
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimple oxime functionalized fluorene polymers for organic solar cells†
Abdullah Adewale
Adeoba
ab,
Xiangyu
Shen
a,
Fuzhen
Bi
 a,
Jianan
Niu
a,
Mingxin
Sun
c,
Shuguang
Wen
*ab and
Xichang
Bao
*ab
a,
Jianan
Niu
a,
Mingxin
Sun
c,
Shuguang
Wen
*ab and
Xichang
Bao
*ab
aCAS Key Laboratory of Bio-Based Materials, Qingdao Institute of Bioenergy and Bioprocess Technology, China. E-mail: wensg@qibebt.ac.cn; baoxc@qibebt.ac.cn
bFunctional Laboratory of Solar Energy, Shandong Energy Institute, Qingdao 266101, China
cQingdao New Energy Shandong Laboratory, Qingdao 266101, China
First published on 12th October 2024
Abstract
Conjugated polymers with simple chemical structures are attractive for the design of high efficiency, wide bandgap (WBG) donor materials used in non-fullerene organic solar cells. Fluorene is one of the most common structures in organic opto-electronic materials. It is herein used, for the first time, functionalized with the oxime group to construct the acceptor unit of D–A polymer donors with a BDT donor unit. The polymers, PBFO-H and PBFO-F, had low-lying HOMOs with a wide bandgap of 2.08 eV and are soluble in chloroform for easy processing. Organic solar cells were constructed using the polymers and a non-fullerene Y6 acceptor. The fluorinated polymer (PBFO-F) had better photovoltaic properties over the non-fluorinated polymer overall, with JSC, VOC, and FF values of 23.17 mA cm−2, 0.84 V, and 55.2%, respectively, and a high PCE of 10.71%. These results demonstrate that fluorene is promising in constructing acceptor units for D–A wide bandgap polymers.
Introduction
Polymer solar cells (PSCs), otherwise known as organic solar cells (OSCs) have drawn scientific and commercial attention because of their numerous advantages.1,2 This class of solar cells is flexible and light-weight, can be processed in low-cost solutions, and is suitable for roll-to-roll mass production.1,3,4 Nowadays, PSCs usually take the structure of a bulk heterojunction (BHJ) in which the conjugated polymer donor is blended with a small molecule acceptor. Fullerene based acceptors were used in the past with a maximum power conversion efficiency (PCE) of around 11%.5 Even though fullerene-based acceptors have good properties such as good solubility, good electron transport and high electron affinity, the fact that they absorb weakly in the visible region of the solar spectrum, have limited energy level tunability, and can easily decompose with long term use, limits their further applications. The discovery of non-fullerene acceptors has led to the improvement of the PCEs of polymer solar cells close to 20%.6–9 This could be attributed to the balance between the open circuit voltage (VOC) and short circuit density (JSC) while the fill factor (FF) has achieved a significant increase when matched against fullerene based solar cells.The conjugated polymer donor used in PSCs usually takes a donor–acceptor (D–A) alternating molecular structure. In order to maximize their performance, the D and A units must be carefully selected so that their absorption spectra and frontier molecular orbital energy levels can be suitably modified to match with the acceptor materials.10–12 Moreover, non-fullerene acceptors (NFAs) usually need a wide band gap (WBG) polymer donor (Eg ≥ 1.8 eV) in order to complement their narrow absorption range.13 Benzodiothiophene (BDT) is commonly used to construct the D building block of the D–A polymer due to its appropriate electron-donating effect which is good for the EHOMO, a large planar structure which helps in hole transport and a face-on molecular orientation of thin films.14,15 On the other hand, the chemical structures which have electron-withdrawing groups such as 3-fluorothieno[3,4-b]thiophene-2-carboxylate (FC-TT), thieno[3,4-c]pyrrole-4,6-dione (TPD), benzo[1,2-c:4,5-c′]dithiophene-4,8-dione (BDD), difluorobenzo[1,2,3]triazole (FBTA), quinoxaline, and benzothiadiazole (BT) have been used as the A building blocks.13,15 The results of the different BDT-acceptor combinations show that the acceptor molecules have noticeable effects on the optical performances of the resulting D–A polymers and by extension, the PCEs of the resulting PSCs.16–20
Fluorene based polymers were known to be initially used for light emitting diodes (LEDs) with absorption at short wavelengths.21 The major advantages of the fluorene compounds include a highly conjugated ring, efficient hole transport, chemical stability, structural rigidity and planarity which give wide band gaps and low HOMO levels, directly leading to a high VOC and JSC.16,22–24 It was later used to design polymer solar cell donor materials by Andersson and co-workers with the resulting PSC having a PCE of 2.2%. After this, many groups have introduced different side-chains into fluorene to improve its various photovoltaic parameters with PCEs of 3.5%,25 4.5%,26 and 6.6%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 27 when used for PSCs. It should be noted that in all of these studies, fluorene is used as the electron-donating unit while its application as the electron-accepting unit in a D–A polymer still remains unreported. Furthermore, fluorene units also feature simple and planar molecular structures, which would be suitable for the construction of efficient polymer donors for OSCs.
27 when used for PSCs. It should be noted that in all of these studies, fluorene is used as the electron-donating unit while its application as the electron-accepting unit in a D–A polymer still remains unreported. Furthermore, fluorene units also feature simple and planar molecular structures, which would be suitable for the construction of efficient polymer donors for OSCs.
In this study, we introduce oxime-functionalized fluorine (FO) as an acceptor unit copolymerized with BDT monomers to synthesize two novel conjugated D–A donor polymers, PBFO-H and PBFO-F, which would be useful for OSCs.28,29 The importance of the oxime functional group is due to its electron withdrawing ability and its usage in conjugated polymers is a fairly new concept, firstly used by He et al. on a thiophene-based monomer.30 Therefore, this work effectively combines the novelty of fluorene as an acceptor unit and oxime as an electron withdrawing functional group. Molecular simulation shows that the fluorene unit is effective as an electron-withdrawing group. The introduction of the fluorine atom is efficient in modulating energy levels and most of its photovoltaic properties. The polymers have a wide bandgap (2.08 eV) and appropriate energy levels and thus were paired with the non-fullerene acceptor Y6 in constructing NFA polymer solar cells. A highest PCE value of 10.71% was obtained for the solar devices with the parameters JSC, VOC and Eloss approaching those of the state-of-the-art systems. These results show that fluorene would be a good choice for constructing the acceptor units in WBG D–A polymer donors for non-fullerene solar cells.
Results and discussion
Materials synthesis and characterization
The oxime-based fluorene monomer was synthesized from scratch as shown in Fig. 1. A hydroxylamine (2) was synthesized using a modification of methods used by Zhu et al.,31 and Aldo et al.32 with about 70% yield. The above was joined as an oxime functional group to a commercial 2,7-dibromo-9H-fluorene-9-one using a method reported by Blake et al.33 A straight-chain alkylthiophene was further introduced through the Stille reaction process. Finally, the thiophenated compound (4) was brominated using N-bromosuccinimide to give the monomer M. The intermediates and final products were confirmed by nuclear magnetic resonance (NMR).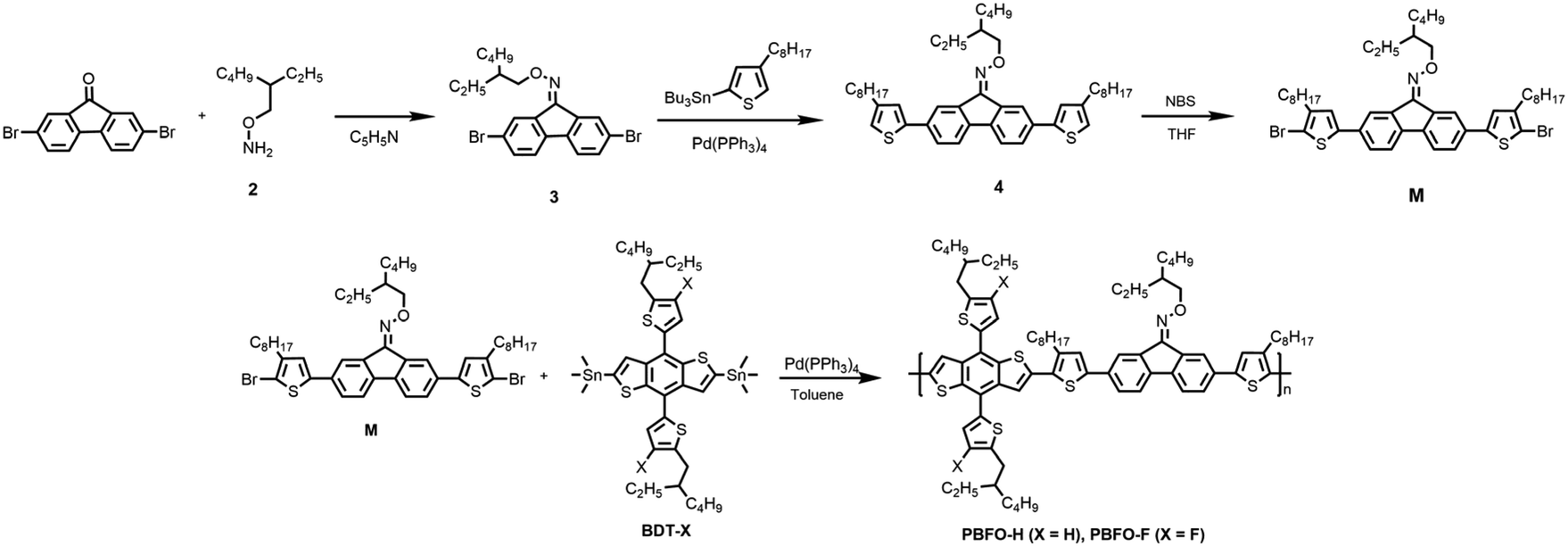 | ||
| Fig. 1 Chemical structure and synthesis routes towards the monomer and polymers PBFO-H and PBFO-F. | ||
To get the polymers, the monomer M was copolymerized with BDT using Pd(PPh3)4 catalysts under Stille conditions. The two polymers synthesized, PBFO-H and PBFO-F, were both soluble in chloroform at room temperature, allowing for easy fabrication of the devices using a simple spin-coating method. The two polymers PBFO-H and PBFO-F have number-average molecular weights (Mn) of 121 and 35 kDa and PDI of 1.71 and 1.64 respectively, measured by high temperature gel permeation chromatography (GPC). The decomposition temperature (Td, 5% weight loss) measured by thermogravimetric analysis (TGA) under a nitrogen atmosphere was 320 °C for PBFO-H and 240 °C for PBFO-F, indicating their good thermal stability for the fabrication of solar cells (Fig. S4†).
Optical properties and energy levels
The optical properties of the pair polymers were studied using ultraviolet-visible (UV-vis) spectroscopy and the results are shown in Fig. 2. The two polymers PBFO-H and PBFO-F in solution showed a low-energy peak of maximum absorption occurring at wavelengths of 477 nm and 505 nm (λmax) respectively. Comparatively, PBFO-F shows a red-shifted absorption peak, and a shoulder peak can also be observed. This behavior indicates that PBFO-F has stronger aggregation tendency in solution than PBFO-H. The absorption spectra for both polymers in thin films are very similar, and PBFO-H has maximum absorption at 507 nm while PBFO-F has maximum absorption at 544 nm, on thin film. The occurrence of shoulder peaks in thin films could be attributed to improved polymer aggregation. The higher shoulder peak of PBFO-F implies its stronger aggregation behavior than PBFO-H, which would be induced by the fluoride incorporated non-covalent bond interaction. The extinction coefficient (εmax) for both polymers PBFO-H and PBFO-F are 3.91 × 104 cm−1 and 3.67 × 104 cm−1 for thin films at ∼505 nm respectively, which are very close values due to their similar backbone. High extinction coefficient values would facilitate light harvesting in thin films (∼100 nm), which is beneficial for efficient charge extraction while avoiding charge recombination. The optical bandgaps of the polymers in film, derived from the absorption onset is 2.08 eV for both polymers which is considered a wide bandgap. In conjunction with the strong absorption range of these polymers (400–544 nm), they are suitable for complementing non-fullerene acceptors (NFAs) due to their narrow bandgap absorption, for example, Y6, which absorbs between 600 and 930 nm and has Eg of 1.33 eV.21 These pairs of polymers showed wider bandgaps when compared to a similar oxime-substituted polymer which used thiophene instead of a fluorene unit (Eg = 2.03 eV) which indicates that fluorene is more suitable for a wider bandgap due to the presence of more conjugation.15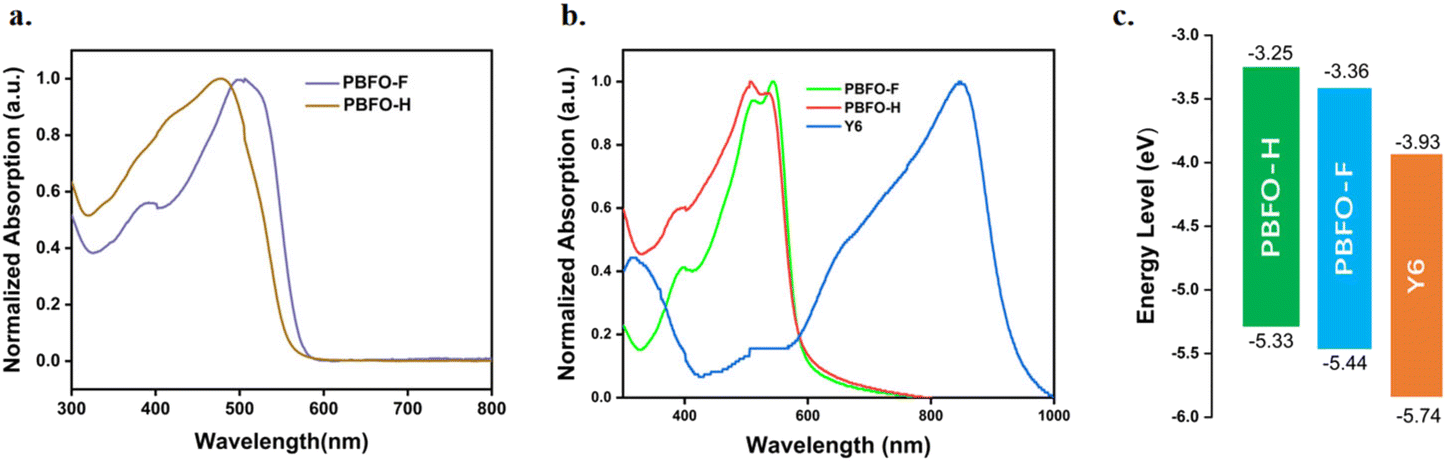 | ||
| Fig. 2 (a) Normalized UV-vis absorption spectra of the polymers in dilute chloroform. (b) Normalized UV-vis absorption spectra of thin films. (c) Energy levels of PBFO-H, PBFO-F and Y6. | ||
Density functional theory (DFT) simulations were carried out at the B3LY/6-31G(d,p) level to understand the molecular geometries and electronic properties of the polymers. The alkyl side chains were replaced with methyl units to minimize computing time (Fig. S1†). The impact of the fluorine atom on the BDT unit is obviously noticed in delocalizing electrons across the backbone of the polymer. The polymer without the fluorine side chain (PBFO-H) has most of its HOMO electron cloud localized on the BDT unit while introducing fluorine helped to spread out the electron cloud throughout the polymer backbone as seen in PBFO-F, making it have a more planar chain which is advantageous for efficient charge transport. As for the LUMO electron cloud, it is majorly situated on the fluorene acceptor side chain which proves it as a novel effective electron-withdrawing unit. Some LUMO electron cloud could also be noticed on the oxime (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–O) functional group which also proves its electron-withdrawing property. The HOMO/LUMO energy levels calculated for PBFO-H and PBFO-F were −4.94/−2.02 and −5.06/−2.09 eV respectively showing that the introduction of fluorine helps to deepen the HOMO level. The dihedral angles between thiophene and BDT units are 22.7° and 21.6° for PBFO-H and PBFO-F respectively, while the thiophene and fluorene molecules are joined at −24.5° and −26.4° for PBFO-H and −24.6° and −26.3° for PBFO-F. The small dihedral angles present in the polymers confirm the planarity which will enable ordered molecular packing and charge transport in devices.
N–O) functional group which also proves its electron-withdrawing property. The HOMO/LUMO energy levels calculated for PBFO-H and PBFO-F were −4.94/−2.02 and −5.06/−2.09 eV respectively showing that the introduction of fluorine helps to deepen the HOMO level. The dihedral angles between thiophene and BDT units are 22.7° and 21.6° for PBFO-H and PBFO-F respectively, while the thiophene and fluorene molecules are joined at −24.5° and −26.4° for PBFO-H and −24.6° and −26.3° for PBFO-F. The small dihedral angles present in the polymers confirm the planarity which will enable ordered molecular packing and charge transport in devices.
The energy levels of the polymers were also investigated using cyclic voltammetry (CV) experiments (Fig. S2†), and the results are shown in Table 1. The HOMO/LUMO energy levels of polymers PBFO-H and PBFO-F were obtained to be −5.33/−3.25 eV and −5.44/−3.36 eV respectively. The effect of introducing fluorine into the polymer is clearly pronounced in reducing the LUMO level and deepening the HOMO level which is useful for it to approach the energy levels of the Y6 acceptor (−5.74/−3.93 eV) in order to reduce energy loss. The difference between the EHOMO of the polymers and ELUMO of Y6 is at least 1.4 eV, which would lead to high VOC when blended together. The HOMO and LUMO energy offsets, ΔEHOMO and ΔELUMO between the PBFO polymers and Y6 is in the least 0.30 eV and 0.57 eV respectively, sufficient enough for driving exciton dissociation.
Photovoltaic and electrical properties
Photovoltaic devices based on the glass/ITO (indium tin oxide)/PEDOT:PSS (poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate))/active layer/PDINN (aliphatic amine-functionalized perylene-diimide)/Ag structure were constructed in order to evaluate the photovoltaic properties of the polymers. Y6 was used as the acceptor layer, blended together with the polymers to form the active layers. The current density–voltage (J–V) graph of the optimized devices with a donor/acceptor ratio of 1:1 is shown in Fig. 3a and the photovoltaic properties are summarized in Table 2. A reference of the detailed device fabrication process is provided in the ESI (Tables S1 and S2†). The devices achieved maximum performances when they were optimized through the addition of 0.2% diiodooctane (DIO). Under these conditions, the PBFO-H:Y6 based device showed an efficiency of 7.26% with VOC of 0.77 V, a fill factor of 47.38% and JSC of 19.94 mA cm−2. With fluorine atom introduced into the polymer donor backbone, the device based on PBFO-F:Y6 had a stellar efficiency of 10.71% with a VOC value of 0.84 V, a JSC value of 23.17 mA cm−2, and an FF value of 55.2%. The simultaneous improvement of all the device parameters with the introduction of fluorine shows that it is an efficient strategy for improving the photovoltaic performance of the devices. The two devices based on PBFO-H and PBFO-F have VOC values of 0.77 V and 0.84 V respectively, which translates to respective Eloss of 0.55 and 0.49 eV, computed from the equation Eloss = Eg − VOC. The Eloss for the PBFO-F:Y6 device, 0.49 eV, is very close to the lowest energy loss found for OSCs.34,35
 | ||
| Fig. 3 (a) J–V curves and (b) the corresponding EQE spectra of the OSCs based on the polymer:Y6 (c) PL spectra of Y6, PBFO-F and PBFO-H (d) PL spectra of Y6:PBFO-F and Y6:PBFO-H devices (e) TRPL spectra of Y6, PBFO-F and PBFO-H and (f) TRPL spectra of PBFO-F:Y6 and PBFO-H:Y6. | ||
The devices have JSC values of 19.94 and 23.17 mA cm−2 respectively with the high JSC and VOC value of the PBFO-F device approaching state-of-the-art values.9,30 The experimental JSC value is also very close to that which was derived from the EQE curve (Jcalc.) (Fig. 3b), which confirms the integrity of the calculation results. The FF values recorded for the devices (0.47 and 0.55) are low compared to the highest reported values (∼0.76) but can be further improved through the optimization of side chain engineering and film morphology with an expectant increase in the PCE.15 To recall from the optical absorption spectra, the donor polymer is responsible for the short wavelength absorption below 600 nm while the longer wavelength above 600 nm reflects the Y6 acceptor absorption. The EQE values in the shorter wavelength region are observed to be conspicuously higher than EQE values in the longer wavelength region which implies that the charges generated through photo-induced electron-transfer from the donor contributes more to the total efficient charges generated in the system.36 This phenomenon brings to light the potency of the fluorine-oxime (FO) acceptor building block used in the construction of the D–A donor polymer.
The exciton diffusion and dissociation properties of the devices were studied using photoluminescence (PL) and time-resolved photoluminescence (TRPL) spectra of neat and blend films with excitation at a wavelength of 400 nm. The results based on Fig. 3c and d show that blending the polymer and acceptor Y6 together reduced the emission of both the polymers and the Y6 acceptor. High quenching efficiencies were achieved for both PBFO-F:Y6 and PBFO-H:Y6 systems (83.3 and 88.3% respectively). It is observed that the PL intensity of PBFO-F:Y6 is more quenched in the donor region and enhanced in the acceptor region compared to that of PBFO-H:Y6, indicating more efficient charge separation between the donor and the acceptor. The fluorescence lifetime (τ) obtained from the TRPL spectra (Fig. 3e) was 1.12 ns for Y6, 1.03 ns for PBFO-F, and 0.71 ns for PBFO-H. On blending the polymers with the acceptor Y6, the lifetime of the PBFO-F:Y6 blend reduced to 0.36 ns while that of PBFO-H decreased to 0.21 ns (Fig. 3f) which is consistent with the PL results. The longer life time of the PBFO-F:Y6 blend is beneficial for excitons to diffuse and dissociate appropriately at the D–A interface in the blend, thus achieving higher JSC and FF values.
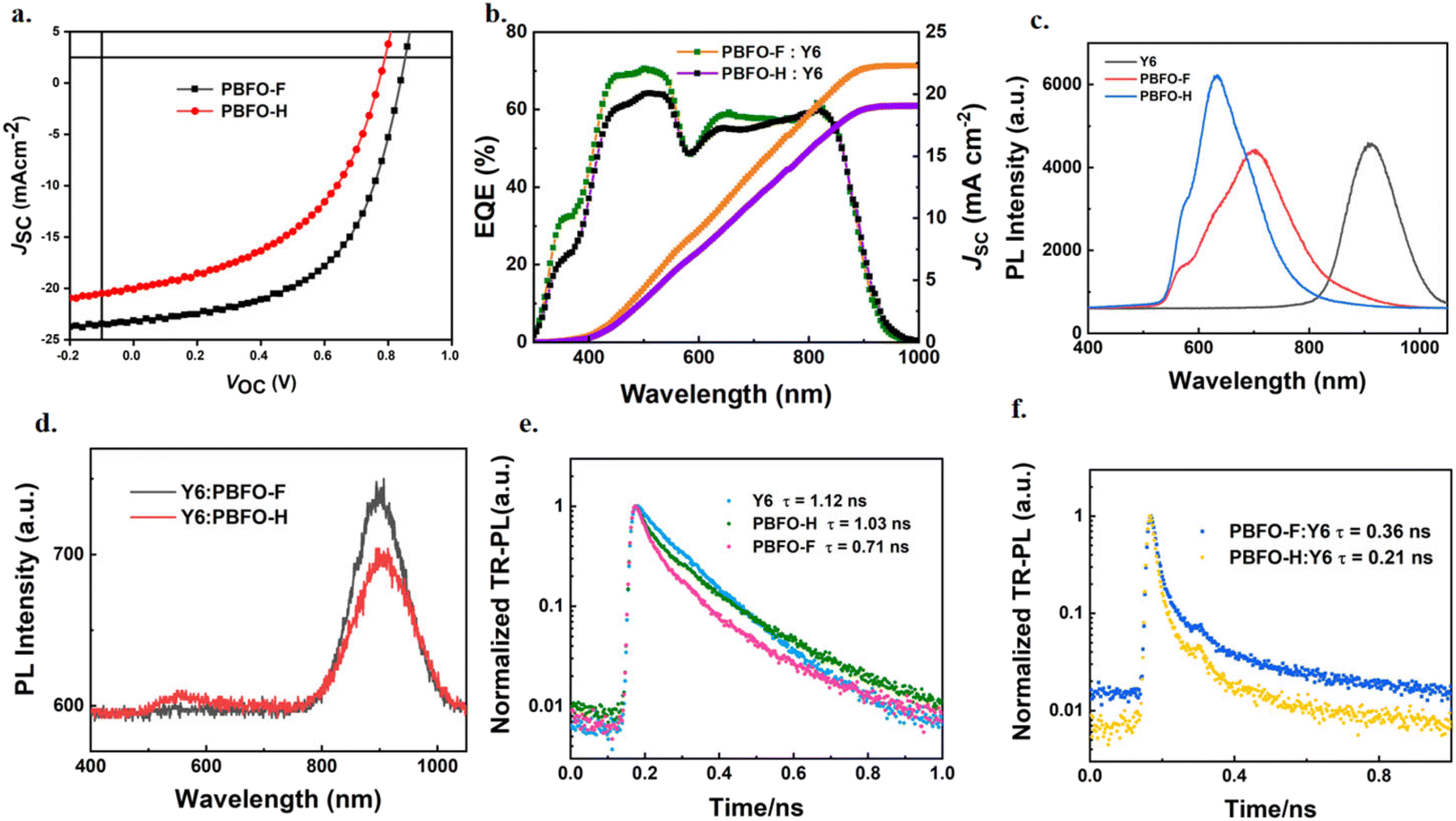
Furthermore, the exciton dissociation and charge collection efficiencies of the system were investigated. The result, shown in Fig. 4a is a plot of photocurrent density (Jph) against the effective applied voltage (Veff). Jph is associated with the current densities under illuminated (JL) and dark conditions (JD) according to the equation Jph = JL − JD. Veff is defined as Veff = V0 − Vappl, where V0 is the voltage at which Jph is 0 while Vappl is the applied voltage. As Veff increases to unity, Jph for PBFO-H:Y6 reaches a maximum value (Jsat) of 21.60 mA cm−2 under lower bias conditions while that of PBFO-F:Y6 reached 21.70 mA cm−2. The exciton dissociation (ηdiss) efficiencies which is calculated from Jph/Jsat at the short-circuit current is found to be 93.9% and 95.5% for PBFO-H and PBFO-F respectively. On the other hand, the charge collection efficiencies (ηcoll) for the devices calculated at the maximum power output point was found to be 63.0% for PBFO-H and 70.0% for PBFO-F. The PBFO-F based device shows higher ηdiss/ηcoll values than PBFO-H, which is consistent with the tendency of the FF values.
 | ||
| Fig. 4 (a) Photocurrent density (Jph) against effective voltage (Veff); dependence of (b) JSC and (c) VOC on Plight of the devices; (d) TPC curves; (e) TPV curves; and (f) charge mobility histogram. | ||
The light intensity (Plight) dependent JSC and VOC of the devices were used in analyzing their charge recombination properties. JSC is proportional to an exponential of the light intensity defined by JSC ∝ Plightα wherein the exponential factor (α) is usually close to 1 under short-circuit conditions since most of the photo-generated charges are evicted from the device and bimolecular recombination would be negligible.37 As shown in Fig. 4b, the exponential factors (α) obtained for PBFO-H and PBFO-F are 0.95 and 0.99 respectively which corroborates with theory. Conversely, the light intensity dependent VOC is usually measured under open circuit conditions, when no current is applied, to ensure that all photo-induced charges effectively recombine. The dependence of VOC on Plight is governed by the equation VOC ∝ (nkT/q) × ln(Plight), where k is the Boltzmann constant, T is the temperature measured in Kelvin, and q is the elementary charge. The n parameter is usually close to 1 when only bimolecular recombination operates in the device but the trap-assisted recombination usually extends it to 1 < n < 2.38 The n value found for PBFO-F:Y6 (1.41) is to our surprise greater that of PBFO-H:Y6 (1.31), which signifies that PBFO-F is more affected by trap-assisted recombination, as shown in Fig. 4c. Space-charge limited current (SCLC) techniques were used to measure the hole-only and electron-only diodes in order to derive their hole and electron mobilities (μh and μe) respectively (Fig. S3 and Table S3†).39 The μh and μe values of PBFO-F:Y6 are high at 5.55 × 10−4 cm2 V−1 s−1 and 4.47 × 10−4 cm2 V−1 s−1, respectively, with a μh/μe ratio of 1.24 (Fig. 4f), suggesting that balanced μh and μe are achieved which is good for obtaining high JSC and FF. On the other hand, the μh and μe values for the PBFO-H:Y6 blend are 4.48 × 10−4 cm2 V−1 s−1 and 2.46 × 10−4 cm2 V−1 s−1 with a μh/μe ratio of 1.82 which is more unbalanced compared with that of PBFO-F and would affect the JSC negatively. These results demonstrate that the conjugated polymer based on oxime functionalized fluorene can achieve good charge mobility. The greater values in PBFO-F show that the addition of fluorine was effective in leading to better electron discharge and mobility.
The transient photocurrent (TPC) technique was used to measure the charge extraction time in order to evaluate the charge extraction rate.40–42 The TPC curves shown in Fig. 4d evidence that the addition of the fluorine to the polymer backbone helps to decrease the charge extraction time (τext) from 0.029 μs in PBFO-H:Y6 to 0.020 μs in PBFO-F which means that there is a faster charge extraction rate in PBFO-F:Y6. Device voltage gradually reduces when there is recombination of holes and electrons.43 The transient photovoltage (TPV) technique is used to analyze the degree of the charge recombination in the devices, and the plot of the gradual charge decomposition of the TPV analysis translates to the photocarrier lifetimes (τrec). The value of τrec was 0.11 μs for both PBFO-H:Y6 and PBFO-F:Y6. The high τrec value shows that charge recombination is comparatively minimized in the both polymers (Fig. 4e).44 The TPV decay lifetime is slower than that of the TPC values similar to others in the literature, which confirms the correctness of the results.45,46 These results confirm that the introduction of fluorine into the polymer is efficient in boosting charge extraction and transfer while reducing charge recombination, and this leads to higher JSC and FF values.
Morphological and packing properties
The miscibility of the donor and acceptor components was derived by measuring the contact angle of each material with water and diiodomethane (Fig. 5). The contact angles were then used to calculate their surface energy values (γ) according to Wu's approach. The γ values were 40.92, 43.41, and 39.44 for PBFO-H, PBFO-F, and Y6 respectively. The Flory–Huggins parameter (χ) which is given by χ = κ(√γ1 −√γ2)2 is used to determine the miscibility between the polymer and acceptor components. The χ values calculated for the PBFO-F:Y6 blend was 0.095k while that of the PBFO-H:Y6 blend was 0.014k. The smaller χ value of the PBFO-H:Y6 blend shows that there is better miscibility between the two materials. Atomic force microscopy (AFM) was used for investigating the surface morphology of the blend films. As can be seen in Fig. 6(a–d), the root-mean-square (RMS) roughness for the blend films are 2.56 and 0.84 nm for PBFO-F:Y6 and PBFO-H:Y6 respectively. The AFM imagery for PBFO-F:Y6 produces a rough image with conspicuous height variability and its high RMS roughness value indicates that the film has a pronounced topography with peaks and valleys which could be due to material aggregation or phase separation.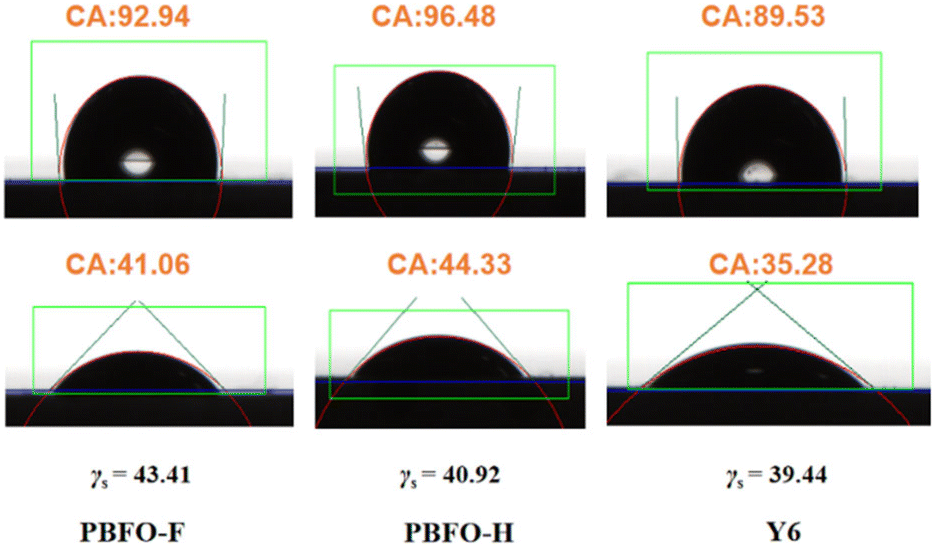 | ||
| Fig. 5 Contact angles of PBFO-F, PBFO-H, and Y6 neat films with water and diiodomethane. | ||
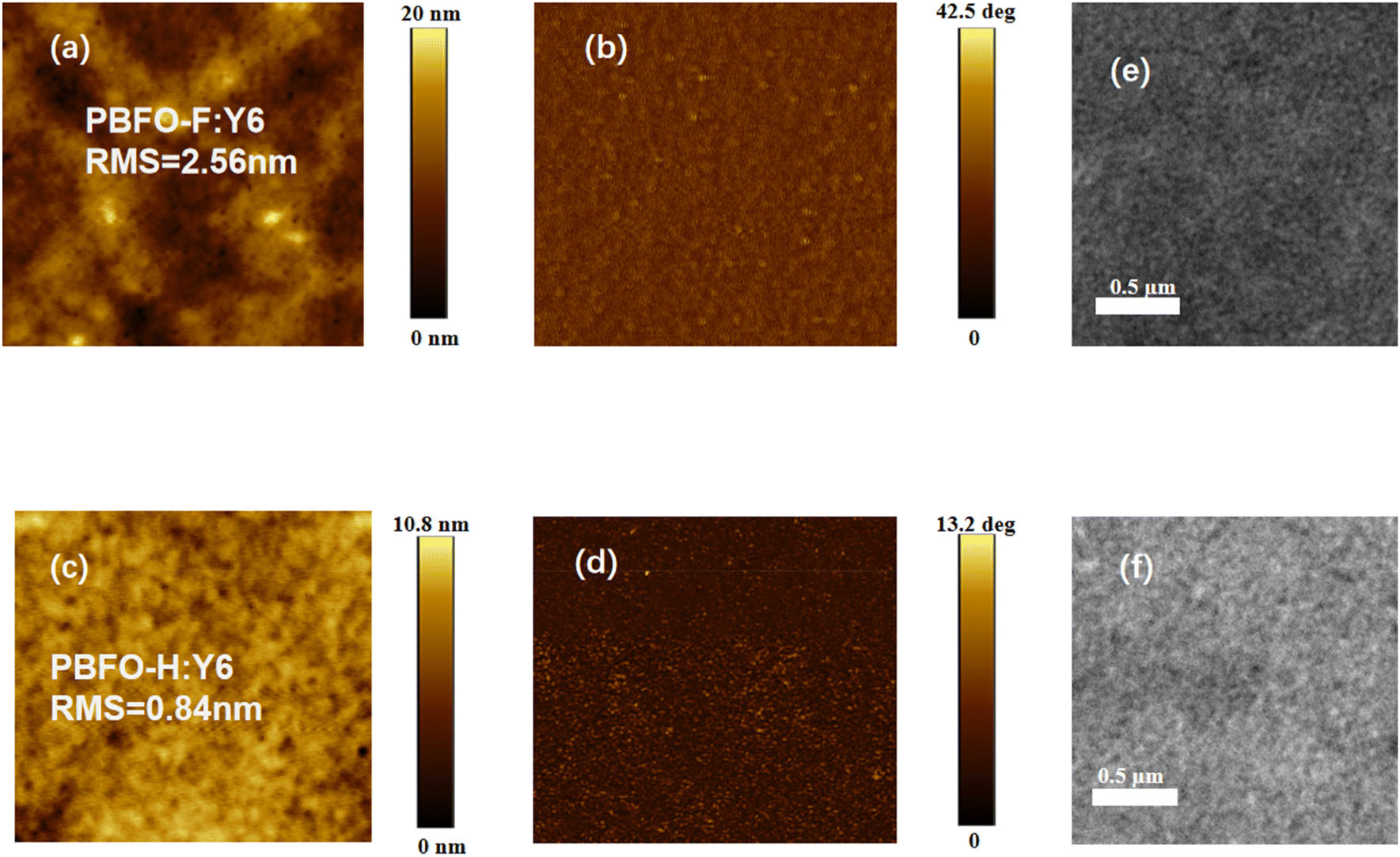 | ||
| Fig. 6 AFM images (3 × 3 μm) of (a) PBFO-F:Y6 height, (b) PBFO-F:Y6 phase, (c) PBFO-H:Y6 height, and (d) PBFO-H:Y6 phase; TEM images of (e) PBFO-F:Y6 and (f) PBFO-H:Y6. | ||
Meanwhile, strong aggregation of PBFO-F would probably be induced by its high crystallinity, which would be beneficial for JSC and FF values in devices. PBFO-H:Y6, on the other hand shows a lower RMS roughness value of 0.84 nm which indicates better miscibility with the acceptor. This further confirms the result obtained from the surface energy analysis. The analysis above is further confirmed with transmission electron microscopy (TEM) measurements, with PBFO-F:Y6 showing a rougher surface than PBFO-H:Y6 (Fig. 6e and f). The phase images of the two blends show uniform phase distribution which suggests that both films have no important phase segregation at the nanoscale level of measurement.
Conclusions
In summary, an oxime-functionalized fluorene (FO) was designed and synthesized as an acceptor unit for a D–A donor copolymerized with benzodithiophene (BDT) to construct novel polymers with a wide bandgap of 2.08 eV used with a Y6 acceptor to construct non-fullerene solar cells. Incorporation of fluorine atoms was beneficial for modulating the energy levels of the polymer as well as aggregation behavior and charge mobilities. As a result, the OSC devices based on PBFO-F:Y6 had a maximum PCE of 10.71% with JSC, VOC and Eloss approaching state-of-the-art values. These findings give confidence that the novel application of oxime functional groups in D–A donor polymers is promising and a brighter future lies with its further exploration.Author contributions
Abdullah Adewale Adeoba: polymer synthesis, investigation and writing – original draft. Xiangyu Shen: device fabrication and characterization. Fuzhen Bi: calculation. Jianan Niu: investigation. Mingxin Sun: formal analysis. Shuguang Wen: conceptualization and writing – review & editing. Xichang Bao: analysis, supervision and writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. U1932118 and 52173291), QIBEBT/SEI/QNESL S202305 and OP202309, Shandong Energy Institute (SEIS202108), and Department of Science and Technology of Shandong Province (ZR2020MB085).Notes and references
- G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153–161 CrossRef CAS.
- H. Sheng, S. Liu, X. Kang, J. Niu, A. A. Adewale, S. Wen, C. Yang, X. Bao and M. Sun, Nano Energy, 2024, 126, 109648 CrossRef CAS.
- J. Hou, O. Inganäs, R. H. Friend and F. Gao, Nat. Mater., 2018, 17, 119–128 CrossRef CAS PubMed.
- M. C. Scharber and N. S. Sariciftci, Prog. Polym. Sci., 2013, 38, 1929–1940 CrossRef CAS PubMed.
- Z. He, C. Zhong, S. Su, M. Xu, H. Wu and Y. Cao, Nat. Photonics, 2012, 6, 591–595 CrossRef.
- S. Li, C.-Z. Li, M. Shi and H. Chen, ACS Energy Lett., 2020, 5, 1554–1567 CrossRef CAS.
- T. H. Lee, S. A. J. Hillman, S. Gonzalez-Carrero, A. Difilippo and J. R. Durrant, Adv. Energy Mater., 2023, 13, 2300400 CrossRef CAS.
- L.-Y. Xu, Y. Gao, W. Wang, Y. Shao, M. Chen, X. Yang, Y. Fu, M. Zhang, X. Lu, R. Sun and J. Min, Energy Environ. Sci., 2023, 16, 3942–3950 RSC.
- Y. Cui, H. Yao, L. Hong, T. Zhang, Y. Xu, K. Xian, B. Gao, J. Qin, J. Zhang, Z. Wei and J. Hou, Adv. Mater., 2019, 31, 1808356 CrossRef PubMed.
- Y. Ma, Z. Kang and Q. Zheng, J. Mater. Chem. A, 2017, 5, 1860–1872 RSC.
- Y.-W. Su, Y.-C. Lin and K.-H. Wei, J. Mater. Chem. A, 2017, 5, 24051–24075 RSC.
- B. Zheng, L. Huo and Y. Li, NPG Asia Mater., 2020, 12, 3 CrossRef CAS.
- K. He, P. Kumar, Y. Yuan and Y. Li, Mater. Adv., 2021, 2, 115–145 RSC.
- H. Yao, L. Ye, H. Zhang, S. Li, S. Zhang and J. Hou, Chem. Rev., 2016, 116, 7397–7457 CrossRef CAS PubMed.
- K. He, P. Kumar, Y. Yuan, Z. Zhang, X. Li, H. Liu, J. Wang and Y. Li, ACS Appl. Mater. Interfaces, 2021, 13, 26441–26450 CrossRef CAS PubMed.
- X. Song, Y. Zhang, Y. Li, F. Li, X. Bao, D. Ding, M. Sun and R. Yang, Macromolecules, 2017, 50, 6880–6887 CrossRef CAS.
- X. Kong, C. Zhu, J. Zhang, L. Meng, S. Qin, J. Zhang, J. Li, Z. Wei and Y. Li, Energy Environ. Sci., 2022, 15, 2011–2020 RSC.
- B. Pang, C. Liao, X. Xu, L. Yu, R. Li and Q. Peng, Adv. Mater., 2023, 23, 2300631 CrossRef PubMed.
- J. Wang, Y. Wang, P. Bi, Z. Chen, J. Qiao, J. Li, W. Wang, Z. Zheng, S. Zhang, X. Hao and J. Hou, Adv. Mater., 2023, 35, 2301583 CrossRef CAS PubMed.
- T. Lin, Y. Hai, Y. Luo, L. Feng, T. Jia, J. Wu, R. Ma, T. A. Dela Peña, Y. Li, Z. Xing, M. Li, M. Wang, B. Xiao, K. S. Wong, S. Liu and G. Li, Adv. Mater., 2024, 36, 2312311 CrossRef CAS PubMed.
- M. Svensson, F. Zhang, S. C. Veenstra, W. J. H. Verhees, J. C. Hummelen, J. M. Kroon, O. Inganäs and M. R. Andersson, Adv. Mater., 2003, 15, 988–991 CrossRef CAS.
- Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS PubMed.
- D. P. Amit Sharma and W. Tomas, J. Optoelectron. Adv. Mater., 2014, 16, 1257–1268 Search PubMed.
- M. Sun, L. Wang, Y. Xia, B. Du, R. Liu and Y. Cao, Front. Chem. Eng. China, 2008, 2, 257–264 CrossRef CAS.
- A. Gadisa, W. Mammo, L. M. Andersson, S. Admassie, F. Zhang, M. R. Andersson and O. Inganäs, Adv. Funct. Mater., 2007, 17, 3836–3842 CrossRef CAS.
- M.-H. Chen, J. Hou, Z. Hong, G. Yang, S. Sista, L.-M. Chen and Y. Yang, Adv. Mater., 2009, 21, 4238–4242 CrossRef CAS.
- W. Lee, H. Cha, Y. J. Kim, J.-E. Jeong, S. Hwang, C. E. Park and H. Y. Woo, ACS Appl. Mater. Interfaces, 2014, 6, 20510–20518 CrossRef CAS PubMed.
- S. Wen, Y. Li, N. Zheng, I. O. Raji, C. Yang and X. Bao, J. Mater. Chem. A, 2020, 8, 13671–13678 RSC.
- L. Ye, X. Jiao, H. Zhang, S. Li, H. Yao, H. Ade and J. Hou, Macromolecules, 2015, 48, 7156–7163 CrossRef CAS.
- K. He, P. Kumar, M. Abd-Ellah, H. Liu, X. Li, Z. Zhang, J. Wang and Y. Li, Macromolecules, 2020, 53, 8796–8808 CrossRef CAS.
- D. Zhu, Q. Zhu, C. Gu, D. Ouyang, M. Qiu, X. Bao and R. Yang, Macromolecules, 2016, 49, 5788–5795 CrossRef CAS.
- B. Aldo, B. Simone, G. Giambattista, L. Annalina, E. O. Susanna Nencettia, R. Simona, R. Armando and G. Soldanib, Eur. J. Med. Chem., 2001, 36, 799–807 CrossRef PubMed.
- A. Jessie, D. A. P. Blake, L. Shuqiong, C. J. Walton, M. Peter and K. U. Ingold, J. Org. Chem., 2004, 69, 3112–3120 CrossRef PubMed.
- H. Sun, T. Liu, J. Yu, T.-K. Lau, G. Zhang, Y. Zhang, M. Su, Y. Tang, R. Ma, B. Liu, J. Liang, K. Feng, X. Lu, X. Guo, F. Gao and H. Yan, Energy Environ. Sci., 2019, 12, 3328–3337 RSC.
- P. Chao, M. Guo, Y. Zhu, H. Chen, M. Pu, H.-H. Huang, H. Meng, C. Yang and F. He, Macromolecules, 2020, 53, 2893–2901 CrossRef CAS.
- Y. Xie, W. Wang, W. Huang, F. Lin, T. Li, S. Liu, X. Zhan, Y. Liang, C. Gao, H. Wu and Y. Cao, Energy Environ. Sci., 2019, 12, 3556–3566 RSC.
- J.-L. Wu, F.-C. Chen, Y.-S. Hsiao, F.-C. Chien, P. Chen, C.-H. Kuo, M. H. Huang and C.-S. Hsu, ACS Nano, 2011, 5, 959–967 CrossRef CAS PubMed.
- L. J. A. Koster, V. D. Mihailetchi, R. Ramaker and P. W. M. Blom, Appl. Phys. Lett., 2005, 86, 123509 CrossRef.
- D. Liu, C. Gu, J. Wang, D. Zhu, Y. Li, X. Bao and R. Yang, J. Mater. Chem. A, 2017, 5, 9141–9147 RSC.
- J. Wang, M. Zhang, J. Lin, Z. Zheng, L. Zhu, P. Bi, H. Liang, X. Guo, J. Wu, Y. Wang, L. Yu, J. Li, J. Lv, X. Liu, F. Liu, J. Hou and Y. Li, Energy Environ. Sci., 2022, 15, 1585–1593 RSC.
- X. Liu, R. Tian, Z. Xiong, Y. Liu and Y. Zhou, Chin. Opt. Lett., 2023, 21, 120031 CrossRef.
- X. Kong, C. Zhu, J. Zhang, L. Meng, S. Qin, J. Zhang, J. Li, Z. Wei and Y. Li, Energy Environ. Sci., 2022, 15, 2011–2020 RSC.
- S. Chen, Y. Liu, L. Zhang, P. C. Y. Chow, Z. Wang, G. Zhang, W. Ma and H. Yan, J. Am. Chem. Soc., 2017, 139, 6298–6301 CrossRef CAS PubMed.
- Y. Cheng, B. Huang, X. Huang, L. Zhan, S. Kim, Q. Xie, C. Liu, T. Heumüller, Z. Liu, Y. Zhang, F. Wu and C. Yang, Angew. Chem., Int. Ed., 2022, 61, e202200329 CrossRef CAS PubMed.
- H. Aldosari, J. Jurado, K. Alkhezaim, S. Alam and F. Laquai, ACS Appl. Energy Mater., 2024, 7(16), 7055–7063 CrossRef CAS.
- Y. Yan, X. Zhou, F. Zhang, J. Zhou, T. Lin, Ya. Zhu, D. Xu, X. Ma, Y. Zou and X. Li, J. Mater. Chem. A, 2022, 10, 23124–23133 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00919c |
| This journal is © The Royal Society of Chemistry 2024 |