
Creep resistance in doubly crosslinked dynamic covalent networks†
Swagata
Mondal
a,
Alexander J.
Wong
 a,
Mahendra A.
Wagh
bc,
Lily
Alperstein
a,
Gangadhar J.
Sanjayan
*bc and
Brent S.
Sumerlin
*a
a,
Mahendra A.
Wagh
bc,
Lily
Alperstein
a,
Gangadhar J.
Sanjayan
*bc and
Brent S.
Sumerlin
*a
aGeorge and Josephine Butler Polymer Research Laboratory, Center of Macromolecular Science, Department of Chemistry, University of Florida, Gainesville, FL 32611, USA. E-mail: sumerlin@chem.ufl.edu
bOrganic Chemistry Division, Council of Scientific and Industrial Research National Chemical Laboratory (CSIR-NCL), Pune 411008, India. E-mail: gj.sanjayan@ncl.res.in
cAcademy of Scientific and Innovative Research (AcSIR), Sector 19, Kamla Nehru Nagar, Ghaziabad, Uttar Pradesh-201002, India
First published on 24th April 2024
Abstract
Vitrimers are a unique class of thermosets that demonstrate reprocessability and recyclability due to dynamic bond exchange at crosslinking sites. However, the same dynamic bond exchange predisposes vitrimers to macroscopic deformation and creep under constant stress, which limits many practical applications. Herein, we demonstrated that the incorporation of Janus-faced guanine–cytosine diamine (GCBDam) functionality within vinylogous urethane vitrimers leads to significant creep resistance due to network reinforcement via hydrogen bonding. The supramolecular associations of the GCBDam groups retarded stress relaxation at temperatures as high as 160 °C. Further, rheological data suggested that the cooperative nature of the bifunctional Janus-faced hydrogen bonding moieties allowed the GCBDam to act as “stickers” within the dynamic covalent networks. These results indicate that incorporating the bifunctional supramolecular moiety improved dimensional stability while conserving the hallmark vitrimer property of reprocessability.
Introduction
Thermosets are a class of robust covalently crosslinked polymers with exceptional thermal, solvent, chemical, and dimensional stability compared to their non-crosslinked thermoplastic counterparts. However, these desirable material properties typically come at the expense of reprocessability, recyclability, and healability. Including dynamic and reversible covalent crosslinking in thermosets can address these limitations by allowing the mobility needed for network rearrangement on heating.1–3 Depending on the nature of the reversible chemistry of the crosslinks, these covalent adaptable networks can be broadly classified into dissociative covalent adaptable networks (CANs)4–6 and associative CANs (i.e., vitrimers).7–9 The crosslink exchange reaction in dissociative CANs comprises two discrete steps wherein crosslinks dissociate and the resulting liberated functional groups form new crosslinks by reacting with other functional moieties within the network. On the other hand, crosslink exchange in vitrimers involves bond dissociation and association in a single reaction, which leads to a unique viscosity–temperature relationship reminiscent of vitreous silica, with network integrity remaining intact at elevated temperatures.10 Various reversible crosslinking chemistries have been developed, including transesterification,11,12 transamination of vinylogous urethanes,13–17 diketoenamine exchange,18 and olefin metathesis.19–26 However, a common disadvantage of materials with dynamic crosslinks is the permanent deformation (i.e., creep) that occurs under mechanical stress at elevated temperatures. Creep is generally undesirable for many practical applications.27Considerable effort has been exerted to reduce creep by controlling the exchange chemistry of the crosslinking reaction. Du Prez et al. demonstrated that amine exchange in vinylogous urethane chemistry was accelerated by acid catalysis and retarded by basic additives.28 Recently, the same group showed that the combination of dicarboxamide dissociation chemistry with the associative transamination of vinylogous urethanes suppressed creep significantly.29,30 We reported catalyst-free vinylogous urethane vitrimers31,32 and later demonstrated that the microphase separation in block copolymer vitrimers led to excellent creep resistance and exceptional thermal reprocessability.33,34
Torkelson et al. adapted the Flory Stockmeyer theory to calculate the limiting fraction of permanent crosslinks in vitrimers. Their calculation aimed to balance reducing creep in a vitrimer while maintaining reprocessability,35 a strategy we have employed in dissociative crosslinked networks.36 Later, Zhang et al. demonstrated significant creep suppression upon reaching optimum crosslinking density in vitrimers.37
Supramolecular reinforcement by metal–ligand coordination has been utilized to suppress creeps in vitrimers. For example, Xu et al. reported that disulfide-cured hydroxyl and pyridyl functionalized polyisoprene block copolymers exhibited excellent creep resistance in the presence of Cu2+ ions through cationic coordination within the network.38 Zhu and coworkers demonstrated that incorporating transition metal catalysts in polyimine vitrimers led to creep suppression due to metal–ligand coordination.39
Physical crosslinking by hydrogen bonds has also been utilized to enhance the properties of networks. Guan et al. demonstrated that the incorporation of sacrificial hydrogen bonding amides in a covalently crosslinked polyolefin network improved the toughness.40 Gels have also been enhanced by incorporating a hydrogen bond secondary network, increasing their toughness and healability.41,42 The utilization of hydrogen bonds to enhance the properties of CANs has been less explored. Yun et al. synthesized “S-vitrimers” involving aromatic disulfide exchange where hydrogen bonds were utilized to increase the healability.43 Sacrificial hydrogen bonds were incorporated into an acrylate vitrimer by Zhang et al. and showed a 4.4 times increase in the tensile strength with only a 20% addition of acrylamide.44 Finally, Du et al.45–47 have explored the integration of hydrogen-bonding moieties into boronate ester vitrimers. Rather than using the hydrogen bonds for sacrificial energy damping, the authors designed the vitrimer backbone around the incorporation of the hydrogen bond-forming pendent group. The resulting dual networks demonstrated enhanced creep resistance up to 130 °C, and increased strength and toughness while remaining processable. While the utility of supplementing vitrimers via secondary reinforcement has been demonstrated, in most cases, a high density of relatively weak supramolecular interactions has been employed.
In a previous report, we introduced a novel Janus-faced guanine-cytosine base (GCB) moiety48 capable of hydrogen bond formation from both sides of the functional group. We proposed that their associating cooperativity was responsible for the remarkable thermal stability of networks containing these groups (Scheme 1A).49 These results suggested that the dynamic bond exchange of GCB controlled the mobility within the dissociative polymer networks to result in predictable viscoelastic flow. When initiating the current work, we hypothesized that the low-density introduction of GCB molecules in a vinylogous urethane vitrimer would result in physical crosslinking that complemented the associative chemistry responsible for network rearrangement. While hydrogen bonding has been shown to decrease creep in vitrimer networks, we believe the effects could be further enhanced by the cooperative association of hydrogen bond moieties that deter network mobility required for crosslink exchange. Nevertheless, the dissociative nature of the GCB bond exchange would result in a rapid decline of viscosity at elevated temperatures to enable fast processing of the networks on heating. Indeed, the enhanced stability of GCB hydrogen bonding, even at temperatures well above the range at which conventional hydrogen bonding moieties remain associated, improved the dimensional stability of vitrimers while maintaining the hallmark vitrimer characteristic of reprocessability.
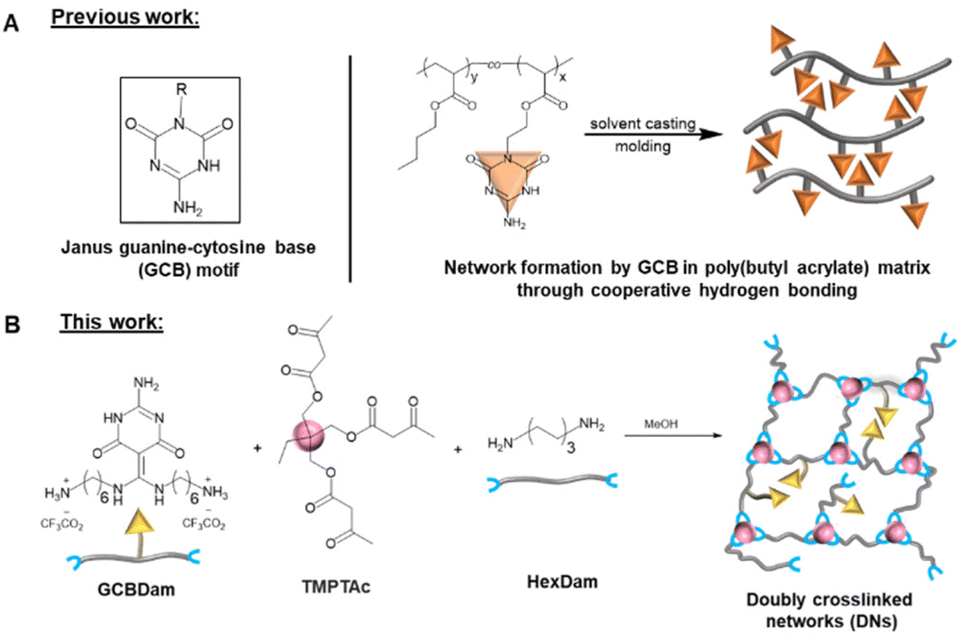 | ||
| Scheme 1 (A) Previous strategy of Janus guanine–cytosine base (GCB) motifs being employed to prepare dissociative dynamic networks. (B) Current strategy of GCB motifs being employed to supplement associative vinylogous urethane. | ||
Results and discussion
To incorporate the GCB moiety into a vinylogous urethane vitrimer, functionalization of the GCB core was performed by the addition of two Boc-protected hexamethylene amine groups (Scheme S1, and Fig. S1–S4†). The protected diamine moiety was then converted to a trifluoroacetate salt following Boc deprotection, resulting in GCBDam (Fig. S5–S8†).50 A control diamine was synthesized via the acidification of hexamethylene diamine with trifluoroacetic acid (HexDam-2TFA) (Scheme S2, and Fig. S9†) to elucidate the role of GCB in the network exchange reactions. Vitrimers were subsequently formed by adding either GCBDam (Scheme 1B) or HexDam-2TFA (Scheme S4†) to trimethylolpropane triacetoacetate (TMPTAc) (Scheme S3, Fig. S10 and S11†) and hexamethylene diamine, ensuring that the total amine content was maintained at a 33% molar excess. Experimental and control networks were investigated at varying incorporations, either 5 or 10 mol%, of GCBDam or HexDam-2TFA. Networks formed with GCBDam were expected to feature both covalent adaptable crosslinking and hydrogen bond-mediated supramolecular crosslinking. As such, we refer to these materials as ‘doubly crosslinked networks’ (DNx%) (Scheme 1B), where x represents the mol% of GCBDam. As a control, materials named ‘single networks’ (SNx%) (Scheme S4†), consist solely of covalent adaptable crosslinks and use x to denote the mol% incorporation of HexDam-2TFA. FT-IR spectroscopy was used to characterize the vitrimers, revealing N–H bending and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching modes at 1655 and 1600 cm−1, respectively (Fig. 1A, C; and S12†). Furthermore, gel swelling experiments were performed to confirm the same extent of network formation in the vitrimers containing 5% and 10% TFA salt (Table S1†). Thermogravimetric analysis (TGA) assessed the thermal stability, revealing that the 5% decomposition temperatures of all four networks were greater than 200 °C (Fig. S13 and 14†). Differential scanning calorimetry (DSC) showed that higher GCBDam content led to decreased glass transition temperature (Tg), an observation attributed to the higher trifluoroacetate salt concentration, which serves as a plasticizer. Conversely, the Tg values for DNs were higher than those for SNs, which we ascribe to the introduced hydrogen bonding moieties;45,46 for example, Tg values for DN5% and DN10% were 71 and 70 °C, while those for SN5% and SN10% were 60 and 52 °C, respectively (Fig. 1B and D).
C stretching modes at 1655 and 1600 cm−1, respectively (Fig. 1A, C; and S12†). Furthermore, gel swelling experiments were performed to confirm the same extent of network formation in the vitrimers containing 5% and 10% TFA salt (Table S1†). Thermogravimetric analysis (TGA) assessed the thermal stability, revealing that the 5% decomposition temperatures of all four networks were greater than 200 °C (Fig. S13 and 14†). Differential scanning calorimetry (DSC) showed that higher GCBDam content led to decreased glass transition temperature (Tg), an observation attributed to the higher trifluoroacetate salt concentration, which serves as a plasticizer. Conversely, the Tg values for DNs were higher than those for SNs, which we ascribe to the introduced hydrogen bonding moieties;45,46 for example, Tg values for DN5% and DN10% were 71 and 70 °C, while those for SN5% and SN10% were 60 and 52 °C, respectively (Fig. 1B and D).
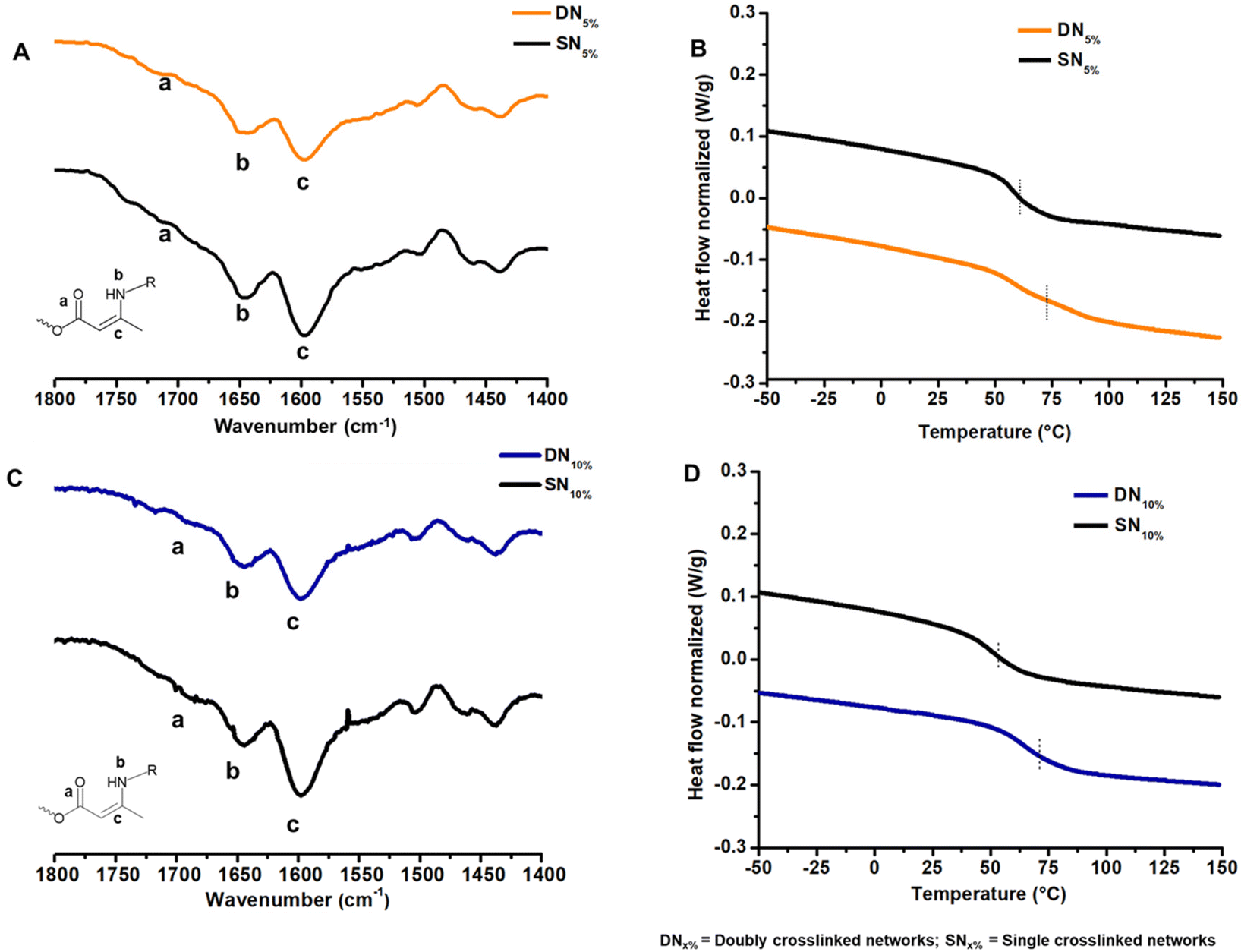 | ||
| Fig. 1 (A) FT-IR spectra of DN5% and SN5%; (B) DSC thermograms of DN5% and SN5%; (C) FT-IR spectra of DN10% and SN10%; (D) DSC thermograms of DN10% and SN10%. (DNx% = doubly crosslinked networks with x% of the GCBDam motif included. SNx% = singly crosslinked networks with x% ditrifluoroacetate salt of hexamethylene diamine). | ||
We first conducted frequency sweeps during oscillatory rheology to investigate the effect of shear on the materials at various temperatures. We observed that both the storage (G′) and loss (G′′) modulus were sensitive to angular frequency changes. Notably, the crossover point, indicative of the characteristic relaxation time, shifted to lower angular frequencies when GCBDam was incorporated into the networks (S15–S18†). This shift is likely due to the additional physical crosslinks formed in the GCBDam-containing networks. To complement these findings, we conducted creep recovery experiments to assess viscoelastic properties. A constant force (stress) was applied to measure material deformation (strain) over time. Upon releasing the stress, we monitored the rate of strain recovery to gauge permanent deformation or ‘creep’. We found that under a stress of 0.01 MPa at 120 °C, DN5% displayed roughly 41% less creep than SN5%. This enhanced performance is attributed to the resilient, orthogonal supramolecular network in the DN5% sample. The GCB units in DN5% resulted in a decrease in creep by up to 32% relative to the creep observed in SN5% at 150 °C (Fig. 2A), though the reduction in creep was diminished as temperature increased due to dissociation of the hydrogen-bonded network.49 Similarly, DN10%, with higher GCBDam content, exhibited a notable 46% reduction in creep at 120 °C in comparison to SN10%, declining to 33% at 150 °C (Fig. 2B). We reasoned that coordinated hydrogen bonding among the GCBDam moieties significantly enhances creep resistance. So far, creep resistance has been observed in lower temperature regions with hydrogen-bonding reinforcement in vitrimers, and recently Du et al. demonstrated creep resistance of boronate ester networks up to 130 °C with a large excess of hydrogen-bonding moieties. These materials set a new standard in creep resistance for vitrimers fortified with only 5 and 10% supramolecular crosslinks at temperature as high as 150 °C (Fig. S19, S20 and Tables S2, S3†).
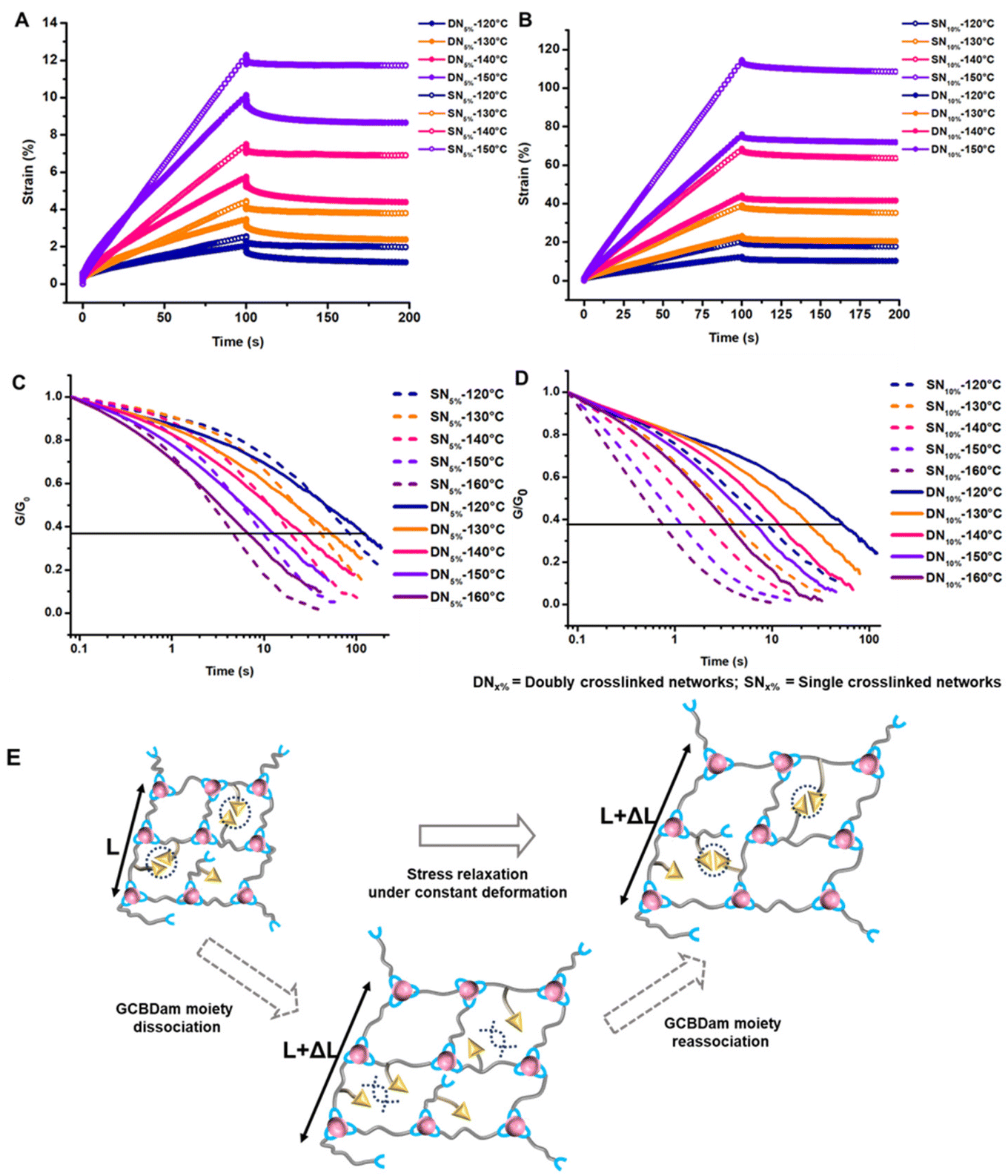 | ||
| Fig. 2 Comparison of creep-recovery experiments at various temperatures for (A) DN5% and SN5%; (B) DN10% and SN10%; comparison of normalized stress–relaxation curves at selected temperatures for (C) DN5% and SN5%; (D) DN10% and SN10%; (E) schematic representation of the two mechanisms of dynamic exchange of vinylogous vitrimers and hydrogen bonding moiety during stress relaxation under constant deformation. (DNx% = doubly crosslinked networks with x% of the GCBDam motif included. SNx% = singly crosslinked networks with x% ditrifluoroacetate salt of hexamethylene diamine). | ||
Finally, we characterized the viscoelastic properties of the networks by conducting stress relaxation experiments over time at a constant deformation of 0.3%. Usually, the characteristic relaxation time (τ) is evaluated assuming relaxation occurs through a single Maxwell mode, where τ corresponds to the time at which stress relaxation reaches (1/e), or 37%, of normalized stress.10,12,25 However, the data discussed herein deviated from a single Maxwell relaxation mode, likely due to multiple complicated relaxation modes. To address this, we employed a three-phase exponential decay model to fit the stress relaxation curves (Fig. S21–S24†). The Arrhenius plot, obtained by varying the temperature within the aforementioned range, displayed a linear relationship between τ and temperature. The activation energy (Ea) for viscous flow was extracted from the slope of this Arrhenius plot (Fig. S25†). Intriguingly, the DNs initially exhibited rapid stress relaxation followed by slower long-term relaxation compared to their corresponding SN controls.51 This effect was notably stronger for DN5% (Fig. 2C) than for DN10% (Fig. 2D). We propose that the early rapid relaxation arises from deterioration of the hydrogen bond network, while the slower long-term relaxation is due to the reorganization of the supramolecular network via hydrogen-bond exchange (Fig. 2E). The variation in relaxation rate of DN5% with respect to SN5% resulted in a crossover point between DN5% and SN5% in stress relaxation plots. The crossover point shifted to shorter times as the temperature increased due to accelerated hydrogen bond exchange at elevated temperatures. For DN10%, the crossover with SN10% was observed only at 120 °C, suggesting instantaneous hydrogen bond network rearrangement due to a higher concentration of the GCBDam bonds (Fig. 2D). Analysis of the Arrhenius plots revealed Ea values around 102 kJ mol−1 for both DN5% and SN5% (Fig. 3A, and Fig. S25A†). As hypothesized,14,15 SN10% had a lower Ea, 88 kJ mol−1, due to a higher concentration of protonated species, which led to faster enaminone exchange reactions (Fig. 3B, and Fig. S25B†). However, upon 10% incorporation of GCBDam, the Ea of DN10% was determined to be 96 kJ mol−1, slightly higher than that of SN10% at 88 kJ mol−1 (Fig. 3B) (Fig. S25B†). The activation energy calculated from the creep-recovery experiments also shows a similar trend (Fig. S26†) as those determined by stress relaxation. This result indicates that in DN5%, the incorporation of GCBDam was insufficient to significantly affect the Ea of the vitrimers. However, further increasing the incorporation of hydrogen bonding moiety to 10% led to a more pronounced change in viscous flow with temperature, resulting in faster reprocessing of DN10%. Interestingly, the increased Ea of DN10% is potentially beneficial for practical application as it increases the energy barrier for network flow while providing a sharp viscosity reduction at elevated temperatures to allow fast reprocessing.
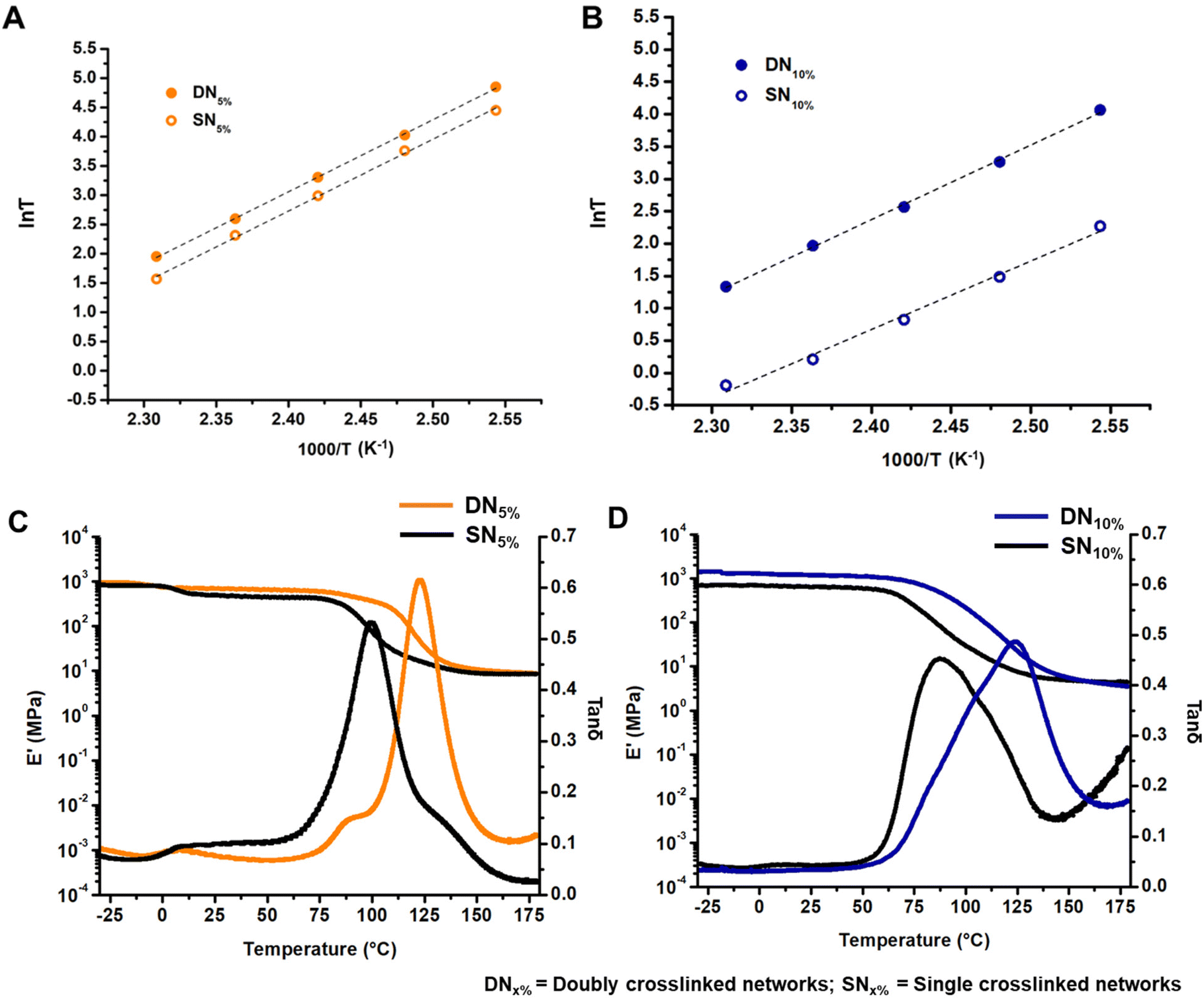 | ||
| Fig. 3 Comparison of Arrhenius plot for (A) DN5% and SN5%; (B) DN10% and SN10% to calculate the activation energy (Ea) of the flow of networks; comparison of the DMA plots for (C) DN5% and SN5%; (D) DN10% and SN10% to exhibit the network integrity at elevated temperatures. (DNx% = doubly crosslinked networks with x% of the GCBDam motif included. SNx% = singly crosslinked networks with x% ditrifluoroacetate salt of hexamethylene diamine). | ||
To investigate the influence of the GCBDam moieties on the network integrity, a temperature ramp was conducted during dynamic mechanical analysis (DMA). Compared to the SNs, both DNs showed an increase in Tg. This was expected and confirms that the chains are further retarded from long-range segmental motion by the GCBDam moieties forming a secondary hydrogen-bond network. Interestingly, the storage moduli (E′) at the elastic rubbery plateau regime of the DNs and SNs were very similar (Fig. 3C and D). These findings suggest that at elevated temperatures, the exchange of the GCBDam moieties occurs more rapidly than the transamination of the vinylogous urethane groups. Consequently, the vinylogous urethane network functions as the permanent network, while the GCBDam moieties act as “stickers” within this temperature range.
To evaluate the impact of GCBDam incorporation on the recyclability and reprocessability of the vitrimers, we subjected both DN5% and SN5% to a series of five reprocessing cycles, following consistent protocols as outlined in Fig. S27.†32 Post-reprocessing, the materials were analyzed using FT-IR spectroscopy, DSC, and DMA to monitor potential changes in their properties and structure. DMA results showed marginal fluctuations in both the Tg and storage modulus (E′) for DN5% across the reprocessing cycles (Table S4†). These slight variations suggest only minor alterations in crosslinking throughout the recycling process (Fig. S28†) due to potential oxidation of the excess amines at high temperatures apparent from the darkening of the reprocessed samples (Fig. S27C†). On the other hand, DSC analysis revealed that the Tg of the recycled material remained remarkably stable, deviating by no more than ±5 °C from the original values (Fig. S30†). Furthermore, FTIR spectroscopic studies confirmed the persistent presence of the characteristic vinylogous urethane peaks, indicating negligible changes in the chemical structure of the networks through multiple cycles of reprocessing (Fig. S32†). Similarly, SN5% demonstrated comparable trends in its Tg, E′, and chemical structure during reprocessing, as substantiated by DMA, DSC, and FTIR results (Fig. S29, S31, and S33†). Further, the chemical degradation study revealed that all the DNs and SNs can be broken down upon mild heating in the presence of excess amines (Fig. S34†). These findings establish that the GCB-containing DNs exhibit superior creep resistance up to 160 °C while retaining reprocessability and degradability—an important material characteristic for potential long-term applications and sustainability.
Conclusions
This study highlights the significant improvement in creep resistance of vitrimers at elevated temperatures achieved by incorporating Janus-faced GCBDam moieties. The hindered mobility of reactive moieties in networks containing GCBDam reduces diffusion and enhances creep resistance with preserved reprocessability. A 10% incorporation of GCBDam resulted in high levels of creep resistance at 150 °C, surpassing previously reported vitrimers reinforced by supramolecular networks. At high temperatures, the bond exchange behavior of GCBDam exhibited faster kinetics compared to the covalent vinylogous urethane exchange chemistry. While DN5% showed insignificant changes in Ea, increasing the GCBDam content to 10% in DN10% resulted in an Ea of 96 kJ mol−1, as compared to 88 kJ mol−1 in SN10%. This increase in the Ea may offer advantages in practical applications by raising the energy barrier for network flow yet allowing for a rapid reduction in viscosity at elevated temperatures. We believe incorporating Janus-faced hydrogen bonding moieties in vitrimers holds great potential for enhancing dimensional stability while maintaining rapid reprocessability at elevated temperatures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the National Science Foundation (NSF EEC-1941529) and the Army Research Office through a MURI Grant (W911NF2310260). A grant to G. J. S. (CSIRSSB-000726) is also gratefully acknowledged. The authors would like to thank Anton Paar for the use of their rheometer through the Anton Paar VIP research program.References
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643–2653 CrossRef CAS PubMed.
- M. Podgorski, B. D. Fairbanks, B. E. Kirkpatrick, M. McBride, A. Martinez, A. Dobson, N. J. Bongiardina and C. N. Bowman, Adv. Mater., 2020, 32, 1–26 Search PubMed.
- C. J. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC.
- A. Jourdain, R. Asbai, O. Anaya, M. M. Chehimi, E. Drockenmuller and D. Montarna, Macromolecules, 2020, 53, 1884–1900 CrossRef CAS.
- Q. Zhou, Z. Sang, K. K. Rajagopalan, Y. Sliozberg, F. Gardea and S. A. Sukhishvili, Macromolecules, 2021, 54, 10510–10519 CrossRef CAS.
- F. Cantamessa, G. Damonte, O. Monticelli, R. Arrigo and A. Fina, ACS Appl. Polym. Mater., 2022, 4, 4796–4807 CrossRef CAS PubMed.
- L. Porath, B. Soman, B. B. Jing and C. M. Evans, ACS Macro Lett., 2022, 11, 475–483 CrossRef CAS PubMed.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- M. Guerre, C. Taplan, J. M. Winne and F. E. Du Prez, Chem. Sci., 2020, 11, 4855–4870 RSC.
- G. M. Scheutz, J. J. Lessard, M. B. Sims and B. S. Sumerlin, J. Am. Chem. Soc., 2019, 141, 16181–16196 CrossRef CAS PubMed.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664–7667 CrossRef CAS PubMed.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Adv. Funct. Mater., 2015, 25, 2451–2457 CrossRef CAS.
- M. Guerre, C. Taplan, R. Nicolay, J. M. Winne and F. E. Du Prez, J. Am. Chem. Soc., 2018, 140, 13272–13284 CrossRef CAS PubMed.
- C. Taplan, M. Guerre, J. M. Winne and F. E. Du Prez, Mater. Horiz., 2020, 7, 104–110 RSC.
- K. A. Stewart, D. P. DeLellis, J. J. Lessard, J. F. Rynk and B. S. Sumerlin, ACS Appl. Mater. Interfaces, 2023, 15, 25212–25223 CrossRef CAS PubMed.
- J. J. Lessard, K. A. Stewart and B. S. Sumerlin, Macromolecules, 2022, 55, 10052–10061 CrossRef CAS.
- P. R. Christensen, A. M. Scheuermann, K. E. Loeffler and B. A. Helms, Nat. Chem., 2019, 11, 442–448 CrossRef CAS PubMed.
- Y. X. G. Lu and Z. Guan, J. Am. Chem. Soc., 2012, 134, 14226–14231 CrossRef CAS PubMed.
- Y. Chen, A. M. Kushner, G. A. Williams and Z. Guan, Nat. Chem., 2012, 4, 467–472 CrossRef CAS PubMed.
- A. J. M. Bañales and M. B. Larsen, ACS Macro Lett., 2020, 9, 937–943 CrossRef PubMed.
- Y. Nishimura, J. Chung, H. Muradyan and Z. Guan, J. Am. Chem. Soc., 2017, 139, 14881–14884 CrossRef CAS PubMed.
- C. A. Tretbar, J. A. Neal and Z. Guan, J. Am. Chem. Soc., 2019, 141, 16595–16599 CrossRef CAS PubMed.
- S. Yu, S. Wu, C. Zhang, Z. Tang, Y. Luo, B. Guo and L. Zhang, ACS Macro Lett., 2020, 9, 1143–1148 CrossRef CAS PubMed.
- M. Röttger, T. Domenech, R. v. d. Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, Science, 2017, 356, 62–65 CrossRef PubMed.
- J. S. A. Ishibashi and J. A. Kalow, ACS Macro Lett., 2018, 7, 482–486 CrossRef CAS PubMed.
- Y. Liu, Z. Tang, D. Wang, S. Wu and B. Guo, J. Mater. Chem. A, 2019, 7, 26867–26876 RSC.
- W. Denissen, M. Droesbeke, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Nat. Commun., 2017, 8, 14857 CrossRef CAS PubMed.
- F. Van Lijsebetten, K. De Bruycker, J. M. Winne and F. E. Du Prez, ACS Macro Lett., 2022, 11, 919–924 CrossRef CAS PubMed.
- F. Van Lijsebetten, K. De Bruycker, Y. Spiesschaert, J. M. Winne and F. E. Du Prez, Angew. Chem., Int. Ed., 2022, 61, 1–8 Search PubMed.
- J. J. Lessard, G. M. Scheutz, R. W. Hughes and B. S. Sumerlin, ACS Appl. Polym. Mater., 2020, 2, 3044–3048 CrossRef CAS.
- J. J. Lessard, L. F. Garcia, C. P. Easterling, M. B. Sims, K. C. Bentz, S. Arencibia, D. A. Savin and B. S. Sumerlin, Macromolecules, 2019, 52, 2105–2111 CrossRef CAS.
- J. J. Lessard, G. M. Scheutz, S. H. Sung, K. A. Lantz, T. H. Epps III and B. S. Sumerlin, J. Am. Chem. Soc., 2020, 142, 283–289 CrossRef CAS PubMed.
- J. S. A. Ishibashi, I. C. Pierce, A. B. Chang, A. Zografos, B. M. El-Zaatari, Y. Fang, S. J. Weigand, F. S. Bates and J. A. Kalow, Macromolecules, 2021, 54, 3972–3986 CrossRef CAS.
- L. Li, X. Chen, K. Jin and J. M. Torkelson, Macromolecules, 2018, 51, 5537–5546 CrossRef CAS.
- J. J. Cash, T. Kubo, D. J. Dobbins and B. S. Sumerlin, Polym. Chem., 2018, 9, 2011–2020 RSC.
- L. Zhong, Y. Hao, J. Zhang, F. Wei, T. Li, M. Miao and D. Zhang, Macromolecules, 2022, 55, 595–607 CrossRef CAS.
- J. Cao, S. Li, C.-C. Wang, R. Xu, M. Tang, X. Ren and Y.-X. Xu, Ind. Eng. Chem. Res., 2022, 61, 13136–13144 CrossRef CAS.
- S. Wang, S. Ma, Q. Li, X. Xu, B. Wang, K. Huang, Y. Liu and J. Zhu, Macromolecules, 2020, 53, 2919–2931 CrossRef CAS.
- J. A. Neal, D. Mozhdehi and Z. Guan, J. Am. Chem. Soc., 2015, 137, 4846–4850 CrossRef CAS PubMed.
- J. Y. Sun, X. Zhao, W. R. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef CAS PubMed.
- A. Phadke, C. Zhang, B. Arman, C. C. Hsu, R. A. Mashelkar, A. K. Lele, M. J. Tauber, G. Arya and S. Varghese, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4383–4388 CrossRef CAS PubMed.
- G. Kim, C. Caglayan and G. J. Yun, ACS Omega, 2022, 7, 44750–44761 CrossRef CAS PubMed.
- H. Gao, Y. Sun, M. Wang, Z. Wang, G. Han, L. Jin, P. Lin, Y. Xia and K. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 1581–1591 CrossRef CAS PubMed.
- L. Wanga, Y. Liu, Y. Qiaoa, Y. Wanga, Z. Cuia, S. Zhua, F. Donga, S. Fanga and A. Du, Polym. Chem., 2022, 13, 4144–4153 RSC.
- L. Wang, Y. Liu, Y. Wei, W. Zeng, Z. Cui and A. Du, Eur. Polym. J., 2023, 193, 112101 CrossRef CAS.
- S. K. Schoustra, V. Asadi, H. Zuilhof and M. M. J. Smulders, Eur. Polym. J., 2023, 195, 112209 CrossRef CAS.
- C. L. Meena, D. Singh, B. Kizhakeetil, M. Prasad, M. George, S. Tothadi and G. J. Sanjayan, J. Org. Chem., 2021, 86, 3186–3195 CrossRef CAS PubMed.
- S. Mondal, J. J. Lessard, C. L. Meena, G. J. Sanjayan and B. S. Sumerlin, J. Am. Chem. Soc., 2022, 144, 845–853 CrossRef CAS PubMed.
- M. A. Wagh, D. R. Shinde, G. R. Gamidi and G. J. Sanjayan, Org. Biomol. Chem., 2023, 21, 6914–6918 RSC.
- A. Schiavi and A. Prato, Polym. Test., 2017, 59, 220–229 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00276h |
| This journal is © The Royal Society of Chemistry 2024 |