
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ferrocene- and ruthenium arene-containing glycomimetics as selective inhibitors of human galectin-1 and -3†
Vojtěch
Hamala
 ab,
Martin
Kurfiřt
ab,
Lucie
Červenková Šťastná
a,
Hedvika
Hujerová
a,
Jana
Bernášková
a,
Kamil
Parkan
cd,
Jakub
Kaminský
d,
Nina
Habanová
d,
Jaroslav
Kozák
d,
Alžběta
Magdolenová
d,
Martin
Zavřel
d,
Tatiana
Staroňová
ef,
Veronika
Ostatná
f,
Lucie
Žaloudková
ef,
Aleš
Daňhel
f,
Jitka
Holčáková
g,
Petr
Voňka
g,
Roman
Hrstka
*g and
Jindřich
Karban
*a
ab,
Martin
Kurfiřt
ab,
Lucie
Červenková Šťastná
a,
Hedvika
Hujerová
a,
Jana
Bernášková
a,
Kamil
Parkan
cd,
Jakub
Kaminský
d,
Nina
Habanová
d,
Jaroslav
Kozák
d,
Alžběta
Magdolenová
d,
Martin
Zavřel
d,
Tatiana
Staroňová
ef,
Veronika
Ostatná
f,
Lucie
Žaloudková
ef,
Aleš
Daňhel
f,
Jitka
Holčáková
g,
Petr
Voňka
g,
Roman
Hrstka
*g and
Jindřich
Karban
*a
aInstitute of Chemical Process Fundamentals of the CAS, v. v. i., Rozvojová 1/135, 165 00 Praha, Czech Republic. E-mail: karban@icpf.cas.cz
bDepartment of Organic Chemistry, University of Chemistry and Technology, Technická 5, 166 28 Praha, Czech Republic
cDepartment of Chemistry of Natural Compounds, University of Chemistry and Technology, Prague, Technická 5, 166 28, Prague, Czech Republic
dInstitute of Organic Chemistry and Biochemistry of the CAS, v. v. i., Flemingovo nám. 542, 160 00 Praha, Czech Republic
eDepartment of Biochemistry, Faculty of Science, Masaryk University, Kotlářská 2, 602 00, Brno, Czech Republic
fInstitute of Biophysics of the CAS, v. v. i., Královopolská 135, 612 00 Brno, Czech Republic
gResearch Centre for Applied Molecular Oncology, Masaryk Memorial Cancer Institute, Žlutý kopec 7, 656 53 Brno, Czech Republic. E-mail: hrstka@mou.cz
First published on 26th September 2024
Abstract
Galectins are a family of β-galactoside-binding proteins with an evolutionarily conserved carbohydrate recognition domain. Their dysregulation has been implicated in physiological and pathological processes, including fibrotic disorders, inflammation, and cancer. For example, elevated levels of galectin-1 contribute to tumor cell migration and immune evasion, whereas overexpression of galectin-3 is associated with increased invasiveness and the formation of metastasis. Pharmacological inhibition of these galectins is a promising therapeutic strategy to counteract their oncogenic effects. In this study, we synthesized a novel series of galectin inhibitors with ferrocene and ruthenium arene motifs attached to lactose, N-acetyllactosamine, or thiodigalactoside scaffolds. We determined their binding affinity toward human galectin-1 (hgal-1) and the CRD domain of human galectin-3 (hgal-3-CRD) using fluorescence polarization, intrinsic fluorescence of galectin tryptophan residues, and isothermal titration calorimetry. The ferrocene analogs exhibited superior affinity for both hgal-1 and hgal-3-CRD compared with ruthenium arenes. In particular, a symmetrical diferrocene thiodigalactoside complex exhibited low nanomolar affinity for hgal-1 and selectivity over hgal-3-CRD. Asymmetrical monoferrocene thiodigalactoside complexes exhibited nanomolar affinity and good selectivity for hgal-3-CRD. Chronopotentiometric stripping analysis demonstrated that the inhibitors stabilized hgal-1 against destabilization by electric field effects. 19F{1H} NMR experiments and molecular dynamics simulations suggested that the incorporation of the ferrocene motif limited the accessible binding modes to hgal-3-CRD whereas binding to hgal-1 remained unrestricted, resulting in attenuated binding affinities to hgal-3-CRD and selectivity for hgal-1. These results open new possibilities for the design and optimization of therapeutic organometallic galectin inhibitors.
Introduction
Lectins are glycan-binding proteins that are abundant in all kingdoms of life.1 In humans, binding of glycans to lectins is essential for many physiological and pathological processes including host–pathogen interactions, inflammation, autoimmune disorders, and cancer progression.2 Although the key roles of lectins in pathologies and their widespread occurrence make them an important target for drug development,2b their pharmacological inhibition by small molecule inhibitors remains underexplored because of the perceived poor druggability of lectins.3 Ligand binding to the shallow water-exposed binding cavity of lectins is associated with a high desolvation penalty, moderate binding affinities of monovalent sugars, and little ability of an inhibitor to form additional productive interactions within the binding site.4 Moreover, carbohydrates have poor pharmacokinetic properties,4d and because lectins form homologous families that bind the same structural motif, it is difficult to achieve selectivity in lectin inhibition. Despite these extraordinary challenges, developments in glycochemistry over the past 20 years have produced potent lectin inhibitors.4b The most successful approach in the design of lectin inhibitors relies on synthetically modified endogenous carbohydrate ligands (glycomimetics).4b Several glycomimetic lectin inhibitors have entered advanced phases of clinical trials,4b,5 but with the exception of GalNAc-siRNA conjugates, none others have yet been marketed.4b Thus, the need for innovative approaches to expand the available chemical space diversity in the design of lectin inhibitors is undeniable.Galectins are a family of animal lectins that non-covalently bind a β-galactopyranoside motif in membrane-bound and extracellular glycans through an evolutionarily conserved binding site located within a carbohydrate recognition domain (CRD).4c,6 Natural disaccharide galectin ligands, such as lactose (Lac) 1 and N-acetyllactosamine (LacNAc) 2 (Fig. 1A), bind the highly conserved region of the CRD termed subsites C and D, with each subsite accommodating one hexopyranose moiety (Fig. 1A).7 The β-galactopyranoside moiety in these disaccharides is recognized by subsite C. Two additional monosaccharides, which can be attached to the 3′-position of the galactopyranoside moiety in endogenous ligands, are recognized by the less conserved extension of the binding groove termed subsites A and B (Fig. 1A and B).7a,b The loosely defined subsite E can interact with the substituents attached to the reducing end of the monosaccharide occupying subsite D (Fig. 1B).7d,8 Galectins are implicated in several pathologies including fibrotic diseases, inflammation, and cancer.9 Among sixteen mammalian galectins,7b human galectin-1 (hgal-1) and -3 (hgal-3) are probably the most studied to date due to their expression in many tissues and their prominent role in diseases.4c,10 The involvement of galectins in pathologies is complex because galectins are multifunctional proteins whose functions depend on their cellular and tissue localization.11 Moreover, different galectins can play opposite roles in disease progression.10 Therefore, the availability of galectin inhibitors that are highly selective for individual galectins is essential for both fundamental research and drug development.
 | ||
| Fig. 1 (A) Structures, numbering, and dissociation constants Kd of natural disaccharide galectin ligands 1 and 2, and the galectin inhibitor TD139 determined by isothermal titration calorimetry (ITC).12a Oxygen functionalities critical for binding are shown in blue, 3-fluorophenyl-triazole moiety of TD139 is shown in red. (B) Binding subsites A–E are shown for the complex of TD139 with hgal-3-CRD (PDB: 5H9P). (C) Structures of thiophene-derived hgal-1 inhibitor 3, thiazole-derived hgal-1 inhibitor GB1908 and their dissociation constants Kd determined by competitive fluorescence polarization (FP),13 (hetero)aromatic moieties shown in red. | ||
Typical glycomimetic small molecule galectin inhibitors comprise a galactose-containing mono- or disaccharide scaffold capable of forming the critical hydrogen bonds found in natural agonists 1 and 2 (Fig. 1A).8 The scaffold is typically decorated with aromatic non-polar substituents that introduce additional interactions within the binding groove.8
The glycomimetic inhibitor TD139 (later renamed GB0139, Fig. 1A) targets four subsites and illustrates this approach.14 The hydroxyl groups at the 3-, 4′- and 6′-positions of natural ligands 1 and 2 (including their stereochemistry), the pyranose oxygen O5′, and the hydrophobic α-side of the galactoside residue are reproduced in the thiodigalactoside scaffold of TD139, and direct the inhibitor into subsites C and D (Fig. 1A) via key interactions with amino acid residues in these subsites. The fluorophenyl-triazole moiety forms guanidinium-arene cation-π interactions with the arginine residues Arg144 and Arg186 in subsites B and E of hgal-3, respectively, while fluorine is involved in orthogonal multipolar interactions15 with the polypeptide backbone, collectively leading to nanomolar affinities toward hgal-3 and approximately 3-fold weaker binding to hgal-1 according to ITC (Fig. 1A).12b Modifications of TD139 and other inhibitors resulted in a series of excellent hgal-3 selective disaccharide8,16 and monosaccharide17 inhibitors. TD139 entered the Phase IIb of clinical trials for the treatment of idiopathic pulmonary fibrosis (IPF), but unfortunately failed due to its low efficacy and its development for IPF treatment was discontinued.18
Although the development of hgal-3 inhibitors has advanced considerably, the development of potent hgal-1 selective inhibitors remains challenging despite their therapeutic potential.19 To the best of our knowledge, the only reported small molecule, single-digit nanomolar inhibitors of hgal-1 are the thien-3-yl derivative 313a,14 (Fig. 1C) and its thiazol-2-yl analog.13a Both had an approximately 10-fold selectivity for hgal-1 over hgal-3 as determined by a competitive fluorescence polarization (FP) assay. However, no further bioactivity of compound 3 has been reported, and it appears that this inhibitor is not being further developed. Very recently, a thiogalactoside-based inhibitor named GB1908 with a two orders of magnitude hgal-1 selectivity has been reported (Fig. 1C).13b
Recent X-ray diffraction and 19F NMR studies have suggested differences between hgal-1 and hgal-3 in the binding mode of TD139 for subsites B and E.12b Inspired by these findings, we hypothesize that lipophilic barrel-shaped organometallic complexes such as ferrocene or cyclopentadienylruthenium arenes may exploit these differences and interact with the binding subsites of galectins more efficiently or selectively than the planar fluorophenyl moiety, leading to higher binding affinity or selectivity. Organotransition metal complexes, due to their three-dimensional coordination sphere, offer structural diversity in protein–ligand interactions that is inaccessible to purely organic compounds.20 For example, improved affinity or selectivity after substituting a ferrocene moiety for a planar arene has been reported for the anticancer drugs paclitaxel,21 tamoxifen,20c the drug candidate plinabulin,22 and the dopamine D3 receptor antagonist BP 897.23 In spite of their advantages, organotransition metal complexes have not been tested as antagonists in lectin inhibition, although multivalent lactose-ferrocene conjugates have been evaluated as electrochemical probes for the detection of galectin-3.24
We prepared a series of lactose, N-acetyllactosamine- and thiodigalactoside-based inhibitors bearing ferrocene or pentamethylcyclopentadienyl ruthenium arene complexes and determined their affinities to hgal-1 and hgal-3 using the FP assay, intrinsic fluorescence of galectin tryptophan residues, and ITC. Here, we report that introducing two ferrocene-triazole moieties into the thiodigalactoside scaffold resulted in an hgal-1 inhibitor with potency comparable to TD139 but with up to 50-fold higher selectivity for hgal-1 over hgal-3, highlighting the potential of organometallic fragments in the design of lectin inhibitors. Subsequent 19F{1H} NMR monitoring of the 3-fluorophenyl-triazole moiety and molecular dynamics simulations were used to explain the differences in the binding mode between hgal-1 and hgal-3. In addition, the electrochemical activity of selected ferrocene derivatives was studied by cyclic voltammetry (CV), and the ability of the synthesized inhibitors to stabilize hgal-1 against electric current was evaluated using chronopotentiometric stripping analysis.
Results
Synthesis of galectin antagonists
The ferrocenyl and ruthenium arene moieties were attached to the selected disaccharide scaffolds via a 1,2,3-triazole linker substituted at the 4-position with an organocomplex (Fig. 2C). The linker was formed by a copper(I)-catalyzed cycloaddition reaction between ferrocenyl- or phenylacetylene and an azido group at the selected positions of the disaccharides (Fig. 2, substituents R1, R2, R3, R3′). These positions were chosen because their modifications generally do not interfere with recognition by galectins and organometallic substituents at these positions enabled us to explore possible attractive interactions with subsites A/B and E flanking the most conserved region of subsites C and D.7b,8,25 For Lac- and LacNAc-derived galectin ligands, we decided to attach the organometallic substituent at the reducing end anomeric position (Fig. 2A, substituent R1) to exploit possible interactions with subsite E. Similar reasoning applies to the 3′-OH hydroxyl of N-acetyllactosamine 2 (Fig. 2A, substituent R3′), which points toward subsites A and B. In addition, the 2-position of the lactosamine scaffold (Fig. 2A, substituent R2) was also modified because this position tolerates modifications with bulky substituents.7b For the symmetrical thiodigalactoside scaffold, we modified the 3/3′-position (Fig. 2B, substituents R3/R3′). Modification at this position with an arene group has previously produced potent galectin inhibitors.8,26 The ferrocene moiety was introduced directly into the disaccharide scaffold by azide–alkyne cycloaddition of the corresponding azido-disaccharides and ethynylferrocene. The (pentamethylcyclopentadienyl)ruthenium arene moiety was introduced by the complexation reaction of a phenyltriazole moiety with the [Cp*Ru(MeCN)3]Cl complex generated in situ.27 Non-metallic compounds bearing a hydroxymethyl, phenyl, or 3-fluorophenyl substituent attached to the disaccharide scaffold via a triazole linker were prepared for comparison (Scheme 1, compounds 4 and 7; Scheme 3, compounds 26, 28 and 29; Scheme 4, compound 37).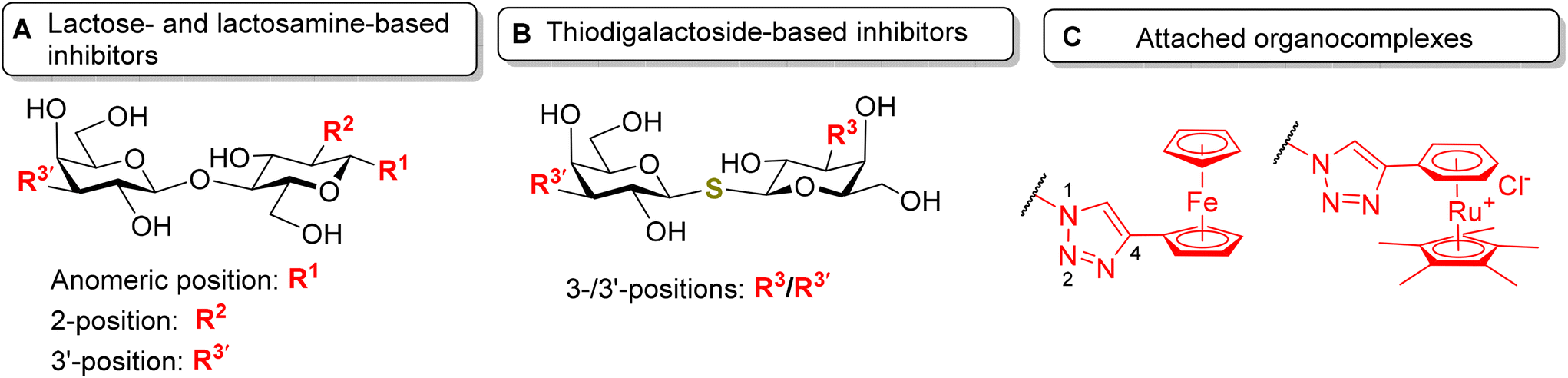 | ||
| Fig. 2 The positions selected for the attachment of modifying substituents to (A) lactose (R2 = OH) or lactosamine (R2 = NH2), (B) thiodigalactoside. (C) Organocomplexes attached to carbohydrate scaffolds and numbering of the 1,2,3-triazole linking moiety. | ||
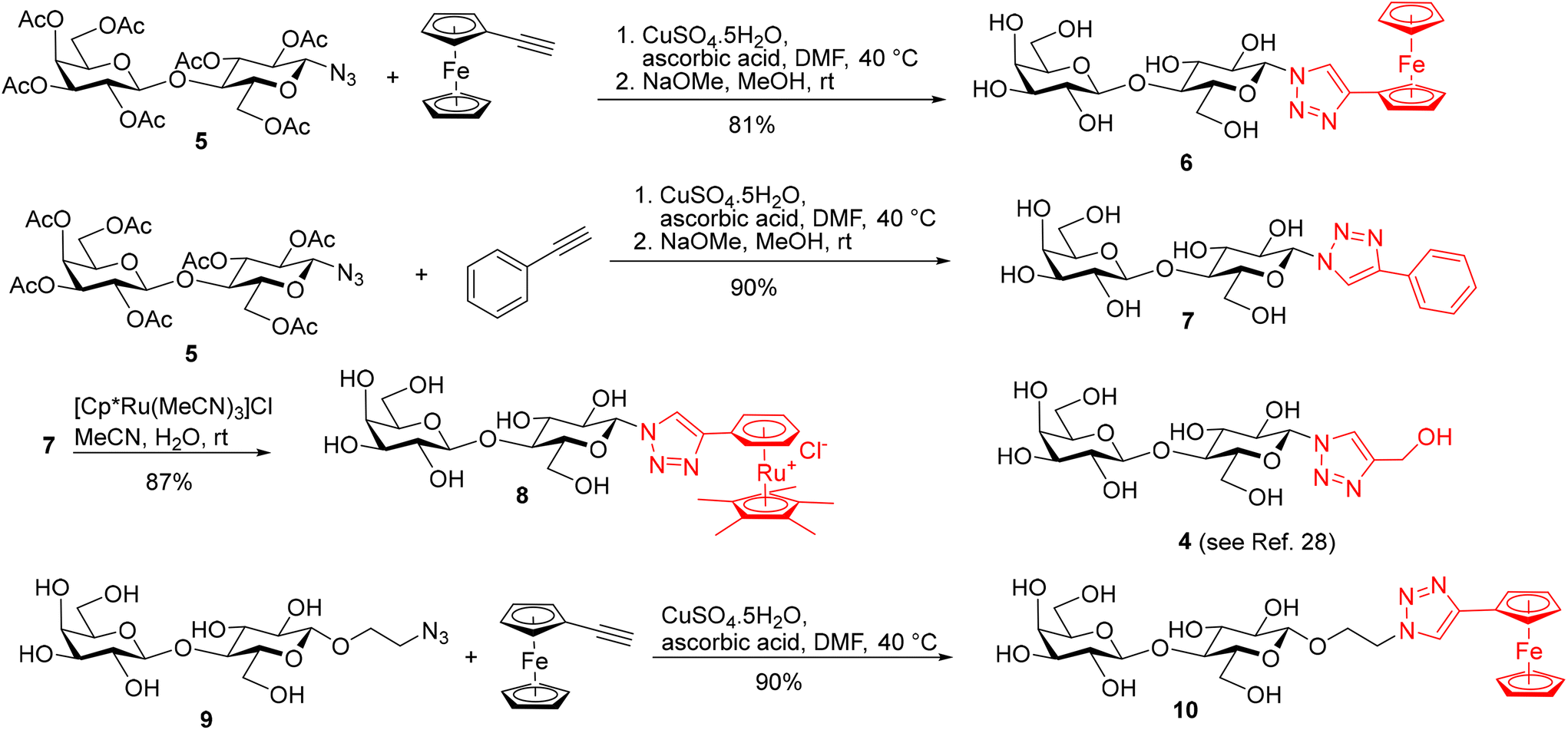 | ||
| Scheme 1 Synthesis of galectin ligands 4, 6–8, and 10 derived from lactose modified at the anomeric position. | ||
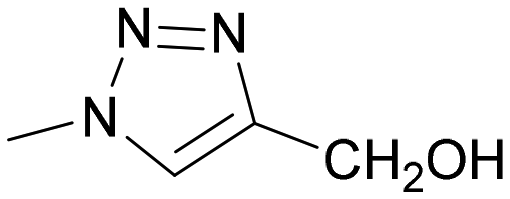
The synthesis of Lac-derived galectin ligands 4, 6–8, and 10 modified at the anomeric position is summarized in Scheme 1. Hydroxymethyl-triazole-substituted lactose 4 was prepared for comparison as reported.28 Starting azido disaccharides 529 and 930 were prepared from lactose 1 as reported and converted to the target ferrocene analogs 6 and 10 using cycloaddition with ethynylferrocene as the key reaction. The phenyl triazole 7 was obtained from azide 5 by reaction with phenyl acetylene followed by Zemplén deacetylation. Phenyl analog 7 was subjected to the complexation reaction with the [Cp*Ru(MeCN)3]Cl complex generated in situ from the tetramer [Cp*RuCl]4 as described previously.27 This complexation reaction afforded the corresponding ruthenium arene 8 (Scheme 1).
The synthesis of LacNAc-derived galectin ligands 16 and 19 modified at the 1- and 2-positions, respectively, is summarized in Scheme 2. Azides 15 and 18 for the copper-catalyzed cycloaddition were prepared from monosaccharide building blocks by chemical glycosylation. N-Acetyllactosaminyl azide 15 was obtained by NIS/TfOH-promoted glycosylation31 of known 2-phthalimido-β-glucosyl-azide 1232 with thiogalactoside 1133 followed by sequential deprotection, using first oxidative de-O-benzylation with the NaBrO3/Na2S2O4 system34 to give compound 14, then treatment with ethylenediamine in refluxing methanol to liberate the 2-amino group,35 and finally N-acetylation (Ac2O, MeOH). Methyl 2-azido-lactoside 18 was obtained by NIS/TfOH-promoted glycosylation of methyl 2-azido-β-glucoside 1736 with thiogalactoside 11 followed by oxidative de-O-benzylation. Copper(I)-catalyzed cycloaddition between azido disaccharides 15, and 18 and ethynylferrocene, followed by Zemplén deacetylation in the case of acetylated disaccharide 18, afforded the target ferrocenyl-triazole-substituted disaccharides 16 and 19 (Scheme 2).
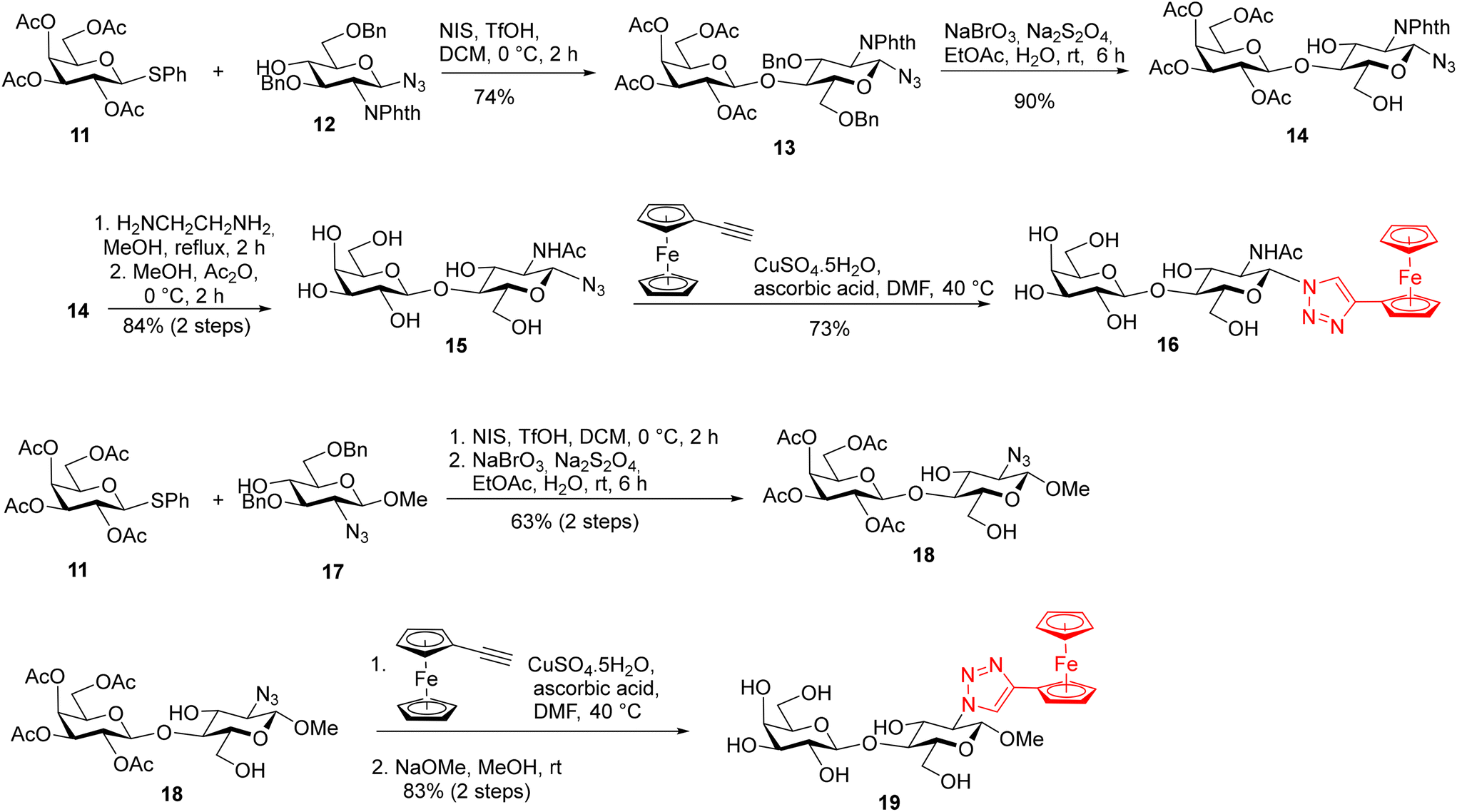 | ||
| Scheme 2 Synthesis of galectin ligands 16, and 19 derived from LacNac modified at the 1- and 2-positions. | ||
The synthesis of LacNAc analogs modified at the 3′-position is described in Scheme 3. The methyl β-glycosides 23 and 24 of 3′-azido-LacNAc required for azide–alkyne cycloaddition were synthesized by glycosylation of methyl 2-phthalimido-β-glucoside 2137 with 3′-azido-thiogalactoside 20.38 The resulting disaccharide 22 was oxidatively de-O-benzylated to yield diol 23 that was subjected to phthalimide deprotection (ethylenediamine in refluxing MeOH) and N-acetylation to afford the 3′-azido-LacNAcβ-OMe 24. Copper(I)-catalyzed cycloaddition between azido disaccharides 23, and 24 and ethynylferrocene, followed by Zemplén deacetylation in the case of acetylated disaccharide 23, afforded the target ferrocenyl-triazole-substituted disaccharides 30 and 25, respectively. The bulky phthalimido group (Phth) was retained at the 2-position of disaccharide 30 because a large aromatic substituent at this position may influence the selectivity in interactions with galectins. For comparison, 3-fluorophenyl-triazole-substituted LacNAc 28 and hydroxymethyl-triazole-substituted LacNAc 29 were prepared from disaccharide 24 by cycloaddition with 3-fluorophenylacetylene and propargyl alcohol, respectively (Scheme 3). The cycloaddition of azide 24 with phenylacetylene afforded disaccharide 26 carrying a phenyl–triazole ligand suitable for the complexation reaction with the [Cp*Ru(MeCN)3]Cl complex, which afforded the corresponding ruthenium arene 27 (Scheme 3). The parent disaccharides 26 along with disaccharide 7 (Scheme 1), both modified with the phenyl–triazole moiety, were also used as the reference compounds to determine how the substitution of the ruthenium arene or ferrocene for a planar phenyl substituent influenced the affinity. All Lac- and LacNAc-based ligands were prepared as β-anomers at the reducing end anomeric position. When this anomeric position did not carry a modifying organometallic substituent, it was protected as a methyl β-glycoside (compounds 19, 25–30) to prevent the formation of a mixture of anomers.
 | ||
| Scheme 3 Synthesis of galectin ligands 25–30 derived from LacNAc modified at the 3′-position. | ||
For the thiodigalactoside-based inhibitors, we attached the modifying substituent at the 3- and 3′-positions of the thiodigalactoside scaffold (Fig. 2B), as previous work has documented that binding to galectins benefits from the attachment of arene-triazole substituents at these positions.14 This produced compounds 34, 36–38, 41, and 42 (Scheme 4). Starting 3-azido-thiodigalactoside 33 was obtained by the tetrabutylammonium fluoride-promoted reaction of 3-azido-galactosyl bromide 3139 with thiogalactoside 3240 in acetonitrile following the published protocol (Scheme 4).40,41 3,3′-Diazido-thiodigalactoside 35,26c,39 3,4,5-trifluorophenyl-analog 39,16 and 3-fluorophenyl analog 4016 were prepared as reported. Cycloaddition of azides 33, 35, 39, and 40 with ethynylferrocene followed by Zemplén deacetylation gave ferrocenyl-triazole-substituted analogs 34, 36, 41, and 42, respectively, whereas azide–alkyne cycloaddition of diazide 35 with phenylacetylene afforded the phenyl–triazole ligand 37 (Scheme 4). The complexation reaction of compound 37 with [Cp*Ru(MeCN)3]Cl generated in situ was expected to afford the bisruthenium arene disaccharide 38. The product produced the expected molecular ion mass in high resolution mass spectra; however, the 1H NMR spectrum showed two signals for the triazole proton and four signals for the methyl protons of the pentamethylcyclopentadienyl ring. We speculate that compound 38 was prepared as a mixture of four inseparable stereoisomers due to hindered rotation at the bond between the triazole and the phenyl ligand. All prepared ferrocenes were obtained as either crystalline or amorphous solids; most of them with limited solubility in water (see ESI for details, section B.2.†), whereas the ruthenium arenes were obtained as white amorphous solids with good water solubility. Both types of compounds exhibited stability in air and in aqueous or DMSO solutions, as confirmed over a period of three weeks.
 | ||
| Scheme 4 Synthesis of galectin ligands 34, 36–38, 41 and 42 derived from thiodigalactoside modified at the 3 and 3′-positions. | ||
Compound characterization
The formation of 1,2,3-triazole was also confirmed by NMR spectroscopy. The proton of the triazole CH group resonated at 8.01–8.90 ppm in 1H NMR spectra, while the corresponding carbon resonated at 119.8–124.7 ppm in 13C NMR spectra, which is characteristic of 1,4-substituted 1,2,3-triazoles.42 The quaternary carbon of the 1,2,3-triazole ring showed a chemical shift in the range of 141.8–148.4 ppm. Ruthenium complexation led to shielding of the corresponding phenyl ring,27 resulting in the decreased values of the proton and carbon chemical shifts (δH = 5.95–6.64 ppm/δC = 84.98–99.3 ppm for the phenyl ring in the ruthenium complexes, δH = 7.30–7.88 ppm/δC = 125.0–131.9 ppm for the phenyl rings in the parent phenyl-substituted disaccharides).
Binding to hgal-1 and hgal-3
| Entry |
|
Compound | K i (hgal-1) (μM) | rp (hgal-1) | K i (hgal-3-CRD) (μM) | rp (hgal-3-CRD) | Selectivity to hgal-1b | ||
|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | R3′ | |||||||
| a Relative potency defined as rp = Ki(Lac 1)/Ki(inhibitor) for Lac-derived inhibitors, rp = Ki(2β-OMe)/Ki(inhibitor) for LacNAc-based inhibitors. b Defined as the ratio Ki(hgal-3-CRD)/Ki(hgal-1). c Fc = ferrocenyl, [Ru] = ruthenium arene moiety, 3F-Ph = 3-fluorophenyl. d 2β-OMe denotes methyl N-acetyl-β-lactosaminide (LacNAc1β-OMe).36 | |||||||||
| Lac-derived inhibitorsc | |||||||||
| 1 | OH | OH | OH | 1 (lactose) | 334 ± 28 | 1.0 | 94 ± 7 | 1.0 | 0.28 |
| 2 |
|
OH | OH | 4 | 157 ± 31 | 2.1 | 55 ± 4 | 1.7 | 0.35 |
| 3 |

|
OH | OH | 7 | 241 ± 36 | 1.4 | 67 ± 7 | 1.4 | 0.28 |
| 4 |
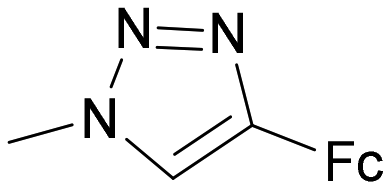
|
OH | OH | 6 | 105 ± 20 | 3.2 | 35 ± 1 | 2.7 | 0.33 |
| 5 |

|
OH | OH | 8 | 262 ± 51 | 1.3 | 132 ± 15 | 0.7 | 0.50 |
| 6 |

|
OH | OH | 10 | 141 ± 6 | 2.4 | 30 ± 1 | 3.1 | 0.21 |
| LacNAc-derived inhibitorsc | |||||||||
| 7 | OMe | NHAc | OH | 2β-OMed | 182 ± 12 | 1.0 | 49 ± 3 | 1.0 | 0.27 |
| 8 |

|
NHAc | OH | 16 | 73 ± 19 | 2.5 | 15 ± 1 | 3.3 | 0.20 |
| 9 | OMe |

|
OH | 19 | 54 ± 2 | 3.4 | 3.9 ± 0.1 | 13 | 0.07 |
| 10 | OMe | NHAc |
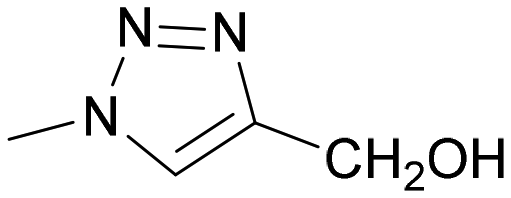
|
29 | 43 ± 8 | 4.2 | 22 ± 5 | 2.2 | 0.51 |
| 11 | OMe | NHAc |
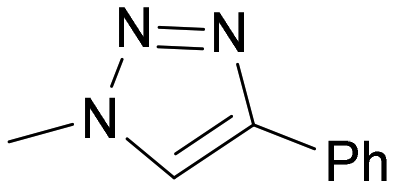
|
26 | 5.2 ± 0.8 | 35 | 2.7 ± 0.7 | 18.1 | 0.52 |
| 12 | OMe | NHAc |
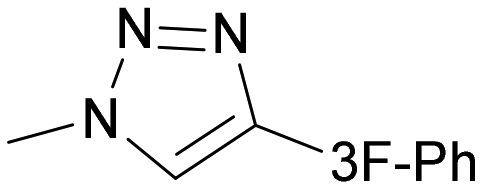
|
28 | 1.7 ± 0.1 | 107 | 0.26 ± 0.01 | 188 | 0.15 |
| 13 | OMe | NHAc |

|
25 | 3.7 ± 1.2 | 49 | 5.0 ± 0.2 | 10 | 1.35 |
| 14 | OMe | NPhth |
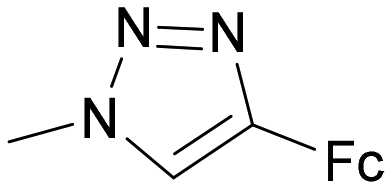
|
30 | 3.4 ± 0.5 | 54 | 1.3 ± 0.1 | 38 | 0.38 |
| 15 | OMe | NHAc |
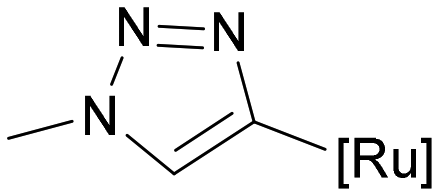
|
27 | 78 ± 11 | 2.3 | 14.8 ± 3.9 | 3.3 | 0.19 |

| Entry |
|
Compound | K i (hgal-1) (μM) | rp (hgal-1) | K i (hgal-3-CRD) (μM) | rp (hgal-3-CRD) | Selectivity to hgal-1c | |
|---|---|---|---|---|---|---|---|---|
| R3 | R3′ | |||||||
| a Relative potency with respect to TD139, one of the most potent inhibitor, defined as rp = Ki(TD139)/Ki(inhibitor). b Fc = ferrocenyl, [Ru] = ruthenium arene moiety. c defined as the ratio Ki(hgal-3-CRD)/Ki(hgal-1). | ||||||||
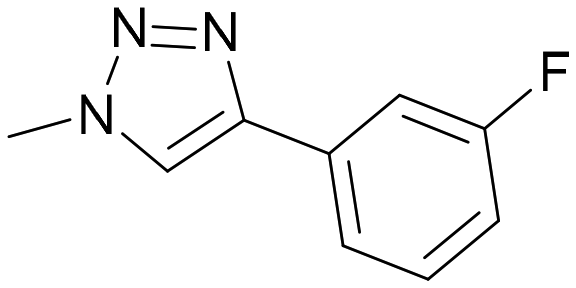
| 1 |

|
|
TD139 | 0.12 ± 0.01 | 1.00 | 0.008 ± 0.001 | 1.00 | 0.07 |
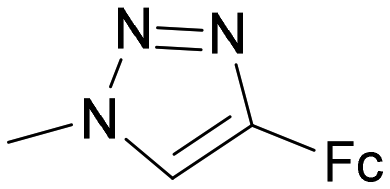
| 2 | OH |
|
34 | 1.9 ± 0.2 | 0.06 | 1.7 ± 0.4 | <0.01 | 0.89 |
| 3 |

|

|
37 | 0.25 ± 0.04 | 0.48 | 0.033 ± 0.002 | 0.24 | 0.13 |
| 4 |
|

|
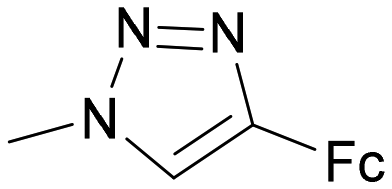
36 | 0.035 ± 0.017 | 3.43 | 0.26 ± 0.066 | 0.03 | 7.43 |
| 5 |
|

|
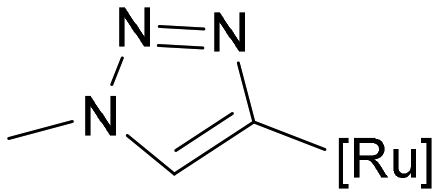
38 | 0.82 ± 0.08 | 0.15 | 0.72 ± 0.16 | 0.01 | 0.88 |
| 6 |

|
|
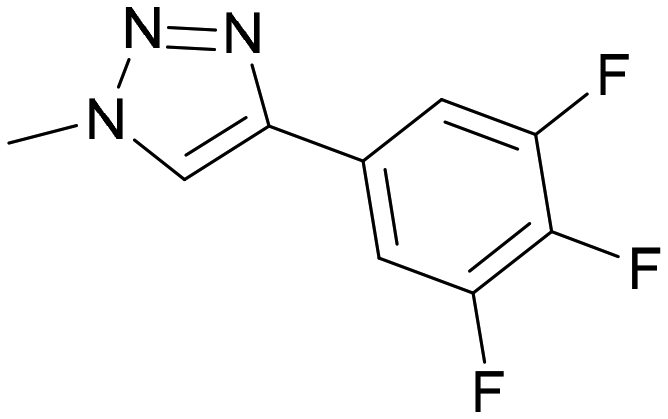
41 | 0.15 ± 0.03 | 0.80 | 0.013 ± 0.004 | 0.62 | 0.09 |
| 7 |

|

|
42 | 0.16 ± 0.01 | 0.75 | 0.026 ± 0.002 | 0.31 | 0.16 |

The attachment of an organometallic moiety to positions 1 and 2 of disaccharides Lac and LacNAc was aimed at the formation of additional interactions with the galectin subsite E.48 Both galectins contain an arginine residue in this subsite (Arg74 in hgal-1 and Arg186 in hgal-3) capable of forming cation-π interactions with arene moieties of the inhibitor.7d,49 On the whole, modifications at these positions mostly resulted in a modest improvement in binding affinity (Table 1, entries 1–9). The selectivity was always in favor of hgal-3-CRD. The best improvement was obtained for compound 19 resulting from the attachment of ferrocenyltriazole to the 2-position of LacNAc. It bound hgal-3-CRD 13-fold more strongly than the parent methyl N-acetyl-β-lactosaminide362β-OMe (entry 9). Complex 19 had affinity and selectivity comparable with that obtained for 5,6,7,8-tetrahydronaphthoate ester 43 (Fig. 3), which was among the most selective hgal-3 binders in a series of previously reported lactose 2-esters.49 The remaining complexes 6, 8, 10, and 16 had at most a two- to threefold improved affinity for both galectins relative to the parent disaccharides Lac (entries 4–6 vs. entry 1) and 2β-OMe (entry 8 vs. entry 7). Ferrocenes 6 and 10 had only marginally better affinity for both hgal-1 and hgal-3-CRD than the non-metallic phenyl and hydroxymethyl compounds 7 and 4 (entries 4 and 6 vs. entries 3 and 2), respectively. Ruthenium arene 8 was a slightly weaker binder than the corresponding phenyl analog 7 (entries 5 and 3).
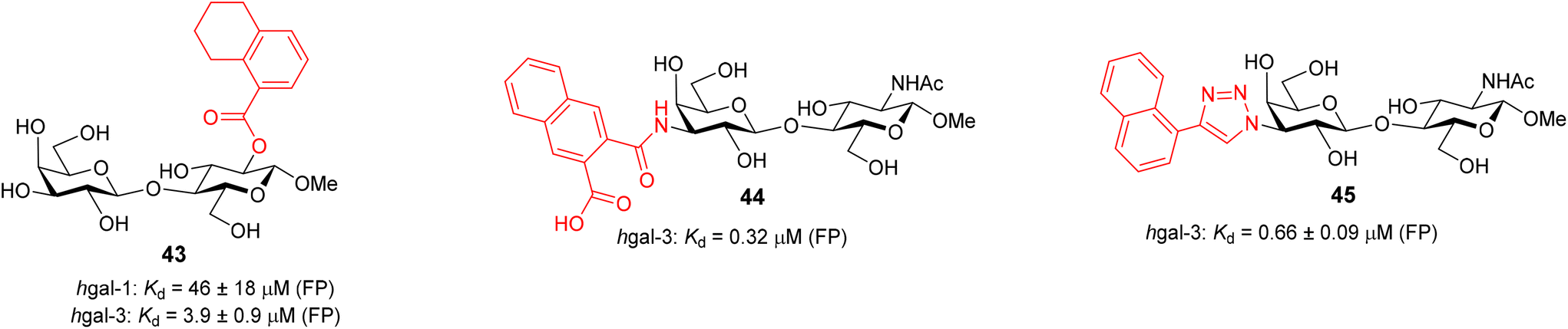 | ||
| Fig. 3 Structures of previously reported galectin inhibitors 43–45 and their affinities to hgal-1 and hgal-3 from FP.26a,49,50 The affinity of 44 and 45 to hgal-1 was not reported. | ||
Next, we modified the 3′-position of the N-acetyllactosamine scaffold (compounds 25–30, Table 1, entries 10–15), with the aim of introducing cation-π interactions between Arg144 in subsite B of the hgal-3 CRD and the ferrocene/ruthenium arene complex attached to the 3′-position.12b,26a,50 A similar approach has been used previously in the preparation of non-organometallic hgal-3 inhibitors7d and resulted in affinities up to the high nanomolar range (see N-acetyllactosaminides 44 and 45 for examples, Fig. 3). Interestingly, the affinity for hgal-1 has not been previously determined for 3′-modified lactosamines. We found that 3-fluorophenyltriazole 28 (entry 12) and ruthenium arene 27 (entry 15) bound hgal-3-CRD approximately 7- and 5-fold more strongly than hgal-1, respectively, whereas the remaining 3′-modified lactosamine analogs displayed comparable or only 2- to 3-fold higher affinities for hgal-3-CRD than for hgal-1 despite the absence of an arginine residue in subsite B of hgal-1.
The 3′-hydroxymethyltriazole 29 (entry 10) bound both galectins better than the unmodified 2β-OMe due to the interaction of the triazole ring with the galectins.7d Substituting the hydroxymethyl group with phenyl in 26 significantly improved the affinity to the single-digit micromolar range. In agreement with the previous observations made for thiodigalactosides,14 substituting fluorine for 3-hydrogen in the phenyl moiety of the analog 28 (entry 12) further increased the affinity, especially for hgal-3-CRD, which reached nanomolar values. Replacing the phenyl with ferrocene (26 → 25, entry 13) resulted in an affinity comparable to that of the phenyl analogue 26, but for the first time in our series, the selectivity was very slightly tilted in favor of hgal-1. Substituting the bulky phthalimide in 30 (entry 14) for the acetamide in 25 (entry 13) further improved binding to hgal-3-CRD, producing the best inhibitor among our LacNAc- and Lac-based metal organocomplexes, probably owing to simultaneous interactions with subsites B and E. Disappointingly, the ruthenium arene complex 27 was a weaker inhibitor of both galectins compared with its ferrocene and phenyl counterparts 25 and 26, respectively (entries 15 vs. 13 and 11).
Some thiodigalactoside-based inhibitors, such as compound TD139, are among the most potent hgal-1 and -3 inhibitors.14 Also in this study, inhibitors resulting from the attachment of an organometallic substituent to the thiodigalactoside scaffold outperformed (Table 2) Lac- and LacNAc-derived inhibitors. Inhibitor TD139 (entry 1) was more selective for hgal-3-CRD in our hands than originally reported. While the affinity for hgal-3-CRD (Ki = 0.008 ± 0.001 μM) was close to the reported value (Ki = 0.014 ± 0.003 μM),14 the affinity for hgal-1 was one order of magnitude lower (Ki = 0.12 ± 0.01 μM) than originally reported (Ki = 0.012 ± 0.003 μM).14 However, a value very close to ours was reported by Zetterberg et al. this year (Ki = 0.109 ± 0.022 μM as determined by FP).13b Symmetrically substituted phenyltriazolyl-thiodigalactoside 37 was a selective hgal-3-CRD inhibitor, although less potent than TD139 because it lacked the fluorine-galectin interactions contributing to the high binding affinity of TD139 (Table 2, entries 1 and 3).14 Substituting both planar phenyls in 37 with ferrocene to give 36 resulted in a significant drop in affinity toward hgal-3-CRD (almost by an order of magnitude, entries 3 and 4) and an improvement in affinity toward hgal-1. As a result, diferrocene 36 was the best and most selective organometallic inhibitor of hgal-1 in the series, because it bound hgal-1 7-fold more strongly than hgal-3-CRD, surpassing not only the diphenyl analog 37 (entries 3 and 4, 7-fold increase in affinity for hgal-1) but also the inhibitor TD139 (entries 1 and 4, 3-fold increase), despite the absence of a fluorine substituent. The asymmetric monoferrocene complex 34 exhibited only micromolar affinity for both galectins (entry 2). The asymmetric ferrocene-phenyltriazole complexes 41 and 42 showed a good hgal-3-CRD selectivity (entries 6 and 7), although 42 bound hgal-3-CRD 3-fold more weakly than TD139. Ruthenium arene complex 38 (entry 5) displayed a significantly lower affinity for both galectins compared with its phenyl and ferrocenyl counterparts 37 and 36, respectively, and was not further investigated.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Compounds | K d (hgal-1) (μM) | K d (hgal-3) (μM) | K d (hgal-3-CRD) (μM) | Selectivity to hgal-1a |
| a Defined as the ratio Kd(hgal-3)/Kd(hgal-1). | |||||
| 1 | 1 (Lac) | 244 ± 29 | 297 ± 28 | 142 ± 23 | 1.2 |
| 2 | 2 (LacNAc) | 84 ± 13 | 80 ± 15 | 45 ± 12 | 0.9 |
| 3 |

|
193 ± 21 | 49 ± 8 | 30 ± 2 | 0.5 |
| 4 |

|
52 ± 4 | 5 ± 1 | 2.9 ± 0.3 | 0.1 |
| 5 |

|
1.8 ± 0.5 | 13 ± 2 | 8 ± 1 | 7.2 |
| 6 |

|
84 ± 12 | 10 ± 1 | 5.2 ± 0.5 | 0.12 |
| 7 |

|
2.2 ± 0.2 | 0.15 ± 0.03 | 0.08 ± 0.02 | 0.07 |
| 8 |

|
0.08 ± 0.01 | 0.0023 ± 0.0004 | 0.0021 ± 0.0004 | 0.03 |
| 9 |

|
0.12 ± 0.02 | 0.031 ± 0.004 | 0.012 ± 0.001 | 0.26 |
| 10 |

|
0.010 ± 0.001 | 0.50 ± 0.04 | 0.40 ± 0.10 | 50 |
| 11 |
|
0.035 ± 0.002 | 0.0067 ± 0.0006 | 0.0040 ± 0.0005 | 0.19 |
| 12 |

|
0.11 ± 0.01 | 0.0044 ± 0.0008 | 0.0038 ± 0.0006 | 0.04 |

In all cases, the measured Kd to hgal-3-CRD was slightly lower than that obtained for full-length hgal-3; however, the difference did not appear significant in most cases, and the affinity trends were similar for both hgal-3 and hgal-3-CRD. Also, the values for disaccharides determined by the intrinsic tryptophan fluorescence assay and by the FP assay were comparable and did not differ by more than a factor of three in most cases. A more significant difference between the two method was found for the potent hgal-1 inhibitor in the series 36 (Table 3, entry 10). The intrinsic tryptophan fluorescence assay indicated a higher affinity and selectivity of diferrocene 36 for hgal-1 than FP: Complex 36 bound hgal-1 50-fold more strongly than hgal-3, and 40-fold more strongly than hgal-3-CRD, whereas the selectivity using the FP assay was lower (Ki(hgal-3-CRD)/Ki(hgal-1) = 7.4). The intrinsic tryptophan fluorescence assay also confirmed the selectivity of disaccharides 19, 27, 28, 37, 41, 42 and TD139 towards hgal-3. In contrast to the FP and ITC assays (see Table 4 for the ITC data), there was practically no difference between disaccharides 41 and 42 in binding to hgal-3, as both had low nanomolar affinity (Table 3, entries 11 and 12). This resulted in the 3-fluorophenyl analog 42 being a more selective hgal-3 inhibitor than the trifluorophenyl analog 41, again in contrast to the FP results (Table 2, entries 6 and 7). Since these potent hgal-3 inhibitors 41 and 42 and hgal-1 inhibitor 36 carry an electroactive ferrocene moiety, we envision their application in electrochemical sensing of galectin inhibition. The commercial inhibitor TD139 showed 4-fold higher hgal-3 affinity and 2-fold higher hgal-3 selectivity in the tryptophan fluorescence assay (Table 3, entry 8) than in the FP assay (Table 2, entry 1). Discrepancies between the two assays can be attributed to the different bioanalytical principles on which the assays are based. Importantly, both assays confirmed that the affinity and selectivity for hgal-1 were significantly improved by insertion of two ferrocene-triazole motifs into the thiodigalactoside scaffold to yield a potent hgal-1 inhibitor 36.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Compound | K d (μM) | ΔG (kcal mol−1) | ΔH (kcal mol−1) | −TΔS (kcal mol−1) | n | Selectivity to hgal-1a |
| a Defined as the ratio Kd(hgal-3)/Kd(hgal-1). b Taken from ref. 12b. | |||||||
| hgal-1 | |||||||
| 1 | TD139 | 0.220 ± 0.05 | −9.1 ± 0.2 | −16.7 ± 0.4 | 7.6 ± 0.4 | 0.96 ± 0.04 | 0.31 |
| 2 | TD139 | 0.279 ± 0.001 | −8.9 ± 0.1 | −16.9 ± 0.4 | 8.0 ± 0.4 | 1.03 ± 0.04 | 0.07 |
| 3 | 37 | 0.288 ± 0.006 | −8.9 ± 0.1 | −9.6 ± 0.4 | 0.7 ± 0.4 | 1.04 ± 0.02 | 0.33 |
| 4 | 36 | 0.258 ± 0.004 | −9.0 ± 0.1 | −12.8 ± 1.4 | 3.8 ± 1.5 | 1.06 ± 0.07 | 3.6 |
| 5 | 41 | 0.340 ± 0.019 | −8.9 ± 0.1 | −10.2 ± 0.1 | 1.3 ± 0.1 | 1.03 ± 0.00 | 0.07 |
| 6 | 42 | 0.366 ± 0.031 | −8.8 ± 0.1 | −9.5 ± 0.1 | 0.6 ± 0.1 | 1.03 ± 0.02 | 0.18 |
| hgal-3-CRD | |||||||
| 7 | TD139 | 0.068 ± 0.01 | −9.9 ± 0.4 | −18.2 ± 0.8 | 8.3 ± 0.9 | 0.96 ± 0.01 | — |
| 8 | TD139 | 0.019 ± 0.003 | −10.6 ± 0.1 | −16.4 ± 0.4 | 5.8 ± 0.3 | 1.08 ± 0.01 | — |
| 9 | 37 | 0.096 ± 0.005 | −9.6 ± 0.1 | −11.2 ± 0.3 | 1.7 ± 0.2 | 1.03 ± 0.03 | — |
| 10 | 36 | 0.923 ± 0.040 | −8.3 ± 0.1 | −12.7 ± 0.2 | 4.4 ± 0.1 | 1.08 ± 0.02 | — |
| 11 | 41 | 0.023 ± 0.006 | −10.5 ± 0.2 | −14.0 ± 1.3 | 3.6 ± 1.5 | 1.03 ± 0.04 | — |
| 12 | 42 | 0.066 ± 0.010 | −9.8 ± 0.1 | −12.0 ± 0.2 | 2.3 ± 0.4 | 1.01 ± 0.00 | — |

 | ||
| Fig. 4 (A) The dependence of peak H area on stripping current ISTR for free hgal-1 (grey) and that in complex with compounds 2 (green), 6 (red), 25 (magenta), and 36 (blue). Inset: peak H height of free hgal-1 measured using different stripping currents. A double peak of structural transition can be seen at −20 μA. (B) ΔISTR1/2 values of hgal-1 complexes normalized52b to free hgal-1. | ||
 | ||
| Fig. 5 Thermograms and binding isotherms obtained by isothermal titration calorimetry for the interaction of 36 with hgal-1 and hgal-3-CRD. | ||
The ITC data confirmed that the binding affinity for hgal-3-CRD decreased when moving from the bis(fluorophenyl) analog TD139 to the diphenyl analog 37 and the differocene analog 36 (Table 4, entries 8–10). This trend was consistent with the FP assay. Compared to diphenyl analog 37 (entry 3), bis(fluorophenyl) inhibitor TD139 (entry 2) displayed a higher binding enthalpy to hgal-1 (−16.9 vs. −9.6 kcal mol−1) accompanied by a significant increase in the entropic penalty (8.0 vs. 0.7 kcal mol−1), leading to a perfect enthalpy-entropy compensation with a negligible change in affinity for hgal-1. Binding of TD139 to hgal-3-CRD also showed a gain in enthalpy relative to diphenyl analog 37 (entries 8 and 9, −16.4 vs. −11.2 kcal mol−1). However, the increased entropic penalty (5.8 vs. 1.7 kcal mol−1) did not fully compensate for this gain, resulting in a higher overall affinity of TD139 to hgal-3-CRD. Thus, the ITC data suggests that while the fluorine substituent in TD139 interacts with the protein backbone in both galectins,12b it produces a significant gain in affinity only in the case of hgal-3-CRD. This is probably due to better protein preorganization.
For the diferrocene complex 36, the higher enthalpy of binding to hgal-1 compared to its diphenyl analog 37 (−12.8 vs. −9.6 kcal mol−1) was completely compensated for by the increased entropic penalty of 36 (3.8 vs. 0.7 kcal mol−1) leading to virtually identical binding affinities of compounds 36 and 37 for hgal-1 (entries 3 and 4). In contrast, the entropic penalty of diferrocene 36 relative to its diphenyl counterpart 37 (4.4 vs. 1.7 kcal mol−1) exceeded the enthalpy gain (−12.7 vs. −11.2 kcal mol−1) in binding to hgal-3-CRD, resulting in a significantly lower affinity of 36 for this galectin (entries 9 and 10), and hence selectivity for hgal-1 over hgal-3. We interpret these ITC data in terms of an entropically unfavorable structural transition that hgal-3-CRD has to undergo to accommodate the bulky ferrocene moiety, which outweighs any attractive interaction. Asymmetrically substituted mono-ferrocene analogs 41 and 42 showed binding behavior similar to TD139, with significantly higher affinity for hgal-3-CRD than for hgal-1 (Table 4, entries 5, 6, 11, and 12), suggesting that hgal-3-CRD accommodates one ferrocene group in inhibitors 41 or 42 more effectively than two ferrocenes in compound 36. This was further investigated by computational modeling and 19F NMR spectroscopy.
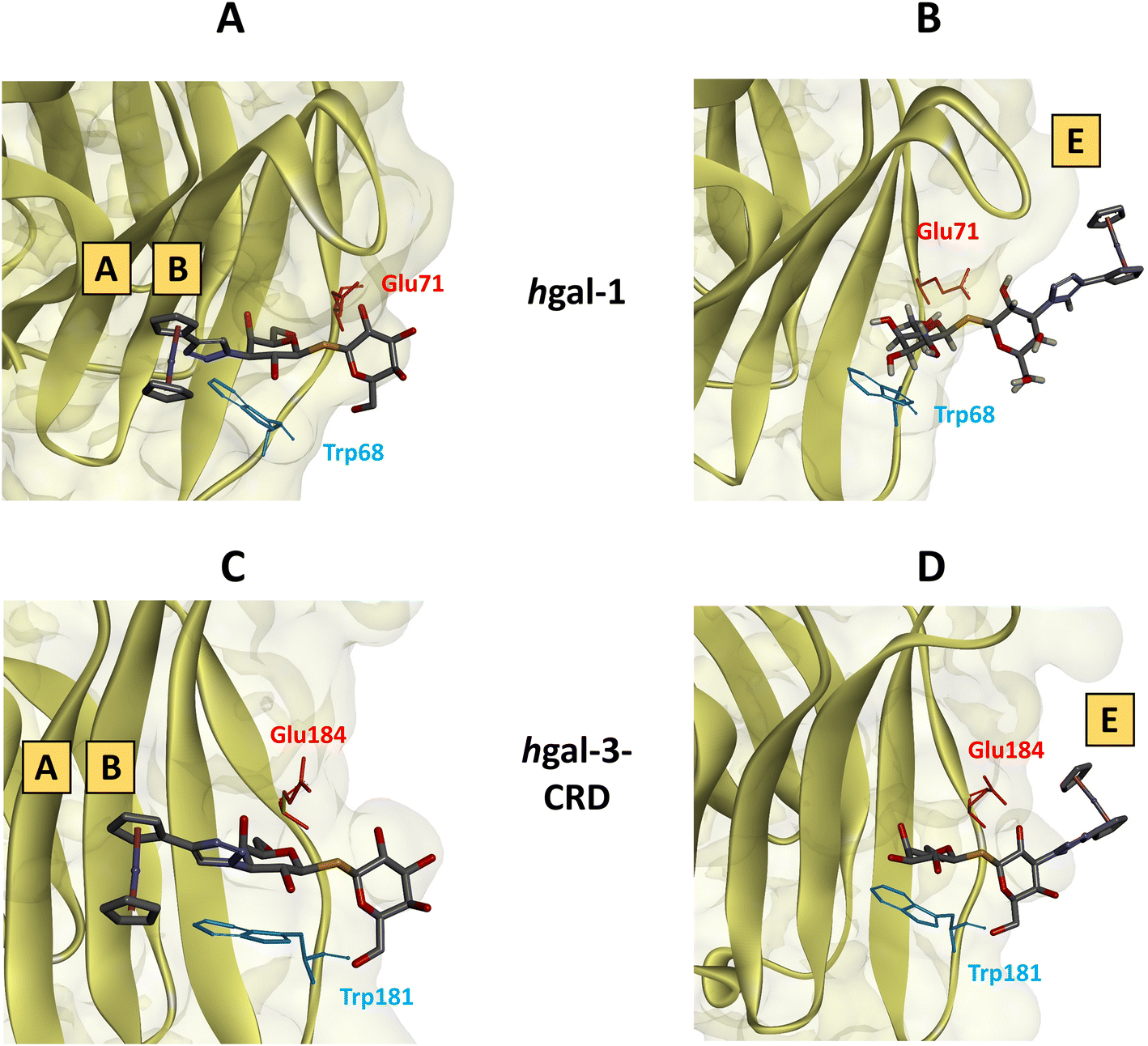 | ||
| Fig. 6 Geometries of compound 34 bound to hgal-1 or -3-CRD derived from molecular dynamics calculations showing characteristic amino acid residues (Glu71 and Trp68 for hgal-1, Glu184 and Trp181 for hgal-3-CRD) interacting with the hexopyranose rings. (A) Ferrocene moiety in subsites A and B of hgal-1. (B) Ferrocene moiety is in subsite E of hgal-1. (C) Ferrocene moiety in subsites A and B of hgal-3-CRD. (D) Ferrocene moiety in subsite E of hgal-3-CRD. | ||
| Entry | Compounds | Binding mode (subsites in which the ferrocene moiety is positioned) | ΔHbind (kcal mol−1) hgal-1 | ΔHbind (kcal mol−1), hgal-3-CRD |
|---|---|---|---|---|
| 1 |

|
A and B (Fig. 6A and C) | −16.9 | −16.3 |
| 2 | E (Fig. 6B and D) | −8.4 | −25.8 | |
| 3 |

|
A and B, and E (Fig. S78–S79 in ESI†) | −34.1 | −30.4 |
| 4 |

|
A and B (Fig. S80 and S82 in ESI†) | −32.3 | −30.4 |
| 5 | E (Fig. S81 and S83 in ESI†) | −27.3 | −32.9 |
Notwithstanding the aforementioned inaccuracies and the absence of the entropic penalty from the calculations, the determined binding enthalpies correlated consistently with the experimentally determined affinities. Accordingly, complex 36 demonstrated the highest binding enthalpy of all calculated disaccharides in binding to hgal-1 (−34.1 kcal mol−1), along with relatively strong binding to hgal-3-CRD (−30.4 kcal mol−1). These results are consistent with the ITC data, where complex 36 displayed a similar enthalpy change in binding to both galectins, whereas the decrease in affinity to hgal-3-CRD stemmed from a higher entropic penalty (Table 4, entries 4 and 10). Notably, complexes 34 and 42 exhibited significantly higher binding enthalpies in binding to hgal-1 when the ferrocene moiety occupied subsites A and B. Conversely, the preferred binding mode of these complexes to hgal-3-CRD positioned the ferrocene moiety in subsite E. Thus, the MD simulation suggests that hgal-1 accommodates the bulky ferrocene moiety in subsites A and B more efficiently than hgal-3-CRD.
 | ||
| Fig. 7 19F{1H} NMR spectra of ligands 28, 42, and TD139 in the presence of hgal-1 or hgal-3-CRD, pink arrows indicate resonances of fluorine in subsite E, purple arrows indicate resonances of fluorine in subsites A/B. Except for the combination 28 with hgal-1, all these spectra were acquired using an excess of the galectin over the ligand. | ||
To facilitate the interpretation of the 19F{1H} NMR spectra of protein-bound TD139, we performed the same NMR experiments with 3′-fluorophenyl N-acetyllactosamine 28. This ligand showed only one 19F resonance of its bound state upon addition of either hgal-1 or hgal-3-CRD (Fig. 7 blue spectra, Fig. S86 and S89 in ESI†). Because disaccharide 28 is an unsymmetrical LacNAc analog, the galactopyranoside ring can only occupy subsite C with the 3′-fluorophenyl ring pointing toward subsites A/B.7b,12a,50,58 Therefore, its 19F resonance was assigned to the fluorophenyl-triazole group located in subsites A/B. The chemical shift of the bound state 19F resonance in compound 28 closely matched one of the two resonances of bound TD139 in the presence of either hgal-1 or hgal-3-CRD (Fig. 7 red vs. blue spectra). Therefore, this resonance of TD139 was also assigned to the fluorophenyl-triazole group positioned in subsites A/B. Consequently, the remaining broad signal at δ = −115.01 ppm of TD139 bound to hgal-1 (Fig. 7, hgal-1, pink label) was assigned to the fluorophenyl moiety occupying subsite E. Similarly, the more downfield-shifted 19F resonance of TD139 bound to the hgal-3-CRD (Fig. 7, hgal-3-CRD, pink label) was also assigned to the fluorophenyl-triazole moiety in subsite E. It should be noted that this assignment is based on the assumption that the differences between the compounds 28 and TD139 do not affect the interactions of the fluorine atom with the binding subsites A/B or E and thus its chemical shift in these subsites.
In principle, the ferrocene moiety in compound 42 can occupy subsites A/B, or subsite E in both galectins, leading to two distinct binding modes (Fig. 6). Indeed, the 19F{1H} NMR spectrum of 42 in the presence of hgal-1 showed two bound state 19F NMR signals (Fig. 7, hgal-1, green spectrum, Fig. S87 in ESI†), whose chemical shifts matched the two bound state resonances of TD139 with hgal-1 (Fig. 7, hgal-1, red spectrum). Therefore, the fluorophenyl group in 42 occupies both subsites A/B and subsite E during recognition by hgal-1, and consequently the ferrocene group is likely to occupy these two subsites as well. Conversely, disaccharide 42 exhibited only a single bound state 19F resonance (Fig. 7, hgal-3-CRD, green spectrum, Fig. S90 in ESI†) in the presence of hgal-3-CRD, similar to compound 28 (Fig. 7, hgal-3-CRD, blue spectrum), suggesting that the fluorophenyl substituent of 42 interacts only with subsites A/B during recognition by hgal-3-CRD. Thus, the bulky ferrocene moiety is unlikely to occupy subsites A/B in hgal-3-CRD and compound 42 prefers to bind hgal-3-CRD through the binding mode with the ferrocene moiety located in subsite E, which is consistent with computational modeling (Table 5, entries 4 and 5).
Cytotoxicity
Ferrocene itself is not significantly toxic.20b Compounds formed by the insertion of ferrocene into bioactive compounds to enhance a particular bioactivity generally showed low toxicity.59 However, we can also find examples of highly cytotoxic ferrocene compounds60 including ferrocene-containing glycomimetics.61 Therefore, we screened the in vitro cytotoxicity of the prepared organometallic hybrid inhibitors against human ovarian cancer cell lines A2780 and SK-OV-3, human triple-negative breast cancer cell line MDA-MB-231, and human embryonic kidney non-malignant cell line HEK-293. A MTT cell viability assay was performed after 72 h of treatment and cell viability was expressed as IC50 values (Table 6). In this initial screening, we did not attempt to correlate the cytotoxicity of inhibitors with galectin expression in the cells, which is reserved for a detailed follow-up study. The tested disaccharides showed minimal or only weak cytotoxicity towards the cancer cell lines SK-OV-3, MDA-MB-231, and the non-malignant HEK-293. Complexes 6, 8 and 34 exhibited moderate cytotoxicity towards A2780 cell line (Table 6, entries 1, 2, and 9). Notably, the potent inhibitors 36, 41 and 42 showed no significant cytotoxicity (entries 10, 12, and 13).|
|
|||||
|---|---|---|---|---|---|
| Entry | Compound | A2780 | SK-OV-3 | MDA-MB-231 | HEK-293 |
| a To facilitate orientation, the structures of compounds are indicated using the abbreviations Lac, LacNAc, and TDG for the disaccharides, and abbreviations Fc′, Ru′, 3FPh, and F3Ph for the triazole-linked moieties as defined below the Table heading. | |||||
| 1 | 6 (Lac1β-Fc′) | 85 ± 8 | >200 | 183 ± 35 | >200 |
| 2 | 8 (Lac1β-Ru′) | 69 ± 12 | >200 | >200 | >200 |
| 3 | 10 (Lac1β-OCH2CH2Fc′) | 116 ± 14 | >200 | 170 ± 5 | 198 ± 35 |
| 4 | 16 (LacNAc1β-Fc′) | >200 | >200 | >200 | >200 |
| 5 | 19 (2-Fc′-Lac1β-OMe) | 159 ± 54 | >200 | >200 | >200 |
| 6 | 25 (3′-Fc′-LacNAc1β-OMe) | >200 | >200 | >200 | >200 |
| 7 | 27 (3′-Ru′-LacNAc1β-OMe) | 161 ± 50 | >200 | >200 | 143 ± 44 |
| 8 | 30 (3′-Fc′-LacNPhth1β-OMe) | >200 | >200 | >200 | >200 |
| 9 | 34 (3′-Fc′-TDG) | 90 ± 5 | >200 | >200 | >200 |
| 10 | 36 (3,3′-diFc′-TDG) | >200 | >200 | >200 | 151 ± 11 |
| 11 | 38 (3,3′-diRu′-TDG) | >200 | >200 | 125 ± 21 | >200 |
| 12 | 41 (3-F3Ph-3′-Fc′-TDG) | 168 ± 8 | >200 | >200 | 152 ± 6 |
| 13 | 42 (3-3FPh-3′-Fc′-TDG) | >200 | >200 | >200 | >200 |
| 14 | cisPt | 1.7 ± 0.3 | 5.6 ± 1.0 | 3.8 ± 0.5 | 3.7 ± 0.6 |

Conclusion
This study reports a novel approach to the design of galectin inhibitors using disaccharides modified with ferrocenes and ruthenium arenes. We have thoroughly investigated their binding to hgal-1 and hgal-3-CRD through a series of binding assays and experiments, including fluorescence anisotropy, intrinsic fluorescence of the tryptophan residues, chronopotentiometric stripping analysis, isothermal titration microcalorimetry, molecular dynamics simulation, and the monitoring of 19F{1H} NMR resonances. Disaccharides containing a ruthenium arene complex were found to be weaker galectin binders than the corresponding ferrocene analogs. The inhibitors interacting with the galectin subsite E via the ferrocene moiety (Lac and LacNAc modified at the 1- or 2-position) exhibited an increase in affinity to both galectins similar to that observed for inhibitors with planar organic arenes at the corresponding disaccharide positions. However, the attachment of ferrocene to positions 3/3′ of the galactopyranoside ring significantly enhanced the affinity for both disaccharide scaffolds tested, N-acetyllactosamine and thiodigalactoside, and in the case of diferrocenyl-thiodigalactoside 36, surpassed the performance of the inhibitors possessing planar arenes, leading to a reversal of the inhibitor selectivity in favor of hgal-1 over hgal-3.Notably, the thiodigalactoside-based diferrocene inhibitor 36 proved to be a potent and selective hgal-1 inhibitor whereas thiodigalactoside analogs 41 and 42, which incorporate only a single ferrocene moiety, showed good affinity and selectivity for hgal-3 comparable to TD139. A preliminary hypothesis based on molecular dynamics simulations and the measurement of 19F{1H} NMR resonances attributed the selectivity of inhibitor 36 to the restrictions imposed by the ferrocene moiety when binding to hgal-3-CRD. Further experiments to elucidate the molecular basis of the unusual affinity and selectivity of this inhibitor are underway in our group. We also envisage that the series of ferrocene-containing inhibitors with varying affinities developed in this study can be used to exploit the electrochemical activity of the ferrocene moiety for the development of an electrochemical galectin binding assay, an avenue currently under active investigation in our group.
Author contributions
Vojtěch Hamala and Martin Kurfiřt contributed equally to this study. Vojtěch Hamala: conceptualization, investigation (synthesis), data curation and writing – original draft. Martin Kurfiřt: investigation (synthesis, 19F NMR experiments) and data curation. Lucie Červenková Šťastná and Jana Bernášková: investigation (NMR and HRMS analysis). Hedvika Hujerová: investigation (synthesis). Kamil Parkan: investigation (fluorescence probe synthesis). Jakub Kaminský: investigation (molecular modelling). Nina Habanová: investigation (19F NMR experiments). Jaroslav Kozák and Alžběta Magdolenová: investigation (fluorescence polarization). Martin Zavřel and Jitka Holčáková: investigation (galectins cloning and purification). Tatiana Staroňová, Veronika Ostatná, Lucie Žaloudková and Aleš Daňhel: investigation (voltammetric analysis, tryptophan fluorescence assay and chronopotentiometric stripping analysis). Petr Voňka: investigation (isothermal titration microcalorimetry). Roman Hrstka: funding acquisition, investigation and supervision. Jindřich Karban: conceptualization, funding acquisition, investigation, project administration, supervision, writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The financial support of The Czech Science Foundation is gratefully acknowledged (Grant No. 23–06115S). This work was financially supported from Specific University Research (grant No. A1 FPBT 2023 003) and by Ministry of Health, Czech Republic – conceptual development of research organization (MMCI, 00209805). The research was also supported by the project National Institute for Cancer Research (Programme EXCELES, ID Project No. LX22NPO5102) – Funded by the European Union – Next Generation EU. We also appreciate the support from Gilead Sciences, Inc., provided under the program ‘Molecules for Life’ at the Gilead Sciences & IOCB Prague Research Centre. Computational resources have been provided by the e-INFRA CZ project (ID: 90254), supported by the Ministry of Education, Youth and Sports of the Czech Republic and by the ELIXIR-CZ project (ID: 90255), part of the international ELIXIR infrastructure. Vojtech Hamala is grateful for the financial support of the Martina Roeselová Memorial Fellowship granted by the IOCB Tech Foundation. The author extends gratitude to Petr Pachl from IOCB Prague for his work on cloning and purifying human galectin-1 for the fluorescence polarization assay.References
- H.-J. Gabius, M. Cudic, T. Diercks, H. Kaltner, J. Kopitz, K. H. Mayo, P. V. Murphy, S. Oscarson, R. Roy, A. Schedlbauer, S. Toegel and A. Romero, What is the Sugar Code?, ChemBioChem, 2022, 23(13), e202100327 CrossRef CAS PubMed.
- (a) H. Rachel and L. Chang-Chun, Chapter 5 - Recent advances toward the development of inhibitors to attenuate tumor metastasis via the interruption of lectin–ligand interactions, in Adv. Carbohydr. Chem. Biochem, ed. D. Horton, Academic Press, 2013, vol. 69, pp. 125–207 Search PubMed; (b) B. A. H. Smith and C. R. Bertozzi, The clinical impact of glycobiology: targeting selectins, Siglecs and mammalian glycans, Nat. Rev. Drug Discovery, 2021, 20(3), 217–243 CrossRef CAS PubMed.
- (a) B. Ernst and J. L. Magnani, From carbohydrate leads to glycomimetic drugs, Nat. Rev. Drug Discovery, 2009, 8(8), 661–677 CrossRef CAS PubMed; (b) J. Aretz, E.-C. Wamhoff, J. Hanske, D. Heymann and C. Rademacher, Computational and Experimental Prediction of Human C-Type Lectin Receptor Druggability, Front. Immunol., 2014, 5, 323 Search PubMed; (c) E. Shanina, S. Kuhaudomlarp, E. Siebs, F. F. Fuchsberger, M. Denis, P. da Silva Figueiredo Celestino Gomes, M. H. Clausen, P. H. Seeberger, D. Rognan, A. Titz, A. Imberty and C. Rademacher, Targeting undruggable carbohydrate recognition sites through focused fragment library design, Commun. Chem., 2022, 5(1), 64 CrossRef CAS PubMed.
- (a) M. Agostino, E. Yuriev and P. A. Ramsland, A Computational Approach for Exploring Carbohydrate Recognition by Lectins in Innate Immunity, Front. Immunol., 2011, 2, 23 Search PubMed; (b) S. Leusmann, P. Menova, E. Shanin, A. Titz and C. Rademacher, Glycomimetics for the inhibition and modulation of lectins, Chem. Soc. Rev., 2023, 52(11), 3663–3740 RSC; (c) C. P. Modenutti, J. I. B. Capurro, S. Di Lella and M. A. Martí, The Structural Biology of Galectin-Ligand Recognition: Current Advances in Modeling Tools, Protein Engineering, and Inhibitor Design, Front. Chem., 2019, 7, 823 CrossRef CAS PubMed; (d) R. Hevey, Strategies for the Development of Glycomimetic Drug Candidates, Pharmaceuticals, 2019, 12(2), 55 CrossRef CAS PubMed.
- V. C. Damalanka, A. R. Maddirala and J. W. Janetka, Novel approaches to glycomimetic design: development of small molecular weight lectin antagonists, Expert Opin. Drug Discovery, 2021, 16(5), 513–536 CrossRef CAS PubMed.
- L. Johannes, R. Jacob and H. Leffler, Galectins at a glance, J. Cell Sci., 2018, 131(9), jcs208884 CrossRef PubMed.
- (a) Y.-C. Chan, H.-Y. Lin, Z. Tu, Y.-H. Kuo, S.-T. D. Hsu and C.-H. Lin, Dissecting the Structure–Activity Relationship of Galectin–Ligand Interactions, Int. J. Mol. Sci., 2018, 19(2), 392 CrossRef PubMed; (b) S. Bertuzzi, J. I. Quintana, A. Ardá, A. Gimeno and J. Jiménez-Barbero, Targeting Galectins With Glycomimetics, Front. Chem., 2020, 8, 593 CrossRef CAS PubMed; (c) C. Porciúncula-González, A. J. Cagnoni, C. Fontana, K. V. Mariño, P. Saenz-Méndez, C. Giacomini and G. Irazoqui, Structural insights in galectin-1-glycan recognition: Relevance of the glycosidic linkage and the N-acetylation pattern of sugar moieties, Biorg. Med. Chem., 2021, 44, 116309 CrossRef PubMed; (d) C. T. Öberg, H. Leffler and U. J. Nilsson, Inhibition of Galectins with Small Molecules, Chim. Int. J. Chem., 2011, 65(1), 18–23 CrossRef PubMed.
- V. Denavit, D. Lainé, T. Tremblay, J. St-Gelais and D. Giguère, Synthetic Inhibitors of Galectins: Structures and Syntheses, Trends Glycosci. Glycotechnol., 2018, 30(172), SE21–SE40 CrossRef.
- (a) R. M. Perrotta, C. A. Bach, M. Salatino and G. A. Rabinovich, Reprogramming the tumor metastasis cascade by targeting galectin-driven networks, Biochem. J., 2021, 478(3), 597–617 CrossRef CAS PubMed; (b) C.-H. Li, Y.-C. Chang, M.-H. Chan, Y.-F. Yang, S.-M. Liang and M. Hsiao, Galectins in Cancer and the Microenvironment: Functional Roles, Therapeutic Developments, and Perspectives, Biomedicines, 2021, 9(9), 1159 CrossRef CAS PubMed; (c) K. V. Mariño, A. J. Cagnoni, D. O. Croci and G. A. Rabinovich, Targeting galectin-driven regulatory circuits in cancer and fibrosis, Nat. Rev. Drug Discovery, 2023, 22(4), 295–316 CrossRef PubMed.
- D. J. Laderach and D. Compagno, Inhibition of galectins in cancer: Biological challenges for their clinical application, Front. Immunol., 2023, 13, 1104625 CrossRef PubMed.
- C. M. Arthur, M. D. Baruffi, R. D. Cummings and S. R. Stowell, Evolving Mechanistic Insights into Galectin Functions, in Galectins: Methods and Protocols, ed. S. R. Stowell, R. D. Cummings, Springer New York, New York, NY, 2015, pp. 1–35 Search PubMed.
- (a) M. F. López-Lucendo, D. Solís, S. André, J. Hirabayashi, K.-i. Kasai, H. Kaltner, H.-J. Gabius and A. Romero, Growth-regulatory Human Galectin-1: Crystallographic Characterisation of the Structural Changes Induced by Single-site Mutations and their Impact on the Thermodynamics of Ligand Binding, J. Mol. Biol., 2004, 343(4), 957–970 CrossRef PubMed; (b) T.-J. Hsieh, H.-Y. Lin, Z. Tu, T.-C. Lin, S.-C. Wu, Y.-Y. Tseng, F.-T. Liu, S.-T. D. Hsu and C.-H. Lin, Dual thio-digalactoside-binding modes of human galectins as the structural basis for the design of potent and selective inhibitors, Sci. Rep., 2016, 6(1), 29457 CrossRef CAS PubMed.
- (a) K. Peterson, P. M. Collins, X. Huang, B. Kahl-Knutsson, S. Essén, F. R. Zetterberg, S. Oredsson, H. Leffler, H. Blanchard and U. J. Nilsson, Aromatic heterocycle galectin-1 interactions for selective single-digit nM affinity ligands, RSC Adv., 2018, 8(44), 24913–24922 RSC; (b) F. R. Zetterberg, K. Peterson, U. J. Nilsson, K. Andreasson Dahlgren, C. Diehl, I. Holyer, M. Hakansson, A. Khabut, B. Kahl-Knutson, H. Leffler, A. C. MacKinnon, J. A. Roper, R. J. Slack, R. Zarrizi and A. Pedersen, Discovery of the Selective and Orally Available Galectin-1 Inhibitor GB1908 as a Potential Treatment for Lung Cancer, J. Med. Chem., 2024, 67(11), 9374–9388 CrossRef CAS PubMed.
- T. Delaine, P. Collins, A. MacKinnon, G. Sharma, J. Stegmayr, V. K. Rajput, S. Mandal, I. Cumpstey, A. Larumbe, B. A. Salameh, B. Kahl-Knutsson, H. van Hattum, M. van Scherpenzeel, R. J. Pieters, T. Sethi, H. Schambye, S. Oredsson, H. Leffler, H. Blanchard and U. J. Nilsson, Galectin-3-Binding Glycomimetics that Strongly Reduce Bleomycin-Induced Lung Fibrosis and Modulate Intracellular Glycan Recognition, ChemBioChem, 2016, 17(18), 1759–1770 CrossRef CAS PubMed.
- R. Paulini, K. Müller and F. Diederich, Orthogonal Multipolar Interactions in Structural Chemistry and Biology, Angew. Chem., Int. Ed., 2005, 44(12), 1788–1805 CrossRef CAS PubMed.
- K. Peterson, R. Kumar, O. Stenström, P. Verma, P. R. Verma, M. Håkansson, B. Kahl-Knutsson, F. Zetterberg, H. Leffler, M. Akke, D. T. Logan and U. J. Nilsson, Systematic Tuning of Fluoro-galectin-3 Interactions Provides Thiodigalactoside Derivatives with Single-Digit nM Affinity and High Selectivity, J. Med. Chem., 2018, 61(3), 1164–1175 CrossRef CAS PubMed.
- (a) F. R. Zetterberg, A. MacKinnon, T. Brimert, L. Gravelle, R. E. Johnsson, B. Kahl-Knutson, H. Leffler, U. J. Nilsson, A. Pedersen, K. Peterson, J. A. Roper, H. Schambye, R. J. Slack and S. Tantawi, Discovery and Optimization of the First Highly Effective and Orally Available Galectin-3 Inhibitors for Treatment of Fibrotic Disease, J. Med. Chem., 2022, 65(19), 12626–12638 CrossRef CAS PubMed; (b) C. Liu, P. R. Jalagam, J. Feng, W. Wang, T. Raja, M. R. Sura, R. K. V. L. P. Manepalli, B. R. Aliphedi, S. Medavarapu, S. K. Nair, V. Muthalagu, R. Natesan, A. Gupta, B. Beno, M. Panda, K. Ghosh, J. K. Shukla, H. Sale, P. Haldar, N. Kalidindi, D. Shah, D. Patel, A. Mathur, B. A. Ellsworth, D. Cheng and A. Regueiro-Ren, Identification of Monosaccharide Derivatives as Potent, Selective, and Orally Bioavailable Inhibitors of Human and Mouse Galectin-3, J. Med. Chem., 2022, 65(16), 11084–11099 CrossRef CAS PubMed; (c) F. R. Zetterberg, K. Peterson, R. E. Johnsson, T. Brimert, M. Håkansson, D. T. Logan, H. Leffler and U. J. Nilsson, Monosaccharide Derivatives with Low-Nanomolar Lectin Affinity and High Selectivity Based on Combined Fluorine–Amide, Phenyl–Arginine, Sulfur–π, and Halogen Bond Interactions, ChemMedChem, 2018, 13(2), 133–137 CrossRef CAS PubMed; (d) F. R. Zetterberg, C. Diehl, M. Håkansson, B. Kahl-Knutson, H. Leffler, U. J. Nilsson, K. Peterson, J. A. Roper and R. J. Slack, Discovery of Selective and Orally Available Galectin-1 Inhibitors, J. Med. Chem., 2023, 66(24), 16980–16990 CrossRef CAS PubMed.
- Galecto Announces Topline Results from Phase 2b GALACTIC-1 Trial of GB0139 for the Treatment of Idiopathic Pulmonary Fibrosis. GLOBE NEWSWIRE: Boston, 2023. https://ir.galecto.com/news-releases/news-release-details/galecto-announces-topline-results-phase-2b-galactic-1-trial/ (accessed August 2024).
- (a) M. Alvala, N. S. Goud, P. S. L. Soukya, M. Ghouse, D. Komal and R. Alvala, Human galectin-1 and its inhibitors: Privileged target for cancer and HIV, Mini-Rev. Med. Chem., 2019, 19(16), 1369–1378 CrossRef PubMed; (b) N. S. Goud and A. Bhattacharya, Human galectin-1 in multiple cancers: A privileged molecular target in oncology, Mini-Rev. Med. Chem., 2021, 21(15), 2169–2186 CrossRef CAS PubMed; (c) I. Camby, M. Le Mercier, F. Lefranc and R. Kiss, Galectin-1: a small protein with major functions, Glycobiology, 2006, 16(11), 137R–157R CrossRef CAS PubMed; (d) L. S. Lau, N. B. B. Mohammed and C. J. Dimitroff, Decoding Strategies to Evade Immunoregulators Galectin-1, -3, and -9 and Their Ligands as Novel Therapeutics in Cancer Immunotherapy, Int. J. Mol. Sci., 2022, 23(24), 15554 CrossRef CAS PubMed; (e) A. Sethi, S. Sanam, R. Alvala and M. Alvala, An updated patent review of galectin-1 and galectin-3 inhibitors and their potential therapeutic applications (2016−present), Expert Opin. Ther. Pat., 2021, 31(8), 709–721 CrossRef CAS PubMed; (f) C. A. Orozco, N. Martinez-Bosch, P. E. Guerrero, J. Vinaixa, T. Dalotto-Moreno, M. Iglesias, M. Moreno, M. Djurec, F. Poirier, H.-J. Gabius, M. E. Fernandez-Zapico, R. F. Hwang, C. Guerra, G. A. Rabinovich and P. Navarro, Targeting galectin-1 inhibits pancreatic cancer progression by modulating tumor–stroma crosstalk, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(16), E3769–E3778 CrossRef CAS PubMed; (g) L. Astorgues-Xerri, M. E. Riveiro, A. Tijeras-Raballand, M. Serova, C. Neuzillet, S. Albert, E. Raymond and S. Faivre, Unraveling galectin-1 as a novel therapeutic target for cancer, Cancer Treat. Rev., 2014, 40(2), 307–319 CrossRef CAS PubMed; (h) K. Ito, K. Stannard, E. Gabutero, A. M. Clark, S.-Y. Neo, S. Onturk, H. Blanchard and S. J. Ralph, Galectin-1 as a potent target for cancer therapy: role in the tumor microenvironment, Cancer Metastasis Rev., 2012, 31(3), 763–778 CrossRef CAS PubMed; (i) M. Salatino, D. O. Croci, G. A. Bianco, J. M. Ilarregui, M. A. Toscano and G. A. Rabinovich, Galectin-1 as a potential therapeutic target in autoimmune disorders and cancer, Expert Opin. Biol. Ther., 2008, 8(1), 45–57 CrossRef CAS PubMed; (j) G. A. Rabinovich, Galectin-1 as a potential cancer target, Br. J. Cancer, 2005, 92(7), 1188–1192 CrossRef CAS PubMed; (k) C. St-Pierre, M. Ouellet, D. Giguère, R. Ohtake, R. Roy, S. Sato and M. J. Tremblay, Galectin-1-Specific Inhibitors as a New Class of Compounds To Treat HIV-1 Infection, Antimicrob. Agents Chemother., 2012, 56(1), 154–162 CrossRef CAS PubMed.
- (a) M. Dörr and E. Meggers, Metal complexes as structural templates for targeting proteins, Curr. Opin. Chem. Biol., 2014, 19, 76–81 CrossRef PubMed; (b) M. Patra and G. Gasser, The medicinal chemistry of ferrocene and its derivatives, Nat. Rev. Chem., 2017, 1(9), 0066 CrossRef CAS; (c) G. Jaouen, A. Vessières and S. Top, Ferrocifen type anti cancer drugs, Chem. Soc. Rev., 2015, 44(24), 8802–8817 RSC.
- A. Wieczorek, A. Błauż, A. Żal, H. J. Arabshahi, J. Reynisson, C. G. Hartinger, B. Rychlik and D. Plażuk, Ferrocenyl Paclitaxel and Docetaxel Derivatives: Impact of an Organometallic Moiety on the Mode of Action of Taxanes, Chem. – Eur. J., 2016, 22(32), 11413–11421 CrossRef CAS PubMed.
- A. Wieczorek, A. Błauż, J. Zakrzewski, B. Rychlik and D. Plażuk, Ferrocenyl 2,5-Piperazinediones as Tubulin-Binding Organometallic ABCB1 and ABCG2 Inhibitors Active against MDR Cells, ACS Med. Chem. Lett., 2016, 7(6), 612–617 CrossRef CAS PubMed.
- K. Schlotter, F. Boeckler, H. Hübner and P. Gmeiner, Fancy Bioisosteres: Metallocene-Derived G-Protein-Coupled Receptor Ligands with Subnanomolar Binding Affinity and Novel Selectivity Profiles, J. Med. Chem., 2005, 48(11), 3696–3699 CrossRef CAS PubMed.
- M. C. Martos-Maldonado, I. Quesada-Soriano, L. García-Fuentes and A. Vargas-Berenguel, Multivalent Lactose–Ferrocene Conjugates Based on Poly (Amido Amine) Dendrimers and Gold Nanoparticles as Electrochemical Probes for Sensing Galectin-3, Nanomaterials, 2020, 10(2), 203 CrossRef CAS PubMed.
- (a) A. Dahlqvist, H. Leffler and U. J. Nilsson, C1-Galactopyranosyl Heterocycle Structure Guides Selectivity: Triazoles Prefer Galectin-1 and Oxazoles Prefer Galectin-3, ACS Omega, 2019, 4(4), 7047–7053 CrossRef CAS; (b) R. Roy, Y. Cao, H. Kaltner, N. Kottari, T. C. Shiao, K. Belkhadem, S. André, J. C. Manning, P. V. Murphy and H.-J. Gabius, Teaming up synthetic chemistry and histochemistry for activity screening in galectin-directed inhibitor design, Histochem. Cell Biol., 2017, 147(2), 285–301 CrossRef CAS PubMed; (c) D. Giguère, S. Sato, C. St-Pierre, S. Sirois and R. Roy, Aryl O- and, S-galactosides and lactosides as specific inhibitors of human galectins-1 and -3: Role of electrostatic potential at O-3, Bioorg. Med. Chem. Lett., 2006, 16(6), 1668–1672 CrossRef PubMed; (d) P. Sörme, Y. Qian, P.-G. Nyholm, H. Leffler and U. J. Nilsson, Low Micromolar Inhibitors of Galectin-3 Based on 3′-Derivatization of N-Acetyllactosamine, ChemBioChem, 2002, 3(2–3), 183–189 CrossRef.
- (a) B. A. Salameh, I. Cumpstey, A. Sundin, H. Leffler and U. J. Nilsson, 1H-1,2,3-Triazol-1-yl thiodigalactoside derivatives as high affinity galectin-3 inhibitors, Biorg. Med. Chem., 2010, 18(14), 5367–5378 CrossRef CAS PubMed; (b) H. Blanchard, K. Bum-Erdene and M. W. Hugo, Inhibitors of Galectins and Implications for Structure-Based Design of Galectin-Specific Therapeutics, Aust. J. Chem., 2014, 67(12), 1763–1779 CrossRef CAS; (c) M. van Scherpenzeel, E. E. Moret, L. Ballell, R. M. J. Liskamp, U. J. Nilsson, H. Leffler and R. J. Pieters, Synthesis and Evaluation of New Thiodigalactoside-Based Chemical Probes to Label Galectin-3, ChemBioChem, 2009, 10(10), 1724–1733 CrossRef CAS PubMed; (d) H. van Hattum, H. M. Branderhorst, E. E. Moret, U. J. Nilsson, H. Leffler and R. J. Pieters, Tuning the Preference of Thiodigalactoside- and Lactosamine-Based Ligands to Galectin-3 over Galectin-1, J. Med. Chem., 2013, 56(3), 1350–1354 CrossRef CAS PubMed.
- M. Lamac, M. Horacek, L. C. St'astna, J. Karban, L. Sommerova, H. Skoupilova, R. Hrstka and J. Pinkas, Harmless glucose-modified ruthenium complexes suppressing cell migration of highly invasive cancer cell lines, Appl. Organomet. Chem., 2020, 34(1), e5318 CrossRef CAS.
- S. R. Rauthu, T. C. Shiao, S. André, M. C. Miller, É Madej, K. H. Mayo, H.-J. Gabius and R. Roy, Defining the Potential of Aglycone Modifications for Affinity/Selectivity Enhancement against Medically Relevant Lectins: Synthesis, Activity Screening, and HSQC-Based NMR Analysis, ChemBioChem, 2015, 16(1), 126–139 CrossRef CAS PubMed.
- T. Machida, K. Lang, L. Xue, J. W. Chin and N. Winssinger, Site-Specific Glycoconjugation of Protein via Bioorthogonal Tetrazine Cycloaddition with a Genetically Encoded trans-Cyclooctene or Bicyclononyne, Bioconjugate Chem., 2015, 26(5), 802–806 CrossRef CAS PubMed.
- R. Šardzík, G. T. Noble, M. J. Weissenborn, A. Martin, S. J. Webb and S. L. Flitsch, Preparation of aminoethyl glycosides for glycoconjugation, Beilstein J. Org. Chem., 2010, 6, 699–703 CrossRef PubMed.
- G. Lian, X. Zhang and B. Yu, Thioglycosides in Carbohydrate Research, Carbohydr. Res., 2015, 403, 13–22 CrossRef CAS PubMed.
- W. Bröder and H. Kunz, A new method of anomeric protection and activation based on the conversion of glycosyl azides into glycosyl fluorides, Carbohydr. Res., 1993, 249(1), 221–241 CrossRef PubMed.
- J. M. Jacobson, P. I. Kitov and D. R. Bundle, The synthesis of a multivalent heterobifunctional ligand for specific interaction with Shiga toxin 2 produced by E. coli O157:H7, Carbohydr. Res., 2013, 378, 4–14 CrossRef CAS PubMed.
- M. Niemietz, L. Perkams, J. Hoffman, S. Eller and C. Unverzagt, Selective oxidative debenzylation of mono- and oligosaccharides in the presence of azides, Chem. Commun., 2011, 47(37), 10485–10487 RSC.
- O. Kanie, S. C. Crawley, M. M. Palcic and O. Hindsgaul, Acceptor-substrate recognition by N-acetylglucosaminyltransferase-V: Critical role of the 4′′-hydroxyl group in β-D-GlcpNAc-(1 → 2)-α-D-Manp(1 → 6)-β-D-Glcp-OR, Carbohydr. Res., 1993, 243(1), 139–164 CrossRef CAS PubMed.
- M. Kurfiřt, M. Dračínský, L. Červenková Šťastná, P. Cuřínová, V. Hamala, M. Hovorková, P. Bojarová and J. Karban, Selectively Deoxyfluorinated N-Acetyllactosamine Analogues as 19F NMR Probes to Study Carbohydrate-Galectin Interactions, Chem. – Eur. J., 2021, 27(51), 13040–13051 CrossRef PubMed.
- J. Alais and S. David, Preparation of disaccharides having a β-D-mannopyranosyl group from N-phthaloyllactosamine derivatives by double or triple SN2 substitution, Carbohydr. Res., 1990, 201(1), 69–77 CrossRef CAS PubMed.
- C. Xia, W. Zhang, Y. Zhang, W. Chen, J. Nadas, R. Severin, R. Woodward, B. Wang, X. Wang, M. Kronenberg and P. G. Wang, The Roles of 3′ and 4′ Hydroxy Groups in α-Galactosylceramide Stimulation of Invariant Natural Killer T
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Cells, ChemMedChem, 2009, 4(11), 1810–1815 CrossRef CAS PubMed.
Cells, ChemMedChem, 2009, 4(11), 1810–1815 CrossRef CAS PubMed. - J. St-Gelais, V. Denavit and D. Giguere, Efficient synthesis of a galectin inhibitor clinical candidate (TD139) using a Payne rearrangement/azidation reaction cascade, Org. Biomol. Chem., 2020, 18(20), 3903–3907 RSC.
- J. St-Gelais, C. Leclerc and D. Giguère, Synthesis of fluorinated thiodigalactoside analogues, Carbohydr. Res., 2022, 511, 108481 CrossRef CAS PubMed.
- S. Mandal and U. J. Nilsson, Tri-isopropylsilyl thioglycosides as masked glycosyl thiol nucleophiles for the synthesis of S-linked glycosides and glyco-conjugates, Org. Biomol. Chem., 2014, 12(27), 4816–4819 RSC.
- X. Creary, A. Anderson, C. Brophy, F. Crowell and Z. Funk, Method for Assigning Structure of 1,2,3-Triazoles, J. Org. Chem., 2012, 77(19), 8756–8761 CrossRef CAS PubMed.
- D. R. van Staveren and N. Metzler-Nolte, Bioorganometallic Chemistry of Ferrocene, Chem. Rev., 2004, 104(12), 5931–5986 CrossRef CAS PubMed.
- J. Špaček, A. Daňhel, S. Hasoň and M. Fojta, Label-free detection of canonical DNA bases, uracil and 5-methylcytosine in DNA oligonucleotides using linear sweep voltammetry at a pyrolytic graphite electrode, Electrochem. Commun., 2017, 82, 34–38 CrossRef.
- S. Ayaz, A. Shah and S. Munir, Investigation of Electron Transfer Mechanistic Pathways of Ferrocene Derivatives in Droplet at Carbon Electrode, C, 2022, 8(3), 45 CAS.
- P. Sörme, B. Kahl-Knutsson, M. Huflejt, U. J. Nilsson and H. Leffler, Fluorescence polarization as an analytical tool to evaluate galectin–ligand interactions, Anal. Biochem., 2004, 334(1), 36–47 CrossRef PubMed.
- J. Dumic, S. Dabelic and M. Flögel, Galectin-3: An open-ended story, Biochim. Biophys. Acta, Gen. Subj., 2006, 1760(4), 616–635 CrossRef CAS PubMed.
- I. Cumpstey, E. Salomonsson, A. Sundin, H. Leffler and U. J. Nilsson, Double Affinity Amplification of Galectin–Ligand Interactions through Arginine–Arene Interactions: Synthetic, Thermodynamic, and Computational Studies with Aromatic Diamido Thiodigalactosides, Chem. – Eur. J., 2008, 14(14), 4233–4245 CrossRef CAS PubMed.
- I. Cumpstey, E. Salomonsson, A. Sundin, H. Leffler and U. J. Nilsson, Studies of Arginine–Arene Interactions through Synthesis and Evaluation of a Series of Galectin-Binding Aromatic Lactose Esters, ChemBioChem, 2007, 8(12), 1389–1398 CrossRef CAS PubMed.
- P. Sörme, P. Arnoux, B. Kahl-Knutsson, H. Leffler, J. M. Rini and U. J. Nilsson, Structural and Thermodynamic Studies on Cation−Π Interactions in Lectin−Ligand Complexes: High-Affinity Galectin-3 Inhibitors through Fine-Tuning of an Arginine−Arene Interaction, J. Am. Chem. Soc., 2005, 127(6), 1737–1743 CrossRef PubMed.
- (a) P. Sindrewicz, X. Li, E. A. Yates, J. E. Turnbull, L.-Y. Lian and L.-G. Yu, Intrinsic tryptophan fluorescence spectroscopy reliably determines galectin-ligand interactions, Sci. Rep., 2019, 9(1), 11851 CrossRef PubMed; (b) A. Göhler, C. Büchner, S. André, S. Doose, H. Kaltner and H. J. Gabius, Sensing ligand binding to a clinically relevant lectin by tryptophan fluorescence anisotropy, Analyst, 2011, 136(24), 5270–5276 RSC.
- (a) E. Paleček, J. Tkáč, M. Bartošík, T. Bertók, V. Ostatná and J. Paleček, Electrochemistry of Nonconjugated Proteins and Glycoproteins. Toward Sensors for Biomedicine and Glycomics, Chem. Rev., 2015, 115(5), 2045–2108 CrossRef PubMed; (b) H. Černocká, P. Vonka, V. Kasalová, L. Sommerova, V. Vandova, R. Hrstka and V. Ostatna, AGR2-AGR3 hetero-oligomeric complexes: Identification and characterization, Bioelectrochemistry, 2021, 140, 107808 CrossRef PubMed.
- V. Ostatná, V. Kasalová-Vargová, L. Kékedy-Nagy, H. Černocká and E. E. Ferapontova, Chronopotentiometric sensing of specific interactions between lysozyme and the DNA aptamer, Bioelectrochemistry, 2017, 114, 42–47 CrossRef PubMed.
- V. Vargová, R. Helma, E. Paleček and V. Ostatná, Electrochemical sensing of concanavalin A and ovalbumin interaction in solution, Anal. Chim. Acta, 2016, 935, 97–103 CrossRef PubMed.
- K. J. Bowers, D. E. Chow, H. Xu, R. O. Dror, M. P. Eastwood, B. A. Gregersen, J. L. Klepeis, I. Kolossvary, M. A. Moraes, F. D. Sacerdoti, J. K. Salmon, Y. Shan and D. E. ShawIn Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters, SC ‘06: Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, 11–17 Nov. 2006; 2006; pp 43–43.
- Schrödinger Release 2024–1, Desmond Molecular Dynamics System, Schrödinger, Inc, New York, 2024 Search PubMed.
- J. J. P. Stewart, MOPAC2016, Stewart Computational Chemistry: Colorado Springs, CO, USA, 2016 Search PubMed.
- (a) C. Atmanene, C. Ronin, S. Téletchéa, F.-M. Gautier, F. Djedaïni-Pilard, F. Ciesielski, V. Vivat and C. Grandjean, Biophysical and structural characterization of mono/di-arylated lactosamine derivatives interaction with human galectin-3, Biochem. Biophys. Res. Commun., 2017, 489(3), 281–286 CrossRef CAS PubMed; (b) J. Seetharaman, A. Kanigsberg, R. Slaaby, H. Leffler, S. H. Barondes and J. M. Rini, X-ray Crystal Structure of the Human Galectin-3 Carbohydrate Recognition Domain at 2.1 Å Resolution*, J. Biol. Chem., 1998, 273(21), 13047–13052 CrossRef CAS PubMed; (c) A. Gimeno, S. Delgado, P. Valverde, S. Bertuzzi, M. A. Berbís, J. Echavarren, A. Lacetera, S. Martín-Santamaría, A. Surolia, F. J. Cañada, J. Jiménez-Barbero and A. Ardá, Minimizing the Entropy Penalty for Ligand Binding: Lessons from the Molecular Recognition of the Histo Blood-Group Antigens by Human Galectin-3, Angew. Chem., Int. Ed., 2019, 58(22), 7268–7272 CrossRef CAS PubMed; (d) S. Bertuzzi, A. Gimeno, R. Núñez-Franco, G. Bernardo-Seisdedos, S. Delgado, G. Jiménez-Osés, O. Millet, J. Jiménez-Barbero and A. Ardá, Unravelling the Time Scale of Conformational Plasticity and Allostery in Glycan Recognition by Human Galectin-1, Chem. – Eur. J., 2020, 26(67), 15643–15653 CrossRef CAS PubMed.
- R. Wang, H. Chen, W. Yan, M. Zheng, T. Zhang and Y. Zhang, Ferrocene-containing hybrids as potential anticancer agents: Current developments, mechanisms of action and structure-activity relationships, Eur. J. Med. Chem., 2020, 190, 112109 CrossRef CAS PubMed.
- B. Sharma and V. Kumar, Has Ferrocene Really Delivered Its Role in Accentuating the Bioactivity of Organic Scaffolds?, J. Med. Chem., 2021, 64(23), 16865–16921 CrossRef CAS PubMed.
- (a) A. Hottin, F. Dubar, A. Steenackers, P. Delannoy, C. Biot and J.-B. Behr, Iminosugar–ferrocene conjugates as potential anticancer agents, Org. Biomol. Chem., 2012, 10(29), 5592–5597 RSC; (b) T. Hodik, M. Lamac, L. C. Stastna, J. Karban, L. Koubkova, R. Hrstka, I. Cisarova and J. Pinkas, Titanocene Dihalides and Ferrocenes Bearing a Pendant alpha-D-Xylofuranos-5-yl or alpha-D-Ribofuranos-5-yl Moiety. Synthesis, Characterization, and Cytotoxic Activity, Organometallics, 2014, 33(8), 2059–2070 CrossRef CAS; (c) T. Hodik, M. Lamac, L. C. Stastna, P. Curinova, J. Karban, H. Skoupilova, R. Hrstka, I. Cisarova, R. Gyepes and J. Pinkas, Improving cytotoxic properties of ferrocenes by incorporation of saturated N-heterocycles, J. Organomet. Chem., 2017, 846, 141–151 CrossRef CAS; (d) R. Trivedi, S. B. Deepthi, L. Giribabu, B. Sridhar, P. Sujitha, C. Ganesh Kumar and K. V. S. Ramakrishna, Synthesis, crystal structure, electronic spectroscopy, electrochemistry and biological studies of carbohydrate containing ferrocene amides, Appl. Organomet. Chem., 2012, 26(7), 369–376 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qi01555j |
| This journal is © the Partner Organisations 2024 |