
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantitative comparison of the copolymerisation kinetics in catalyst-transfer copolymerisation to synthesise polythiophenes†
Yifei
He
a and
Christine K.
Luscombe
 *b
*b
aDepartment of Materials Science and Engineering, University of Washington, Seattle, USA
bPi-Conjugated Polymers Unit, Okinawa Institute of Science and Technology, Okinawa, Japan. E-mail: christine.luscombe@oist.jp
First published on 10th June 2024
Abstract
Polythiophenes are one of the most widely studied conjugated polymers. With the discovery of the chain mechanism of Kumada catalyst-transfer polymerisation (KCTP), various polythiophene copolymer structures, such as random, block, and gradient copolymers, have been synthesized via batch or semi-batch (sequential addition) methods. However, the lack of quantitative kinetic data for thiophene monomers brings challenges to experimental design and structure prediction when synthesizing the copolymers. In this study, the reactivity ratios and the polymerisation rate constants of 3-hexylthiophene with 4 thiophene comonomers in KCTP are measured by adapting the Mayo–Lewis equation and the first-order kinetic behaviour of chain polymerisation. The obtained kinetic information highlights the impact of the monomer structure on the reactivity in the copolymerisations. The kinetic data are used to predict the copolymer structure of equimolar batch copolymerisations of the 4 thiophene derivatives with 3-hexylthiophene, with the experimental data agreeing well with the predictions. 3-Dodecylthiophene and 3-(6-bromo)hexylthiophene, which have higher structural similarity to 3-hexylthiophene, show nearly equivalent reactivity to 3-hexylthiophene and give random copolymers in the batch copolymerisation. 3-(2-Ethylhexyl)thiophene with a branched side chain is less reactive compared to 3-hexylthiophene and failed to homopolymerize at room temperature, but produced gradient copolymers with 3-hexylthiophene. Finally, the bulkiest 3-(4-octylphenyl)thiophene, despite its ability to homopolymerize, failed to maintain chain polymerisation in the copolymerisation with 3-hexylthiophene, possibly due to the large steric hindrance caused by the phenyl ring directly attached to the thiophene center. This study highlights the importance of monomer structures in copolymerisations and the need for accurate kinetic data.
Introduction
Conjugated polymers (CPs) are a strong candidate for use in a variety of electronic devices, including organic field effect transistors (OFETs),1 organic photovoltaics (OPVs),2 and organic electrochemical transistors (OECTs)3 because of their solution processability,4,5 mechanical flexibility6,7 and tuneable electronic properties.8,9 To achieve good device performance with CPs, the control of the morphology and microstructure is important.10 In addition to the efforts in optimizing the film formation conditions to control the morphology,11 synthetic chemistry strategies have been applied to modify the molecular structures of CPs and thus fundamentally tune the chain assembly behaviour. Such modifications have been vigorously practiced on the backbone12 and the side chain.3 Since the discovery of the chain mechanism of KCTP by Yokoyama et al.13 and Sheina et al.,14 the monomer sequence has become a new structural factor that could be modified to tune the polymer morphology.Poly(3-hexylthiophene) (P3HT) and its derivatives are the most extensively studied model systems synthesized by KCTP. Various polythiophene copolymers have been developed to reveal the structure–property relationship in CPs. For example, random,15,16 block,15–19 and gradient polythiophene copolymers15,16,20,21 have shown different solid-state morphologies. Compared to that of the random copolymers of the same composition, the morphology of the block copolymers shows further microphase separation, in which the size of the crystalline and the amorphous domains depends on the feed ratio of the comonomers.16,17 In between the random and block distribution patterns, gradient copolymers tend to have more ordered and separated domains than the random copolymers, but with different properties than the pure block copolymers.16 In the design of copolymers with more than two components, the sequence of each segment also plays a role in controlling the morphology.18,19 An important lesson revealed by the preceding studies in copolymer design is the necessity of more precise control over the monomer sequence in tuning the morphology and thus the properties.
Understanding the effect of the monomer structure on the kinetics of polymerisation can lay the foundation for the precise placement of comonomers in KCTP. There is currently no systematic and quantitative study about the kinetic behaviour of thiophene monomers. The lack of kinetic information leads to laborious tuning of experimental parameters for copolymerisation and a misunderstanding of the structure of the final product. A contradiction between the measured monomer reactivity and the predicted copolymer structure due to insufficient kinetic information is observed in the literature. For example, Schmode et al. have qualitatively measured that the reactivity of 2,5-dibromo-3-(6-bromohexyl)thiophene is higher than 2,5-dibromo-3-hexylthiophene, and thus the batch copolymerisation method was adopted to obtain a gradient copolymer.22,23 However, Palermo et al. obtained similar reactivity of 2-bromo-3-(6-bromohexyl)-5-iodothiophene (3BrHT) and 2-bromo-3-hexyl-5-iodothiophene (3HT) and obtained a random copolymer instead of a gradient copolymer in the batch copolymerization.20
With the above inspiration, this study examines the reactivity of the 3HT monomer and a series of thiophene comonomers (Fig. 1(a)–(d)) having different degrees of structural similarity with 3HT in KCTP for one particular concentration. The chosen comonomers vary by the side chains at their 3 positions and have been shown to be compatible with KCTP in either homopolymerisation or copolymerisation.17,24–26
 | ||
| Fig. 1 Thiophene monomer structures. (a) 2-Bromo-3-dodecyl-5-iodothiophene (3DDT), (b) 2-bromo-3-(2-ethylhexyl)-5-iodothiophene (2EHT), (c) 2-bromo-3-(4-octylphenyl)-5-iodothiophene (3OPT), and (d) 2-bromo-3-(6-bromohexyl)-5-iodothiophene (3BrHT). | ||
In addition to the reactivity ratios commonly applied to describe the relative kinetics of comonomers,21,27,28 we further calculated the four rate constants, k11, k12, k22, and k21 that define the ratios. The more specific kinetic information will help determine the kinetic difference related to the order of monomer addition, which is helpful in the synthesis of more complex copolymer architectures such as triblock copolymers. We also characterize the monomer consumption rate and the copolymer structure in the equimolar batch copolymerisation to verify the obtained reactivity values.
Experiments
Materials
2-Bromo-3-hexyl-5-iodothiophene (3HT), 2-bromo-3-dodecyl-5-iodothiophene (3DDT), and 2-bromo-3-(2-ethylhexyl)-5-iodothiophene (3EHT) were purchased from TCI America and filtered using silica plugs with hexane before use. Isopropylmagnesium chloride [i-PrMgCl, 2.0 M in tetrahydrofuran (THF)], [1,3-bis(diphenylphosphino)-propane]-dichloronickel(II) [Ni(dppp)Cl2] and 1,3,5-trimethoxybenzene (TMB) were purchased from Sigma Aldrich and directly used. All the organic solvents used for the polymerisation and monomer synthesis process were obtained from PureSolv dry stills. 2-Bromo-3-(4-octylphenyl)-5-iodothiophene (3OPT) and 2-bromo-3-(6-bromohexyl)-5-iodothiophene (3BrHT) were synthesized based on the previously reported literature.25,26Homopolymerisation kinetics studies via NMR
3HT monomer (0.373 g, 1 mmol) with TMB (16.8 mg, 0.1 mmol) as the reference compound was added to an acid-washed and oven-dried 50 mL round bottom flask under a N2 environment of the Schlenk line. Anhydrous THF (10 mL) obtained from the solvent still was added to dissolve the monomer after the monomer was degassed in vacuo for 30 minutes. After the mixture was stirred at 0 °C for 5 min, 2 M i-PrMgCl solution in THF (0.475 mL, 0.95 mmol) was added dropwise to the monomer solution. Subsequently, the reaction mixture was allowed to return to room temperature and react for an hour. A time 0 aliquot (0.1 mL) was extracted before the initiation step. Ni(dppp)Cl2 (9.03 mg, 0.0167 mmol) solid was directly added to the reaction mixture. The polymerisation was allowed to proceed for 120 min at room temperature. During the polymerisation stage, aliquots (0.1 mL) were taken from the mixture at times 0, 1, 3, 5, 10, 15, 20, 30, 45, 60, 90, and 120 min. The obtained aliquots were quenched with 5 M HCl (1 mL) and extracted with chloroform (CHCl3). After passing through a layer of anhydrous Na2SO4, the solvent of the extracted organic layer was removed in vacuo and the residuals were redissolved in d-CHCl3 for 1H-NMR characterisation. The DP of homopolymers was estimated using previously reported methods.29 The same experimental procedure was triplicated for all the thiophene monomers.Copolymerisation kinetics studies via GC-MS
In two 50 mL acid-washed and oven-dried round bottom flasks, 3HT (0.373 g, 1 mmol) and 3DDT (0.457 g, 1 mmol) were added respectively. Anhydrous THF (10 mL) was added to each monomer flask after the monomers were degassed in vacuo for 30 minutes. In another dry and acid-washed round bottom flask, tetradecane (TDC) (50 μL, 0.192 mmol) as the reference compound was added and degassed under vacuum. Different ratios of 3HT and 3DDT solutions were added to the TDC loaded flask to sum up to 2.5 mL. Once the mixture was stirred at 0 °C for 5 min, 2 M i-PrMgCl solution in THF (0.119 mL, 0.238 mmol) was added dropwise to the monomer solution and stirred for an hour at room temperature. Before the addition of the catalyst, a time 0 aliquot was taken from the activated monomer mixture. Ni(dppp)Cl2 (1.13 mg, 0.00208 mmol) was added to the monomer mixture. After the reaction proceeded for 1 min and 2.5 min, two aliquots (0.1 mL) were extracted respectively at these time points. The whole reaction was then quenched with 5 M HCl (1 mL). The three aliquots were diluted in MeOH to make GC-MS aliquots. The calibration procedure of each quenched comonomer and the calibration curve are summarized in Fig. S1.†Equimolar batch copolymerisation
Monomer distribution characterisation was performed to verify the accuracy of copolymer structure prediction, which could be reflected by the trend of copolymer fraction with respect to the chain growth.20 In an acid-washed and oven-dried 50 mL round bottom flask, 3HT (0.187 g, 0.05 mmol) and 3DDT (0.229 g, 0.05 mmol) with TDC (50 μL, 0.192 mmol) as the reference were added. After degassing under vacuum for 30 min and switching to the N2 environment, anhydrous THF (10 mL) was added. The mixture was allowed to stir on an ice bath for 5 min before the dropwise addition of 2 M i-PrMgCl solution in THF (0.475 mL, 0.95 mmol). After the addition of the Grignard reagent, the mixture was stirred under dark at room temperature for an hour. A time 0 aliquot was extracted before the initiation step. Ni(dppp)Cl2 (9.03 mg, 0.0167 mmol) solid was directly added to the reaction mixture as one batch. A series of aliquots (0.8 mL) were taken out of the reaction at different time points. Aliquots were quenched with 5 M HCl (1 mL) first before being extracted by CHCl3. The extracted organic layer was later precipitated in MeOH. Polymer precipitates were separated from the solution and washed with extra MeOH. The eluent was further diluted for the GC-MS characterisation. The procedure is the same for the other three monomer combinations. The DP of copolymers was estimated using methods that were previously reported.29Copolymerisation of diblock P(3HT-b-3OPT) and P(3OPT-b-3HT)
In two 50 mL acid-washed and oven-dried round bottom flasks, 3HT monomer (0.373 g, 1 mmol) and 3OPT monomer (0.477 g, 1 mmol) were added respectively and degassed in vacuo for 30 min. The environment was switched to N2 once degassing was performed. 10 mL of anhydrous THF directly from the solvent still was added to the flasks. The 3HT mixture was stirred on an ice bath for 5 min and added with 2 M i-PrMgCl solution in THF (0.475 mL, 0.95 mmol) dropwise. The mixture was then allowed to warm up to room temperature and reacted for an hour. After 30 min of the exchange of 3HT monomer, the same procedure was performed with the 3OPT monomer. Once the 3HT monomer completed the exchange process, Ni(dppp)Cl2 (27.1 mg, 0.05 mmol) solid was added. The mixture reacted at room temperature for 30 min to achieve the completion of the growth of 3HT block. 10 mL of activated 3OPT solution was subsequently added in one shot. The mixture was further stirred for an hour at room temperature and quenched with 5 M HCl (1 mL). Polymers were extracted with CHCl3 and precipitated in MeOH. The synthesis of P(3OPT-b-3HT) was attempted using the same experiment procedure with different monomer addition order.Results and discussion
Homopolymerisation kinetic studies
The conversion of the monomer with respect to the internal standard, TMB, reveals the homopolymerisation kinetics of each monomer. As previously studied, KCTP follows the chain mechanism, which indicates that its kinetic behaviour is first-order.13,14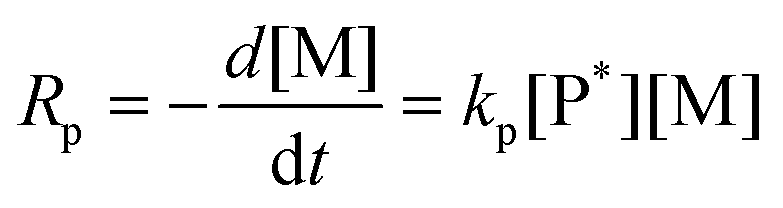

k appp of each monomer can thus be calculated from the slope of the linear regime of the semi-logarithmic conversion plot in Fig. 2 from triplicated polymerisation of each monomer. The NMR characterization and estimated Mn of each homopolymer are summarized in Fig. S2–S5.† We see that increasing side chain steric hindrance lowers the homopolymerisation rate constants. The three thiophene monomers bearing linear side chains, 3HT, 3DDT and 3BrHT, possess similar homopolymerisation rate constants, while the reactivity of 3OPT homopolymerisation is significantly lower. 3EHT failed to homopolymerize at room temperature using our experimental conditions. The synthesis of P3EHT homopolymers or blocks in copolymers via KCTP using the same Grignard and monomer has been commonly performed at temperatures above 35 °C.17,30,31 The required temperature elevation is due to the increased steric bulk of 3EHT that impedes the coupling of incoming monomers and chain ends.32
 | ||
| Fig. 2 The monomer consumption behaviour in homopolymerisation. The homopolymerisation of 3EHT failed at room temperature. | ||
To ensure the strict control of all other factors that may also impact the reaction kinetics, we approximate the reactivity of P3EHT homopolymerisation as infinitesimal in the later quantitative analysis rather than perform the synthesis at an elevated temperature.
Copolymerisation kinetics study
A couple of models have been discussed for the binary chain copolymerisation system, among which the terminal model is widely used for its ease of experimental data collection.33–35 In a binary copolymerisation system, the kinetic behaviour during the propagation stage could be described by 4 rate constants based on the identity of the chain end and the added monomer: k11, k12, k22 and k21, which are summarized in Table 1.| Growing chain end | Adding monomer | Rate constant | Reaction product |
|---|---|---|---|

|
M1 | k 11 |

|

|
M2 | k 12 |

|

|
M1 | k 21 |

|

|
M2 | k 22 |

|
Mayo et al. further correlated the four governing parameters in the forms of reactivity ratios as shown below.35 The experiment design and the calculation of the reactivity ratio using Mayo–Lewis equation were based on previous study.21 [M1] and [M2] represent the initial feed of the comonomers measured by t0 aliquots. d[M1] and d[M2] were approximated by the monomer consumption between 1 min and 2.5 min after the initiation of copolymerisation. A series of 6 initial feed ratios covering 2 orders of magnitude of concentration were prepared for each comonomer combination. Each series was then triplicated to generate 15–18 valid datasets for the non-linear fitting. k11 and k22 were approximated by the rate constants measured from the homopolymerisation kinetic studies of each monomer as described in section 3.1.36 Thus, the heteropolymerisation constants k12 and k21 were calculated based on the corresponding reactivity ratio and homopolymerisation constants. The fitted reactivity ratios and calculated rate constants of each pair of comonomer are summarized in Table 2.

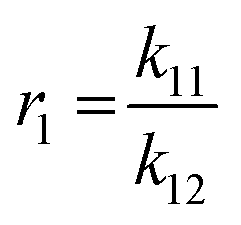
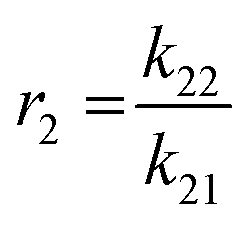
| M 1 | M 2 | r 1 | M 2 | R 2 | k 11 [min−1] | k 22 [min−1] | k 12 [min−1] | k 21 [min−1] |
|---|---|---|---|---|---|---|---|---|
| 3HT | 3DDT | 0.89 ± 0.07 | 0.73 ± 0.23 | 0.952 | 0.023 ± 0.002 | 0.018 ± 0.007 | 0.026 ± 0.003 | 0.025 ± 0.01 |
| 3HT | 3EHT | 2.15 ± 0.22 | 0.42 ± 0.23 | 0.939 | 0.023 ± 0.002 | Infinitesimal | 0.011 ± 0.001 | Infinitesimal |
| 3HT | 3OPT | 1.47 ± 0.50 | 1.47 × 10–18 ± 0.46 | 0.217 | 0.023 ± 0.002 | 0.010 ± 0.002 | 0.016 ± 0.006 | 6.80 × 1015 ± 5 × 1015 |
| 3HT | 3BrHT | 1.08 ± 0.32 | 1.24 ± 0.84 | 0.783 | 0.023 ± 0.002 | 0.027 ± 0.003 | 0.022 ± 0.007 | 0.022 ± 0.01 |
The trend of reactivity ratios also correlates with the dissimilarity of the chemical structures of comonomers with respect to 3HT. 3DDT and 3BrHT both have reactivity ratios close to 1 with respect to 3HT, which suggests that these two monomers are kinetically equivalent in the copolymerisation with 3HT. The result is consistent with their chemical structures, where both 3DDT and 3BrHT possess simple and linear side chains. Based on this, the final products of 3HT with either of these two monomers are expected to be random copolymers. In the batch copolymerisation of 3EHT and 3HT, r1 is greater than 1 and r2 is smaller than 1, which suggests that the 3HT monomer is preferably consumed compared to 3EHT. In addition, the product of the reactivity ratios is close to 1, which predicts that the final copolymer will have gradient monomer distribution along the chain. Moreover, by setting kapp22 as infinitesimal based on the failure of homopolymerisation of 3EHT, kapp21 becomes infinitesimal as well. The four rate constants suggest the necessity to build up a 3HT block first before the 3EHT block in the semi-batch copolymerisation of block copolymers. The reactivity ratio of the 3HT and 3OPT monomer pair also indicates the faster consumption of the 3HT monomer in batch copolymerisation, as r1 is greater than 1 and r2 is close to zero. This pair of reactivity ratios predicts that the final product of the batch copolymerisation will have primary 3HT blocks along the chain. Additionally, in the data fitting of 3HT and 3OPT reactivity ratio, the accuracy of the model decreases as shown by the R2 value. The decrease in the accuracy is likely the result of direct attachment of the phenyl ring to the thiophene, which leads to the loss of the catalyst transfer feature under the current experimental conditions thus preventing a chain polymerisation.
Characterisation of comonomer distribution
To validate the information provided by the kinetics parameters, the equimolar copolymerisation of each pair was assessed through monomer consumption behaviour and the resultant copolymer structure. The copolymer products were characterized via NMR and summarized in Fig. S6–S9.† The reactivity ratio of 3HT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3DDT predicts that in batch copolymerisation, the final polymer will be a random copolymer with the composition ratio equal to the monomer feed ratio. Given the high similarity of the chemical structures of the two comonomers, the copolymer composition is hard to distinguish solely by NMR signals. Thus, we combined monomer consumption behaviour via GC-MS and polymer weight distribution via MALDI-TOF to characterize the copolymer composition as presented in Fig. 3. 3HT and 3DDT were consumed at a similar rate in the copolymerisation as shown in Fig. 3(a). Fig. 3(b) and (c) presents MALDI-TOF spectra of aliquots from the growing and plateau stages. Both aliquots contain mass peaks that could only represent copolymers as labeled on the spectra. In addition, the ratios of calculated units of 3HT and 3DDT from the signature peaks are close to 1:1. This result indicates that the 3DDT composition is present in the copolymer throughout the polymerisation and holds a composition around 50%. The resulting product is a copolymer with random comonomer distribution, which is aligned with the prediction from the reactivity ratio.
:3DDT predicts that in batch copolymerisation, the final polymer will be a random copolymer with the composition ratio equal to the monomer feed ratio. Given the high similarity of the chemical structures of the two comonomers, the copolymer composition is hard to distinguish solely by NMR signals. Thus, we combined monomer consumption behaviour via GC-MS and polymer weight distribution via MALDI-TOF to characterize the copolymer composition as presented in Fig. 3. 3HT and 3DDT were consumed at a similar rate in the copolymerisation as shown in Fig. 3(a). Fig. 3(b) and (c) presents MALDI-TOF spectra of aliquots from the growing and plateau stages. Both aliquots contain mass peaks that could only represent copolymers as labeled on the spectra. In addition, the ratios of calculated units of 3HT and 3DDT from the signature peaks are close to 1:1. This result indicates that the 3DDT composition is present in the copolymer throughout the polymerisation and holds a composition around 50%. The resulting product is a copolymer with random comonomer distribution, which is aligned with the prediction from the reactivity ratio.
 | ||
| Fig. 3 (a) Respective monomer consumption behaviour of 3HT and 3DDT, (b) MALDI-TOF spectrum of P(3HT-co-3DDT) at t = 4 min, and (c) MALDI-TOF spectrum of P(3HT-co-3DDT) at t = 60 min. | ||
The reactivity ratio of 3HT:3EHT predicts that the batch copolymerisation of this combination will produce copolymers with the monomer composition gradually shifting from 3HT to 3EHT. The prediction was supported by the monomer consumption rate and NMR characterisation over a series of polymer aliquots taken throughout the reaction. 3EHT was less reactive than 3HT, as indicated by the slower monomer consumption rate shown in Fig. 4(a). As shown in Fig. S7,† the 3EHT composition could be represented by the singlet appearing at 6.94 ppm while 3HT signal shows up at 6.98 ppm. By taking the ratio of the two thiophene hydrogens, we obtain the composition change plot shown in Fig. 4(b). The linear increment of the 3EHT composition in the copolymer demonstrates the gradient distribution of monomers in the final product. We hypothesize that the different initiating dimers of 3EHT formed in copolymerisation and homopolymerisation may lead to contrasting results of the two polymerisations. Ni(dppp)Cl2 as the catalyst leads to the in situ formation of tail-to-tail dimers as the intiator.14 In homopolymerisation, only 3EHT-3EHT dimers would be formed as the initiation point of polymer chains. While in the copolymerisation, less sterically hindered dimers such as 3HT–3HT and 3HT–3EHT offer more efficient paths to add incoming monomers, which results in the successful copolymerisation of P(3HT-co-3EHT) at room temperature.
 | ||
| Fig. 4 (a) Respective monomer consumption behaviour of 3HT and 3EHT and (b) compositional change of 3EHT in P(3HT-co-3EHT). | ||
The close values of the reactivity ratios of 3HT and 3BrHT predict a random copolymer structure with the comonomer composition equal to the feed ratio. The consumption rate of both comonomers is close to each other as expected by the kinetic values. In Fig. S9,† the characteristic triplet at around 3.42 ppm represents the fraction of 3BrHT in the polymer. Taking the ratio of the triplet over the alpha methylene signal, we obtain the composition plot as shown in Fig. 5(b) with a stable 3BrHT composition throughout the synthesis. The result shows that the final copolymer has random monomer distribution.
 | ||
| Fig. 5 (a) Respective monomer consumption behaviour of 3HT and 3BrHT and (b) compositional change of 3BrHT in P(3HT-co-3BrHT) (initial feed ratio was 0.65:0.48). | ||
Examination of failed chain polymerisation of 3HT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :3OPT
:3OPT
Since the Mayo–Lewis fitting for 3HT:3OPT has shown difficulty given the decreased R2 value, we hypothesized that the deviation from the model resulted from the failed chain polymerisation for this pair. We validate the hypothesis by evaluating the dispersity change of 3OPT homopolymers and random copolymers as shown in Fig. 6. The homopolymerisation of P3OPT gives steady dispersity values of around 1.4 throughout the reaction and linear growth of the molecular weight with monomer conversion, as shown in Fig. 6(a). On the other hand, the dispersities of the equimolar random copolymerisation of 3HT and 3OPT have increased significantly with the apparent loss of linear relationship between MW and monomer conversion [Fig. 6(a) and (b)]. The other combinations showed clear linearity between DP and monomer conversion (Fig. S10–S12†). This implies that the loss of the chain polymerisation comes from the difference in the structures of the two comonomers.
 | ||
| Fig. 6 (a) Dispersities of P3OPT and P(3HT-co-3OPT) vs. the monomer conversion and (b) Mn change of P3OPT and P(3HT-co-3OPT) vs. the monomer conversion. Here, Mn values were obtained from GPC measurements. | ||
Further qualitative investigation was performed to understand the kinetics of hetero-monomer addition in the failed KCTP of 3HT:3OPT. The syntheses of diblock copolymers of P(3HT-b-3OPT) and P(3OPT-b-3HT) were attempted and characterized with MALDI-TOF. As shown in Fig. 7(a), m/z values of the most abundant peaks reflect the growth of 3OPT blocks from the 3HT chain ends. However, in the case of P(3OPT-b-3HT), we observed very limited growth of 3HT segments from the 3OPT chain ends (Fig. 7(b)). Moreover, the MALDI spectrum of P(3HT-b-3OPT) also implies the loss of end group control as Br/Br chain ends become predominant, as indicated in Fig. 7(a). These observations possibly result from the interfered association of the 3OPT monomer with the Ni catalyst due to the phenyl group.37
 | ||
| Fig. 7 (a) MALDI-TOF spectra and the comonomer unit calculation of characteristic peaks of P(3HT-co-3OPT). Values marked in red indicate Br/Br chain ends. (b) MALDI-TOF spectra and the comonomer unit calculation of characteristic peaks of P(3OPT-co-3HT) with the comonomer unit calculation of characteristic peaks. Values marked in blue indicate the loss of C8H17 radicals from 3OPT during characterization. | ||
As a final point, we address the differences observed in the polymerization of 3EHT and 3OPT. Both are bulkier than the other monomers. 3EHT fails to homopolymerize, while 3OPT does. 3EHT successfully copolymerizes while 3OPT does not. We speculate that these differences may arise from the differences in reactive species present due to the Schlenk equilibrium, where the alkyl thiophenes may form more dimers exasperating steric issues for 3EHT,38 or because the additional phenyl unit on 3OPT alters the association of the Ni catalyst.37 These issues may also affect the differences in monomer conversion, as shown in Fig. 3–5(a). Teasing out these details will be a topic of future study.
Conclusions
In this study, we compared the copolymerisation reactivity of 3HT with four thiophene comonomers in KCTP with different degrees of chemical structure similarity to explore the impact of the comonomer identity on reaction kinetics. In addition to the traditional reactivity ratios, we further investigated the four rate constants derived based on the terminal model in binary copolymerisation. Among the four comonomers, 3DDT and 3BrHT that possess higher similarity to 3HT show very similar reactivity with the major comonomer. The product from batch copolymerisation provides random copolymers with composition equal to the monomer feed ratio, which is consistent with the prediction of the kinetic values. On the other hand, the 3EHT monomer is less reactive than 3HT in the copolymerisation due to the steric hindrance caused by the bulky side chain. The product of their reactivity ratio is close to 1, which leads to copolymers with the gradient feature in batch copolymerisation. Finally, while being able to undergo KCTP of homopolymers, the most sterically hindered 3OPT failed to follow the chain polymerisation with 3HT under the current experimental conditions. Further examination via switching the chain end and incoming monomer identities qualitatively revealed that different kinetics exist for adding hetero-monomers to either type of chain end in the case of 3HT:3OPT. Our systematic study thus indicates the need for special handling of experimental conditions in the cases of bulky comonomers, such as temperature, catalyst ligands,37 and choice of Grignard reagents.39 Understanding the impact of the monomer structure on the reaction kinetics allows the efficient selection of appropriate comonomers and experimental conditions without tremendous commitment to the trial-and-error process.
Conflicts of interest
The authors have no conflicts of interest.Acknowledgements
Work by Y. H. was supported by the National Science Foundation (NSF) under DMR-2104234. 1H NMR characterisation of all the homopolymers and copolymers was supported by instrumentation from NIH S10 OD030224-01A1. C. L. was supported by the Okinawa Institute of Science and Technology Graduate University and JSPS KAKENHI Grant Number JP22K20547. We sincerely thank Viktoria Pakhnyuk for her assistance in 3BrHT monomer synthesis of this work and James Movius for his assistance in acquiring MALDI spectra of P(3HT-b-3OPT) and P(3OPT-b-3HT).References
- J. Yang, Z. Zhao, S. Wang, Y. Guo and Y. Liu, Chem, 2018, 4, 2748–2785 CAS
.
- X. Zhan and D. Zhu, Polym. Chem., 2010, 1, 409–419 RSC
- Y. He, N. A. Kukhta, A. Marks and C. K. Luscombe, J. Mater. Chem. C, 2022, 10, 2314–2332 RSC
- J. Mei and Z. Bao, Chem. Mater., 2014, 26, 604–615 CrossRef CAS
- G. Li, W. H. Chang and Y. Yang, Nat. Rev. Mater., 2017, 2, 17043 CrossRef CAS
- M. Ashizawa, Y. Zheng, H. Tran and Z. Bao, Prog. Polym. Sci., 2020, 100, 101181 CrossRef CAS
- J. Onorato, V. Pakhnyuk and C. K. Luscombe, Polym. J., 2017, 49, 41–60 CrossRef CAS
- Y. Liu, X. Zhan and Y. Leu, Macromol. Chem. Phys., 2011, 212, 428–443 CrossRef CAS
- Y. Yang, Z. Liu, G. Zhang, X. Zhang and D. Zhang, Adv. Mater., 2019, 31, 1903104 CrossRef CAS PubMed
- N. A. Kukhta and C. K. Luscombe, Chem. Commun., 2022, 58, 6982–6997 RSC
- Y. Yao, H. Dong and W. Hu, Polym. Chem., 2013, 4, 5197–5205 RSC
- X. Guo, M. Baumgarten and K. Müllen, Prog. Polym. Sci., 2013, 38, 1832–1908 CrossRef CAS
- A. Yokoyama, R. Miyakoshi and T. Yokozawa, Macromolecules, 2004, 37, 1169–1171 CrossRef CAS
- E. E. Sheina, J. Liu, M. C. Lovu, D. W. Laird and R. D. McCullough, Macromolecules, 2004, 37, 3526–3528 CrossRef CAS
- C. Kong, B. Song, E. A. Mueller, J. Kim and A. J. McNeil, Adv. Funct. Mater., 2019, 29, 1900467 CrossRef
- E. F. Palermo and A. J. McNeil, Macromolecules, 2012, 45, 5948–5955 CrossRef CAS
- Y. Zhang, K. Tajima and K. Hashimoto, Macromolecules, 2009, 42, 7008–7015 CrossRef CAS
- D. Zhai, M. Zhu, S. Chen, Y. Yin, X. Shang, L. Li, G. Zhou and J. Peng, Macromolecules, 2020, 53, 5775–5786 CrossRef CAS
- L. Li, H. Zhan, S. Chen, Q. Zhao and J. Peng, Macromolecules, 2022, 55, 7834–7844 CrossRef CAS
- E. F. Palermo, H. L. Van Der Laan and A. J. McNeil, Polym. Chem., 2013, 4, 4606–4611 RSC
- J. R. Locke and A. J. McNeil, Macromolecules, 2010, 43, 8709–8710 CrossRef CAS
- P. Schmode, D. Ohayon, P. M. Reichstein, A. Savva, S. Inal and M. Thelakkat, Chem. Mater., 2019, 31, 5286–5295 CrossRef CAS
- P. Schmode, K. Schötz, O. Dolynchuk, F. Panzer, A. Köhler, T. Thurn-Albrecht and M. Thelakkat, Macromolecules, 2020, 53, 2474–2484 CrossRef CAS
- S. Li, R. Cranston, B. H. Lessard and D. S. Seferos, ACS Appl. Polym. Mater., 2022, 4, 6030–6037 CrossRef
- T. W. Holcombe, C. H. Woo, D. F. J. Kavulak, B. C. Thompson and J. M. J. Fréchet, J. Am. Chem. Soc., 2009, 131, 14160–14161 CrossRef CAS PubMed
- V. Pakhnyuk, J. W. Onorato, E. J. Steiner, T. A. Cohen and C. K. Luscombe, Polym. Int., 2020, 69, 308–316 CrossRef CAS
- H. Abdollahi, V. Najafi and F. Amiri, Polym. Bull., 2021, 78, 493–511 CrossRef CAS
- I. S. Fazakas-Anca, A. Modrea and S. Vlase, Polymers, 2021, 13, 3811 CrossRef CAS PubMed
- M. A. Ansari, S. Mohiuddin, F. Kandemirli and M. I. Malik, RSC Adv., 2018, 8, 8319–8328 RSC
- Y. Zhang, K. Tajima, K. Hirota and K. Hashimoto, J. Am. Chem. Soc., 2008, 130, 7812–7813 CrossRef CAS PubMed
- L. Li, L. Li, X. Liu and J. Peng, ACS Appl. Polym. Mater., 2022, 4, 8461–8470 CrossRef CAS
- S. Ye, M. Steube, E. I. Carrera and D. S. Seferos, Macromolecules, 2016, 49, 1704–1711 CrossRef CAS
- S. K. Fierens, P. H. M. Van Steenberge, M. F. Reyniers, D. R. D'Hooge and G. B. Marin, React. Chem. Eng., 2018, 3, 128–145 RSC
- B. S. Beckingham, G. E. Sanoja and N. A. Lynd, Macromolecules, 2015, 48, 6922–6930 CrossRef CAS
- F. R. Mayo and F. M. Lewis, J. Am. Chem. Soc., 1941, 147, 1594–1601 Search PubMed
- P. Von Tiedemann, J. Blankenburg, K. Maciol, T. Johann, A. H. E. Müller and H. Frey, Macromolecules, 2019, 52, 796–806 CrossRef CAS
- S. Hameury, C. Gourlaouen and M. Sommer, Polym. Chem., 2018, 9, 3398–3405 RSC
- E. R. Curtis, M. D. Hannigan, A. K. Vitek and P. M. Zimmerman, J. Phys. Chem. A, 2020, 124, 1480–1488 CrossRef CAS PubMed
- B. S. Beckingham, V. Ho and R. A. Segalman, Macromolecules, 2014, 47, 8305–8310 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00009a |
| This journal is © The Royal Society of Chemistry 2024 |