
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phase change material nanocapsules for latent function thermal fluids with tuneable thermal energy storage profiles†
Joshua R.
Booth
 ,
Joshua D.
Davies
and
Stefan A. F.
Bon
*
,
Joshua D.
Davies
and
Stefan A. F.
Bon
*
Department of Chemistry, The University of Warwick, Coventry CV4 7AL, UK. E-mail: S.Bon@warwick.ac.uk; Web: https://bonlab.info
First published on 30th July 2024
Abstract
Phase change materials (PCMs) can capture and release thermal energy in the form of latent heat and PCMs as liquid dispersions are known as latent function thermal fluids. For these dispersions, the PCMs are encapsulated to warrant colloidal stability. Here, capsule formation of mini-emulsions of various methacrylates in the presence of trimethylolpropane trimethacrylate (TMA) as a crosslinker, and n-hexadecane (HD), n-octadecane (OCT), and n-docosane (DOC) as PCM was investigated. An ω-unsaturated poly(n-butyl methacrylate-b-[(methacrylic acid)-co-(methyl methacrylate)]) macromonomer was used as a reactive macromolecular emulsifier. The mini-emulsion polymerizations, capsule fabrication and performance were optimized. The resulting latent function thermal fluids and their dried equivalents were studied with differential scanning calorimetry (DSC). A challenge of these materials is matching the temperature range of the application to that of their phase change. The performance of a thermal fluid of DOC nanocapsules was tested against the base fluid water with promising results. As a tunability concept, crosslinked poly(methyl methacrylate) nanocapsules of n-octadecane (OCT) and n-docosane (DOC) were blended as a tuneable latent function thermal fluid.
Introduction
The global climate and energy crises and the desire to reach net-zero emissions drive the importance of the capture, storage, and (re)use of energy. Phase change materials (PCMs) offer a viable option in helping to address these issues.1–4 PCMs rely on a phase transition, commonly a crystalline solid-to-liquid transition or vice versa. During the phase transition of such PCMs, external energy is captured (heating cycle) or released (cooling cycle) by the material in the form of latent heat.5 Materials with high latent heat suitable for temperature regulation are typically fatty acids, alkyl esters, n-alkanes and paraffin waxes, metals, and salt hydrates.6 PCMs for thermal management and storage are often stored as bulk materials in stainless steel and polyolefin containers,4 or manufactured as metallic and organic composites.7By segregating bulk PCMs into micron-sized or even nano-sized domains, heat transfer rates are increased, and the ease of product formulation and handling can improve.8 One way to do this is to fabricate core–shell capsules, the core being the PCM and the shell a mechanically robust layer. PCM capsules can be made by various methods including suspension polymerization,9–13 interfacial polymerization,14,15in situ phase separation polymerization,16,17 solvent evaporation,18–20 coacervation,21 spray drying22 and by use of the dynamic swelling method.23,24
Dried powders of encapsulated PCMs are frequently used as direct replacements for bulk materials. However, this limits their use to applications that are rigid or immobile.25,26 By dispersing the PCM capsules in a carrier fluid, the benefits of latent energy storage and release are now combined with the fluid's ability to flow. In addition, the specific heat capacity of the fluid itself and thermal conductivity can be optimised. This subclass of PCM materials is known as latent function thermal fluids or PCM slurries. Thermal fluids can be composed of surfactant-stabilised PCM emulsions.27,28 Encapsulation of the PCM in a (polymeric) shell improves stability and can potentially allow for dry powder storage and redispersion. Micro-encapsulated PCM slurries have been used in air conditioning,29 solar heat collectors30 and solar photovoltaic systems.31 However, the use of nano-capsule PCM fluids for these applications remains sparse. The surface area to volume ratio of encapsulated PCMs plays a crucial role in determining their thermal conductivity. For spherical capsules, the area-to-volume ratio equals 3/r, where r is the radius of the sphere. By decreasing the size of the capsules to the nanoscale, the thermal performance of PCM fluids can be increased significantly. Moreover, miniaturizing the dimensions of the capsules brings rheological benefits when used as dispersions, with clustering and jamming of capsules less likely once colloidal stability has been optimized. Encapsulation is essential as the crystallization of dispersions of bare PCM mini-emulsion droplets at high overall volume fractions upon cooling can lead to fusion where a crystallized droplet penetrates an adjacent droplet, triggering crystal growth.
PCM nanocapsule-based fluids, therefore, have great potential. Petrovic and co-workers evaluated the performance of encapsulated n-octadecane aqueous dispersions for use in heat sinks.32 In microchannels of 0.3 mm in diameter, it was found that 2 and 3% v/v slurries outperformed pure water by demonstrating higher heat transfer coefficients and lower substrate temperatures. The best results were at low temperatures due to the temperature dependence of heat capacity. Rehman and co-workers used a computational model to evaluate the thermal and hydrodynamic performance of a microchannel with hydrofoil-shaped walls.33 The PCM dispersion combined with the ribbed walls significantly outperformed pure water in smooth microchannels. However, for particles of 100 nm in diameter, the performance was reduced at solid contents higher than 10% w/w due to, as they claimed, increased turbulence. Doruk and co-workers investigated using aqueous n-nonadecane nanocapsule slurries in a heat exchanger.34 The double pipe design had a central tube inside a larger pipe. The PCM slurry was pumped through the central tube, absorbing heat from the hot water in the outer pipe. The slurries with the lowest concentrations of PCM capsules (0.42 and 0.84% w/v) showed no improvement over water. However, a 10% increase in heat transfer coefficient was obtained when the concentration was increased to 10% w/w.
To produce PCM nanocapsules, mini-emulsion polymerization is an ideally-suited technique owing to (1) the long-term stability of the small droplets against coarsening, also known as Ostwald-ripening, and (2) the direct droplet nucleation mechanism. Nano-encapsulation by mini-emulsion encompasses multiple polymerization techniques (polycondensation,35,36 sol–gel,37,38 phase-inversion,39etc.) and core materials.40–44
The fundamentals of the encapsulation process have been studied throughout the last few decades, elucidating the interplay of thermodynamic and kinetic forces to achieve complete encapsulation. Due to the classical use of n-hexadecane (HD) as an agent to resist emulsion droplet coarsening (Ostwald ripening),45,46 much of the mini-emulsion research employs HD or other long-chain n-alkanes as model compounds for encapsulation. One of the early studies on encapsulation of HD by mini-emulsion polymerisation was carried out by Landfester and co-workers in 2001.47 They studied the effect of monomer type, monomer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HD ratio and amount of surfactant on the morphology of the nanocapsules/particles. The authors demonstrated that the difference in hydrophilicity between the core HD material and polymer shell drove nanocapsule formation. Capsule morphology predictions were also made by Mahdavian and co-workers, who assessed the impact of shell polarity.48 In mini-emulsions which contain HD as the core material, methyl methacrylate (MMA) was copolymerised with 2-ethylhexyl acrylate (EHA) or methacrylic acid (MA). The phase-separated morphologies were compared by electron microscopy and DSC against configurations predicted by Torza and Mason's thermodynamic theory, and overall, good agreement was found.49
:HD ratio and amount of surfactant on the morphology of the nanocapsules/particles. The authors demonstrated that the difference in hydrophilicity between the core HD material and polymer shell drove nanocapsule formation. Capsule morphology predictions were also made by Mahdavian and co-workers, who assessed the impact of shell polarity.48 In mini-emulsions which contain HD as the core material, methyl methacrylate (MMA) was copolymerised with 2-ethylhexyl acrylate (EHA) or methacrylic acid (MA). The phase-separated morphologies were compared by electron microscopy and DSC against configurations predicted by Torza and Mason's thermodynamic theory, and overall, good agreement was found.49
Thermodynamic factors influencing the formation of polystyrene-paraffin capsules were researched by Lou and Zhou.50 The authors investigated the thermodynamic effect of increasing the SDS concentration and observed a shift from capsules to bowl-shaped particles. This aligns with the spreading coefficient, where lowering only the polymer-aqueous and HD-aqueous interfacial tensions shifts the predicted equilibrium morphology from capsules to partial encapsulation and eventually non-engulfment. Similar to the research by Mahdavian, the authors adjusted the shell polarity by copolymerising 1–3% w/w of MAA with styrene. Yet, adding a hydrophilic monomer disfavours capsule formation based on Torza and Mason's spreading coefficient equations. Given that they report capsule formation, it is more likely a kinetic factor in which the ionised MAA restricts polymerisation to the interface.
This kinetic factor was investigated by Klumperman and co-workers who demonstrated that capsule formation could be driven by the choice of initiator.51 Initiating the mini-emulsion polymerisation of n-butyl acrylate (BA) and HD with AIBN resulted in solid particles (total dissociation) as predicted thermodynamically. However, when water-soluble anionic initiators or oil-soluble redox initiators were used, capsules were synthesised. Using an anionic initiator, such as potassium persulfate, may influence capsule morphology thermodynamically by forming amphiphilic oligomers. However, this is not true for the cumyl hydroperoxide redox initiator, whose influence on particle morphology is purely kinetic. This result highlighted that capsule formation by mini-emulsion polymerisation can be directed by fixing the locus of polymerisation to the interface.
It is feasible that these findings inspired the development of interface-confined reversible-deactivation radical mini-emulsion polymerisation. In this sub-category, amphiphilic RAFT or ATRP agents reside at the droplets’ surface and restrict polymerisation to the interface, controllably encapsulating the core material.52–58 It has been shown by Lou and Hawkett that the choice of stabiliser block length is crucial to ensure a high percentage of capsules are synthesised.53,59 If the stabiliser is too water-soluble, desorption can occur, triggering secondary nucleation. With the ability to tailor the stabiliser's surface activity, Lou and Chen synthesised HD capsules with crosslinked fluorinated shells.56 This was only made possible using an amphiphilic poly(dodecafluoroheptyl acrylate-co-methacrylic acid) RAFT agent.
We decided to use ω-unsaturated methacrylate-based macromonomers as a reactive macromolecular stabilizer, as not only will these provide superior colloidal stability through grafting but also have the potential to graft more exotic core materials and polymer shells in the future.60 Polymeric stabilisers also benefit from favouring the engulfment morphology thermodynamically. They are able to provide considerable colloidal stability without drastically lowering the interfacial tensions between the aqueous phase and the polymer or core. Inversely, low molecular weight anionic surfactants such as SDS favour non-engulfment at high concentrations.61–63
The use of crosslinking monomers can have a substantial effect on capsule formation. Crosslinking reduces the mobility of chains, allowing for the access of non-equilibrium morphologies. Though it has been proven for styrene-divinyl benzene shells in particular, high amounts of the crosslinker lead to porous shells. The effect of divinyl benzene (DVB) on the morphology of alkane capsules was studied by Landfester and co-workers.64 Using polymerizable surfactants in various amounts, they found that when using styrene to encapsulate HD, there was a 50% split of capsules and particles. Swapping styrene entirely for DVB produced a much higher percentage of capsules. Research by Li and co-workers synthesised isooctane capsules by balancing the styrene:DVB and core:shell ratios.65 In accordance with the thermodynamic theory developed by Sundberg,66 the most thermodynamically favourable morphology depends on the surface area of the phases, as well as the interfacial tensions. Capsules with styrene-DVB shells, containing 30% mol/mol DVB, were synthesised by increasing the mass fraction of isooctane to monomer. The authors also studied the effect of DVB concentration, claiming from TEM analysis that the capsule shells become porous when >70% mol/mol DVB was used. The speculation of shell porosity was confirmed by work from Yingwu and co-workers as they attempted to synthesize non-collapsible hollow capsules.57 The authors measured the porosity of polystyrene hollow capsules crosslinked with 60% mol/mol DVB by nitrogen adsorption and desorption and found them exceedingly porous. The 110 nm diameter capsules, with a shell thickness of 11 nm, had a substantial pore volume of 2.74 mL g−1 and an approximate pore diameter of 9 nm.
As an aqueous capsule dispersion, the presence of pores is less of an issue due to capillary forces, preventing leakage of the hydrophobic core into the aqueous phase. However, if the capsules were used as dried PCMs and stored as a powder, the release of the core material, especially if it is volatile, is likely to occur.
In characterising PCM capsules, dynamic scanning calorimetry (DSC) is commonly used to inspect and compare the thermal properties of the materials with a particular focus on latent heat. To do so, the enthalpies of fusion (ΔHf) and crystallisation (ΔHc) are measured during temperature ramps. Due to the additional mass of the shell material, the ΔHf of the PCM capsules will generally be lower than the bulk material. The ratio-percentage of capsule enthalpy of fusion (ΔHf,cap) compared to the bulk value (ΔHf,bulk) is often presented as eqn (1):
 | (1) |
This, however, is of little use without knowing the mass fraction of PCM core to the overall capsule mass shell material, w, as shown in eqn (2) where Mcore and Mshell are the masses of the PCM core and shell monomer and pM is monomer conversion:
 | (2) |
Its percentage value we can also refer to as ‘PCM loading’. By combining eqn (1) and (2), a measure of the percentage latent heat of fusion of the PCM capsules with respect to the theoretical maximum based on PCM loading and referred to as the thermal storage efficiency (TSE) then becomes:8
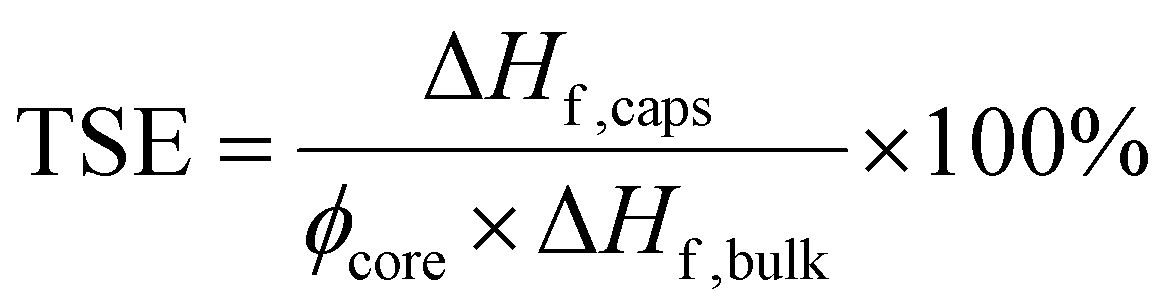 | (3) |
However, as Okubo and co-workers have pointed out,67 the equation is not always used correctly. Efficiency implies that if the value is lower than 100%, not all the PCM was encapsulated. Yet, even if all PCM were encapsulated, if a portion of it did not crystalise on cooling, a lower-than-expected ΔHf,cap would be recorded. This partial crystallisation is likely, as studies by NMR and neutron scanning have revealed that molecules near the wall of a confined geometry behave differently than their corresponding bulk counterparts.68,69 Furthermore, Amanuel and co-workers have proposed a method to estimate the thickness of non-freezing organic solvents in silica nanopores.70,71 The use of eqn (1) to measure efficiency also assumes that the latent heat of the confined material is equal to its bulk value. This is a poor assumption, as ΔHf has been shown to decrease with confinement diameter at very small radii.72,73 However, the relationship between ΔHf and geometry radii is not yet fully understood, with a conflicting relationship determined by Dorner.74
Table 1 shows a selection of literature data for PCM nanocapsules synthesised by mini-emulsion polymerization. Due to the variance of published latent heat equations, data was extracted from the publications and used to recalculate the TSE using eqn (3).
| Shell material | PCM | w /wt/wt% | ΔHf,caps/kJ kg−1 | TSE/% | Ref. |
|---|---|---|---|---|---|
| a Calculated using monomer conversion. b Monomer conversion not available. Shell materials methyl methacrylate (MMA), 2-ethylhexyl acrylate (EHA), methacrylic acid (MAA), ethyl methacrylate (EMA) and octadecyl methacrylate (ODMA). Values ΔHf,HD and ΔHf,OCT were 235 and 237 kJ kg−1. | |||||
| PMMA | HD | 28.9 | 74.0 | 108.9 | 75 |
| PMMA | HD | 51.3 | 75.3 | 62.5 | 75 |
| PMMA | HD | 77.5 | 96.8 | 53.1 | 75 |
| PMMA | HD | 50.4 | 72.9 | 61.5 | 48 |
| P(MMA + 15% EHA) | HD | 51.3 | 79.9 | 66.3 | 48 |
| P(MMA + 20% EHA) | HD | 51.1 | 40.2 | 33.5 | 48 |
| P(MMA + 30% EHA) | HD | 50.9 | 18.4 | 15.4 | 48 |
| P(MMA + 15% MAA) | HD | 50.7 | 77.8 | 65.4 | 48 |
| P(MMA + 20% MAA) | HD | 51.2 | 48.2 | 40.0 | 48 |
| PMMA | OCT | 86.7 | 208.7 | 101.5 | 76 |
| PEMA | OCT | 88.9 | 198.5 | 94.2 | 76 |
| P(MMA + 2% ODMA) | OCT | 28b | 102.0 | 154.9 | 77 |
| P(MMA + 20% ODMA) | OCT | 30b | 85.0 | 119.2 | 77 |
| P(MMA + 40% ODMA) | OCT | 31b | 84.0 | 115.0 | 77 |
Regarding the top three entries in Table 1, Mahdavian and co-workers studied the nanoencapsulation of HD in PMMA shells.75 They demonstrated that although increasing the core:shell ratio did lead to an increase in the enthalpy of fusion of the capsules, this was not reflected in the thermal storage efficiency (TSE). Another study by Mahdavian and co-workers investigated the influence of comonomer polarity and is reported in the second set of entries in Table 1.48 Adding 15% w/w of hydrophobic or hydrophilic comonomers increased the efficiency marginally, however adding more caused a significant decrease. This drop is caused by the thermodynamically favoured particle morphology shifting from encapsulated to partially encapsulated due to changes in interfacial tensions. Zhao and co-workers used a high core/shell weight ratio of 80/20 to encapsulate n-octadecane in poly(methyl methacrylate) and poly(ethyl methacrylate) shells.76 The authors reported TSE levels close to unity. However, it is difficult to confirm the morphology from the SEM images provided. The final three entries of Table 1 by Tang and co-workers highlight the importance of reporting monomer conversion when calculating TSE values.77 Clearly, there cannot be more PCM in the system than added, and the only answer is that there is less shell material than expected.
The concept of this paper is to design a tuneable latent function thermal fluid by mixing dispersions of polymer nanocapsules, each loaded with a bespoke n-alkane PCM. We will show that our prototype water-based thermal fluid with multiple latent heat transitions has potential. The capsules are made by mini-emulsion radical polymerization. The effects of crosslinker and shell materials on shell morphology and PCM performance are investigated. As mentioned earlier, ω-unsaturated methacrylate-based macromonomers will be used as a reactive macromolecular stabilizer.60 The covalent attachment of the emulsifiers to the particles ensures robust colloidal stability in the end application and aids capsule formation during polymerization.
Experimental
Materials
The monomers benzyl methacrylate (BzMA, 96%), n-butyl methacrylate (nBMA, ≥99%), isobornyl methacrylate (IBMA, technical grade), trimethylolpropane trimethacrylate (TMA, technical grade) and methyl methacrylate (MMA, 99%) were purchased from Sigma-Aldrich and filtered through basic activated alumina to remove radical inhibitors before use. Methacrylic acid (MAA, 99%, Sigma-Aldrich) was filtered through a column of neutral activated alumina. n-Hexadecane (HD, ≥99%) was purchased from Alfa Aesar. Ammonia solution (aqueous, 35% w/w) and sodium hydroxide (NaOH) were purchased from Fisher Scientific. Ammonium persulphate (APS, ≥98%), azobisisobutyronitrile (AIBN), n-docosane (DOC, 99%), n-octadecane (OCT, 99%), potassium persulfate (≥99%) and sodium dodecyl sulfate (SDS, ≥98.5%) were purchased from Sigma-Aldrich. AIBN was recrystallized from methanol and stored at −18 °C prior to use. Bis[(difluoroboryl)diethylglyoximato]cobalt(II) (CoEtBF) was synthesized using an analogous procedure as described for CoBF in the literature.78Methods
120 g of the above synthesised P(MAA-co-MMA) macromonomer latex (12.5% w/w, MM 15 g, 5.21 mmol) was added to a jacketed 250 mL reactor. Deionised water (30.0 g) was added to reduce the solid content to 10% w/w. The reactor was purged with nitrogen for 30 minutes with stirring at 250 rpm. n-Butyl methacrylate (11.21 g, 12.54 mL, 78.83 mmol) and an aqueous potassium persulfate solution (70.2 mg, 0.26 mmol, in 12.54 mL) were purged with nitrogen for 30 minutes. On heating the reactor to 85 °C, the n-butyl methacrylate and potassium persulfate solution were both fed into the reactor at a rate of 6.27 mL h−1. Feeding monomer ensures starved conditions, that is a high instantaneous conversion and thus low monomer concentration in the particles, essential for control of propagation to form the BMA block. After 2 hours, the pumps were stopped and the reactor temperature maintained for a further 1 hour. The latex was then removed from the reactor, cooled and quenched with bubbling of air. Solid content 16.0%, DLS: Z-average diameter 218.8 ± 3.1 nm, PDI 0.123 ± 0.004. GPC (THF): Mn = 5700, Mw = 8000, Đ = 1.41. 1H NMR (400 MHz, 2:1 CDCl3:DMSO-d6) δ: 6.01 (m, 1H), 5.41 (m, 1H), 3.84 (br, 2H), 3.50 (s, 3H), 2.44 (m, 2H), 2.08–1.24 (m, 6H, m 1H), 1.16–0.42 (m, 6H). DPn (1H NMR) = 43.
000 rpm. The coarse emulsion was then emulsified further using a Branson 250 Digital Sonifier for 15 minutes with pulses of 10 seconds at 60% strength, during which the mixture was cooled in a water bath regulated to 20 °C. The mini-emulsion was transferred to a round-bottom flask and purged with nitrogen gas for 30 min. The mini-emulsion was heated for 3 h at 70 °C under a positive pressure of nitrogen, with stirring at 350 rpm. Batch mini-emulsion polymerizations of methacrylate monomers under similar conditions take approximately 1 h. We ran for 3 hours as the presence of alkanes and crosslinks lowers the effective monomer concentration at the polymerization sites in each particles. 3 h was sufficiently long to reach near complete monomer conversion. Polymerization at 70 °C with AIBN means that throughout the reaction a steady flux of radicals is produced as t1/2 = 6 h.
000 rpm at 50 °C. The coarse emulsion was then emulsified further using a Branson 250 Digital Sonifier for 15 minutes with pulses of 10 seconds at 60% strength, during which the mixture temperature was regulated at 50 °C (DOC) or 35 °C (OCT). The mini-emulsion was transferred to a round-bottom flask. The RBF and APS solutions were purged with nitrogen gas for 30 min at room temperature. After purging, the mini-emulsion was heated at 70 °C for 10 minutes, whilst stirring at 350 rpm, after which 1 mL of the APS stock was added. The reaction was run for 3 h at 70 °C under a positive pressure of nitrogen, with stirring at 350 rpm.
Characterisation
Results & discussion
A series of ω-unsaturated amphiphilic macromonomers, poly(n-butyl methacrylate-b-[methacrylic acid-co-methyl methacrylate]) P(BMA-b-[MAA-co-MMA]), were synthesised by catalytic chain transfer emulsion polymerisation (CCTEP) and subsequent seeded sulfur-free RAFT emulsion polymerisation.60An initial series of mini-emulsion polymerisations was carried out at 20 wt% solids content, the dispersed phase being half (wt%) hexadecane and half monomer (benzyl methacrylate and trimethylolpropane trimethacrylate (TMA) as crosslinker) and using 2.5 wt% macromonomer stabiliser with respect to the dispersed phase. The reaction without the crosslinker formed, as expected, bowl-shaped particles. The introduction of crosslinker (10–30 wt% of total monomer) resulted in capsule formation. An amount of 20 wt% TMA (mole fraction of 0.115) was seen as a good compromise between the mechanical strength of the capsules and the inner surface roughness of the capsule walls.
Choice of methacrylate shell monomer
An important question is the effect different shell materials have on the morphology and thermal properties of the PCM capsules. A wide range of methacrylate monomers are available. Methyl (MMA), benzyl (BzMA), n-butyl (nBMA) and isobornyl (IBMA) methacrylate were selected as they display a range of thermophysical properties and hydrophobicities (Table 2).| Water solubilitya/g L−1 |
T
g,∞b/°C |
|
|---|---|---|
| a Literature data measured at 20 °C. MMA from Handbook of Aqueous Solubility Data86 and others supplied by Evonik GPS Safety Summaries. b Glass transition temperatures obtained from the Polymer Handbook.87 | ||
| Methyl methacrylate | 15.70 | 105 |
| Benzyl methacrylate | 0.19 | 54 |
| n-Butyl methacrylate | 0.20 | 20 |
| Isobornyl methacrylate | 5.44 × 10−3 | 110 |
Mini-emulsions of HD, TMA (approximately 20% w/w to monomer) and each methacrylate monomer were prepared using a sonicator probe. The mass ratio of HD to monomer was fixed at 2:3 (see Table S1† for all experimental quantities). The mini-emulsions were stabilised by P(BMA-b-[MAA-co-MMA]) macromonomers (Table S2†), using 2.5 wt% with respect to the dispersed phase. An oil-soluble initiator, AIBN (1.25 g L−1 in the dispersed phase), was used to discourage secondary nucleation and added to the HD monomer mixture before emulsification.
The size distributions of mini-emulsion droplets before polymerization and the corresponding latex particles were measured by DLS (Table 3 and Fig. 1).
 | ||
| Fig. 1 Size analysis by DLS. Correlograms (left) and intensity weighted particle size distributions (right) for hexadecane crosslinked capsules made with four methacrylate monomers. Mini-emulsion droplets are dashed lines, post-polymerization capsules are represented by solid lines. | ||
| Reaction | Z-Avg. droplet diameter/nm | Droplet PDI | Z-Avg. capsule diameter/nm | Capsule PDI |
|---|---|---|---|---|
| a Droplet and capsule size were recorded on a Malvern Zetasizer Ultra at 25 °C and scattering angle of 173°. b Droplet and capsule size were recorded on an Anton Paar Litesizer Litesizer 500 25 °C and scattering angle of 175°. For the Z-average diameter and PDI results, three DLS scans were recorded and an average taken for each sample. | ||||
| PMMA_HDa | 150.2 ± 2.3 | 14.27 ± 1.20% | 158.1 ± 3.4 | 6.52 ± 5.96% |
| PBzMA_HDa | 152.2 ± 2.5 | 14.52 ± 0.21% | 155.4 ± 2.38 | 9.87 ± 5.70% |
| PnBMA_HDb | 160.2 ± 0.1 | 0.130 ± 0.018 | 161.2 ± 1.4 | 0.055 ± 0.030 |
| PIBMA_HDb | 174.1 ± 1.0 | 0.080 ± 0.027 | 176.8 ± 0.8 | 0.098 ± 0.016 |
Consistency in size distributions from droplets to particles indicated that stable mini-emulsions were formed in all cases and that particle formation and growth occurred predominantly by droplet nucleation. There is a sizeable difference of 9.5% and 8.8% between the average droplet and particle diameters of IBMA to the other three monomers. As IBMA has a much lower water solubility (Table 2), and therefore is more hydrophobic, the larger droplet and particles may be a result of P(BMA-b-[MAA-co-MMA]) stabiliser incompatibility. Although not explored in this work, the versatility of the macromonomer stabilisers allows the user to tune the hydrophobicity of the blocks to suit the intended application best.
To investigate the morphologies of the methacrylate series, the latexes were imaged by SEM and STEM, as shown in Fig. 2. For all samples in this paper, transmission electron micrographs were obtained using the ZEISS Gemini Field Emission Scanning Electron Microscope in TEM mode. This was chosen as the electron beam (15–20 kV) is much lower than conventional TEM (∼200 kV), and this reduces the degradation of soft polymer samples.88 However, even with this precaution, imaging the capsules proved difficult, especially at high magnification. When focusing on samples for more than a few seconds, the capsules visibly deformed, comparable to melting. This effect is illustrated for crosslinked PMMA octadecane capsules in the ESI (Fig. S1†).
 | ||
| Fig. 2 Electron microscope images of hexadecane crosslinked capsules synthesised using different methacrylate monomers. Images from SEM (left) and STEM (right). SEM scale bars are 200 nm, TEM scale bars are 500 nm. | ||
The SEM images of reaction PBzMA_HD show a mixture of spheres and erupted spheres, whereas crumpled spheres are present in PMMA_HD, PnBMA_HD and PIBMA_HD reactions. All structures are a result of hexadecane evaporation and give an indication for capsule formation. The STEM images give more substantial evidence for encapsulation, showing a contrast difference across the hollow capsules compared to the few solid particles present. These solid particles may be formed through limited particle nucleation by other means than the intended droplet nucleation, most likely homogeneous nucleation. Comparing the grey value profile across a capsule and particle (Fig. 3) highlights the different morphologies, the y-axis has been inverted to aid with visualisation. In Fig. 3, the grey value is a measure of how light/dark each pixel is, with absolute black being 0 and absolute white being 255. As the profile runs across the capsule and particle, the pixels become darker as the polymer wall scatters the electron beam. For the particle, there is a consistently dark pixel profile across the particle and pixels are darker, as indicated by a lower grey value. In contrast, as the centre of the hollow capsule is thinner, the grey value tends towards a lighter shade because more electrons reach the STEM detector.
 | ||
| Fig. 3 (Left) STEM micrograph showing line profile across a PMMA capsule (dark blue) and PMMA particle (light blue). (Right) Grey value profile plotted against distance of a line traced across a PMMA capsule (dark blue) and PMMA particle (light blue). | ||
With capsule formation confirmed by electron microscopy, the thermal properties of the capsules were studied using DSC. The latexes were dried at ambient conditions to remove water before analysis. For each dry powder sample, 3 cool and heating scans between −15 and 25 °C were conducted at a rate of 1 °C min−1. A low rate of temperature change was chosen to resolve multiple crystalline phase transitions.
There is a reduction of the crystallisation and melting temperatures in all cases, caused by confinement of the HD. There are three matters to discuss for the crystallisation of HD in each sample: (i) the shape of the exotherms and multiple peaks, (ii) the large differences of supercooling (ΔTc) and (iii) the small exothermic peak at 9.8 °C.
(i) Due to the molecular structure of linear n-alkanes, when cooled below the melting point, the molecules can undergo multiple transitions before reaching their most stable crystal structure.89–93 These metastable/transient phases are known as rotator phases, named so because the molecules stack in lamella crystals and have almost free rotation around their axis.94–96 There are five known rotator phases97 and for even n-alkanes (C12 to C26) in bulk, the generally reported rotator phase is hexagonal and this stable phase is triclinic.98,99 HD transient and metastable rotator phases were studied by Sirota and Herhold who determined the thermodynamic stability of the phases.100 These solid–solid transitions are observed for bulk HD when cooled at 1 °C min−1 using the TA Instruments DSC 2500 (Fig. S2†). However, it has been shown that under confinement, n-alkanes exhibit changes in their crystal properties.101 Montengro and Landfester observed additional rotator phases transitions in nanodroplets and a size dependence of the stable crystal structure.102 From the spectra in Fig. 4 and 5, a mixture of many transitional phases is observed, but the elucidation of them all is beyond the scope of this work. Rotator phase transitions may also be responsible for the twin melting peaks for reaction PIBMA_HD, but more work using X-ray scattering is required to comment further.
 | ||
| Fig. 4 Dynamic scanning calorimetry (DSC) curves for dried n-hexadecane crosslinked capsules synthesised with four different methacrylate monomers. Cooling curve for scans 1, 2 and 3 are coloured dark blue, light blue and green. Heating curves for scans 1, 2 and 3 are coloured yellow, pink and purple. Heat and cooling scans of bulk HD are shown as grey dashed line. | ||
 | ||
| Fig. 5 Comparison of DSC curves for different methacrylate HD capsules. Measured using a TA Instruments DSC 2500, under nitrogen atmosphere, at 1 °C min−1. The final scan of three temperature cycles is shown, cooling curves and heating curves are blue and red, respectively. Solid line (PMMA_HD), dashed line (PBzMA_HD), short-dashed line (PnBMA_HD), dashed-dotted line (PIBMA_HD). | ||
(ii) As well as a significant difference in the shape of the crystallisation peaks, there are also considerable differences of supercooling (ΔTc) depending on shell material. Reactions PMMA_HD and PBzMA_HD have a ΔTc of 11.4 and 11.7 °C, whereas PnBMA_HD and PIBMA_HD crystalise at lower temperatures with a ΔTc of 14.0 and 15.2 °C. The reasons for this are not fully understood at this moment in time. Nevertheless, it is possible to theorise potential reasons. Supercooling occurs due to the requirement of a nucleation event occurring in each capsule. As the capsules are <500 nm in diameter, the probability of having a foreign object (dust, etc.) to trigger heterogeneous crystal nucleation is extremely small, if not zero.103 However, it may be possible that the capsule wall may act as a nucleation surface if it is significantly rough, in the same way scratching smooth glass triggers heterogeneous crystal nucleation. The inner surface of crosslinked PBzMA HD capsules was found to be considerably rough (Fig. S3†). The high inner shell roughness of PBzMA matches with the lower extent of ΔTc. However, we do not know the inner surface topography of PMMA_HD, PnBMA_HD and PIBMA_HD capsules. Yet, for PnBMA and PIBMA, given the closer likeness of the monomer side groups to HD, the more favourable solvent–polymer wetting during shell formation may lead to a smoother inner surface. This smooth surface would reduce the number of nucleation sites and further suppress the crystallisation point. Beside this, molecular dynamic studies on how different alkanes react to a specific polymer surface may provide insights.
(iii) A crystallisation peak at approximately 9.8 °C eventually appears for all capsule samples. For PMMA_HD and PBzMA_HD the peak does not appear in the first cooling scan and its appearance coincides with a shift in the main Tc to higher temperatures. The peak is present in all scans for reactions PnBMA_HD and PIBMA_HD. The event is kinetically driven as the melting transition for the four samples are identical. Fette and co-workers report a similar shift in aqueous capsule dispersions but, in their work, the entire peak shifts to the bulk temperature.104 The authors proposed two hypotheses, the first explained the shift due to the exclusion of dissolved impurities in the HD over repeated cycles. These soluble impurities lowered Tc and are not to be confused with impurities that enable heterogeneous nucleation. Alternatively, over repeated cycles the shell walls become deformed producing a solid-phase nucleation site for crystallisation. The first theory does not fit as the impurity would also affect the melting transition. The shell deformation theory is also not in line with Fig. 4 as the peak at 9.8 °C remains stable after the first cycle in all samples. Nevertheless, the lower extent of supercooling does indicate a change in environment for the HD. It is possible that appearance of the peak may be caused by diffusion of HD out of the shell. For PnBMA_HD and PIBMA_HD the rate of diffusion through the shell was found to be fast (Fig. 6) and will be discussed later. It is likely the HD already moves through the shell upon drying and this matches with the observed peak during the first cycle. For PMMA_HD and PBZMA_HD, the rate of HD diffusion through the shell is restricted, but repeated crystallisation and melting appear to encourage a certain fraction to change the environment. In addition, the small peak appearing at 9.8 °C may be the result of a surface freezing phenomenon.105
 | ||
| Fig. 6 Semi-log plot of percentage mass loss for crosslinked capsules containing hexadecane heated at 150 °C for 6 hours. Crosslinked capsule polymer used was poly(methyl methacrylate) (dark blue), poly(benzyl methacrylate) (light blue), poly(n-butyl methacrylate) (green) and poly(isobornyl methacrylate) (pink). Theoretical percentage masses after complete evaporation of HD are shown as dashed lines. Evaporation of bulk HD is shown as a black dashed line. | ||
Regarding the melting points, there also appears to be the same split between the PMMA_HD, PBzMA_HD and PnBMA_HD, PIBMA_HD. The melting point depression (ΔTm) of PMMA_HD and PBzMA_HD is 4.4 and 4.7 °C whereas ΔTm of PnBMA_HD and PIBMA_HD are 5.2 and 6.6 °C. However, this trend appears to go against the thermodynamic theory of the Gibbs–Thomson equation (eqn (2)).73 The Gibbs–Thomson equation describes the extent of melting point depression for a given capsule/pore size.73,106 The equation relates the energetic cost required to melt in a confined geometry due to the increase in Laplace pressure (ΔP = 2γijr).
 | (4) |
Regardless, given the likeness of the side group of IBMA to HD, the solvent may interact strongly with the shell, resulting in non-freezing layers,71 which would alter the crystal-shell interfacial tensions and complicate the thermodynamic prediction. Furthermore, the PIBMA shell material has a significant effect on the shape of the IBMA_HD endotherm. The theme of PIBMA as an outlier also carries over when comparing the measured enthalpy changes of the dried capsules in Table 4. Regarding the calculated percentage thermal storage efficiency (TSE, eqn (1)), reaction PIBMA_HD has a significantly lower value than the other three samples.
| Reaction | Monomer conversion/% | Theory | Measured | TSEc/% |
|---|---|---|---|---|
| PCM loadinga/% w/w | Capsule ΔHf ratiob/% | |||
| a Calculated using eqn (2), monomer conversion measured by gravimetry. b Calculated using eqn (1). c Calculated using eqn (3). | ||||
| PMMA_HD | 88.0 | 44.3 | 33.1 | 74.9 |
| PBzMA_HD | 100.0 | 39.0 | 29.5 | 75.7 |
| PnBMA_HD | 98.8 | 39.5 | 28.1 | 71.1 |
| PIBMA_HD | 98.6 | 39.6 | 18.2 | 45.9 |
The interaction of HD with the shell may result in non-freezing layers which result in a deficit between the theoretical and measure ΔHf.71
Given the hydrophobic nature of PnBMA alkane side chain and the bicyclic side group of PIBMA, a significant decrease in TSE would be expected as they are more compatible with HD.48 Although the trend in Table 4 does not follow the theory exactly, there is a large decrease for IBMA_HD.
To further investigate the interaction of HD with the shell, the release profile of HD at elevated temperatures was measured by thermogravimetric analysis (TGA). It should be restated that TMA crosslinker was added to direct capsule formation. However, others report that crosslinking PS capsules with DVB results in highly porous shells.57 For the TGA experiment, 50 μL of latex (20% w/w solids) was transferred into a 40 μL aluminium DSC/TGA pan. The surface tension of the liquid allowed for the pan to be overfilled. The samples were dried overnight at room temperature and the water evaporated leaving a ring of dried capsules in the bottom of each pan. Considering the boiling point of HD is 287 °C any loss overnight would be negligible. The dried samples were prepared this way to ensure comparable data, as the surface area of the material may influence evaporation rate.107 The pans were sealed with pierced, crimped lids and the mass loss was recorded at 150 °C for 6 hours (Fig. 6).
For reference, a pan of pure HD was measured and is shown as the black dashed line. The coloured dashed lines represent the theoretical maximum mass loss for each sample based on the HD mass ratio. From Fig. 6 there is a clear split in evaporation rate between the capsules. For PnBMA and PIBMA, the rate of HD evaporation is unaffected, and it freely diffuses through the shell. For PMMA and PBzMA capsules, there is a two-stage release profile. HD evaporates at the bulk rate until at 15% mass loss, then the rate slows dramatically to −0.90% h−1 for PBzMA and −0.40% h−1 for PMMA. Okubo and co-workers demonstrated from PS/DVB capsules swollen with toluene, the mass loss of residual solvent in the shell was dependent on crosslink density and shell thickness.108 However, as the capsules in the four-monomer series had the same molar ratio of TMA crosslinker and ratio of HD to monomer, crosslinking and shell thickness cannot explain the observed difference in release rate. Alternatively, Taden and co-workers found a direct correlation between the capsule diffusion barrier and the interaction of the core and shell materials.109 This argument appears much more likely, especially given the lower thermal storage efficiency of PIBMA_HD (Table 4), which was hypothesised to be due to more favourable HD–shell interaction.
Although the four polymers are immiscible in HD, and precipitate during capsule formation, the HD may act as a plasticizer. Plasticizers are a class of low-molecular-weight organic molecules that increase the flexibility, decrease tensile strength and lower the glass transition of polymers.110 The effect of HD on the glass transition of the four methacrylate polymers was probed by DSC. For this study, linear polymers were synthesised by solution polymerization. The glass transitions of the linear methacrylate polymers were measured dry and after soaking in HD for 48 hours (Table 5 and Fig. S4†).
| M n/×104 g mol−1 | M w/×104 g mol−1 | Đ | T g cooling/°C | T g heating/°C | |||
|---|---|---|---|---|---|---|---|
| Dry | HD soaked | Dry | HD soaked | ||||
| Glass transitions were measure using a TA Instruments DSC 2500 at a heating/cooling rate of 10 °C min−1 under an atmosphere of nitrogen. Data was taken from the final of three temperature cycles. Midpoint glass transition temperatures were measured following ASTM D3418-21 standards.82 | |||||||
| PMMA | 2.8 | 5.2 | 1.85 | 106.0 | 92.2 | 110.6 | 94.4 |
| PBzMA | 6.6 | 14.8 | 2.24 | 62.3 | 51.2 | 64.1 | 55.4 |
| PnBMA | 3.3 | 6.9 | 2.11 | 24.2 | n.d | 31.3 | n.d |
| PIBMA | 3.5 | 7.8 | 2.21 | 184.8 | n.d | 195.3 | n.d |
Comparing the dry cooling cycle Tg values to literature (Table 5) PMMA, PBzMA and PnBMA are all close to reported data. Remarkably the value for PIBMA is 75 °C higher than the reported temperature in the polymer handbook. Although there is also a discrepancy in the literature, a higher value of 150 °C was reported by Zhang and Wang for PIBM, Mw 2.1 × 105 g mol−1.85
For PMMA and PBzMA, which both were soaked in hexadecane, the Tg of PMMA decreased by 14.0 and 16.2 °C measured during the heating and cooling scan, whereas PBzMA experienced a smaller decrease of 11.0 and 8.7 °C, during cooling and heating respectively. From the structures of these two polymers, it would be logical to assume they have a low compatibility with the aliphatic hydrocarbon HD. Plasticizers have the greatest effect when their structural properties are a close match to the polymers that they are interacting with.110
For PnBMA it would be expected that the plasticization effect is much greater. When cooled to −150 °C a sharp increase in specific heat was observed that shifted to lower temperatures with each scan (Fig. S4†). In the first scan (purple), a small step at −87.9 °C; in the second scan, a step change with a midpoint of −93.5 °C (light blue); and a shift to −101.9 °C in the third scan (dark blue). Unfortunately, the Tg of HD soak PnBMA remains inconclusive due to the shape of the transition on cooling and lack of transition on heating. It should be noted that the mass of polymer analysed was substantial and should display a clear transition under normal conditions. 19.11 mg of sample was weighed and the calculated ΔHf of HD was 20% of the bulk value, estimating ≈15 mg of polymer is present.
For HD soaked PIBMA, the sample was initially analysed between 20 and 230 °C (not shown). The glass transition for the dried polymer was determined in the cooling and heating scans to be 184.8 and 195.3 °C, respectively. However, for the HD-soaked polymer, no transition was observed between 20–230 °C. In response to this, the polymer was analysed down to −150 °C. Similar to HD soaked PnBMA, no discernible glass transition was observed in either cooling or heating scans. A weak transition in the heating scan that resembles the shape of glass transition is observed at −40.0 °C, however this contradicts the somewhat glassy physical appearance of the soaked polymer. Again, approximately 10 mg of polymer was analysed based on the measured enthalpy HD. Furthermore, for both PnBMA and PIBMA, if the transition was to occur below the crystallisation point of HD, the crystal lattice may also to interfere with the dynamic polymer glass formation process.111
Once again there is a split in the observed properties of PMMA and PBzMA and of PnBMA and PIBMA when interacting with HD. Although the exact nature of the interaction could not be determined, to synthesise n-alkane PCM nanocapsules for use in thermal fluids, PMMA and PBzMA show the most potential, with PMMA having a slight edge looking at HD TGA release.
Encapsulation of n-octadecane and n-docosane for use as thermal fluid
To produce an aqueous PCM slurry, HD is no longer suitable due to the reduced crystallisation temperature (<0 °C) when encapsulated. In response to this, two higher molecular weight n-alkanes, n-octadecane (OCT, C18) and n-docosane (DOC, C22), with bulk crystallisation temperatures of 25.8 and 42.0 °C, were selected. We assume that the knowledge gained using HD as a model compound is transferable to OCT and DOC. Regarding capsule formation by phase separation, any minor changes to the γPol/PCM and γAqu/PCM values will be overcome by the kinetic force of restricted polymer mobility due to cross-linking.To encapsulate OCT and DOC, MMA was chosen as methacrylate shell monomer. Firstly, the size distributions of the mini-emulsion droplets and polymerised capsules were measured by DLS and presented alongside correlograms in Fig. 7. The spectra of the PMMA_OCT reaction droplets and capsules appears as expected for a typical mini-emulsion polymerization. The droplet and particle distribution are near identical with droplet Z-average diameter 186.5 ± 0.9 nm and PDI 0.109 ± 0.018 and capsule Z-average diameter 181.1 ± 1.3 nm and PDI 0.141 ± 0.031. By contrast, there appears to be some irregularities in the correlogram and fitted distribution for the PMMA_DOC reaction. The correlation decay for capsules (solid line) is broader than droplets (dashed line) and when the data is fit using the cumulants method, a second distribution appears. For reaction PMMA_DOC, the droplet had a Z-average diameter of 175.0 ± 0.7 nm and PDI of 0.127 ± 0.021. After polymerization, DLS recorded mean diameters of 213.3 ± 1.8 nm and 4891 ± 291 nm for the two populations. The percentage of larger objects by DLS intensity was 6.4%, by volume 15.8% and the peak disappeared in the number distribution. Additionally, the size distributions of PMMA_OCT and PMMA_DOC appear much broader than those recorded for the methacrylate monomer series (Fig. 1). To investigate this further the polymerised capsule latexes were imaged by SEM and STEM techniques (Fig. 8). As indicated by the DLS distribution, for PMMA_OCT, a very larger span of sizes can be seen in the SEM image, suggesting the smallest particles have formed via secondary nucleation. This assumption is confirmed in the STEM image as the smallest particles are much darker, as explained previously (Fig. 3). A drawback of selecting MMA as the shell monomer is the higher water solubility (Table 2), which increases the probability of secondary nucleation. For the capsules, a difference in grey value across their profile is observed as n-octadecane is sublimed under EM vacuum, leaving hollow capsules.
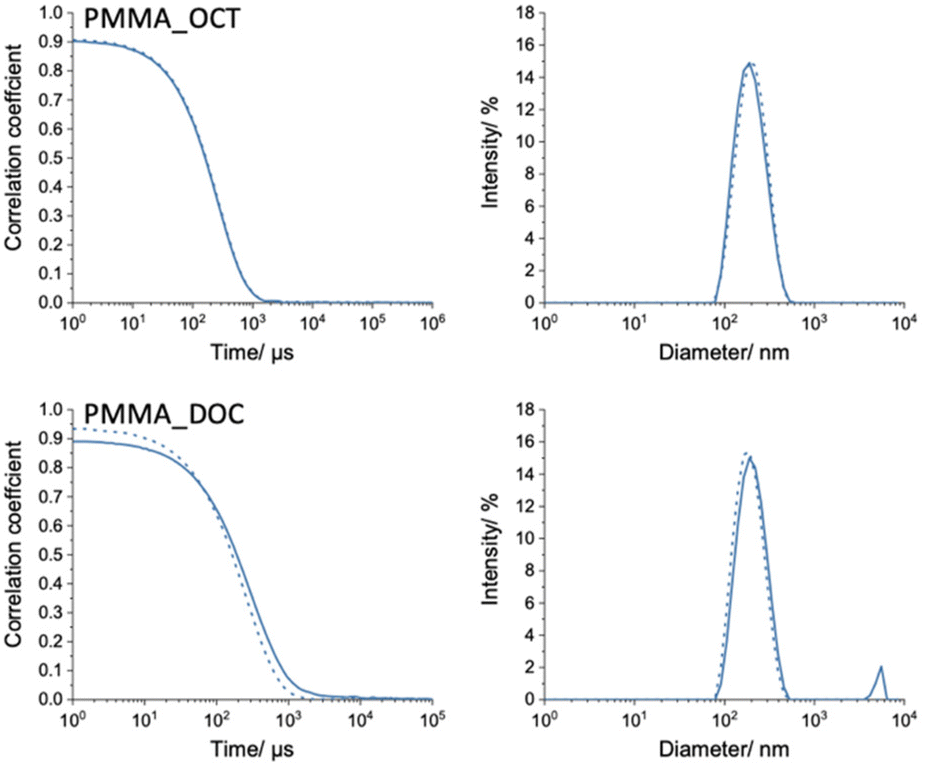 | ||
| Fig. 7 Particle size analysis by DLS of crosslinked poly(methyl methacrylate) n-octadecane and n-docosane capsules. Correlograms (left) and normalized intensity weighted particle size distributions (right). Miniemulsion droplets are dashed lines, capsules are represented by solid lines. | ||
 | ||
| Fig. 8 Electron microscope images of crosslinked poly(methyl methacrylate) n-octadecane or n-docosane capsules. Scanning electron microscope (left) and scanning transmission electron microscope (right). | ||
For PMMA_DOC, the spread of sizes for the main population is like PMMA_OCT with some solid particles also found. Even with a melting point of 42 °C the docosane is sublimed under the extremely low pressures of the EM. This causes capsules to buckle, and a contrast difference is observed across the hollow capsules, identical to HD capsules (Fig. 3). However, unlike any of the capsule recipes discussed so far, the existence of micron-sized hollow structures was noted, a comparison of PMMA_OCT and PMMA_DOC imaged by light microscope in dark field mode is shown in Fig. 9.
 | ||
| Fig. 9 Images of PMMA_OCT and PMMA_DOC latexes taken using a light microscope in dark field mode. | ||
The left image shows an image of PMMA_OCT latex, but the nanoparticles are unresolved. This is because the resolution of a light microscope is limited to half the wavelength of visible light and the particles by DLS are <500 nm in diameter. Several brighter particles are shown in the right image of PMMA_DOC, these represent the larger distribution seen in DLS after polymerization (Z-average 4891 ± 291 nm). To investigate this further the larger particles of PMMA_DOC were imaged by SEM and TEM, as shown in Fig. 10.
 | ||
| Fig. 10 Low magnification electron microscope images of poly(methyl methacrylate) n-docosane micro-sized objects. Scanning electron microscope (left) and scanning transmission electron microscope (right). Scale bars are both 2 μm. | ||
The large hollow objects appear to be decorated by smaller capsules and their structure draws similarities to Pickering stabilised emulsions.112 As well as stabilising dispersed liquid, Pickering stabiliser have also been shown to stabile air droplets in the formation of stable foams.113 As DOC sublimes under EM conditions, it is inconclusive whether the large capsules are filled with DOC or air at ambient conditions. To probe this, the capsules as a latex were imaged on a heated light microscope stage above their melting point (Fig. 11). The latex was sandwiched between a microscope slide and a cover slip.
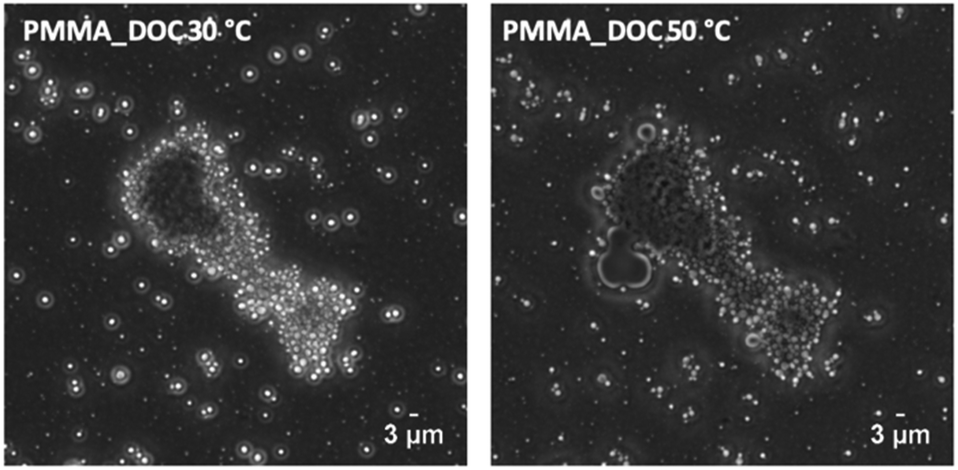 | ||
| Fig. 11 Light microscope images of PMMA_DOC latex at 30 °C and 50 °C, DOC bulk Mp is 41.9 °C. | ||
The left image shows a collection of large objects in the PMMA_DOC latex, the objects with a halo effect are out of focus due to diffusion in the z-plane. The collection of large objects is fixed in place due to the pressure of the glass cover slide. On heating above the melting point of bulk DOC, the expulsion of liquid can be seen in the right image. The evidence proves the capsules contain DOC, but that they also have a shell; although some liquid DOC is present at 50 °C, the majority of large capsules retained their core.
Their formation is not fully understood but it can be explained by investigating the reaction preparation steps. The mini-emulsion was prepared and emulsified 10 °C above DOC melting point. However, the mini-emulsion was not actively heated during 30 min of purging with nitrogen gas. On cooling, the monomer DOC mixture would have become supersaturated allowing for DOC to crystalise. When not encapsulated, this can lead to mini-emulsion destabilisation and fusion of droplets leading to further crystal growth. Upon polymerization above the melting point, the now large droplets of DOC can heterocoagulate with mini-emulsion-derived DOC capsules, leading to the observed Pickering effect. To overcome this, the mini-emulsion should be heated above the DOC Mp at all times and not be allowed to cool.
Nevertheless, a substantial fraction of DOC was encapsulated into nanocapsules, as shown by DLS (Fig. 7) and therefore their thermal properties were measured. To investigate the phase change temperatures and latent heats of OCT and DOC capsules, the 20% w/w latexes were dried at ambient conditions to remove water and analysed using DSC. The dried powders were heated and cooled at a rate of 1 °C min−1.
From the DSC curves of dried OCT PMMA capsules (Table 6 and Fig. 12), suppression of crystallisation and melting onset, as well as rotator phase transitions are observed. The main crystallisation peak for PMMA_OCT has a ΔTc of 5.7 °C, this reduces to 4.4 °C after the first cooling/heating cycle as an addition peak appears, similar to the DSC spectra of PMMA_HD and PBzMA_HD (Fig. 5). The extent of supercooling is much less for PMMA_OCT than for HD PMMA capsules. The reason for this is not fully understood but given the similarities of the ΔTm, it would appear the cause is a kinetic factor and not thermodynamic. A second large peak (onset of 9.3 °C) with an extended tail is also present. It is likely that this is a transition from rotator phase to the stable crystal phase as observed by in the bulk spectra (see later Fig. 15) and by Montenegro and Landfester for OCT in confinement.102
 | ||
| Fig. 12 Dynamic scanning calorimetry spectra of dried PMMA crosslinked capsules containing n-octadecane (left) and n-docosane (right). Phase transitions were measured using a TA Instruments DSC 2500, under nitrogen atmosphere, at a rate of 1 °C min−1. Cooling scans 1, 2 and 3 shown as dark blue, light blue and green. Heating scans 1, 2 and 3 shown as yellow, pink and purple. Bulk n-octadecane and n-docosane are shown as dashed lines. | ||
| Sample | Crystallisation | Melting | ||||
|---|---|---|---|---|---|---|
| T c,i/°C | T c,ii/°C | ΔHc/J g−1 | T m,i/°C | T m,ii/°C | ΔHf/J g−1 | |
| T c,i and Tc,ii are onset temperatures of crystallisation, ΔHc enthalpy of crystallisation, Tm,i and Tm,ii are onset temperatures of melting, ΔHf enthalpy of fusion.a Crystallisation phase transition peak is not present in the first cooling scan. | ||||||
| OCT (bulk) | 25.4 | — | 208.9 | 26.0 | — | 209.7 |
| PMMA_OCT | 21.0a | 19.7 | 68.4 | 19.9 | 23.3 | 67.5 |
| DOC (bulk) | 42.8 | — | 234.1 | 41.9 | 43.3 | 232.5 |
| PMMA_DOC | 39.1 | — | 111.4 | 31.5 | 40.4 | 110.3 |
Unlike PMMA_HD, the melting transition of PMMA_OCT shows a clear step change due to the solid–solid rotator phase melting transition with two onset temperatures 19.9 and 23.3 °C, both onsets are lower than the bulk temperature, ΔTm 6.1 and 2.7 °C, due to nanocapsule confinement.114–116 The extent of melting point suppression for reaction PMMA_OCT is comparable to the 4.4 °C ΔTm for the PMMA_HD nanocapsules. This result is in line with thermodynamic theory as the capsules are of similar size and will have similar core–shell interfacial tensions.
Inspecting the crystallisation behaviour of PMMA_DOC, three transitions occur during the broad transition with onset temperatures of 39.1, 29.5 and 26.1 °C. The large tailing peak at 39.1 °C may represent the micron-sized objects observed in DLS and EM imaging as the magnitude of ΔTc is decreased on increasing capsule radius. On the other hand, the peak may represent a rotator phase transition. Further analysis using crystallographic techniques is required to confirm these theories. For PMMA_DOC the melting transition reflects the melting behaviour of the bulk material as both display secondary peaks during the transition due to the presence metastable crystal structures. The determination ΔTm is complicated by the rotator transitions as they are likely to alter under confinement.101 Furthermore, the main melting transition of PMMA_DOC has three different gradients. In contrast to the sharp transition of bulk DOC, the process of melting in nanoconfinement is much more complex.
As with the previous capsules, the enthalpy of fusion was measured by DSC and compared to the theoretical value, this comparison is presented as the thermal storage efficiency (eqn (3) and Table 7). The TSE of PMMA_OCT is comparable to PMMA_HD (74.9%, Table 4) and this lower-than-expected value can be explained by favourable PCM–shell interaction causing non-freezing OCT. Assuming all the OCT is encapsulated, a TSE of 71.6% accounts for 14.5% by mass of the OCT that does not freeze. A further experiment to test this would be to vary the core:shell ratio. If this theory was correct, reducing the amount of shell material would reduce the amount of non-freezing OCT and a higher TSE would be obtained. Interestingly, in line with the theory, Zhao and co-workers encapsulated OCT with PMMA (no crosslinker) using a PCM loading of 86.7% and achieved a TSE of 101.5.
Although the core loading percentage of PMMA_OCT and PMMA_DOC is similar, PMMA_DOC has a much higher TSE. This cannot be explained by shell ratio but may be caused by shell interaction. Longer alkanes are less compatible with PMMA and it possible that the low interaction encourages all of the DOC to freeze when cooled. TSE of 100% for PMMA capsules of a commercial paraffin wax were also reported by Okubo and co-workers.67
As the PCM nanocapsule are to be used as a latent function thermal fluid, their thermal properties as a 20% w/w solids latex were also analysed by DSC. Approximately 35 μL of the latexes were transferred into hermetically sealed pans and the phase transition were measured as low as 0 °C and as high as 55 °C at a rate of 1 °C min−1 (Fig. 13). As before the phase transitions were characterised and are presented in Table 8.
 | ||
| Fig. 13 Dynamic scanning calorimetry spectra of 20% w/w latexes of PMMA crosslinked capsules containing n-octadecane (left) and n-docosane (right). Phase transitions were measured using a TA Instruments DSC 2500, under nitrogen atmosphere, at a rate of 1 °C min−1. Cooling scans 1, 2 and 3 shown as dark blue, light blue and green. Heating scans 1, 2 and 3 shown as yellow, pink and purple. Bulk n-octadecane and n-docosane are shown as dashed lines. | ||
| Sample | Crystallisation | Melting | ||||
|---|---|---|---|---|---|---|
| T c,i/°C | T c,ii/°C | ΔHc/J g−1 | T m,i/°C | T m,ii/°C | ΔHf/J g−1 | |
| T c,i and Tc,ii are onset temperatures of crystallisation, ΔHc enthalpy of crystallisation, Tm,i and Tm,ii is onset temperatures of melting, ΔHf enthalpy of fusion. | ||||||
| OCT (bulk) | 25.4 | — | 208.9 | 26.0 | — | 209.7 |
| LATEX_OCT | 12.4 | — | 13.0 | 24.4 | — | 12.5 |
| DOC (bulk) | 42.8 | — | 234.1 | 41.9 | 43.3 | 232.5 |
| LATEX_DOC | 37.8 | 30.2 | 18.6 | 32.3 | 41.3 | 17.4 |
Interestingly, the crystallisation transitions are significantly altered when the nanocapsules are analysed as an aqueous dispersion. For LATEX_OCT (Fig. 13, left) the crystallisation phase transition is simplified, with a single tailing peak with a crystallisation suppression ΔTc of 13.0 °C. This is a much greater suppression than recorded for dried capsules in which the main peak was supercooled by 5.7 °C. Regarding the DSC spectra of LATEX_DOC, there is a main crystallisation peak with a small shoulder as well as a smaller peak closer to the bulk transition temperature. Interestingly, the smaller peak, with onset 37.8 °C matches the onset of large exotherm in PMMA_DOC at 39.1 °C, whereas the main peak for the latex at 30.2 °C is close to the dried second peak of 29.5 °C. There is a striking difference in intensity of OCT crystallisation around 38 °C when dried or dispersed and the similarities of the values cannot be ignored.
The significant difference in supercooling between the dried capsules and the aqueous dispersion of capsule may be caused by shell diffusion. To begin each DSC experiment, the PCM nanocapsule sample is heated above its melting point, then the three cooling–heating cycles begin. If during this initial heating stage, leakage of OCT and DOC occurred, crystallisation of this material would happen at much higher temperatures, this is more possible when dried as the capsules are in close contact in a packed film. As an aqueous dispersion the nanocapsules are continuously separated and only experience momentary collisions undergoing Brownian motion. This enhanced separation prevents crosstalk between capsules and leads to greater compartmentalisation and supercooling.
For the melting transitions, the endotherm for LATEX_OCT is also simplified; compared to the dried capsules (Fig. 12), the rotator transition at 19.9 °C is no longer observed. For the degree of suppression in main melting transitions, there is a slight difference with ΔTm for LATEX_OCT calculated as 1.6 °C and PMMA_OCT as 2.7 °C. The melting transition of LATEX_DOC is a similar complexity to the dried sample with a small rotator phase transition occurring at 32.3 °C (31.5 °C for PMMA_DOC) and at least four changes in gradient during the main transition. Multiple gradients complicate the melting point depression calculation, but it is estimated to be 0.8–2 °C (onset between 41.3–42.5 °C) depending on the region of analysis. The difference of melting transitions may be influenced by the enhanced thermal regulation of water.
Proof of concept for a binary thermal fluid with multiple tuneable transitions
To demonstrate the feasibility of the OCT and DOC capsules as latent thermal fluid, an experiment was designed to evaluate the thermal performance of PCM capsule slurry against the base fluid. The designed experiment is similar to a method published by Yang for determining the thermal properties of bulk PCMs.116 A flat-based glass vial was partially filled with either PMMA_DOC latex or water. A temperature probe was submerged and the vial sealed with a PTFE temperature probe guide. The vial was placed on a thermoelectric module and heated at a constant heat flux for 800 seconds.PMMA_DOC was chosen for the feasibility study due to the high TSE. To test the performance of the thermal fluid the sample was concentrated to 31.3% w/w under reduced pressure, this was carried out to increase the latent heat response of the fluid. However, high capsule solid contents should be avoided due to the loss of performance from the lower ratio of water and its high heat capacity (Table 9).
| Pure water | Capsule solid content/% w/w | |||
|---|---|---|---|---|
| 20 | 30 | 50 | ||
| The temperature-dependent specific heat capacity and transition enthalpy data found in the ESI (Table S4†). | ||||
| Energy required/kJ | 168.4 | 164.4 | 162.4 | 158.4 |
The temperature change over time was recorded in triplicate for the 31% w/w PMMA_DOC fluid and water and is plotted in Fig. 14. It should be noted that this is a study of first principles and further development of the experimental procedure is required. Due to the simplified experimental design, three repeats of each sample were carried out. Comparison of the 30% w/w PMMA_DOC latex and water base fluid heating rates implies the greater performance of the PCM dispersion. The high variance in the rates is most likely due to the heat loss through the glass vial sidewalls. An expected step change in temperature gradient during DOC melting, as proposed for PCMs by Yang, is not observed.117 This is due to the fast temperature ramp during the experiment which will lengthen the melting transition. Nevertheless, the initial results are encouraging for the application of the thermal fluid.
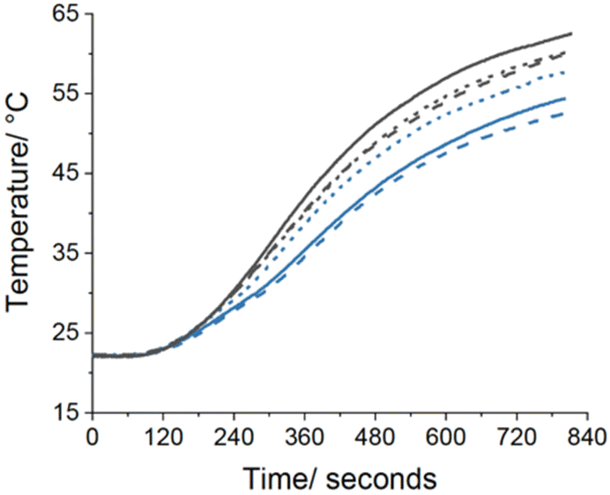 | ||
| Fig. 14 Plot of fluid temperature during heating from a thermoelectric module at a constant heat flux. Water shown in black, DOC latex at 30% w/w solids shown in blue. Solid, dashed and dotted lines are repeat measurements. | ||
The compartmentalisation of OCT and DOC PCMs into nanocapsules enables the creation of a thermal fluid with multiple transitions. The current proposed system is 1:1 binary mixture but the possible combinations of different encapsulated PCMs and capsule mass fractions are vast. A branch of research exists that focuses on the study PCM mixtures known as eutectic PCMs.118 A eutectic system is a mixture of substances at a specific ratio that has a single melting point, the melting temperature at the eutectic point is lower than the individual components.119 The eutectic PCM is generally chosen to alter the thermophysical properties of the system to match a specific application.120,121
For the design of a binary thermal fluid, the encapsulation of waxes and prevention of core mixing is essential. To demonstrate the importance of PCM segregation, the phase transitions of bulk OCT and DOC, analysed separately and as a 1:1 mixture, are compared to a 1:1 dispersion of PMMA_OCT and PMMA_DOC capsules. Three cycles between −20 and 60 °C for the bulk n-alkanes were conducted, cycles between 0 and 60 °C were carried out for the latex mixture due to the presence of water. All scans were conducted at a rate of 1 °C min−1. The phase changes of the n-alkanes when cooled and heated as a 1:1 mixture (Fig. 16) are changed dramatically compared to their separate bulk spectra (Fig. 15). There is a sharp onset of crystallisation for the mixture at 33.0 °C but the heat flow continuous to change until 0 °C indicating during this period a stable crystal structure was not reached. A similar reflection of this is observed in the melting transition of the bulk mixture.
 | ||
| Fig. 15 DSC scans of n-octadecane and n-docosane. X-axes of temperature and time are shown to visual rotator transitions. Cooling and heating scans shown in blue and red, respectively. Cooling and heating rate was 1 °C min−1 under an atmosphere of nitrogen. | ||
 | ||
| Fig. 16 Dynamic scanning calorimetry spectra of 1:1 pure wax mixture of n-octadecane and n-dodecane. Phase transitions were measured using a TA Instruments DSC 2500, under nitrogen atmosphere, at a rate of 1 °C min−1. Cooling and heating shown as blue and red, respectively. | ||
The benefits of encapsulation are clear when analysing the 1:1 mixture of OCT and DOC nanocapsules (Fig. 17). Blending the capsule dispersions produces a combinatorial spectrum of the two PCMs from Fig. 13. The identical phase transition onset temperatures in the latex blend compared to individual latexes demonstrates that no cross-talk occurs and the different PCMs remain compartmentalised throughout.
 | ||
| Fig. 17 Dynamic scanning calorimetry spectra of 1:1 capsule mixture of n-octadecane and n-dodecane waxes. Phase transitions were measured using a TA Instruments DSC 2500, under nitrogen atmosphere, at a rate of 1 °C min−1. Cooling and heating shown as blue and red, respectively. | ||
To this end, through the combination of both OCT and DOC capsules in varying proportions, it is possible to afford tunability to the thermal energy storage properties of a latent thermal fluid. That is, we can choose whether to have one or two latent heats present and we can vary the magnitude of each by simply varying the relative proportions of each capsule dispersion added to the system. Theoretical calculations were carried out to model the thermal energy storage profile of each of the DOC and OCT dispersions individually and of blends of the two capsule dispersions thereof. The total thermal energy stored by a material can be viewed as the summation of the sensible/specific heat (related to increasing the temperature of the material) and the latent heat (related to a material phase change) and is given by eqn (5) and (6).
| Q = ∑(sensible + latent) | (5) |
| Q = mcpΔT + ΔHtransition | (6) |
This is made more complex by the temperature-dependent nature of the specific heat capacity of a material and can be expressed as:
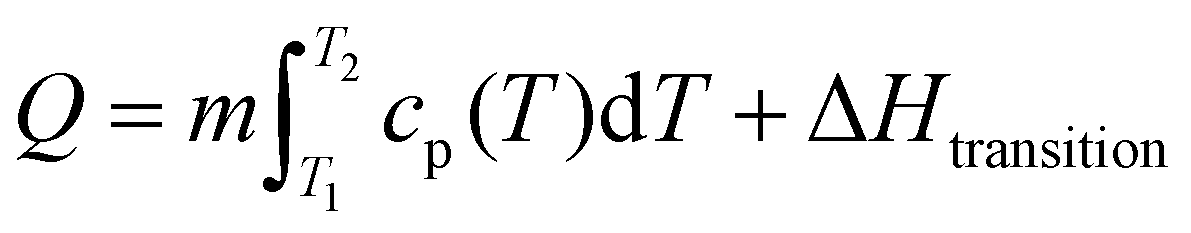 | (7) |
Given that a phase transition may occur anywhere during a given temperature range the total thermal energy stored can be split into the sensible heat before and after the phase transition and the enthalpy of the given transition:
 | (8) |
Using eqn (8) and the temperature-dependent specific heat capacity and transition enthalpy data found in the ESI (Table S4†) the thermal storage profiles for heating from 25 to 60 °C can be calculated (Fig. 18 and 19). The latent heats utilised in these calculations were taken as those for the confined wax obtained via DSC (see Table 6).
 | ||
| Fig. 18 Thermal energy profile for PMMA_OCT (left) and PMMA_DOC (right) capsule dispersions at varying capsule wt%:30 wt% (blue), 50 wt% (pink), 70 wt% (yellow). | ||
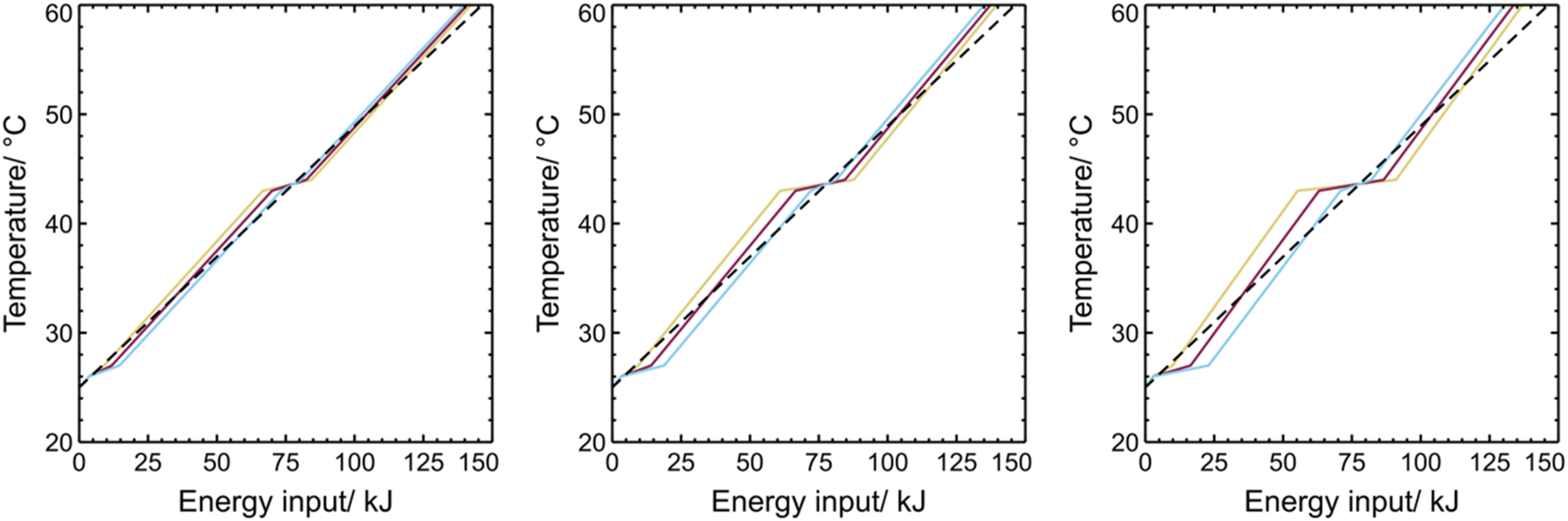 | ||
| Fig. 19 Thermal energy profile for linear combinations of PMMA_OCT and PMMA_DOC capsule dispersions at varying capsule wt%. 30 wt% (left), 50 wt% (centre) and 70 wt% (right). DOC:OCT ratio in each case is represented as 20:80 (blue), 50:50 (pink), 80:20 (yellow). | ||
Conclusions
A latent functional thermal fluid with multiple, separate, endothermic and exothermic transitions has been developed. This was accomplished by nanoencapsulation of OCT and DOC PCMs in a crosslinked PMMA shell via mini-emulsion polymerization. SEM and STEM were used to show the encapsulation of both PCMs was successful. However, a population of micron-sized DOC capsules were also observed which were a result of DOC crystallisation in monomer droplets before polymerization. It was found the extent of supercooling was much greater for the fluid dispersion than for dried capsules. This was theorised to be caused by capsule leakage in the dried state, which was restricted when dispersed. This observation also strengthens the argument for the usefulness of the PCM fluid. A deep understanding of the encapsulation process and shell–core compatibility was gained in this study, and this helped to drive the development of a binary thermal fluid. The proof-of-concept study was successful. The performance of the DOC nanocapsule dispersion was evaluated as a cooling fluid against water by itself and demonstrated promising results. Furthermore, we showed that, indeed, dispersions of PCM capsules containing n-octadecane and n-dodecane could be blended into a bespoke thermal fluid dispersion. Calculations show that such mixtures bring benefits to performance and, therefore, that the concept of tuneability has been realised. From the overall results, it becomes clear that nanocapsule PCM dispersions can behave superior to water in energy storage and release, in certain temperature windows. The challenges are not only to optimize this efficiency but also to look at the effects on the thermal conductivity and durability of the system.Author contributions
Joshua R. Booth: conceptualization, formal analysis, investigation, methodology, project administration, resources validation, visualization, writing – original draft. Joshua D. Davies: formal analysis, investigation, visualization, writing – original draft. Stefan A. F. Bon: conceptualization, supervision, project administration, methodology, formal analysis, resources, funding acquisition, writing – review and editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the University of Warwick Polymer Characterization Research Technology Platform (RTP), specifically Dr Daniel Lester and Dr James Town, for maintenance and access to GPC thermal characterisation equipment. The authors would also like to thank the University of Warwick electron microscopy RTP for the use of EM equipment and providing specialist training.References
- A. Pasupathy and R. Velraj, Energy Build., 2008, 40, 193–203 CrossRef.
- K. Lafdi, O. Mesalhy and A. Elgafy, J. Electron. Packag., 2008, 130, 1–8 CrossRef.
- R. Kandasamy, X.-Q. Wang and A. S. Mujumdar, Appl. Therm. Eng., 2007, 27, 2822–2832 CrossRef.
- M. Kenisarin and K. Mahkamov, Renewable Sustainable Energy Rev., 2007, 11, 1913–1965 CrossRef CAS.
- S. Raoux, Annu. Rev. Mater. Res., 2009, 39, 25–48 CrossRef CAS.
- K. Pielichowska and K. Pielichowski, Prog. Mater. Sci., 2014, 65, 67–123 CrossRef CAS.
- J. Yang, L.-S. Tang, L. Bai, R.-Y. Bao, Z.-Y. Liu, B.-H. Xie, M.-B. Yang and W. Yang, Mater. Horiz., 2019, 6, 250–273 RSC.
- G. Peng, G. Dou, Y. Hu, Y. Sun and Z. Chen, Adv. Polym. Technol., 2020, 1–20 CrossRef.
- L. Sánchez, P. Sánchez, M. Carmona, A. de Lucas and J. F. Rodríguez, Colloid Polym. Sci., 2008, 286, 1019–1027 CrossRef.
- L. Sánchez-Silva, J. F. Rodríguez, A. Romero, A. M. Borreguero, M. Carmona and P. Sánchez, Chem. Eng. J., 2010, 157, 216–222 CrossRef.
- P. Chaiyasat, Y. Ogino, T. Suzuki, H. Minami and M. Okubo, Colloid Polym. Sci., 2008, 286, 217–223 CrossRef CAS.
- P. Chaiyasat, S. Noppalit, M. Okubo and A. Chaiyasat, Sol. Energy Mater. Sol. Cells, 2016, 157, 996–1003 CrossRef CAS.
- P. Chaiyasat, Y. Ogino, T. Suzuki and M. Okubo, Colloid Polym. Sci., 2008, 286, 753–759 CrossRef CAS.
- C. Liang, X. Lingling, S. Hongbo and Z. Zhibin, Energy Convers. Manage., 2009, 50, 723–729 CrossRef.
- J.-S. Cho, A. Kwon and C.-G. Cho, Colloid Polym. Sci., 2002, 280, 260–266 CrossRef CAS.
- W. Li, X.-X. Zhang, X.-C. Wang and J.-J. Niu, Mater. Chem. Phys., 2007, 106, 437–442 CrossRef CAS.
- H. Zhang and X. Wang, Colloids Surf., A, 2009, 332, 129–138 CrossRef CAS.
- T. Y. Wang and J. Huang, J. Appl. Polym. Sci., 2013, 130, 1516–1523 CrossRef CAS.
- T. Feczkó, A. F. Kardos, B. Németh, L. Trif and J. Gyenis, Polym. Bull., 2014, 71, 3289–3304 CrossRef.
- Y. Lin, C. Zhu, G. Alva and G. Fang, Appl. Energy, 2018, 231, 494–501 CrossRef CAS.
- E. Onder, N. Sarier and E. Cimen, Thermochim. Acta, 2008, 467, 63–72 CrossRef CAS.
- A. M. Borreguero, J. L. Valverde, J. F. Rodríguez, A. H. Barber, J. J. Cubillo and M. Carmona, Chem. Eng. J., 2011, 166, 384–390 CrossRef CAS.
- M. Okubo, M. Shiozaki, M. Tsujihiro and Y. Tsukuda, Colloid Polym. Sci., 1991, 269, 222–226 CrossRef CAS.
- M. Okubo and H. Minami, Colloid Polym. Sci., 1997, 275, 992–997 CrossRef CAS.
- Y. Konuklu, M. Ostry, H. O. Paksoy and P. Charvat, Energy Build., 2015, 106, 134–155 CrossRef.
- J. Singh, S. Parvate, P. Dixit and S. Chattopadhyay, Energy Fuels, 2020, 34, 8919–8930 CrossRef CAS.
- F. Wang, W. Lin, Z. Ling and X. Fang, Sol. Energy Mater. Sol. Cells, 2019, 191, 218–234 CrossRef CAS.
- M. Delgado, A. Lázaro, J. Mazo and B. Zalba, Renewable Sustainable Energy Rev., 2012, 16, 253–273 CrossRef CAS.
- X. Wang, J. Niu and A. H. C. van Paassen, Energy Build., 2008, 40, 1691–1698 CrossRef.
- F. Ma and P. Zhang, Appl. Therm. Eng., 2019, 162, 114244 CrossRef.
- L. Liu, Y. Jia, Y. Lin, G. Alva and G. Fang, Energy Convers. Manage., 2017, 145, 30–40 CrossRef CAS.
- A. Petrovic, D. Lelea and I. Laza, Int. Commun. Heat Mass Transfer, 2016, 79, 39–45 CrossRef CAS.
- M. M. U. Rehman, T. A. Cheema, F. Ahmad, M. Khan and A. Abbas, J. Thermophys. Heat Transfer, 2020, 34, 243–254 CrossRef CAS.
- S. Doruk, O. N. Şara, A. Karaipekli and S. Yapıcı, Heat Mass Transfer, 2017, 53, 3399–3408 CrossRef CAS.
- L. Torini, J. F. Argillier and N. Zydowicz, Macromolecules, 2005, 38, 3225–3236 CrossRef CAS.
- H. Johnsen and R. B. Schmid, J. Microencapsulation, 2007, 24, 731–742 CrossRef CAS PubMed.
- K.-F. Ni, G.-R. Shan and Z.-X. Weng, Macromolecules, 2006, 39, 2529–2535 CrossRef CAS.
- T. P. Doan-Nguyen, S. Jiang, K. Koynov, K. Landfester and D. Crespy, Angew. Chem., Int. Ed., 2021, 60, 18094–18102 CrossRef CAS PubMed.
- N. Anton, P. Gayet, J.-P. Benoit and P. Saulnier, Int. J. Pharm., 2007, 344, 44–52 CrossRef CAS PubMed.
- I. Hofmeister, K. Landfester and A. Taden, Macromolecules, 2014, 47, 5768–5773 CrossRef CAS.
- M. B. Bannwarth, S. Ebert, M. Lauck, U. Ziener, S. Tomcin, G. Jakob, K. Münnemann, V. Mailänder, A. Musyanovych and K. Landfester, Macromol. Biosci., 2014, 14, 1205–1214 CrossRef CAS PubMed.
- Y. Zhao, J. Fickert, K. Landfester and D. Crespy, Small, 2012, 8, 2954–2958 CrossRef CAS PubMed.
- X. Liang, Y. Hu, Z. Cao, L. Xiao, J. Lou, L. Liu, Y. Wang, Z. Zhao, D. Qi and Q. Cui, Dyes Pigm., 2019, 163, 371–380 CrossRef CAS.
- X. Liang, M. Tao, D. Wu, B. Yu, Y. Mi, Z. Cao, Z. Zhao and D. Chen, Dyes Pigm., 2020, 177, 108287 CrossRef CAS.
- W. Ostwald, Lehrbuch der allgemeinen chemie, W. Engelmann, Leipzig, 1885 Search PubMed.
- J. M. Asua, Prog. Polym. Sci., 2002, 27, 1283–1346 CrossRef CAS.
- F. Tiarks, K. Landfester and M. Antonietti, Langmuir, 2001, 17, 908–918 CrossRef CAS.
- A. R. Shirin-Abadi, S. Khoee, M. M. Rahim-Abadi and A. R. Mahdavian, Colloids Surf., A, 2014, 462, 18–26 CrossRef CAS.
- S. Torza and S. G. Mason, Science, 1969, 163, 813–814 CrossRef CAS PubMed.
- Y. Luo and X. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 2145–2154 CrossRef CAS.
- A. J. P. van Zyl, R. D. Sanderson, D. de Wet-Roos and B. Klumperman, Macromolecules, 2003, 36, 8621–8629 CrossRef CAS.
- Y. Luo and H. Gu, Macromol. Rapid Commun., 2006, 27, 21–25 CrossRef CAS.
- F. Lu, Y. Luo and B. Li, Ind. Eng. Chem. Res., 2010, 49, 2206–2212 CrossRef CAS.
- Y. Luo and H. Gu, Polymer, 2007, 48, 3262–3272 CrossRef CAS.
- F. Lu, Y. Luo and B. Li, Macromol. Rapid Commun., 2007, 28, 868–874 CrossRef CAS.
- H. Chen and Y. Luo, Macromol. Chem. Phys., 2011, 212, 737–743 CrossRef CAS.
- Y. Changhuai, L. Yingwu and L. Xuesong, Polymer, 2011, 52, 683–693 CrossRef.
- Z. Luo, Y. Li and B. Liu, Chem. Commun., 2017, 53, 8649–8652 RSC.
- B. T. T. Pham, H. Zondanos, C. H. Such, G. G. Warr and B. S. Hawkett, Macromolecules, 2010, 43, 7950–7957 CrossRef CAS.
- J. R. Booth, J. D. Davies and S. A. F. Bon, Polym. Chem., 2022, 13, 1335–1349 RSC.
- N. Saito, Y. Kagari and M. Okubo, Langmuir, 2007, 23, 5914–5919 CrossRef CAS PubMed.
- N. Saito, Y. Kagari and M. Okubo, Langmuir, 2006, 22, 9397–9402 CrossRef CAS PubMed.
- Y. Wang, B.-H. Guo, X. Wan, J. Xu, X. Wang and Y.-P. Zhang, Polymer, 2009, 50, 3361–3369 CrossRef CAS.
- D. Crespy, A. Musyanovych and K. Landfester, Colloid Polym. Sci., 2006, 284, 780–787 CrossRef CAS.
- Y. Li, Z. Wang, X. Kong and G. Xue, Colloids Surf., A, 2010, 363, 141–145 CrossRef CAS.
- D. C. Sundberg, A. P. Casassa, J. Pantazopoulos, M. R. Muscato, B. Kronberg and J. Berg, J. Appl. Polym. Sci., 1990, 41, 1425–1442 CrossRef CAS.
- P. Chaiyasat, S. Noppalit, M. Okubo and A. Chaiyasat, Phys. Chem. Chem. Phys., 2014, 17, 1053–1059 RSC.
- D. W. Aksnes and L. Gjerdåker, J. Mol. Struct., 1999, 475, 27–34 CrossRef CAS.
- R. M. Dimeo, D. A. Neumann, Y. Glanville and D. B. Minor, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 104201 CrossRef.
- S. Amanuel, H. Bauer, P. Bonventre and D. Lasher, J. Phys. Chem. C, 2009, 113, 18983–18986 CrossRef CAS.
- H. C. Bauer, A. D. Safiq, J. Dulmaa, A. S. Khraisat and S. Amanuel, Mater. Res. Soc. Symp. Proc., 1212, 1423, 30–35 Search PubMed.
- G. G. Litvan, Can. J. Chem., 1966, 44, 2617–2622 CrossRef CAS.
- C. L. Jackson and G. B. McKenna, J. Chem. Phys., 1990, 93, 9002–9011 CrossRef CAS.
- H. W. Dorner, Cem. Concr. Res., 1984, 14, 807–815 CrossRef CAS.
- A. R. Shirin-Abadi, A. R. Mahdavian and S. Khoee, Macromolecules, 2011, 44, 7405–7414 CrossRef CAS.
- G. H. Zhang, S. A. F. Bon and C. Y. Zhao, Sol. Energy, 2012, 86, 1149–1154 CrossRef CAS.
- X. Tang, W. Li, H. Shi, X. Wang, J. Wang and X. Zhang, Colloid Polym. Sci., 2013, 291, 1705–1712 CrossRef CAS.
- A. Bakac, M. E. Brynildson and J. H. Espenson, Inorg. Chem., 1986, 25, 4108–4114 CrossRef CAS.
- J. Krstina, G. Moad, E. Rizzardo, C. L. Winzor, C. T. Berge and M. Fryd, Macromolecules, 1995, 28, 5381–5385 CrossRef CAS.
- C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1996, 29, 7717–7726 CrossRef CAS.
- K. G. Suddaby, D. M. Haddleton, J. J. Hastings, S. N. Richards and J. P. O'Donnell, Macromolecules, 1996, 29, 8083–8091 CrossRef CAS.
- ASTM International, ASTM D3418-21 Standard Test Method for Transition Temperatures and Enthalpies of Fusion and Crystallization of Polymers by Differential Scanning Calorimetry, 2021 Search PubMed.
- T. Gruendling, T. Junkers, M. Guilhaus and C. Barner-Kowollik, Macromol. Chem. Phys., 2010, 211, 520–528 CrossRef CAS.
- M. D. Zammit, M. L. Coote, T. P. Davis and G. D. Willett, Macromolecules, 1998, 31, 955–963 CrossRef CAS.
- X. Q. Zhang and C. H. Wang, J. Polym. Sci., Part B: Polym. Phys., 1994, 32, 1951–1956 CrossRef CAS.
- S. H. Yalkowsky, Y. He and P. Jain, Handbook of Aqueous Solubility Data, CRC Press, 2016 Search PubMed.
- J. Brandrup, E. H. Immergut and E. A. Grulke, Polymer Handbook, Wiley, 2003 Search PubMed.
- X. Geng, M. X. Zhai, T. Sun and G. Meyers, Microsc. Microanal., 2013, 19, 319–326 CrossRef CAS PubMed.
- A. Müller and W. H. Bragg, Proc. R. Soc. London, Ser. A, 1932, 138, 514–530 Search PubMed.
- W. M. Mazee, Recl. Trav. Chim. Pays-Bas, 1948, 67, 197–213 CrossRef CAS.
- K. Larsson, Nature, 1967, 213, 383–384 CrossRef CAS.
- G. Ungar and N. Masic, J. Phys. Chem., 1985, 89, 1036–1042 CrossRef CAS.
- G. Ungar, J. Phys. Chem., 1983, 87, 689–695 CrossRef CAS.
- P. J. Flory, Statistical Mechanics of Chain Molecules, Hanser Publishers, 1989 Search PubMed.
- J. B. Hendrickson, J. Am. Chem. Soc., 1961, 83, 4537–4547 CrossRef CAS.
- R. A. Scott and H. A. Scheraga, J. Chem. Phys., 1965, 42, 2209–2215 CrossRef CAS PubMed.
- P. K. Mukherjee, Phys. Rep., 2015, 588, 1–54 CrossRef CAS.
- J. Doucet, I. Denicolò, A. F. Craievich and C. Germain, J. Chem. Phys., 1984, 80, 1647–1651 CrossRef CAS.
- A. M. Taggart, F. Voogt, G. Clydesdale and K. J. Roberts, Langmuir, 1996, 12, 5722–5728 CrossRef CAS.
- E. B. Sirota and A. B. Herhold, Science, 1999, 283, 529–532 CrossRef CAS PubMed.
- D. Cholakova and N. Denkov, Adv. Colloid Interface Sci., 2019, 269, 7–42 CrossRef CAS PubMed.
- R. Montenegro and K. Landfester, Langmuir, 2003, 19, 5996–6003 CrossRef CAS.
- P. Walstra and E. van Beresteyn, Neth. Milk Dairy J., 1975, 29, 35–65 CAS.
- E. v Fette, A. Pham and T. Adalsteinsson, J. Phys. Chem. B, 2008, 112, 5403–5411 CrossRef CAS PubMed.
- S. Wang, K. Tozaki, H. Hayashi, S. Hosaka and H. Inaba, Thermochim. Acta, 2003, 408, 31–38 CrossRef CAS.
- R. Defay and I. Prigogine, Surface tension and adsorption, Wiley, New York, 1966 Search PubMed.
- M. Okubo, H. Minami and Y. Yamamoto, Colloids Surf., A, 1999, 153, 405–411 CrossRef CAS.
- M. Okubo, H. Minami and Y. Yamamoto, Colloid Polym. Sci., 2001, 279, 77–81 CrossRef CAS.
- I. Hofmeister, K. Landfester and A. Taden, Angew. Chem., Int. Ed., 2015, 54, 327–330 CrossRef CAS PubMed.
- G. Wypych, Handbook of Plasticizers, ChemTec Publishing, 2004 Search PubMed.
- R. D. Priestley, D. Cangialosi and S. Napolitano, J. Non-Cryst. Solids, 2015, 407, 288–295 CrossRef CAS.
- T. Ngai and S. A. F. Bon, Particle-Stabilized Emulsions and Colloids: Formation and Applications, Royal Society of Chemistry, 2014 Search PubMed.
- B. P. Binks and T. S. Horozov, Angew. Chem., Int. Ed., 2005, 44, 3722–3725 CrossRef CAS PubMed.
- M. Alcoutlabi and G. B. McKenna, J. Phys.: Condens. Matter, 2005, 17, 461–524 CrossRef.
- K. Lee, G. Yu, E. Woo, S. Hwang and K. Shin, in Adsorption and Phase Behaviour in Nanochannels and Nanotubes, ed. L. J. Dunne and G. Manos, Springer Netherlands, Dordrecht, 2010, pp. 257–272 Search PubMed.
- Q. Qin and G. B. McKenna, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 3475–3486 CrossRef CAS.
- X.-H. Yang and J. Liu, Int. J. Heat Mass Transfer, 2018, 127, 457–468 CrossRef CAS.
- V. v Tyagi, K. Chopra, R. K. Sharma, A. K. Pandey, S. K. Tyagi, M. S. Ahmad, A. Sarı and R. Kothari, Sol. Energy Mater. Sol. Cells, 2022, 234, 111392 CrossRef CAS.
- F. Guthrie, London, Edinburgh Dublin Philos. Mag. J. Sci., 1884, 17, 462–482 CrossRef.
- H. Nazir, M. Batool, M. Ali and A. M. Kannan, Appl. Therm. Eng., 2018, 142, 466–475 CrossRef CAS.
- A. Sarı, C. Alkan and C. Bilgin, Appl. Energy, 2014, 136, 217–227 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Series of recipes for n-hexadecane capsules made with various methacrylates, details of the reactive macromolecular emulsifier, transition onset temperatures and enthalpies for crystallisation and fusion for hexadecane (HD) containging capsules, scanning transmission microscopy data of crosslinked PMMA-OCT capsules, DSC data for n-hexadecane and polymer n-hexadecane blends, electron microscope images of poly(benzyl methacrylate)-hexadecane capsules synthesised using different amounts of trimethylolpropane trimethacrylate (TMA) crosslinking monomer, temperature specific heat capacity data. See DOI: https://doi.org/10.1039/d4py00789a |
| This journal is © The Royal Society of Chemistry 2024 |