
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible-light-mediated Diels–Alder reactions under single-chain polymer confinement: investigating the role of the crosslinking moiety on catalyst activity†
Michael
Spicuzza
a,
Shweta Prakesh
Gaikwad
a,
Steven
Huss
a,
Annemarie A.
Lee
b,
Cristina V.
Craescu
a,
Anna
Griggs
a,
Joshmi
Joseph
a,
Mark
Puthenpurayil
a,
Wilson
Lin
a,
Christopher
Matarazzo
a,
Stanley
Baldwin
ac,
Victoria
Perez
b,
Diego Alejandro
Rodriguez-Acevedo
a,
John R.
Swierk
 b and
Elizabeth
Elacqua
*a
b and
Elizabeth
Elacqua
*a
aDepartment of Chemistry, The Pennsylvania State University, University Park PA, 16802, USA. E-mail: elizabeth.elacqua@psu.edu
bDepartment of Chemistry, State University of New York at Binghamton, Vestal, NY 13850, USA
cDepartment of Natural, Health, and Mathematical Sciences, MidAmerica Nazarene University, Olathe, KS 66062, USA
First published on 10th April 2024
Abstract
Macromolecular scaffolds are rapidly emerging in catalysis owing to the ability to control catalyst placement at precise locations. This spatial proximity allows for enhanced catalyst activity that may not be observed using small molecules. Herein, we describe a triphenylpyrylium (TPT)-based visible-light active single-chain polymer nanoparticle (SCNP) that facilitates the radical cation [4 + 2]-cycloaddition. We find that the catalytic activity is highly dependent on the styrylarene comonomer used, wherein it can act as a redox mediator under confinement, increasing the catalytic turnover (TON) by up to 30 times in comparison to free TPT in solution. Mechanistic studies indicate that TPT excited states are quenched by the acene, with the resultant radical cation formed from naphthalene-based SCNPs able to proceed in oxidizing the dienophile in the elementary step of the reaction, while leading to near quantitative yields of the cycloadduct. The TPT-SCNP demonstrates enhanced photocatalyst efficiency compared to molecular TPT, and is able to be recycled and reused in three iterations of the reaction prior to decreased performance from photobleaching. Our results overall suggest that the confined nature of the SCNP and spatial proximity of acene-based pendants enforces their participation as cocatalytic redox mediators that impart enhanced photoredox catalysis under confinement.
Introduction
Over the last decade, the design and use of single-chain polymer nanoparticles (SCNPs) as homogeneous catalysts has garnered much interest.1–11 SCNPs constitute confined macromolecular architectures that arise through the controlled folding of individual polymer chains.12–15 Multiple compaction approaches have been reported based primarily on the use of reversible supramolecular, dynamic covalent, and/or irreversible covalent crosslinking chemistries. To date, the majority of these folding strategies have installed catalytically-relevant groups through judicious selection of comonomers. For instance, Roesky and coworkers incorporated diphenylphosphinostyrene into a linear poly(styrene) that, in the presence of Pt[cod]Cl2, formed SCNPs featuring crosslinks that mimicked traditional cross-coupling catalysts (Scheme 1).16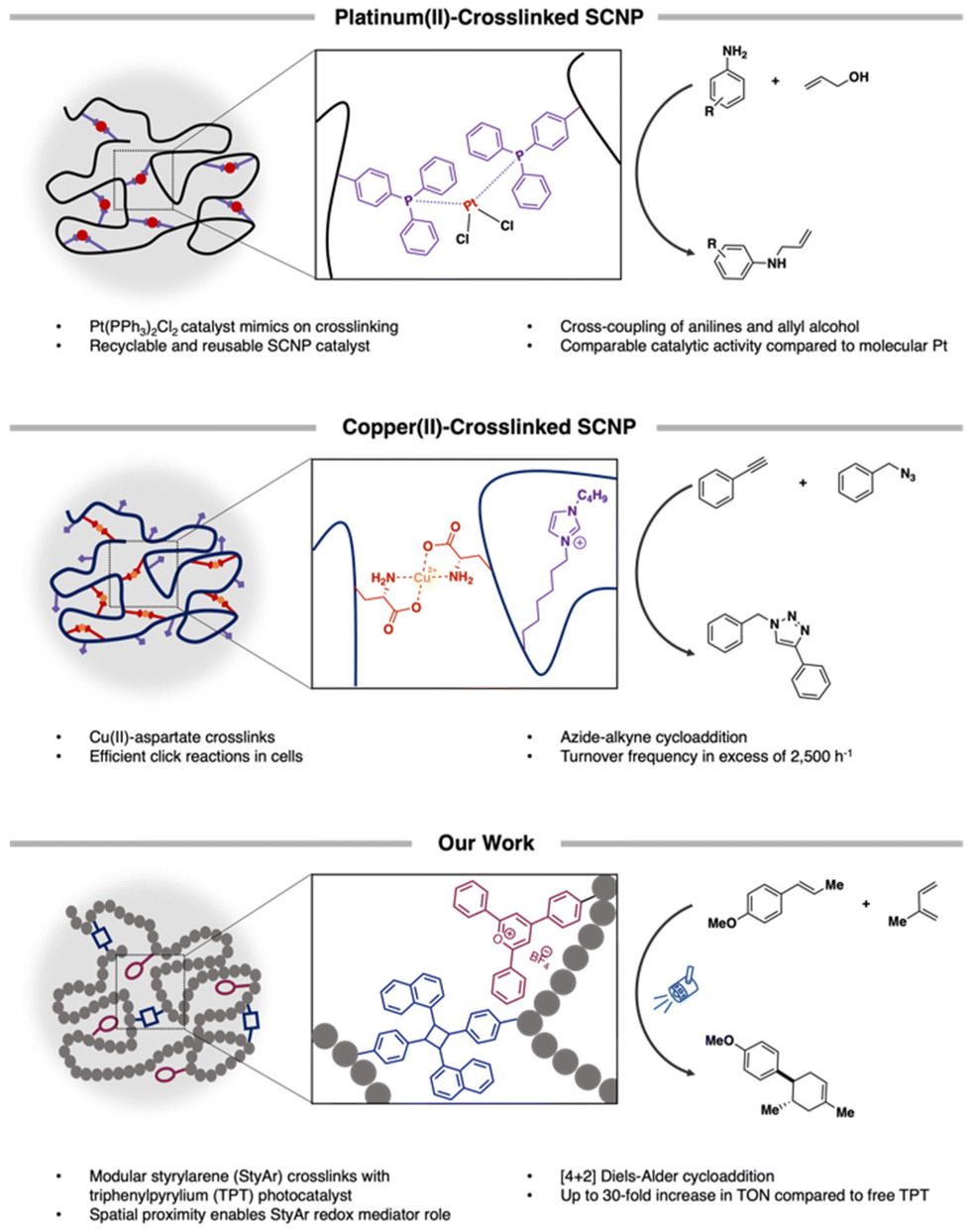 | ||
| Scheme 1 Overview and conceptual designs for SCNP-based catalysis: (top) Traditional cross-coupling mimicked through the PtII-mediated crosslinking of pendant -PPh3 groups, leading to recyclable SCNPs; (middle) CuII-based SCNPs that, after reduction, enable reactant diffusion into the interior, accelerating aqueous click reactions; and (bottom) triarylpyrylium-based SCNPs wherein spatial proximity of acene-based pendants enforces their participation as cocatalytic redox mediators that accelerate Diels–Alder reactions. | ||
The compact nature of SCNPs, best described as resembling intrinsically-disordered proteins (IDPs) with locally-compact portions connected by flexible chains,17 has led to confinement-aided catalysis. For example, Lemcoff reported an Ir/Rh-bimetallic poly(cyclooctadiene) wherein close proximity of the catalytic centers led to enhanced cross-coupling reactions.18 Further, CuII-based SCNPs achieved selective Glaser-Hay couplings,10 where confinement controlled selectivity and circumvented homocoupling. Zimmerman also demonstrated that CuI-SCNPs facilitated highly efficient click reactions in small molecules19 and cells; key to this was the diffusion of azides and alkynes into the polymer interior, proximal to Cu-binding sites (Scheme 1).6,20 In these combined studies, key designs have enabled product selectivity and/or enhanced reactivity compared to molecular analogues.
Recent efforts,1,3,4,21 have utilized SCNPs to investigate photoredox catalysis. Photoredox methods are potent means of constructing challenging bonds, particularly when using dual catalysis.22–28 Despite the widespread application of this strategy, the full potency can only be realized if the transient radical is generated in close proximity to the second catalytic species, thus circumventing off-cycle events. Moreover, these processes are inherently diffusion limited, with high local concentrations of both catalysts rarely being achieved. The need for close proximity and restricted catalyst rotation has been recognized by the materials community.29–34
While SCNPs have been used often in a biomimetic manner, fine-tuning their designs can enable enhanced photocatalysis.13 We recently reported triarylpyrylium-based SCNPs that achieved the dimerization of electron-rich styrenics.3 The SCNP design enabled high local concentrations of the photocatalyst and requisite electron relay,35 resulting in more efficient single-electron transfer (SET) and higher product yields. Recent work by the Barner-Kowollik team demonstrated that SCNPs containing Rose Bengal can enable more efficient photooxidation of oleic acid owing to spatial proximity of photosensitizers.13 Both investigations designed SCNPs that enforce proximity-guided synergy between catalytic components and/or their surroundings.
Herein, we disclose a single-chain polymer nanoparticle (SCNP) design that enables significant photochemical reaction enhancements under confinement. We demonstrate that triphenylpyrylium tetrafluoroborate (TPT), an organic photoredox catalyst with a typically short-lived excited state, can be rendered efficient through synergistic effects arising from the folded SCNP. In our design, a more rigid styrylarene (StyAr) unit serves to modulate photocatalyst efficiency, with the identity of the β-styryl unit being a critical design element. Specifically, we find that pendant styrylnaphthalene (SNap) groups increase the photocatalyst activity, relative to those featuring styrylpyrene (SP) and styrylphenanthrene (SPhen) motifs (Scheme 2), in the model Diels–Alder reaction. Moreover, the collective series of SCNPs increases the catalytic turnover (TON) up to 30-fold, rendering the SCNP highly effective in promoting the [4 + 2]-cycloaddition of trans-anethole and isoprene compared to free TPT in solution. We attribute these results to the SCNP's ability to enforce participation of the acenes as cocatalytic redox mediators owing to spatial proximity under confinement. The mechanism was probed with quenching studies, where the acenes were found to quench the majority of TPT photoexcited states, enabling the formation of a charged-separated state with the respective acene˙+. We find that this, combined with the ability of naphthalene˙+ and phenanthrene˙+ to efficiently oxidize trans-anethole, results in an overall more efficient TPT photocatalyst.
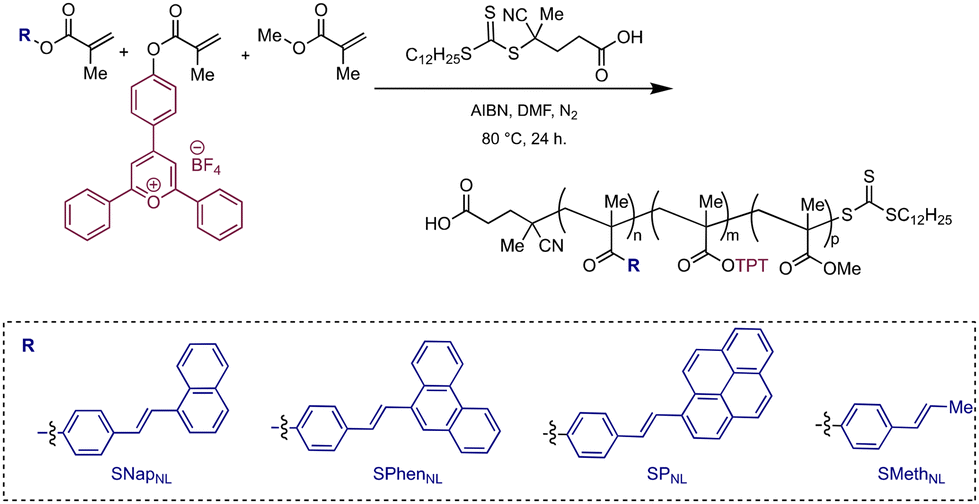 | ||
| Scheme 2 Triphenylpyrylium-based polymers that are studied in the radical cation Diels–Alder reaction. | ||
Results and discussion
We designed a poly(methyl methacrylate) backbone featuring TPT and StyAr-based pendant groups (bearing pyrene, phenanthrene, or naphthalene). TPT and StyAr monomers without flexible linkers were intentionally selected, along with targeting 5–10% crosslinking, hypothesizing that higher compactions may also support enhanced photocatalyst bleaching. The target polymers were formed using reversible addition–fragmentation chain-transfer (RAFT) polymerization (Scheme 2), resulting in statistical copolymers with molecular weights in the 10–20 kDa range. 1H- and 19F-NMR spectroscopy were utilized to determine incorporation ratios in all polymers (Table 1). The StyAr groups were subsequently crosslinked (see ESI† for details), with SCNP formation being evidenced by comparison of molecular weights before and after compaction using size exclusion chromatography (SEC, Table 1 and Fig. S17–20†).| Polymer | % TPTa | %StyAra | M n (kDa) | M w (kDa) | Đ |
|---|---|---|---|---|---|
| a Determined using 1H- and/or 19F-NMR spectroscopy. | |||||
| TPT-co-SPNL-co-MMA | 1.7 | 5.5 | 9.1 | 11.4 | 1.25 |
| TPT-SPNL-SCNP | — | — | 3.9 | 7.7 | 1.20 |
| TPT-co-SPhenNL-co-MMA | 2.3 | 5.6 | 10.7 | 13.1 | 1.23 |
| TPT-SPhenNL-SCNP | — | — | 6.8 | 8.1 | 1.20 |
| TPT-co-SNapNL-co-MMA | 2.4 | 5.5 | 8.4 | 11.5 | 1.38 |
| TPT-SNapNL-SCNP | — | — | 7.0 | 10.1 | 1.22 |
| TPT-co-SMethNL-co-MMA | 2.5 | 9.8 | 11.2 | 14.3 | 1.27 |
| TPT-SMethNL-SCNP | — | — | 6.5 | 7.8 | 1.20 |
| TPT-co-MMA | 1.8 | N/A | 15.2 | 17.9 | 1.17 |
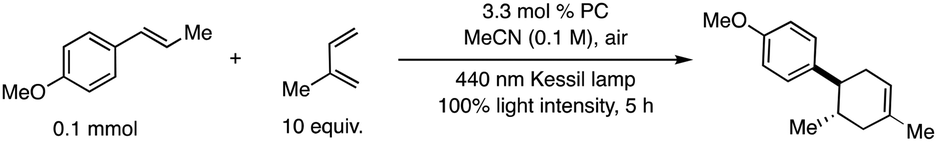
The photoredox properties of the resultant SCNPs were investigated using the [4 + 2]-cycloaddition of trans-anethole and isoprene as a model; this cycloaddition proceeds in the presence of both metallaphotoredox and organic photoredox systems.36–38 In a typical experiment, trans-anethole and isoprene were added to a solution of the desired catalyst, followed by irradiation with 440 nm LEDs (see ESI† for details). Conversions were monitored using 1H-NMR spectroscopy. We first sought to interrogate the model reaction using molecular TPT with our light setup. When using 3.3 mol% of TPT, we obtained <5% of the desired cyclohexene in a period of 5 hours; attempts to extend the reaction to 24 hours did not afford more of the cycloadduct. Similar results were observed using a linear TPT-co-MMA polymer,3 suggesting that the polymeric pyrylium does not significantly alter the photophysics of the dye.
When using the SCNPs, we observed that reaction of trans-anethole and isoprene in the presence of TPT-SNapNL-SCNP achieved 99% of the desired cycloadduct within a five-hour period, while TPT-SPhenNL-SCNP and TPT-SPNL-SCNP afforded 68% and 26% of the cycloadduct, respectively (Table 2). For comparison, reactions conducted without crosslinking the polymer (i.e., using TPT-co-SNapNL-co-MMA, wherein the photocatalyst and acene are present in the same ratio and distribution as the compartmentalized SCNP) furnished only 46% of the cycloadduct and demonstrated poor mass balance (Table 2, entry 4). The combined results confirm that the catalyst activity was enhanced within the SCNPs, owing to colocalization of the two species. While the absence of light or photocatalyst resulted in no product, the lack of oxygen had deleterious effects on yield. The observed differences in reactivity suggested that the spatial confinement inherent to the SCNP modulated catalytic activity, likely driving chemical reactivity.
|
|
|||
|---|---|---|---|
| Entry | Photocatalyst | %Anethole remaining | Yield (%), [4 + 2]-cycloadduct |
| a equivalents added relative to trans-anethole. | |||
| 1 | TPT-SNapNL-SCNP | 0% | 99% |
| 2 | TPT-SPhenNL-SCNP | 0% | 68% |
| 3 | TPT-SPNL-SCNP | 20% | 26% |
| 4 | TPT-co-SNapNL-co-MMA | 38% | 45% |
| 5 | TPT | 89% | 2% |
| 6 | TPT-co-MMA | 90% | 5% |
| 7 | TPT-SPNL-SCNP + 0.5 eq. naphthalenea | 0% | 62% |
| 8 | TPT-SPhenNL-SCNP + 0.5 eq. naphthalenea | 0% | 88% |
| 9 | TPT-SNapNL-SCNP + 0.5 eq. pyrenea | 0% | 86% |
| 10 | TPT-SNapNLSCNP + 1.0 eq. pyrenea | 15% | 68% |
We attributed differences in the catalyst activity to the redox properties of the acenes, which may act to oxidize trans-anethole (under the reaction conditions). Observing that TPT-SPNL-SCNP demonstrated the lowest catalyst activity suggested that generation of a putative pyrene radical cation suppresses the reaction, leading to low degrees of cycloadduct formation. Our experimental results are consistent with the redox potentials for the expected acene˙+/acene reductive event:39 the pyrene radical cation (Ered1/2 = +1.34 V vs. S.C.E.) is less likely to be reduced, whereas the radical cations of phenanthrene (Ered1/2 = +1.78 V vs. S.C.E.) and naphthalene (Ered1/2 = +1.80 V vs. S.C.E.) can still engage with trans-anethole (+1.35 V vs. S.C.E.).40 This mechanistic insight was further corroborated when adding naphthalene to the reaction using TPT-SPNL-SCNP, resulting in the likelihood of unquenched TPT oxidizing naphthalene to afford naphthalene˙+, which may oxidize trans-anethole and/or accelerate any chain propagation steps. Accordingly, we observe a significant increase in cycloadduct yield (62%; Table 2, Entry 7). In similar experiments using TPT-SPhenNL-SCNP, we observe modest increases in cycloadduct formation. Finally, addition of pyrene to the optimal catalyst, TPT-SNapNL-SCNP, results in deleterious effects on yield.
We further probed the role of the acene in modulating photocatalyst efficiency, synthesizing an analogous polymer featuring a propenylbenzene pendant (denoted TPT-co-SMethNL-co-MMA). The absence of groups that could participate as redox mediators was hypothesized to aid in comprehending how catalytic activity is altered within the SCNP. Emission experiments demonstrated that TPT-SMethNL-SCNP was roughly six times more emissive than those functionalized with acenes, further confirming the role of the acene unit in quenching the TPT excited state. Emission quenching with trans-anethole indicated that at reaction conditions, roughly 90% of the emission intensity from TPT-SMethNL-SCNP was quenched (see ESI† for details).
We investigated the role of the acene pendants in facilitating [4 + 2]-cycloadditions under confinement. In related [2 + 2]-cycloadditions, acenes were viewed as electron relays that facilitate SET between the excited state TPT and trans-anethole,3,35 leading to the intermediate radical cation. We thus investigated the role of the acene as a quencher of the TPT excited state. We compared emission spectra of the acene-containing SCNPs to TPT-SMethNL-SCNP, keeping the TPT concentration constant (Fig. 1). Compared to TPT-SMethNL-SCNP, the emission from all other SCNPs was greatly reduced, indicating quenching by the acenes. There were no notable differences in the presence or absence of oxygen, despite purging with nitrogen resulting in minimal formation of the cycloadduct (see ESI†). Oxygen is likely involved in oxidizing the reduced form of the photocatalyst, regenerating TPT to turn over the catalytic cycle.
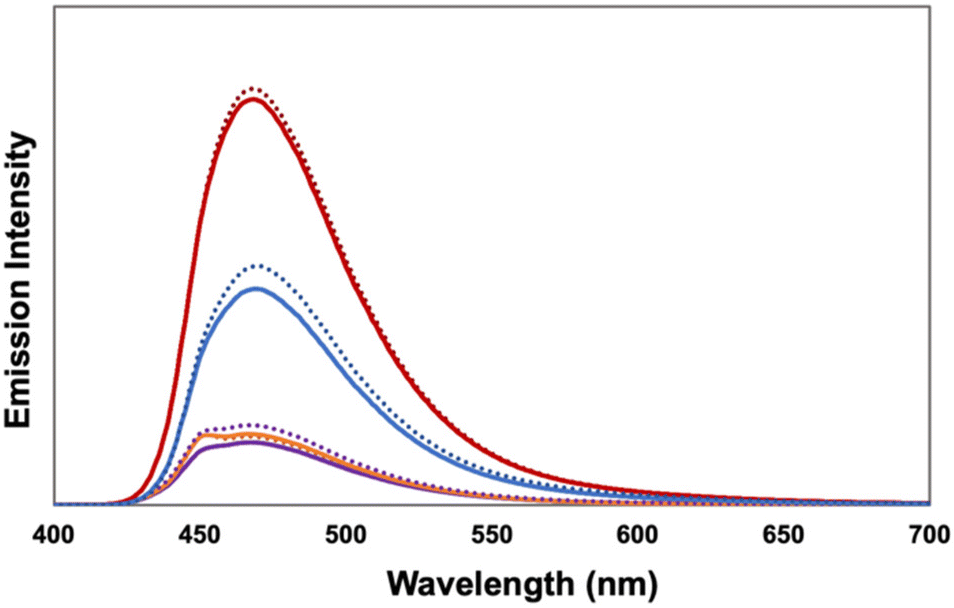 | ||
| Fig. 1 Emission of TPT-SMethNL-SCNP (red), TPT-SPhenNL-SCNP (blue), TPT-SNapNL-SCNP (purple), and TPT-SPNL-SCNP (orange) in air (solid line) and under N2 (dotted line). | ||
We sought to further comprehend the mechanism, conducting quenching experiments with trans-anethole. We noted that even in the presence of the acene, additional quenching of the excited state of the TPT photocatalyst was observed (see ESI†). Different additives were also introduced to identify the oxygen species generated (Table S7†).38 When the [4 + 2]-cycloaddition was attempted in the presence of CuCl2 or 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO), the reaction pathway was completely suppressed, confirming the involvement of SET processes and the presence of a radical pathway, respectively. Addition of sodium azide also demonstrated a significantly lower yield, indicating the involvement of activated oxygen species.
Our overall observations are consistent with the first elementary step in the SCNP-mediated [4 + 2]-cycloaddition consisting of photoexcitation of TPT (Scheme 3). The TPT excited state can then undergo photoinduced electron transfer from either the acene or the trans-anethole. On the basis of quenching experiments (see ESI† for details) we estimate that in the case of pyrene or naphthalene, approximately 75% of the excited states are quenched by the acene, another 12.5% oxidize trans-anethole, and another 12.5% relax back to the ground state. With phenanthrene, only 28% of the excited states are quenched by the acene, with another 36% quenched by anethole. In the case of the naphthalene or phenanthrene, there is a large driving force for the oxidized acene to oxidize trans-anethole,39,41 while in the case of pyrene there is a much smaller driving force to oxidize trans-anethole. Following oxidation of the acene or trans-anethole, we suggest that a dark electron transfer from the reduced TPT to oxygen occurs. This presumably suppresses back electron transfer to the acene or trans-anethole radical cation allowing for cycloaddition with isoprene to occur. Finally, reduction of the cycloadduct radical cation by superoxide generates the final product. In the case of TPT-SPNL-SCNP, the pyrene˙+ is slower to oxidize anethole, allowing reactive oxygen species generated by the reduced TPT to react directly with anethole. The combined data suggested TPT-SNapNL-SCNP had the best performance for the Diels–Alder reaction. We note that cannot exclude an unproductive triplet energy transfer pathway between the excited TPT and the pyrene.
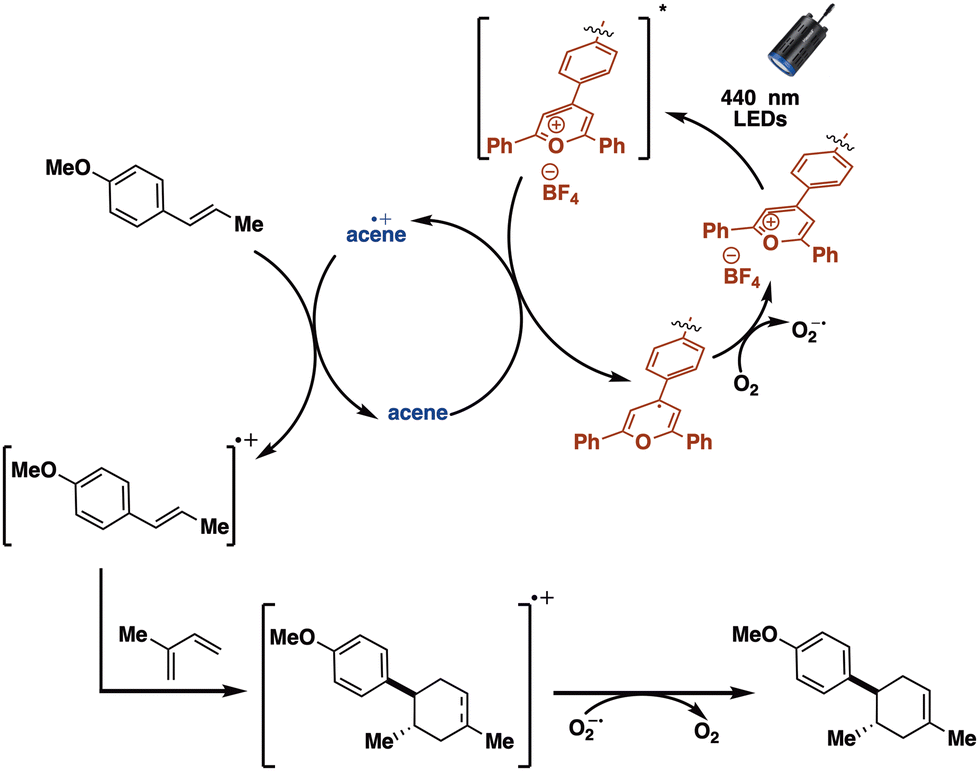 | ||
| Scheme 3 Schematic depicting the proposed mechanism for the TPT-SCNP-mediated [4 + 2]-cycloaddition under confinement. | ||
Using the optimized conditions, we investigated the reactivity of various dienophiles in the presence of TPT-SNapNL-SCNP (Fig. 2). We found that a range of dienes and dienophiles were successful reaction partners. Symmetrical dienes such as 2,3-dimethylbutadiene and hindered dienes like 2,4-dimethyl-1,3-pentadiene gave successful results, while cyclic dienes did not. Dienophiles with moderately electron-rich alkoxy (compounds 1, 6, 8) or silyl ether groups para-substituted on the styryl group (5) proved to be effective coupling partners with butadienes. Notably, the highly electron-rich dienophile, asarone, had lower yields of the cycloaddition product (9), likely due to competing oxidation that affords an aldehyde as the major product. Electron-deficient dienophiles (compounds 2–4) also exhibited a range of reactivities in the presence of TPT-SNapNL-SCNP. However, electron-rich N-vinyl carbazole (7) afforded the cycloadduct in poor yields, owing to competitive polymerization of the vinyl group. Polymerization was also observed when using 4-methoxystyrene as a dienophile.42 Overall, styryl groups without electron-donating substituents did not react well, likely owing to greater difficulty in oxidizing these substrates. Competitive photoisomerization was observed with cinnamates and stilbenes. These transformations validate the generality of the TPT-based SCNP photoredox catalyst.
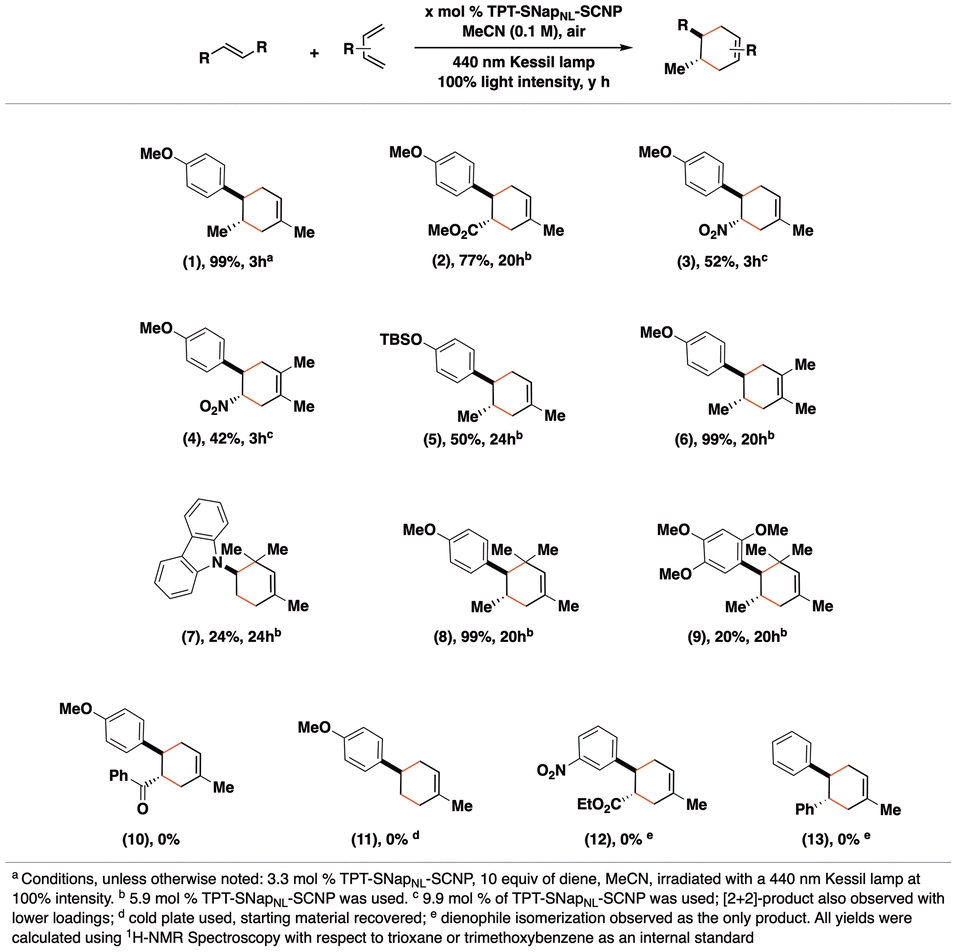 | ||
| Fig. 2 Scope of products accessible by the TPT-SNapNL-SCNP-catalyzed Diels–Alder reaction. | ||
Given the inherent solubility differences between the SCNPs and small molecule reactants and/or products, we sought to investigate the possibility that the TPT-SNapNL-SCNP could be recycled and reused. In recyclability studies, we observe that two additional cycles can be completed, prior to a substantial loss in cycloadduct yield (54%; see ESI† for details), although that yield still outperforms that of small molecule TPT. Arguably, this polymer system would suffer from pyranyl radical coupling43 over time, leading to lower catalyst loadings in subsequent iterations. This photobleaching can be observed during the collection of UV-Vis spectra before re-use of the SCNP catalyst (Fig. S35†). Regardless, the ability to recycle the SCNP multiple times prior to complete catalyst degradation bodes well for the development of further potent polymer-based catalysts for use in methods development and other synthetic applications.
Conclusions
Herein, we demonstrated that a dual catalytic SCNP bearing visible-light activated triphenylpyrylium groups can be fine-tuned to facilitate the radical cation Diels–Alder cycloaddition under confinement. In our studies, we find that the identity of the pendant StyAr plays an integral role in fine-tuning catalytic activity, leading to a 30-fold increase in TON compared to free TPT and near quantitative yields when the β-styryl unit is naphthalene. Moreover, we observe that the majority of photocatalyst excited states are quenched by the acene, with the resultant radical cation formed from naphthalene-based SCNPs able to efficiently oxidize trans-anethole, thus confirming the acene's role as a redox mediator. Indeed, this is not observed in the reactions of molecular TPT and the acene, where negligible product yield is observed under both analogous reaction conditions. The combined suggests that the confined nature of the SCNP and spatial proximity to acene-based pendants enforces their participation as cocatalytic redox mediators to enable chemical reactivity. Looking forward, our SCNP framework has several implications for future catalyst design, as the principles of confinement might favor reaction pathways that were previously inaccessible.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation Early CAREER program (CHE-2046470 and CHE-2047492) and Research Experience for Undergraduates Program (CHE-2050927). V. P. thanks the Bridges to Baccalaureate (NIH 5 R25 GM056637) for a summer research fellowship. The authors also acknowledge support from the Alfred P. Sloan Foundation (E.E., FG-2021-15490) and start-up funds generously provided by the Pennsylvania State University. We acknowledge Sarah Freeburne and Christian Pester for assistance with GPC data collection and Morgan Murphy for assistance with graphics.References
- J. J. Piane, S. Huss, L. T. Alameda, S. J. Koehler, L. E. Chamberlain, M. J. Schubach, A. C. Hoover and E. Elacqua, J. Polym. Sci., 2021, 59, 2867–2877 CrossRef CAS.
- R. Zeng, L. Chen and Q. Yan, Angew. Chem., Int. Ed., 2020, 59, 18418–18422 CrossRef CAS PubMed.
- J. J. Piane, L. E. Chamberlain, S. Huss, L. T. Alameda, A. C. Hoover and E. Elacqua, ACS Catal., 2020, 10, 13251–13256 CrossRef CAS.
- F. Eisenreich, E. W. Meijer and A. R. A. Palmans, Chem. – Eur. J., 2020, 26, 10355–10361 CrossRef CAS PubMed.
- J. Chen, K. Li, J. S. L. Shon and S. C. Zimmerman, J. Am. Chem. Soc., 2020, 142, 4565–4569 CrossRef CAS PubMed.
- J. Chen, E. S. Garcia and S. C. Zimmerman, Acc. Chem. Res., 2020, 53, 1244–1256 CrossRef CAS PubMed.
- H. Rothfuss, N. D. Knöfel, P. W. Roesky and C. Barner-Kowollik, J. Am. Chem. Soc., 2018, 140, 5875–5881 CrossRef CAS PubMed.
- J. Rubio-Cervilla, E. González and J. A. Pomposo, Nanomaterials, 2017, 7, 341 CrossRef PubMed.
- J. Willenbacher, O. Altintas, V. Trouillet, N. Knöfel, M. J. Monteiro, P. W. Roesky and C. Barner-Kowollik, Polym. Chem., 2015, 6, 4358–4365 RSC.
- A. Sanchez-Sanchez, A. Arbe, J. Colmenero and J. A. Pomposo, ACS Macro Lett., 2014, 3, 439–443 CrossRef CAS PubMed.
- S. Huss, A. R. Walsh, A. Griggs, D. A. Rodriguez-Acevedo, D. M. Arias-Rotondo and E. Elacqua, Polym. Chem., 2023, 14, 4560–4568 RSC.
- Z. Zhao, M. Liu, K. Zhou, L. Guo, Y. Shen, D. Lu, X. Hong, Z. Bao, Q. Yang, Q. Ren, P. R. Schreiner and Z. Zhang, ACS Appl. Mater. Interfaces, 2023, 15, 6982–6989 CrossRef CAS PubMed.
- K. Mundsinger, B. T. Tuten, L. Wang, K. Neubauer, C. Kropf, M. L. O'Mara and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2023, 62, e202302995 CrossRef CAS PubMed.
- K. Mundsinger, A. Izuagbe, B. Tuten, P. Roesky and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2023, 63, e202311734 CrossRef PubMed.
- E. B. Berda, E. J. Foster and E. W. Meijer, Macromolecules, 2010, 43, 1430–1437 CrossRef CAS.
- N. D. Knöfel, H. Rothfuss, J. Willenbacher, C. Barner-Kowollik and P. W. Roesky, Angew. Chem., Int. Ed., 2017, 56, 4950–4954 CrossRef PubMed.
- J. A. Pomposo, I. Perez-Baena, F. Lo Verso, A. J. Moreno, A. Arbe and J. Colmenero, ACS Macro Lett., 2014, 3, 767–772 CrossRef CAS PubMed.
- S. Mavila, I. Rozenberg and N. G. Lemcoff, Chem. Sci., 2014, 5, 4196–4203 RSC.
- Y. Bai, X. Feng, H. Xing, Y. Xu, B. K. Kim, N. Baig, T. Zhou, A. A. Gewirth, Y. Lu, E. Oldfield and S. C. Zimmerman, J. Am. Chem. Soc., 2016, 138, 11077–11080 CrossRef CAS PubMed.
- J. Chen, J. Wang, K. Li, Y. Wang, M. Gruebele, A. L. Ferguson and S. C. Zimmerman, J. Am. Chem. Soc., 2019, 141, 9693–9700 CrossRef CAS PubMed.
- F. Eisenreich and A. R. A. Palmans, Chem. – Eur. J., 2022, 28, e202201322 CrossRef CAS PubMed.
- A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed.
- A. Almansa, D. Jardel, S. Massip, T. Tassaing, C. Schatz, J. Domergue, F. Molton, C. Duboc and J.-M. Vincent, J. Org. Chem., 2022, 87, 11172–11184 CrossRef CAS PubMed.
- M. Yuan, Z. Song, S. O. Badir, G. A. Molander and O. Gutierrez, J. Am. Chem. Soc., 2020, 142, 7225–7234 CrossRef CAS PubMed.
- E. B. McLean, V. Gauchot, S. Brunen, D. J. Burns and A.-L. Lee, Chem. Commun., 2019, 55, 4238–4241 RSC.
- M. J. González and B. Breit, Chem. – Eur. J., 2019, 25, 15746–15750 CrossRef PubMed.
- J. C. Tellis, C. B. Kelly, D. N. Primer, M. Jouffroy, N. R. Patel and G. A. Molander, Acc. Chem. Res., 2016, 49, 1429–1439 CrossRef CAS.
- K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed.
- J. M. Mouat, J. K. Widness, D. G. Enny, M. T. Meidenbauer, F. Awan, T. D. Krauss and D. J. Weix, ACS Catal., 2023, 13, 9018–9024 CrossRef CAS PubMed.
- D. Meng, J. Xue, Y. Zhang, T. Liu, C. Chen, W. Song and J. Zhao, Catal. Sci. Technol., 2023, 13, 1518–1526 RSC.
- J. T. Bryant, M. W. Logan, Z. Chen, M. Djokic, D. R. Cairnie, D. A. Vazquez-Molina, A. Nijamudheen, K. R. Langlois, M. J. Markley, G. Pombar, A. A. Holland, J. D. Caranto, J. K. Harper, A. J. Morris, J. L. Mendoza-Cortes, T. Jurca, K. W. Chapman and F. J. Uribe-Romo, J. Am. Chem. Soc., 2023, 145, 4589–4600 CrossRef CAS PubMed.
- J. Liu, T. A. Goetjen, Q. Wang, J. G. Knapp, M. C. Wasson, Y. Yang, Z. H. Syed, M. Delferro, J. M. Notestein, O. K. Farha and J. T. Hupp, Chem. Soc. Rev., 2022, 51, 1045–1097 RSC.
- A. Jati, K. Dey, M. Nurhuda, M. A. Addicoat, R. Banerjee and B. Maji, J. Am. Chem. Soc., 2022, 144, 7822–7833 CrossRef CAS PubMed.
- Y.-Y. Zhu, G. Lan, Y. Fan, S. S. Veroneau, Y. Song, D. Micheroni and W. Lin, Angew. Chem., Int. Ed., 2018, 57, 14090–14094 CrossRef CAS PubMed.
- M. Riener and D. A. Nicewicz, Chem. Sci., 2013, 4, 2625–2629 RSC.
- S. Lin, M. A. Ischay, C. G. Fry and T. P. Yoon, J. Am. Chem. Soc., 2011, 133, 19350–19353 CrossRef CAS PubMed.
- S. M. Stevenson, R. F. Higgins, M. P. Shores and E. M. Ferreira, Chem. Sci., 2017, 8, 654–660 RSC.
- J. Kollmann, Y. Zhang, W. Schilling, T. Zhang, D. Riemer and S. Das, Green Chem., 2019, 21, 1916–1920 RSC.
- A. P. Davis and A. J. Fry, J. Phys. Chem. A, 2010, 114, 12299–12304 CrossRef CAS PubMed.
- C. Demaille and A. J. Bard, Acta Chem. Scand., 1999, 53, 842–848 CrossRef CAS.
- H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 27, 714–723 CAS.
- A. J. Perkowski, W. You and D. A. Nicewicz, J. Am. Chem. Soc., 2015, 137, 7580–7583 CrossRef CAS PubMed.
- H. Kawata and S. Niizuma, Bull. Chem. Soc. Jpn., 1989, 62, 2279–2283 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures for monomer and polymer synthesis and characterization; photoredox catalysis setup and reactions; catalysis optimization, quenching and recyclability studies. See DOI: https://doi.org/10.1039/d4py00282b |
| This journal is © The Royal Society of Chemistry 2024 |