
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel albumin-binding macrocyclic Gd-HPDO3A complex bearing a deoxycholic acid residue: the role of the hydration state, water exchange and local dynamics in the observed relaxivity†
Ferdeze
Hasallari
ab,
Carla
Carrera
 a,
Eleonora
Cavallari
a,
Eliana
Gianolio
*a and
Silvio
Aime
c
a,
Eleonora
Cavallari
a,
Eliana
Gianolio
*a and
Silvio
Aime
c
aDepartment of Molecular Biotechnology and Health Sciences, Molecular Imaging Centre, University of Torino Via Nizza 52, 10126 Torino, Italy. E-mail: eliana.gianolio@unito.it
bDiSIT – Department of Science and Technological Innovation, Università del Piemonte Orientale “A. Avogadro”, Viale T. Michel 11, 15121, Alessandria, Italy
cIRCCS SDN SynLab, Via Gianturco 113, Naples, Italy
First published on 19th August 2024
Abstract
Gd-HPDO3A (ProHance, Bracco) appears to be one of the MRI contrast agents with the lowest level of gadolinium retention in the body. Recently, we reported the synthesis and characterization of a Gd-HPDO3A derivative with good binding affinity to human serum albumin whose MR contrast efficiency was limited by the slow exchange rate of the coordinated water molecule and reduced hydration state of the albumin-bound complex. Here, we report the results on a strictly related analogue, Gd-HIBDO3A-DCA, which contains an additional methyl group on the hydroxyl arm. The minor structural modification resulted in the stabilization of the TSAP isomer (85%) (as clearly assessed by means of the 1H-NMR spectroscopy of the europium complex). Gd-HIBDO3A-DCA displayed an enhanced water exchange rate (τM = 53 ns), by maintaining the stability of the parent Gd-HPDO3A complex. The expected high relaxivity of the supramolecular adduct with HSA is partly “quenched” by the flexibility of the spacer joining the Gd(III) chelate and the deoxycholic acid moiety. The high HSA binding capacity (three bound complexes) yields an excellent agent with good contrasting ability as shown in in vivo images acquired on a preclinical MRI scanner operating at 1T in a murine breast tumor model. Gd-HIBDO3A-DCA, administered at a half-dose (0.05 mmol kg−1) with respect to the conventional clinical dose, yielded more than two fold signal enhancement compared to ProHance in the tumor region. Moreover, the very high liver signal enhancement makes this complex a promising candidate for liver imaging in hepatic diseases.
Introduction
The use of gadolinium-based contrast agents (GBCAs) to enhance diagnostic accuracy in Magnetic Resonance Imaging (MRI) remains a highly consolidated procedure in clinical practice, as they are considered highly useful tools to support clinicians in a wide range of pathologies, such as cancer, infections, and neurological disorders. In the last two decades, some concerns about their use have been raised in relation to, first, the discovery of an association between kidney failure and contrast-enhanced MRI in the development of Nephrogenic Systemic Fibrosis (NSF), and, then years later, following findings indicating the presence of residual gadolinium in the brain and other organs of patients with normal renal functions. These reports prompted the spreading of a new class warning for all GBCAs and restrictions on the use, or suspension of authorization, for certain linear GBCAs by the European Medicines Agency (EMA). To date, nearly ten years after the first reports of MRI signal hyper-intensity on unenhanced T1-weighted brain images of patients with repeated exposure to GBCAs, there is no evidence that the retained gadolinium can lead to the development of pathological conditions. Nevertheless, the findings from both preclinical and clinical studies regarding the deposition of GBCAs in different tissues1–4 suggest that the search for systems endowed with enhanced relaxivity is still of interest in the light of applying significantly lower doses to attain the same contrast. First and foremost, it is evident how crucial it is to have Gd-complexes with high thermodynamic and kinetic stability to minimize the retention of this exogenous metal in tissues. From clinical and preclinical studies, in fact, it was ascertained that highly stable Gd-complexes with macrocyclic ligands lead to far less retained Gd than those with linear ones, and, among the commercial macrocyclic complexes, Gd-HPDO3A (ProHance, gadoteridol) appears to be one of the agents with a lower level of retention in the body.5–7The goal of achieving higher relaxivity can be pursued through various approaches. The most straightforward approach deals with design systems with increased hydration of the Gd ion, thereby scaling the contribution of the inner sphere to relaxivity. This strategy has been pursued in the development of a new q = 2 (q = inner sphere water molecules) contrast agent, gadopiclenol, whose relaxivity (12.5 mM−1 s−1 at 0.47T in water at 37 °C) is nearly three times higher than the one reported for the previous generation of clinically approved GBCAs characterized by having a single inner sphere water molecule.8–10 However, although this is not the case for gadopiclenol, increasing the hydration of the metal in a complex may carry the intrinsic limitation of providing coordination compounds with lower stability due to the reduced denticity of the used ligands.
Another strategy to increase the relaxivity is to slow down the reorientation time of the paramagnetic complex by forming larger systems. A recently reported case is represented by gadoquatrane, a tetramer of Gd-DOTA monoamide whose relaxivity per Gd ion is 11.8 mM−1 s−1 in human plasma at 37 °C and 1.41T.11 The lengthening of the molecular re-orientational time has been pursued in the past in the design of several Gd-complexes. Particularly relevant are systems whose structure incorporates hydrophobic residues capable of providing specific interactions with blood proteins, most notably human serum albumin (HSA).12–20 However, in many cases, the theoretically achievable relaxation enhancement has been hampered by a non-optimal exchange lifetime of the coordinated water molecule, which resulted in slowing down of the efficient transfer of the paramagnetic information from the Gd ion to all water molecules of its surrounding. When rotation is slowed down upon formation of a high molecular weight system, as in the case of albumin binding, the exchange rate of the coordinated water molecule can exert a limiting effect on relaxivity if optimal values are out of range (10–50 ns at 0.47T).21
Very recently,22 two new α-aryl substituted Gd-DOTA derivatives have been reported where several of the key parameters used to obtain the theoretical very high relaxivity were simultaneously optimized. One of these derivatives, Gd-DOTFA, demonstrates rapid water exchange, improved electronic relaxation, and moderate binding affinity to serum albumin. Calculations suggest a remarkable relaxivity of 110 mM−1 s−1 (at 20 MHz and 25 °C) when the complex is fully bound to the protein. However, despite these promising characteristics, the binding affinity to human serum albumin (HSA) falls short of ensuring the anticipated very high relaxivity when the complex is dissolved in human serum at MRI acquisition doses.
Now, in order to provide a GBCA capable of generating good SE% at a reduced administered dose, maintaining the high stability required for safe use, our research was focused on the development of new Gd(III)-complexes based on the modified HPDO3A structure.23–27
The design of the herein reported system was based on the findings obtained from two previous studies. On one hand, we discovered a way to modify the structure of HPDO3A to obtain a Gd complex with a fast water exchange rate without compromising its stability. Indeed, the introduction of an additional methyl group on the hydroxyl-propyl arm of HPDO3A, which increased the steric bulk around the OH moiety, resulted in the formation of almost exclusively the TSAP isomer (95%). As a consequence, the gadolinium analogue of this complex (Gd-HIBDO3A) exhibited a very fast water exchange rate, but with no detectable loss of kinetic stability, demonstrating a notable improvement over Gd-HPDO3A.23
In another study, the good HSA binding properties of a Gd-HPDO3A derivative functionalized with a deoxycholic acid moiety (Gd-HPDO3A-DCA) were reported. However, the relaxivity of the supramolecular Gd-HPDO3A-DCA/HSA adduct was hindered by the occurrence of an apparently lower hydration state, slow water exchange and fast internal motions.24
Now, with this study, we have combined the knowledge acquired from two previous studies to synthesize a new HPDO3A derivative (Gd-HIBDO3A-DCA) where the introduction of an additional methyl group in the hydroxyl arm is expected to favour the formation of the TSAP structure endowed with rapid exchange of inner sphere water. The presence of the deoxycholic acid residue guarantees increased HSA binding and a long blood circulation time (Fig. 1).
 | ||
| Fig. 1 Chemical structures of the new Gd-HIBDO3A-DCA complex and its parent complexes Gd-HPDO3A-DCA and Gd-HIBDO3A. | ||
Results and discussion
Synthesis of Gd/Eu-HIBDO3A-DCA
The synthetic procedure to obtain Gd-HIBDO3A-DCA and Eu-HIBDO3A-DCA is reported in Scheme 1 and described in the Experimental section. Briefly, tri-tert-butyl 2,2′,2′′-(10-(3-amino-2-hydroxy-2-methylpropyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl) triacetate (A) was prepared through a four step synthesis method: i. introduction of a Z group for the protection of 2-methyl allylamine; ii. oxidation of the double bond to epoxide with m-chloroperbenzoic acid; iii. its reaction with the 1,4,7,10-tetraazacyclododecan-1,4,7-triacetic acid tri-tert-butyl ester in the presence of DIPEA;28 iv. deprotection of the amine via hydrogenative reduction using palladium on carbon as a catalyst. The methyl ester of (3β,5β,12α)-3-(3-carboxypropionyl)amino-12-hydroxycholan-24-oic acid (product B) was synthesized according to literature procedures.29 Standard amide coupling between products A and B, using HATU/DIPEA in DMF, gave the protected conjugate, which was then cleaved through protonolysis by TFA and successive alkaline hydrolysis of the methyl ester. | ||
| Scheme 1 Synthetic procedure of Gd-HIBDO3A-DCA and Eu-HIBDO3A-DCA. 1. Benzyl chloroformate 95%, pH 9; 2. MCPBA, RT, 1 day; 3. DIPEA, 60 °C, 6 h; 4. H2/Pd, RT; 5. DIPEA, HATU, 24 h, RT; 6. (i) TFA, 24 h, RT, (ii) NaOH 2N (pH 12), 24 h RT, 3 h 50 °C; 7. GdCl3 or EuCl3. | ||
Gd- and Eu-complexes were obtained by adding stoichiometric amounts of GdCl3 or EuCl3 to an aqueous solution of the ligand and maintaining the pH at 7 at room temperature.
High resolution 1H-NMR of Eu-HIBDO3A-DCA
The coordination structure of Ln(III) complexes with octadentate ligands based on the tetra-aza-cyclododecane macrocycle, such as DOTA or HPDO3A, can yield four different diasteroisomers, namely two Square Anti Prismatic (SAP1 and SAP2) and two Twisted Square Anti Prismatic (TSAP1 and TSAP2) isomers. Basically they differ for the torsion angle of the macrocyclic ring and the orientation of the ligand arms. Moreover, Ln-HPDO3A (as its derivatives) has a chiral carbon which can be either R or S and this further yields the possibility of having a total of 8 isomers. The dominant diastereoisomers in a solution of Eu-HPDO3A have been identified in SAP2-R and TSAP1-R and their enantiomeric pairs SAP1-S and TSAP2-S.30One commonly used method to obtain structural information about europium complexes relies on the acquisition of their 1H-NMR spectra. By this method, one can determine the ratio of isomers in aqueous solution, and apply this information to the corresponding Gd-complexes. For Eu-complexes, the axial ring proton region (5–40 ppm) is highly diagnostic for assessing the SAP/TSAP isomeric ratio, with the signals of SAP isomers being more shifted than those ascribable to TSAP ones, and therefore easily distinguishable from each other. Fig. 2 shows the 1H-NMR spectrum of Eu-HIBDO3A-DCA. The assignment of the protons for each isomer has been done with the aid of a 2D EXSY-NMR acquired to identify the exchange between isomers (Fig. S13†). From the inspection of the axial peak region, two interesting considerations can be made: (i) contrary to the precursors Eu-HIBDO3A23 and Eu-HPDO3A-DCA,24 both major (SAP2 and TSAP1) and minor (SAP1 and TSAP2) isomers are populated and (ii) a marked prevalence (85%) of TSAP isomers (TSAP1 + TSAP2) over SAP ones (SAP2 + SAP1) is present. The EXSY 2D-NMR spectrum (Fig. S12†) indicates that both ring inversion (box A) and arm rotation (boxes B and C) exchange between the isomers occurs.
 | ||
| Fig. 2 1H-NMR spectrum of Eu-HIBDO3A-DCA (and enlargement of the axial region) acquired in D2O at pH 7.4, 600 MHz and 275 K. The peaks are labelled as follows: SAP2-R/1-S = green circle, SAP1-R/2-S = red circle, TSAP1-R/2-S = green square, and TSAP2-R/1-S = red square. | ||
As for the parent Gd-HIBDO3A complex, the TSAP conformation dominates most likely due to the increased steric bulkiness around the bound metal, which limits isomerization and forces a specific conformation. Actually, the less abundant (15%) SAP isomers display an exchanging scheme that suggests arm rotation and ring inversion toward TSAP conformation. It appears reasonable that the exchange between the two TSAP enantiomers occurs through either direct exchange of enantiomers or via the SAP isomers, which do not remain substantially populated.
The large occurrence of the TSAP conformation makes highly likely the fast exchange of the coordinated water, as reported in the literature for numerous examples of DOTA and HPDO3A derivatives with a prevalent TSAP structure.22,23,31–33
Water exchange dynamics of Gd-HIBDO3A-DCA
The water exchange lifetime (τM) of the coordinated water molecule was investigated by measuring the transverse 17O relaxation rate (R2) of H217O as a function of temperature. Fig. 3a shows the 17O-NMR R2 profiles for aqueous solutions of Gd-HIBDO3A-DCA compared to those obtained for the parent Gd-HIBDO3A and Gd-HPDO3A-DCA complexes. All three profiles display the typical trend observed in the presence of two isomers/species in solution. In the case of Gd-HPDO3A derivatives this behaviour is associated with the TSAP and SAP isomers, with the faster and slower exchange being associated with the TSAP and SAP conformations, respectively. By fitting the experimental data with a two-species model, it was possible to calculate a residence lifetime of the water molecule in the inner coordination sphere (τM) of 6.5 ns for the TSAP isomer and 316 ns for the SAP one. Taking into account the relative abundances of each isomer, it was possible to calculate an average exchange time of 53 ns for Gd-HIBDO3A-DCA. The close similarity of Gd-HIBDO3A-DCA and Gd-HIBDO3A profiles suggests that the fast water exchange has to be associated with the prevalence in the solution of the TSAP isomer which is characterized by a short τM value. | ||
| Fig. 3 17O-NMR transverse relaxation rate measured at variable temperatures of solutions of (a) Gd-HIBDO3A, Gd-HPDO3A-DCA and Gd-HIBDO3A-DCA complexes in water and (b) Gd-HIBDO3A-DCA in water and in the presence of 3 mM HSA, pH 7.4 and 14.1T. Data were acquired for the 10 mM Gd(III)-complex in (a) and the 3.3 mM Gd(III)-complex in (b) and normalized to 20 mM. | ||
The increase of the exchange of the coordinated water with a τM value in the optimal range between 10 and 50 ns, allows one to expect it to reach high relaxivity values once the complex is bound to a macromolecule.22 Since the Gd-HIBDO3A-DCA complex was designed to bind to human serum albumin (HSA), it was deemed of interest to acquire the 17O-R2 profile of the complex in the presence of HSA. As shown in Fig. 3b the condition of rapid exchange of the coordinated water is maintained after the formation of the adduct with the protein.
The relationship between the exchange rate and the structure/dynamics of the binding interaction with albumin could be quite complex and, in most of the reported cases, it results in an elongation of the lifetime of the coordinated water molecule.14,24,34,35 In principle the exchange lifetime is determined essentially from the enthalpy of the Gd–O(water) bond, a property that, for macrocyclic GBCAs, is related to the occurrence of a TSAP or SAP coordination polyhedron. In principle, one may expect that the relative populations of the two isomers can also be affected by the constraint induced on the surface of the chelating moiety upon the setup of the interaction at the protein binding sites.
Actually, in this case, the binding to HSA seems to exert a stabilizing effect toward TSAP conformation as the small contribution relative to the SAP isomer disappeared from the profile when the adduct with the protein is formed. A good fitting of experimental data was obtained considering the occurrence of 100% TSAP isomer with an exchange lifetime of 18 ns.
Binding to HSA
The relaxivity (r1) of Gd-HIBDO3A-DCA measured in water, in phosphate buffer and in human serum at 298 K and 21 MHz resulted to be 7.90 ± 0.17, 8.15 ± 0.38 and 26.8 ± 1.12 mM−1 s−1, respectively. As anticipated above, the higher relaxivity value measured in human serum with respect to pure water and PBS is likely to be associated with binding to serum albumin expected on the basis of enhanced lipophilicity (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P = −1.6 ± 0.09) with respect to the parent Gd-HPDO3A complex (logP = −2.09 ± 0.11).
P = −1.6 ± 0.09) with respect to the parent Gd-HPDO3A complex (logP = −2.09 ± 0.11).
By exploiting the increase in relaxivity obtained when a paramagnetic complex binds to a macromolecular substrate (PRE – Paramagnetic Relaxation Enhancement), the binding parameters of Gd-HIBDO3A-DCA towards HSA were determined. The number of binding sites on the protein can be derived by performing a titration in which the relaxation rate (R1obs) of solutions of a fixed concentration of HSA (0.6 mM) and increasing concentrations of the gadolinium complex is measured (Fig. 4A). The inflection point of the graph indicates the concentration of the complex required to saturate the strongest interaction sites on the protein. In this case, the inflection was achieved for a complex concentration of 1.8 mM which is three times the concentration of HSA used, indicating that the number of binding sites is 3.
 | ||
| Fig. 4 (A) Proton relaxation enhancement titration of the HSA containing solution (0.6 mM) with increasing concentrations of Gd-HIBDO3A-DCA, in the phosphate buffer, 298 K, 0.47T. (B) Proton relaxation enhancement titration of Gd-HIBDO3A-DCA (0.1 mM) with increasing concentrations of HSA, in the phosphate buffer, 298 K, 0.47T. | ||
On the other hand, the association constant (Ka) and relaxivity of the supramolecular Gd-HIBDO3A-DCA-HSA (r1b) adduct were determined by measuring the R1obs values of solutions of a constant concentration of the Gd-complex with increasing concentrations of HSA (Fig. 4B). Interpolation of the experimental data allowed the calculation of KA = 3.03 ± 1.2 × 104 M−1 with three equivalent interaction sites (apparent nKA = 9.09 ± 3.8 × 104 M−1) and an adduct relaxivity of 27.9 ± 0.41 mM−1 s−1. Both the affinity constant and the relaxivity of the adduct appear considerably higher than those reported in our previous study for Gd-HPDO3A-DCA.23 The higher protein affinity might be ascribed to the increased lipophilicity of Gd-HIBDO3A-DCA with respect to Gd-HPDO3A-DCA due to the introduction of a methyl group on the arm.
Next, competitive relaxometry experiments were performed in order to gain more insights into the characterization of the binding sites. The binding on HSA was measured in the presence of competitive ligands whose recognition abilities for specific binding sites on HSA are well known.36–40 The typical substrates used for each binding site were ibuprofen for subdomain IIIA (Sudlow site II), iodipamide for subdomain IIA (Sudlow site I), and methyl orange for subdomain IB, respectively. Fig. 5a shows the variation of the relaxation rate of the Gd-HIBDO3A-DCA/HSA adduct in the presence of an increasing concentration of competitors. The use of any of the three competitor drugs results in a notable reduction in the relaxation rate of the solution. This suggests that all three competitors are able to displace Gd-HIBDO3A-DCA from albumin, indicating that the three principal binding locations of Gd-HIBDO3A-DCA on the protein are subdomains IIA, IIIA, and IB. This finding is not consistent with the observations we reported for the closely related Gd-HPDO3A-DCA complex for which only two HSA binding sites (IIA and IIIA) were reported.24 Now we think that the discrepancy relies on the fact that in the previous work the test for assessing the involvement of site IB was done with the mitoxantrone ligand instead of methyl orange. The addition of mitoxantrone to HSA up to a ratio of 35:1 did not induce any decrease in the relaxivity of Gd-HPDO3A-DCA bound to the protein, suggesting the absence of direct competition between the two molecules for site IB. In contrast, the addition of methyl orange to the HSA-bound Gd-HIBDO3A-DCA led to a significant reduction in relaxivity, indicating its binding to site IB, in addition to the classical IIA and IIIA Sudlow's sites.
 | ||
| Fig. 5 (a) Change in the relaxivity of a solution containing 0.6 mM Gd-HIB-DO3A-DCA and 0.2 mM HSA as a function of increasing concentrations of ibuprofen, iodipamide or methyl orange (298 K, 0.47T, in the phosphate buffer). Data are reported as a variation of R1/R1° i.e. by normalizing the R1 values observed at different competitor/Gd-HIBDO3A-DCA ratios with respect to the initial value (in the absence of any inhibitor). (b) Proton relaxation enhancement titration of Gd-HIBDO3A-DCA (0.53 mM) with increasing concentrations of HSA, in the presence of couples of competitors (as detailed in the legend) at a ratio of 1.5:1 with respect to HSA (298 K, 0.47T, in the phosphate buffer). | ||
Indeed, the affinity of methyl orange for albumin is much higher (by 1 order of magnitude) than that of mitoxantrone,38 partially explaining the observed different competition behavior for the two Gd-complexes. Considering that methyl orange can be definitively considered a better competitor for the IB site, it is worth concluding that contrary to what was reported in our previous study, secondary binding of Gd-HPDO3A-DCA at site IB cannot be excluded.
This conclusion is fully consistent with the earlier findings supporting the view that bile acids (i.e. chenodeoxycholic acid) can bind to subdomain IB.38,40
Next, additional titrations were performed to estimate the binding constant of Gd-HIBDO3A-DCA for each binding site on albumin. Three titrations of Gd-HIBDO3A-DCA were carried out with increasing albumin concentrations in the simultaneous presence of two of the specific competitors for the different main binding sites (in a ratio of 1.5:1 with HSA) and leaving only one site free for the interaction with Gd-HIBDO3A-DCA (Fig. 5b). By applying the same set of PRE equations as used above (Fig. 4B), the binding constant and relaxivity of the bound complex for the unoccupied binding site could be calculated. Thus for the determination of the binding constant, for site IB a combination of ibuprofen and iodipamide, for site IIA, ibuprofen and methyl orange, and for site IIIA, iodipamide and methyl orange were used, respectively (Table 1).
| nK a (M−1) | r 1 b (mM−1 s−1) | |
|---|---|---|
| Overall | 9.09 ± 3.8 × 104 | 27.9 ± 0.41 |
| Site IB | 3.83 ± 1.8 × 104 | 25.1 ± 0.36 |
| Site IIA | 7.66 ± 3.2 × 104 | 28.2 ± 0.63 |
| Site IIIA | 2.11 ± 1.2 × 104 | 27.3 ± 0.61 |
From this set of experiments, it can be concluded that the IIA site on albumin is both the strongest for Gd-HIBDO3A-DCA binding and the one yielding the highest relaxivity.
Proton longitudinal relaxivity determinants
The proton longitudinal relaxivity (r1), i.e. the property that is directly related to the efficiency of a paramagnetic complex to be considered as a potential MRI contrast agent, of Gd-HIBDO3A-DCA was investigated over an extended range of pH at a fixed proton Larmor frequency of 21.5 MHz at 298 K and compared with those of the parent complexes Gd-HPDO3A and Gd-HIBDO3A (Fig. 6a). Gd-HIBDO3A-DCA showed a less pronounced increase in relaxivity (+13%) at basic pH, due to the base-catalyzed mobilization of the coordinated hydroxyl proton compared to Gd-HPDO3A (+29%) and Gd-HIBDO3A (+35%).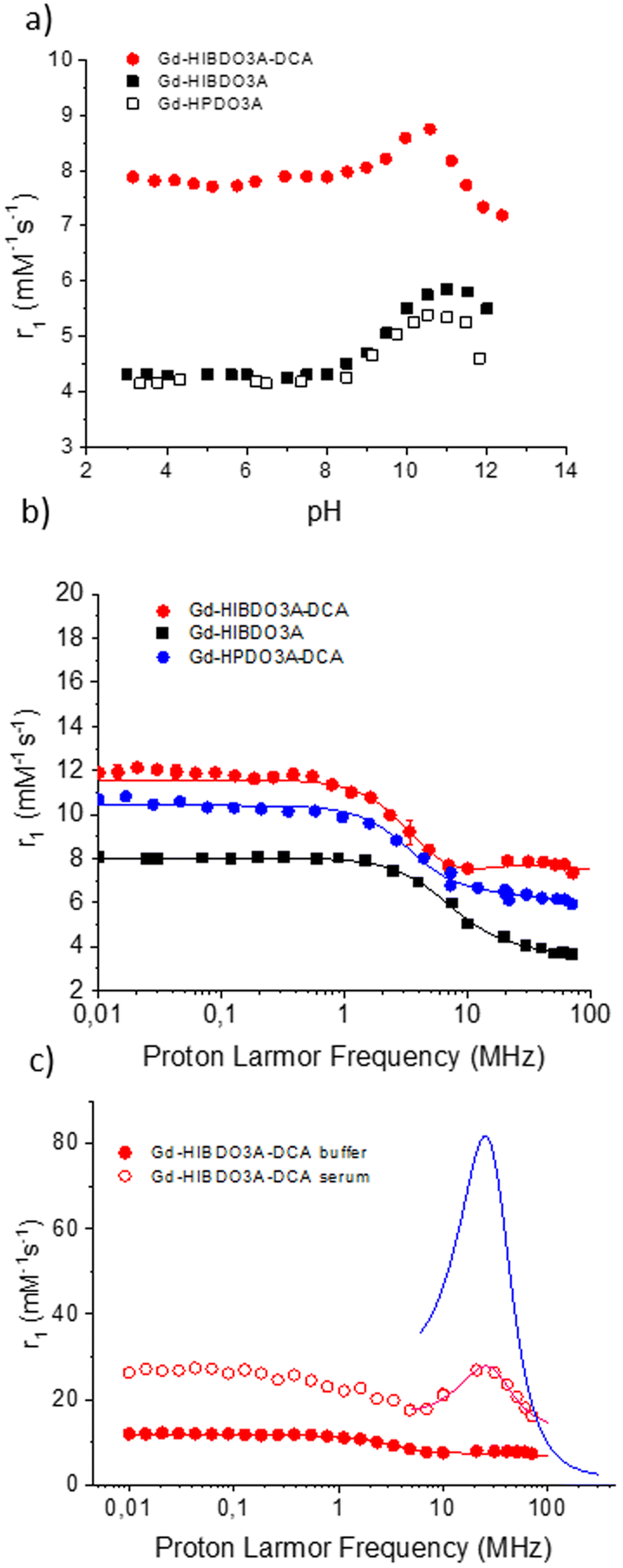 | ||
| Fig. 6 (a) Variation of the millimolar relaxivity of Gd-HIBDO3A-DCA, Gd-HIBDO3A and Gd-HPDO3A measured at 0.47T and 298 K as a function of solution pH; (b) 1H-NMRD profiles of Gd-HIBDO3A-DCA, Gd-HIBDO3A and Gd-HPDO3A-DCA measured at 298 K in phosphate buffer; (c) 1H-NMRD profiles of Gd-HIBDO3A-DCA in phosphate buffer (PBS) and in human serum at 298 K and pH 7.4. The blue line is the simulated profile obtained by maintaining all the fitting parameters equal to those reported in Table 2 but with a τR value equal to 30 ns. All data were normalized to 1 mM concentration of the Gd(III)-complex. | ||
This behaviour appears in agreement with the occurrence of partial intramolecular catalysis of the prototropic exchange of the coordinated alcoholic group, as previously shown for similar systems.25–27,41,42 The catalysis likely involves the amide proton on the linker connecting HPDO3A to the deoxycholic acid moiety making the contribution of the OH moiety already evident at neutral pH values. In the case of Gd-HPDO3A, the contribution from the coordinated alcoholic proton (considered as 0.5 water molecules) accounts for a ca. 30% increase in relaxivity and it occurs only at basic pH values. For Gd-HIBDO3A-DCA, the increase of the solution pH resulted only in a 13% increase of the observed relaxivity, and on this basis we assume that the contribution from the prototropic exchange is markedly present already at physiological pH. On this basis it has been concluded that this contribution, due to an intramolecular catalysis of the prototropic exchange, accounts for a gain of ca. 17% of the relaxivity at neutral pH (i.e. contributing as ca. 0.3 water molecules). This leads us to assume an actual total hydration value of q = 1.3 for the successive analysis of the NMRD profiles.
In Fig. 6b, the NMRD profile, in the proton Larmor frequency range of 0.01–80 MHz, obtained for Gd-HIBDO3A-DCA in PBS at 25 °C and neutral pH is shown and compared with the profiles of the parent complexes Gd-HPDO3A-DCA and Gd-HIBDO3A. The experimental data were fitted using the Solomon Bloembergen Morgan (SBM) equations to extract the relevant relaxometry determinants, such as the rotational correlation time (τR) and the electronic relaxation parameters (Δ2 and τv). The best fitting parameters are reported in Table 2. During the fitting procedure, τM was fixed to the mean value (53 ns) determined from the 17O-R2vs. T profiles. The number of inner sphere water molecules (q) was fixed to 1.3 according to the value calculated above from the relaxivity pH dependence experiment. The distance between the gadolinium ion and the water protons (rGd–H) was fixed to 3.2 Å, i.e. ca. 5% longer than the values normally measured for analogous gadolinium complexes (3.0–3.1 Å). This assumption was based on the previously reported evidence that for Gd-complexes where the TSAP conformation is the preferred one, such as in the case of Gd-DOTMA, the coordinated water molecule is pushed further away from the metal ion, weakening the metal–ion interaction and favouring faster exchange.32,43
| Parameters | Gd-HIBDO3Aa | Gd-HPDO3A-DCAb | Gd-HIBDO3A-DCA | |
|---|---|---|---|---|
| PBS | Human serum | |||
| NMRD profiles used for fitting were acquired at 298 K, with the following parameters fixed during the fitting procedure: q = 1.3, aGdH = 3.8 Å, and DGdH = 2.24 × 10−5 cm2 s−1. A two-isomer model was used for fitting of the 17O-NMR data with fixed parameters: Er = 10 kJ mol−1, Ev = 10 kJ mol−1, and A/ħ = −3.5 × 106 rad s−1.a From ref. 23.b From ref. 19. | ||||
| 298 r 1 (mM−1 s−1) | 4.3 | 6.8 | 8.1 | 26.8 |
| Mol. Fracn TSAP | 0.95 | 0.3 | 0.85 | 1 |
| Weighted avg. τ m (ns) | 16.1 | 570 | 53 | 18 |
| Δ 2 (1019 s−2) | 5.4 ± 1.0 | 0.95 ± 0.2 | 3.0 ± 0.5 | 0.73 ± 0.04 |
| τ v (ps) | 12.9 ± 1.8 | 44.9 ± 8.9 | 24.1 ± 0.4 | 13.7 ± 1.7 |
| τ R (ps) | 62.1 ± 2.7 | 140 ± 4.6 | 170 ± 5.1 | τ RL = 527 ± 27 |
|
τ
RG = 30000 |
||||
| K | 0.41 | |||
| r GdH (Å) | 3.1 | 3.1 | 3.2 | 3.2 |
| r GdO (Å) | 2.5 | 2.5 | 2.5 | 2.65 |
The higher relaxivity of Gd-HIBDO3A-DCA measured in PBS over all the investigated range of Larmor frequencies with respect to the related Gd-HIB-DO3A and Gd-HPDO3A-DCA complexes appears to be associated with: (i) a higher inner sphere contribution and (ii) a slight elongation of the reorientational correlation time (τR). The fastened water exchange dynamics appears to affect the NMRD profile between 10 and 80 MHz where a small hump of relaxivity is visible for Gd-HIBDO3A-DCA. A direct correlation can be observed between the values of τR obtained from fitting the NMRD profiles and the molecular weight of the corresponding Gd-complexes, as demonstrated in Fig. S14.†
Fig. 6c shows the comparison between the profiles obtained for Gd-HIBDO3A-DCA in the buffer and in human serum. In serum, the expected relaxivity at 20–30 MHz for a system with a short τM value (18 ns) and a long τR value (the value reported for albumin is about 30 ns), as is the case for Gd-HIBDO3A-DCA bound to HSA, should be higher than the obtained experimental value (see simulated curve in Fig. 6c and previously reported Gd-complexes12,22,44). It can be hypothesized that in the formation of the supramolecular adduct, Gd-HIBDO3A-DCA still has a faster internal motion than the protein it binds to, which limits its relaxivity. Support to this hypothesis was found by fitting the experimental data (the fitting was limited to the high-field region (6–80 MHz) due to the limitation of the SBM theory at low magnetic fields for paramagnetic systems characterized by slow rotation) using the Lipari–Szabo model-free approach.45 This model considers both an internal rotation, characterized by a correlation time τRL, and a global motion described by τRG. The correlation between these two motions is quantified by the parameter K2, which varies in the range 0–1, where a value of zero indicates complete independence between the motions, and a value of 1 suggests immobilization in the absence of local fluctuations. τRG was set at 30 ns to represent the global reorientation of the protein, while τRL and S were treated as variable parameters. The fitting parameters are listed in Table 2. Indeed, a high degree of local flexibility is observed as witnessed by the quite small value of τRL, which increases only roughly three times with respect to the free Gd-complex. Despite this limit on the Gd-L/HSA adduct attainable relaxivity, the very high affinity of Gd-HIBDO3A-DCA toward albumin ensures its complete binding up to a ratio of 3:1 with respect to the protein, thus leading to a high overall relaxivity in human serum.
Apart from being highly efficient, a GBCA with ambition for clinical translation has to demonstrate to be highly stable under in vivo conditions. One notable benefit of macrocyclic contrast agents, such as Gd-HPDO3A, is their enhanced thermodynamic and kinetic stability, which results in a significantly reduced likelihood of Gd(III) detachment from the chelate. Indeed, recent literature witnessed the efforts to further improve the already very high stability of macrocyclic chelates, by introducing chiral groups to the macrocycle and pendant arms on the Gd-DOTA structure where the increased steric hindrance prevents the interactions between the chelated metal ions and outer species, contributing to the enhancement of both the thermodynamic stability (logK of up to 27) and the kinetic inertness of the complex (up to 750 days of incubation in HCl 1 M at room temperature).46,47
To confirm the kinetic inertness of Gd-HIBDO3A-DCA under acidic conditions and for transmetallation two relaxometry experiments were carried out to follow acid catalyzed dissociation in 1 M HCl solution at room temperature and the competition with ZnCl2 over several days at a temperature of 310 K (Fig. S15†). The results demonstrated that the complex displayed no signs of transmetalation when competing with 1 eq. of zinc in 50 mM phosphate buffer for a duration of 7 days. The rate of decomplexation under strongly acidic conditions resulted to be slightly slower than that of the parent complex Gd-HPDO3A (ca. 2 hours vs. ca. 1 hour).
In vivo MRI in a breast cancer murine model
Due to its ability to form a non-covalent adduct with albumin, resulting in a high relaxivity system, Gd-HIBDO3A-DCA holds great potential as an outstanding contrast system for various applications. These include its use as a blood pool agent in angiography, for liver imaging in hepatic pathologies, and for enhanced visualization and characterization of tumors. In this work, we decided to test Gd-HIBDO3A-DCA, in comparison with the parent commercial ProHance (gadoteridol, Gd-HPDO3A), to evaluate its efficiency in increasing the contrast in magnetic resonance images recorded at 7T and 1T in a mouse tumor model. In particular, the aim was to assess whether binding with HSA could result in a longer washout from the extracellular region of the tumor. Furthermore, the changes in the signal intensity in the kidneys and liver (Fig. S16†) provided information on the biodistribution and excretion pathways following in vivo administration. As already reported for other HSA-binding contrast agents, Gd-HIBDO3A-DCA displayed very high uptake in the liver (Fig. 7b and Fig. S15C†) and in the gallbladder (Fig. S16†), indicating the occurrence of the hepatobiliary excretion pathway. | ||
| Fig. 7 Percentage MR signal enhancement in the kidneys (a), livers (b) and tumors (c) of mice administered with 0.05 mmol kg−1 of Gd-HIBDO3A-DCA or gadoteridol (ProHance and Gd-HPDO3A) on MRI scanners operating at 1T and 7T. The curves were obtained by averaging the data collected from different mice (n = 4 per group). The error bars represent the standard deviation. | ||
The diagnosis of hepatic malignancies is a crucial application of MRI. Currently, the two hepatospecific GBCAs available in clinical practice are both linear complexes, namely Gd-EOB-DTPA and Gd-BOPTA. Therefore, the search for alternatives, either using Mn(II)48–50 instead of Gd(III) or employing hepatospecific macrocyclic complexes,46,47,51 has been highly prolific in recent years. In this context, the observed high and persistent hyperintensity in the liver suggests that Gd-HIBDO3A-DCA could likely find application to detect metastases arising from tumors developed in other anatomical regions or other malignancies in the liver.
The observed hyperintensity in the kidneys (Fig. 7a and Fig. S16A†) suggests partial excretion of Gd-HIBDO3A-DCA via the renal route, which is the excretion pathway for the parent complex Gd-HPDO3A.
The biodistribution of Gd-HIBDO3A-DCA in healthy BALB/c mice at 4 h after intravenous injection at a dose of 0.05 mmol Gd kg−1 is reported in Fig. S18† and compared to the results obtained with Gd-HPDO3A. The highest amount of Gd(III) was found in the liver and in the spleen, underlying a preferential hepato-biliary elimination route, as anticipated by MRI investigation. Only negligible amounts of the resulting Gd were retained in the other analysed organs for both agents. In the tumour region, the amount of accumulated Gd is significantly higher than in the case where Gd-HPDO3A is administered.
Fig. 7 and 8 illustrate that by acquiring images at 1T (which corresponds to a Proton Larmor Frequency of 40 MHz), it is possible to fully exploit the signal enhancement generated by the binding of Gd-HIBDO3A-DCA to albumin. This led to a marked increase of the signal intensity when compared to the results obtained with the hydrophilic Gd-HPDO3A. In contrast, at 7T (300 MHz), the contrast enhancements induced by the two contrast agents appear comparable (Fig. 7), except in the case of the liver where the hepatobiliary elimination route preferred by the albumin-binding agent leads to a larger signal enhancement, in spite of the fact that its relaxivity at this field is markedly lower than that at 1T. The outcome achieved in the tumor at 1T (Fig. 7c) is very encouraging, as it shows a signal increase of over two fold the one generated by Gd-HPDO3A, with 70% signal enhancement even at 1 hour post-administration.
 | ||
| Fig. 8 Representative T1-weighted MSME images at 1T of (a) solutions of Gd-HPDO3A and Gd-HIBDO3A-DCA of 0.1 and 0.5 mM in the phosphate buffer and in human serum; (b) TSA tumor xenograft mice obtained pre- and post-injection with ProHance or Gd-HIBDO3A-DCA at a dose of 0.05 mmol kg−1. Images show left to right pre-contrast, and post-contrast at 10 and 50 min. | ||
This feature appears favourable for potential clinical translation of this agent as its efficiency is optimized at a field strength close to that of clinical scanners (1.5–3T). The significantly higher signal enhancement efficiency of Gd-HIBDO3A-DCA compared to the parent complex Gd-HPDO3A can be appreciated in Fig. 8 where the images acquired on a 1T scanner in vitro (Fig. 8a), for solutions of the complexes in the buffer and in serum, and in vivo (Fig. 8b), in a murine breast tumor model, are reported. The tumor region results were far more contrasted, up to 50 min after intravenous injection, when Gd-HIBDO3A-DCA was used. The signal was particularly hyperintense in the rim of the tumor, where the vascularization is higher. This demonstrates a particularly valuable capability of this system to highlight tumor heterogeneity.
Finally, the blood elimination curve was measured and the obtained results are reported in ESI Fig. S19.† The time dependent blood Gd concentrations (measured by ICP-MS) following a single bolus injection through the tail vein at a dose of 0.05 mmol Gd per kg were measured and fitted on the basis of a two compartment pharmacokinetic model. Analysis of the data revealed its very rapid elimination from blood (t1/2β = 4.75 min) comparable to or even faster than that of hydrophilic GBCAs. A similar behavior was previously reported for MS-325 in rats, but not in higher mammals, where plasma pharmacokinetics was virtually indistinguishable from that of extracellular agents.55
Moreover, the whole-body coronal maximum intensity projections (MIPs) of 3D FLASH images were obtained following intravenous administration of Gd-HIBDO3A-DCA at a dose of 0.05 mmol Gd per kg as reported in Fig. S20.† A significant blood pool enhancement was visible only at 2 min post-injection, with the signal that gradually faded away due to the clearance of the agent from the blood, resulting in a strong enhancement of the liver. Indeed, this result closely recalls what was previously observed for B22956 and B25716, two Gd-complexes functionalized with the deoxycholic acid moiety.15 Both experiments allowed us to conclude that Gd-HIBDO3A-DCA is quickly eliminated from blood due to the high liver uptake and hepatobiliary elimination route leading to decreased plasma concentration. On this basis, one draws the conclusion that the use of Gd-HIBDO3A-DCA as a blood pool contrast agent implies that image acquisitions have to be made in a very short time after injection.
Conclusions
The synthesis of Gd-HIBDO3A-DCA was pursued on the basis of a rational design aimed at optimizing the determinants of the relaxivity in serum. Overall the results are satisfactory as the obtained complex displays good relaxation enhancement properties when compared with related Gd-HPDO3A-based agents. The successful shift towards a dominant TSAP structure, while confirming the expected effect on the enhanced exchange lifetime of the coordinated water molecule, outlines a slight elongation of the distance between the paramagnetic Gd(III) ion and the protons of the coordinated water molecule. Moreover, the intramolecular activation of the prototropic exchange of the OH moiety enhances the inner sphere contribution at neutral pH. Unfortunately, the optimized water exchange dynamics cannot be fully exploited in the formation of a supramolecular adduct with serum albumin due to the occurrence of local flexibility of the protein-bound complex. This finding outlines the importance of introducing rigid spacers in the design of GBCAs that bind to proteins. Nevertheless, due to its very high stability and albumin affinity, we think that Gd-HIBDO3A-DCA has great potential as a contrast agent for various applications. These include its use for liver imaging in hepatic pathologies, and for enhanced visualization and characterization of tumors.Experimental
General
Reagents and chemicals were purchased from Sigma Aldrich (unless otherwise stated) and used without further purification. 1H spectra were recorded at 14.1T using a Bruker Avance 600 spectrometer or at 9.4T using a Bruker 400 Ultra Shield, both equipped with a 5 mm probe and a temperature control system. Mass spectra were acquired using a Waters ACQUITY UPLC H-Class system coupled with a QDa detector (direct infusion with H2O 0.1% TFA/ACN 0.1% TFA 50:50). pH measurements were made using an AS instruments pH meter equipped with a glass electrode. HPLC was performed on a LiChrosorb RP Select B 250 × 4 mm, 5 μm column, with mobile phase A: KH2PO4 0.01 M/H3PO4 0.017 M; mobile phase B: ACN; gradient: 0 min 5% B, 30 min 80% B, 45 min 80% B; injection 10 μl; and revelator: UV 210 nm.
UPLC was performed using a Waters ACQUITY UPLC H-Class system coupled with a QDa detector on an ACQUITY UPLC BEH C18 column (2.1 × 50 mm) with mobile phase A: H2O 0.1% TFA; mobile phase B: CAN 0.1% TFA; flow 0.4 ml min−1; gradient: 0 min 5% B, 10 min 100% B, 15 min 100% B; injection 20 μl; and revelator: UV 210 nm. The purifications were performed using an AKTA pure chromatography system equipped with a UV-900 detector, P-900 pump and Frac-920 fraction collector. 1,4,7,10-Tetraazacyclododecan-1,4,7-triacetic acid tri-tert-butyl ester was synthesized according to literature procedures.52
Synthesis
Benzyl (2-methylallyl) carbamate. To a solution of 2-methyl allylamine hydrochloride (10 g; 93 mmol) in dioxane (30 ml) and water (8 ml), cooled with an ice bath, was slowly added dropwise a solution of benzyl chloroformate 95% (15.5 g; 86 mmol) in dioxane (50 ml). pH 9 was maintained with 2N NaOH (58 ml) until stability, and then 1N HCl was added until pH 7 to quench the reaction. The dioxane was evaporated from the mixture; the product was extracted with dichloromethane (4 × 50 ml) and evaporated to residue (14.95 g, yield 84%). MS ESI: m/z calcd 205.26, found 206.26 [M + H]+. HPLC: tR 23.66 min.
Benzyl ((2-methyloxiran-2-yl) methyl)carbamate. A solution of m-chloroperbenzoic acid [MCPBA] (21.5 g; 87.6 mmol) in chloroform (90 ml) was added dropwise to a solution of product 1 (14.9 g; 73 mmol) in chloroform (15 ml). After 1 day, the precipitate was filtered and the reaction mixture was washed with NaHCO3 5% (120 ml + 3 × 50 ml), water (50 ml) and brine (25 ml). The solution was dried over Na2SO4 and evaporated to obtain an oily residue (23 g). The crude product was purified by gravimetric chromatography on silica gel 70–230 mesh with a DCM/ethyl acetate gradient. The pure fractions were combined and concentrated into an oily residue (12.3 g, yield 76%). MS ESI: m/z calcd 221.26, found 222.02 [M + Na]+. HPLC: tR 23.5 min. TLC: mobile phase DCM – riv. UV 254 nm: Rf 0.14.
1H NMR (600 MHz, CDCl3, 298 K) δ 1.35 (s, 3H), 2.6–2.71 (d, 2H), 3.4 (m, 2H), 5.1 (s, 2H), 7.35 (m, 5H) ppm (Fig. S1†).
13C NMR (600 MHz, CDCl3, 298 K) δ 19.3, 45.1, 51.6, 56.0, 67.0, 128.2, 128.3, 128.7, 136.5, 156.7 ppm (Fig. S2†).
Tri-tert-butyl 2,2′,2′′-(10-(3-(((benzyloxy)carbonyl) amino)-2-hydroxy-2-methylpropyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl)triacetate. To a solution of 1,4,7,10-tetraazacyclododecan-1,4,7-triacetic acid tri-tert-butyl ester (5.8 g; 11.3 mmol) and DIPEA (11.2 ml; 65 mmol) in ACN (10 ml), compound 2 (4.5 g; 17 mmol) was added by dissolving in ACN (20 ml). The reaction mixture was heated at 60 °C for 6 h and RT overnight. The mixture was then evaporated and dissolved in ethyl acetate (40 ml), washed with water (4 × 40 ml) and brine (4 × 40 ml) and evaporated to residue. The product was purified by flash chromatography (silica gel 230–400 mesh, DCM/MeOH 95
:5 as an eluent) and evaporated (6.75 g; yield 81%) MS ESI: m/z calcd 735.96, found 736.55 [M + H]+. UPLC: tR 4.67 min.
1H NMR (600 MHz, CDCl3, 298 K) δ 1.4–1.44 (m, 30H), 2.15–3.5 (br m, 26H), 5.03 (m, 2H), 7.2–7.31 (m, 5H) ppm (Fig. S3†).
13C NMR (600 MHz, CDCl3, 298 K) δ 25.2 (CH3), 28.1 (CH3), 49.4–58.2 (CH2), 66.4 (CH2), 66.6 (CH), 72.2 (CH2), 82.3–82.9 (C), 127.9–128.4 (CH), 137.2 (C), 158.1 (CO), 171.7–172.7 (CO) ppm (Fig. S4†)
Tri-tert-butyl 2,2′,2′′-(10-(3-amino-2-hydroxy-2-methylpropyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl) triacetate. Compound 3 (6.75 g; 9.17 mmol) was dissolved in MeOH (50 ml) together with palladium on carbon 5% (50% water) (400 mg) and was hydrogenated at RT and 1 atm; the reaction was followed up by HPLC and MS/ESI until complete. The catalyst was then filtered and the solution was evaporated to residue (5.35 g, yield 97%). MS ESI: m/z calcd 601.83, found 602.35 [M + H]+.
1H NMR (600 MHz, CDCl3, 298 K) δ 1.4–1.44 (m, 30H), 2.0–3.7 (br m, 26H), ppm (Fig. S5†).
13C NMR (600 MHz, CDCl3, 298 K) δ 27.97 (CH3), 28.07–28.26 (CH3), 49.0–56.9 (CH2), 69.3 (CH2), 71.06 (CH), 72.14 (CH2), 82.3–82.9 (C), 171.8–172.8 (CO) ppm (Fig. S6†)
Tri-tert-butyl 2,2′,2′′-(10-(2-hydroxy-3-(4-(((3S,5R,10S,12S,13R,17R)-12-hydroxy-17-((R)-5-methoxy-5-oxopentan-2-yl)-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-3-yl)amino)-4-oxobutanamido)-2-methylpropyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl)triacetate. Compound A (930 mg; 1.55 mmol) and compound B (780 mg; 1.55 mmol) were dissolved in DMF (10 ml); DIPEA (900 mg; 1.21 ml; 7 mmol) and HATU (648 mg; 1.7 mmol) were added to the reaction mixture. After 24 h of stirring at RT, the reaction was complete and the product precipitated by adding 15 ml of water. The solid was isolated and dissolved in ethyl acetate (25 ml), washed with Na2CO3 5% (2 × 7 ml), H2O (5 × 7 ml) and brine (7 ml). The organic solution was dried over Na2SO4 and evaporated to residue (1.87 g). The crude product was used in the next reaction without further purification. MS ESI: m/z calcd 1089.51, found 1089.68 [M + H]+. HPLC tR 26 min.
1H NMR (600 MHz, CDCl3, 298 K) δ 0.66 (s, 3H), 0.95 (m, 6H), 1–2.19 (br m, 62H; steroidal protons), 2.1–3.61 (br m, 26H), 3.64 (s, 3H) ppm (Fig. S7†).
13C NMR (600 MHz, CDCl3, 298 K) δ 12.86 (CH3), 14.30 (CH3), 17.44 (CH3), 23.7 (CH2), 24.9 (CH2), 25.7 (CH2), 26.0 (CH2), 26.8 (CH3), 28.1 (CH3), 29.0 (CH2), 30.6 (CH2), 30.1 (CH2), 32.5 (CH2), 32.7 (CH2), 33.1 (CH2), 34.6 (CH), 35.2 (C), 35.9 (CH), 37.4 (CH), 45.5 (CH), 47.4 (CH), 48.5 (CH), 50.5 (CH3), 51.6 (CH2), 55.5 (CH2), 56.7 (CH2), 60.5 (CH2), 61.8 (CH2), 72.4 (CH), 73.3 (CH), 82.5 (C), 82.9 (C), 171.5 (CO), 172.1 (CO), 174.8 (CO) ppm (Fig. S8†).
2,2′,2′′-(10-(3-(4-(((3S,5R,10S,12S,13R,17R)-17-((R)-4-carboxybutan-2-yl)-12-hydroxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-3-yl) amino)-4-oxobutanamido)-2-hydroxy-2-methylpropyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl)triacetic acid (HIBDO3A-DCA). The crude product of reaction 5 (1.8 g; 1.55 mmol) was dissolved in DCM (10 ml) and TFA (2 ml) was added; after evaporation, fresh TFA (7 ml) and 2 drops of triisopropylsilane were added. After one night at RT, the product was precipitated with diethyl ether, isolated and dried under vacuum (1.56 g).
The solid was dissolved in water (10 ml) and basified to pH 12 with 2N NaOH. After overnight incubation at RT and 3 hours at 50 °C, the deprotection was complete. Product 6 was then purified with an AKTA purifier using an Amberchrome CG161M resin column and eluted with a water/ACN gradient. The fractions containing the pure product were evaporated and freeze-dried (938 mg, yield 67%, and purity >91%). MS ESI: m/z calcd 907.16, found 907.53 [M + H]+. UPLC tR 4.57 min (Fig. S9†).
1H NMR (600 MHz, DMSO d6, 298 K) δ 0.59 (s, 3H), 0.9 (m, 6H), 0.94–1.99 (br m, 26H; steroidal protons), 2.04–2.26 (m, 2H); 2.27–2.47 (6H); 2.53–3.20 (br m, 18H), 3.22–3.70 (br m, 8H); 3.78 (s, 1H); 3.95 (s, 1H); 7.75 (s, 1H); 7.87 (d, 1H) ppm (Fig. S10†).
13C NMR (600 MHz, DMSO d6, 298 K) δ 13.03 (CH3), 17.52 (CH3), 23.84 (CH3), 24.05 (CH2), 24.36 (CH3) 25.16 (CH2), 26.33 (CH2), 27.04 (CH2), 27.72 (CH2), 28.41 (CH), 29.30 (CH2), 30.87 (CH2), 30.99 (CH2), 31.35 (CH2), 31.44 (CH2), 31.85 (CH2), 32.06 (CH2), 32.75 (CH), 34.73 (C), 35.52 (CH), 36.03 (CH), 36.9 (CH), 44.85 (CH), 46.63 (C), 46.79 (CH), 48.06 (CH), 48.15 (CH2), 49.87 (CH2), 51.88 (CH2), 52.48 (CH2), 52.66 (CH2), 55.78 (CH2), 56.38 (CH2), 63.52 (CH2), 71.72 (CH), 73.34 (C), 170.24 (CO), 171.34 (CO),172.07 (CO), 172.63 (CO), 175.53 (CO) ppm (Fig. S11†).
Lanthanide complexation. Ligand 6 was dissolved in water and 0.1 N NaOH was added to adjust the pH to 7. Then, a slight excess of the metal salt was added, using a solution of GdCl3 or EuCl3 in water, until the Xylenol Orange assay53 showed free metal in solution. The mixture was filtered and purified over an AmberChrom CG161 resin (AKTA purifier) with a water/ACN gradient: the pure product was eluted with 25% ACN. The fractions were evaporated and freeze-dried. For the Gd complex: yield 40%, purity >99%. MS ESI: m/z calcd 1061.39, found 1062.57 [M + H]+. UPLC tR 4.61 min (Fig. S12†).
High resolution 1H-NMR
Eu-HIBDO3A-DCA was dissolved in D2O (20 mM) and the pH was adjusted by addition of DCl or KOD and checked with a glass electrode connected to an AsInstruments pH-meter. 1H-NMR monodimensional and 2D-EXSY spectra were recorded at 14.1T on a Bruker Avance 600 spectrometer at 298 K. The temperature was controlled with Bruker thermostat units.The gadolinium concentration of the complexes was determined as follows: gadolinium complex solutions were mixed in equal volumes with 37% HCl and heated in sealed vials at 120 °C overnight to solubilize the free Gd3+ aqua ion. The R1 of the solution was measured at 25 °C and 21.5 MHz and the concentration was determined using the equation: R1 = R1d + r1P × [Gd], where R1d is the diamagnetic contribution (0.5 s−1) and r1P is the relaxivity of free Gd(III) aqua ions (13.5 mM−1 s−1) under the same experimental conditions.
The acid catalyzed Gd-complex dissociation was measured as follows: Gd-HIBDO3A-DCA and Gd-HPDO3A were dissolved in water and HCl was added to final concentrations of 1 M HCl and 0.2 mM Gd complexes. The solutions were incubated at room temperature and the longitudinal relaxation rate (R1) was measured at 21 MHz and 298 K for 6 days. Two samples were measured for each complex and the average of the observed R1 values was calculated. The ratios between R1 after a certain time of incubation and its original value [R1(t)/R1(0)] were plotted versus time.
Transmetallation experiments were performed as follows: gadolinium complexes were dissolved in a phosphate buffer (26 mM KH2PO4 and 41 mM Na2HPO4) at pH 7. ZnCl2 solution (250 mM ZnCl2 in H2O) was added to give an equimolar Gd:Zn solution. The solutions were incubated at 310 K and the longitudinal relaxation rate (R1) was measured at 21 MHz and 298 K for 7 days. Two samples were measured for each complex and the average of the observed R1 values was calculated. The ratios between R1 after a certain time of incubation and its original value [R1(t)/R1(0)] were plotted versus time.
The interaction of Gd-HIBDO3A-DCA with HSA was studied using the well-established proton relaxation enhancement (PRE) method (fitting model in the ESI†).54 Namely, the apparent binding constant (Ka) and the relaxivity of the resulting adduct (r1b) were determined by measuring R1 values of Gd-HIBDO3A-DCA solutions at a fixed Gd concentration, as a function of increasing concentrations of protein, in PBS at 298 K, 21.5 MHz, and pH 7.4. The number of independent binding sites (n) was calculated by measuring the R1 values of HSA solutions at a fixed concentration as a function of increasing concentrations of Gd-HIBDO3A-DCA (in PBS at 298 K, 21.5 MHz, pH 7.4).
The relaxometry competition tests for the hydrophobic sites of HSA were done by measuring the R1 values of solutions containing Gd-HIBDO3A-DCA (0.6 mM), HSA (0.2 mM) and increasing concentrations of the HSA-binders in PBS at 298 K, 21.5 MHz, and pH 7.4. Iodipamide was used for Sudlow site I (subdomain IIA), ibuprofen for Sudlow site II (subdomain IIIA) and methyl orange for subdomain IB.
NMRD profiles were recorded using a Stelar SpinMaster Fast Field-Cycling (FFC) relaxometer at a continuum of proton frequencies from 0.01 MHz to 20 MHz; additional points were obtained between 21.5 MHz and 80 MHz with a Bruker WP80 electromagnet coupled to a Stelar SpinMaster spectrometer. Both systems were equipped with Stelar VTC-91 temperature control and the internal temperature was checked with a calibrated RS PRO RS55-11 digital thermometer. The samples consisted of 1 mM Gd(III) complex in the phosphate buffer and in human serum. Data were analysed and fitted using the SBM and Freed's equations (fitting model in the ESI†). The weighted average of τm, determined from the fitting of the 17O NMR data, was applied as a fixed parameter in the fitting of the NMRD profiles of Gd-HIBDO3A-DCA in the buffer and in human serum.
Octanol–water partition coefficient (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) P)
P)
Octanol–water partition coefficients were obtained by the addition of 500 μl of 1-octanol to 500 μl of a 0.5 mM water solution of Gd-HIBDO3A-DCA or Gd-HPDO3A. The resulting mixture was vigorously shaken for 2 h using a horizontal mechanical shaker. The separation of the two phases was then achieved by centrifugation for 10 minutes at 10.000 rpm and 25 °C. Aliquots of 200 μl were withdrawn from the aqueous layer and the amount of Gd-complex was determined through the relaxometry method as reported above. The quantity of Gd-complexes present in the organic layer was calculated as a difference from the total quantity originally introduced. P values were calculated as the ratio between the quantity of the Gd-complex in 1-octanol and in water layers, respectively, and reported as log values.
Assessment of blood elimination by ICP-MS
The pharmacokinetics of intravenously administered Gd-HIBDO3A-DCA was assessed by ICP-MS quantification of the Gd content in plasma. For this purpose, after the intravenous injection of 0.05 mmol kg−1 Gd-HIBDO3A-DCA to healthy BALB/c mice (n = 3, each group), blood was collected from mouse tail veins at variable time points (t = 3 min, 7 min, 15 min, 30 min, 1 h and 4 h). Before ICP-MS analysis, blood samples were digested with concentrated HNO3 (70%) under microwave heating (Milestone MicroSYNTH Microwave lab-station, Balgach, Switzerland, equipped with an optical fiber temperature control and HPR-1000/6M high-pressure reactor, Milestone, Bergamo, Italy). After digestion, 2 mL of ultrapure water were added to each sample. The specimens were then subjected to ICP-MS analysis (Element-2; Thermo-Finnigan, Rodano (MI), Italy) to measure the concentration of Gd with respect to standard curves. The results were reported as the Gd micromolar concentration as a function of the collection time. A two-compartment pharmacokinetic model was used to analyze the data and to calculate the pharmacokinetic parameters.55Biodistribution study
BALB/c mice were used for the biodistribution study of Gd-HIBDO3A-DCA and ProHance. The mice were euthanized 4 h after the intravenous injection of complexes (0.05 mmol kg−1), and the tissues (liver, spleen, kidneys, brain, and tumors) were collected for Gd analysis. Organs were weighed, mixed with 0.5 mL of concentrated HNO3 (70%) and digested under microwave heating. After digestion, 3 mL of ultrapure water were added to each sample. The specimens were then subjected to ICP-MS analysis. The experiments were performed in triplicate.All mice were maintained under environmentally controlled conditions, including 12-hour light/dark cycles, a temperature range of 20–23 °C, and a relative humidity of 50%. They had access to food and water ad libitum. The mice were group-housed in well-ventilated cages with appropriate cage enrichment. Each mouse was assigned an ear tag for identification purposes, and randomization was applied to mitigate measurement bias.
All procedures involving the animals adhered to national and international regulations concerning experimental animals (L.D. 26/2014; Directive 2010/63/EU) and received approval from the Committee on Animal Care.
MRI in a mouse tumor model. MR images were obtained 10–13 days after the inoculation of the tumor, with tumor dimensions ranging from 80 to 200 mm3. To administer the contrast agent, an intravenous catheter was inserted into the tail vein of the animal while it was under anaesthesia, prior to positioning it inside the MR scanner.
The animals were anesthetized by intramuscular injection of a combination of Tiletamine/Zolazepam (Zoletil 100, Virbac, Milan, Italy) at a dosage of 20 mmol kg−1 and xylazine (Rompun; Bayer, Milan, Italy) at a dosage of 5 mmol kg−1. To maintain the body temperature of the mice, a heated pad was used in addition to monitoring the breathing rate using an air pillow placed beneath the animals (SA Instruments, Stony Brook, NY, USA).
Mice were injected with either gadoteridol (ProHance, Bracco) or Gd-HIBDO3A-DCA at a dose of 0.05 mmol kg−1. MR images were acquired both at 7T using a Bruker Pharmascan MRI system equipped with a 35 mm 1H/1H volume coil and at 1T using a Bruker ICON MRI system equipped with a 35 mm 1H volume coil.
1H sequences (RARE T2-weighted) were acquired on each animal to obtain an accurate anatomical reference using the following parameters: TR = 4000 ms, TE = 35 ms, RARE factor = 24, flip angle = 180°, number of averages = 2, FOV = 30 mm × 30 mm, slice thickness = 1 mm, matrix size 128 × 128, and spatial resolution = 0.234 mm per pixel × 0.234 mm per pixel.
A series of T1-weighted MSME scans were acquired before and after (for 50 minutes) the intravenous administration of the gadolinium complex to track the kinetics of the contrast agent in the diseased mass. The scans were performed with the following parameters: TR = 221.714 ms, TE = 8 ms, number of averages = 6, FOV = 30 mm × 30 mm, slice thickness = 1 mm, matrix size 128 × 128, spatial resolution = 0.234 mm per pixel × 0.234 mm per pixel, and acquisition time = 2 minutes and 50 seconds. A 5 mm NMR glass tube containing 0.5 mM ProHance in water was inserted near the mouse body as a reference.
After acquiring the images, the T1 contrast enhancement (SE%) was calculated using the following formula: SE% = [(SI)post − (SI)pre]/(SI)pre × 100, where (SI)post and (SI)pre represent the signal intensities (normalized by dividing for the external standard reference) after and before the injection of both Gd(III)-contrast agents, respectively. Regions of Interest (ROIs) were manually delineated within the reference standard sample, tumors, kidneys, and various regions of the liver. The SE% was calculated in all the ROIs as described above.
MRI angiography. Healthy female BALB/c mice weighing approximately 20 g were used in contrast enhanced MR blood pool imaging. The mouse was anesthetized by injecting a mixture of 20 mg kg−1 tiletamine/zolazepam (Zoletil 100; Virbac, Milan, Italy) and 5 mg kg−1 xylazine (Rompun; Bayer, Milan, Italy) and placed in a solenoid Tx/Rx coil with an inner diameter of 3.5 cm. The contrast agent Gd-HIBDO3A-DCA was injected via the tail vein at a dose of 0.05 mmol kg−1.
MR images were acquired before and at 2, 5, 10, 15, 30, 45 and 60 min post-injection of the contrast agents on a 1T MRI Bruker Icon™ system (Bruker BioSpin MRI, Ettlingen, Germany). A 35 mm Tx/Rx mouse solenoid whole body coil was used for both RF excitation and reception of the MR signal. A 3D GRE fast low angle shot (FLASH) pulse sequence (TR 10 ms; TE 4.1 ms; flip angle 30°; FOV 80 × 40 × 40 mm; MTX 192 × 96 × 96; NEX 2; temporal resolution 3 min 4 s per image) with an isotropic spatial resolution of 0.417 mm. Three dimensional maximum intensity projection (MIP) images were reconstructed using the ImageJ program by subtracting pre-contrast images from post-contrast images.
MRI in vitro. A phantom containing 8 cuvettes was prepared to evaluate and compare the MRI signal of ProHance and Gd-HIBDO3A-DCA at two different concentrations (0.1 and 0.5 mM). The compounds were dissolved in both PBS and serum, creating a scenario that mimics the presence of albumin.
MR image was acquired at 1T using a Bruker ICON MRI system equipped with a 35 mm 1H volume coil.
A T1-weighted MSME scan was acquired with the following parameters: TR = 70 ms, TE = 9.24 ms, number of averages = 16, FOV = 30 mm × 30 mm, slice thickness = 2 mm, matrix size 96 × 96, spatial resolution = 0.3125 mm per pixel × 0.3125 mm per pixel, and acquisition time = 1 minute and 48 seconds.
Author contributions
F. H. carried out the in vitro physicochemical characterization of the Gd-HIBDO3A-DCA complex. C. C. carried out the synthesis of the Gd-HIBDO3A-DCA complex. E. C. carried out the MRI studies in vitro and on murine tumor models. S. A. and E. G. conceived the original project plan and E. G. coordinated the activities of the project team. All authors contributed to the preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
Data are available on request from the authors.Acknowledgements
The authors acknowledge the Italian Ministry of Research for FOE contribution to the Euro-BioImaging MultiModal Molecular Imaging Italian Node (https://www.mmmi.unito.it).References
- J. Starekova, A. Pirasteh and S. B. Reeder, Update on Gadolinium Based Contrast Agent Safety, From the AJR Special Series on Contrast Media, Am. J. Roentgenol., 2023 DOI:10.2214/AJR.23.30036.
- A. J. van der Molen, C. C. Quattrocchi, C. A. Mallio and I. A. Dekkers, Ten years of gadolinium retention and deposition: ESMRMB-GREC looks backward and forward, Eur. Radiol., 2024, 34, 600 CrossRef PubMed.
- N. Lyad, M. S. S. Ahmad, S. G. Alkhatib and M. Hjouj, Gadolinium contrast agents- challenges and opportunities of a multidisciplinary approach: Literature review, Eur. J. Radiol. Open, 2023, 11, 100503 CrossRef PubMed.
- Q. N. Do, R. E. Lenkinski, G. Tircso and Z. Kovacs, How the Chemical Properties of GBCAs Influence Their Safety Profiles In Vivo, Molecules, 2021, 27, 58 CrossRef PubMed.
- J. W. Choi and W. J. Moon, Gadolinium Deposition in the Brain: Current Updates, Korean J. Radiol., 2019, 20, 134 CrossRef PubMed.
- S. Bussi, A. Coppo, C. Botteron, V. Fraimbault, A. Fanizzi, E. De Laurentiis, S. Colombo Serra, M. A. Kirchin, F. Tedoldi and F. Maisano, Differences in gadolinium retention after repeated injections of macrocyclic MR contrast agents to rats, J. Magn. Reson. Imaging, 2018, 47, 746 CrossRef PubMed.
- S. Bussi, A. Coppo, R. Celeste, A. Fanizzi, A. Fringuello Mingo, A. Ferraris, C. Botteron, M. A. Kirchin, F. Tedoldi and F. Maisano, Macrocyclic MR contrast agents: Evaluation of multiple-organ gadolinium retention in healthy rats, Insights Imaging, 2020, 11, 1 CrossRef PubMed.
- C. Robic, M. Port, O. Rousseaux, S. Louguet, N. Fretellier, S. Catoen, C. Factor, S. Le Greneur, C. Medina, P. Bourrinet, I. Raynal, J. M. Idée and C. Corot, Physicochemical and Pharmacokinetic Profiles of Gadopiclenol: A New Macrocyclic Gadolinium Chelate With High T1 Relaxivity, Invest. Radiol., 2019, 54, 475 CrossRef CAS PubMed.
- N. Fretellier, M. Rasschaert, J. Bocanegra, P. Robert, C. Factor, A. Seron, J. M. Idée and C. Corot, Safety and Gadolinium Distribution of the New High-Relaxivity Gadolinium Chelate Gadopiclenol in a Rat Model of Severe Renal Failure, Invest. Radiol., 2021, 56, 826 CrossRef CAS PubMed.
- P. Robert, V. Vives, M. Rasschaert, J. Hao, M. Soares, M. Lemaître, A. Dencausse and S. Catoen, Detection of Brain Metastases by Contrast-Enhanced MRI: Comparison of Gadopiclenol and Gadobenate in a Mouse Model, Invest. Radiol., 2024, 59, 131 CrossRef CAS PubMed.
- J. Lohrke, M. Berger, T. Frenzel, C. S. Hilger, G. Jost, O. Panknin, M. Bauser, W. Ebert and H. Pietsch, Preclinical Profile of Gadoquatrane: A Novel Tetrameric, Macrocyclic High Relaxivity Gadolinium-Based Contrast Agent, Invest. Radiol., 2022, 57, 629–638 CrossRef CAS PubMed.
- R. B. Lauffer, D. J. Parmelee, S. U. Dunham, H. S. Ouellet, R. P. Dolan, S. Witte, T. J. McMurry and R. C. Walovitch, MS-325: albumin-targeted contrast agent for MR angiography, Radiology, 1998, 207, 529 CrossRef CAS PubMed.
- C. De Haën, P. L. Anelli, V. Lorusso, A. Morisetti, F. Maggioni, J. Zheng, F. Uggeri and F. M. Cavagna, Gadocoletic acid trisodium salt (B22956/1) - A new blood pool magnetic resonance contrast agent with application in coronary angiography, Invest. Radiol., 2006, 41, 279 CrossRef PubMed.
- E. Gianolio, C. Cabella, S. Colombo Serra, G. Valbusa, F. Arena, A. Maiocchi, L. Miragoli, F. Tedoldi, F. Uggeri and M. Visigalli, et al. B25716/1: a novel albumin-binding GdAAZTA MRI contrast agent with improved properties in tumor imaging, J. Biol. Inorg. Chem., 2014, 19, 715 CrossRef CAS PubMed.
- D. L. Longo, F. Arena, L. Consolino, P. Minazzi, S. Geninatti-Crich, G. B. Giovenzana and S. Aime, Gd-AAZTA-MADEC, an improved blood pool agent for DCE-MRI studies on mice on 1T scanners, Biomaterials, 2016, 75, 47 CrossRef CAS PubMed.
- V. Henrotte, L. Vander Elst, S. Laurent and R. N. Muller, Comprehensive investigation of the non-covalent binding of MRI contrast agents with human serum albumin, J. Biol. Inorg. Chem., 2007, 12, 929 CrossRef CAS PubMed.
- A. R. Baek, H. K. Kim, S. Kim, J. Yang, M. K. Kang, J. J. Lee, B. Sung, H. Lee, M. Kim, A. E. Cho, J. A. Park and Y. Chang, Effect of Structural Fine-Tuning on Chelate Stability and Liver Uptake of Anionic MRI Contrast Agents, J. Med. Chem., 2022, 65, 6313–6324 CrossRef CAS PubMed.
- M. K. Islam, A. R. Baek, B. W. Yang, S. Kim, D. W. Hwang, S. W. Nam, G. H. Lee and Y. Chang, Manganese(II) Complex of 1,4,7-Triazacyclononane-1,4,7-Triacetic Acid (NOTA) as a Hepatobiliary MRI Contrast Agent, Pharmaceuticals, 2023, 16, 602 CrossRef CAS PubMed.
- R. C. Hall, J. Qin, V. Laney, N. Ayat and Z. R. Lu, Manganese(II) EOB-Pyclen Diacetate for Liver-Specific MRI, ACS Appl. Bio Mater., 2022, 5, 451–458 CrossRef CAS PubMed.
- Z. Cai, L. Jiang, Y. Cao, S. Fu, S. Wang, Y. Jiang, H. Gu, N. Li, X. Fu, S. Tang, J. Zhu, W. Cao, L. Zhong, Z. Cheng, C. Xia, S. Lui, B. Song, Q. Gong and H. Ai, Lipophilic Group-Modified Manganese(II)-Based Contrast Agents for Vascular and Hepatobiliary Magnetic Resonance Imaging, J. Med. Chem., 2024, 67, 5011–5031 CrossRef CAS PubMed.
- J. Wahsner, E. M. Gale, A. Rodríguez-Rodríguez and P. Caravan, Chemistry of MRI Contrast Agents: Current Challenges and New Frontiers, Chem. Rev., 2019, 119, 957 CrossRef CAS PubMed.
- K. B. Maier, L. N. Rust, F. Carniato, M. Botta and M. Woods, α-Aryl substituted GdDOTA derivatives, the perfect contrast agents for MRI?, Chem. Commun., 2024, 60, 2898–2901 RSC.
- L. R. Tear, C. Carrera, E. Gianolio and S. Aime, Towards an Improved Design of MRI Contrast Agents: Synthesis and Relaxometric Characterisation of Gd-HPDO3A Analogues, Chem. – Eur. J., 2020, 26, 6056 CrossRef CAS PubMed.
- L. R. Tear, C. Carrera, C. B. Dhakan, E. Cavallari, F. Travagin, C. Calcagno, S. Aime and E. Gianolio, An albumin-binding Gd-HPDO3A contrast agent for improved intravascular retention, Inorg. Chem. Front., 2021, 8, 4014 RSC.
- S. Aime, S. Baroni, D. Delli Castelli, E. Brücher, I. Fábián, S. Colombo Serra, A. Fringuello Mingo, R. Napolitano, L. Lattuada, F. Tedoldi and Z. Baranyai, Exploiting the Proton Exchange as an Additional Route to Enhance the Relaxivity of Paramagnetic MRI Contrast Agents, Inorg. Chem., 2018, 57, 5567 CrossRef CAS PubMed.
- L. Lattuada, D. Horváth, S. Colombo Serra, A. Fringuello Mingo, P. Minazzi, A. Bényei, A. Forgács, F. Fedeli, E. Gianolio, S. Aime, G. B. Giovenzana and Z. Baranyai, Enhanced Relaxivity of Gd III -Complexes with HP-DO3A-like Ligands upon the Activation of the Intramolecular Catalysis of the Prototropic Exchange, Inorg. Chem. Front., 2021, 8, 1500 RSC.
- R. Stefania, L. Palagi, E. Di Gregorio, G. Ferrauto, V. Dinatale, S. Aime and E. Gianolio, Seeking for Innovation with Magnetic Resonance Imaging Paramagnetic Contrast Agents: Relaxation Enhancement via Weak and Dynamic Electrostatic Interactions with Positively Charged Groups on Endogenous Macromolecules, J. Am. Chem. Soc., 2024, 146, 134 CrossRef CAS PubMed.
- W. Dastrù, V. Menchise, G. Ferrauto, S. Fabretto, C. Carrera, E. Terreno, S. Aime and D. Delli Castelli, Modulation of the Prototropic Exchange Rate in pH-Responsive Yb-HPDO3A Derivatives as ParaCEST Agents, ChemistrySelect, 2018, 3, 6035 CrossRef.
- P. L. Anelli, L. Lattuada and F. Uggeri, One-pot Mitsunobu-Staudinger preparation of 3-aminocholan-24-oic acid esters from 3-hydroxycholan-24-oic acid esters, Synth. Commun., 1998, 28, 109 CrossRef CAS.
- D. Delli Castelli, M. C. Caligara, M. Botta, E. Terreno and S. Aime, Combined High Resolution NMR and 1H and 17O Relaxometric Study Sheds Light on the Solution Structure and Dynamics of the Lanthanide(III) Complexes of HPDO3A, Inorg. Chem., 2013, 52, 7130 CrossRef CAS PubMed.
- G. Ferrauto, D. Delli Castelli, L. Leone, M. Botta, S. Aime, Z. Baranyai and L. Tei, Modifying LnHPDO3A Chelates for Improved T 1 and CEST MRI Applications, Chem. – Eur. J., 2019, 25, 4184 CrossRef CAS PubMed.
- S. Aime, M. Botta, Z. Garda, B. E. Kucera, G. Tircso, V. G. Young and M. Woods, Properties, Solution State Behavior, and Crystal Structures of Chelates of DOTMA, Inorg. Chem., 2011, 50, 7955 CrossRef CAS PubMed.
- K. B. Maier, L. N. Rust, C. I. Kupara and M. Woods, Chem. – Eur. J., 2023, 29, e202301887 CrossRef CAS PubMed.
- S. Aime, M. Chiaussa, G. Digilio, E. Gianolio and E. Terreno, Contrast agents for magnetic resonance angiographic applications: 1H and 17O NMR relaxometric investigations on two gadolinium(III) DTPA-like chelates endowed with high binding affinity to human serum albumin, J. Biol. Inorg. Chem., 1999, 4, 766 CrossRef CAS PubMed.
- É. Tóth, F. Connac, L. Helm, K. Adzamli and A. E. Merbach, Direct assessment of water exchange on a Gd(III) chelate bound to a protein, J. Biol. Inorg. Chem., 1998, 3, 606 CrossRef.
- G. Sudlow, D. J. Birkett and D. N. Wade, The Characterization of Two Specific Drug Binding Sites on Human Serum Albumin, Mol. Pharmacol., 1975, 11, 824 CAS.
- D. Sudlow, D. J. Birkett and D. N. Wade, Further Characterization of Specific Drug Binding Sites on Human Serum Albumin, Mol. Pharmacol., 1976, 12, 1052 Search PubMed.
- F. Zsila, Subdomain IB Is the Third Major Drug Binding Region of Human Serum Albumin: Toward the Three-Sites Model, Mol. Pharmaceutics, 2013, 10, 1668 CrossRef CAS PubMed.
- G. Fanali, A. di Masi, V. Trezza, M. Marino, M. Fasano and P. Ascenzi, Human Serum Albumin: From Bench to Bedside, Mol. Aspects Med., 2012, 33, 209 CrossRef CAS PubMed.
- S. Curry, Lessons from the crystallographic analysis of small molecule binding to human serum albumin, Drug Metab. Pharmacokinet., 2009, 24, 342 CrossRef CAS PubMed.
- S. Aime and Z. Baranyai, How the Catalysis of the Prototropic Exchange Affects the Properties of Lanthanide(III) Complexes in Their Applications as MRI Contrast Agents, Inorg. Chim. Acta, 2022, 532, 120730 CrossRef CAS.
- E. Boros, R. Srinivas, H. K. Kim, A. M. Raitsimring, A. V. Astashkin, O. G. Poluektov, J. Niklas, A. D. Horning, B. Tidor and P. Caravan, Intramolecular Hydrogen Bonding Restricts Gd–Aqua-Ligand Dynamics, Angew. Chem., Int. Ed., 2017, 56, 5603 CrossRef CAS PubMed.
- M. Woods, M. Botta, S. Avedano, J. Wang and A. D. Sherry, Towards the rational design of MRI contrast agents: a practical approach to the synthesis of gadolinium complexes that exhibit optimal water exchange, Dalton Trans., 2005, 3829 RSC.
- S. Avedano, L. Tei, A. Lombardi, G. B. Giovenzana, S. Aime, D. Longo and M. Botta, Maximizing the relaxivity of HSA-bound gadolinium complexes by simultaneous optimization of rotation and water exchange, J. Chem. Soc., Chem. Commun., 2007, 4726–4728 RSC.
- (a) G. Lipari and A. Szabo, Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity, J. Am. Chem. Soc., 1982, 104, 4546–4559 CrossRef CAS; (b) G. Lipari and A. Szabo, Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 2. Analysis of experimental results, J. Am. Chem. Soc., 1982, 104, 4559–4570 CrossRef CAS.
- W. Xu, Y. Lu, J. Xu, H. Li, R. Lan, R. Gao, Y. Ding, X. Ye, K. Shu, F. Ye, Z. Yan and L. Dai, Rational Design of Gd-DOTA-Type Contrast Agents for Hepatobiliary Magnetic Resonance Imaging, J. Med. Chem., 2023, 66, 8993–9005 CrossRef CAS PubMed.
- W. Xu, X. Ye, M. Wu, X. Jiang, L. Hang Hugo TSE, Y. Gu, K. Shu, L. Xu, Y. Jian, G. Mo, J. Xu, Y. Ding, R. Gao, J. Shen, F. Ye, Z. Yan and L. Dai, J. Med. Chem., 2023, 66, 14669–14682 CrossRef CAS PubMed.
- J. Wang, H. Wang, I. A. Ramsay, D. J. Erstad, B. C. Fuchs, K. K. Tanabe, P. Caravan and E. M. Gale, Manganese-Based Contrast Agents for Magnetic Resonance Imaging of Liver Tumors: Structure-Activity Relationships and Lead Candidate Evaluation, J. Med. Chem., 2018, 61, 8811–8824 CrossRef CAS PubMed.
- Y. Xue, B. Xiao, Z. Xia, L. Dai, Q. Xia, L. Zhong, C. Zhu and J. A. Zhu, New OATP-Mediated Hepatobiliary-Specific Mn(II)-Based MRI Contrast Agent for Hepatocellular Carcinoma in Mice: A Comparison With Gd-EOB-DTPA, J. Magn. Reson. Imaging, 2023, 58, 926–933 CrossRef PubMed.
- Z. Cai, L. Jiang, Y. Cao, S. Fu, S. Wang, Y. Jiang, H. Gu, N. Li, X. Fu, S. Tang, J. Zhu, W. Cao, L. Zhong, Z. Cheng, C. Xia, S. Lui, B. Song, Q. Gong and H. Ai, Lipophilic Group-Modified Manganese(II)-Based Contrast Agents for Vascular and Hepatobiliary Magnetic Resonance Imaging, J. Med. Chem., 2024, 67, 5011–5031 CrossRef CAS PubMed.
- A. R. Baek, H. K. Kim, S. Kim, J. U. Yang, M. K. Kang, J. J. Lee, B. Sung, H. Lee, M. Kim, A. E. Cho, J. A. Park and Y. Chang, Effect of Structural Fine-Tuning on Chelate Stability and Liver Uptake of Anionic MRI Contrast Agents, J. Med. Chem., 2022, 65, 6313–6324 CrossRef CAS PubMed.
- H. O. Axelsson and M. A. Olsson, Synthesis of Cyclen Derivatives, WO2006/112723, 2006.
- A. Barge, G. Cravotto, E. Gianolio and F. Fedeli, How to determine free Gd and free ligand in solution of Gd chelates. A technical note, Contrast Media Mol. Imaging, 2006, 1, 184 CrossRef PubMed.
- S. Aime, M. Botta, M. Fasano, S. Geninatti Crich and E. Terreno, Gd(III) Complexes as Contrast Agents for Magnetic Resonance Imaging: A Proton Relaxation Enhancement Study of the Interaction with Human Serum Albumin, J. Biol. Inorg. Chem., 1996, 1, 312 CrossRef CAS.
- D. J. Parmelee, R. C. Walovitch, H. S. Ouellet and R. B. Lauffer, Preclinical Evaluation of the Pharmacokinetics, Biodistribution, and Elimination of MS-325, a Blood Pool Agent for Magnetic Resonance Imaging, Invest. Radiol., 1997, 32, 741 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qi00894d |
| This journal is © the Partner Organisations 2024 |