
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phenoxy imine NOON complexes of heavy alkaline earth ions for the ring-opening polymerisation of cyclic esters†
Rebecca L.
Jones
 ,
Zoë R.
Turner
,
Jean-Charles
Buffet
and
Dermot
O'Hare
*
,
Zoë R.
Turner
,
Jean-Charles
Buffet
and
Dermot
O'Hare
*
Chemistry Research Laboratory, Department of Chemistry, University of Oxford, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: dermot.ohare@chem.ox.ac.uk
First published on 11th April 2024
Abstract
The polymerisation of lactide monomers by heavier alkaline earth complexes supported by a phenoxy-imine NOON ligand, DippLM(thf)x (DippL = [2,7-(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NDipp)-1,8-O-C10H4]2− where Dipp = 2,6-iPr-C6H3; M = Ca (1), Sr (2) and Ba (3)) is described. Complexes 1–3 all proved active initiators for the ROP of LA converting over 500 eq. at ambient temperatures. The dependency on monomer concentration was found to vary depending on the alkaline-earth metal used; calcium-based initiator 1 exhibited first-order dependency with respect to monomer in all cases, while there was a second order dependancy for the heavier strontium analogues. Addition of benzyl alcohol as a co-initiator resulted in an ∼8-fold and ∼6-fold increase in the observed rate for 1 and 2 respectively. In these systems, presence of the expected –OCH2Ph/–OH end groups, formed via the activated monomer pathway, was confirmed by 1H and 13C{1H} NMR spectra of the isolated polymers. Further investigations with complex 2 suggested that without addition of co-initiator, the ROP could proceed via an anionic mechanism in which the initiating step would be monomer deprotonation. In all cases, poor polymerisation control was observed. GPC analysis of the polymers indicated the presence of different initiating species, which may be due to the complex speciation of complexes 1–3 or the formation of a mixture of linear and cyclic PLA.
NDipp)-1,8-O-C10H4]2− where Dipp = 2,6-iPr-C6H3; M = Ca (1), Sr (2) and Ba (3)) is described. Complexes 1–3 all proved active initiators for the ROP of LA converting over 500 eq. at ambient temperatures. The dependency on monomer concentration was found to vary depending on the alkaline-earth metal used; calcium-based initiator 1 exhibited first-order dependency with respect to monomer in all cases, while there was a second order dependancy for the heavier strontium analogues. Addition of benzyl alcohol as a co-initiator resulted in an ∼8-fold and ∼6-fold increase in the observed rate for 1 and 2 respectively. In these systems, presence of the expected –OCH2Ph/–OH end groups, formed via the activated monomer pathway, was confirmed by 1H and 13C{1H} NMR spectra of the isolated polymers. Further investigations with complex 2 suggested that without addition of co-initiator, the ROP could proceed via an anionic mechanism in which the initiating step would be monomer deprotonation. In all cases, poor polymerisation control was observed. GPC analysis of the polymers indicated the presence of different initiating species, which may be due to the complex speciation of complexes 1–3 or the formation of a mixture of linear and cyclic PLA.
Introduction
Polylactide (PLA) is a biodegradable, aliphatic polyester that can be derived from renewable resources (corn, beets) thus making it a sustainable alternative to polyolefins.1 It also has relatively improved end of life options which include hydrolysis and subsequent enzyme-catalysed degradation into CO2 and H2O.2,3Recent life cycle analyses suggest that the formation of PLA results in reductions in both GHG emissions (∼40%) and non-renewable energy usage (∼25%) when compared to petrochemically derived plastics,4–6 though the true life cycle of all such polymers should be carefully considered in the quest for a more sustainable future and a circular economy.7
Polylactide is one of the most widely studied “green polymers” due to its already proven versatile applications in packaging, films, pharmaceuticals and even biomedical engineering (it has been utilised in 3D printing techniques to fabricate tissue scaffolds).28,10 As a result, its global production capacity has almost doubled, and it now accounts for 20.7% of all bioplastics.11 Market analysis reports suggest that this will continue to increase with an estimated compound annual growth rate of 21.4% from 2023 to 2030.9
To date, the most successful route for the production of high molecular weight PLA is the ring-opening polymerisation (ROP) of lactide (LA). This method also facilitates enhanced control over other polymer macromolecular properties including polydispersity, stereoselectivity and nature of end-groups.12
Previously reported initiators for this process include (single-site) metal complexes,13–23 organic species24–26 and even enzymes.27–29
The two most common mechanisms reported for the ROP of LA are the coordination–insertion and activated monomer pathways.15,30 The majority of neutral (single-site) metal-mediated polymerisations are thought to operate via coordination–insertion whereas the activated monomer route is more prevalent for cationic and organocatalytic systems.24,31 The anionic mechanism for the ROP of LA is less widely studied. It typically involves ionic, nucleophilic catalysts such as alkali metal alkyls/alkoxides.10,32–37 It is proposed to proceed via one of two initiation steps: monomer deprotonation or direct nucleophilic attack.32 The active anionic chain ends, produced in both cases, are commonly observed to cause unwanted racemisation and/or transesterification reactions. As a result, poor polymerisation control, as indicated by broad molecular weight distributions, is a characteristic feature of anionic ROP.33
In the context of lactide polymerisation, there have been numerous reports of well-defined bimetallic M2+ systems with N/O-donating ligands; the majority are magnesium-based and incorporate highly active amide/alkoxide initiating groups.38–40 Investigations of analogous heavier bis(group 2) systems are much less common due to their propensity to redistribute in solution.41 There are however, examples of monometallic Ca, Sr and Ba LA ROP catalysts; the coordination sphere of these complexes is typically filled with donating solvent molecules as opposed to dedicated initiating groups.13,42–44 This precludes polymerisation activity via the typical coordination–insertion and, in the absence of a co-initiator, activated monomer pathways.
Bimetallic calcium and strontium 2,4-di-tert-butyl phenolates (DBP) have been studied, by Mountford et al., as initiators for the ring-opening polymerisation of lactide.41 The tethered calcium system (Chart 1a) was capable of converting 89% of 100 eq. of rac-LA in 150 minutes.41 During the same time, 86% conversion was achieved with the dinuclear strontium catalyst (Chart 1b).41
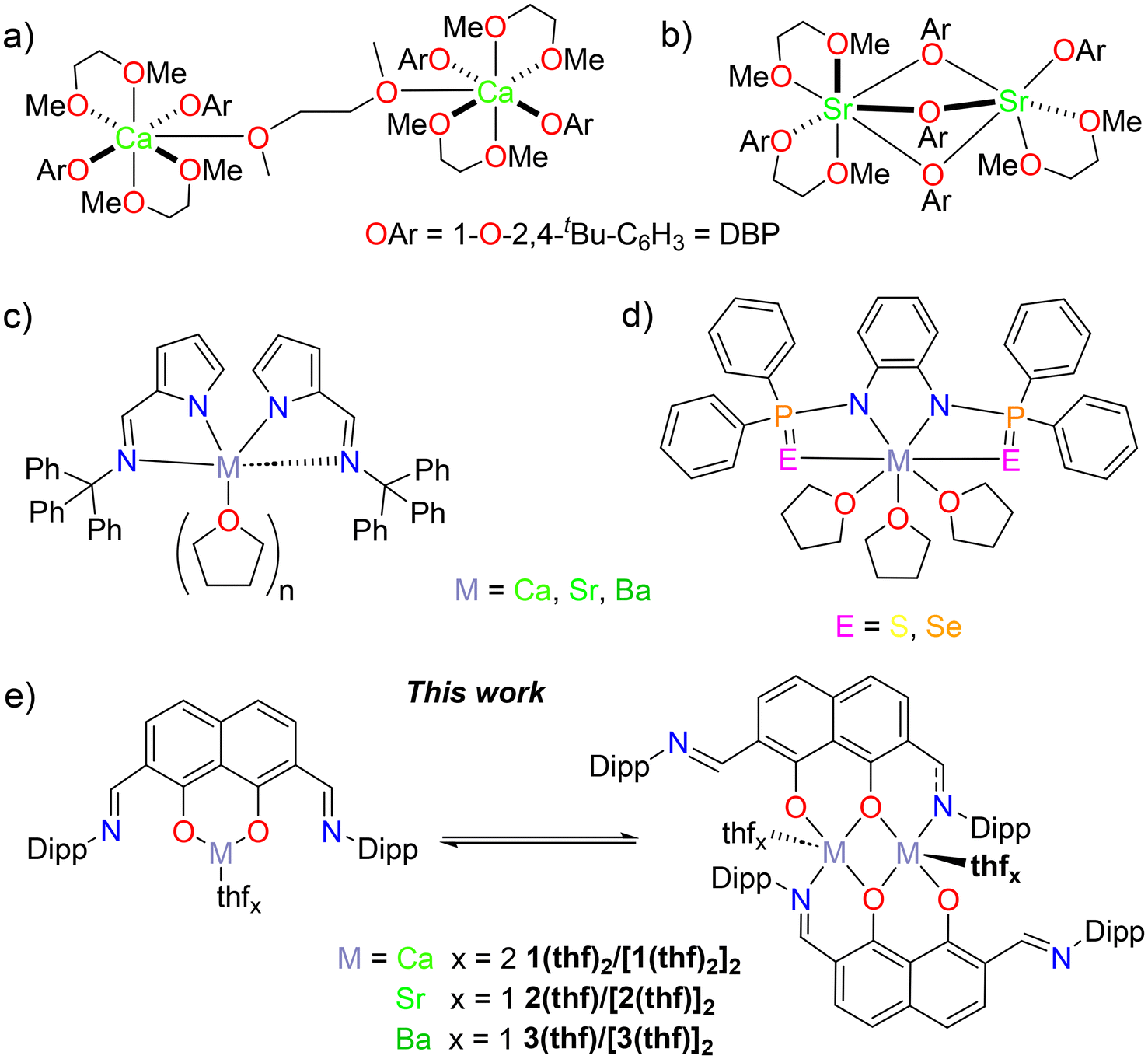 | ||
Chart 1 (a) and (b) Bimetallic calcium and strontium phenolates, reported by Mountford et al., for the ring-opening polymerisation of lactide;41 (c) and (d) previously reported monomeric, heavier alkaline-earth initiators for the ring-opening polymerisation of lactide,42–44 and (e) this work, DippLM(thf)x ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) [DippLM(thf)x]2 (M = Ca; 1(thf)2/[1(thf)2]2, M = Sr; 2(thf)/[2(thf)]2 and M = Ba; 3(thf)/[3(thf)]2).45 [DippLM(thf)x]2 (M = Ca; 1(thf)2/[1(thf)2]2, M = Sr; 2(thf)/[2(thf)]2 and M = Ba; 3(thf)/[3(thf)]2).45 | ||
A series of group two iminopyrrolyl compounds, with no dedicated initiating groups, have been reported as efficient catalysts for the ring-opening polymerisation of the cyclic ester ε-caprolactone (Chart 1c).42,43 The calcium analogue showed comparable activity to that of the magnesium variants.43 In contrast, the strontium and barium systems showed improved reactivity (600 eq. were converted within 5–10 minutes at 25 °C) without compromising polymerisation control (Mw/Mn = Đ < 1.59).43 The authors report that this is due to the larger ionic radii of the Sr2+ and Ba2+ cations increasing the availability of space for initiator-monomer interactions.43,44
A similar trend in polymerisation activity (Ca: 0.137 < Sr: 0.149 < Ba: 0.839 M−1 h−1; E = S; Đ < 1.60) was observed in related chalcogen containing systems (Chart 1d).44
We have recently reported the synthesis of alkaline earth complexes supported by a phenoxy-imine NOON ligand, DippL ([2,7-(CNDipp)-1,8-O-C10H4]2− where Dipp = 2,6-iPr-C6H3) and found the oxophilic M2+ cations to preferably bind to the central [O−,O−] coordination site.45 As such, a well-defined dimeric magnesium complex [DippLMg(thf)]2 was isolated. For calcium, strontium and barium, equilibrium mixtures of monomeric and dimeric species were formed; DippLM(thf)x [DippLM(thf)x]2 (M = Ca; 1(thf)2/[1(thf)2]2, M = Sr; 2(thf)/[2(thf)]2 and M = Ba; 3(thf)/[3(thf)]2); these equilibrium mixtures will be referred to as 1 (Ca), 2 (Sr) and 3 (Ba) hereafter for simplicity (Chart 1e).
In light of the paucity of heavier alkaline earth complexes utilised for the ring-opening polymerisation of lactide monomers; we report studies into the capabilities of heavier alkaline earth complexes 1–3 to polymerise lactide monomers, exploring the kinetics of the reaction for complex 2 in depth.
Results and discussion
Preliminary lactide polymerisation studies using DippLM(thf)x; M = Ca (1), Sr (2) and Ba (3)
The speciation of complexes 1–3 is not well-defined; these complexes are dimeric in the solid-state but exist as an equilibrium mixture of monomer and dimer in solution. In order to collect consistently comparable results, the stoichiometries in the following polymerisation studies were calculated with respect to the molecular weight of the monomeric, monometallic alkaline-earth systems. For simplicity, the initiators will be referred to as 1 (Ca), 2 (Sr) and 3 (Ba) throughout.Initial testing of complexes 1, 2 and 3 as initiators in the ring-opening polymerisation of lactide showed that all three systems were capable of polymerising 500 eq. of L-LA at ambient temperatures (Fig. S1† and Table 1).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[M]0 = 500:1 in benzene-d6 at 40 °Ca
:[M]0 = 500:1 in benzene-d6 at 40 °Ca
| M | t (h) | Conv.b (%) |
k
obsc (h−1 or M−1 h−1) |
R
2c |
M n (GPC)e | M n (calcd)f | Đ |
|---|---|---|---|---|---|---|---|
|
a Conditions: [L-LA]0:[M]0 = 500:1, [L-LA]0 = 0.5 M in 0.6 mL benzene-d6 at 40 °C.
b Measured by 1H NMR spectroscopic analyses; an average of polymerisation runs.
c First-order rate constant (kobs; h−1) and R2 were obtained from plots of ln([L-LA]0/[L-LA]t) vs. time.
d Second-order rate constant (kobs; M−1 h−1) and R2 were obtained from plots of 1/[L-LA]tvs. time.
e Determined by GPC in thf against PS standards using the appropriate Mark–Houwink corrections.46.
f Calculated Mn for PLA synthesised = (conv.(%) × [L-LA]0/[M]0) × 144.13.
|
|||||||
| 1 | 118 | 88 | 0.018 ± 0.02c | 0.999 | 55300 |
60705 |
1.59 |
| 2 | 5 | 66 | 0.82 ± 0.2d | 0.971 | 34400 |
54784 |
1.69 |
| 3 | 1 | 94 | — | — | 40600 |
67590 |
1.55 |
The activity of the catalysts followed the trend: Ba > Sr > Ca with 94%, 29% and 4% conversion after 1 h for 3, 2 and 1 respectively. Similar patterns have been widely reported and are rationalised by the larger cations being less sterically congested and having more labile bonding – both of which result in lower kinetic barriers towards monomer coordination/insertion.43,44,47,48 The reaction catalysed by 3 was considerably faster, and accurate acquisition of kinetic data was therefore not obtained (even at higher [L-LA]0/[Ba]0 ratios).
In contrast, plots of ln([L-LA]0/[L-LA]t) vs. time for the Ca (1) and Sr (2) initiated polymerisations, enabled experimental rate constants to be determined (Fig. S1†). For 1, the first-order rate constant was calculated to be 0.018 h−1; this value aligns with others reported for heavier alkaline-earth bis(diphenylphosphinothioic/selenoic) amine catalysts (kobs range from 0.15–0.43 h−1 for Ca; 0.15–1.21 h−1 for Sr and 0.906–1.54 h−1 for Ba for [LA]0/[M]0 = 200).44,48 The kinetic data for the analogous strontium system had a much poorer correlation with the first-order linear fit (R2 = 0.971). This suggested that the polymerisation may be second-order with respect to monomer concentration and thus the true rate constant is 0.82 M−1 h−1 in these conditions. The second-order nature of the Sr-catalysed polymerisations was confirmed, via plots of 1/[LA]tvs. time, in further experiments (Fig. 1).
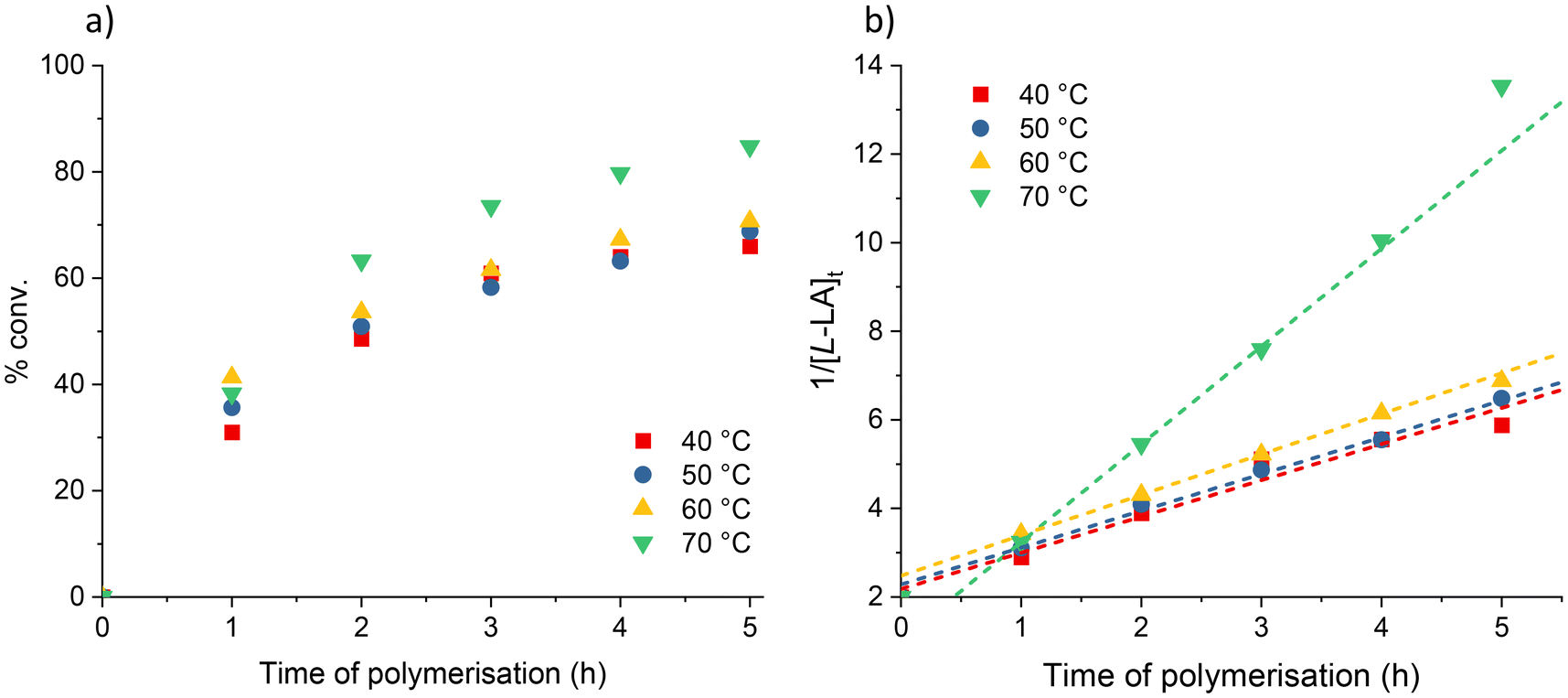 | ||
| Fig. 1 (a) Plots of percentage conversion vs. time. (b) Plots of 1/[L-LA]tvs. time for L-LA polymerisation using 2. Red squares: 40 °C (kobs = 0.82 M−1 h−1. R2 = 0.963); blue circles: 50 °C (kobs = 0.83 M−1 h−1. R2 = 0.993); yellow up triangles: 60 °C (kobs = 0.91 M−1 h−1. R2 = 0.992); green down triangles: 70 °C (kobs = 2.20 M−1 h−1. R2 = 0.999). Conditions: [L-LA]0:[Sr]0 = 500:1, [L-LA]0 = 0.5 M in 0.6 mL benzene-d6 at stated temperature. | ||
The polymerisations using 1, 2 and 3 all proceeded with moderate control; this was reflected in broad molecular weight distributions which were bimodal for 2 and 3 (Fig. S2†), and indicated by intermediate dispersity values (Đ = 1.55–1.69; Table 1). These values are comparable to those reported for other heavier alkaline-earth, monometallic initiators: Đ = 1.01–1.57.44,48 The bimodal nature of the molecular weight distributions can be rationalised by the presence of both linear and cyclic PLA and the presence of different initiating species as a consequence of catalyst speciation (vide infra).41,49,50 The experimental molecular weights however, are slightly lower than the previously documented range for the polymerisation of 500 eq. using Ca, Sr and Ba systems (67000–74100 gmol−1).44,48 There is a relatively good agreement between the experimental and calculated molecular weights, based on the monometallic initiating unit propagating a single chain, in the case of 1 (55300 vs. 60705 gmol−1). However, as the size of the cation increases, the agreement between these values becomes poorer (Δ% = 37 for 2 and Δ% = 40 for 3); this is most likely due to the larger ions being able to host multiple propagating chains. In fact, chain count calculations (eqn (S1)†) confirm that, on average, more than one polymer chain was growing per monometallic initiator in both the strontium (2, av. 1.59) and barium (3, av. 1.67) systems. The non-integer averages reflect the nature of the speciation of the active species.
For calcium and strontium DBP systems, the recorded molecular weights seemed consistent with one poly(lactide) chain per terminal Ae-DBP unit (4 for Ca: Mn,(GPC): 4050 vs. Mn,(calcd): 3200 gmol−1; 1 for Sr: Mn,(GPC): 13200 vs. Mn,(calcd): 12400 gmol−1) (Chart 1a and b respectively).41
Kinetic studies revealed that the dependency on monomer concentration varied with respect to which alkaline-earth metal was employed. In the case of the binary calcium-based system (1/BnOH), linear plots of ln([L-LA]0/[L-LA]t) vs. time indicated a first-order dependence on [L-LA] (kobs = 0.14 h−1–8-fold increase vs. no co-initiator; Fig. S7a†). Analogous plots for the strontium system (2) revealed non-linear relationships implying that the order with respect to monomer concentration exceeds one (Fig. S6†). In this case, a rare, yet not unprecedented, second-order dependency was confirmed via linear plots of 1/[L-LA]tvs. time (Fig. S7b†).41,52–58 This suggests that two monomers are required per active site for the reaction to proceed.54 Such a monomer-triggering polymerisation mechanism has been postulated by Ystenes and involves a monomer-insertion transition state which can only be by-passed if a second monomer molecule enters the coordination sphere.59
In the presence of BnOH, the second-order rate constant is ∼6-fold faster than in the absence of co-initiator (kobs = 5.30 vs. 0.82 M−1 h−1; Table 2). The same second-order dependency on monomer concentration and comparable experimental rate constant (kobs = 6.6 M−1 h−1) were observed for [(SalenMe)Mg(μ-OCH2Ph)]2 at 25 °C with [LA]0/[Mg]0 = 42 (SalenMe = [(2-O-3,5-tBu-C6H2CH=N)(2-OMe-C6H4CH=N)C6H10]−).53 A much higher second-order rate constant was recorded however for a trimetallic β-diketiminato (BDI) zinc alkoxide species [{(BDI-OMe)Zn(μ-OCH2Ph)}2Zn(μ-OCH2Ph)2] – kobs = 118.8 M−1 h−1 at 25 °C with [LA]0/[Zn]0 = 200 (BDI-OMe = [HC{C(Me)N(2-OMe-C6H4)}2]−).58
:[M]0:[BnOH]0 = 500:1:1 in benzene-d6 at 40 °Ca
| M | t (h) | Conv.b (%) |
k
obs
(h−1 or M−1 h−1) |
R
2c |
M
n
(GPC) |
M
n
(calcd) |
Đ |
|---|---|---|---|---|---|---|---|
|
a Conditions: [L-LA]0:[M]0:[BnOH]0 = 500:1:1, [L-LA]0 = 0.5 M in 0.6 mL benzene-d6 at 40 °C.
b Average reported; measured by 1H NMR spectroscopic analyses.
c First-order rate constant (kobs: h−1) and R2 were obtained from average plots of ln([L-LA]0/[L-LA]t) vs. time.
d Second-order rate constant (kobs: M−1 h−1) and R2 were obtained from average plots of 1/[L-LA]tvs. time.
e Determined by GPC in thf against PS standards using the appropriate Mark–Houwink corrections.46
f Calculated Mn for PLA synthesised = (conv.(%) × [L-LA]0/[M]0) × 144.13 + (Mw of end groups = OCH2Ph/–OH).
g Calculated assuming that two polymer chains propagate per metal centre.
|
|||||||
| 1 | 18 | 91 | 0.14 ± 0.003c | 0.992 | 20700 |
52809 |
1.23 |
| 2 | 2 | 85 | 0.78 ± 0.04c | 0.933 | 31200 |
30736g |
1.38 |
| 5.30 ± 0.03d | 0.999d | ||||||
When using initiator 2 with BnOH, there is a good agreement between experimental and theoretical molecular weights assuming that two polymer chains grow per monometallic initiator centre (31200 vs. 30736 gmol−1; Fig. S8† and Table 2).
Conversely, when catalyst 1 was employed with BnOH, a large disparity in experimental and theoretical molecular weights was recorded (20800 vs. 52809 gmol−1), assuming that the smaller metal centre still hosts only a single polymer chain. If benzyl alcohol has a significant effect on the speciation of the catalyst in this case, the agreement between these values can be anticipated to be poor. The reduction in experimental molecular weight may be due to free benzyl alcohol acting as a chain transfer agent.52,60 However, since this behaviour was not observed for 2/BnOH, it is alternatively postulated that the addition of co-initiator to 1 results in a change in mechanistic regime to allow two polymer chains to grow per monometallic unit (i.e. it now behaves more similarly to 2). If this is the case, the experimental and calculated values are more comparable: 20700 vs. 32790 gmol−1.
Further lactide polymerisation studies using 2
:[Sr]0 = 500:1 in benzene-d6 at stated temperaturea
| Temp. (°C) | Time (h) | Conv.b (%) |
k
obs
(M−1 h−1) |
R
2c |
M n (GPC)d | M n (calcd)g | Đ |
H:Lh |
||
|---|---|---|---|---|---|---|---|---|---|---|
| ie | iif | ie | iif | |||||||
|
a Conditions: [L-LA]0:[Sr]0 = 500:1, [L-LA]0 = 0.5 M in 0.6 mL benzene-d6 at stated temperature.
b Average reported; measured by 1H NMR spectroscopic analyses.
c Second-order rate constant (kobs) and R2 were obtained from average plots of 1/[L-LA]tvs. time.
d Determined by GPC in thf against PS standards using the appropriate Mark–Houwink corrections.46.
e Data corresponding to the higher molecular weight fraction.
f Data corresponding to the lower molecular weight fraction.
g Calculated Mn for PLA synthesised (assuming two polymer chains propagate per metal centre) = [(conv.(%) × [L-LA]0/[Sr]0) × 144.13]/2.
h Average higher:lower (H:L) molecular weight fraction ratio determined from area of peaks in average GPC trace.
|
||||||||||
| 40 | 5 | 66 | 0.82 ± 0.08 | 0.963 | 23800 |
2400 | 27392 |
1.52 | 1.39 | 85:15 |
| 50 | 5 | 75 | 0.83 ± 0.03 | 0.993 | 30000 |
2000 | 26405 |
1.68 | 1.61 | 60:40 |
| 60 | 5 | 71 | 0.91 ± 0.04 | 0.992 | 26500 |
1890 | 24560 |
1.59 | 1.67 | 58:42 |
| 70 | 5 | 85 | 2.20 ± 0.01 | 0.999 | 31800 |
1950 | 31478 |
1.56 | 1.70 | 56:44 |
The nature of the rate determining step, which is responsible for the polymerisation activation barriers, depends on the mechanism of the reaction. As this catalyst system contains no direct initiating group(s) or co-initiator, the conventional coordination–insertion or activated monomer mechanisms could be disregarded. It is therefore possible that the polymerisation propagates via an anionic mechanism which is initiated by deprotonation of the monomer by the aryloxyimine ligand (Fig. 2).
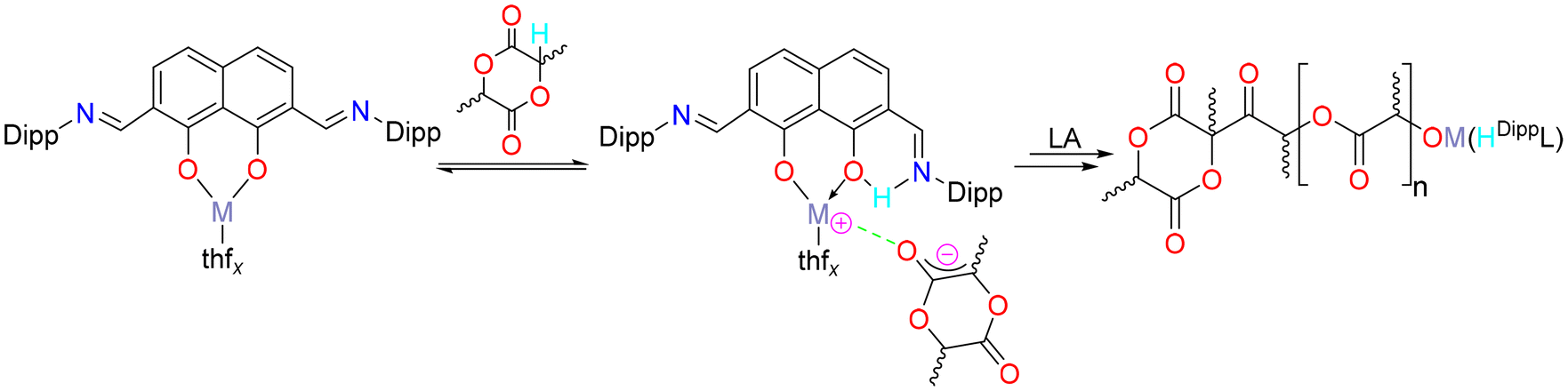 | ||
| Fig. 2 A possible anionic mechanism for the ring-opening polymerisation of lactide catalysed by complexes 1–3. | ||
MALDI-TOF mass spectrometric and 1H NMR spectroscopic end group analysis were carried out. Anionic mechanisms are typical for simple alkali metal initiators (nBuLi, ROK (R = Me, tBu, C6H5C(O)O)),32–37 but reports exist of sodium and potassium complexes supported by multidentate phenolate ligands initiating the polymerisation of lactide via the metal-phenolate bond in a coordination–insertion fashion. The anionic mechanism has also been shown for the magnesium initiator nBu2Mg.36,37 In this mechanism, the ligand scaffold deprotonates the monomer (R−M+ + LA-H → RH + M+LA−). In the case of initiator 2, residual H2DippL was detected in the 1H NMR spectrum of the polymerisation mixtures (Fig. S9†). It was considered that these pro-ligand signals were an indication of catalyst decomposition as it is difficult to categorically eliminate the possibility of a low concentration of protic impurities, however, they were observed not to increase over the course of the polymerisation and were therefore interpreted as being consistent with a possible anionic mechanism.
At all temperatures, the GPC traces of the PLA produced using complex 2 were bimodal; similar observations were made by Schubert and co-workers when employing strontium iso-propoxide as a catalyst,49 and also Mountford et al. for the bimetallic strontium DBP system.41 This bimodality may result from different initiating species in solution (i.e.2/[2]2) or, as suggested by Chisholm et al., the formation of different polymer structures.50 In their study of lactide polymerisation using an amido tin (Ph3SnNMe2) catalyst, the authors propose that the two GPC curves correspond to linear and cyclic PLA.50 This was rationalised by the fact that polymeric rings elute later than their straight chained analogues as a result of their smaller hydrodynamic volume.51
End group analysis does provide evidence to support the different polymer structure theory but, as the catalyst is not well-defined in solution (2/[2]2), the presence of different initiating species, or a combination of the two factors, likely contributes to the observed multimodality. As a result, the different GPC curves will be referred to as the higher/lower molecular weight fractions throughout (i and ii respectively in the Figures depicting GPC traces and results Tables).
The proportion of lower molecular weight products increases with temperature, as assessed by comparison of GPC peak areas (Fig. S10†). This implies poorer polymerisation control at higher temperatures, possibly leading to unwanted back-biting reactions. This is reflected in the increased molar-mass dispersity values (Đ) for peak ii with temperature: 1.39, 1.59, 1.67 and 1.70 for 40, 50, 60 and 70 °C respectively (Table 3). Additionally, the molecular weights of the heavier fraction are larger than the theoretical values (e.g. at 70 °C – Mn,(GPC): 31800 vs. Mn,(calcd): 31478 gmol−1) which assume 100% of monomers are incorporated into two propagating chains per metal centre. This suggests that not all metal centres are active during polymerisation.
:[Mg] = 42, in chloroform-d1 at 20 °C) and [Sr2(DBP)(μ-DBP)3(DME)3] (kobs = 2.28 M−1 h−1 for [rac-LA]:[Sr]:[BnOH] = 400:1:2.5 in thf at room temperature).8 In contrast to these two examples, catalyst 2 did not require the use of a co-initiator/pre-formed alkoxide initiating group to achieve relatively high polymerisation rates.
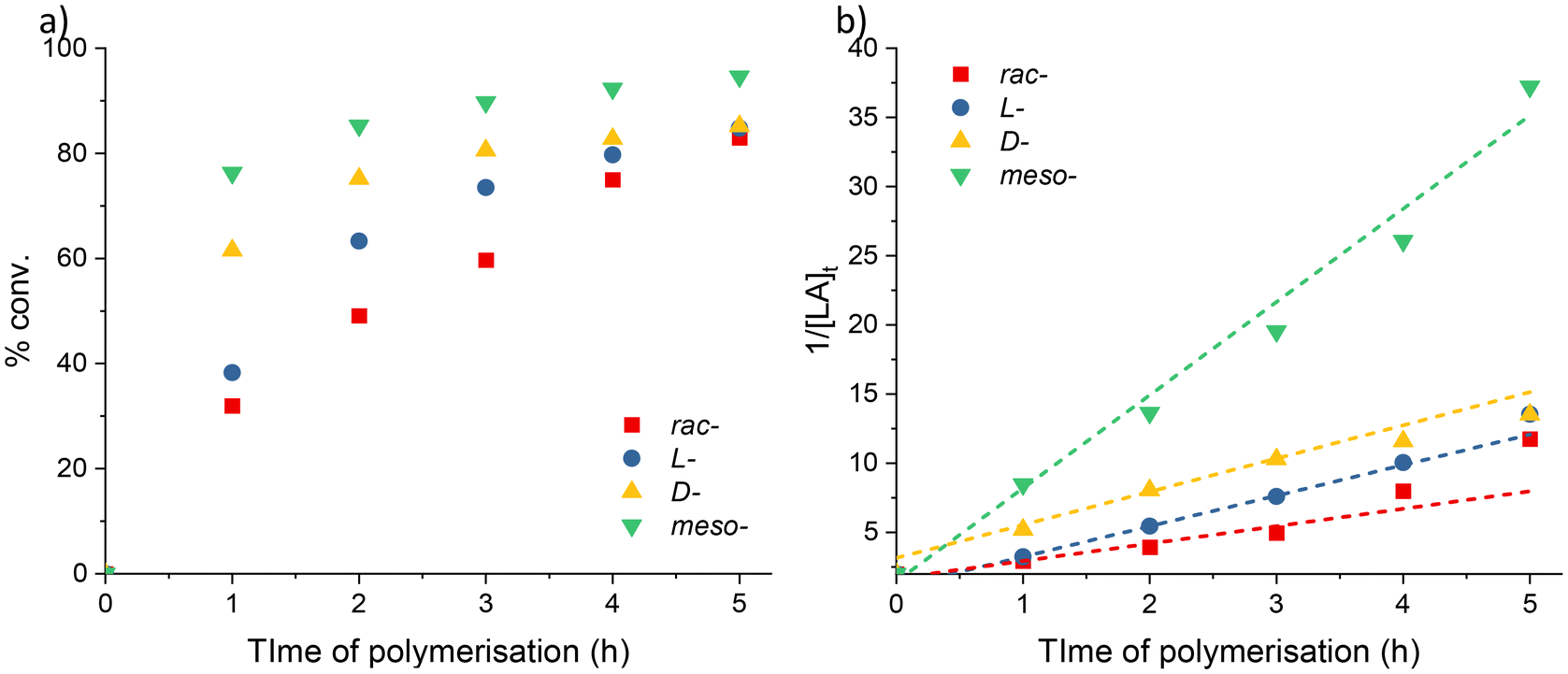 | ||
| Fig. 3 (a) Plots of percentage conversion vs. time. (b) Plots of 1/[LA]tvs. time for LA polymerisation using 2. Red squares: rac-LA (kobs = 1.30 M−1 h−1. R2 = 0.961); blue circles: L-LA (kobs = 2.20 M−1 h−1. R2 = 0.999); yellow up triangles: D-LA (kobs = 2.40 M−1 h−1. R2 = 0.982); green down triangles: meso-LA (kobs = 6.70 M−1 h−1. R2 = 0.987). Conditions: [LA]0:[Sr]0 = 500:1, [LA]0 = 0.5 M in 0.6 mL benzene-d6 at 70 °C. | ||
:[Sr]0 = 500:1 in benzene-d6 at 70 °Ca
| LA | Time (h) | Conv.b (%) |
k
obs
(M−1 h−1) |
R
2c |
M n (GPC)d | M n (calcd)g | Đ |
H:Lh |
||
|---|---|---|---|---|---|---|---|---|---|---|
| ie | iif | ie | iif | |||||||
|
a Conditions: [LA]0:[Sr]0 = 500:1, [LA]0 = 0.5 M in 0.6 mL benzene-d6 at 70 °C.
b Average reported; measured by 1H NMR spectroscopic analyses.
c Second-order rate constant (kobs) and R2 were obtained from average plots of 1/[LA]tvs. time.
d Determined by GPC in THF against PS standards using the appropriate Mark–Houwink corrections.46.
e Data corresponding to the higher molecular weight fraction.
f Data corresponding to the lower molecular weight fraction.
g Calculated Mn for PLA synthesised (assuming two polymer chains propagate per metal centre) = [(conv.(%) × [LA]0/[Sr]0) × 144.13]/2.
h Average higher:lower (H:L) molecular weight fraction ratio determined from area of peaks in average GPC trace.
|
||||||||||
| rac- | 5 | 83 | 1.30 ± 0.2 | 0.961 | 20900 |
1630 | 30177 |
1.59 | 1.54 | 44:56 |
| L- | 5 | 85 | 2.20 ± 0.01 | 0.999 | 31800 |
1950 | 31478 |
1.52 | 1.70 | 56:44 |
| D- | 5 | 85 | 2.40 ± 0.2 | 0.982 | 27000 |
1860 | 30487 |
1.63 | 1.56 | 58:42 |
| meso- | 5 | 95 | 6.70 ± 0.4 | 0.987 | 19300 |
1250 | 34231 |
1.46 | 1.73 | 33:67 |
Mixtures of higher and lower molecular weight fractions of PLA were formed with all the different isomers as highlighted by the observed bimodal GPC traces (Fig. S11a†). The proportion of lower molecular weight products formed during the reaction was higher for rac- and meso- vs. L- and D- (av. 62% vs. 43%); this implies that the formation of racemic (–RS– or –SR–) linkages in the polymer chain facilitated increased transesterification behaviour.
Homonuclear decoupled 1H{1H} NMR spectra of the polymeric products were used to determine their microstructure (Fig. S11b and S12†). The results showed that atactic PLA (Pr = 0.48) is formed from rac-LA. The lack of stereocontrol in complex 2 is most likely due to insufficient (directed) shielding of the metal centre. When meso-LA is polymerised, the resultant PLA is syndiotactic enriched (Pr = 0.74). This indicates that the polymerisations catalysed by 2 favour racemic enchainment.62 PLA with comparable –SRSR-microstructures has previously been synthesised by Coates and co-workers using a chiral, BINAP-based aluminium calalyst.63,64 Epimerisation was detected in the polymerisations of L- and D-lactide. This lack of reaction control was confirmed by the observation of multiple resonances in the methine region of the polymer 1H{1H} NMR spectra (Fig. S11b†), and is known to affect the physical properties (i.e. lower the melting point) of the resultant PLA.65,66
Initiator concentration dependency.
In order to determine the kinetic order dependence on catalyst concentration, polymerisations of L-LA with 2 were carried out at 70 °C using various monomer to catalyst ratios. The concentration of L-LA was held constant at 0.5 M while that of the catalyst was varied to provide the ratio of [L-LA]0:[Sr]0 = 500, 750 and 1000. In all cases, linear plots of 1/[L-LA]tvs. time were observed, reflecting the second-order dependence on monomer concentration (Fig. S13†). The observed second-order rate constants were found to increase with initiator concentration (kobs = 1000:1:1.10 < 750:1:1.90 < 500:1:2.20 M−1 h−1). This is due to the higher catalyst concentration having a greater number of active sites present during the polymerisation.67,68
At all ratios, the molecular weights of the heavier fraction is consistent (31800–32800 gmol−1; Δ% = 3; Table S1†) suggesting that the number of chain transfer events increases with the [L-LA]0/[Sr]0 ratio. This is most likely due to the longer reaction times resulting in more opportunities for these side reactions to occur, and is highlighted by the increasing of the higher molecular weight molar-mass dispersity values (Đ = 1.52, 1.60, 1.73 for 500:1, 750:1 and 1000:1 respectively; Fig. S14†). In contrast, the results show that higher catalyst concentrations, and subsequent higher reaction rates, favour (possible) back-biting transesterifications; this is reflected in the higher proportion of lower molecular weight PLA being produced at smaller [L-LA]0/[Sr]0 ratios (500:1 = 56:44; 750:1 = 63:37; 1000:1 = 67:33).
A linear plot of −ln(kobs) vs. −ln[Sr]0 determined the order with respect to catalyst to be 0.96 ± 0.4 (Fig. S15a†). This value and its associated error, imply a first-order dependency on catalyst concentration. Assuming this remains constant throughout the polymerisation, the following rate law can be derived for the ROP of LA catalysed by 2:
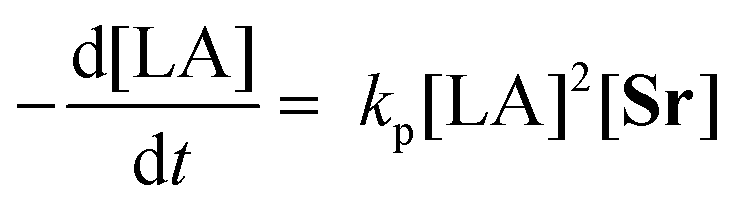 | (1) |
The propagation rate constant (kp), was calculated via a plot of kobsvs. [Sr]0, to be 2025 ± 970 M−1 h−1 (Fig. S15b†). This is comparable to that of a magnesium benzyloxide complex, [(SalenMe)Mg(μ-OCH2Ph)]2, that follows the same rate equation (kp = 1711 M−2 h−1).53 It is, however, much lower than the values reported for bimetallic calcium and strontium phenolates in the presence of a BnNH2 co-initiator (kp = 276 × 107 M−4.5 h−1 and 156 × 107 M−4.5 h−1 respectively).41
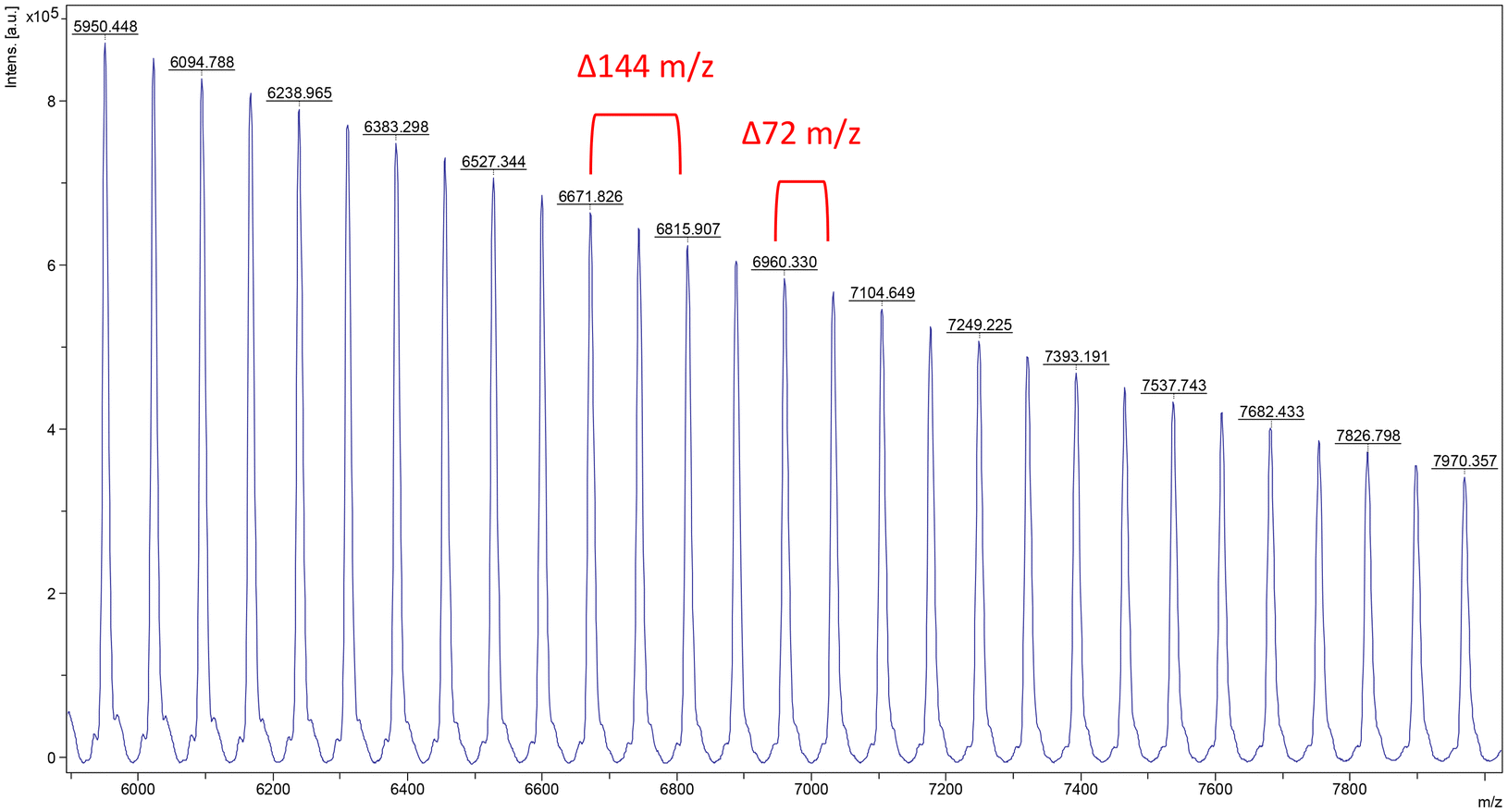 | ||
| Fig. 4 Expanded MALDI-TOF mass (m/z) spectrum of PLA produced using the 2 system. Conditions: [L-LA]0:[Sr]0 = 50:1, [L-LA]0 = 0.5 M, 0.6 mL benzene-d6, 40 °C. | ||
In all cases, the 1H NMR spectra of the resultant polymers revealed broad signals (Fig. S16†). It is postulated that this could be the result of the overlapping of resonances associated with different repeating structures (i.e. linear and cyclic).69 The carbon signals of the same lactide-derived end-group appear as shoulders to those of the main polymer chain, though we note that the process of epimerisation could also result in these features being observed (Fig. S17†).
Conclusions
Complexes 1–3 all proved active initiators for the ROP of LA converting over 500 eq. at ambient temperatures. The dependency on monomer concentration was found to vary depending on the alkaline-earth metal used. Calcium (1) exhibited first-order dependency with respect to monomer in all cases; the proposed number of metal chains propagating per monomeric initiator molecule however, depended on whether a co-initiator was added. Under all conditions, the strontium system (2) displayed second-order monomer dependency, via linear plots of 1/[LA]t, with two chains growing per monometallic initiator (confirmed by chain count calculations). Although kinetics for the barium complex 3 were too rapid for quantitative analysis, the propagation of more than one polymer chain per metal centre is also likely in this case.Addition of benzyl alcohol as a co-initiator resulted in a ∼8-fold and ∼6-fold increase in the observed rate for 1 (0.0018 h−1vs. 0.14 h−1) and 2 (0.82 M−1 h−1vs. 5.3 M−1 h−1) respectively. In these systems, presence of the expected –OCH2Ph/–OH end groups, formed via the activated monomer pathway, were confirmed by 1H and 13C{1H} NMR spectra of the isolated polymers.
Further investigations with complex 2 demonstrated that without addition of co-initiator, poor polymerisation control was observed. This was highlighted by broad, bimodal molecular weight distributions (1.23 < Đ < 1.73) and 72 m/z split of the peaks in the MALDI-TOF mass spectra of the isolated polymers.
The separate peaks in the GPC traces indicated the presence of different initiating species and/or the formation of a mixture of linear and cyclic PLA.50 It is proposed that the broad signals present in the polymer 1H and 13C{1H} NMR spectra confirm the formation of the different polymeric structures. It remains inconclusive however, as to whether this is the sole cause of the GPC bimodality because the ill-defined nature of the catalyst (2/[2]2) in solution may also result in a variety of simultaneous initiating species. The formation of lower molecular weight cyclic polylactide, was found to be favoured by higher reaction temperatures, lower [L-LA]0/[Sr]0 ratios and the formation of racemic-linkages within the polymer chain. Under any of the tested conditions however, the exclusive formation of either the higher or lower molecular weight species was not observed.50
Variation of initiator concentration revealed a first-order dependency on catalyst concentration; the overall rate law for the ROP using system 2 is therefore described by eqn (1).
Complexes 1–3 add to the exploration of heavier alkaline earths for the ring-opening polymerisation of lactide monomers, and were found to be active initiators. Unfortunately, the complex speciation of the NOON system does not lend itself well to the synthesis of polyesters with good control. As a result, we are currently exploring related, well-defined NON systems as initiators.
Experimental details
Materials and methods
Author contributions
R.L.J: investigation, formal analysis, writing – original draft, review and editing. Z.R.T: writing – original draft, review and editing, supervision, project administration. J.-C. B. supervision, writing – review and editing, project administration. D. O. H. – supervision, project administration, funding acquisition, resources, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All authors would like to thank SCG Chemicals Co. Ltd (Thailand) for funding.References
- C. K. Williams, N. R. Brooks, M. A. Hillmyer and W. B. Tolman, Chem. Commun., 2002, 2132–2133 RSC.
- R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef CAS.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS PubMed.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
- W. J. Groot and T. Borén, Int. J. Life Cycle Assess., 2010, 15, 970–984 CrossRef CAS.
- M. F. Cosate de Andrade, P. M. S. Souza, O. Cavalett and A. R. Morales, J. Polym. Environ., 2016, 24, 372–384 CrossRef CAS.
- T. Keijer, V. Bakker and J. C. Slootweg, Nat. Chem., 2019, 11, 190–195 CrossRef CAS PubMed.
- J. Wu, T.-L. Yu, C.-T. Chen and C.-C. Lin, Coord. Chem. Rev., 2006, 250, 602–626 CrossRef CAS.
- Grand-View-Research, https://www.grandviewresearch.com/industry-analysis/polylactic-acid-pla-market, (accessed 20/06/2023).
- P. Dubois, O. Coulembier and J. M. Raquez, Handbook of Ring–Opening Polymerization, Wiley, 1st edn, 2009 Search PubMed.
- European-Bioplastics, https://docs.european-bioplastics.org/publications/EUBP_Facts_and_figures.pdf, (accessed 20/06/2023).
- E. Balla, V. Daniilidis, G. Karlioti, T. Kalamas, M. Stefanidou, N. D. Bikiaris, A. Vlachopoulos, I. Koumentakou and D. N. Bikiaris, Polymers, 2021, 13, 1822 CrossRef CAS PubMed.
- B. J. O'Keefe, M. A. Hillmyer and W. B. Tolman, J. Chem. Soc., Dalton Trans., 2001, 2215–2224, 10.1039/B104197P.
- R. H. Platel, L. M. Hodgson and C. K. Williams, Polym. Rev., 2008, 48, 11–63 CrossRef CAS.
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC.
- P. J. Dijkstra, H. Du and J. Feijen, Polym. Chem., 2011, 2, 520–527 RSC.
- J.-C. Buffet and J. Okuda, Polym. Chem., 2011, 2, 2758–2763 RSC.
- N. Ajellal, J.-F. Carpentier, C. Guillaume, S. M. Guillaume, M. Helou, V. Poirier, Y. Sarazin and A. Trifonov, Dalton Trans., 2010, 39, 8363–8376 RSC.
- A. B. Kremer and P. Mehrkhodavandi, Coord. Chem. Rev., 2019, 380, 35–57 CrossRef CAS.
- H. A. Brown and R. M. Waymouth, Acc. Chem. Res., 2013, 46, 2585–2596 CrossRef CAS PubMed.
- Y. Sarazin and J.-F. Carpentier, Chem. Rev., 2015, 115, 3564–3614 CrossRef CAS PubMed.
- D. M. Lyubov, A. O. Tolpygin and A. A. Trifonov, Coord. Chem. Rev., 2019, 392, 83–145 CrossRef CAS.
- E. Fazekas, P. A. Lowy, M. Abdul Rahman, A. Lykkeberg, Y. Zhou, R. Chambenahalli and J. A. Garden, Chem. Soc. Rev., 2022, 51, 8793–8814 RSC.
- E. F. Connor, G. W. Nyce, M. Myers, A. Möck and J. L. Hedrick, J. Am. Chem. Soc., 2002, 124, 914–915 CrossRef CAS PubMed.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS PubMed.
- M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093–2107 CrossRef CAS.
- J. Engel, A. Cordellier, L. Huang and S. Kara, ChemCatChem, 2019, 11, 4983–4997 CrossRef CAS.
- H. Ö. Düşkünkorur, A. Bégué, E. Pollet, V. Phalip, Y. Güvenilir and L. Avérous, J. Mol. Catal. B: Enzym., 2015, 115, 20–28 CrossRef.
- H. Zhao, G. A. Nathaniel and P. C. Merenini, RSC Adv., 2017, 7, 48639–48648 RSC.
- J.-F. Carpentier and Y. Sarazin, in Alkaline-Earth Metal Compounds: Oddities and Applications, ed. S. Harder, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, pp. 141–189 Search PubMed.
- H. R. Kricheldorf, I. Kreiser-Saunders, C. Jürgens and D. Wolter, Macromol. Symp., 1996, 103, 85–102 CrossRef CAS.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed.
- A. Bhaw-Luximon, D. Jhurry, N. Spassky, S. Pensec and J. Belleney, Polymer, 2001, 42, 9651–9656 CrossRef CAS.
- H. R. Kricheldorf and I. Kreiser-Saunders, Makromol. Chem., 1990, 191, 1057–1066 CrossRef CAS.
- Z. Jedliński, W. Wałach, P. Kurcok and G. y. Adamus, Makromol. Chem., 1991, 192, 2051–2057 CrossRef.
- H. R. Kricheldorf and S.-R. Lee, Polymer, 1995, 36, 2995–3003 CrossRef CAS.
- J. Kasperczyk and M. Bero, Polymer, 2000, 41, 391–395 CrossRef CAS.
- S. Range, D. F.-J. Piesik and S. Harder, Eur. J. Inorg. Chem., 2008, 3442–3451 CrossRef CAS.
- J. Wu, Y.-Z. Chen, W.-C. Hung and C.-C. Lin, Organometallics, 2008, 27, 4970–4978 CrossRef CAS.
- A. Pilone, M. Lamberti, M. Mazzeo, S. Milione and C. Pellecchia, Dalton Trans., 2013, 42, 13036–13047 RSC.
- L. Clark, G. B. Deacon, C. M. Forsyth, P. C. Junk, P. Mountford, J. P. Townley and J. Wang, Dalton Trans., 2013, 42, 9294–9312 RSC.
- J. Bhattacharjee, A. Sarkar and T. K. Panda, Chem. Rec., 2021, 21, 1898–1911 CrossRef CAS PubMed.
- R. K. Kottalanka, A. Harinath, S. Rej and T. K. Panda, Dalton Trans., 2015, 44, 19865–19879 RSC.
- J. Bhattacharjee, A. Harinath, H. P. Nayek, A. Sarkar and T. K. Panda, Chem. – Eur. J., 2017, 23, 9319–9331 CrossRef CAS PubMed.
- R. L. Jones, Z. R. Turner, J.-C. Buffet and D. O’Hare, Organometallics, 2024, 43(3), 414–426 CrossRef CAS , Accepted.
- J. R. Dorgan, J. Janzen, D. M. Knauss, S. B. Hait, B. R. Limoges and M. H. Hutchinson, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 3100–3111 CrossRef CAS.
- R. K. Kottalanka, A. Harinath and T. K. Panda, RSC Adv., 2015, 5, 37755–37767 RSC.
- A. Harinath, J. Bhattacharjee, A. Sarkar, H. P. Nayek and T. K. Panda, Inorg. Chem., 2018, 57, 2503–2516 CrossRef CAS PubMed.
- D. Bandelli, C. Weber and U. S. Schubert, Macromol. Rapid Commun., 2019, 40, 1900306 CrossRef CAS PubMed.
- M. H. Chisholm and E. E. Delbridge, New J. Chem., 2003, 27, 1167–1176 RSC.
- P. Wongmahasirikun, P. Prom-on, P. Sangtrirutnugul, P. Kongsaeree and K. Phomphrai, Dalton Trans., 2015, 44, 12357–12364 RSC.
- J. V. Lamb, J.-C. Buffet, J. E. Matley, C. M. R. Wright, Z. R. Turner and D. O'Hare, Dalton Trans., 2019, 48, 2510–2520 RSC.
- J.-C. Wu, B.-H. Huang, M.-L. Hsueh, S.-L. Lai and C.-C. Lin, Polymer, 2005, 46, 9784–9792 CrossRef CAS.
- P. I. Binda and E. E. Delbridge, Dalton Trans., 2007, 4685–4692 RSC.
- M. P. Blake, A. D. Schwarz and P. Mountford, Organometallics, 2011, 30, 1202–1214 CrossRef CAS.
- T.-P.-A. Cao, A. Buchard, X. F. Le Goff, A. Auffrant and C. K. Williams, Inorg. Chem., 2012, 51, 2157–2169 CrossRef CAS PubMed.
- L. Clark, M. G. Cushion, H. E. Dyer, A. D. Schwarz, R. Duchateau and P. Mountford, Chem. Commun., 2010, 46, 273–275 RSC.
- H.-Y. Chen, B.-H. Huang and C.-C. Lin, Macromolecules, 2005, 38, 5400–5405 CrossRef CAS.
- M. Ystenes, J. Catal., 1991, 129, 383–401 CrossRef CAS.
- N. Diteepeng, J.-C. Buffet, Z. R. Turner and D. O'Hare, Dalton Trans., 2019, 48, 16099–16107 RSC.
- M. H. Chisholm, N. W. Eilerts, J. C. Huffman, S. S. Iyer, M. Pacold and K. Phomphrai, J. Am. Chem. Soc., 2000, 122, 11845–11854 CrossRef CAS.
- B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229–3238 CrossRef CAS PubMed.
- T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 1999, 121, 4072–4073 CrossRef CAS.
- M. C. D'Alterio, C. De Rosa and G. Talarico, Chem. Commun., 2021, 57, 1611–1614 RSC.
- N. Nomura, J. Hasegawa and R. Ishii, Macromolecules, 2009, 42, 4907–4909 CrossRef CAS.
- C. Goonesinghe, H.-J. Jung, H. Roshandel, C. Diaz, H. A. Baalbaki, K. Nyamayaro, M. Ezhova, K. Hosseini and P. Mehrkhodavandi, ACS Catal., 2022, 12, 7677–7686 CrossRef CAS.
- Z. R. Turner, J.-C. Buffet and D. O'Hare, Organometallics, 2014, 33, 3891–3903 CrossRef CAS.
- R. Dattatray Karande, V. K. Abitha, A. Vasudeo Rane and R. K. Mishra, J. Mater. Sci. Eng. Adv. Tech., 2015, 12, 1–37 Search PubMed.
- C. Rentero, J. Damián, A. Medel, M. Fernández-Millán, Y. Rusconi, G. Talarico, T. Cuenca, V. Sessini and M. E. G. Mosquera, Polymers, 2022, 14, 2982 CrossRef CAS PubMed.
- Z. R. Turner, J. V. Lamb, T. P. Robinson, D. Mandal, J.-C. Buffet and D. O′Hare, Dalton Trans., 2021, 50, 4805–4818 RSC.
- N. Diteepeng, I. A. P. Wilson, J.-C. Buffet, Z. R. Turner and D. O'Hare, Polym. Chem., 2020, 11, 6308–6318 RSC.
- R. L. Jones, Z. R. Turner, C. Collins Rice and D. O’Hare, Organometallics, 2024 DOI:10.1021/acs.organomet.4c00028.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00103f |
| This journal is © The Royal Society of Chemistry 2024 |