
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Degradable branched and cross-linked polyesters from a bis(1,3-dioxolan-4-one) core†
Orla
Buensoz
ab,
Christina A. R.
Picken
ab,
Paul
Price
c,
Christopher
Fidge
c and
Michael P.
Shaver
 *ab
*ab
aDepartment of Materials, Engineering Building A, University of Manchester, Oxford Road, M13 9PL, UK. E-mail: michael.shaver@manchester.ac.uk
bSustainable Materials Innovation Hub, Henry Royce Institute, University of Manchester, Manchester, M13 9PL, UK
cUnilever, Port Sunlight, Wood St, Birkenhead, Wirral CH62 4U, UK
First published on 6th September 2024
Abstract
The control of macromolecular architecture is key to tailoring polymers, with cross-linked and branched topologies conferring useful bulk properties. However, conventional methods to produce topologically diverse polymers typically rely on fossil-fuel derived starting materials and non-degradable backbones. This work reports a facile approach to produce both cross-linked and branched polyesters, using a bio-derived bis(1,3-dioxolan-4-one) (bisDOX) core. Through the copolymerisation of bisDOX with readily available diols, we demonstrate the synthesis of cross-linked polyesters with diverse thermal properties and understand their structure–property relationships. Additionally, branched polyesters are synthesised via the introduction of a tri-functional alcohol monomer with branched structures characterised by detailed NMR spectroscopy. Model compound studies reveal the reactivity of bisDOX and identify preferential, but not exclusive, reactivity at primary vs. secondary hydroxyls. Moreover, multiple end-of-life fates are investigated, with both reprocessability and degradability of the cross-linked polyesters explored. This work offers insights into the synthesis of topologically diverse polyesters and highlights information that could inform the future design of more sustainable materials.
Introduction
Molecular topology plays a key role in determining polymer properties, and the ability to control macromolecular architecture to produce non-linear polymers is an important aspect of polymer science.1 Polymer topologies include branched, star-polymers and cross-linked networks, and these all possess markedly different material properties as a result of their architectures. Highly branched polymers typically possess low intrinsic viscosities and good solubilities thanks to their large number of functional end groups.2,3 Cross-linked polymers possess excellent mechanical and thermal properties as a result of their network structure.4Both classes of polymer suffer from individual sustainability challenges. Branched polymers, used in formulations such a paints and shampoos, are often not (bio)degradable and derive from fossil resources. If not recovered, they can end up in wastewater or leach into the environment.5 Cross-linked polymers are generally used in diverse applications requiring robust performance, such as tyres and wind-turbines. These thermosets have cross-links that preclude mechanical recycling, meaning their end-of-life fate is limited to landfill or pyrolysis.6 Polyesters have been widely studied as possible degradable and sustainable alternatives, owing to the labile esters bonds present in their backbone.7,8 Cross-linked polyesters can degrade back to their constituent monomers and, if vitrimeric, can be reprocessed and facilitate mechanical recycling.9–11 Branched polyesters have found use in formulations such as inks, coatings and fabric conditioners.12–15
Altering the topology of a polymer typically requires changing the entire monomer/polymer system. It would be advantageous to have a system in which topology is controlled by the reaction conditions and facile swapping of co-monomers, to provide a simple platform from which more diverse polyesters could be made. This is enabled by a detailed understanding of reactivity for the platform to facilitate the rational design of materials for specific applications.
1,3-Dioxolan-4-one (DOX) monomers can be used to form linear polyesters via ring-opening polymerisation and elimination of a small molecule (such as formaldehyde, acetaldehyde or acetone) (Scheme 1a).16,17 We previously showed that degradable polyester resins can be synthesised by combining a bis(1,3-dioxolan-4-one) (bisDOX) monomer with cyclic ester comonomers (Scheme 1b).18 BisDOX is synthesised in a single step from L-(+)-tartaric acid, a renewable, non-toxic by-product from food production. In these systems, it is assumed that bisDOX acts as a tetrafunctional cross-linker, reacting with the cyclic ester functionalities as well as the secondary hydroxyl groups formed upon ring-opening. However, the precise reactivity of bisDOX is unknown.
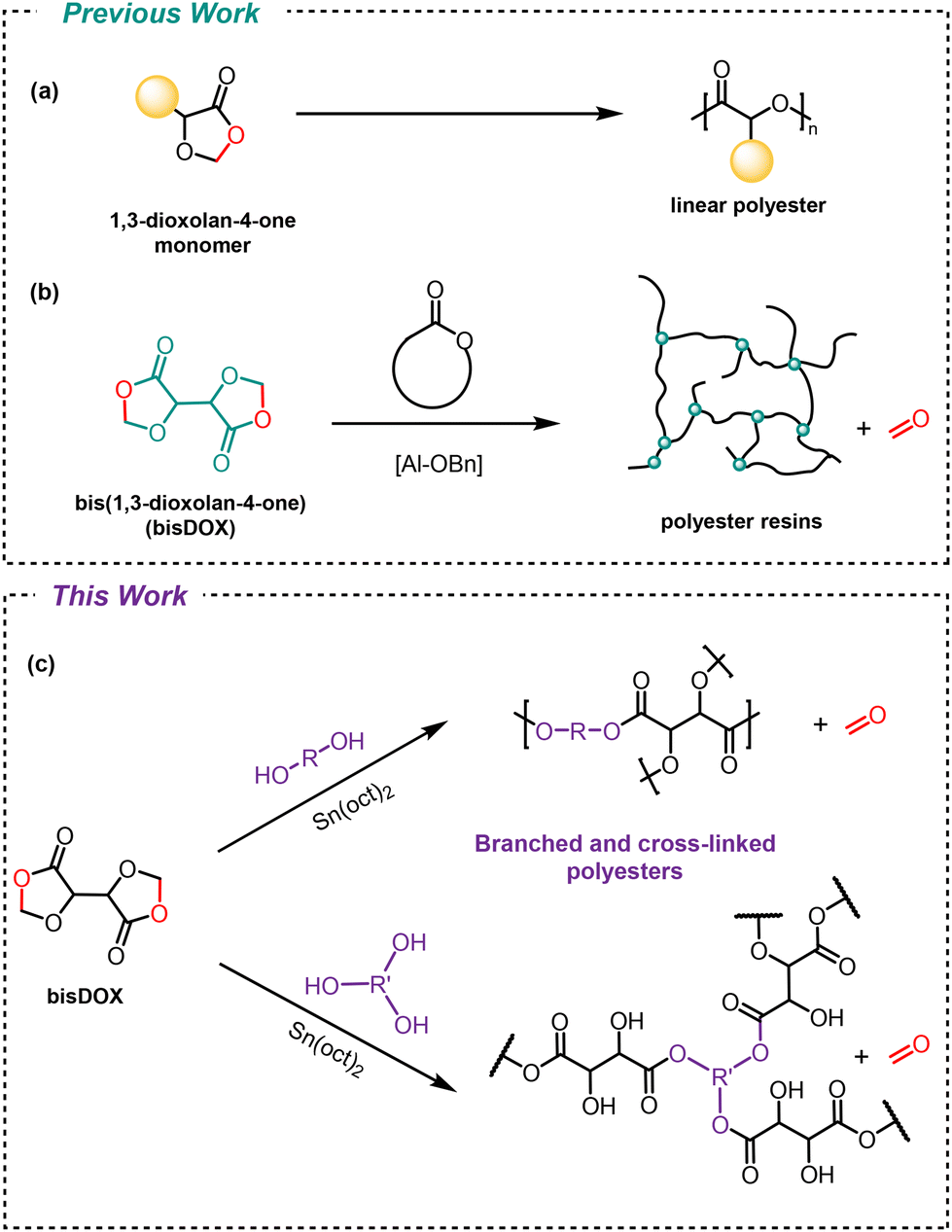 | ||
| Scheme 1 (a) Linear polyesters synthesised by ring-opening polymerisation of 1,3-dioxolan-4one monomers (b) cross-linked polyester resins using bisDOX as a cross-linker with cyclic esters. (c) Synthesis of cross-linked and branched polyesters via copolymerisation of bisDOX with diol and triols showing simplified structures. | ||
Condensation of diols with bisDOX offers the opportunity to both target topologically diverse structures and unequivocally understand the reactivity of this system. Structurally diverse alcohols, diols and triols can model reactivity and serve as levers to balance branching and cross-linking reactions.
The reactivity of bisDOX is explored using tin-catalysed transesterification with inexpensive comonomers. Diols predominantly afford cross-linked systems with a range of thermal properties dependent on the structure of the diol. Switching to a trifunctional alcohol co-monomer and carefully controlling reaction conditions, the synthesis of hyperbranched polyesters is achieved. The reactivity observed in these two systems is corroborated and explained with complementary model compound studies. Multiple potential end-of-life fates, including depolymerisation and reprocessing, are evaluated.
Results and discussion
Model compound studies
In order to understand the reactivity of bisDOX, a model compound study was performed with 2-(benzyloxy)ethanol (2.0 eq.) and bisDOX (1.0 eq.) (functional group ratio (r) of ester![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :primary alcohol, r = 1) (Scheme 2). These compounds were reacted in the presence of Sn(oct)2 (0.03 eq.) at 120 °C and monitored by 1H NMR spectroscopy over 18 h. The major product after 18 h was identified as the tartaric diester formed by ring opening of bisDOX by 2-(benzyloxy)ethanol.9 When the ester:alcohol ratio was decreased to 1:0.5, (r = 0.5) the 1H NMR spectrum of the reaction mixture was significantly more complicated, suggesting the formation of multiple products alongside the tartaric diester. Products present in the reaction mixture were identified as the asymmetric tartaric mono-ester, formed by ring-opening of one DOX ring, along with dimers/trimers/oligomers of branched bisDOX units. The peaks associated methylene protons in the branched units are similar in chemical shift as those in the tartaric diester product (3.62, 4.33 and 4.47 ppm in Fig. S6†); broadening of the signal is visible in the branched polymer peaks from multiple microenvironments.
:primary alcohol, r = 1) (Scheme 2). These compounds were reacted in the presence of Sn(oct)2 (0.03 eq.) at 120 °C and monitored by 1H NMR spectroscopy over 18 h. The major product after 18 h was identified as the tartaric diester formed by ring opening of bisDOX by 2-(benzyloxy)ethanol.9 When the ester:alcohol ratio was decreased to 1:0.5, (r = 0.5) the 1H NMR spectrum of the reaction mixture was significantly more complicated, suggesting the formation of multiple products alongside the tartaric diester. Products present in the reaction mixture were identified as the asymmetric tartaric mono-ester, formed by ring-opening of one DOX ring, along with dimers/trimers/oligomers of branched bisDOX units. The peaks associated methylene protons in the branched units are similar in chemical shift as those in the tartaric diester product (3.62, 4.33 and 4.47 ppm in Fig. S6†); broadening of the signal is visible in the branched polymer peaks from multiple microenvironments.
 | ||
| Scheme 2 Model compound reactions between bisDOX and 2-(benzyloxy)ethanol at different ratios, showing the products formed. | ||
These model compound studies suggest that, in the presence of Sn(oct)2, bisDOX will preferentially react with 1° OHs, however, when there is a stoichiometric imbalance, bisDOX can react with the 2° OHs formed on ring-opening. In the copolymerisation of bisDOX and 1,4-butanediol, gel formation is observed even with a 1:1 functional group ratio. This suggests a slower rate of reaction of bisDOX with the diol alcohols after initial ring opening, likely due to entropic effects or diffusion control.
Copolymerisation of bisDOX and diols
To investigate the reactivity of bisDOX, copolymerisation with 1,4-butanediol was carried out. BisDOX and 1,4-butanediol were mixed with catalyst at a ratio of 1:1 in toluene at 120 °C for 20 h. Initial catalyst screens using aluminium salen catalysts (Scheme S3†), which are known to be active towards DOX ring-opening, showed limited polymerization, evidenced by very low gel contents (<10%, Table S1†) while lower polymerisation temperatures (80 °C) did not promote productive polymerisations. An alternative metal alkoxide catalyst, tin(II) 2-ethylhexanoate (Sn(oct)2), was tested. Copolymerisation of bisDOX and 1,4-butanediol with Sn(oct)2 yielded a cross-linked polyester, BD-4 (Scheme 3a). The resultant thermoset was insoluble in all common organic solvents and water, but swelled in organic solvents such as dichloromethane, acetonitrile, chloroform. Sn(oct)2 was hypothesised to be more effective than the Al catalyst due to its greater propensity for transesterification across polymer chains, which may be key to forming network polymers.19 FT-IR spectroscopic analysis of BD-4 showed the disappearance of the bisDOX carbonyl resonance at 1790 cm−1 and the appearance of a new carbonyl stretching frequency at 1730 cm−1, supporting the formation of the target polyester (Scheme 3b). Deposition of extruded oligomeric formaldehyde within the reaction vessel, a characteristic by-product of DOX monomer polymerisations,16,17 suggested no poly(ester acetal) retention. The FT-IR spectrum also shows a small carbonyl peak at 1800 cm−1, tentatively assigned to a small number of pendant bisDOX rings in which only one of the cyclic esters has undergone ring opening. The normalised FT-IR spectra of all BD-X samples (Fig. S6†) shows the lowest intensity for the peak at 1800 cm−1 in BD-4, suggesting that this sample has the highest density of cross-links as fewer unopened bisDOX rings remain. The broad peak at around 3400 cm−1 corresponded to the free hydroxyls of the ring-opened bisDOX units.
 | ||
Scheme 3 (a) Synthesis of cross-linked polyesters using bisDOX and diols. (b) FT-IR spectra of bisDOX showing its characteristic carbonyl stretching at 1790 cm−1 and BD-4 showing the appearance of a new C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching peak at 1733 cm−1. O stretching peak at 1733 cm−1. | ||
The observation of a cross-linked product in the copolymerisation bisDOX with a difunctional alcohol necessitates activation of the secondary alcohols produced from ring opening to reveal the tartaric acid substructure. This is key to understanding reactivity, primarily that it acts as a tetrafunctional monomer (functionality, f = 4). If bisDOX were to only react at the ester groups, we would expect to see a linear soluble polymer. The observation of an insoluble cross-linked polyester corroborates this tetrafunctionality, but the presence of unreacted DOX rings and free hydroxyl units suggests that the system retains latent reactivity. BisDOX does not always react at all four possible reactive moieties which may be due to the limited mobility of the chains in a kinetically trapped gel or different reactivity of the primary and secondary alcohols towards ring-opening and inter-chain transesterification.
To understand the structure–property relationships of the cross-linked polymers, the diol monomer chain length was varied using 1,6-hexanediol and 1,10-decanediol. In all cases, translucent polyester samples were formed with gel contents between 78–93%, indicating the formation of networks with a high degree of cross-linking (Table 1). The thermal properties were assessed by differential scanning calorimetry (DSC) (Fig. S1†) and thermogravimetric analysis (TGA) (Fig. S2†).


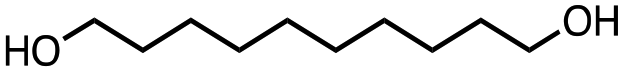
All products were amorphous and displayed glass transitions temperatures (Tg) between −11 and 23 °C (Table 1), with an increasing number of carbons in the diol co-monomer correlating to a decreasing Tg, with the lower density of cross-linking in BD-10 arising from the longer alkyl chains creating a larger free volume within the polymer and therefore greater chain mobility compared to BD-4. The thermal stability of the samples, however, increases with increasing carbon chain length. The Td,5% for BD-4 is 167 °C was lower than that of BD-10 (250 °C), which could result from the former possessing a greater degree of cross-linking, with more ester bonds creating internal stress and degrading at a lower temperature. Moreover, the relative weight of the ester linkages decreases with increasing alkyl chain length, providing additional thermal stability to BD-10 compared to BD-4. The variation in material properties provided by simply altering the diol co-monomer offers a way to rationally design polyester network properties.
Branched polyester synthesis
While numerous cross-linked polyester systems exist,18 extending to more challenging polymer topologies, such as those featuring in formulation polymers, is important to enable degradation in these systems. The copolymerisation of bisDOX with a tri-functional alcohol, 1,1,1-tris(hydroxymethyl)ethane (TME) over 18 h at a concentration of 5 M in toluene at 120 °C led to gel formation and release of paraformaldehyde within the reaction vessel. Drying the gels afforded a sticky solid insoluble in common organic solvents or water. Initial characterisation suggested a similar structure to diol reaction, with the disappearance of the bisDOX carbonyl at 1790 cm−1 and appearance of a new carbonyl peak at 1740 cm−1 in FT-IR spectra supporting the formation of a crosslinked polyester. The additional triol functionality could cross-link without invoking the secondary alcohols from the tartaric acid, however gelation can be prevented in such systems by carefully selecting reaction parameters such as the concentration or extent of reaction.20 A systematic approach to altering alter the reaction conditions in order to produce a branched polyester rather than a cross-linked sample was undertaken.We hypothesised that decreasing monomer concentration and reaction time would avoid gelation by entropically disfavouring network formation and quenching the reaction before its gel point. When reaction times were decreased (18 h–2 h) and monomer concentration reduced to 0.2 M, the gel vs. soluble fractions could be tuned. Reaction times were incrementally decreased until optimised at 2 hours; only soluble polymer was formed and no gel fraction observed (Table 2). The polymer was isolated by removal of toluene under reduced pressure, dissolution in the minimum amount of tetrahydrofuran (THF) and precipitation into cold diethyl ether to give white, tacky solids that formed brittle solids when fully dried under vacuum.
| Entry | [M]0 (mol L−1) | Time (h) |
M
na (g mol−1) |
Đ
Ma |
T
gd (°C) |
Gel (%) |
|---|---|---|---|---|---|---|
|
a
M
n and ĐM were determined by size exclusion chromatography in THF using triple detection methods to obtain absolute molecular weights.
b Denotes polymer was insoluble so SEC analysis could not be performed.
c Denotes sol and gel fractions were formed and SEC analysis was carried out on sol fraction.
d Glass transition temperature obtained by differential scanning calorimetry, taken from second heating curve. Polymerisations were conducted in toluene at 120 °C where [bisDOX]:[TME]:[C] = 50:50:1.
|
||||||
| 1 | 5 | 18 | Gelb | 33 | 46 | |
| 2 | 0.2 | 18 | Gelb | 46 | 35 | |
| 3 | 0.2 | 8 | 450c | 2.5c | — | — |
| 4 | 0.2 | 2 | 22000 |
1.2 | 38 | 0 |
NMR spectroscopy was used to confirm the branched nature of the polymer (entry 4, Table 2). The complex 1H spectrum (Fig. 1) was deconvoluted and assigned with the help of 13C and 2D NMR experiments (Fig. S3–S5†). The peaks at 0.8–1.03 ppm correspond to the –CH3 protons of the TME unit and have different chemical shift values, dependent on whether 1, 2 or 3 alcohols in the TME have reacted.21 The broad peak at 3.4 ppm represents unreacted –CH2OH protons on TME cores. Peaks appearing between 4.01–4.26 ppm correspond to the –CH2 protons of substituted TME arms, i.e., those that are adjacent to an ester group. The methylene proton assignment is corroborated by the 1H–13C HMBC spectrum (Fig. S5†), in which a cross-resonance corresponding to the coupling of the esterified –CH2 protons with a carbonyl proton was observed. Accordingly, a cross-resonance peak corresponding to the coupling of the alcohol-adjacent methylene protons to a carbonyl peak was not observed. The peaks corresponding to the methine protons of the ring-opened bisDOX are seen at 4.5–4.84 ppm. A degree of branching for the branched bisDOX-TME polymer was calculated using the relative integrations of peaks a′ and a′′ (see ESI† for more detail) and found to be 0.66, indicative of a hyperbranched polymer. These results verified the branched structure and showcased the diverse structural units possible within the polymer network.
 | ||
| Fig. 1 1H NMR spectrum (THF-d8, 298 K, 500 MHz) of branched bisDOX and TME co-polymer showing possible different structural units. | ||
End of life opportunities
Polymers with all-carbon backbones persist in the environment, resisting degradation even under harsh conditions.22 The labile ester bonds present in polyesters should render them susceptible to hydrolytic degradation.23,24 Degradability in both cross-linked polymers and branched structures is important, as recovery and recycling at end of life is challenging for both thermosets and formulation polymers. We explored hydrolytic degradation of this system through an accelerated degradation study. Thin-film samples of BD-10 (10 × 5 × 0.8 mm) were immersed in 1.0 M NaOH at 25 °C. Complete dissolution was observed after 24 h, confirming that these polymers are highly susceptible to hydrolytic degradation.23,24Chemical depolymerisation is another possible end-of-life fate for polyesters, converting back polymers to monomers for re-polymerisation. This offers a complementary solution to circularising the linear plastics economy,25 especially for complex multi-materials.26,27 Polyesters can be readily depolymerised by a variety of catalysts,28 with base-catalysed depolymerisation by 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), a common choice.29 A thin film sample of BD-10 (10 × 5 × 0.8 mm) was immersed in a solution of 1.0 M DBU in acetonitrile. Complete dissolution of the sample was observed within 20 m; 1H NMR spectroscopy revealed the presence of L-tartaric acid and the starting monomer, 1,10-decanediol. These results are a proof-of-concept for the ability to chemically recycle these cross-linked polyesters.
Cross-linked polymers are typically not mechanically recyclable, as their permanent network structure precludes reprocessing. However, networks containing dynamic covalent bonds are able to rearrange their topology, and therefore can be reshaped upon application of a stimuli such as heat.30
Such dynamic covalent networks have been reported based on a number of exchangeable linkages including polyesters.31–41
It was hypothesised that the ester bonds and residual catalyst in the synthesised samples would facilitate the required transesterification reactions.30 Indeed, these systems are readily reprocessable and display vitrimeric behaviour. A sample of BD-10 was cut in half and hot pressed at 120 °C for 2 hours, after which the healing of the sample was observed (Fig. 2a). Stress-relaxation analyses, in which the sample is subjected to a set strain and the resultant stress relaxation modulus is measured over time as it dissipates, is often used to quantify reprocessability. The characteristic relaxation time, τ* is measured at the point when the relaxation modulus has relaxed to 1/e of its original value.31 Stress-relaxation experiments were carried out on BD-10 between 90–120 °C, with an applied strain of 2%. τ* was found to be approximately 1100 s at 90 °C (Fig. 2b), and decreased as the temperature increased, to a value of 110 s at 120 °C. To determine the activation energy (Ea) for the transesterification reaction, ln(τ*) and 1/T were fitted to an Arrhenius plot (Fig. 2c). Ea was determined to be 89 kJ mol−1, which is similar to the reported value for Sn(oct)2-catalysed dynamic exchange in bulk materials (83 kJ mol−1).42
 | ||
| Fig. 2 (a) Pictures showing BD-10 before and after reprocessing by hot-pressing (b) stress-relaxation analysis for BD-10 indicating the characteristic relaxation time, τ*, when the relaxation modulus reaches 37% of its original value. (c) Arrhenius plot relating τ* to the inverse of temperature for BD-10. | ||
Conclusions
The synthesis of polyesters using a bio-based bis(1,4-dioxolan-4-one) core can be expanded through copolymerisation of bisDOX with diols and a triol. Tuning comonomer and reaction conditions can tailor architectures and tune material properties. The reactivity of bisDOX, elucidated through model compound studies, shows that the preferential reactivity of bisDOX with primary versus secondary hydroxyl moieties enables control over polymer architecture, enhancing the versatility of this monomer in the synthesis of polyesters. End-of-life opportunities for these systems are diverse, with the products susceptible to hydrolytic degradation, chemical depolymerisation, and reprocessing through dynamic transesterification reactions. Together these systems represent promising avenues for moving towards a more circular polymer economy, especially in polymer applications where material recovery or performance are not met by traditional linear topologies.Experimental
General considerations
All air-sensitive manipulations were performed in an MBraun LABmaster sp glovebox under nitrogen or using conventional Schlenk techniques under dry nitrogen.Materials
The following chemicals were used as they were received, L-(+)-tartaric acid (>99.5%, Sigma-Aldrich) paraformaldehyde (Fischer Scientific), p-toluenesulfonic acid monohydrate (98.5%, Sigma-Aldrich), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (98%, Sigma Aldrich), tetrahydrofuran-d8 (≥99.5, Sigma Aldrich), chloroform-d. 1,4-Butanediol (99% Sigma-Aldrich) and 2-(benzyloxy)ethanol (98%, FluoroChem) were dried over CaH2 and distilled under vacuum. Tin(II) 2-ethylhexanoate (92.5–100%, Sigma Aldrich) was dried over CaH2 and fractionally distilled at 120 °C under vacuum, followed by vacuum distillation twice more. 1,6-Hexanediol (99% Sigma-Aldrich) and 1,10-decanediol (99%, Thermo Scientific) – were recrystallised from DCM. 1,1,1-Tris(hydroxymethyl)ethane (97%, Acros Organics) was re-crystallised from THF and hexane. Anhydrous toluene was obtained from an Mbraun 7 Solvent Purification System containing alumina and copper catalysts and degassed by three successive freeze–pump–thaw cycles.Synthetic procedures
BisDOX and aluminium salen catalysts were synthesised according to literature.18,43Representative procedure for the copolymerisation of bisDOX and diol
In a glovebox under a nitrogen atmosphere, Sn(oct)2, bisDOX, and diol monomer were weighed out directly into a vial. The vial was sealed, removed from the glovebox and either placed in a pre-heated oil bath at 120 °C for 18 h or placed into a glovebag. The glovebag was degassed and filled with nitrogen before the mixture was placed in a pre-heated Petri dish sprayed with PTFE mould-release spray (Rocol). The samples were heated at 120 °C on a hot plate for 18 h. The samples were cooled and removed from the vial or Petri dish.Representative procedure for the copolymerisation of BisDOX and TME
In a glovebox under a nitrogen atmosphere, an ampoule was charged with BisDOX, TME, catalyst and toluene. The ampoule was sealed, removed from the glovebox and heated to 120 °C with stirring. The reaction was quenched by the addition by a few drops of MeOH and the toluene was removed in vacuo. The residue was dissolved in the minimum amount of THF and an aliquot was taken for crude 1H NMR spectroscopic analysis. The polymer was isolated by precipitation into a swirling vortex of cold Et2O, filtered and then dried overnight in a vacuum oven at 40 °C.Gel fraction studies
Gel fraction studies were performed using THF or CHCl3. A small amount of each pre-weighed sample was immersed in solvent for 24 h before the solvent was decanted. The swollen sample was then dried under vacuum for 24 h or until constant weight before the final mass was measured. The gel fraction was calculated by taking the ratio of the final mass to the initial mass.Depolymerisation of BD-10
Thin film samples of BD-10 were placed in a 1.0 M solution of diazabicyclo[5.4.0]undec-7-ene (DBU) in acetonitrile and stirred at room temperature until visible dissolution the polymer had occurred.Characterisation
All NMR experiments were performed at 298 K on either a 400 MHz Bruker AVIII spectrometer or a 500 MHz Bruker AVIII HD spectrometer. Chemical shifts are reported as δ in parts per million (ppm) and referenced to the chemical shift of the residual solvent resonances (CDCl31H: δ = 7.26 ppm, 13C: δ = 77.16 ppm, THF-d81H: δ = 1.72, 3.58 ppm, 13C: δ = 67.21, 25.31) The resonance multiplicities are described as s (singlet), d (doublet), t (triplet), q (quartet) or m (multiplet).Gel permeation chromatography (GPC) was carried our using an Agilent 1260 Infinity II Multi-Detector Gel Permeation Chromatography (GPC)/Size Exclusion Chromatography System through Plgel 5 μm columns packed with PSDVB beads. The GPC was run with THF at a flow rate of 1.00 ml min−1 at 35 °C.
Molecular weights were obtained using triple detection based on a calibration prepared with narrow dispersity polystyrene standards.
Differential scanning calorimetry (DSC) analysis was performed on a TA instruments DSC 2500. All samples were prepared to weigh between 3–10 mg and were placed in Tzero pans and fitted with Tzero lids. Samples were subject to an initial equilibration step before initial thermal ramp at 10 °C min−1 up until 180 °C to erase the samples thermal history and then cooled to −50 or −20 °C at 10 °C min−1 and then heated at 10 °C min−1 until 180 °C. Nitrogen was used as a purge gas at 50 mL min−1.
Thermogravimetric analysis (TGA) was performed using a TA Simultaneous Thermal Analyser SDT650 instrument. The samples (10–25 mg) were placed in alumina pans (90 μL) and heated in nitrogen from room temperature to 600 °C at a rate of 10 °C min−1. The onset of thermal degradation temperature is defined as the temperature at which the material loses 5% of its mass (Td,5%) and this was reported for all materials. All experiments were performed in triplicate.
Fourier transform infrared (FT-IR) spectra were produced on a Bruker INVENIO. Scans were performed between 400 cm−1 and 4000 cm−1 and peaks manually picked on the INVENIO software.
Stress-relaxation analyses were performed on a stress-controlled TA DH2 rheometer with a 20 mm parallel plate geometry and a peltier plate. The samples were equilibrated at the test temperature for 15 minutes, after which they were subjected to an instantaneous strain of 2%. The stress decay was monitiored until the stress relaxation modulus had relaxed to at least 37% (1/e) of its initial value.
Author contributions
Orla Buensoz: Investigation, methodology, writing – original draft, review and editing. Christina A. R. Picken: Writing – review and editing. Paul D. Price: Funding acquisition, supervision. Christopher Fidge: Funding acquisition, supervision. Michael P. Shaver: Conceptualization, funding acquisition, supervision, writing – review and editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
Unilever provided sponsorship for this project as part of their efforts to improve product and process sustainability.Acknowledgements
The authors would like to thank Unilever and the University of Manchester for their financial support. This work was also supported by the Henry Royce Institute for Advanced Materials, funded through EPSRC grants EP/R00661X/1, EP/S019367/1, EP/P025021/1, EP/P025498/1 and the Sustainable Materials Innovation Hub, funded through the European Regional Development Fund OC15R19P. The authors wish to thank Dr Theona Şucu for advice and helpful discussions, and Dr Peng Huang for assistance in scale-up of bisDOX synthesis.References
- G. Odian, Principles of Polymerization, John Wiley & Sons, Inc, Hoboken, New Jersey, 4th edn, 2004 Search PubMed.
- C. Gao, D. Yan and H. Frey, in Hyperbranched Polymers: Synthesis, Properties, and Applications, ed. D. Yan, John Wiley & Sons, 2011 Search PubMed.
- X. Zhu, Y. Zhou and D. Yan, in Hyperbranched Polymers: Synthesis, Properties, and Applications, John Wiley & Sons, 2011, pp. 317–331, DOI:10.1002/9780470929001.ch12.
- T. Şucu and M. P. Shaver, Polym. Chem., 2020, 11, 6397–6412 RSC.
- RSC, Royal Society of Chemistry Polymers in liquid formulations Technical report, 2021 Search PubMed.
- D. J. Fortman, J. P. Brutman, G. X. De Hoe, R. L. Snyder, W. R. Dichtel and M. A. Hillmyer, ACS Sustainable Chem. Eng., 2018, 6, 11145–11159 CrossRef CAS.
- J. Rydz, W. Sikorska, M. Kyulavska and D. Christova, Int. J. Mol. Sci., 2015, 16, 564–596 CrossRef PubMed.
- Y. Wang, R. J. van Putten, A. Tietema, J. R. Parsons and G. M. Gruter, Green Chem., 2024, 26, 3698–3716 RSC.
- S. Debnath, S. Kaushal and U. Ojha, ACS Appl. Polym. Mater., 2020, 2, 1006–1013 CrossRef CAS.
- J. L. Self, N. D. Dolinski, M. S. Zayas, J. Read de Alaniz and C. M. Bates, ACS Macro Lett., 2018, 7, 817–821 CrossRef CAS PubMed.
- J. P. Brutman, P. A. Delgado and M. A. Hillmyer, ACS Macro Lett., 2014, 3, 607–610 CrossRef CAS PubMed.
- V. Shpotya, A. Perepukhov, A. Maksimychev, V. Gomzyak, N. Sedush and S. Chvalun, Polym. Bull., 2023, 80, 4523–4534 CrossRef CAS.
- T. P. Haider, C. Völker, J. Kramm, K. Landfester and F. R. Wurm, Angew. Chem. Int. Ed., 2019, 58, 50–62 CrossRef CAS.
- Z. Żołek-Tryznowska and J. Izdebska, Dyes Pigm., 2013, 96, 602–608 CrossRef.
- C. Picken, O. Buensoz, P. Price, C. Fidge, L. Points and M. P. Shaver, Chem. Sci., 2023, 14, 12926–12940 RSC.
- S. A. Cairns, A. Schultheiss and M. P. Shaver, Polym. Chem., 2017, 8, 2990–2996 RSC.
- Y. Xu, M. R. Perry, S. A. Cairns and M. P. Shaver, Polym. Chem., 2019, 10, 3048–3054 RSC.
- T. Şucu, M. Wang and M. P. Shaver, Macromolecules, 2023, 56, 1625–1632 CrossRef.
- M. Bero, B. Czapla, P. Dobrzyński, H. Janeczek and J. Kasperczyk, Macromol. Chem. Phys., 1999, 200, 911–916 CrossRef CAS.
- T. Biela and I. Polanczyk, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4214–4221 CrossRef CAS.
- T. Emrick, H. T. Chang and J. M. J. Fréchet, Macromolecules, 1999, 32, 6380–6382 CrossRef CAS.
- A. Chamas, H. Moon, J. Zheng, Y. Qiu, T. Tabassum, J. H. Jang, M. Abu-Omar, S. L. Scott and S. Suh, ACS Sustainable Chem. Eng., 2020, 8, 3494–3511 CrossRef CAS.
- M. Vert, S. Li, H. Garreau, J. Mauduit, M. Boustta, G. Schwach, R. Engel and J. Coudane, Angew. Makromol. Chem., 2003, 247, 239–253 CrossRef.
- A. Göpferich, Biomaterials, 1996, 17, 103–114 CrossRef PubMed.
- R. A. Clark and M. P. Shaver, Chem. Rev., 2024, 124, 2617–2650 CrossRef CAS PubMed.
- P. Huang, J. Pitcher, A. Mushing, F. Lourenço and M. P. Shaver, Resour., Conserv. Recycl., 2023, 190, 106854 CrossRef CAS.
- P. Huang, A. Ahamed, R. Sun, G. X. De Hoe, J. Pitcher, A. Mushing, F. Lourenço and M. P. Shaver, ACS Sustainable Chem. Eng., 2023, 11, 15328–15337 CrossRef CAS.
- Y. Weng, C.-B. Hong, Y. Zhang and H. Liu, Green Chem., 2024, 26, 571–592 RSC.
- K. Fukushima, D. J. Coady, G. O. Jones, H. A. Almegren, A. M. Alabdulrahman, F. D. Alsewailem, H. W. Horn, J. E. Rice and J. L. Hedrick, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1606–1611 CrossRef CAS.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- R. L. Snyder, D. J. Fortman, G. X. De Hoe, M. A. Hillmyer and W. R. Dichtel, Macromolecules, 2018, 51, 389–397 CrossRef CAS.
- R. L. Snyder, C. A. L. Lidston, G. X. De Hoe, M. J. S. Parvulescu, M. A. Hillmyer and G. W. Coates, Polym. Chem., 2020, 11, 5346–5355 RSC.
- D. J. Fortman, J. P. Brutman, C. J. Cramer, M. A. Hillmyer and W. R. Dichtel, J. Am. Chem. Soc., 2015, 137, 14019–14022 CrossRef CAS.
- M. I. Lawton, K. R. Tillman, H. S. Mohammed, W. Kuang, D. A. Shipp and P. T. Mather, ACS Macro Lett., 2016, 5, 203–207 CrossRef CAS PubMed.
- O. R. Cromwell, J. Chung and Z. Guan, J. Am. Chem. Soc., 2015, 137, 6492–6495 CrossRef CAS PubMed.
- M. Röttger, T. Domenech, R. van der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, Science, 2017, 356, 62–65 CrossRef PubMed.
- N. Zheng, Z. Fang, W. Zou, Q. Zhao and T. Xie, Angew. Chem., Int. Ed., 2016, 55, 11421–11425 CrossRef CAS.
- X. Yang, L. Guo, X. Xu, S. Shang and H. Liu, Mater. Des., 2020, 186, 108248 CrossRef CAS.
- T. Liu, C. Hao, S. Zhang, X. Yang, L. Wang, J. Han, Y. Li, J. Xin and J. Zhang, Macromolecules, 2018, 51, 5577–5585 CrossRef CAS.
- M. Hayashi and R. Yano, Macromolecules, 2019, 53, 182–189 CrossRef.
- A. Duval, W. Benali and L. Avérous, Green Chem., 2024, 26, 8414–8427 RSC.
- Y. Yu, G. Storti and M. Morbidelli, Ind. Eng. Chem. Res., 2011, 50, 7927–7940 CrossRef CAS.
- P. Hormnirun, E. L. Marshall, V. C. Gibson, R. I. Pught and A. J. P. White, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15343–15348 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00551a |
| This journal is © The Royal Society of Chemistry 2024 |