Band Alignment Properties of BaZrS3 Chalcogenide Perovskite for Photovoltaics, ACS Appl. Energy Mater., 2023, 6(11), 5729–5738, DOI:10.1021/acsaem.3c00103.
- S. Sharma, Z. D. Ward, K. Bhimani, M. Sharma, J. Quinton, T. D. Rhone, S. Shi, H. Terrones and N. Koratkar, Machine Learning-Aided Band Gap Engineering of BaZrS3 Chalcogenide Perovskite, ACS Appl. Mater. Interfaces, 2023, 15(15), 18962–18972, DOI:10.1021/acsami.3c00618.
- N. Gross, Y. Y. Sun, S. Perera, H. Hui, X. Wei, S. Zhang, H. Zeng and B. A. Weinstein, Stability and Band-Gap Tuning of the Chalcogenide Perovskite BaZrS3 in Raman and Optical Investigations at High Pressures, Phys. Rev. Appl., 2017, 8(4), 18–21, DOI:10.1103/PhysRevApplied.8.044014.
- Y. Han, J. Xu, Y. Liang, X. Chen, M. Jia, J. Zhang, L. Lian, Y. Liu, X. Li and Z. Shi, P-Type Conductive BaZrS3 Thin Film and Its Band Gap Tunning via Ruddlesden-Popper Ba3Zr2S7 and Titanium Alloying, Chem. Eng. J., 2023, 473, 145351, DOI:10.1016/j.cej.2023.145351.
- Z. Yu, H. Hui, D. West, H. Zhang, Y. Sun, S. Kong, Y. Zhang, C. Deng, S. Yang, S. Zhang and H. Zeng, Chalcogenide Perovskite Thin Films with Controlled Phases for Optoelectronics, Adv. Funct. Mater., 2023, 2309514, 1–9, DOI:10.1002/adfm.202309514.
- D. Liu, H. Peng, J. He and R. Sa, Alloy Engineering to Tune the Optoelectronic Properties and Photovoltaic Performance for Hf-Based Chalcogenide Perovskites, Mater. Sci. Semicond. Process., 2024, 169, 107919, DOI:10.1016/j.mssp.2023.107919.
- S. Niu, J. Milam-Guerrero, Y. Zhou, K. Ye, B. Zhao, B. C. Melot and J. Ravichandran, Thermal Stability Study of Transition Metal Perovskite Sulfides, J. Mater. Res., 2018, 33(24), 4135–4143, DOI:10.1557/jmr.2018.419.
- Y. Han, J. Xu, Y. Liang, X. Chen, M. Jia, J. Zhang, L. Lian, Y. Liu, X. Li and Z. Shi, P-Type Conductive BaZrS3 Thin Film and Its Band Gap Tunning via Ruddlesden-Popper Ba3Zr2S7 and Titanium Alloying, Chem. Eng. J., 2023, 473, 145351, DOI:10.1016/j.cej.2023.145351.
- R. Bystrický, S. K. Tiwari, P. Hutár and M. Sýkora, Thermal Stability of Chalcogenide Perovskites, Inorg. Chem., 2024, 63(28), 12826–12838, DOI:10.1021/acs.inorgchem.4c01308.
- D. Zilevu and S. E. Creutz, Shape-Controlled Synthesis of Colloidal Nanorods and Nanoparticles of Barium Titanium Sulfide, Chem. Mater., 2021, 33(13), 5137–5146, DOI:10.1021/acs.chemmater.1c01193.
- D. Zilevu, O. O. Parks and S. E. Creutz, Solution-Phase Synthesis of the Chalcogenide Perovskite Barium Zirconium Sulfide as Colloidal Nanomaterials, Chem. Commun., 2022, 58(75), 10512–10515, 10.1039/D2CC03494H.
- A. K. Jena, A. Kulkarni and T. Miyasaka, Halide Perovskite Photovoltaics: Background, Status, and Future Prospects, Chem. Rev., 2019, 119(5), 3036–3103, DOI:10.1021/acs.chemrev.8b00539.
- S. Suresh and A. R. Uhl, Present Status of Solution-Processing Routes for Cu(In,Ga)(S,Se)2 Solar Cell Absorbers, Adv. Energy Mater., 2021, 11(14), 2003743, DOI:10.1002/aenm.202003743.
- W. Li, J. M. R. Tan, S. W. Leow, S. Lie, S. Magdassi and L. H. Wong, Recent Progress in Solution-Processed Copper-Chalcogenide Thin-Film Solar Cells, Energy Technol., 2018, 46–59, DOI:10.1002/ente.201700734.
- S. Suresh, D. J. Rokke, A. A. Drew, E. Alruqobah, R. Agrawal and A. R. Uhl, Extrinsic Doping of Ink-Based Cu(In,Ga)(S,Se)2 -Absorbers for Photovoltaic Applications, Adv. Energy Mater., 2022, 2103961, DOI:10.1002/aenm.202103961.
- T. Todorov, H. W. Hillhouse, S. Aazou, Z. Sekkat, O. Vigil-Galán, S. D. Deshmukh, R. Agrawal, S. Bourdais, M. Valdés, P. Arnou, D. B. Mitzi and P. J. Dale, Solution-Based Synthesis of Kesterite Thin Film Semiconductors, J. Phys.: Energy, 2020, 2(1), 012003, DOI:10.1088/2515-7655/ab3a81.
- J. W. Turnley and R. Agrawal, Solution Processed Metal Chalcogenide Semiconductors for Inorganic Thin Film Photovoltaics, Chem. Commun., 2024, 60(40), 5245–5269, 10.1039/D4CC01057D.
- S. D. Deshmukh, R. G. Ellis, D. S. Sutandar, D. J. Rokke and R. Agrawal, Versatile Colloidal Syntheses of Metal Chalcogenide Nanoparticles from Elemental Precursors Using Amine-Thiol Chemistry, Chem. Mater., 2019, 2, DOI:10.1021/acs.chemmater.9b03401.
- X. Zhao, S. D. Deshmukh, D. J. Rokke, G. Zhang, Z. Wu, J. T. Miller and R. Agrawal, Investigating Chemistry of Metal Dissolution in Amine−Thiol Mixtures and Exploiting It toward Benign Ink Formulation for Metal Chalcogenide Thin Films, Chem. Mater., 2019, 31(15), 5674–5682, DOI:10.1021/acs.chemmater.9b01566.
- S. D. Deshmukh, L. F. Easterling, J. M. Manheim, N. J. LiBretto, K. G. Weideman, J. T. Miller, H. I. Kenttämaa and R. Agrawal, Analyzing and Tuning the Chalcogen–Amine–Thiol Complexes for Tailoring of Chalcogenide Syntheses, Inorg. Chem., 2020, 59(12), 8240–8250, DOI:10.1021/acs.inorgchem.0c00597.
- C. L. McCarthy and R. L. Brutchey, Solution Processing of Chalcogenide Materials Using Thiol-Amine “Alkahest” Solvent Systems, Chem. Commun., 2017, 53(36), 4888–4902, 10.1039/c7cc02226c.
- S. D. Deshmukh, C. K. Miskin, A. A. Pradhan, K. Kisslinger and R. Agrawal, Solution Processed Fabrication of Se-Te Alloy Thin Films for Application in PV Devices, ACS Appl. Energy Mater., 2022, 5(3), 3275–3281, DOI:10.1021/acsaem.1c03896.
- J. W. Turnley, S. D. Deshmukh, V. M. Boulos, R. Spilker, C. J. Breckner, K. Ng, J. K. Liu, J. T. Miller, H. I. Kenttämaa and R. Agrawal, A Selenium-Based “Alkahest”: Reactive Dissolutions of Metals and Metal Compounds with n -Alkylammonium Polyselenide Solutions, Inorg. Chem. Front., 2023, 10(20), 6032–6044, 10.1039/D3QI01632C.
- R. Zhang, S. Cho, D. G. Lim, X. Hu, E. A. Stach, C. A. Handwerker and R. Agrawal, Metal–Metal Chalcogenide Molecular Precursors to Binary, Ternary, and Quaternary Metal Chalcogenide Thin Films for Electronic Devices, Chem. Commun., 2016, 52(28), 5007–5010, 10.1039/C5CC09915C.
- A. A. Pradhan, C. Yao, S. A. McClary, K. G. Weideman, D. D. Blach, S. Khandelwal, J. Andler, D. J. Rokke, L. Huang, C. Handwerker, Y. Yan and R. Agrawal, Tuning the Optoelectronic Properties of Enargite (Cu3AsS4) Solar Cells by Ag Alloying: A DFT-Informed Synthesis, Appl. Phys. Lett., 2023, 123(19), 193301, DOI:10.1063/5.0170314.
- D. B. Mitzi, M. Yuan, W. Liu, A. J. Kellock, S. J. Chey, V. Deline and A. G. Schrott, A High-Efficiency Solution-Deposited Thin-Film Photovoltaic Device, Adv. Mater., 2008, 20(19), 3657–3662, DOI:10.1002/adma.200800555.
- J. Kim, H. Hiroi, T. K. Todorov, O. Gunawan, M. Kuwahara, T. Gokmen, D. Nair, M. Hopstaken, B. Shin, Y. S. Lee, W. Wang, H. Sugimoto and D. B. Mitzi, High Efficiency Cu2ZnSn(S,Se)4 Solar Cells by Applying a Double In2S3/CdS Emitter, Adv. Mater., 2014, 26(44), 7427–7431, DOI:10.1002/adma.201402373.
- W. Ki and H. W. Hillhouse, Earth-Abundant Element Photovoltaics Directly from Soluble Precursors with High Yield Using a Non-Toxic Solvent, Adv. Energy Mater., 2011, 1(5), 732–735, DOI:10.1002/aenm.201100140.
- S. Agarwal, K. Weideman, D. Rokke, K. C. Vincent, D. Zemlyanov and R. Agrawal, Enhancing the Optoelectronic Properties of Solution-Processed AgInSe2 Thin Films for Application in Photovoltaics, J. Mater. Chem. C, 2024, 12(1), 325–336, 10.1039/D3TC03540A.
- D. Zilevu, O. O. Parks and S. E. Creutz, Solution-Phase Synthesis of the Chalcogenide Perovskite Barium Zirconium Sulfide as Colloidal Nanomaterials, Chem. Commun., 2022, 58(75), 10512–10515, 10.1039/D2CC03494H.
- R. Yang, A. D. Jess, C. Fai and C. J. Hages, Low-Temperature, Solution-Based Synthesis of Luminescent Chalcogenide Perovskite BaZrS3 Nanoparticles, J. Am. Chem. Soc., 2022, 144(35), 15928–15931, DOI:10.1021/jacs.2c06168.
- J. W. Turnley, K. C. Vincent, A. A. Pradhan, I. Panicker, R. Swope, M. C. Uible, S. C. Bart and R. Agrawal, Solution Deposition for Chalcogenide Perovskites: A Low-Temperature Route to BaMS3 Materials (M = Ti, Zr, Hf), J. Am. Chem. Soc., 2022, 144(40), 18234–18239, DOI:10.1021/jacs.2c06985.
- A. A. Pradhan, M. C. Uible, S. Agarwal, J. W. Turnley, S. Khandelwal, J. M. Peterson, D. D. Blach, R. N. Swope, L. Huang, S. C. Bart and R. Agrawal, Synthesis of BaZrS3 and BaHfS3 Chalcogenide Perovskite Films Using Single-Phase Molecular Precursors at Moderate Temperatures, Angew. Chem., Int. Ed., 2023, 62(15), e202301049, DOI:10.1002/anie.202301049.
- K. C. Vincent, S. Agarwal, Z. Fan, A. S. M. Canizales and R. Agrawal, Expanding the Horizons for Viable Precursors and Liquid Fluxes for the Synthesis of BaZrS3 and Related Compounds, J. Mater. Chem. C, 2024, 12, 12521–12534, 10.1039/D4TC02287D.
- K. C. Vincent, S. Agarwal, J. W. Turnley and R. Agrawal, Liquid Flux–Assisted Mechanism for Modest Temperature Synthesis of Large-Grain BaZrS3 and BaHfS3 Chalcogenide Perovskites, Adv. Energy Sustainability Res., 2023, 4(5), 2300010, DOI:10.1002/aesr.202300010.
- R. Yang, J. Nelson, C. Fai, H. A. Yetkin, C. Werner, M. Tervil, A. D. Jess, P. J. Dale and C. J. Hages, A Low-Temperature Growth Mechanism for Chalcogenide Perovskites, Chem. Mater., 2023, 35(12), 4743–4750, DOI:10.1021/acs.chemmater.3c00494.
- D. C. Hayes, S. A. Langdon, R. M. Spilker and R. Agrawal, Carbon Impurity Minimization of Solution-Processed, Thin-Film Photovoltaics via Ligand Engineering of CuInS2 Nanoparticles, ACS Appl. Energy Mater., 2024, 7(3), 885–895, DOI:10.1021/acsaem.3c01922.
- J. R. Thompson Development of Single-Source CVD Precursors for Group IV, V and VI Metal Disulfides, PhD thesis, University of Bath, 2016 Search PubMed.
- D. C. Bradley and I. M. Thomas, Metallo-Organic Compounds Containing Metal–Nitrogen Bonds. Part I. Some Dialkylamino-Derivatives of Titanium and Zirconium, J. Chem. Soc., 1960, 878, 3857–3861, 10.1039/JR9600003857.
- D. C. Bradley and M. H. Gitlitz, Preparation and Properties of NN-Dialkyldithiocarbamates of Early Transition Elements, J. Chem. Soc. A, 1969, 1152, 10.1039/j19690001152.
- S. Agarwal, J. W. Turnley, A. A. Pradhan and R. Agrawal, Moderate Temperature Sulfurization and Selenization of Highly Stable Metal Oxides: An Opportunity for Chalcogenide Perovskites, J. Mater. Chem. C, 2023, 11(45), 15817–15823, 10.1039/D3TC02716C.
- P. Kayastha; G. Longo and L. D. Whalley A First-Principles Thermodynamic Model for the Ba–Zr–S System in Equilibrium with Sulfur Vapour. 2024.
- H. Rau, T. R. N. Kutty and J. R. F. Guedes De Carvalho, Thermodynamics of Sulphur Vapour, J. Chem. Thermodyn., 1973, 5(6), 833–844, DOI:10.1016/S0021-9614(73)80045-X.
- W. Li, S. Niu, B. Zhao, R. Haiges, Z. Zhang, J. Ravichandran and A. Janotti, Band Gap Evolution in Ruddlesden-Popper Phases, Phys. Rev. Mater., 2019, 3(10), 101601, DOI:10.1103/PhysRevMaterials.3.101601.
- P. Kayastha, D. Tiwari, A. Holland, O. S. Hutter, K. Durose, L. D. Whalley and G. Longo, High-Temperature Equilibrium of 3D and 2D Chalcogenide Perovskites, Solar RRL, 2023, 2201078, DOI:10.1002/solr.202201078.
- Y. Nishigaki, T. Nagai, M. Nishiwaki, T. Aizawa, M. Kozawa, K. Hanzawa, Y. Kato, H. Sai, H. Hiramatsu, H. Hosono and H. Fujiwara, Extraordinary Strong Band-Edge Absorption in Distorted Chalcogenide Perovskites, Solar RRL, 2020, 4(5), 1900555, DOI:10.1002/solr.201900555.
- H. I. Eya and N. Y. Dzade, Density Functional Theory Insights into the Structural, Electronic, Optical, Surface, and Band Alignment Properties of BaZrS3 Chalcogenide Perovskite for Photovoltaics, ACS Appl. Energy Mater., 2023, 6(11), 5729–5738, DOI:10.1021/acsaem.3c00103.
- X. Wu, W. Gao, J. Chai, C. Ming, M. Chen, H. Zeng, P. Zhang, S. Zhang and Y.-Y. Sun, Defect Tolerance in Chalcogenide Perovskite Photovoltaic Material BaZrS3, Sci. China Mater., 2021, 64(12), 2976–2986, DOI:10.1007/s40843-021-1683-0.
- S. Mukherjee, S. Riva, C. Comparotto, F. O. L. Johansson, G. J. Man, D. Phuyal, K. A. Simonov, J. Just, K. Klementiev, S. M. Butorin, J. J. S. Scragg and H. Rensmo, Interplay between Growth Mechanism, Materials Chemistry, and Band Gap Characteristics in Sputtered Thin Films of Chalcogenide Perovskite BaZrS3, ACS Appl. Energy Mater., 2023, 6(22), 11642–11653, DOI:10.1021/acsaem.3c02075.
- C. J. Hages, A. Redinger, S. Levcenko, H. Hempel, M. J. Koeper, R. Agrawal, D. Greiner, C. A. Kaufmann and T. Unold, Identifying the Real Minority Carrier Lifetime in Nonideal Semiconductors: A Case Study of Kesterite Materials, Adv. Energy Mater., 2017, 7(18), 1–10, DOI:10.1002/aenm.201700167.
- T. A. O’Brien, P. D. Rack, P. H. Holloway and M. C. Zerner, Crystal Field and Molecular Orbital Calculation of the Optical Transitions in Ce Doped Alkaline Earth Sulfide (MgS, CaS, SrS, and BaS) Phosphors, J. Lumin., 1998, 78(4), 245–257, DOI:10.1016/S0022-2313(98)00011-8.
- V. Kavitha and M. P. Rani, Fabrication of Red-Emitting Eu3 + -Induced CaS Phosphors: A View of Optical, in Vitro, Lifetime, Structural and Morphological Studies for Biomedical Applications, Eur. Phys. J. Plus, 2022, 137(11), 1210, DOI:10.1140/epjp/s13360-022-03381-4.
- B. L. Rawat and J. D. Ranade, Photoluminescence of CaS: Zr Phosphors, J. Lumin., 1976, 14(5–6), 417–422, DOI:10.1016/0022-2313(76)90010-7.
- C. Avril, V. Malavergne, R. Caracas, B. Zanda, B. Reynard, E. Charon, E. Bobocioiu, F. Brunet, S. Borensztajn, S. Pont, M. Tarrida and F. Guyot, Raman Spectroscopic Properties and Raman Identification of CaS-MgS-MnS-FeS-Cr2FeS4 Sulfides in Meteorites and Reduced Sulfur-Rich Systems, Meteorit. Planet. Sci., 2013, 48(8), 1415–1426, DOI:10.1111/maps.12145.
- P. M. P. Salomé, H. Rodriguez-Alvarez and S. Sadewasser, Incorporation of Alkali Metals in Chalcogenide Solar Cells, Sol. Energy Mater. Sol. Cells, 2015, 143, 9–20, DOI:10.1016/j.solmat.2015.06.011.
- D. Jia, R. S. Meltzer and W. M. Yen, Ce3+ Energy Levels Relative to the Band Structure in CaS: Evidence from Photoionization and Electron Trapping, J. Lumin., 2002, 99(1), 1–6, DOI:10.1016/S0022-2313(02)00326-5.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qm00441h |
| ‡ Equal contribution. |
| This journal is © the Partner Organisations 2024 |