
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of megadalton stereoregular ring-substituted poly(phenylacetylene)s by a rhodium(I) catalyst with a N-functionalized hemilabile phosphine ligand†
Marta
Angoy
,
M. Victoria
Jiménez
 *,
Eugenio
Vispe
and
Jesús J.
Pérez-Torrente
*
*,
Eugenio
Vispe
and
Jesús J.
Pérez-Torrente
*
Departamento de Química Inorgánica, Instituto de Síntesis Química y Catálisis Homogénea–ISQCH, Universidad de Zaragoza–CSIC, Facultad de Ciencias, C/Pedro Cerbuna, 12, 50009 Zaragoza, Spain. E-mail: perez@unizar.es; vjimenez@unizar.es
First published on 19th April 2024
Abstract
The cationic compound [Rh(nbd){κ2P,N-Ph2P(CH2)3NMe2}][BF4] efficiently catalyzes the polymerization of a series of ring-substituted phenylacetylene derivatives, R-C6H4–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH with groups of different electronic and steric properties at the para (R = F, CF3, Me, Bu, tBu, OMe, OBu) and meta (R = OMe) positions to give highly stereoregular ring-substituted poly(phenylacetylene)s with a cis-transoidal configuration of very high molar mass and moderate dispersities. The polymers have been characterized by size exclusion chromatography (SEC-MALS), NMR, DSC and TGA. The polymerization of phenylacetylene and 1-ethynyl-3-methoxybenzene gives megadalton poly(phenylacetylene)s, while the polymerization of 1-ethynyl-4-methoxybenzene and 1-(tert-butyl)-4-ethynylbenzene gives ultra-high molecular weight poly(phenylacetylene)s with Mn of 1.70 × 106 and 2.72 × 106, respectively. The electronic effect of the substituent strongly influences the catalytic activity. Phenylacetylene derivatives with an electron-withdrawing substituent in para position polymerize faster than those with an electron-donating substituent.
CH with groups of different electronic and steric properties at the para (R = F, CF3, Me, Bu, tBu, OMe, OBu) and meta (R = OMe) positions to give highly stereoregular ring-substituted poly(phenylacetylene)s with a cis-transoidal configuration of very high molar mass and moderate dispersities. The polymers have been characterized by size exclusion chromatography (SEC-MALS), NMR, DSC and TGA. The polymerization of phenylacetylene and 1-ethynyl-3-methoxybenzene gives megadalton poly(phenylacetylene)s, while the polymerization of 1-ethynyl-4-methoxybenzene and 1-(tert-butyl)-4-ethynylbenzene gives ultra-high molecular weight poly(phenylacetylene)s with Mn of 1.70 × 106 and 2.72 × 106, respectively. The electronic effect of the substituent strongly influences the catalytic activity. Phenylacetylene derivatives with an electron-withdrawing substituent in para position polymerize faster than those with an electron-donating substituent.
Introduction
Conjugated polymers have attracted significant attention due to their use as conductive polymers for optoelectronics and energy applications.1–3 In particular, poly(phenylacetylene) (PPA) has been intensively studied due to its stability in air, high solubility in common organic solvents, excellent processability and semiconducting properties.4,5 The wide choice of substituted phenylacetylene (PA) derivatives has provided access to functional polymers with applications in electronics, photoelectronics, optics, and membrane separation.6–8 Furthermore, a range of supramolecular assemblies derived from PPAs have been prepared, including nanoparticles, nanotubes, gels, liquid crystals, fibers, and composites.9The processing behavior and many of the end-use properties of polymers are influenced by the molecular weight and molecular weight distribution (MWD). Therefore, control of both parameters is essential to tailor polymers to meet specific application requirements.10 Ultrahigh-molecular weight (UHMW) polymer materials offer improved physical and mechanical properties. Consequently, the development of synthetic methods for UHMW polymers, i.e. polymers with a number-average molecular weight (Mn) greater than 106 g mol−1, is an interesting research topic in polymer chemistry.11–14 In addition to molecular weight, the stereochemistry and conformation of PPAs also influence their chemical and physical properties.15
Although a number of transition metal catalysts are available for the polymerization of phenylacetylene derivatives, those based on late transition metals such as ruthenium, rhodium, iridium and palladium have attracted considerable attention due to their high activity, stability to air and moisture, and high tolerance to many of the heteroatoms in alkyne-functionalized monomers.16 In particular, rhodium catalysts efficiently catalyze the polymerization of phenylacetylene derivatives to give highly stereoregular polymers with cis-transoidal conformation.17,18 Recently, significant progress has been made in the design of rhodium(I) catalysts for the living polymerization of alkyne-based monomers.19–21 Well-defined Rh-alkyny,22,23 Rh–vinyl,24–26 and Rh–aryl,27–29 complexes allow the controlled (co)polymerization of PA derivatives to give highly stereoregular (co)polymers with narrow MWD and very high initiation efficiencies. However, moderate to high molecular weight PPAs have typically been obtained.
The number of rhodium-based catalytic systems leading to ultrahigh-molecular weight PPAs is rather limited (Chart 1). Some time ago, Tabata et al. reported that the PA polymerization activity of the rhodium dimer [Rh(μ-Cl)(nbd)]2 (nbd = 2,5-norbornadiene) could be significantly enhanced in polar solvents such as triethylamine, alcohol, water, and THF.30 Indeed, PA polymerization in triethylamine afforded stereoregular ultrahigh-molecular weight PPA, Mw up to 4.43 × 106, albeit in low yield. More recently, Bielawski et al. reported a heterogeneous catalyst based on a rhodium(I) polymer, [Rh2(μ-Cl)2(cyclooctatetraene)]n, supported on silica. In the presence of an external base, triethylamine, ultrahigh-molecular weight PPA, Mn up to 1.29 × 106, was produced in a stereoselective manner.31 Our research group has developed a complementary strategy for the design of efficient rhodium(I) PA polymerization catalysts based on the utilization of functionalized ligands with the ability to act as an internal base. In particular, the mononuclear phosphino-anilido [Rh(nbd){κ2P,N-Ph2P(C6H4)NMe}] complex efficiently catalyzed the polymerization of PA to yield ultrahigh-molecular weight PPA with Mn up to 1.94 × 106.32
 | ||
| Chart 1 Catalytic systems for phenylacetylene polymerization leading to ultrahigh-molecular weight (UHMW) PPA. | ||
Following this strategy, we have prepared a series of rhodium(I) catalysts based on N-functionalized phosphine ligands of hemilabile character (Chart 2). Cationic rhodium(I) complexes containing amino-functionalized phosphine ligands, such as [Rh(nbd){κ2P,N-Ph2P(CH2)3NR2}]+ (R = H, Me) and [Rh(nbd){κ2P,N-Ph2P(C6H4)NHMe}]+, efficiently catalyzed PA polymerization leading to stereoregular megadalton PPAs (Mn in the range 1.1–1.5 × 106) with moderate dispersity.32,33 Similarly, neutral complexes with amino-functionalized N-heterocyclic carbene ligands, [RhCl(nbd){κC-MeIm(CH2)3NMe2}] and [RhBr(nbd){κC-MeIm(CH2)3NH2}], or pyridine-functionalized phosphino ligands, such as [RhCl(nbd){κP-Ph2P(CH2)2Py}], also yielded megadalton PPAs with Mn of about 1.3 × 106.34
 | ||
| Chart 2 Molecular catalysts for phenylacetylene polymerization leading to megadalton PPAs (Mn = 1.1–1.5 × 106). | ||
The aim of this work is to evaluate the potential of the catalyst [Rh(nbd){κ2P,N-Ph2P(CH2)3NMe2}][BF4].35 in the polymerization of a series of ring-substituted phenylacetylene derivatives with different electronic and steric properties for the preparation of very high molecular weight PPAs. The choice of the 3-(dimethylamino-propyl)diphenylphosphine ligand is based on both the ease of its preparation36 and the catalytic performance of cationic rhodium-diene catalysts with this hemilabile ligand, which exhibit excellent PA conversions in short reaction times, resulting in the formation of megadalton PPA polymers.
Experimental section
Materials
All experiments were carried out under an atmosphere of argon using Schlenk techniques or glovebox. Solvents were distilled immediately prior to use from the appropriate drying agents or obtained from a Solvent Purification System (Innovative Technologies). The terminal arylacetylenes: phenylacetylene (PAa), 1-ethynyl-3-methoxybenzene (PAf), 1-butoxy-4-ethynylbenzene (PAh) (Merck), and 1-ethynyl-4-fluorobenzene (PAb), 1-ethynyl-4-methylbenzene (PAc), 1-ethynyl-4-(trifluoromethyl)benzene (PAd), 1-ethynyl-4-methoxybenzene (PAe), 1-butyl-4-ethynylbenzene (PAg), 1-(tert-butyl)-4-ethynylbenzene (PAi) (Acros Organics), were distilled over CaH2 under reduced pressure and stored over molecular sieves.Methods
The absolute molecular weight averages (Mn and Mw), dispersity (Đ, Mw/Mn) and molecular weight distribution were determined by SEC-MALS at the Chromatography and Spectroscopy Service of the ISQCH. SEC-MALS analyses were carried out using a Waters 2695 instrument, equipped with three PL-Gel Mixed B LS columns fitted to a MALS detector (MiniDawn Treos, Wyatt) and a differential refractive index detector (Optilab Rex, Wyatt). The polymer solutions in THF (≈2.0 mg mL−1) were filtered through a 0.45 μm PTFE membrane filter before being injected in the GPC systems. The analyses were carried out immediately after the dissolution of the polymer sample in THF to minimize sample degradation.37,38 Data analysis was performed with ASTRA Software from Wyatt. Polymer samples were eluted at 25 °C with THF at a flow rate of 1.0 mL min−1. The dn/dc parameter in THF at 658 nm was determined for each polymer using the full mass recovery method, which assumes 100% recovery of the injected mass of a polymer sample of known concentration,39 except for PPAe and PPAh which exhibited poor mass recovery due to insoluble species and therefore the dilution method was used (five samples from 0.1 mg mL−1 to 10 mg mL−1 were analyzed). The polymers PPAa, PPAb and PPAi were completely soluble. Other polymers were difficult to dissolve, resulting in mass recoveries of 90% for PPAg and PPAf, 65% for PPAe and 75% for PPAh. The dn/dc values of PAa, PAb, PAe, PAf, PAg, PAh, and PAi, were determined to be 0.286, 0.210, 0.258, 0.265, 0.231, 0.261, and 0.224 mL g−1, respectively. The poor solubility of PAc and PAd precluded dn/dc determination and molar mass measurement.1H NMR spectra were recorded on a Bruker Avance 300 or 400 spectrometers. NMR chemical shifts are reported in ppm relative to tetramethylsilane and referenced to partially deuterated solvent resonances. Coupling constants (J) are given in Hertz. Thermal gravimetric analyses were carried out on a TA Q-5000 TGA apparatus (TA Instruments). Samples were heated under nitrogen from room temperature to 600 °C at a rate of 10 °C min−1, and then up to 750 °C on air at the same rate. Differential scanning calorimetry experiments were carried out on a DSC Q-20 apparatus (TA Instruments). Samples were heated under nitrogen from 20 °C to 200 °C at a rate of 10 °C min−1 and cooled to the initial temperature at the same rate. In some experiments this cycle was repeated several times. DSC plots are shown in exo-down mode. Chromatographic analysis was performed on an HP 5890 Series II gas chromatograph with ionization detector and an HP-5 column (25 m × 0.32 mm i.d. × 0.17 μm). Calibration of the detector response to the different alkynes was carried out using n-octane as internal standard.
Synthesis of catalyst [Rh(nbd){κ2P,N-Ph2P(CH2)3NMe2}][BF4]35
AgBF4 (169 mg, 0.868 mmol) was added to a suspension of [Rh(μ-Cl)(nbd)]2 (200 mg, 0.434 mmol) in tetrahydrofuran (10 mL). The resulting suspension was stirred for 30 min and the formed AgCl filtered off and washed with 3 tetrahydrofuran (3 × 1 mL). The volume of the resulting yellow solution, containing the solvated species [Rh(nbd)(THF)n]+, was reduced to about 5 mL and then a solution of the phosphine Ph2P(CH2)3NMe2 (235 mg, 0.868 mmol) in tetrahydrofuran (2 mL) was slowly added at 0 °C to give an orange solution. Concentration of the solution to about 0.2 mL and slow addition of diethyl ether (5 mL) afforded the compound as an orange solid which was washed with diethyl ether (3 × 2 mL) and dried under vacuum. Yield: 61%. 1H NMR (298 K, CDCl3): δ 7.58–7.42 (m, 10H, Ph), 5.31 (br, 2H,![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH cod), 3.14 (br, 2H, CH cod), 2.86 (br, 2H, CH2N), 2.40 (m, 6H; 4H CH2 cod, 2H CH2), 2.34 (s, 6H, NMe2), 1.94 (m, 6H; 4H CH2 cod, 2H CH2). 31P{1H} NMR (298 K, CDCl3): δ 17.92 (d, JP–Rh = 155.7).
CH cod), 3.14 (br, 2H, CH cod), 2.86 (br, 2H, CH2N), 2.40 (m, 6H; 4H CH2 cod, 2H CH2), 2.34 (s, 6H, NMe2), 1.94 (m, 6H; 4H CH2 cod, 2H CH2). 31P{1H} NMR (298 K, CDCl3): δ 17.92 (d, JP–Rh = 155.7). 
Polymer synthesis and characterization
The polymerization reactions were carried out in round bottom flasks with efficient stirring. A typical polymerization procedure is as follows: terminal aryl alkyne (0.64 mmol) was added to a THF solution (2.5 mL) of the catalysts (6.4 μmol) and the mixture stirred at 293 K in the absence of light. The consumption of monomer was monitored by GC using octane as internal standard. The polymer solutions were transferred into vigorously stirred cold methanol (25 mL, 273 K) using a cannula under argon. The polymers were filtered, washed with methanol (3 × 5 mL) and dried under vacuum to constant weight. The polymers were obtained as yellow-orange solids in good yields according to the attained conversion values.NMR data
PPAa (R = H). 1H NMR (298 K, 300 MHz, CD2Cl2): δ 6.98 (d, 3H), 6.67 (t, 2H), 5.85 (s, 1H,CH). 13C{1H} NMR (298 K, 300 MHz, CD2Cl2): δ 142.9, 139.4 (Cq), 131.8 (CH), 127.8, 127.6, 126.7 (CH). PPAb (R = p-F). 1H NMR (298 K, 300 MHz, CDCl3): δ 6.67 (m, 4H), 5.72 (s, 1H, CH). 13C{1H} NMR (298 K, 400 MHz, CDCl3): δ 163.9, 161.5, 139.3 (Cq), 131.5 (CH), 129.3 (CH, JC–F = 8 Hz), 113.3 (CH, JC–F = 48 Hz). PPAe (R = p-OMe). 1H NMR (298 K, 300 MHz, CDCl3): δ 6.96 (m, 2H), 6.70 (d, 2H), 5.91 (s, 1H, CH), 3.59 (s, 3H, OMe). 1H NMR (298 K, 400 MHz, CD2Cl2): δ 6.65 (d, 2H), 6.48 (d, 2H), 5.76 (s, 1H, CH), 3.58 (s, 3H, OMe). 13C{1H} NMR (298 K, 400 MHz, CD2Cl2): δ 158.9, 139.1, 136.0 (Cq), 128.9 (CH), 127.8, 113.3 (CH), 55.32 (OMe). PPAf (R = m-OMe). 1H NMR (298 K, 300 MHz, CDCl3): δ 6.83 (t, 1H), 6.52 (d, 1H), 6.29 (d, 2H), 6.26 (s, 1H), 5.85 (s, 1H, CH), 3.56 (s, 3H, OMe). 1H NMR (298 K, 300 MHz, CD2Cl2): δ 6.85 (t, 1H), 6.55 (d, 1H), 6.30 (d, 1H), 6.28 (s, 1H), 5.85 (s, 1H, CH), 3.56 (s, 3H, OMe). 13C{1H} NMR (298 K, 300 MHz, CD2Cl2): δ 159.1, 144.2, 139.3 (Cq), 131.6 (CH), 128.6, 120.4, 112.7, 112.3 (CH), 55.0 (OMe). PPAg (R = p-Bu). 1H NMR (298 K, 300 MHz, CDCl3): δ 6.70 (d, 2H), 6.54 (d, 2H), 5.73 (s, 1H, CH), 3.69 (m, 2H, –OCH2), 1.63 (m, 2H, >CH2), 1.39 (m, 2H, >CH2), 0.91 (t, 3H, –CH3). 13C{1H} NMR (298 K, 400 MHz, CDCl3): δ 142.0, 138.5 (Cq), 130.1 (CH), 128.7, 127.0 (CH), 35.2, 33.5, 22.1 (>CH2), 13.7 (–CH3). PPAh (R = p-OBu). 1H NMR (298 K, 300 MHz, CD2Cl2): δ 6.60 (d, 2H), 6.43 (d, 2H), 5.73 (s, 1H, CH), 2.39 (m, 2H, >CH2), 1.44 (m, 2H, >CH2), 1.27 (m, 2H, >CH2), 0.89 (t, 3H, –CH3). 13C{1H} NMR (298 K, 300 MHz, CD2Cl2): δ 158.2, 138.8, 135.7 (Cq), 130.1 (CH), 128.7, 113.5 (CH), 67.7, 31.4, 19.3 (>CH2), 13.7 (–CH3). PPAi (R = p-tBu). 1H NMR (298 K, 300 MHz, CDCl3): δ 6.97 (d, 2H), 6.54 (d, 2H), 5.81 (s, 1H, CH), 1.14 (s, 9H, tBu). 13C{1H} NMR (298 K, 400 MHz, CDCl3): δ 148.8, 140.4, 138.8 (Cq), 127.1 (CH), 126.8, 124.3 (CH), 40.6 (CqtBu) 31.2 (tBu).
Results and discussion
Polymerization of ring-substituted phenylacetylene derivatives catalyzed [Rh(nbd){κ2P,N-Ph2P(CH2)3NMe2}][BF4]
The cationic compound [Rh(nbd){κ2P,N-Ph2P(CH2)3NMe2}][BF4]35 efficiently catalyzed the polymerization of a series of para- and meta-substituted phenylacetylene derivatives with groups of different electronic and steric properties to afford megadalton PPAs (Fig. 1).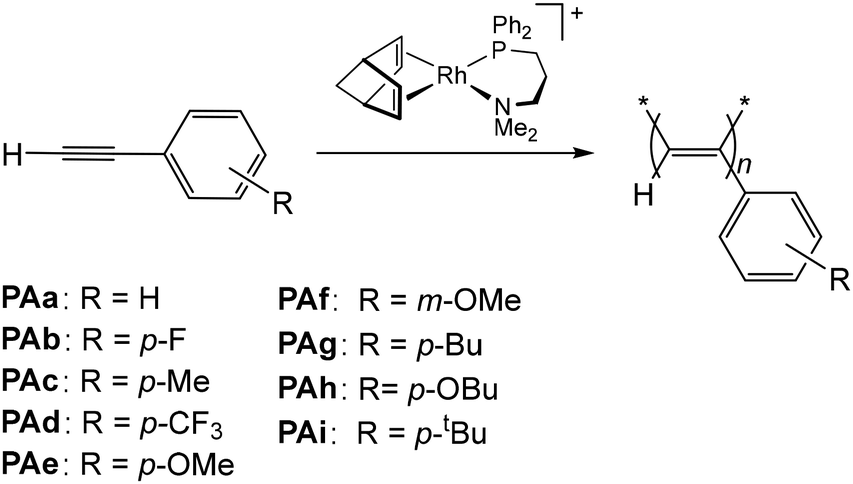 | ||
| Fig. 1 Polymerization of ring-substituted phenylacetylene derivatives (PAa–PAi) catalyzed by [Rh(nbd){κ2P,N-Ph2P(CH2)3NMe2}][BF4]. | ||
The polymerization reactions were carried out in THF at 20 °C in the absence of light with a monomer to rhodium, [PA]o/[Rh], ratio of 100. The ring-substituted PPAs were isolated as yellow solids in practically quantitative yields according to the conversion values. The 1H NMR spectra of the polymers in CDCl3 or CD2Cl2 showed a sharp singlet corresponding to the vinylic protons of the chain in the range δ 5.85–5.72 ppm, indicating a highly stereoregular structure with a cis-transoidal configuration.40,41 The PPAs were characterized by size exclusion chromatography (SEC) using light-scattering (MALS) and refractive index (DRI) detectors. In general, full PA conversion was achieved in 5–90 min, except in the case of PAh (R = p-OBu), to give ring-substituted poly(phenylacetylene)s with very high molar mass (MM) (Table 1).
| Polymer | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Mon. | t (min) | Conv.b (%) |
M
w![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c c |
Đ |
M
nd |
IEe (%) |
| a Reaction conditions: [PA]o = 0.25 M, [PA]o/[Rh] = 100, in THF at 293 K. b Determined by GC (octane as internal standard). c Estimated by SEC-MALS in THF, Mw = weight-average molecular weight (g mol−1), Đ = dispersity (Mw/Mn, Mn = number-average molecular weight). d Calculated from Mw and Đ, in g mol−1. e Initiation efficiency, IE = Mtheor/Mn × 100; where Mtheor = [PA]o/[Rh]nMWPA × polymer yield. f Ref. 33. | |||||||
| 1 |
PAaf |
60 | 100 | 2.18 × 106 | 2.00 | 1.09 × 106 | 0.9 |
| 2 | PAb | 30 | 100 | 1.35 × 106 | 2.39 | 5.65 × 105 | 1.8 |
| 3 | PAc | 60 | 100 | Insoluble | |||
| 4 | PAd | 5 | 100 | Insoluble | |||
| 5 | PAe | 45 | 100 | 2.36 × 106 | 1.39 | 1.70 × 106 | 0.6 |
| 6 | PAf | 90 | 100 | 2.11 × 106 | 1.77 | 1.19 × 106 | 0.9 |
| 7 | PAg | 75 | 100 | 9.94 × 105 | 1.53 | 6.50 × 105 | 1.4 |
| 8 | PAh | 60 | 40 | 3.12 × 105 | 1.53 | 2.04 × 105 | 2.0 |
| 9 | PAi | 30 | 100 | 4.84 × 106 | 1.78 | 2.72 × 106 | 0.4 |
The obtained ring-substituted PPAs exhibit very high weight-average molar masses, Mw in the range 3.1 × 105–4.8 × 106, and moderate dispersities, Đ in the range 1.4–2.4 (Table 1). A graphical representation of the MMs of the PPA polymers is shown in Fig. 2. Polymerization of the PAi (R = p-tBu) monomer was completed in 30 min yielding a fully soluble polymer with a surprisingly high molar mass, Mw of 4.84 × 106, and an initiation efficiency as low as 0.4%. Polymerization of the methoxy-substituted monomers PAe (R = p-OMe) and PAf (R = m-OMe) also gave soluble polymers of very high MMs (Mw ≈ 2.0 × 106). The polymers derived from PAb (R = p-F) and PAg (R = p-Bu) showed MMs of around 1.0 × 106. Polymerization of the monomer PAh (R = p-OBu) was much slower, with only 40% conversion in one hour, yielding the polymer with the lowest molar mass, Mw of 3.12 × 105. The narrowest dispersities were obtained for the polymers PPAe (R = p-OMe), PPAg (R = p-Bu) and PPAh (R = p-OBu) with Đ values of 1.39 and 1.53, respectively. The number-average molecular weight of the polymers (Mn), calculated from the weight-average molecular weight (Mw) and Đ, showed the formation of megadalton PPAa (R = H) and PPAf (R = m-OMe) (Mn ≈ 1.1 × 106), and UHMW PPAe (R = p-OMe) and PPAi (R = p-tBu) polymers, with Mn of 1.70 × 106 and 2.72 × 106, respectively (Table 1).
 | ||
| Fig. 2 Molar mass (Mw) of the polymer samples obtained by polymerization of ring-substituted phenylacetylene derivatives catalyzed by [Rh(nbd){κ2P,N-Ph2P(CH2)3NMe2}][BF4]. | ||
The polymerization of 1-ethynyl-4-methylbenzene (PAc) and 1-ethynyl-4-trifluoromethylbenzene (PAd) gave polymers that were insoluble in common solvents such as THF, CHCl3 or toluene, so that their Mw could not be determined. In this context, it has been reported that the polymerization of ortho-substituted phenylacetylenes is slower than that of meta- and para-substituted derivatives and usually leads to insoluble polymers. Analysis of some of these materials by high-angle X-ray scattering (WAXS) revealed highly crystalline materials with a pseudohexagonal network consisting of macromolecules arranged as rods.42 However, some insoluble polymers derived from para-substituted monomers, such as 1-ethynyl-4-chlorobenzene and 1-ethynyl-4-iodobenzene, were found to be almost amorphous materials due to the strong aggregation between the polymer chains.43 On the other hand, it has already been described that the polymerization of 1-ethynyl-4-methylbenzene leads to the formation of insoluble materials.44
The polymers prepared from the ring-substituted phenylacetylene monomers showed a unimodal molecular weight distribution as determined by SEC-MALS analysis (see the ESI†). The ring-substituted PPAs were found to be linear regardless of their molar mass or the characteristics of the substituent. As an example, Fig. 3 shows the MM vs. elution volume plot and the log–log plot of the radius of gyration (rg) vs. MM for a PPAe sample prepared from PAe (R = p-OMe). The linearity of the MM plot over the elution volume ranges where both MALS and DRI detectors have detectable intensities, and the linear relationship of the radius of gyration (rg) vs. the molar mass (MM) in the high molar mass region, is typical of a linear polymer. The slopes of the linear part of the conformation plots, in the range of 0.51–0.63, deviate from the expected value of about 0.58 for a linear polymer reflecting the complex behavior of PPA in dilute solutions due to changes in solvent–polymer and polymer–polymer interactions, as well as the process of σ-trans to σ-cis isomerization.45 In sharp contrast, the analysis of the PPAa samples obtained by polymerization of phenylacetylene showed the presence of branched polymer with high-MM.33 In this case, both the deviation from linear behavior in the high-MM region of the log–log plot of rgvs. MM and the detectable increase in the MM in the high-MM region of the MM vs. elution volume plot are consistent with branching (see the ESI†).
 | ||
| Fig. 3 (a) SEC chromatograms: light scattering detector response (90 degrees) (red) and differential refractometer response (blue), MM (molar mass) vs. elution volume plot for a PPAe sample prepared from PAe (R = p-OMe). (b) Log–log plot of the radius of gyration (rg) vs. MM. | ||
Light scattering chromatograms and cumulative weight fraction vs. molar mass plots for representative polymers are shown in Fig. 4 showing the increase in Mw along the series PPAh (R = p-OBu, Mw = 3.12 × 105), PPAe (R = p-OMe, Mw = 2.36 × 106) and PPAi (R = p-tBu, Mw = 4.84 × 106).
 | ||
| Fig. 4 (a) SEC chromatograms: light scattering detector response (90 degrees) and (b) Cumulative molar mass distribution plots for polymers: PPAi (green), PPAe (red) and PPAh (blue). | ||
Time-conversion profiles of PAa–PAi polymerization reactions catalyzed by [Rh(nbd){κP,N-Ph2P(CH2)3NMe2}][BF4]
The polymerization reactions were monitored by gas chromatography at regular time intervals, using octane as internal standard, which allowed to obtain time-conversion profiles showing the conversion of the corresponding monomer as a function of time. The influence of the electronic effects due to the substituent in para position can be analyzed in Fig. 5, which shows the time-conversion profiles for the polymerization of PAa, PAb, PAc, PAd and PAe. Phenylacetylene derivatives with an electron-withdrawing substituent in para position polymerized faster than phenylacetylene (PAa). The fastest polymerizing substituted phenylacetylene derivatives were PAd (R = p-CF3) and PAb (R = p-F), which reached full conversion in 5 and 30 min, respectively. Monomer PAc (R = p-Me), with an electro-donating substituent, showed lower activity than phenylacetylene (PAa) although both reached full conversion in 60 min. The slowest polymerizing alkyne in this series was PAe (R = p-OMe) with lower conversions at short reaction times. | ||
| Fig. 5 Time-conversion plots for the polymerization of ring-substituted phenylacetylene derivatives. Reaction conditions: THF, 20 °C, [PA]o = 0.25 M, [PA]o/[Rh] = 100. | ||
In principle, the presence of electron-withdrawing substituents on the aromatic ring should increase the acidity of the alkyne, favoring the initiation step by facilitating its deprotonation in route to the active alkynyl species (see below).46 Indeed, the following pKa values: p-nitrophenylacetylene, 17.98; p-fluorophenylacetylene, 18.14; phenylacetylene, 18.50; and p-methylphenylacetylene, 18.60; are fully consistent with the inductive effect predictions.47,48 Accordingly, the calculated initiation efficiency in the polymerization of PAb (R = p-F), 1.8%, is slightly higher than that calculated for phenylacetylene (PAa, 0.9%) and 1-ethynyl-4-methoxybenzene (PAe, 0.6%). However, the influence of electronic effects on the chain propagation step, due to the reduced coordinative ability of the triple bond because of the attracting character of the substituent, has also been proposed.25 The key role of acidity for homogeneous Rh(diene)-type catalysts is also evidenced by the lack of activity in polymerization of alkyl-acetylenes, which are much less acidic than the aryl-acetylene derivatives.49 The catalyst also has a decisive influence on the activity in the polymerization of ring-substituted phenylacetylenes. In the case of the [Rh(tfb)(PPh3)2]+/iPrNH2 catalytic system, electron-withdrawing groups decrease the catalytic activity and renders living polymerization difficult.50 Interestingly, a positive correlation between the acidity of the ethynyl group, the catalytic activity and the polymer molar mass has been observed in the polymerization of ring-substituted phenylacetylene derivatives with para-substituted aromatic Schiff bases as pendant groups using the molecular catalyst [Rh(μ-OMe)(cod)]2 or Rh(I)/MCM41 supported catalysts, which suggests that the monomer acidity might play a substantial role also in the propagation step.51,52
The time-conversion profiles for the polymerization of monomers PAc, PAg and PAi, have enable the analysis of the influence of steric effects resulting from the substituent, as they all contain electron-donating alkyl groups at the para position (Fig. 6). PAc (R = p-Me) polymerized faster than PAg (R = p-Bu), as would be expected for the smaller methyl substituent. PAc achieved 33% conversion after 4 min, whereas PAg required 15 min to achieve 30% conversion. The polymerization of both alkynes was slower than that of phenylacetylene (PAa), which is consistent with the presence of electron-donating groups in the para position and the bulkiness of the n-butyl group. Unexpectedly, the polymerization of PAi (R = p-tBu) proceeded very quickly, despite having a very bulky substituent, achieving full conversion in just 30 min. In contrast, full conversion of PAg (R = p-Bu) took 95 min. It is worth noting that the spherical shape of tert-butyl compared to the tail shape of the flexible n-butyl substituent may facilitate the alkyne insertion throughout the propagation step of the polymerization process, resulting in faster polymerization rate. Also, the compactness plots representing the ratio between the hydrodynamic radius (rh) and radius of gyration (rg) versus elution volume,53 showed that PPAi (R = p-tBu) has a compact spherical shape with a much denser core (rh(v)/rg < 0.72) than PPAg (R = p-Bu) and PPAa (R = H) (rh(v)/rg < 0.77) (see the ESI†). The lower value of rh(v)/rg for PPAi, indicates a more compact polymer with a smaller volume, which is expected to facilitate the polymer propagation step. In this context, it should be noted that effective Rh(I) catalysts have been described for the polymerization of PAi (R = p-tBu),25,50 among which the catalyst [(η6-C6H5-BPh3)Rh(tfb)] stands out, achieving full conversion in 16 min using a [PA]o/[Rh] ratio of 500.54 However, the molar mass of the polymer, Mn = 6.3 × 104, was much lower than that obtained with our catalyst [Rh(nbd){κ2P,N-Ph2P(CH2)3NMe2}]+.
 | ||
| Fig. 6 Time-conversion plots for the polymerization of ring-substituted phenylacetylene derivatives. Reaction conditions: THF, 20 °C, [PA]o = 0.25 M, [PA]o/[Rh] = 100. | ||
On the other hand, ring-substituted phenylacetylene derivatives with an alkyl (–R) substituent polymerized faster than the corresponding alkoxy (–OR) derivatives, consistent with the strong electron-donating ability of the later. The observed order of activity was: PAc (R = p-Me) > PAe (R = p-OMe) > PAf (R = m-OMe). Similarly, PAg (R = p-Bu) polymerized faster than PAh (R = p-OBu), although the latter exhibited a very limited activity, reaching only 40% conversion in 60 min (see the ESI†). On the other hand, the slow polymerization rate of PAc at high conversions (Fig. 5) should be a consequence of the insolubility of PPAc, which precipitates in the reaction medium, making the polymerization more difficult.
The first-order kinetic plots for the polymerizations of PA derivatives showed a linear dependence up to almost quantitative conversion in most cases, indicating that the concentration of the propagating species does not change during the polymerization, which indicates the absence of significant termination reactions,55,56 as illustrated for PAg (R = p-Bu) in Fig. 7a. However, in some cases such as PAc (R = p-Me) and PAh (R = p-OBu), the line slightly deviates from the origin due to a relatively fast polymerization rate in the early stage of the reaction.57 This is particularly relevant in the case of PAa, for which a 40% conversion was achieved after 1 min, with a decrease in activity after 5 min. It should be noted that for PAb (R = p-F) and PAi (R = p-tBu) the plots deviate from linearity after reaching around 80% conversion due to the slowing down of the polymerization reaction (see the ESI†). Finally, a slight induction period of approximately 1 min was observed in the polymerization of the monomers PAe (R = p-OMe) (Fig. 7b) and PAf (R = m-OMe) for which negligible active species are present.
 | ||
| Fig. 7 Pseudo first-order kinetic plots for the polymerization of substituted phenylacetylene derivatives: (a) PAg (R = p-Bu) and (b) PAe (R = p-OMe). Reaction conditions: THF, T = 20 °C, [PA]o = 0.25 M, [PA]o/[Rh] = 100. | ||
Thermal properties of polymers
Thermal properties of the PPA polymers were investigated by differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA). The DSC thermograms (exo-down plots) showed an exothermic peak in the first heating cycle that was not detected in the subsequent scan. As no thermal decomposition was observed in the TGA curve within this temperature range (see below), this irreversible peak could be attributed to exothermic polymer reactions such as cis–trans isomerization or the cross-linking of polymer chains. This peak, centered between 160–170 °C, is similar to that observed in other PPA samples and was ascribed to cis–trans isomerization of the main chain structure.58 The isomerization temperature is not strongly influenced by either the nature or the position of the substituent, as it was observed slightly above 160 °C for all the PPA samples (Table 2), similar to poly(phenylacetylene). The exothermic peak is sharp in the case of PPAa (R = H), PPAb (R = p-F), PPAe (R = p-OMe) and PPAf (R = m-OMe), but this peak is broader for PPAg (R = p-Bu) and PPAi (R = p-tBu) (Fig. 8). This result suggests that polymers with larger groups have stiffer chains, making thermal geometric isomerization more difficult.59 | ||
| Fig. 8 DSC thermograms of polymer samples PPAf (R = m-OMe) and PPAg (R = p-Bu) in two consecutive heating/cooling cycles (10 °C min−1). | ||
| Polymer |
T
5%a (°C) |
T
onsetb (°C) |
T
maxc (°C) |
Other (°C)d | Exothermic procces (°C) |
|---|---|---|---|---|---|
| a T 5%: temperature for which the weight loss is 5%. b T onset: onset of the decomposition processes. c T max: temperature of the maximum in the weight loss rate. d Other maxima in the derivative thermogram. | |||||
| PPAb (R = p-F) | 273 | 286 | 374 | — | 167 |
| PPAe (R = p-OMe) | 297 | 325 | 424 | 369 | 163 |
| PPAf (R = m-OMe) | 304 | 325 | 408 | 368 | 161 |
| PPAg (R = p-Bu) | 314 | 351 | 432 | 393 | 161 |
| PPAi (R = p-tBu) | 338 | 383 | 430 | — | 165 |
Thermal degradation of the polymers also occurs in solution. 1H NMR monitoring of a solution of PPAe (R = p-OMe) in toluene-d8 showed evidence of polymer degradation after heating at 100 °C. In fact, a decrease in the intensity of the signals corresponding to the protons of the polymer main chain was observed after heating the solution for 10 min. In addition, analysis of the aromatic region of the spectrum recorded after heating at 100 °C for 90 min revealed the formation of a small amount of 1,3,5-triphenylbenzene (see the ESI†).60
The TGA curves of selected ring-substituted PPA polymers are shown in Fig. 9. The temperatures corresponding to 5% weight losses are in the range 270–340 °C (T5%). The most thermally stable polymer was PPAi (R = p-tBu), whose degradation temperature is 383 °C, while the least stable was PPAb (R = p-F) with a temperature of 286 °C (Tonset). On the other hand, polymer PPAg (R = p-Bu), also with a bulky substituent, showed a high decomposition temperature of 351 °C. Considering the temperature of the maximum weight loss rate (Tmax), corresponding to a turning point of the weight loss curve, the PPAs with bulkier substituents, PPAi (R = p-tBu) and PPAg (R = p-Bu), showed the highest temperatures, together with PPAe (R = p-OMe). Polymers PPAe–PPAg also exhibited local maxima at slightly lower temperatures (Table 2).
 | ||
| Fig. 9 TGA thermograms of PPA samples obtained from different monosubstituted phenylacetylenes. | ||
The thermal decomposition process produces a significant weight loss up to about 500 °C with slight weight losses at higher temperature. At temperatures below 600 °C polymers PPAi (R = p-tBu) and PPAe (R = p-OMe) showed the highest residue (24%) while polymer PPAg (R = p-Bu) was completely degraded at this temperature. The polymers degrade above 600 °C on air generating residues of less than 5% by weight. The thermal behavior of substituted PPAs is comparable to that exhibited by poly(phenylacetylene).35 However, bulky substituents have been found to improve the thermal stability of the polymers, as evidenced by the higher decomposition temperature.
Mechanistic considerations: the role of the hemilabile ligand
Studies of PA polymerization by rhodium(I) initiators have shown that the presence of an external base as co-catalyst, such as 4-(dimethylamino)pyridine (DMPA) or iPrNH2, often improves catalyst performance by increasing initiation efficiency22,50,61 or inhibiting catalyst deactivation.62 Mechanistic investigations on PA polymerization by the catalyst precursor [Rh(cod){κ2P,N-Ph2P(CH2)3NMe2}]+ led us to observe the alkynyl species [Rh(CC–Ph)(cod){κP-Ph2P(CH2)3NHMe2}]+ formed by the intramolecular proton transfer from a η2-alkyne ligand to the –NMe2 group, which acts as an internal base (Fig. 10a). In addition, mechanistic studies on PA polymerization by 2-diphenylphosphinopyridine-based rhodium(I) catalysts have allowed us to disclose the key role of rhodium-alkynyl species such as [Rh(CCPh)(cod)(κP-Ph2PPy)] in the polymerization reaction.23
 | ||
| Fig. 10 (a) Formation of a Rh-alkynyl species triggered by a hemilabile phosphine ligand. (b) Plausible mechanism for the polymerization of ring-substituted PAs. | ||
This cationic alkynyl intermediates may be involved in the initiation step, which likely entails the PA insertion into the Rh–alkynyl bond, through a four-membered transition state (not shown) to afford a stable rhodium-vinyl species responsible for the propagation step by successive PA coordination–insertion reactions with 2,1-regioselectivity (Fig. 10b). In this regard, related alkynyl species have been suggested to be the initiating species likely involved in the generation of stable rhodium–vinyl species, responsible for the propagation step.32,50,63 Theoretical studies by Morokuma et al. with catalyst [Rh(nbd)(CC–Ph)(PA)] have shown that PA insertion into the Rh–alkynyl bond is possible as initiation step. In fact, the energy barrier for the PA insertion into the Rh–alkynyl bond (initiation step) is almost 4 kcal mol−1 higher than the barrier for the insertion into the Rh–vinyl bond (propagation step), which explains the low initiation efficiency (0.6–2%) observed in the polymerization of ring-substituted phenylacetylene derivatives.64
The low initiation efficiencies for the polymerization of ring-substituted phenylacetylene derivatives suggest that the activation of the catalysts to the alkynyl active species (Fig. 10a) does not determine the differences in the polymerization activity. Therefore, the electronic effects imparted by the substituent seem to influence the chain propagation step, which is faster for phenylacetylene derivatives with electron-withdrawing substituents on the aromatic ring.
Conclusions
The cationic compound [Rh(nbd){κ2P,N-Ph2P(CH2)3NMe2}][BF4] efficiently catalyzes the polymerization of a series of para- and meta-substituted phenylacetylene derivatives with groups of different electronic and steric properties to afford highly stereoregular ring-substituted poly(phenylacetylene)s with a cis-transoidal configuration of very high molar mass, Mw in the range 3.1 × 105–4.8 × 106, and moderate dispersities. Megadalton PPAs have been obtained in the polymerization of phenylacetylene and 1-ethynyl-3-methoxybenzene (Mn ≈ 1.1 × 106). Notably, the polymerization of 1-ethynyl-4-methoxybenzene and 1-(tert-butyl)-4-ethynylbenzene yields ultra-high molecular weight PPAs with Mn of 1.70 × 106 and 2.72 × 106, respectively. SEC-MALS analysis of the ring-substituted PPAs evidences the formation of linear PPAs unlike poly(phenylacetylene) whose conformational plot is consistent with the presence of high molar mass branched material. A correlation between the electronic properties of the substituent and the polymerizing activity has been observed. Phenylacetylene derivatives with an electron-withdrawing substituent in para position polymerize faster than phenylacetylene, whereas derivatives with an electron-donating substituent polymerize more slowly. Since the initiation efficiency is very low for both types of ring-substituted alkynes (<2%), the difference in activity should be related to the electronic effects in the chain propagation step. Phenylacetylene derivatives with bulky long chain substituents in para position polymerize at a slower rate than phenylacetyene. In contrast, the polymerization of 1-(tert-butyl)-4-ethynylbenzene is very fast and gives an ultra-high molecular weight PPA.Author contributions
Marta Angoy: Investigation, methodology. M. Victoria Jiménez: Conceptualization, supervision, validation. Eugenio Vispe: Investigation, validation, visualization. Jesús J. Pérez-Torrente: Funding acquisition, conceptualization, supervision, writing – original draft, writing – review & editing.Data availability
Data available upon request from the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Spanish Ministry of Science and Innovation MCIN/AEI/10.13039/501100011033, under the Projects PID2019-103965GB-I00 and PID2022-137208NB-I00, and the Departamento de Educación, Ciencia y Universidades del Gobierno de Aragón (group E42_23R) is gratefully acknowledged.References
- R. M. Pankow and B. C. Thompson, The development of conjugated polymers as the cornerstone of organic electronics, Polymer, 2020, 207, 122874 CrossRef CAS.
- P. Palania and S. Karpagam, Conjugated polymers – a versatile platform for various photophysical, electrochemical and biomedical applications: a comprehensive review, New J. Chem., 2021, 45, 19182–19209 RSC.
- H. Lu, X. Li and Q. Lei, Conjugated Conductive Polymer Materials and its Applications: A Mini-Review, Front. Chem., 2021, 9, 732132 CrossRef CAS PubMed.
- J. W. Lam and B. Z. Tang, Functional polyacetylenes, Acc. Chem. Res., 2005, 38, 745–754 CrossRef CAS PubMed.
- T. Masuda, Substituted Polyacetylenes: Synthesis, Properties, and Functions, Polym. Rev., 2017, 57, 1–14 CrossRef CAS.
- L. Liu, Y. Zang, H. Jia, T. Aoki, T. Kaneko, S. Hadano, M. Teraguchi, M. Miyata, G. Zhang and T. Namikoshi, Helix-Sense-Selective Polymerization of Achiral Phenylacetylenes and Unique Properties of the Resulting Cis-cisoidal Polymers, Polym. Rev., 2017, 57, 89–118 CrossRef CAS.
- A. Xu, T. Masuda and A. Zhang, Stimuli-Responsive Polyacetylenes and Dendronized Poly(phenylacetylene)s, Polym. Rev., 2017, 57, 138–158 CrossRef CAS.
- R. Sakai, T. Satoh and T. Kakuchi, Polyacetylenes as Colorimetric and Fluorescent Chemosensor for Anions, Polym. Rev., 2017, 57, 159–174 CrossRef CAS.
- F. Freire, E. Quiñoá and R. Riguera, Supramolecular Assemblies from Poly(phenylacetylene)s, Chem. Rev., 2016, 116, 1242–1271 CrossRef CAS PubMed.
- D. T. Gentekos, R. J. Sifri and B. P. Fors, Controlling polymer properties through the shape of the molecular-weight distribution, Nat. Rev. Mater., 2019, 4, 761–774 CrossRef.
- K. Nomura, S. Pengoubol and W. Apisuk, Synthesis of ultrahigh molecular weight polymers by homopolymerisation of higher α-olefins catalysed by aryloxo-modified half-titanocenes, RSC Adv., 2016, 6, 16203–16207 RSC.
- A. Reyhani, S. Allison-Logan, H. Ranji-Burachaloo, T. G. McKenzie, G. Bryant and G. G. Qiao, Synthesis of ultra-high molecular weight polymers by controlled production of initiating radicals, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1922–1930 CrossRef CAS.
- L. Zhang, X. Ren, Y. Zhang and K. Zhang, Step-Growth Polymerization Method for Ultrahigh Molecular Weight Polymers, ACS Macro Lett., 2019, 8, 948–954 CrossRef CAS PubMed.
- K. Patel, S. H. Chikkali and S. Sivaram, Ultrahigh molecular weight polyethylene: Catalysis, structure, properties, processing and applications, Prog. Polym. Sci., 2020, 109, 101290 CrossRef CAS.
- J. Liu, J. W. Lam and B. Z. Tang, Acetylenic polymers: syntheses, structures, and functions, Chem. Rev., 2009, 109, 5799–5867 CrossRef CAS PubMed.
- M. Shiotsuki, F. Sanda and T. Masuda, Polymerization of substituted acetylenes and features of the formed polymers, Polym. Chem., 2011, 2, 1044–1058 RSC.
- M. A. Casado, A. Fazal and L. A. Oro, Rhodium-Catalyzed Polymerization of Phenylacetylene and its Derivatives, Arabian J. Sci. Eng., 2013, 38, 1631–1646 CrossRef CAS.
- J. Sedláček and H. Balcar, Substituted Polyacetylenes Prepared with Rh Catalysts: From Linear to Network-Type Conjugated Polymers, Polym. Rev., 2017, 57, 31–51 CrossRef.
- M. Isomura, Y. Misumi and T. Masuda, Living polymerization and block copolymerization of various ring-substituted phenylacetylenes by rhodium-based ternary catalyst, Polym. Bull., 2000, 45, 335–339 CrossRef CAS.
- J. Sedláček and J. Vohlídal, Controlled and living polymerizations induced with rhodium catalysts. A review, Collect. Czech. Chem. Commun., 2003, 68, 1745–1790 CrossRef.
- N. S. L. Tan and A. B. Lowe, Polymerizations Mediated by Well-Defined Rhodium Complexes, Angew. Chem., Int. Ed., 2020, 59, 5008–5021 CrossRef CAS PubMed.
- Y. Kishimoto, P. Eckerle, T. Miyatake, M. Kainosho, A. Ono, T. Ikariya and R. Noyori, Well-controlled polymerization of phenylacetylenes with organorhodium(I) complexes: Mechanism and structure of the polyenes, J. Am. Chem. Soc., 1999, 121, 12035–12044 CrossRef CAS.
- M. Angoy, M. V. Jiménez, F. J. Modrego, L. A. Oro, V. Passarelli and J. J. Pérez-Torrente, Mechanistic Investigation on the Polymerization of Phenylacetylene by 2-Diphenyl-phosphinopyridine Rhodium(I) Catalysts: Understanding the Role of the Cocatalyst and Alkynyl Intermediates, Organometallics, 2018, 37, 2778–2794 CrossRef CAS.
- I. Saeed, M. Shiotsuki and T. Masuda, Living Polymerization of Phenylacetylene with Tetrafluorobenzobarrelene Ligand-Containing Rhodium Catalyst Systems Featuring the Synthesis of High Molecular Weight Polymer, Macromolecules, 2006, 39, 8567–8573 CrossRef CAS.
- N. Onishi, M. Shiotsuki, T. Masuda, N. Sano and F. Sanda, Polymerization of Phenylacetylenes Using Rhodium Catalysts Coordinated by Norbornadiene Linked to a Phosphino or Amino Group, Organometallics, 2013, 32, 846–853 CrossRef CAS.
- N. S. L. Tan, P. V. Simpson, G. L. Nealon, A. N. Sobolev, P. Raiteri, M. Massi, M. I. Ogden and A. B. Lowe, Rhodium(I)-α-Phenylvinylfluorenyl Complexes: Synthesis, Characterization, and Evaluation as Initiators in the Stereospecific Polymerization of Phenylacetylene, Eur. J. Inorg. Chem., 2019, 592–601 CrossRef CAS.
- N. S. L. Tan, G. L. Nealon, J. M. Lynam, A. N. Sobolev, M. R. Rowless, M. I. Ogden, M. Massi and A. B. Lowe, A (2-(naphthalen-2-yl)phenyl)rhodium(I) complex formed by a proposed intramolecular 1,4-ortho-to-ortho′ Rh metal-atom migration and its efficacy as an initiator in the controlled stereospecific polymerisation of phenylacetylene, Dalton Trans., 2019, 48, 16437–16447 RSC.
- N. S. L. Tan, G. L. Nealon, G. F. Turner, S. A. Moggach, M. I. Ogden, M. Massi and A. B. Lowe, Rh(I)(2,5-norbornadiene)(biphenyl)(tris(4-fluorophenyl)phosphine): Synthesis, Characterization, and Application as an Initiator in the Stereoregular (Co)Polymerization of Phenylacetylenes, ACS Macro Lett., 2020, 9, 56–60 CrossRef PubMed.
- T. Taniguchi, T. Yoshida, K. Echizen, K. Takayama, T. Nishimura and K. Maeda, Facile and Versatile Synthesis of End-Functionalized Poly(phenylacetylene)s: A Multicomponent Catalytic System for Well-Controlled Living Polymerization of Phenylacetylenes, Angew. Chem., Int. Ed., 2020, 59, 8670–8680 CrossRef CAS PubMed.
- W. Yang, M. Tabata, S. Kobayashi, K. Yokota and A. Shimizu, Synthesis of Ultra-High-Molecular-Weight Aromatic Polyacetylenes with [Rh(norbornadiene)Cl]2-Triethylamine and Solvent-Induced Crystallization of the Obtained Amorphous Polyacetylenes, Polym. J., 1991, 23, 1135–1138 CrossRef CAS.
- T. Tang, S. J. Lu, G. Ahumada and C. W. Bielawski, Megadalton Macromolecules Made-to-Order in Minutes: A Highly Active Nanosphere Catalys for Preparing High-Molecular Weight Polymers, Macromolecules, 2022, 55, 9943–9950 CrossRef CAS.
- M. Angoy, M. V. Jiménez, P. García-Orduña, L. A. Oro, E. Vispe and J. J. Pérez-Torrente, Dinuclear Phosphine-Amido [Rh2(diene){μ-NH(CH2)3PPh2}2] Complexes as Efficient Catalyst Precursors for Phenylacetylene Polymerization, Organometallics, 2019, 38, 1991–2006 CrossRef CAS.
- M. V. Jiménez, J. J. Pérez-Torrente, M. I. Bartolomé, E. Vispe, F. J. Lahoz and L. A. Oro, Branched Poly(phenylacetylene), Macromolecules, 2010, 43, 6278–6283 CrossRef.
- M. Angoy, M. V. Jiménez, F. J. Lahoz, E. Vispe and J. J. Pérez-Torrente, Polymerization of phenylacetylene catalyzed by rhodium(I) complexes with N-functionalized N-heterocyclic carbene ligands, Polym. Chem., 2022, 13, 1411–1421 RSC.
- M. V. Jiménez, J. J. Pérez-Torrente, M. I. Bartolomé, E. Vispe, F. J. Lahoz and L. A. Oro, Cationic rhodium complexes with hemilabile phosphine ligands as polymerization catalyst for high molecular weight stereoregular poly(phenylacetylene), Macromolecules, 2009, 42, 8146–8156 CrossRef.
- M. V. Jiménez, J. J. Pérez-Torrente, M. I. Bartolomé and L. A. Oro, Convenient Methods for the Synthesis of a Library of Hemilabile Phosphines, Synthesis, 2009, 1916–1922 CrossRef.
- J. Sedlácek, J. Vohlídal and Z. Grubisic-Gallot, Molecular–weight determination of poly(phenylacetylene) by size–exclusion chromatography/low–angle laser light scattering. influence of polymer degradation, Makromol. Chem., Rapid Commun., 1993, 14, 51–53 CrossRef.
- V. Percec and J. G. G. Rudick, Independent electrocyclization and oxidative chain cleavage along the backbone of cis-poly(phenylacetylene), Macromolecules, 2005, 38, 7241–7250 CrossRef CAS.
- S. Podzimek, Light Scattering, Size Exclusion Chromatography and Asymmetric Flow Field Flow Fractionation, John Wiley and Sons, Hoboken, New Jersey, 2011, pp. 65–72 Search PubMed.
- A. Furlani, C. Napoletano, M. V. Russo and W. Feast, Stereoregular polyphenylacetylene, J. Polym. Bull., 1986, 16, 311–317 CAS.
- A. Furlani, C. Napoletano, M. V. Russo, A. Camus and N. J. Marsich, The influence of the ligands on the catalytic activity of a series of RhI complexes in reactions with phenylacetylene: Synthesis of stereoregular poly(phenyl) acetylene, J. Polym. Sci., Part A: Polym. Chem., 1989, 27, 75–86 CrossRef CAS.
- Y. Kishimoto, M. Itou, T. Miyatake, T. Ikariya and R. Noyori, Polymerization of monosubstituted acetylenes with a zwitterionic rhodium(I) complex, Rh+(2,5-norbornadiene)[η6-C6H5)B(C6H5)3], Macromolecules, 1995, 28, 6662–6666 CrossRef CAS.
- A. Miyasaka, T. Sone, Y. Mawatari, S. Setayesh, K. Müllen and M. Tabata, Poly[(para-halogenated)phenylacetylene]s Prepared with a [Rh(norbornadiene)Cl]2 Catalyst: Syntheses and Structure Elucidation, Macromol. Chem. Phys., 2006, 207, 1938–1944 CrossRef CAS.
- W. Yun-Hua and F. Tsai, Reusable Rhodium(I)/Cationic Bipyridyl-catalyzed Polymerization of Phenylacetylenes in Water under Aerobic Conditions, Chem. Lett., 2007, 36, 1492–1493 CrossRef.
- C. Cametti, P. Codastefano, R. D'Amato, A. Furlani and M. V. Russo, Static and dynamic light scattering measurements of polyphenylacetylene (PPA) in different organic solvents (tetrahydrofuran, toluene and chloroform), Synth. Met., 2000, 114, 173–179 CrossRef CAS.
- A. Escudero, R. Vilar, R. Salcedo and T. Ogawa, Effects of substituent groups and substituted benzenes on the polymerization of phenylacetylenes initiated by di-μ-pentafluorothiophenolatebis(1,5-cyclooctadiene)-rhodium(I), Eur. Polym. J., 1995, 31, 1135–1138 CrossRef CAS.
- J. O. Stoffer, D. R. Strait, D. L. Filger, E. T. Lloyd and C. Crain, A novel method for direct measurement of the pKa's of weakly acidic hydrocarbons, J. Org. Chem., 1978, 43, 1812–1813 CrossRef CAS.
- E. T. Lloyd, Part I: Thermodynamic acidities of substituted phenylacetylenes, Missouri University of Science and Technology, USA, 1976. https://scholarsmine.mst.edu/20masters_theses/5998 Search PubMed.
- O. Trhlíková, J. Zedník, H. Balcar, J. Brus and J. Sedláček, [Rh(cycloolefin)(acac)] complexes as catalysts of polymerization of aryl- and alkylacetylenes: Influence of cycloolefin ligand and reaction conditions, J. Mol. Catal. A: Chem., 2013, 378, 57–66 CrossRef.
- M. Shiotsuki, N. Onishi, F. Sanda and T. Masuda, Living polymerization of phenylacetylenes catalyzed by cationic rhodium complexes bearing tetrafluorobenzobarrelene, Polym. J., 2011, 43, 51–57 CrossRef CAS.
- H. Balcar, J. Sedláček, J. Zedník, V. Blechta, P. Kubát and J. Vohlídal, Polymerization of isomeric N-(4-substituted benzylidene)-4-ethynylanilines and 4-substituted N-(4- ethynylbenzylidene)anilines by transition metal catalysts: preparation and characterization of new substituted polyacetylenes with aromatic Schiff base type pendant groups, Polymer, 2001, 42, 6709–6721 CrossRef CAS.
- H. Balcar, J. Sedláček, J. Čejka and J. Vohlídal, MCM–41−Immobilized [Rh(cod)OCH3]2 Complex – A Hybrid Catalyst for the Polymerization of Phenylacetylene and Its Ring–Substituted Derivatives, Makromol. Chem., Rapid Commun., 2002, 23, 32–37 CrossRef CAS.
- A. K. Brewer and A. M. Striegel, Characterizing the Size, Shape, and Compactness of a Polydisperse Prolate Ellipsoidal Particle via Quadruple-Detector Hydrodynamic Chromatography, Analyst, 2011, 136, 515–519 RSC.
- N. Onishi, M. Shiotsuki, F. Sanda and T. Masuda, Polymerization of Phenylacetylenes with Rhodium Zwitterionic Complexes: Enhanced Catalytic Activity by π-Acidic Diene Ligands, Macromolecules, 2009, 42, 4071–4076 CrossRef CAS.
- Y. Misumi, K. Kanki, M. Miyake and T. Masuda, Living polymerization of phenylacetylene by rhodium-based ternary catalysts, (diene)Rh(I) complex/vinyllithium/phosphorus ligand. Effects of catalyst components, Macromol. Chem. Phys., 2000, 201, 2239–2244 CrossRef CAS.
- N. S. L. Tan, G. L. Nealon, S. A. Moggach, J. M. Lynam, M. I. Ogden, M. Massi and A. B. Lowe, (η4−Tetrafluoro-benzobarrelene)–η1−((tri-4-fluorophenyl)phosphine)–η1−(2-phenylphenyl)rhodium(I): A Catalyst for the Living Polymerization of Phenylacetylenes, Macromolecules, 2021, 54, 6191–6203 CrossRef.
- M. Shiotsuki, N. Onishi, F. Sanda and T. Masuda, Living Polymerization of Phenylacetylene Catalyzed by a Cationic Rh Complex Bearing Tetrafluorobenzobarrelene, Chem. Lett., 2010, 39, 244–245 CrossRef CAS.
- M. Goto, M. Minami, H. Sogawaa and F. Sanda, Reinvestigation of thermal isomerization of cis-stereoregulated poly(phenylacetylene) by spectroscopic study and DFT calculation, Polymer, 2021, 229, 124013 CrossRef CAS.
- Y. Fujita, Y. Misumi, M. Tabata and T. Masuda, Synthesis, geometric structure, and properties of poly(phenylacetylenes) with bulky para-substituents, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 3157–3163 CrossRef CAS.
- O. Trhlíková, J. Zedník, J. Vohlídal and J. Sedlácek, Molecular weight and configurational stability of poly(phenylacetylene) prepared with Rh catalyst, Polym. Degrad. Stab., 2011, 96, 1310–1320 CrossRef.
- Y. Kishimoto, T. Miyatake, T. Ikariya and R. Noyori, An efficient rhodium(I) initiator for stereospecific living polymerization of phenylacetylenes, Macromolecules, 1996, 29, 5054–5055 CrossRef CAS.
- H. Komatsu, Y. Suzuki and H. Yamazaki, Unprecedented rhodium-mediated tetramerization of bulky terminal alkynes leading to hydropentalenylrhodium complexes, Chem. Lett., 2001, 30, 998–999 CrossRef.
- Y. Tian, X. Li, J. Shi, B. Tonga and Y. Dong, Monomer-induced switching of stereoselectivity and limitation of chain growth in the polymerization of amine-containing para-substituted phenylacetylenes by [Rh(norbornadiene)Cl]2, Polym. Chem., 2017, 8, 5761–5768 RSC.
- Z. Ke, S. Abe, T. Ueno and K. Morokuma, Rh-catalyzed Polymerization of Phenylacetylene: Theoretical Studies of the Reaction Mechanism, Regioselectivity, and Stereoregularity, J. Am. Chem. Soc., 2011, 133, 792–7941 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, SEC-MALS chromatograms and DSC thermograms of PPAs. Kinetic data for the PA polymerization reactions. See DOI: https://doi.org/10.1039/d4py00259h |
| This journal is © The Royal Society of Chemistry 2024 |