
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePost-synthetic modification of bis-iron(III)-μ-oxo-porphyrin prisms to enhance oxygen reduction electrocatalysis†
Daoyang
Zhang
 ,
Lauren E.
Rosch
,
Matthew R.
Crawley
and
Timothy R.
Cook
*
,
Lauren E.
Rosch
,
Matthew R.
Crawley
and
Timothy R.
Cook
*
Department of Chemistry, University at Buffalo, The State University of New York, Buffalo, New York 14260, USA. E-mail: trcook@buffalo.edu
First published on 17th July 2024
Abstract
Bis-iron(III)-μ-oxo porphyrins are known electrocatalysts for Oxygen Reduction Reaction (ORR) that favor four-electron four-proton chemistry. The facile formation of the μ-oxo motif makes forming cofacial structures straightforward unlike for other metalloporphyrins where the face-to-face geometry must be enforced by tethering groups. We have pioneered the use of coordination chemistry to obtain cofacial porphyrin ORR catalysts containing Co(II) ions. Here, we adapt our use of molecular clips for the post-synthetic modification of μ-oxo porphyrin dimers rather than as self-assembly building blocks. Although the cofacial geometry is inherent to the μ-oxo core, under ORR catalysis conditions, the dimer is rapidly cleaved, resulting in reactivity differences between traditional unclipped bis-iron(III)-μ-oxo porphyrins and our post-synthetically tethered architectures. We demonstrate our approach using two molecular clips that differ in length. The bis-iron(III)-μ-oxo precatalysts were characterized by 1H NMR, ESI-MS, and molecular modeling to support the formation of cofacial prisms. Catalytic activity was studied electrochemically using cyclic voltammetry and hydrodynamic voltammetry. Whereas untethered bis-iron(III)-μ-oxo porphyrins are limited to heterogenous catalysis so that the dimeric structure is not lost upon the removal of the oxo bridge, our clipped architectures show significant catalytic current response under homogeneous conditions. Furthermore, when a shorter molecular clip is used as a post-synthetic tether, the selectivity under heterogeneous conditions is significantly enhanced: monomeric Fe(III) tetraphenylporphyrin (TPhP) generates 64.3% H2O2. When this same porphyrin is templated into a cofacial environment (Fe2O TPhP) the selectivity improves to 15.8% H2O2. When a tetrapyridyl porphyrin analogue is post-synthetically tethered through a oxalate-bridged Rh2 molecular clip, the selectivity improves further to 7.2% H2O2. In contrast, when a longer bis-hydroxybenzoquinato bridge is used in the clip, the selectivity drops back to 14.5% H2O2. Our most selective system also shows the highest current response and therefore our post-synthetic modification provides a kinetic enhancement as well. These results establish that straightforward coordination chemistry can be used for post-synthetic tuning of molecular ORR catalysts and when proper molecular clips are selected, marked enhancements to both selectivity and activity may be realized.
Introduction
Many metal enzymes feature Fe-containing sites as critical functional elements, such as the heme/Cu-His active site of cytochrome c oxidase;1 the Ni–Fe sulfur cluster in carbon monoxide dehydrogenase;2 the Fe–Mo cluster cofactor in nitrogenase3 and the heme active sites of cytochrome P450s that often act as monooxygenases.4 A common factor between all those enzymes is that they are highly active and selective for small molecule activations and furthermore, many invoke the involvement of two or more metals during catalysis. Biomimetic inspiration from these enzymes has long been the basis for the design of small molecule catalysts as structural and/or functional models.5,6Iron porphyrins have been well-studied for their ability to catalytically activate small molecules such as H2,7,8 O2,9–13 and CO2.14–19 Iron(III) porphyrins typically possess an axial anionic ligand or a μ-oxo bridging two metal sites, as shown in Fig. 1.9,20–23 Fe porphyrins have been extensively studied for the Oxygen Reduction Reaction (ORR). Homogeneous ORR catalyzed by monomeric species has shown wildly different selectivities that vary by condition and research group. For example, Fe(II) 5,10,15,20-tetrakis(4-N-methylpyridyl)porphyrin has been reported to quantitatively form H2O2, though other studies report 100% H2O. This inconsistent selectivity in aqueous conditions can be explained by a 2 + 2 mechanism, where a second catalytic cycle reduces the hydrogen peroxide further. Alternatively, an H2O2 dismutase mechanism may be active. In DMF, Mayer and coworkers have provided a detailed mechanistic analysis of ORR by Fe tetraphenyl porphyrin in which the first proton transfer is rate-limiting and selectivity for H2O is high as a result.24 In contrast, when a cofacial environment is present, molecular oxygen can undergo authentic four-electron four-proton chemistry and as such, existing bis-iron(III)-μ-oxo porphyrins are most commonly studied as heterogeneous catalysts in aqueous conditions. In solution, these dimers dissociate to monomeric species during turnover but in the solid-state, hindered molecular motion maintains the necessary cofacial clefts.
 | ||
| Fig. 1 Select examples of dinuclear iron porphyrins. | ||
One strategy to maintain a cofacial cleft independent of a μ-oxo bridge or heterogeneous conditions is to formally tether the two porphyrins. This approach has been most extensively used to increase the selectivity and activity of Co-based ORR catalysts, for example Co FTF4, which contains four amide-based covalent linkers.25–27 We have used coordination-driven self-assembly for the synthesis and systematic tuning of multi-porphyrin architectures based on pioneering designs by Guo-Xin Jin,28,29 Bruno Therrien,30,31 and Jonathan Nitschke,32,33 adapting them for ORR catalysis using Co(II) while controlling the length of the tethers,34–36 nuclearity,37 symmetry and stoichiometry,38 and rigidity.39
Studies of analogous diiron architectures are underdeveloped. Nocera and coworkers investigated O-atom transfer chemistry using so-called Pacman porphyrins where a reactive Fe(IV)-oxo could be exposed upon photolysis of the μ-oxo bridge,21,22,40–43 building upon Balch's work on untethered μ-oxo dimers that established this reactivity.42,43 Naruta and coworkers explored CO2 reduction electrocatalysis using a similar Pacman structure.23 To the best of our knowledge, there are no reports of tethered cofacial iron porphyrins as ORR catalysts despite the high selectivity of untethered Fe(III) μ-oxo dimers under heterogeneous conditions. In 2017, Tanaka and coworkers reported a cofacial porphyrin/phthalocyanine architecture that was ∼98% selective for H2O as an electrocatalyst for ORR.44
Herein, we adapt our coordination-driven self-assembly approach as a method to post-synthetically modify bis-iron(III)-μ-oxo-porphyrin prisms. There is a growing interest in externally controlling catalysis via post-synthetic coordination that does not alter the primary environment of active metal sites. For example, binding cations to crown ethers can promote phase transfer,45,46 can gate substrate access,47 and can impart conformation control.48 We now show how dinuclear molecular clips can be used for conformational control of tetrapyridylporphyrins to enforce cofacial geometries independent of μ-oxo bridges. We have fully characterized these novel architectures and established their ORR reactivity against monomeric and unclipped μ-oxo analogues under homogenous (acetonitrile) and heterogeneous (aqueous) conditions. The selectivities and kinetics of ORR were sensitive to the lengths of the molecular clips. Notably, when an oxalate-bridged molecular clip was used, four-electron four-proton chemistry was favored (92.8% H2O). This work establishes that diiron porphyrin ORR catalysts may be tuned and enhanced using straightforward post-synthetic modifications via coordination chemistry, enforcing well-defined cofacial cavities both in solution and in the solid state that are highly active and selective.
Experimental
Materials
Chemicals were purchased from commercial sources and used as received unless otherwise noted. The porphyrins were prepared following a literature procedure.21,24,49,50 Solvents were purified using a solvent-drying system (Pure Process Technology). 1H NMR spectra were acquired on a Varian Inova 500 MHz spectrometer. Chemical shifts (δ) are reported in parts per million (ppm) and referenced against the residual proton resonances of the deuterated solvent. Mass spectra were recorded on an electrospray ionization (ESI) linear ion trap (LTQ)-Orbitrap XL mass spectrometer (Thermo Fisher Scientific) or by electrospray ionization using a home-built sprayer into a Bruker SolariX 12 T FT ion cyclotron resonance mass spectrometer (12 T FT-ICR) equipped with a dual source. No precautions were taken to exclude air (O2 or water) from self-assembly reactions. UV-vis spectroelectrochemistry experiments were measured using a Pine Research WaveDriver 40 and an Avantes AvaSpec UV-vis spectrophotometer. A Pine honeycomb UV-vis spectrochemical cell consisting of a built-in platinum honeycomb working electrode and a platinum counter electrode was used. Homogeneous CV experiments were carried out in dry acetonitrile solution with 100 mM TBAPF6 as the supporting electrolyte. During the electrochemistry measurements, Ag/AgNO3 was used as a pseudo-reference electrode. At the end of measurements, a spatula tip of ferrocene was added to the solution to convert the potentials to the ferrocene/ferrocenium couple (FcH/FcH+). Heterogeneous electrochemistry experiments were carried out in DI water on catalyst inks with 0.5 M H2SO4 as the electrolyte and proton source. Ag/AgCl was used as a reference electrode. Catalyst ink film preparation: in a 2-dram vial, 1.0 μmol of dimer (Fe2O Rhoxo, Fe2O Rh-benzo, (FeTPhP)2O) or 2.0 μmol of monomer (FeTPhPCl), 5.0 mg of carbon black, 100 μL of ethanol, and 500 μL methanol were sonicated for 1 hour. The solvent was then evaporated in vacuo, followed by the addition of a 70 μL Nafion solution (5% w/w in isopropanol) and 600 μL of ethanol. The resulting suspension was sonicated for 1 hour. The Nafion catalyst ink was then pipetted onto the working electrode. When CV were acquired using a 3 mm working electrode, 2 μL was drop-cast onto the surface and allowed to stand in air until dry. In the case of the larger glassy carbon disk for the ring-disk electrode, 3 μL were used to completely cover the surface. UV-vis spectroelectrochemistry experiments were carried out in dry DMF solution with 100 mM TBABF4 as electrolyte. Ag/AgNO3 was used as a reference electrode.51,52Synthetic procedures
Results and discussion
Synthesis and characterization
The iron(III) tetrapyridyl porphyrin (FeTPyPCl) was synthesized by refluxing FeCl2 with tetrapyridyl porphyrin in DMF; the dimerization occurred within a biphasic mixture of FeTPyPCl in DCM and 1 M NaOH in H2O that was stirred at room temperature to form (FeTPyP)2O. A post-assembly modification of the dimer core with rhodium-based molecular clips prepared by following a literature procedure53 (Rhoxo clip or Rh-benzo clip) produced clipped Fe2O prisms (Scheme 1). No precautions were taken to exclude air (O2 or water) from the post-assembly reactions. | ||
| Scheme 1 | ||
NMR analysis
Monomeric Fe(III) porphyrins are paramagnetic and their 1H NMR spectra often have broad features and significant downfield shifts relative to diamagnetic species. Metallated tetrapyridyl porphyrins have three types of proton resonances arising from the pyrrolic protons, and the α- and β-pyridyl protons. The pyrrolic signal for FeTPyPCl is found at 83 ppm, as shown in Fig. S3 and S4.† Once a dimer is formed with a μ-oxo bridge, the chemical shifts are more straightforward as the paramagnetic effects are lessened due to coupling between the two Fe(III) centers. For Fe2O Rhoxo, the pyrrolic peaks shift back to ∼14 ppm and appear as a broad singlet (Fig. 2). The resonances corresponding to protons on the Cp* occur at ∼2 ppm. The resonances of the pyridyl protons occur between 7.5 and 9.5 ppm. The resonances associated with pyrrolic protons occur around ∼14.5 ppm and are broadened due to proximity to the iron(III) center. The more distant α-protons of the pyridyl moiety occur between 8.5 and 9.5 ppm and are significantly sharper than that of the β-protons at 7.5–8.5 ppm (Fig. 2). The integrations of these peaks match the expectation of 120 protons for the Cp* groups, 32 total pyridyl protons, and 16 pyrrolic protons (Fig. 2) for our proposed structures containing two porphyrins and four molecular clips. | ||
| Fig. 2 1H NMR (CD3OD, 500 MHz) of Fe2O Rhoxo Prism (top) and Fe2O Rh-benzo Prism (bottom). Pyridyl protons are shown in purple and blue in the insets. Pyrrolic protons are shown in green in the insets. Cp* protons are shown in orange in the full spectra. | ||
Although many of the features of Fe2O Rh-benzo are like its shorter-clipped counterpart, there are a few important distinctions. In this prism, the three types of protons on the porphyrin all split into six resolvable peaks. There are two pyrrolic proton environments, two α-proton environments, and two β-proton environments, in contrast to monomeric metalated tetrapyridyl porphyrin where these features would collapse to three total signals. This splitting to six peaks is consistent with our previous studies of a diamagnetic Zn Rhoxo prism,38 and is rationalized by a twisting of the porphyrin faces that breaks the D4h symmetry. This twisting occurs across all our family of clipped cofacial porphyrin prisms,35,36,38 however in the case of these Fe prisms, the porphyrin–porphyrin separation is strongly enforced because of the μ-oxo bridge.
Crystal structures of unclipped Fe2O dimers show metal–metal separations of ∼3.5 Å.54 Since this distance is shorter than the metal–metal separation in the molecular clips, the porphyrins must twist so that the pyridyl groups properly match the Rh–Rh separation. For Fe2O Rhoxo the twist is small and the pyrrolic proton environment is similar, resulting in a single broad feature that corresponds to both protons; however, for Fe2O Rh-benzo this twist is significant and results in one of the pyrrolic protons being oriented towards the clipped cleft where it can be shielded by the π-cloud of the bridging 2,5-dihydroxy-1,4-benzoquinato group, while the other is directed to a clip-free cleft (see H and H′ Fig. 3). Because the environments are so different, the two unique peaks are easily resolved, with one shifting upfield due to the shielding.
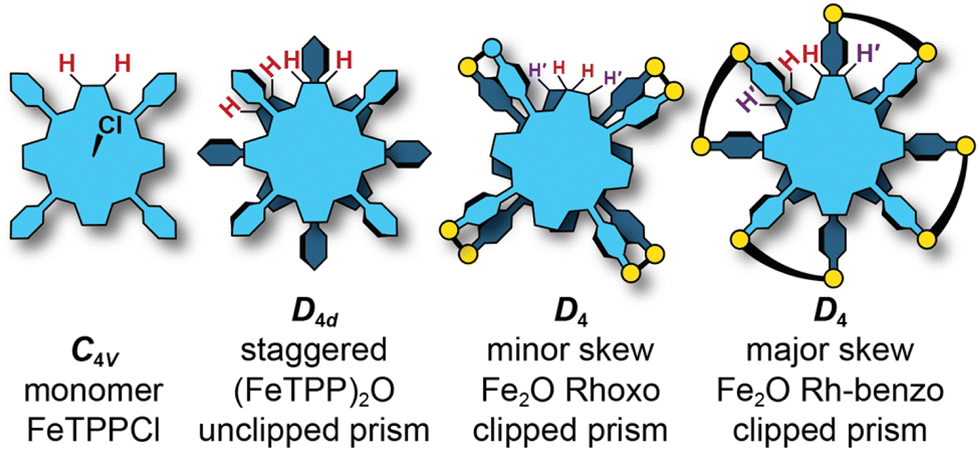 | ||
| Fig. 3 Examples of different symmetries for porphyrin architectures depending on the extent of twisting. (a) Monomeric FeTPhPCl. (b) Unclipped Fe2O porphyrin dimer. (c) Fe2O Rhoxo. (d) Fe2O Rh-benzo. | ||
The structures of both clipped prisms were optimized using ORCA 5.0.3 at the r2SCAN-3c level of theory with the def2-mTZVPP basis set and are consistent with the conclusions drawn from the NMR data that the twisting of the porphyrin faces in Fe2O Rh-benzo is greater than that of Fe2O Rhoxo (Fig. 4).55,56
 | ||
| Fig. 4 Optimized structures of Fe2O Rh-benzo and Fe2O Rhoxo. | ||
Mass spectrometry
High-resolution mass spectrometry (HR-MS) was used to verify the stoichiometry and elemental composition of the self-assembled cofacial structures. As shown in Fig. 5, the top red of the Fe2O Rhoxo contains a 3+ peak at 1453.7 and a 4+ peak at 1053.1, arising from fully intact cores (iron porphyrin dimer clipped by four Rhoxo clips) that were ionized by the loss of three or four outer sphere triflate counterions, respectively. A few other peaks can be assigned that result from further fragmentation. For instance, a 3+ peak at 1166.419 is consistent with the loss of a single Rhoxo clip as well as three triflates. The bottom green trace in Fig. 5 is the HR-MS of Fe2O Rh-benzo, with a 3+ peak at 1521.1 and a 4+ peak at 1103.6 due to fully intact cores (two porphyrins, four molecular clips) that have lost three and four triflate counterions, respectively. The dominant peaks for both prisms consistent with two porphyrins and four molecular clips, along with the high symmetry NMR that show distinct pyrrolic environments strongly support our structural assignment of bis-iron(III)-μ-oxo porphyrins where the molecular clips span both macrocycles. It is possible for clipped porphyrins to form bow-tie structures where the bridge spans two pyridyl groups of the same macrocycle.57 In our prisms, the molecular clips are too rigid and short to accommodate the severe distortions that would be required for bowtie structures (see Fig. S29† for a simple molecular model showing). Examples where these clips can bridge two pyridyl sites on the same molecule have N–N separations of ∼6.7 Å.58 In TPyP, the separation is 10.9 Å. | ||
| Fig. 5 High resolution mass spectra of Fe2O Rhoxo (top) and Fe2O Rh-benzo (bottom). “Prism” accounts for the mass of two porphyrins and four molecular clips. The number of counterions remaining are listed for each peak. Bolded labels correspond to peaks for intact prisms while non-bolded labels refer to fragments peaks that have lost molecular clips in addition to counterions. | ||
Paramagnetic DOSY NMR were acquired (Fig. S5 and S6†) to further support the formation of singular species. Although some broadened peaks could not be resolved, all signals for Fe2O Rhoxo and Fe2O Rh-benzo appear with the same diffusion coefficients that also line up well with the signal from the Cp*, supporting that the porphyrin and clip are found in a single molecular species. The diffusion coefficients associated with these prisms is similar to what was observed for our diamagnetic Zn Rhoxo prism,38 as expected for structurally related cofacial porphyrins.
Electrochemical analysis
ORR catalyzed by metallated porphyrins or related complexes often proceed by one of two pathways: a four-proton/four-electron route to generate H2O, or a two-proton/two-electron path to form H2O2. To investigate the catalytic activities of these iron complexes, we conducted homogenous cyclic voltammetry experiments under four different conditions as summarized by different color traces in Fig. 6: blue (N2, catalyst), red (O2, catalyst), purple (N2, trifluoroacetic acid (TFA), catalyst), and green (O2, TFA, catalyst). Under N2 atmosphere, there was no catalytic response. Under O2, a reversible superoxide formation at the glassy carbon working electrode was observed with an E1/2 value of −1.2 V versus FcH+/FcH. With N2 and TFA present as proton source, a catalytic wave was seen with an onset of −1 V vs. FcH+/FcH that we ascribe to the hydrogen evolution reaction (HER). Finally, under O2 atmosphere with TFA present, an appreciable current response was observed with an onset far more positive than that for HER, which we attributed to the oxygen reduction reaction, consistent with other cofacial porphyrins we have studied.35,36 | ||
| Fig. 6 CVs of Fe catalysts under homogeneous conditions. Blue: 0.2 mM monomeric porphyrin, or 0.1 mM of iron dimer (0.2 mM iron sites) under N2 atmosphere. Red: 0.2 mM iron sites, O2 atmosphere. Purple: 0.2 mM iron sites, N2 atmosphere, 100 mM TFA. Green: 0.2 mM iron sites, O2 atmosphere, 100 mM TFA. In all cases, dry acetonitrile with 100 mM TBAPF6 was used. Scan rate: 100 mV s−1, scan direction: reduction first. Ag/AgNO3 pseudo reference electrode, data later referenced to FcH+/FcH upon addition of ferrocene. | ||
By comparing the current responses of monomer, unclipped dimer, and clipped dimers, we can gain some insight about the influence that post-assembly modification can have on the ORR chemistry of bis-iron(III)-μ-oxo porphyrins. The (FeTPhP)2O, Fe2O Rhoxo and Fe2O Rh-benzo all have onset potentials that are more positive than that of monomeric FeTPhPCl; the onset potentials of all three bis-iron(III)-μ-oxo porphyrins (unclipped and two clipped dimers) are similar, suggesting that the monomer proceeds through a different mechanism than the dimers. Compared to our previously reported Co-based cofacial porphyrins, these prisms have a ∼200 mV higher overpotential for ORR under identical conditions, as shown in Fig. S19.† Other metrics for ORR catalysis by the Fe systems studied here and the analogous Co2 Rhoxo are summarized in Table 1.


To further interrogate ORR reactivity, faradaic efficiencies, numbers of electrons transferred, and kinetic rate constants were determined under heterogeneous conditions wherein the catalysts were immobilized along with carbon black and Nafion. The catalyst inks were cast on the surface of glassy carbon electrodes (GC), either for three-electrode measurements or hydrodynamic voltammetry using a ring disk electrode (RDE, GC disk, platinum ring). A detailed procedure is described in the ESI.† Cyclic voltammograms in 0.5 M H2SO4 were collected under both N2 and O2 atmospheres. Under the potential window of interest, no background HER was observed and all current response was attributed to ORR (Fig. S11–S14†).
Linear sweep voltammetry (LSV) was carried out with catalyst inks immobilized on the disk of an RRDE (Fig. 7). For FeTPhPCl, (FeTPhP)2O, and Fe2O Rh-benzo, over the potential window of interest, catalysis was kinetically limited, and no plateaus were observed to signal mass-transfer limitations. In contrast, over the same potential window Fe2O Rhoxo began to exhibit a plateau in current, consistent with a depletion of O2 at the electrode, serving as a qualitative indicator that this catalyst promotes faster ORR than the other species. By comparing disk and ring current responses, where H2O2 generated at the disk was oxidized at the ring, we extracted selectivity information. The monomeric FeTPhPCl was the least selective system for H2O, favoring H2O2. All three bis-iron(III)-μ-oxo structures were significantly more selective. However, the impact of post-assembly modification with a properly selected clip is significant. For the unclipped (FeTPhP)2O, the selectivity improved to 15.8% H2O2. Clipping with the larger benzo-spaced bridge showed only a modest improvement to 14.5%, which matched the selectivity of our previously studied Co2 Rhoxo. When the shorter clip was used for post-assembly modification, the selectivity was greatly enhanced, with only 7.2% H2O2 generated.
 | ||
| Fig. 7 RRDE voltammograms at scan rates of 20 mV s−1. In all cases, the catalysts were immobilized in Nafion inks with carbon black and immersed in 0.5 M H2SO4. The ring potential was held at 1 V. Scan direction: reducing. (a) FeTPhPCl, (b) (FeTPhP)2O, (c) Fe2O Rhoxo, (d) Fe2O Rh-benzo. | ||
A Koutecký–Levich (KL) analysis was carried out for the Fe2O Rhoxo (Fig. S20–S25†). Since a plateau region is needed for this analysis, kinetic parameters were not extracted for FeTPhPCl, (FeTPhP)2O, and Fe2O Rh-benzo as scanning more negative would introduce current response from HER. The standard rate constant measured for Fe2O Rhoxo was 2.3 × 102 M−1 s−1, which is on the same order as that measured for Co2 Rhoxo (2.6 × 102 M−1 s−1).38
UV-vis spectroelectrochemistry
To probe the mechanism of catalysis by Fe2O Rhoxo, spectroelectrochemical experiments were performed. Fig. 8 shows the UV-vis spectra recorded at different applied potentials to a honeycomb electrode placed in a solution of the cofacial porphyrin in DMF with 100 mM TBABF4. The inset of Fig. 8 shows the spectral response when the Fe2O Rhoxo is placed in the acidic DMF solution without any applied potential. The well-defined Soret band splits into two peaks, which is consistent with the reactivity of Fe2O cores with acid wherein the oxo is protonated to yield an Fe(III)–OH and a solvated second Fe(III) center; thus, two distinct porphyrin absorptions are observed.9 Upon reduction, the two Soret bands at 416 nm and 398 nm collapse back to a single peak at 426 nm while maintaining an isosbestic point, indicative of a clean transformation of the Fe(III)–OH and Fe(III)-solvated porphyrins to identical Fe(II) centers. | ||
| Fig. 8 Spectroelectrochemical measurements of 0.016 mM of Fe2O Rhoxo, 100 mM TBABF4, 100 mM TFA, DMF. Initial potential: −100 mV vs. Ag/Ag+, final potential: −0.500 V vs. Ag/AgNO3. Step amplitude: 0.050 V, step period: 30 seconds. Inset: Absorbance spectra of Fe2O Rhoxo in DMF (green dashed) and with 100 mM TFA (black solid). | ||
Proposed mechanism
Prior descriptions of the mechanism of Fe2O dimers as ORR catalysts conclude that the oxo-bridge templates a cofacial cleft that is maintained under heterogeneous conditions where the porphyrin sites are largely immobile. In other words, even though the oxo bridge is lost upon acidification and there is no intramolecular bridge between the two porphyrin sites, the porphyrin–porphyrin separation is already fixed. Fe2O dimers are overwhelmingly studied in the solid state because the loss of the oxo bridge under homogenous conditions means that under the conditions of catalysis (i.e. acid present) only monomers will persist.When the cofacial arrangement is maintained either by immobilization or, in our case, by post-assembly clipping the two porphyrins, a subsequent reduction will provide a bis-iron(II) cavity poised for ORR. This Fe(II)2 state represents an entry into the catalytic cycle shown in Scheme 2, which favors four proton/four electron chemistry. The Fe(II)2 binds O2 to give a μ-peroxo Fe(III)2 species and both iron centers can be converted to Fe(III)–OH upon the addition of two more protons and electrons. A second two-proton two-electron step releases water and restores the Fe(II)2 active species. In contrast, mononuclear iron porphyrins favor two proton/two electron chemistry because O2 binds to furnish an Fe(III)-superoxo with no second metal to assist in breaking the O–O bond.
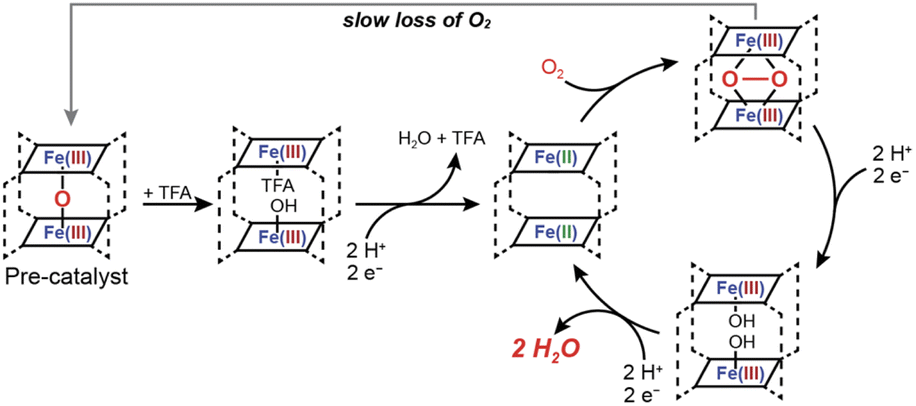 | ||
| Scheme 2 Proposed mechanism of Oxygen Reduction Reaction catalyzed by Fe2O prism. | ||
This mechanism is operative in the solid state for (FeTPhP)2O, Fe2O Rhoxo, and Fe2O Rh-benzo, yet all three systems do not display the same kinetics and selectivity. From our NMR data and computational results, the Fe2O Rh-benzo exhibits a more significant twist that will impact orbital overlap associated with interactions with substrate and metal–metal electronic communication. Furthermore, once the μ-oxo is removed under acidic conditions, the dynamics of the two clipped prisms will differ. As with our Co2 prisms, when shorter molecular clips are used, selectivities and kinetics are enhanced.38 In addition, since both Fe2O Rhoxo, and Fe2O Rh-benzo preserve a cofacial arrangement in solution under acidic conditions despite the loss of the μ-oxo, our prisms are a rare example of dimeric ORR electrocatalysts that operate in homogenous conditions, which results in markedly different current responses. The selective and highly active Fe2O Rhoxo operates in the “K region” of homogenous electrocatalysis wherein substrate is depleted.51 The current response for Fe2O Rhoxo does show evidence for some substrate depletion but more closely resembles the KS region prior to the onset of the plateau. The current response for (FeTPhP)2O is actually due to monomeric FeTPhP that is generated by the acidic conditions and thus its curve is significantly different.
Conclusions
We have repurposed dinuclear molecular clips that have previously been used to drive coordination-driven self-assembly reactions to instead enable the post-assembly modification of cofacial porphyrins. Although bis-iron(III)-μ-oxo porphyrins already adopt a cofacial geometry, the use of molecular clips can have a significant impact on the reactivity of such species. Under acidic conditions, the μ-oxo bridge is rapidly severed and unclipped architectures quickly transform into two monomeric porphyrins. Our post-assembly modification preserves the cofacial arrangement under such conditions. A dinuclear cavity can be maintained via solid-state immobilization, yet even here the selectivity and activity of such species as electrocatalysts for ORR is still impacted by the introduction of molecular clips. We demonstrate that these effects are dependent on the length of the molecular clips. Our catalyst containing the shortest bridge, Fe2O Rhoxo, cuts down the production of H2O2 by half relative to the longer Fe2O Rh-benzo and unclipped (FeTPhP)2O (7.2% versus ∼15%). Thus, the design principles used for coordination self-assembly, where metal–ligand bond formation is the basis for the construction of complex polynuclear architectures, can readily be applied to tuning catalytic centers to enhance stability and reactivity.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
DZ spearheaded experimental work including synthesis, characterization, and reactivity studies and wrote the paper. LER conducted spectroelectrochemistry measurements. MRC carried out the calculations. TRC designed the research and wrote the paper. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Alan Friedman for helpful discussions in collecting ESI-MS data. This work was supported by NSF CAREER Award #1847950 (TRC). Characterization work was performed in part in the Chemistry Instrument Center (CIC), University at Buffalo, SUNY, Buffalo, NY. This work used the 12T Bruker SolariXR 12 Hybrid FTMS with Imaging MALDI and Nano-LC supported by NSF S10RR029517.References
- S. Yoshikawa and A. Shimada, Chem. Rev., 2015, 115, 1936–1989 CrossRef CAS.
- S. W. Ragsdale, Chem. Rev., 2006, 106, 3317–3337 CrossRef CAS.
- J. B. Howard and D. C. Rees, Chem. Rev., 1996, 96, 2965–2982 CrossRef CAS PubMed.
- J. T. Groves, in Cytochrome P450: Structure, Mechanism, and Biochemistry, ed. P. R. Ortiz de Montellano, Springer US, Boston, MA, 2005, pp. 1–43. DOI:10.1007/0-387-27447-2_1.
- E. Y. Tshuva and S. J. Lippard, Chem. Rev., 2004, 104, 987–1012 CrossRef CAS.
- K.-Y. Wang, J. Zhang, Y.-C. Hsu, H. Lin, Z. Han, J. Pang, Z. Yang, R.-R. Liang, W. Shi and H.-C. Zhou, Chem. Rev., 2023, 123, 5347–5420 CrossRef CAS PubMed.
- I. Bhugun, D. Lexa and J.-M. Savéant, J. Am. Chem. Soc., 1996, 118, 3982–3983 CrossRef CAS.
- X. Li, H. Lei, L. Xie, N. Wang, W. Zhang and R. Cao, Acc. Chem. Res., 2022, 55, 878–892 CrossRef CAS.
- O. Kenichi, H. Agus, N. Junichiro, S. Hiroshi and T. Eishun, Bull. Chem. Soc. Jpn., 2000, 73, 1153–1163 CrossRef.
- L. Xie, X.-P. Zhang, B. Zhao, P. Li, J. Qi, X. Guo, B. Wang, H. Lei, W. Zhang, U.-P. Apfel and R. Cao, Angew. Chem., Int. Ed., 2021, 60, 7576–7581 CrossRef CAS PubMed.
- S. Bhunia, A. Ghatak, A. Rana and A. Dey, J. Am. Chem. Soc., 2023, 145, 3812–3825 CrossRef CAS.
- A. Ghatak, S. Bhunia and A. Dey, ACS Catal., 2020, 10, 13136–13148 CrossRef CAS.
- S. Dey, B. Mondal, S. Chatterjee, A. Rana, S. Amanullah and A. Dey, Nat. Rev. Chem., 2017, 1, 0098 CrossRef CAS.
- B. Mondal, P. Sen, A. Rana, D. Saha, P. Das and A. Dey, ACS Catal., 2019, 9, 3895–3899 CrossRef CAS.
- D. J. Martin and J. M. Mayer, J. Am. Chem. Soc., 2021, 143, 11423–11434 CrossRef CAS PubMed.
- K. Guo, X. Li, H. Lei, H. Guo, X. Jin, X.-P. Zhang, W. Zhang, U.-P. Apfel and R. Cao, Angew. Chem., Int. Ed., 2022, 61, e202209602 CrossRef CAS PubMed.
- K. Guo, H. Lei, X. Li, Z. Zhang, Y. Wang, H. Guo, W. Zhang and R. Cao, Chin. J. Catal., 2021, 42, 1439–1444 CrossRef CAS.
- P. T. Smith, B. P. Benke, Z. Cao, Y. Kim, E. M. Nichols, K. Kim and C. J. Chang, Angew. Chem., Int. Ed., 2018, 57, 9684–9688 CrossRef CAS PubMed.
- J. S. Derrick, M. Loipersberger, S. K. Nistanaki, A. V. Rothweiler, M. Head-Gordon, E. M. Nichols and C. J. Chang, J. Am. Chem. Soc., 2022, 144, 11656–11663 CrossRef CAS PubMed.
- K. M. Kadish, G. Larson, D. Lexa and M. Momenteau, J. Am. Chem. Soc., 1975, 97, 282–288 CrossRef CAS.
- J. Rosenthal, B. J. Pistorio, L. L. Chng and D. G. Nocera, J. Org. Chem., 2005, 70, 1885–1888 CrossRef CAS PubMed.
- J. Rosenthal, T. D. Luckett, J. M. Hodgkiss and D. G. Nocera, J. Am. Chem. Soc., 2006, 128, 6546–6547 CrossRef CAS PubMed.
- E. A. Mohamed, Z. N. Zahran and Y. Naruta, Chem. Commun., 2015, 51, 16900–16903 RSC.
- M. L. Pegis, D. J. Martin, C. F. Wise, A. C. Brezny, S. I. Johnson, L. E. Johnson, N. Kumar, S. Raugei and J. M. Mayer, J. Am. Chem. Soc., 2019, 141, 8315–8326 CrossRef CAS.
- J. P. Collman, C. M. Elliott, T. R. Halbert and B. S. Tovrog, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 18–22 CrossRef CAS.
- J. P. Collman, M. Marrocco, P. Denisevich, C. Koval and F. C. Anson, J. Electroanal. Chem. Interfacial Electrochem., 1979, 101, 117–122 CrossRef CAS.
- J. P. Collman, P. Denisevich, Y. Konai, M. Marrocco, C. Koval and F. C. Anson, J. Am. Chem. Soc., 1980, 102, 6027–6036 CrossRef CAS.
- Y.-J. Lin, Y.-F. Han and G.-X. Jin, J. Organomet. Chem., 2012, 708–709, 31–36 CrossRef CAS.
- W.-X. Gao, H.-N. Zhang and G.-X. Jin, Coord. Chem. Rev., 2019, 386, 69–84 CrossRef CAS.
- N. P. E. Barry, P. Govindaswamy, J. Furrer, G. Süss-Fink and B. Therrien, Inorg. Chem. Commun., 2008, 11, 1300–1303 CrossRef CAS.
- N. P. E. Barry, M. Austeri, J. Lacour and B. Therrien, Organometallics, 2009, 28, 4894–4897 CrossRef CAS.
- S. P. Black, D. M. Wood, F. B. Schwarz, T. K. Ronson, J. J. Holstein, A. R. Stefankiewicz, C. A. Schalley, J. K. M. Sanders and J. R. Nitschke, Chem. Sci., 2016, 7, 2614–2620 RSC.
- W. Meng, B. Breiner, K. Rissanen, J. D. Thoburn, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2011, 50, 3479–3483 CrossRef CAS.
- A. N. Oldacre, A. E. Friedman and T. R. Cook, J. Am. Chem. Soc., 2017, 139, 1424–1427 CrossRef CAS PubMed.
- A. N. Oldacre, M. R. Crawley, A. E. Friedman and T. R. Cook, Chem. – Eur. J., 2018, 24, 10984–10987 CrossRef CAS PubMed.
- M. R. Crawley, D. Zhang, A. N. Oldacre, C. M. Beavers, A. E. Friedman and T. R. Cook, J. Am. Chem. Soc., 2021, 143, 1098–1106 CrossRef CAS.
- M. R. Crawley, D. Zhang and T. R. Cook, Inorg. Chem. Front., 2023, 10, 316–324 RSC.
- D. Zhang, M. R. Crawley, A. N. Oldacre, L. J. Kyle, S. N. MacMillan and T. R. Cook, Inorg. Chem., 2023, 62, 1766–1775 CrossRef CAS.
- D. Zhang, M. R. Crawley, M. Fang, L. J. Kyle and T. R. Cook, Dalton Trans., 2022, 51, 18373–18377 RSC.
- C. J. Chang, E. A. Baker, B. J. Pistorio, Y. Deng, Z.-H. Loh, S. E. Miller, S. D. Carpenter and D. G. Nocera, Inorg. Chem., 2002, 41, 3102–3109 CrossRef CAS PubMed.
- B. J. Pistorio, C. J. Chang and D. G. Nocera, J. Am. Chem. Soc., 2002, 124, 7884–7885 CrossRef CAS.
- D.-H. Chin, G. N. La Mar and A. L. Balch, J. Am. Chem. Soc., 1980, 102, 4344–4350 CrossRef CAS.
- D.-H. Chin, G. N. La Mar and A. L. Balch, J. Am. Chem. Soc., 1980, 102, 5945–5947 CrossRef CAS.
- N. Mihara, Y. Yamada, H. Takaya, Y. Kitagawa, S. Aoyama, K. Igawa, K. Tomooka and K. Tanaka, Chem. – Eur. J., 2017, 23, 7508–7514 CrossRef CAS.
- O. Tamon, I. Masahiro, S. Takayoshi, K. Hisatoshi and K. Jitsuo, Chem. Lett., 1986, 15, 1467–1470 CrossRef.
- O. Tamon, Y. Manabu, N. Takeo, K. Hisatoshi and K. Jitsuo, Chem. Lett., 1982, 11, 977–980 CrossRef.
- S. Acosta-Calle and A. J. M. Miller, Acc. Chem. Res., 2023, 56, 971–981 CrossRef CAS.
- M.-N. Birkholz, Z. Freixa and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2009, 38, 1099–1118 RSC.
- D. W. Thomas and A. E. Martell, J. Am. Chem. Soc., 1959, 81, 5111–5119 CrossRef CAS.
- E. B. Fleischer, Inorg. Chem., 1962, 1, 493–495 CrossRef CAS.
- E. S. Rountree, B. D. McCarthy, T. T. Eisenhart and J. L. Dempsey, Inorg. Chem., 2014, 53, 9983–10002 CrossRef CAS PubMed.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
- Y.-F. Han, Y.-J. Lin, L.-H. Weng, H. Berke and G.-X. Jin, Chem. Commun., 2008, 350–352, 10.1039/B711809K.
- T. Schuh, O. Kataeva and H.-J. Knölker, Chem. Sci., 2023, 14, 257–265 RSC.
- S. Grimme, A. Hansen, S. Ehlert and J.-M. Mewes, J. Chem. Phys., 2021, 154, 064103 CrossRef CAS.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS.
- P. A. Benavides, M. A. Gordillo, A. Yadav, M. A. Joaqui-Joaqui and S. Saha, Chem. Sci., 2022, 13, 4070–4081 RSC.
- V. Vajpayee, S. Bivaud, S. Goeb, V. Croué, M. Allain, B. V. Popp, A. Garci, B. Therrien and M. Sallé, Organometallics, 2014, 33, 1651–1658 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qi01219d |
| This journal is © the Partner Organisations 2024 |