
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of luminescent coumarin-substituted phosphinoamide-bridged polynuclear gold(I) metallacycles and reactivity studies†
Vanitha R.
Naina
 a,
Akhil K.
Singh
a,
Shubham
a,
Julia
Krämer
a,
Mohd
Iqbal
a and
Peter W.
Roesky
*ab
a,
Akhil K.
Singh
a,
Shubham
a,
Julia
Krämer
a,
Mohd
Iqbal
a and
Peter W.
Roesky
*ab
aInstitute of Inorganic Chemistry, Karlsruhe Institute of Technology (KIT), Kaiserstr. 12, 76131, Karlsruhe, Germany. E-mail: roesky@kit.edu
bInstitute for Nanotechnology, Karlsruhe Institute of Technology (KIT), Kaiserstr. 12, 76131, Karlsruhe, Germany
First published on 5th August 2024
Abstract
Herein, we present the first report of coumarin-based phosphinoamine ligands [CoumPNH] and their gold(I) complexes. The deprotonation and chloride abstraction from phosphinoamine gold(I) complexes [(CoumPNH)AuICl] led to the unexpected formation of polynuclear gold(I) metallacycles. The size of the cycles depends on the steric bulk of the phosphine groups. The gold(I) metallacycles feature only weak intramolecular aurophilic interactions and do not exhibit intermolecular interactions. Additionally, these metallacycles show significant luminescent properties. Quantum chemical calculations support the photophysical properties of the complexes. Notably, these compounds are the first polynuclear cyclic rings bridged by phosphinoamide ligands. Moreover, the reactivity of the cyclic compounds was explored in this study.
Introduction
Cyclic gold(I) complexes are a unique class of metallacycles featuring intra- and intermolecular metallophilic interactions.1–5 These molecular architectures have garnered significant interest in various research fields, including supramolecular assemblies, optoelectronics and acid–base chemistry.6–11 Cyclic motifs containing AuI cations can be constructed of different nuclearity, varying from trinuclear to hexanuclear complexes.12–16 Such complexes were synthesized using several angular ditopic anionic bridging ligands, which include, e.g., pyrazolate, carbeniate, imidazolate, pyridinate, and 1,2,4-triazolate ligands (Fig. 1).6–8,17–19 Additionally, cyclic gold(I) rings can also be constructed from phosphanyl ligands and phosphorus ylides (Fig. 1).20–22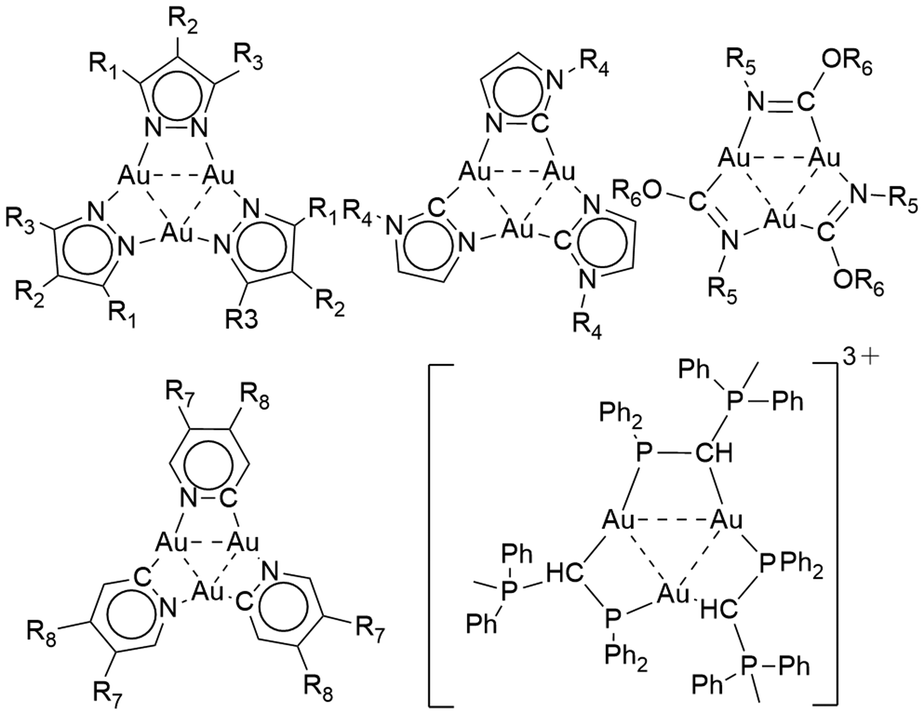 | ||
| Fig. 1 Previously reported cyclic gold(I) metallacycles. | ||
Phosphinoamides ([PN]) are another such class of bridging ligands containing PN− units,23 which are analogous to pyrazolates and 1,2,4-triazolates containing NN− units.24 Phosphinoamide metal complexes are synthesized by deprotonating the respective phosphinoamine ligand, followed by its reaction with metal precursors or by transmetallation of the alkali metal phosphinoamides.24–27 These ligands have been used as both chelating and bridging ligands to access several monometallic and heterobimetallic complexes with applications in the field of catalysis.27–31 However, only a handful of [PN] based coinage metal complexes have been reported (Fig. 2).32–35 Even though this ligand motif is isoelectronic to pyrazolates, until now, only bimetallic complexes have been reported with phosphinoamides.36,37 Specifically, Braunstein and co-workers reported a phosphanyl iminolate-bridged Au–Pd monomer (Fig. 2a) and an unprecedented Ag–Pd coordination polymer (Fig. 2b).38 Later, they also reported a N-(diphenylphosphino)-thiazolin-2-amine-bridged Au–Pt bimetallic complex (Fig. 2c).39 Breher and co-workers reported an unusual dimeric AgI complex bridged by a PN motif (Fig. 2d).40 Nagashima and co-workers used a phosphinoamide ligand to synthesize heterobimetallic complexes, yielding dimer complexes (Fig. 2e).41 However, these complexes were not explored for their photophysical properties.
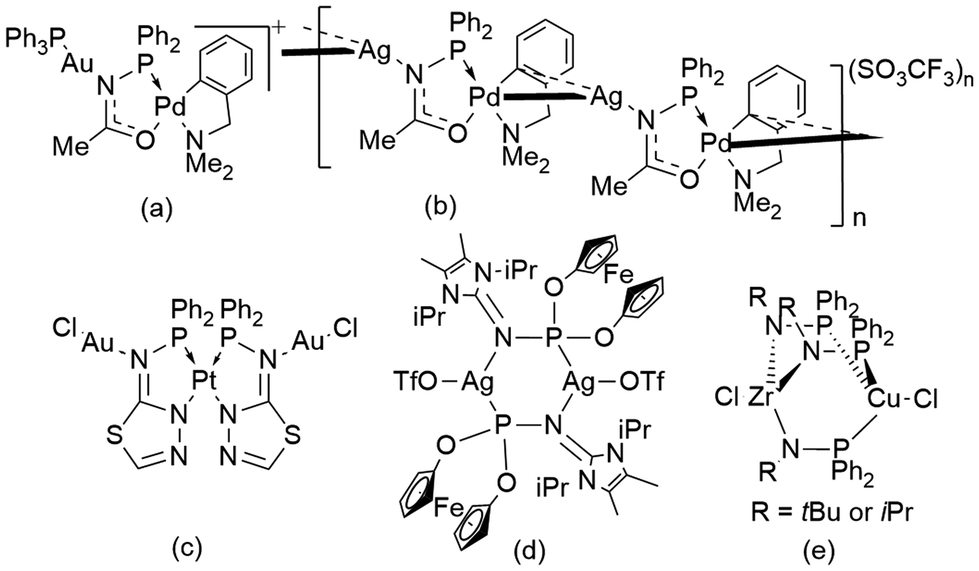 | ||
| Fig. 2 Previously reported [PN]-bridged coinage metal-containing complexes. | ||
Recently, we have shown that incorporating 7-amino-4-methylcoumarin (dye) in the ligand backbone can significantly affect the photophysical properties of bisphosphinoamine and their copper(I) and silver(I) complexes.9,42 The complexes exhibited visually visible ultralong phosphorescence at 77 K. The properties of these class of compounds were strongly phase-dependent.42
Inspired by the promising luminescent nature of the coumarin-based metal complexes43–46 and the lack of [PN]-bridged gold(I) complexes, herein, we designed phosphinoamine ligands with 7-amino-4-methylcoumarin in the ligand backbone to access luminescent gold(I) complexes. The attempts to synthesize [PN]-bridged gold(I) complexes led to the isolation of polynuclear metallacycles with significant photoluminescence properties. Further, the effect of substituents on the phosphine group and their reactivity was investigated. To the best of our knowledge, the complexes reported in this study are the first examples of luminescent polynuclear cyclic gold(I) complexes supported by phosphinoamide ligands.
Results and discussion
For the synthesis of dye functionalized luminescent polynuclear gold(I) complexes, we first designed and isolated a phosphinoamine ligand 1 featuring 7-amino-4-methylcoumarin in the backbone. 7-Amino-4-methylcoumarin was reacted with bis(4-(tert-butyl)phenyl)chlorophosphine in an equimolar ratio in the presence of triethylamine as a base. The reaction yielded the desired ligand 1 as colorless solid, which was crystallized by layering a tetrahydrofuran (THF) solution with n-pentane. Compound 1 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , and its molecular structure in the solid state is depicted in Fig. 3a. The P–N bond distance is 1.7099(10) Å, indicating the single bond character.47 The deprotonation of the N–H proton was confirmed by the integration of the N–H signal in the 1H NMR spectrum, and it appears as a doublet at δ 5.91 ppm (compound 1). The phosphine resonance of the ligand 1 appears as a singlet at δ 24.4 ppm in the 31P{1H} NMR spectrum.
, and its molecular structure in the solid state is depicted in Fig. 3a. The P–N bond distance is 1.7099(10) Å, indicating the single bond character.47 The deprotonation of the N–H proton was confirmed by the integration of the N–H signal in the 1H NMR spectrum, and it appears as a doublet at δ 5.91 ppm (compound 1). The phosphine resonance of the ligand 1 appears as a singlet at δ 24.4 ppm in the 31P{1H} NMR spectrum.
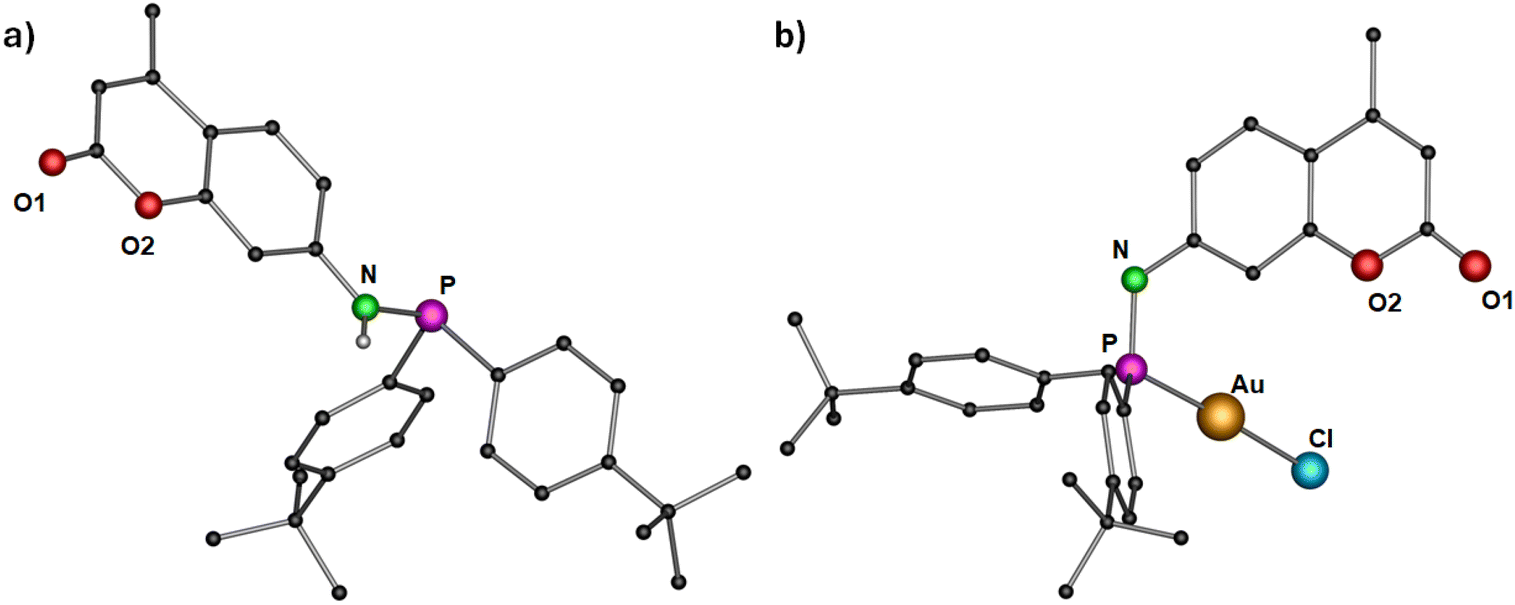 | ||
| Fig. 3 Molecular structure of (a) ligand 1 and (b) complex 2 in the solid state. Hydrogen atoms (except N–H for ligand 1) and non-coordinating solvents are removed for clarity. Selected bond distances (Å) and angles (°): 1: N–P 1.7099(10); 2: Au–Cl 2.2892(10), Au–P 2.2181(11), P–N 1.685(4); P–Au–Cl 178.85(4).43 | ||
The synthesized ligand 1 was reacted with [Au(tht)Cl] to obtain its corresponding gold(I) complex 2 (Scheme 1). The molecular structure of the complex is displayed in Fig. 3b. As expected, the gold(I) cation is coordinated by a phosphorous atom and a chloride ion, and therefore, it adopts a linear geometry with the P–Au–Cl angle of 178.85(4)°. The P–N and Au–P bond distances are 1.685(4) Å and 2.2181(11) Å, respectively, comparable to similar reported molecules.48 The complex features a singlet at δ 57.2 ppm in the 31P{1H} NMR spectrum.
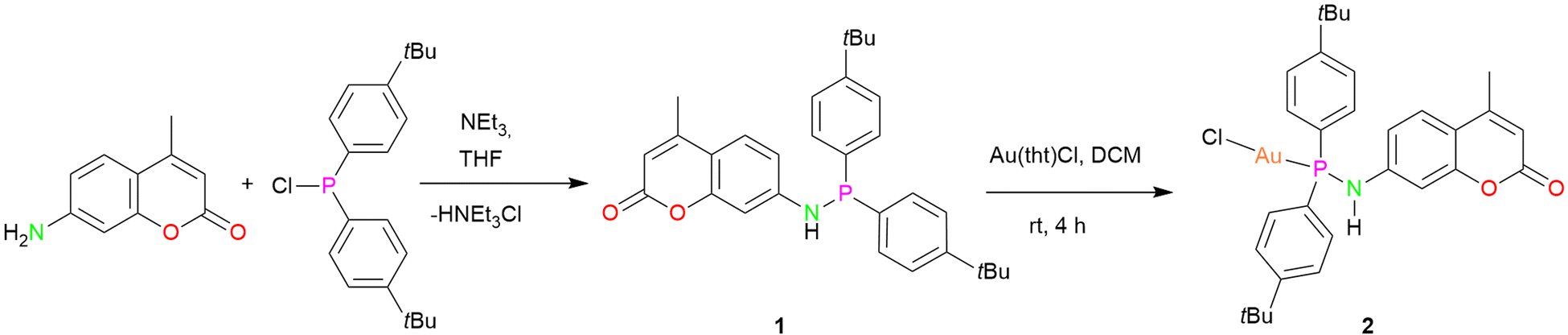 | ||
| Scheme 1 Synthesis of ligand 1 and complex 2. | ||
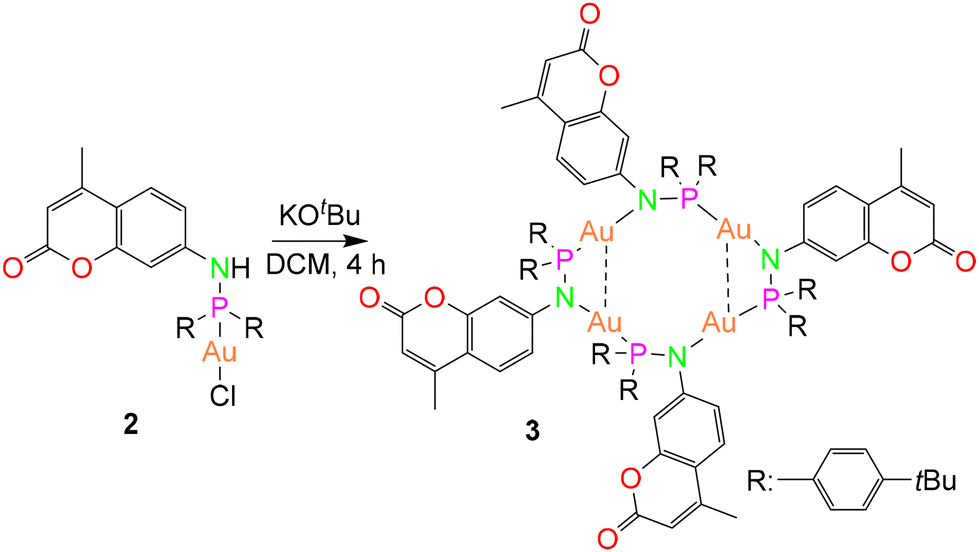
Subsequently, the deprotonation of the gold(I) complex 2 with potassium tert-butoxide resulted in the isolation of an metallacycle complex 3 (Scheme 2), featuring four gold(I) ions in the core, supported by four bridging phosphinoamide ligand moieties (Fig. 4a).
 | ||
| Scheme 2 Synthesis of the polynuclear complex 3.43 | ||
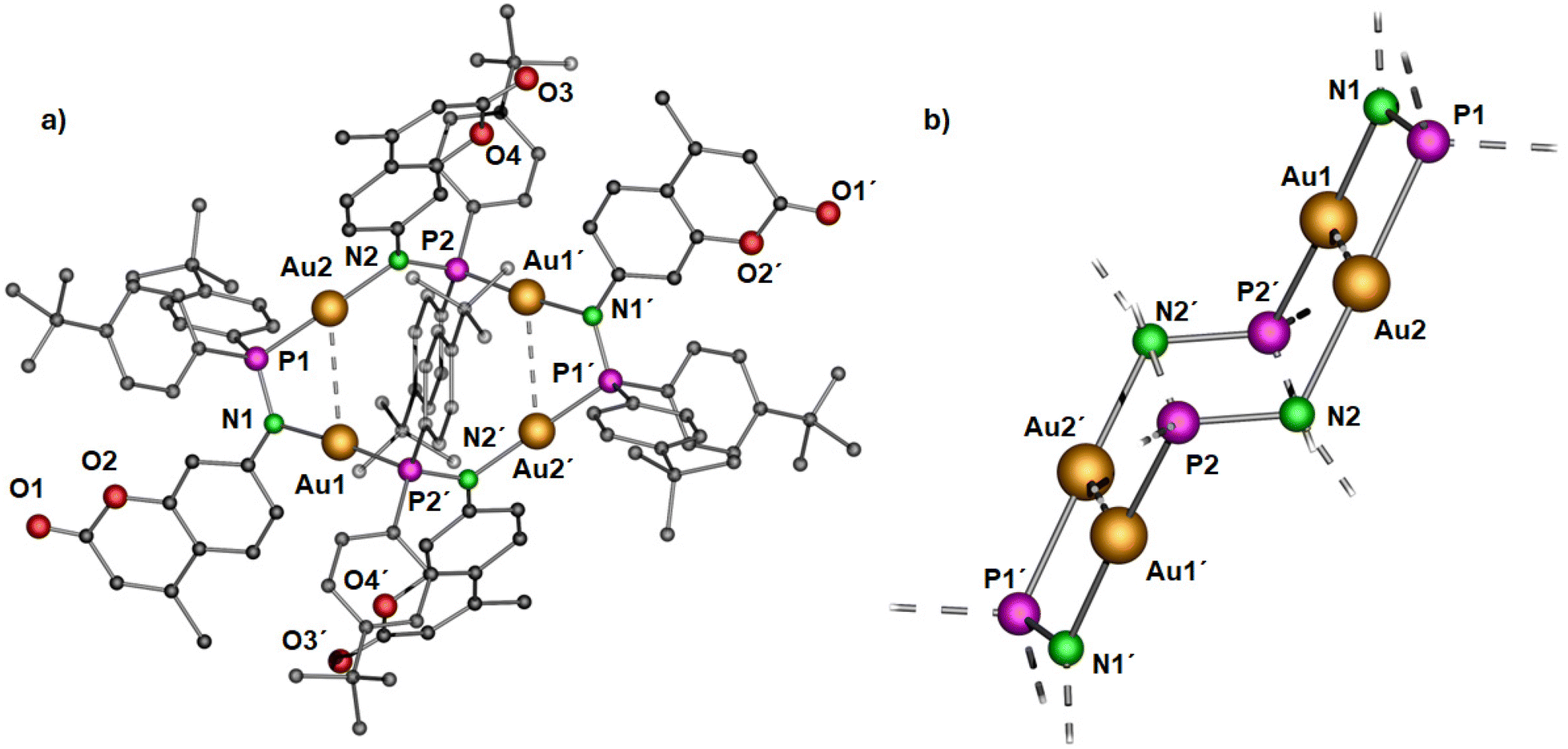 | ||
| Fig. 4 Molecular structure of (a) polynuclear gold(I) complex 3 in the solid state and (b) simplified view of the core structure without the coumarin and para-(tert-butyl)phenyl rings featuring in a ladder-like configuration (right). Hydrogen atoms and non-coordinating solvents are removed for clarity. Selected bond distances (Å) and angles (°): Au1–Au2 3.2718(4), Au1–P2′ 2.2253(15), P1–Au2 2.2195(15), Au1–N1 2.055(5), Au2–N2 2.072(4), N1–P1 1.632(5), N2–P2 1.655(6); P2–N2–Au2 119.7(3), P1–Au2–N2 177.4(2), P1–N1–Au1 113.8(3), N1–Au1–P2′ 175.04(14).43 | ||
Yellow colored crystals of complex 3 were grown from a concentrated toluene solution and X-ray crystallographic studies show that the asymmetric unit is comprised of only half of the molecule with two gold(I) cations. Unlike the pyrazolate coordinated saddle-shaped tetramer [AuAg(μ-pzR)(BF4)]4 (pzR = 3,5-bis(organothiomethyl)-pyrazolate),12 in complex 3, the ring adopts a ladder-like arrangement (Fig. 4b). The four gold(I) ions in unique twelve membered ring structure are bridged by a N and P atom of the phosphinoamide motif in a typical linear geometry (∠P–Au–N: >175°). In the ladder-like tetramer structure, two gold(I) ions and two phosphine groups are situated in a spatially different environment, leading to two distinct intramolecular Au–Au distances. The shortest Au1⋯Au2 distance of 3.2718(4) Å is indicative of weak intramolecular aurophilic interactions.49–52 The Au⋯Au distance is also in close agreement with the values obtained from optimized geometry (Fig. 10). The longest distance between the gold(I) ions is greater than 4.5 Å, suggesting no metallophilic interactions between Au1′ and Au2. The Au–N and Au–P bond distances are ∼2.1 Å and 2.2 Å, respectively, comparable to reported compounds with similar bonding situations.48 The P–N bond distances are ∼1.6 Å, slightly shorter than those in the protonated ligand (1.71 Å) and the precursor complex (1.68 Å).
The characteristic resonances of two non-equivalent phosphorous atoms in complex 3 appear at δ 54.9 and δ 60.9 ppm in the 31P{1H} NMR spectrum. An additional singlet is observed at δ 57.9 ppm in the 31P{1H} NMR spectrum. Even after ten days, there were no changes in the relative intensities among the peaks, which indicates that the additional singlet doesn't appear due to decomposition. Hence, we speculated that solution dynamics might exist between the tetramer and trimer molecules. The singlet at δ 57.9 ppm in the 31P{1H} NMR spectrum might belong to the latter compound (based on the resonance observed for the trimer complex 6, discussed below), which is present in approximately 13% ratio upon the dissolution of crystals of complex 3.
Therefore, we performed variable temperature 31P{1H} NMR studies to gain insights into the solution dynamics. From the variable temperature NMR experiments, we noticed that the intensity of the resonance corresponding to the trimer decreases on lowering the temperature, and it disappears below 243 K. These results confirm our assumptions concerning the dynamics between the trimer and tetramer structure in the solution. Additionally, NMR shielding tensors were calculated using the Gauge-Independent Atomic Orbital (GIAO) method at the CAM-B3LYP level with the def2TZVP basis set, showing consistency with experimental observations.
In order to establish the generality of the reaction shown in Scheme 2, another ligand 4, with two tert-butyl groups at the meta-position of the phenyl rings, was designed. Compound 4 was synthesized by following a similar procedure as that of ligand 1 except that bis(3,5-(di-tert-butyl)phenyl)chlorophosphine was used instead of bis(4-(tert-butyl)phenyl)chlorophosphine (Scheme 3). The molecular structure of the ligand with expected planar N and pyramidal phosphorous geometry is displayed in Fig. 5a. From the single crystal X-ray diffraction studies, the bond distances and angles of the ligand are similar to that of compound 1. The N–H resonance appears as a doublet at δ 4.89 ppm in the 1H NMR spectrum. The phosphine resonance of the ligand appears as a singlet at δ 32.7 ppm in the 31P{1H} NMR spectrum, which is downfield-shifted compared to ligand 1. In the next step, its corresponding gold(I) chloride complex was synthesized by reacting it with [Au(tht)Cl] in a stoichiometric ratio to give the gold(I) complex 5. Similar to complex 2, the gold(I) ion is coordinated to a phosphine P atom and chloride ion with a Au–P bond distance of 2.287(13) Å and P–Au–Cl angle of 179.26(6)° (Fig. 5b), in accordance with similar reported structures.53 The complex exhibits a singlet at δ 60.1 ppm in the 31P{1H} NMR spectrum, which is downfield-shifted compared to the free ligand 4 (δ 32.7 ppm) upon coordination. As expected, the introduction of an additional t-butyl group on the backbone of phenyl rings of ligand 4 resulted in enhanced solubility of the gold(I) complexes.
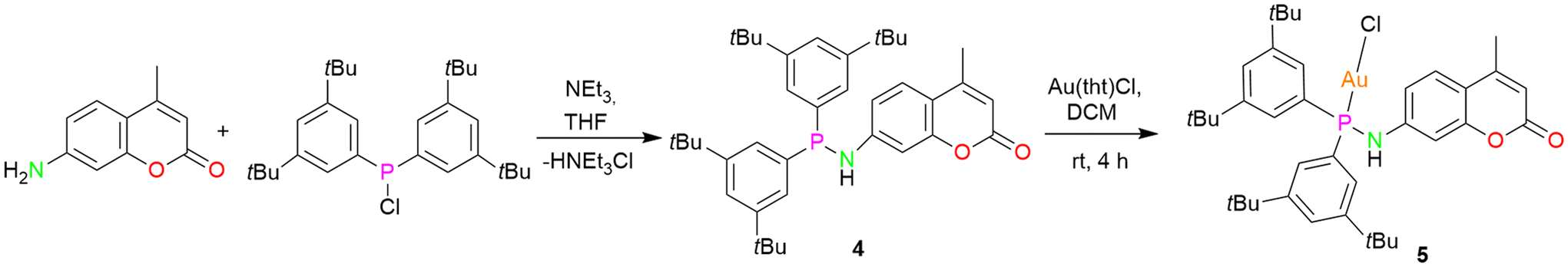 | ||
| Scheme 3 Synthesis of the ligand 4 and the gold(I) complex 5. | ||
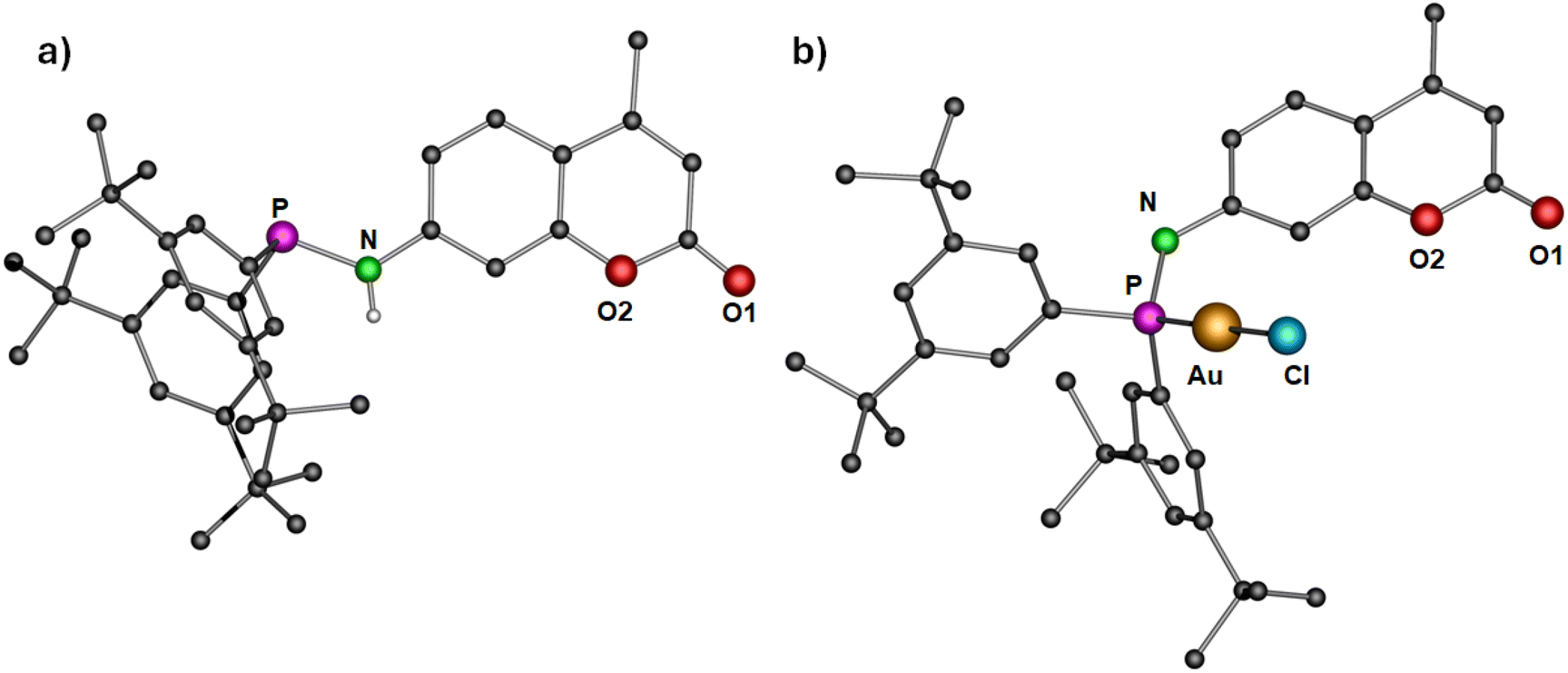 | ||
| Fig. 5 Molecular structure of (a) ligand 4 and (b) complex 5 in the solid state. Hydrogen atoms (except N–H of the ligand 4) and non-coordinating solvents are removed for clarity. Selected bond distances (Å) and angles (°): 4: N–P 1.705(3); 5: Au–Cl 2.2742(14), Au–P 2.287(13), N–P 1.682(5); P–Au–Cl 179.26(6). | ||
The deprotonation and chloride abstraction of the complex 5 using KOtBu resulted in the macrocyclic complex 6 (Scheme 4). X-ray crystallographic studies revealed that complex 6 exists as a trinuclear phosphinoamide-bridged motif [Au(μ-PN)]3 (Fig. 6) rather than the tetramer seen in compound 3. The formation of a trimeric complex rather than a tetramer could result from the increased steric bulk of the substituents in the ligand 4.20 The central [Au(μ-PN)]3 is almost planar with Au⋯Au distances of ∼3.5 Å, indicating the absence of notably intramolecular aurophilic interactions. These distances are significantly longer than that of complex 3 and other similar pyrazolate-bridged trimers. In the nine-membered ring containing three gold(I) ions, each metal center is bridged by a N and P atom of the phosphinoamide ligand moiety 4 nearly in a linear coordination geometry with ∠P–Au–N: ∼173°. The P–N bond distances are ∼1.65 Å, comparatively shorter than the precursor complex 5 (1.68 Å).
 | ||
| Scheme 4 Synthesis of the trimer gold(I) complex 6 and its reactivity with HCl. | ||
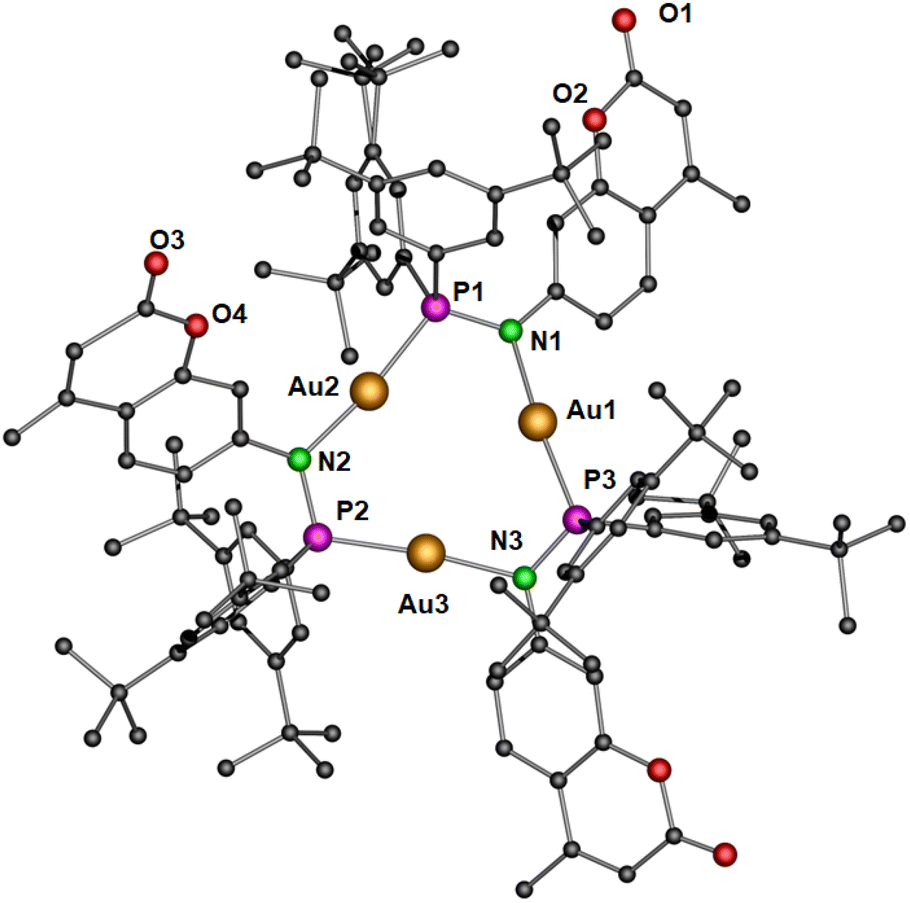 | ||
| Fig. 6 Molecular structure of trimeric gold(I) complex 6 in the solid state. Hydrogen atoms and non-coordinating solvents are removed for clarity. Selected bond distances (Å) and angles (°): Au1–N1 2.057(4), Au1–P3 2.2325(12), Au2–N2 2.082(4), Au2–P1 2.2414(13), Au3–N3 2.067(4), Au3–P2 2.2417(12), N1–P1 1.653(4), N2–P2 1.660(4), N3–P3 1.651(4); N1–Au1–P3 172.47(12), Au1–P3–N3 112.80(14), P3–N3–Au3 115.6(2), N3–Au3–P2 173.26(11), Au3–P2–N2 109.80(15), P2–N2–Au2 115.7, N2–Au2–P1 173.39(10), Au2–P1–N1 111.41(2), P1–N1–Au1 117.4(2). | ||
The trimer compound exhibits a single set of resonances in the 1H NMR spectrum and a singlet at δ 59.4 ppm in the 31P{1H} NMR spectrum which corroborates the structural integrity of the solid state structure in solution. The phosphine resonance is upfield shifted compared to the complex 5 on formation of the metallacycle 6. The absence of additional signals confirms the existence of only one trimeric species in the solution. It is noteworthy that the metallacycles reported in this work are not photosensitive.
To investigate the π-acidity of the trimer compound, we reacted complex 6 with ethylene.54 The trimer didn't show any changes in the NMR and only crystals of the trimer complex were isolated upon reaction with ethylene. Further, to probe the reactivity of the trimer 6, we reacted the complex with phenylacetylene (Scheme 5). The reaction yielded a monomer complex 7, wherein the acidic proton of the phenylacetylene migrated to the N atom of the coumarin ring, and the phenylacetylene moiety was coordinated to the AuI ion as an ancillary anionic ligand. Low quality single crystals of 7 only provided the connectivity owing to the low precision of the data quality. Hence, only a pictorial representation of the structure is presented in Fig. S38.† However, the complex was fully characterized by NMR and IR spectroscopy as well as combustion analysis.
 | ||
| Scheme 5 Reactivity of the trimer 6 with phenylacetylene. | ||
The 1H NMR resonances of complex 7 unambiguously corroborate the chemical structure shown in Scheme 5, with a characteristic N–H resonance as a doublet at δ 4.98 ppm. The complex exhibits a singlet in the 31P{1H} NMR spectrum at δ 74.7 ppm, which is downfield shifted compared to the precursor complex 6 (δ 59.4 ppm). The presence of N–H group was further confirmed by IR spectroscopy showing characteristic absorption at 3265 cm−1.
Based on the observations made in this reaction (migration of acidic proton and the formation of monomer), we planned to react the trimer complex with HCl to yield its precursor complex 5. As expected, upon addition of HCl to the yellow-colored solution of the trimer (Scheme 4), the reaction mixture immediately turned colorless and the NMR studies confirm complete conversion of the trimer 6 to complex 5 within 15 min.
Photophysical properties
The absorption spectra of the precursor gold(I) complexes (2 and 5) and polynuclear gold(I) metallacycles (3 and 6) are depicted in Fig. 7. UV-Vis spectra of all the complexes in dichloromethane show strong absorption band centered at ∼348 nm (molar absorptivity of order 104 M−1 cm−1). The coumarin motif in the ligand backbone mainly contributes to the absorption band between 340–350 nm.42,55,56 The high energy absorption band below 300 nm is caused by ligand centered π–π* transition. The absorption spectra of the polynuclear complexes 3 and 6 differ from the precursor complexes 2 and 5 due to the substantial contribution from the metal centers as corroborated by the theoretical studies.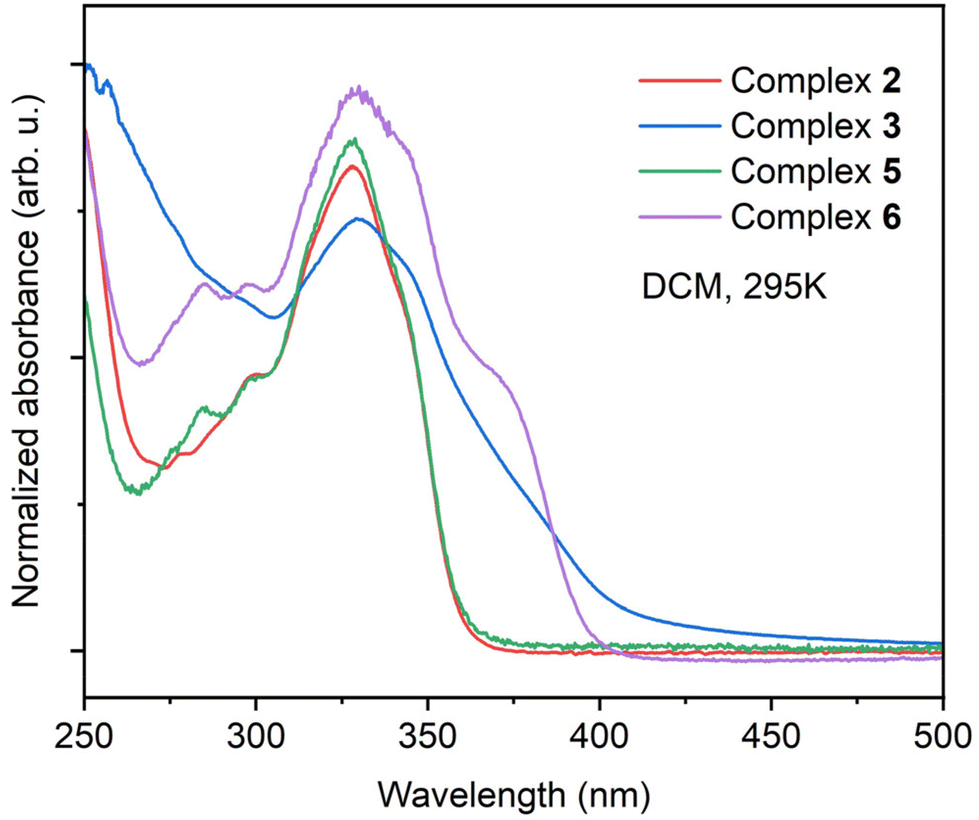 | ||
| Fig. 7 Normalized absorption spectra of complexes 2, 3, 5 and 6 in dichloromethane at ambient temperature. | ||
For the tetranuclear complex 3, the low-energy absorption band is broad, and the absorption onset is shifted to the visible region. The trinuclear complex 6 features an additional shoulder centered at 375 nm, with absorption onset shifted to the visible region. These observations indicate that the formation of the polynuclear complex significantly affects the optical properties.
Further, the synthesized metallacycles were investigated for their photophysical properties. The photoluminescence spectra of gold(I) precursor complex 2 at 77 K and 295 K in the solid state is shown in Fig. 8. At low temperatures, the complex exhibits a PL band at 400 nm (contributed by the coumarin motif) and minor bands at 495 nm and 530 nm. The PL band decay rapidly, and the luminescence lifetimes are below our detector's detection limit. At room temperature, only the high-energy emission band is observed in the PL spectra.
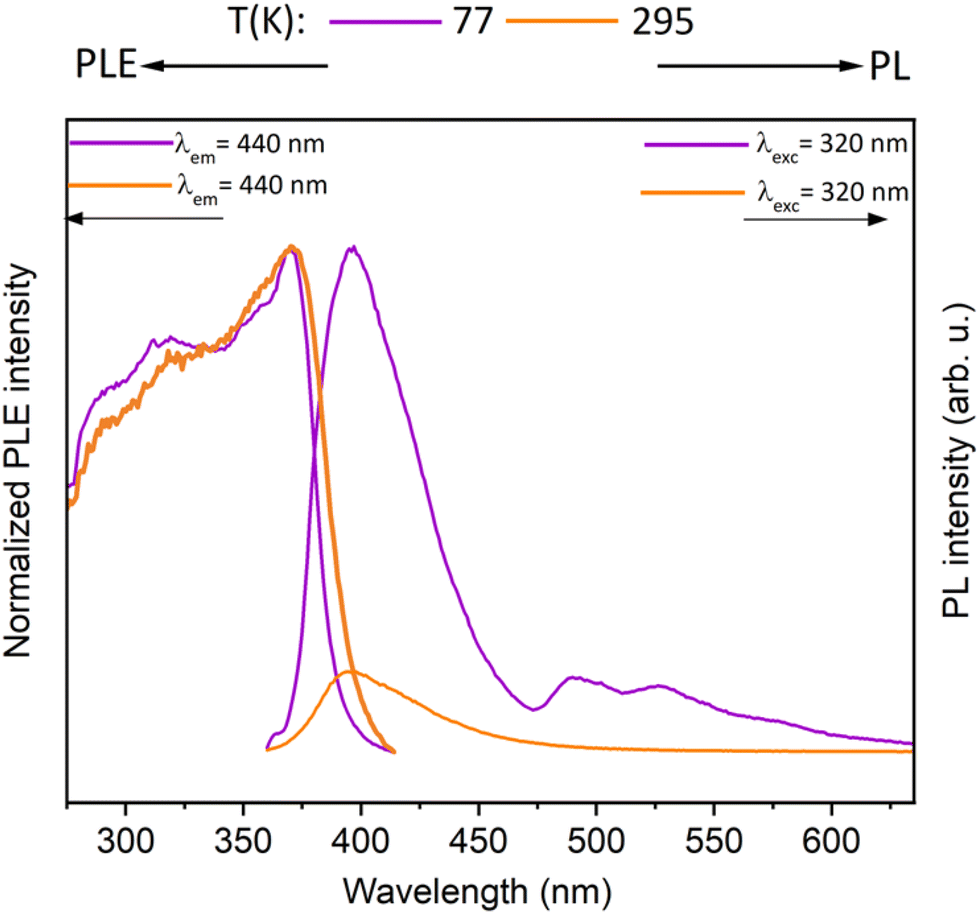 | ||
| Fig. 8 Normalized photoluminescence excitation (PLE) and emission (PL) spectra of polycrystalline sample of 2 in the solid state at room temperature and 77 K. PLE and PL spectra were recorded at the depicted wavelengths (λem and λex). | ||
In contrast to the precursor complex 2, the tetramer 3 exhibits a broad and structureless band centered at ∼590 nm in the solid state at 77 K (Fig. 9a). The formation of the tetramer led to a shift of the emission band to lower energy by ∼1.03 eV, and the excited states now decay biexponentially with lifetimes in the scale of microseconds, indicating phosphorescence (τ1 = 5.5 μs and τ2 = 32.8 μs). This could be in good agreement with a contribution of the metal d orbitals to the electronic transitions involved in the excited states. Complex 3 features a broad band at room temperature with λmax = 585 nm. The excited states now decay rapidly with lifetime less than 6 μs.
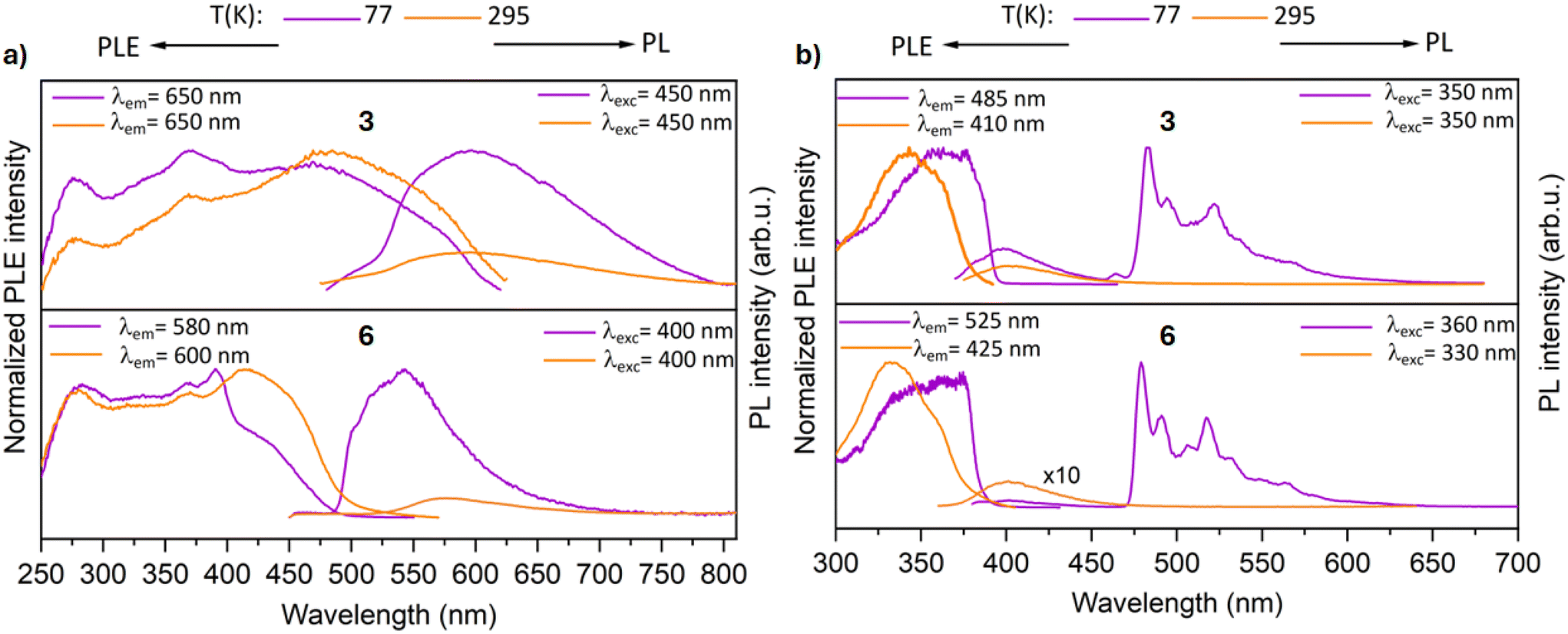 | ||
| Fig. 9 Normalized photoluminescence excitation (PLE) and emission (PL) spectra of (a) polycrystalline samples (b) Me-THF solutions of complexes 3 and 6 at room temperature and 77 K. PLE and PL spectra were recorded at the depicted wavelengths (λem and λex). | ||
In the solid state at 77 K, the trimer complex 6 features also a broad band centered at 550 nm upon excitation at 400 nm (Fig. 9a). The luminescence decay kinetics of complex 6 is also biexponential and in the order of milliseconds (τ1 = 7.5 μs and τ2 = 1.06 ms). Such long lifetimes are rarely reported for analogous pyrazolate-bridged cyclic trinuclear complexes lacking intermolecular interactions.57,58 On increasing the temperature, the luminescence intensity rapidly decreases and features an emission band at λmax = 590 nm (with a lifetime of 7.8 μs). Even though, the metallaycles exhibit long lifetimes, their PL efficiency (ΦPL) is very poor (1–2%) in the solid state. In contrast, their precursor AuI complexes have comparatively high ΦPL values of 21% and 9% for complexes 2 and 5, respectively.
The polynuclear compounds were also investigated for their luminescence properties in solution (Fig. 9b). The measurements were carried out in a frozen 2-methyl-THF glass matrix at 77 K. The complexes 3 and 6 feature a complicated PL spectra with several decay processes. A fluorescent band of low intensity was observed at 400 nm with lifetime of 4 ns. Additionally, the complexes feature a strong phosphorescent band at ∼480 nm. The decay kinetics in the milliseconds range are clearly non-monoexponential for both the tetramer 3 (τ1 = 6.0 μs and τ2 = 3.45 ms) and trimer 6 (τ1 = 4.4 μs and τ2 = 4.01 ms). It is noteworthy that such long phosphorescence was not observed for the gold(I) chloride precursors.
The excitation spectra of the compounds align with their respective absorption spectrum, which strongly supports our notion that the polynuclear compounds are indeed the emissive species. At 295 K in 2-methyl-THF, both the complexes (3 and 6) feature only one emission band centered at 400 nm, which is attributed to the coumarin moiety. The excited states decay rapidly with a lifetime of ∼3 ns, indicative of emission from a singlet excited state.
This work can further be extended to synthesize analogous copper(I) and silver(I) complexes. Copper(I) complexes can potentially feature visible light absorption and low-energy emission band, which can be promising compounds for photocatalysis.59 Further, the silver(I) complexes may show reactivity with ethylene,54 which was not observed with the gold(I) complexes in this study.
Quantum chemical calculations
For a deeper understanding of the electronic structure of complexes, we conducted density functional theory (DFT) calculations and time-dependent density functional theory (TDDFT) calculations (for detailed methodology, refer to the ESI†). We optimized the geometries of complexes 3 and 6 in the gas phase without constraints (Fig. 10), and the calculation reveals that they maintain their original structural motifs. Frontier molecular orbitals of the gold complexes are depicted in Fig. S43–S46.†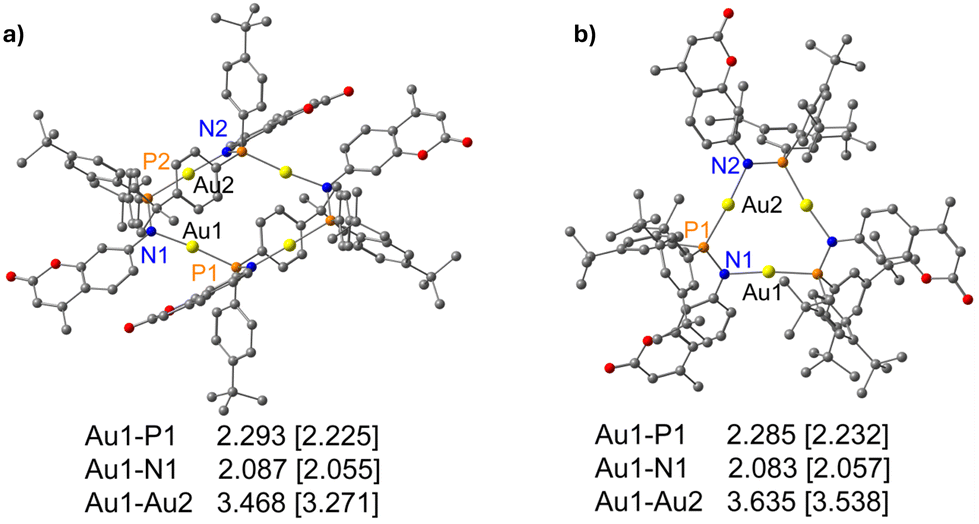 | ||
| Fig. 10 CAM-B3LYP optimized geometries of (a) complex 3 and (b) complex 6 with bond lengths in Å as calculated using def2TZVP basis set for gold atom and 6-31G** for all other atoms. Parentheses contain the experimental value obtained from the respective X-ray structures. | ||
Across all complexes, the highest occupied molecular orbitals (HOMOs) primarily originate from coumarin-centered orbitals with minimal contribution from gold atoms and none from coordinating phosphorus atoms, while the lowest unoccupied molecular orbitals (LUMOs) are predominantly ligand-localized. In tri- and tetra-metallic complexes, LUMOs exhibit significant contributions from the [Au3(PN)3] and [Au4(PN)4] cores, respectively. Absorption and emission properties of precursor gold(I) complexes (2 and 5) primarily arise from intraligand π to π* transitions. However, polynuclear gold(I) metallacycles (3 and 6) display distinct luminescent properties compared to their precursor complexes. Selected excited state energy levels along with main orbital configurations are summarized in Tables S2 and S3;† however, it's worth noting that these values tend to underestimate experimental results, particularly for states with significant charge transfer character. TDDFT calculation explains the broad and red-shifted absorption band in complexes 3 and 6. Specifically, the red-shifted shoulder at 375 nm in complex 6 originates from transitions from coumarin-centered HOMOs to the [Au3(PN)3] core, including transitions from HOMO to LUMO+1, HOMO−2 to LUMO, and HOMO−1 to LUMO (Table S3†). Similarly, the broad absorption features for complex 3 are attributed to transitions from HOMO−2 to LUMO, HOMO−3 to LUMO, and HOMO−1 to LUMO+1.
Complexes 3 and 6 feature favourable triplet states energetically proximal to S1, with energy differences typically ≤0.4 eV, and exhibit similar orbital configurations between singlet (S1) and triplet (Tn) states.42 These properties position them as promising candidates for intersystem crossing (ISC), contributing to their observed red-shifted phosphorescence attributed to the [Au(PN)] core. Additionally, NMR shielding tensors were calculated using the Gauge-Independent Atomic Orbital (GIAO) method at the B3LYP level with the def2TZVP basis set, showing consistency with experimental observations.
Conclusions
In this work, we have reported the synthesis of new coumarin based phosphinoamine ligands 1 and 3, and their respective gold(I) chloride complexes 2 and 5. The reaction of the gold(I) chloride complexes led to the formation of unexpected cyclic structures containing four or three gold(I) ions. The number of gold(I) ions is dependent on the steric bulk of the organic substituents on the phosphine moiety. The cyclic structures show yellow-orange emission in the solid state and coumarin centered emission in solution both at room temperature and 77 K. At low temperatures in frozen Me-THF matrix, the excited states of the cyclic compounds decay on a timescale of milliseconds. The quantum chemical calculations reveal that in the cyclic complexes, the HOMOs primarily originate from the coumarin-centered orbitals, while the LUMOs show significant contributions from [Aux(PN)x] cores. The absorption and emission properties are altered compared to the precursor complexes. This work showcases the possibility to alter the optical properties of 7-amino-4-methyl coumarin based phosphinoamine ligands. Furthermore, the trimer compound was explored for its reactivity towards phenylacetylene and HCl.Author contributions
V. R. N. and J. K. performed the synthesis, characterization, photophysical studies and analysed the data with the help of S. and M. I. A. K. S. performed the quantum chemical calculations. P. W. R. conceived the idea and supervised the work. All authors provided suggestions and comments on the manuscript.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for 1–6 has been deposited at the Cambridge Crystallographic Data Centre as a supplementary publication no. 2362899–2362904.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
V. R. N., S., and P. W. R. acknowledges GRK 2039 Molecular Architectures for Fluorescent Cell Imaging for the financial support. P. W. R. acknowledges Deutsche Forschungsgemeinschaft for support within the project 540378534, RO 2008/22-1.Notes and references
- J. Zheng, Z. Lu, K. Wu, G.-H. Ning and D. Li, Coinage-Metal-Based Cyclic Trinuclear Complexes with Metal–Metal Interactions: Theories to Experiments and Structures to Functions, Chem. Rev., 2020, 120, 9675–9742 CrossRef CAS PubMed.
- T. P. Seifert, V. R. Naina, T. J. Feuerstein, N. D. Knöfel and P. W. Roesky, Molecular gold strings: aurophilicity, luminescence and structure–property correlations, Nanoscale, 2020, 12, 20065–20088 RSC.
- H. Schmidbaur and A. Schier, Aurophilic interactions as a subject of current research: an update, Chem. Soc. Rev., 2012, 41, 370–412 RSC.
- P. Braunstein and A. A. Danopoulos, Transition Metal Chain Complexes Supported by Soft Donor Assembling Ligands, Chem. Rev., 2021, 121, 7346–7397 CrossRef CAS PubMed.
- S. Sculfort and P. Braunstein, Intramolecular d10–d10 interactions in heterometallic clusters of the transition metals, Chem. Soc. Rev., 2011, 40, 2741 RSC.
- A. A. Titov, O. A. Filippov, L. M. Epstein, N. V. Belkova and E. S. Shubina, Macrocyclic copper(I) and silver(I) pyrazolates: Principles of supramolecular assemblies with Lewis bases, Inorg. Chim. Acta, 2018, 470, 22–35 CrossRef CAS.
- J. Zheng, H. Yang, M. Xie and D. Li, The π-acidity/basicity of cyclic trinuclear units (CTUs): from a theoretical perspective to potential applications, Chem. Commun., 2019, 55, 7134–7146 RSC.
- O. Elbjeirami, M. A. Rawashdeh-Omary and M. A. Omary, Phosphorescence sensitization via heavy-atom effects in d10 complexes, Res. Chem. Intermed., 2011, 37, 691–703 CrossRef CAS.
- V. R. Naina, F. Krätschmer and P. W. Roesky, Selective coordination of coinage metals using orthogonal ligand scaffolds, Chem. Commun., 2022, 58, 5332–5346 RSC.
- Shubham, V. R. Naina and P. W. Roesky, Luminescent Tetranuclear Copper(I) and Gold(I) Heterobimetallic Complexes: A Phosphine Acetylide Amidinate Orthogonal Ligand Framework for Selective Complexation, Chem. – Eur. J., 2024, e202401696 CrossRef CAS PubMed.
- R. Yadav, M. Dahlen, A. K. Singh, X. Sun, M. T. Gamer and P. W. Roesky, Nonanuclear zinc–gold [Zn3Au6] heterobimetallic complexes, Dalton Trans., 2021, 50, 8558–8566 RSC.
- A. C. Jahnke, K. Pröpper, C. Bronner, J. Teichgräber, S. Dechert, M. John, O. S. Wenger and F. Meyer, A New Dimension in Cyclic Coinage Metal Pyrazolates: Decoration with a Second Ring of Coinage Metals Supported by Inter-ring Metallophilic Interactions, J. Am. Chem. Soc., 2012, 134, 2938–2941 CrossRef CAS PubMed.
- R. A. Smith, R. Kulmaczewski and M. A. Halcrow, Ligand-Directed Metalation of a Gold Pyrazolate Cluster, Inorg. Chem., 2023, 62, 9300–9305 CrossRef CAS PubMed.
- H. Murray, R. G. Raptis and J. P. Fackler Jr, Syntheses and X-ray structures of group 11 pyrazole and pyrazolate complexes. X-ray crystal structures of bis(3,5-diphenylpyrazole)copper(II) dibromide, tris(μ-3,5-diphenylpyrazolato-N,N′)trisilver(I)-2-tetrahydrofuran, tris(μ-3,5-diphenylpyrazolato-N,N′)trigold(I), and hexakis(μ-3,5-diphenylpyrazolato-N,N′)hexagold(I), Inorg. Chem., 1988, 27, 26–33 CrossRef CAS.
- K. Fujisawa, Y. Ishikawa, Y. Miyashita and K.-i. Okamoto, Pyrazolate-bridged group 11 metal(I) complexes: Substituent effects on the supramolecular structures and physicochemical properties, Inorg. Chim. Acta, 2010, 363, 2977–2989 CrossRef CAS.
- H. E. Abdou, A. A. Mohamed and J. P. Fackler, Syntheses of Mixed-Ligand Tetranuclear Gold(I)–Nitrogen Clusters by Ligand Exchange Reactions with the Dinuclear Gold(I) Formamidinate Complex Au2(2,6-Me2Ph-form)2, Inorg. Chem., 2007, 46, 141–146 CrossRef CAS PubMed.
- M. A. Rawashdeh-Omary, Remarkable alteration of photophysical properties of cyclic trinuclear complexes of monovalent coinage metals upon interactions with small organic molecules, Comments Inorg. Chem., 2012, 33, 88–101 CrossRef.
- R. Galassi, M. A. Rawashdeh-Omary, H. V. R. Dias and M. A. Omary, Homoleptic Cyclic Trinuclear d10 Complexes: from Self-Association via Metallophilic and Excimeric Bonding to the Breakage Thereof via Oxidative Addition, Dative Bonding, Quadrupolar, and Heterometal Bonding Interactions, Comments Inorg. Chem., 2019, 39, 287–348 CrossRef CAS.
- M. A. Omary, A. A. Mohamed, M. A. Rawashdeh-Omary and J. P. Fackler, Photophysics of supramolecular binary stacks consisting of electron-rich trinuclear Au(I) complexes and organic electrophiles, Coord. Chem. Rev., 2005, 249, 1372–1381 CrossRef CAS.
- D. M. Stefanescu, H. F. Yuen, D. S. Glueck, J. A. Golen and A. L. Rheingold, Synthesis and Structure of Cyclic Gold(I) Phosphanyl Complexes [{Au(PR2)}n], Angew. Chem., Int. Ed., 2003, 42, 1046–1048 CrossRef CAS PubMed.
- R. Visbal, N. Rosado, J. Zapata-Rivera and M. C. Gimeno, Isolation of a Cyclic Trinuclear Gold(I) Complex with Metalated Phosphorus Ylides: Synthesis and Structural Properties, Inorg. Chem., 2024, 63, 6589–6599 CrossRef CAS PubMed.
- S. Bhargava, K. Kitadai, T. Masashi, D. W. Drumm, S. P. Russo, V. W.-W. Yam, T. K.-M. Lee, J. Wagler and N. Mirzadeh, Synthesis and structures of cyclic gold complexes containing diphosphine ligands and luminescent properties of the high nuclearity species, Dalton Trans., 2012, 41, 4789–4798 RSC.
- P. W. Roesky, P-N ligands in lanthanide chemistry, Heteroat. Chem., 2002, 13, 514–520 CrossRef CAS.
- Z. Fei and P. J. Dyson, The chemistry of phosphinoamides and related compounds, Coord. Chem. Rev., 2005, 249, 2056–2074 CrossRef CAS.
- M. T. Gamer and P. W. Roesky, Lanthanide–Potassium Wheels, Inorg. Chem., 2005, 44, 5963–5965 CrossRef CAS PubMed.
- P. W. Roesky, M. T. Gamer and N. Marinos, Yttrium and Lanthanide Diphosphanylamides: Syntheses and Structures of Complexes with one {(Ph2P)2N}− ligand in the Coordination Sphere, Chem. – Eur. J., 2004, 10, 3537–3542 CrossRef CAS PubMed.
- F. Völcker, F. M. Mück, K. D. Vogiatzis, K. Fink and P. W. Roesky, Bi- and trimetallic rare-earth–palladium complexes ligated by phosphinoamides, Chem. Commun., 2015, 51, 11761–11764 RSC.
- K. M. Gramigna, D. A. Dickie, B. M. Foxman and C. M. Thomas, Cooperative H2 Activation across a Metal–Metal Multiple Bond and Hydrogenation Reactions Catalyzed by a Zr/Co Heterobimetallic Complex, ACS Catal., 2019, 9, 3153–3164 CrossRef CAS.
- H. Zhang, G. P. Hatzis, C. E. Moore, D. A. Dickie, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, O2 Activation by a Heterobimetallic Zr/Co Complex, J. Am. Chem. Soc., 2019, 141, 9516–9520 CrossRef CAS PubMed.
- S. L. Marquard, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Stoichiometric C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O Bond Oxidative Addition of Benzophenone by a Discrete Radical Intermediate To Form a Cobalt(I) Carbene, J. Am. Chem. Soc., 2013, 135, 6018–6021 CrossRef CAS PubMed.
O Bond Oxidative Addition of Benzophenone by a Discrete Radical Intermediate To Form a Cobalt(I) Carbene, J. Am. Chem. Soc., 2013, 135, 6018–6021 CrossRef CAS PubMed. - C. Fliedel, A. Ghisolfi and P. Braunstein, Functional Short-Bite Ligands: Synthesis, Coordination Chemistry, and Applications of N-Functionalized Bis(diaryl/dialkylphosphino)amine-type Ligands, Chem. Rev., 2016, 116, 9237–9304 CrossRef CAS PubMed.
- S. Kuppuswamy, M. W. Bezpalko, T. M. Powers, M. J. T. Wilding, C. K. Brozek, B. M. Foxman and C. M. Thomas, A series of C3-symmetric heterobimetallic Cr–M (M = Fe, Co and Cu) complexes, Chem. Sci., 2014, 5, 1617–1626 RSC.
- J. Du, Y. Zhang, Z. Huang, S. Zhou, H. Fang and P. Cui, Heterobimetallic Pd(0) complexes with Pd→Ln (Ln = Sc, Y, Yb, Lu) dative bonds: rare-earth metal-dominated frustrated Lewis pair-like reactivity, Dalton Trans., 2020, 49, 12311–12318 RSC.
- F. Völcker and P. W. Roesky, Bimetallic rare-earth/platinum complexes ligated by phosphinoamides, Dalton Trans., 2016, 45, 9429–9435 RSC.
- S. Maggini, Classification of P,N-binucleating ligands for hetero- and homobimetallic complexes, Coord. Chem. Rev., 2009, 253, 1793–1832 CrossRef CAS.
- C. Voß, R. Pattacini and P. Braunstein, Intramolecular d10–d10 interactions in neutral, dinuclear Au(I) complexes supported by amino-thiazoline- and -thiazole-based P,N-phosphine ligands, C. R. Chim., 2012, 15, 229–236 CrossRef.
- P. A. Bella, O. Crespo, E. J. Fernández, A. K. Fischer, P. G. Jones, A. Laguna, J. M. López-De-Luzuriaga and M. Monge, Gold complexes of 3,4-bis(diphenylphosphinoamino)toluene and 1,2-bis(diphenylphosphinoamino)benzene. A comparative study, J. Chem. Soc., Dalton Trans., 1999, 4009–4017, 10.1039/a905857e.
- P. Braunstein, C. Frison, N. Oberbeckmann-Winter, X. Morise, A. Messaoudi, M. Bénard, M.-M. Rohmer and R. Welter, An Oriented 1D Coordination/Organometallic Dimetallic Molecular Wire with Ag-Pd Metal–Metal Bonds, Angew. Chem., Int. Ed., 2004, 43, 6120–6125 CrossRef CAS PubMed.
- S. Zhang, R. Pattacini and P. Braunstein, Chelating or bridging Pd(II) and Pt(II) metalloligands from the functional phosphine ligand N-(diphenylphosphino)-1,3,4-thiadiazol-2-amine. New heterometallic Pd(II)/Pt(II) and Pt(II)/Au(I) complexes, Dalton Trans., 2011, 40, 5711–5719 RSC.
- B. S. Birenheide, F. Krämer, L. Bayer, P. Mehlmann, F. Dielmann and F. Breher, Multistimuli-Responsive [3]Dioxaphosphaferrocenophanes with Orthogonal Switches, Chem. – Eur. J., 2021, 27, 15067–15074 CrossRef PubMed.
- T. Sue, Y. Sunada and H. Nagashima, Zirconium(IV) Tris(phosphinoamide) Complexes as a Tripodal-Type Metalloligand: A Route to Zr–M (M = Cu, Mo, Pt) Heterodimetallic Complexes, Eur. J. Inorg. Chem., 2007, 2897–2908 CrossRef CAS.
- V. R. Naina, A. K. Singh, P. Rauthe, S. Lebedkin, M. T. Gamer, M. M. Kappes, A.-N. Unterreiner and P. W. Roesky, Phase-Dependent Long Persistent Phosphorescence in Coumarin-Phosphine-Based Coinage Metal Complexes, Chem. – Eur. J., 2023, 29, e202300497 CrossRef CAS PubMed.
- V. R. Naina, Coumarin-based Coinage Metal Complexes: Synthesis, Luminescence and Cytotoxicity Studies, Cuvillier Verlag, 2023 Search PubMed.
- A. Trommenschlager, F. Chotard, B. Bertrand, S. Amor, P. Richard, A. Bettaïeb, C. Paul, J.-L. Connat, P. Le Gendre and E. Bodio, Gold(I)–Coumarin–Caffeine-Based Complexes as New Potential Anti-Inflammatory and Anticancer Trackable Agents, ChemMedChem, 2018, 13, 2408–2414 CrossRef CAS PubMed.
- C. Sobrerroca, I. Angurell, A. de Aquino, G. Romo, C. Jubert and L. Rodríguez, Mono- and Dinuclear Gold(I) Coumarin Complexes: Luminescence Studies and Singlet Oxygen Production, ChemPlusChem, 2023, 88, e202300020 CrossRef CAS PubMed.
- M. Ali, L. Dondaine, A. Adolle, C. Sampaio, F. Chotard, P. Richard, F. Denat, A. Bettaieb, P. Le Gendre, V. Laurens, C. Goze, C. Paul and E. Bodio, Anticancer Agents: Does a Phosphonium Behave Like a Gold(I) Phosphine Complex? Let a “Smart” Probe Answer!, J. Med. Chem., 2015, 58, 4521–4528 CrossRef CAS PubMed.
- T. Böttcher and C. Jones, Extremely bulky secondary phosphinoamines as substituents for sterically hindered aminosilanes, Dalton Trans., 2015, 44, 14842–14853 RSC.
- S. G. Rachor, R. Müller, P. Wittwer, M. Kaupp and T. Braun, Synthesis, Reactivity, and Bonding of Gold(I) Fluorido–Phosphine Complexes, Inorg. Chem., 2022, 61, 357–367 CrossRef CAS PubMed.
- H. E. Abdou, A. A. Mohamed, J. M. López-de-Luzuriaga, M. Monge and J. P. Fackler, Fine-Tuning the Luminescence and HOMO–LUMO Energy Levels in Tetranuclear Gold(I) Fluorinated Amidinate Complexes, Inorg. Chem., 2012, 51, 2010–2015 CrossRef CAS PubMed.
- H. E. Abdou, A. A. Mohamed, J. M. López-de-Luzuriaga and J. P. Fackler, Tetranuclear Gold(I) Clusters with Nitrogen Donor Ligands: Luminescence and X-Ray Structure of Gold(I) Naphthyl Amidinate Complex, J. Cluster Sci., 2004, 15, 397–411 CrossRef CAS.
- T. J. Feuerstein, M. Poß, T. P. Seifert, S. Bestgen, C. Feldmann and P. W. Roesky, A highly luminescent octanuclear gold(I) carbide cluster, Chem. Commun., 2017, 53, 9012–9015 RSC.
- S. Bestgen, M. T. Gamer, S. Lebedkin, M. M. Kappes and P. W. Roesky, Di- and Trinuclear Gold Complexes of Diphenylphosphinoethyl-Functionalised Imidazolium Salts and their N-Heterocyclic Carbenes: Synthesis and Photophysical Properties, Chem. – Eur. J., 2015, 21, 601–614 CrossRef CAS PubMed.
- M. Navrátil, I. Císařová and P. Štěpnička, Intermolecular interactions in the crystal structures of chlorogold(I) complexes with N-phosphinoamide ligands, Inorg. Chim. Acta, 2021, 516, 120138 CrossRef.
- H. V. R. Dias, D. Parasar, A. A. Yakovenko, P. W. Stephens, Á. Muñoz-Castro, M. Vanga, P. Mykhailiuk and E. Slobodyanyuk, In situ studies of reversible solid–gas reactions of ethylene responsive silver pyrazolates, Chem. Sci., 2024, 15, 2019–2025 RSC.
- C. Cunha, A. Pinto, A. Galvão, L. Rodríguez and J. S. Seixas de Melo, Aggregation-Induced Emission with Alkynylcoumarin Dinuclear Gold(I) Complexes: Photophysical, Dynamic Light Scattering, and Time-Dependent Density Functional Theory Studies, Inorg. Chem., 2022, 61, 6964–6976 CrossRef CAS PubMed.
- A. Pinto, C. Cunha, G. Aullón, J. C. Lima, L. Rodríguez and J. S. Seixas de Melo, Comprehensive Investigation of the Photophysical Properties of Alkynylcoumarin Gold(I) Complexes, J. Phys. Chem. B, 2021, 125, 11751–11760 CrossRef CAS PubMed.
- R. Galassi, M. M. Ghimire, B. M. Otten, S. Ricci, R. N. McDougald, R. M. Almotawa, D. Alhmoud, J. F. Ivy, A.-M. M. Rawashdeh, V. N. Nesterov, E. W. Reinheimer, L. M. Daniels, A. Burini and M. A. Omary, Cupriphication of gold to sensitize d10–d10 metal–metal bonds and near-unity phosphorescence quantum yields, Proc. Natl. Acad. Sci. U. S. A., 2017, 201700890, DOI:10.1073/pnas.1700890114.
- J. C. Vickery, M. M. Olmstead, E. Y. Fung and A. L. Balch, Solvent-Stimulated Luminescence from the Supramolecular Aggregation of a Trinuclear Gold(I) Complex that Displays Extensive Intermolecular AuċAu Interactions, Angew. Chem., Int. Ed. Engl., 1997, 36, 1179–1181 CrossRef CAS.
- A. Hossain, A. Bhattacharyya and O. Reiser, Copper's rapid ascent in visible-light photoredox catalysis, Science, 2019, 364, eaav9713 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2362899–2362904 (1–6). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qi01747a |
| This journal is © the Partner Organisations 2024 |