
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fast relaxing sustainable soft vitrimer with enhanced recyclability†
Sargun Singh
Rohewal
ab,
Nihal
Kanbargi
b,
Rebecca
Young
c,
Logan T.
Kearney
b,
Joshua T.
Damron
b,
Holly
Hinton
b,
Laurene
Tetard
 c and
Amit K.
Naskar
*ab
c and
Amit K.
Naskar
*ab
aBredesen Center for Interdisciplinary Research and Graduate Education, University of Tennessee, Knoxville, Tennessee 37996, USA
bChemical Sciences Division, Oak Ridge National Laboratory, 1 Bethel Valley Rd, Oak Ridge, TN 37831-6053, USA. E-mail: naskarak@ornl.gov
cNanoScience Technology Center and Department of Physics, University of Central Florida, 4353 Scorpius St, Orlando, Fl 32816, USA
First published on 29th January 2024
Abstract
Vitrimers have been introduced to circumvent the lack of recyclability of traditional thermosets with permanent cross-linked structures, while preserving the advantages of structural stability and mechanical properties. The success of this lies in the successful incorporation of a robust networked structure to achieve reversible extensibility and toughness while preserving processability akin to thermoplastics. In this study, we report the synthesis of vitrimers utilizing 100% renewable and plant-based building blocks that exhibit transesterification exchange reaction (TER). The vitrimer was synthesized by solvent-free, high-shear reactive mixing of a biomass-derived lignin fraction enriched with carboxyl functionality and an epoxidized polyisoprene from natural rubber. The oxirane functionality in rubber reacts catalytically (zinc acetylacetonate) with carboxyl moieties in lignin to form esters at 180 °C. The ester linkages in the networked matrix undergo topological rearrangement upon heating above 180 °C, thus enabling (re)processability similar to thermoplastics. The material exhibits fast stress relaxation (characteristic relaxation time of <10 seconds) above 200 °C, which indicates the material's potential for use in rapid manufacturing of components and their recycling. This approach provides a pathway for circular and value-added utilization of lignin and subsequent use as a matrix for reinforced composites.
In the past few decades, the growing recognition of the connection between greenhouse gas emissions and global warming has significantly heightened awareness regarding the crucial role of bio-based feedstocks in the advancement of emerging high-performance materials.1–6 Replacing fossil-derived feedstocks, either partially or entirely, with bio-based feedstocks in the production of engineering and commodity plastics shows great promise in enhancing their sustainability. This approach has the potential to reduce or even eliminate reliance on non-renewable fossil resources. Extensive efforts have been dedicated to developing sustainable alternatives to petrochemical-based plastics, specifically tailored to possess desired characteristics like chemical circularity of building blocks and inherent reprocessability.7–9 However, research in this field is still in its early stages and faces various challenges, including scalability, compliance and competitiveness when compared to fossil-fuel derived counterparts. Addressing these challenges is crucial to drive the commercialization of biomass-derived sustainable materials.
Thermosetting polymers are extensively utilized in various applications due to their remarkable chemical resilience, stability, and superior mechanical performance arising from their inherent permanently crosslinked network. Nevertheless, a primary challenge in thermoset materials, particularly those with low glass transition temperature (Tg), lies in the successful incorporation of robust networked structure to achieve reversible extensibility and toughness while preserving processability akin to thermoplastics. By convention, the inherent permanence of cross-linkages within thermoset materials constitutes a significant barrier to their processability using conventional methods (e.g., thermal shearing). Utilizing dynamic covalent bonds in crosslinked networks affords specific bond exchange mechanisms when exposed to external stimuli (e.g., light, heat, pH). The required temperatures for bond exchange often fall well below the degradation temperature of the polymer backbone, which enables the networked structures to be processed and reprocessed similarly to thermoplastics. Although networked polymers with dynamic covalent linkages are being investigated for high Tg recyclable polymers (or vitrimers) as suitable substitutes for industrial thermosets, fast-relaxing, low Tg, networked yet recyclable rubber products as alternatives to traditional vulcanizates or elastomers are rare.10 Fast relaxing, cleavable networks are desired for rapid manufacturing of components via injection molding. Faster relaxation allows shorter time for part manufacturing cycle without causing shrinkage induced defects.11 For a Maxwell viscoelastic fluid, characteristic relaxation time, τ* is experimentally measured as the time required for a material to decay its initial applied stress value to 37% (1/e fraction). Materials with fast relaxation can maintain low viscosity.
Conventional rubber products require sulfur, peroxides, or exotic crosslinking agents to create permanent covalent networks which induce very high viscosity, prohibiting thermal reprocessing potential. Formulating recyclable yet networked rubber compositions containing only renewable feedstocks poses two key challenges: (1) decoupling the use of common crosslinking or vulcanizing agents and (2) tailoring functionalities in the building blocks that enable dynamic covalent bonding at an appropriate thermal condition. Successful manufacturing of these novel fast relaxing elastomer equivalents from renewable building blocks can alleviate issues pertaining to carbon emission and post-use environmental pollution.12 Vitrimeric products13–16 from renewables are sought-after not only for their recyclability and contribution to circular economy but also for their potential to exhibit additional functionalities such as self-healing and shape recovery.17 Recent reports on multiple vitrimeric systems, designed exclusively from renewable building blocks are encouraging18,19 and therefore, we focus on a combination of a soft and rigid segments from renewables that meet fast relaxation criteria along with thermal (re)processability.
We considered lignin, a natural polyphenol that is abundant, low-cost and readily available from sustainable sources of non-food plant biomass, as a rigid building block in this work. Lignin constitutes around 30% of the non-fossil carbon on Earth,20 but its utilization for materials manufacturing remains comparatively sparse. Rather, it is utilized as an inexpensive fuel in biomass processing mills.21,22 Structurally, isolated lignin in oligomeric form possesses a highly branched, aromatic architecture with a variety of naturally occurring functional groups, such as hydroxyl, aldehydes, and carboxylic acids, which can be exploited as reactive sites to design materials with dynamic exchange chemistries that proceed via transesterification, transacetylation, transimination, and more.23,24 When formulated with a combination of lignin in a soft matrix, phase separated morphologies prevail, depending on the miscibility criteria, leading to a broad range of mechanical properties.25–27 Moreover, lignin's inherently complex macromolecular architecture together with its high polydispersity contributes to performance variations that limit its use as a sustainable building block for value-added applications.28,29 Another dominant factor that limits lignin valorization is its recalcitrance towards chemical modification due to the strong self-assembly potential through physical interactions between its polar functional groups.30–32 In recent years, numerous innovative techniques like ozonolysis and catalytic conversions have been employed to improve reactivity and to tailor its physio-chemical properties. However, most of these procedures are very energy intensive, requiring multi-step synthesis routes that are difficult to control, specifically for technical lignin extracted from black liquors produced during wood pulping.23 Recently, we demonstrated that a fraction of Kraft softwood lignin, obtained via solvent extraction, consists of functionally enriched lignin oligomer with improved reactivity and narrower polydispersity.33 We presumed that a highly polar solvent would enhance compatibility and the integration of the solvent fractionated lignin phase within a polar rubber matrix. To test this, we utilized acetonitrile, which can be manufactured from renewables, for fractionation of the Kraft lignin.34
Our earlier report by Cui et al., showed that the degree of substitution (DS) in lignin aromatic ring for phenolic, alcoholic, and aldehyde groups were almost identical in acetonitrile extracted (ACN-lignin) and as-received lignin. However, there is almost a five-fold increase in the DS of carboxylic acid groups on extracted ACN-lignin as compared to the as-received lignin.33 Secondly, the DS of methoxy groups was reduced by half in the extracted lignin as compared to as-received lignin. These factors synergistically contributed to making ACN-lignin highly polar and more reactive than as-received lignin. Furthermore, the ACN-lignin obtained displayed a clear and distinct Tg of 75 °C, accompanied by a significantly narrower polydispersity of 1.16. In contrast, the as received lignin sample exhibited no noticeable Tg and a higher polydispersity of 1.83. It is surmised that the isolated lignin, being a heterogeneous mixture of molecules, can be thoroughly mixed with a specific solvent that will selectively separate and fractionate only those molecules that are highly interactive and soluble. We hypothesized that this polar lignin fraction could be used as a building block to enable better network forming ability, as well as to enhance dispersion within a polymer matrix during reactive mixing and extrusion of the resulting multiphase polymer. The carboxylic acid groups from lignin are reactive (in presence of a catalyst) with oxirane ring in epoxidized building blocks to yield binary mixtures with lignin.
In this work, the reactive carboxylic acid, phenolic, and alcoholic groups on lignin were exploited for reaction with the epoxy groups of an epoxidized natural rubber containing 25 mol% epoxy content (ENR-25). The covalent interactions between the reactive functional groups on lignin and epoxy groups of ENR-25, in presence of zinc(II) catalyst, yield ester and ether linkages (Fig. 1). Conventional catalysts, such as Zn(II) catalyst, are commonly used to facilitate the opening of epoxy rings in the presence of –COOH and –OH groups. Notably, catalysts employed in the epoxy-acid polymerization, including Zn(acac)2, PPh3, and triazabicyclodecane (TBD), inherently exhibit efficiency in transesterification reactions as well.35,36 It is well understood that ester linkages undergo TER in the presence of surrounding –COOH and –OH groups at elevated temperatures, making such products behave like vitrimers.37 As the –COOH group exists as a much stronger acid than corresponding alcohols and phenolic groups due to the presence of resonance stabilized carboxylate ion, the reactivity of –COOH groups with epoxy groups of ENR-25 is expected to be higher than that of the alcohols and phenolic groups.23,38 Thus, the lignin fraction having higher –COOH content is expected to be a better building block as compared to as-received technical lignin, where the average –COOH content is low.
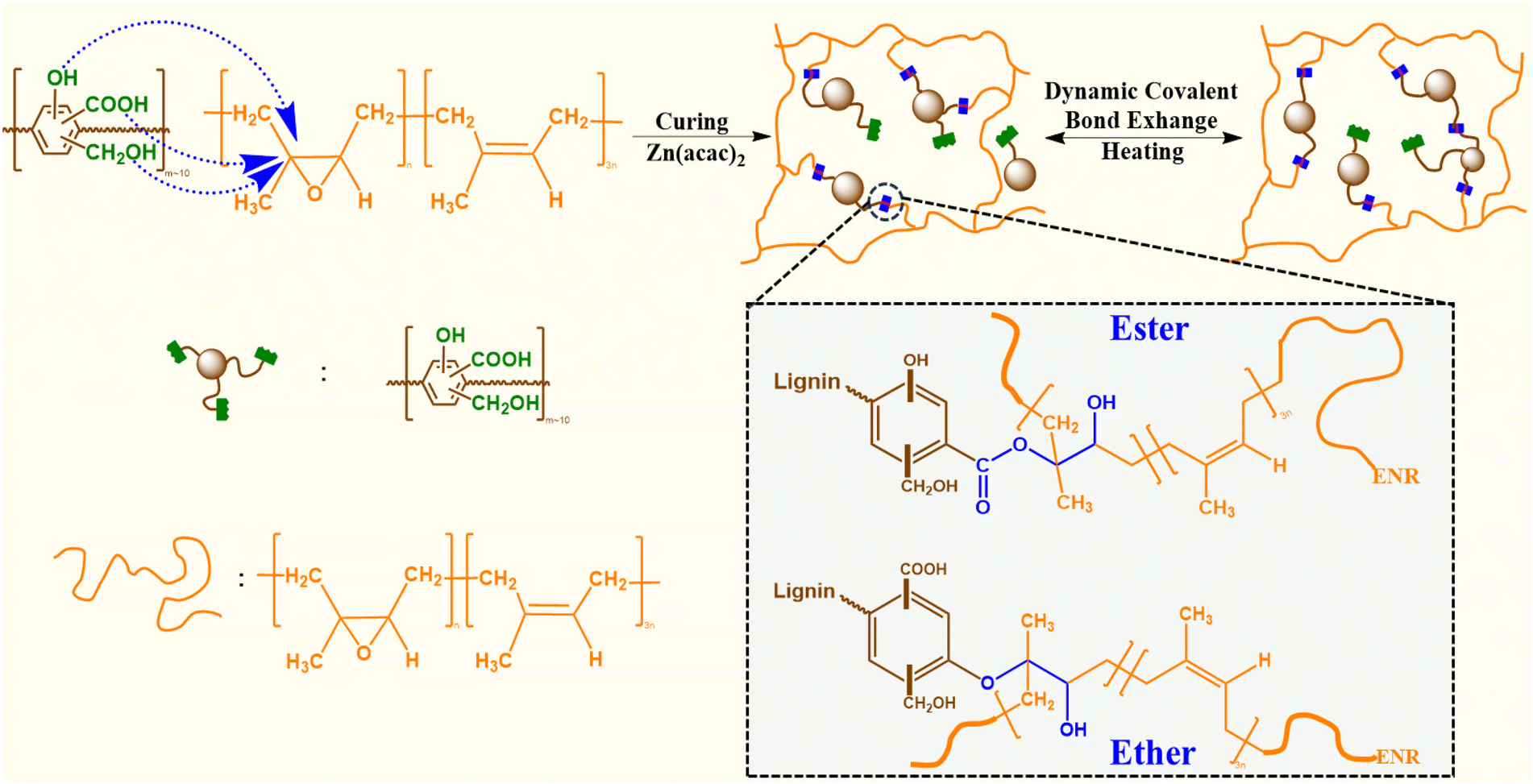 | ||
| Fig. 1 Schematic of ACN-lignin/ENR vitrimer synthesis and potential dynamic covalent bond exchange via TERs. | ||
Physical characterization of the vitrimers
Three vitrimers samples with different ACN-lignin to ENR-25 ratios were prepared by using a catalytic, solvent-free high temperature shear-mixing procedure (described in ESI†) followed by molding in a hot-press at 180 °C for 2 h to ensure complete equilibrium crosslinking. The FTIR spectra (see Fig. S1, ESI†) confirmed the crosslinking reaction between the reactive functional groups of lignin and epoxy groups of the ENR. The covalent interaction between the epoxy groups of ENR-25 with carboxyl and alcoholic functionalities on lignin lead to formation of β-hydroxy ester and ether linkages which was evidenced by appearance of additional peaks in the vibration band of carbonyl bonds at 1700–1750 cm−1 and aromatic ether bonds at 1250 cm−1 upon curing. The higher content of lignin in the reaction matrix leads to higher concentration of formed ester linkage (1700–1750 cm−1) along with an increased concentration of β-hydroxy groups at 3400 cm−1 (Fig. S1, ESI†). The increased loading of ACN-lignin enabled formation of material with higher crosslink density due to more reactive functional groups being engaged with epoxy groups of the ENR-25. Formation of products with higher concentration of ester and hydroxyl groups as function of lignin loading is also evident from increased relative peak intensity of respective groups (Fig. S1b†). This was evidenced by a systematic increase in the gel content on increasing the weight fraction of ACN-lignin in the overall matrix (see Table 1 below; Methods in ESI†). The decrease in swelling ratio and increase in gel content is often correlated with an increase in the crosslink density of the material.39 Both ACN-lignin and ENR-25 can dissolve well in tetrahydrofuran (THF), thus it was used as a solvent for gel content and swelling experiments.| Sample | ACN-lignina (wt%) |
T
g![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (°C)
(°C) |
T
g
(°C) |
T
d5%
(°C) |
T
se (°C) |
Gel Contentf (%) |
E
ag (kJ mol−1) |
τ
*
210 °Ch (s) |
T
vi (°C) |
Tensile strength (MPa) | Elongation at break (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Weight fraction of ACN-lignin in the sample. b Glass transition temperature (Tg) from DSC thermogram curves (Fig. S3†). c T g from loss tangent spectra (DMA data) (Fig. S3†). d Temperature corresponding to 5% mass loss. e Static heat resistant temperature (Ts) calculated from equation A1 (shown in ESI†). f Gel content of the samples were measured by extraction with THF for 48hrs at room temperature. g Activation energy calculated from Arrhenius plots for each sample. h Characteristic relaxation time (τ*) at 210 °C obtained from stress relaxation plots. i Theoretical topology freezing temperature (Tv) calculated by extrapolation for a viscosity of 1012 Pa s. | |||||||||||
| ENR25 | 0 | −47 | — | 328 | 172.0 | 0 | — | — | — | 0.341 | >1000 |
| 40 phr | 28.5 | −41.8 | −18 | 292 | 166.5 | 75.14 | 210.22 | 95.3 | 119.0 | 5.4 ± 0.7 | 1334 ± 87 |
| 50 phr | 33.3 | −40.1 | −15.5 | 291 | 164.8 | 81.23 | 198.44 | 18.9 | 120.5 | 7.4 ± 0.2 | 1213 ± 226 |
| 60 phr | 37.5 | −37.4 | −5 | 286 | 167.4 | 83.31 | 169.30 | 6.7 | 112.9 | 9.5 ± 0.4 | 1342 ± 132 |
| ACN-lignin | 100 | 72.4 | — | 184 | 151.3 | 0 | — | — | — | — | — |
All the lignin-derived vitrimeric elastomers exhibited excellent thermostability. The thermal degradation onset starting at 5% weight loss, from thermogravimetry data (see Fig. S2, ESI†), was detected around 290 °C for 40–50 phr lignin loaded compositions (Table 1). Thermal degradation onset temperature (at 5% weight loss) dropped to 286 °C for composition with 60 phr lignin. The lignin-based vitrimeric compound presented here exhibits considerably higher thermal stability than the previously reported lignin-derived vitrimeric systems and thermoset materials that showed thermal degradation onset at 190°–220 °C.24,40 This suggests robust reprocessing potentials of ACN-lignin/ENR25 compositions without significant material degradation (see Fig. S2, ESI†). Dynamic mechanical analysis (DMA) data, specifically, the peak temperature of loss tangent profile (tanδ) and differential scanning calorimetry (DSC) thermograms revealed an enhancement in the Tg with higher lignin loading (see Fig. S3, ESI†). The lignin-derived vitrimer with higher lignin content (e.g., 70 phr) acts like a stiff and brittle plastic with an ultimate elongation at break of only ∼8%, however, with higher tensile strength of 15.1 MPa. Such brittle glassy plastic-like materials have not been thoroughly characterized in this work.
As schematically shown in Fig. 1, the crosslinking reaction between ACN-lignin and ENR-25 yields ester and ether linkages along with β-hydroxy bonds. The β-hydroxy bonds further participate in transesterification exchange reactions (TERs) with adjacent ester linkages at elevated temperatures. In turn, these thermally stimulated TERs result in the topological rearrangement of the overall matrix resulting in changes in the viscoelastic properties of the vitrimer. Usually, thermal stress relaxation experiments assist in revealing the viscoelastic behavior of a vitrimer, where a constant external strain is applied to a sample and the stress response is followed as a function of time at different temperatures. The viscoelastic characteristics of the ACN Lignin/ENR vitrimers were investigated through a stress relaxation experiment. In this procedure, a consistent strain of 0.667% was applied, and the relaxation modulus was tracked over the course of one hour. All the ACN Lignin/ENR vitrimer samples exhibited a clear stress relaxation trend. As the temperature surpassed 180 °C, a systematic decrease in relaxation times indicated an accelerated rate of TERs, pointing to the vitrimeric nature of the material. Notably, at lower temperatures (25 °C, 70 °C), all vitrimer materials exhibited conventional thermoset-like (networked) behavior, observable through the insignificant stress relaxation (see Fig. 2 and Fig. S5 and 6 in ESI†). The 60 phr composition (ACN-lignin/ENR25 = 60/100), with the highest lignin content, showed the fastest relaxation rate with a τ* of a mere 6.7 s at 210 °C. Zn(acac)2, the catalyst used in this work, is known to play a dual role in epoxy-acid polymerization. Not only does it facilitate crosslinking between epoxy and acids, but it also aids in promoting bond exchange between ester linkages and neighboring β-hydroxy groups within the matrix. To comprehend the impact of TERs as a driving force for stress relaxation in the vitrimer, we synthesized a 60 phr ACN Lignin/ENR control sample without the catalyst. In absence of the catalyst, the control sample not only possesses fewer ester cross-linkages but also undergo sparce TERs. During stress relaxation experiments on both samples, which contain identical amounts of lignin and ENR, the sample with the catalyst exhibits instantaneous stress relaxation (τ < 10 seconds) at 210 °C. In contrast, the sample without the catalyst exhibits insignificant relaxation even after one hour, underscoring the crucial role of TERs in driving stress relaxation phenomena at elevated temperatures (Fig. S5†). This result suggests that the rapid stress relaxation observed in the vitrimer samples can be attributed to TERs facilitated by Zn(II) catalyst.
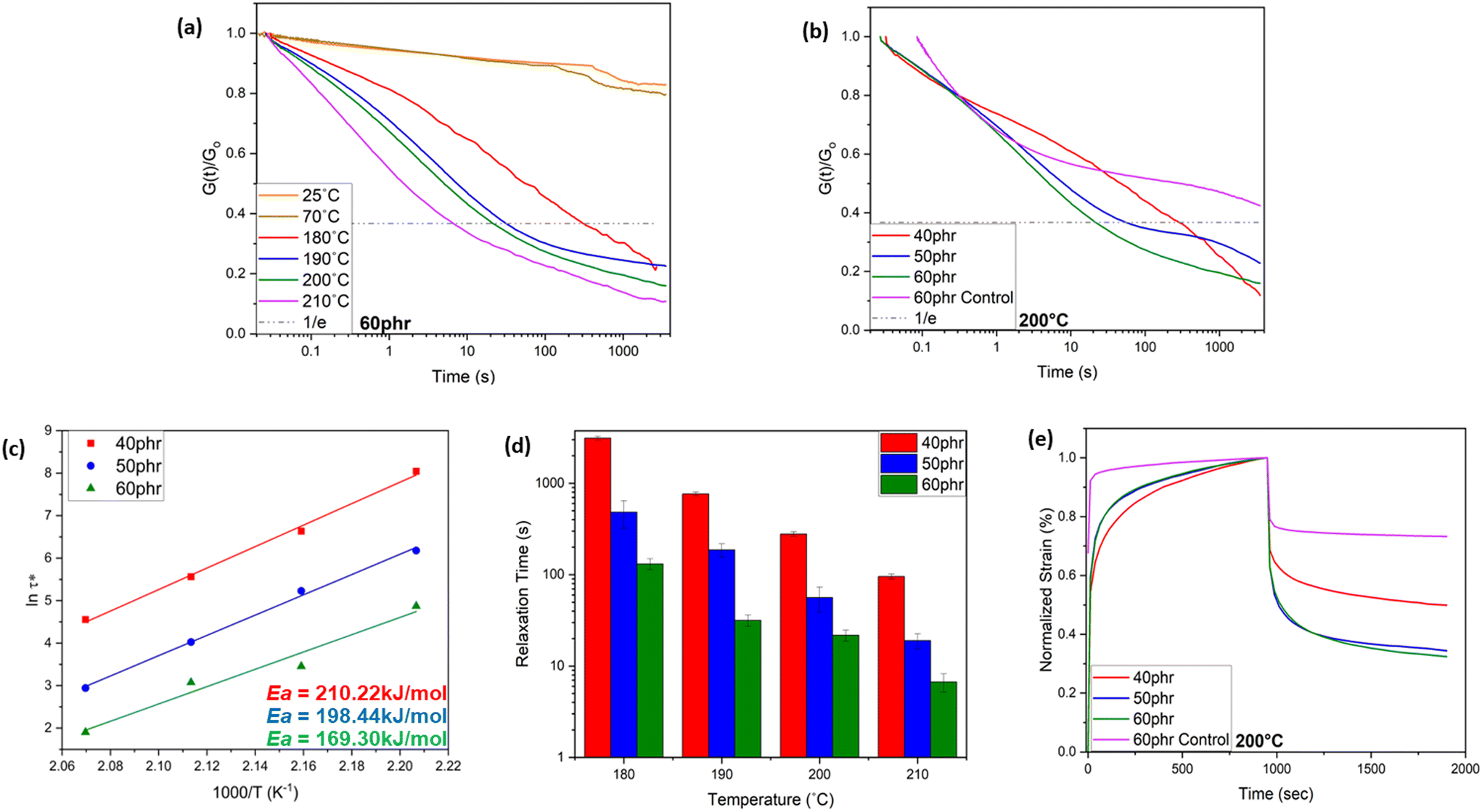 | ||
| Fig. 2 Rheological characterization of ACN-lignin/ENR25 vitrimer: (a) stress relaxation curves of 60 phr vitrimer at different temperatures (b) stress relaxation curves of vitrimer with different lignin content and 60 phr control sample without catalyst at 200 °C. (c) Fitting of the experimental relaxation times to the Arrhenius equation. (d) Relaxation times of vitrimers with different lignin content at different temperatures. (e) Creep and creep recovery behavior of vitrimers with different lignin content and 60 phr control sample without catalyst. | ||
Typically, most bio-based vitrimeric systems exhibit longer relaxation times (>100 s), thereby limiting applications in continuous processing like extrusion and injection molding,42–45 but are of interest for processing via compression molding. Apart from the dynamic character of covalently adaptable networks, the macromolecular architecture and spatial orientation of bonds also play a crucial role in governing the exchange kinetics.46–48 The formation of hydroxyl bonds upon ring opening of epoxy due to the nucleophilic attack of the carboxylic acid group from ACN-lignin plays a pivotal role in the fast topological rearrangement. The presence of a hydroxyl nucleophile (β-hydroxy) in the vicinity of an ester moiety increases the likelihood of a productive exchange between the reaction partners, thus accelerating the exchange rate. Reducing the weight fraction of lignin in the system resulted in a slower relaxation rate of the system (Fig. 2d). The 50 phr and 40 phr ACN-lignin/ENR25 samples demonstrated τ* of 18.9 s and 95.3 s, respectively, at 210 °C. This is likely due to decreased ester cross-linkages and unreacted carboxylic acid and alcoholic moieties, which result in lower site availability for effective chemical exchange within the system. The experimental relaxation times follow the Arrhenius law, allowing determination of the activation energy from the plots of ln(τ*) against 1000/T (Fig. 2c; see ESI† for activation energy data assessment). The activation energy for relaxation via topological rearrangement dropped from 210.22 kJ mol−1 to 169.30 kJ mol−1 upon increasing the lignin content from 40 phr to 60 phr, causing a faster relaxation (Fig. 2d). Furthermore, the theoretical topology freezing temperature (Tv) calculated by extrapolation for viscosity of 1012 Pa s (see ESI†) was observed to decrease with higher lignin content as well, indicating a faster topological rearrangement (Table 1). To understand the creep resistance behavior of the vitrimers and a representative control, creep experiments were conducted at 200 °C (see ESI† for Methods). While the vitrimer demonstrates a capacity to undergo reversible breaking and reforming of bonds which enables a stress relaxation phenomenon, it still maintains a networked structure crucial for imparting creep recovery (Fig. 2e). Our observation revealed an augmentation in creep recovery with increasing lignin content, attributed to an elevated crosslink density in the material (see earlier discussion on solvent swelling and gel content). A vitrimer consisting of 60 phr lignin shows nearly 70% creep recovery at 200 °C. Presence of a consistent networked structure (even though bonds are exchanging) at elevated temperature enables storage of deformation energy; this is a reason why a fast-relaxing vitrimer does not achieve complete relaxation even at elevated temperatures. In contrast, the 60 phr control sample (without catalyst) exhibits instantaneous creep behavior with significantly lower degree of creep recovery (nearly 25%) due to limited network structure formation in the absence of Zn(II) catalyst.
Atomic force microscopy (AFM) coupled with nano-IR spectroscopy
To probe the morphology and interfacial characteristics, we conducted AFM and nano-IR experiments. First, we developed a protocol to determine the transition temperature of a Lignin/ENR sample using polycarbonate as a reference material for calibration (see ESI† for methods, calibration, and analysis of structure by AFM coupled with nano-IR spectroscopy; Fig. S8 and 9†). Topography images of the sample film revealed morphological features, as shown in Fig. 3a. Lignin particulate features protruding up to 4 μm above the surface of the rubber matrix are distributed in the material. The spectral fingerprints of the two main regions of the films were acquired using nano-IR spectroscopy in contact mode. The contact resonance of the cantilever was first acquired at each point immediately before collecting each nanoscale infrared spectrum. The contact resonance in the 260–320 kHz range was used for the spectral analysis, as it was the strongest resonance of the cantilever used for the study. The nano-IR spectra of the two different regions, acquired by monitoring the amplitude of the resonance as a function of illumination wavenumber, are shown in Fig. 3b. Before normalization, the signal acquired from the regions denoted by point A exhibited much lower photothermal expansion (15 times lower) compared to the spectra of the protruding particulate morphological features. For comparison of the fingerprint and position of the peaks, we normalized and smoothed the data (see Methods). At room temperature, the rubber-rich regions (point A, green curve) and of the lignin-rich regions (point B, blue curve) exhibit notably different fingerprints in the 1800–1300 cm−1 range. The spectrum acquired at point A exhibits absorption peaks at 1715 cm−1 (rubber, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), 1660 cm−1 (rubber, CC stretching), 1610 cm−1 (epoxy, CC stretching in aromatic), 1508 cm−1 (epoxy, CC stretching in aromatic), and 1448 cm−1 (CH2 deformation).49 The spectrum acquired at point B exhibits strong peaks at 1660 cm−1 (CO stretching), 1600 cm−1 (aromatic skeleton vibration), 1555 cm−1, and 1456 cm−1 (C–H bending) which are attributed to lignin.50 The strong signal of the carboxylic group at point B agrees with previous demonstration that the degree of substitution for carboxylic acid is high in ACN-lignin.
O stretching), 1660 cm−1 (rubber, CC stretching), 1610 cm−1 (epoxy, CC stretching in aromatic), 1508 cm−1 (epoxy, CC stretching in aromatic), and 1448 cm−1 (CH2 deformation).49 The spectrum acquired at point B exhibits strong peaks at 1660 cm−1 (CO stretching), 1600 cm−1 (aromatic skeleton vibration), 1555 cm−1, and 1456 cm−1 (C–H bending) which are attributed to lignin.50 The strong signal of the carboxylic group at point B agrees with previous demonstration that the degree of substitution for carboxylic acid is high in ACN-lignin.
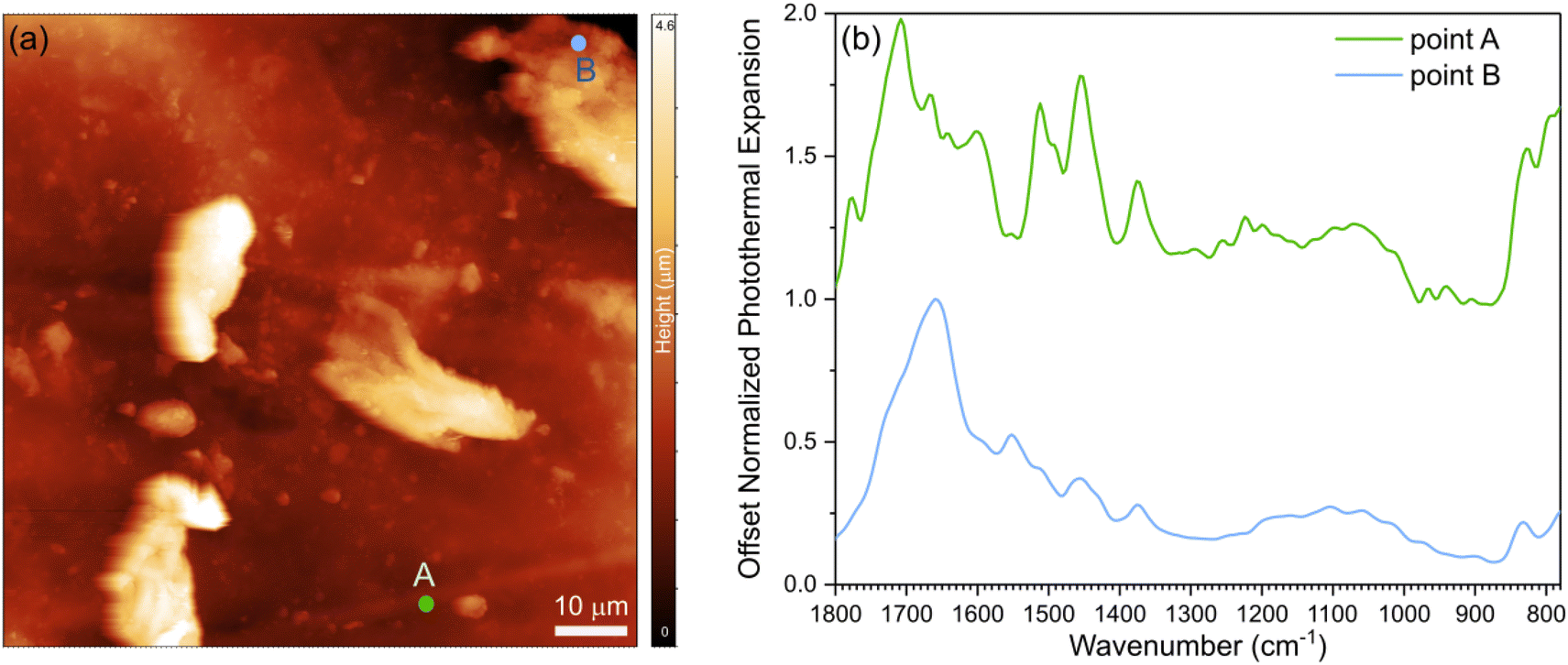 | ||
| Fig. 3 (a) AFM topography image of the lignin-ENR25 50 phr film with (b) corresponding nanoscale infrared spectra collected at point A (green curve) representative of a rubber-rich material and point B (blue curve) representative of a lignin-rich material. | ||
Based on these fingerprints, the illumination parameters were set at a power (∼130 mW) under illumination at 1670 cm−1, which is expected to heat up the material by exciting the vibrational mode of lignin to reach ∼150 °C. The topography images of the films at increasing laser power up to 130 mW are presented in Fig. S10a–e.† The images were acquired in tapping mode using the first resonance mode of the cantilever (∼60 kHz) to record the topography information, with the infrared laser pulse frequency set at 300 kHz. Comparison of the same region imaged under increasing laser power makes it possible to visualize the selective changes taking place in the material, with very slight changes in morphology (width, shape, and height) of the lignin-rich features (Fig. S10b and c†). At 130 mW the rubber region was found to expand and become more unstable for imaging (Fig. S10d†), which was accentuated when increasing the pulse width from 200 ns to 400 ns (Fig. S10e†). Under these conditions, the sample surface became too unstable for high resolution imaging, but the response was found to be preferentially from the lignin features, with an effect on the flatness of the rubber matrix. Force curves measurements acquired before illumination and during illumination at 130 mW confirms notable changes in the behavior of the material. Before heating, the rubber exhibited a significantly larger adhesion to the AFM tip compared to lignin, while the contact indentation was comparable between lignin and rubber. Upon heating, the indentation of rubber increased and the adhesion force with the tip decreased. In addition, the slope of the approach and withdraw curve transitioned from linear to nonlinear when probing the heated rubber matrix. The work of adhesion between the AFM tip and lignin increased upon heating the material, while the indentation and slope of the fore-indentation curve remained linear, though with a lower slope than before heating.
To better probe topological exchange reaction via transesterification, spectral analysis was done by acquiring nano-IR spectra at increasing laser power (Fig. 4). Four spectra were collected across the transition from the lignin feature to the rubber region at room temperature (Fig. 4a) and at ∼150 °C (Fig. 4b), as marked on the AFM images. The spectra acquired at room temperature correspond to the fingerprints presented in Fig. 3, namely three points exhibit the fingerprint of lignin while the last point corresponds to rubber. The main IR bands were marked with dashed lines for reference. When the sample was heated, the infrared fingerprint revealed important changes in the chemical bonds of the material, including in the lignin region (red curve), in the rubber region (green curve), and in the transition between the two regions (blue and yellow curves). Variations in the peak position and width in carbonyl region (1800–1700 cm−1) suggest a change in the ester linkages in the material. A strong peak at ∼1224 cm−1 at the interface and in the rubbery region suggests changes related to the ether bonds in the system. At high temperature the interface region displays strong presence of ether group, likely due to the extended esterification reaction at the interface. During transesterification some non-resonating carbonyls are formed, exhibiting strong blue-shift in carbonyl intensity. The variations in the spectra indicate that the chemical changes depend on the phases and morphological structures of the vitrimer considered.
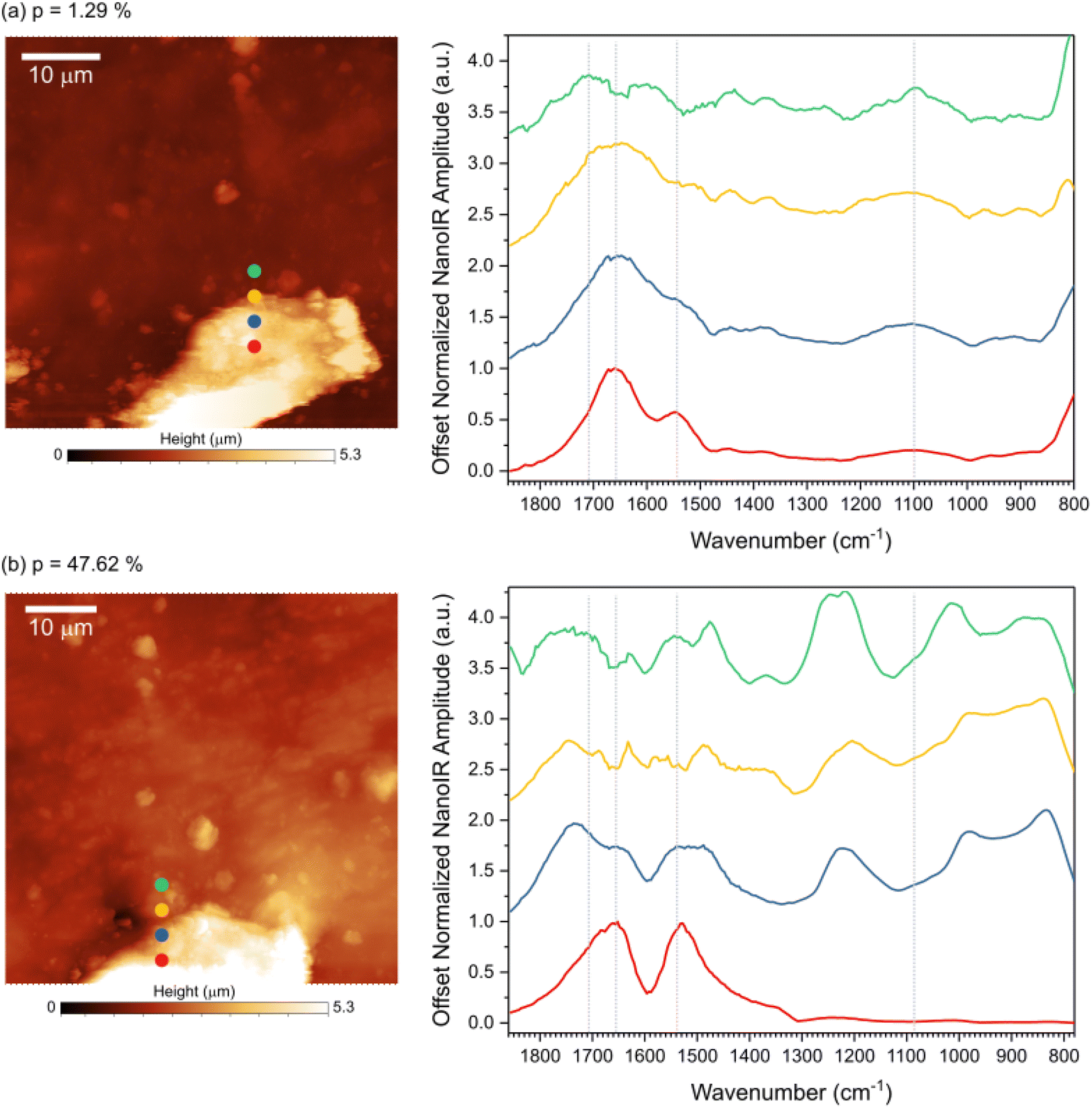 | ||
| Fig. 4 Evolution of the nanoscale infrared fingerprints as a function of temperature. (a) AFM topography image indicating the 4 points at which the nanoscale infrared spectra were acquired at room temperature. (b) AFM topography image collected after illumination, indicating the 4 points at which the nanoscale infrared spectra were acquired at p = 47.62%. | ||
1H time domain NMR spectroscopy
The dynamics of soft and rigid components of a representative vitrimer at various temperature range was investigated by low-field NMR relaxometry. 1H time domain NMR was collected on the vitrimer containing 60 phr lignin and its corresponding control without use of catalyst. The analyzed data (see Method in ESI†) are summarized in Fig. 5(a–d). Overall, the mobile phase with the catalyst has reduced mobility compared to the control group over the entire temperature range Fig. 5(b). This indicates better networking throughout the system and better compatibility between rubber and lignin phases. The rigid lignin phase relaxations are similar until higher temperatures where the control starts to dramatically increase at 175 °C, while this increase is subdued/delayed until 200 °C in the catalyst system (Fig. 5a). The phase fraction behavior of the catalyst added system has a sharper, more linear response to temperature until ∼125 °C and then seemingly plateaus (Fig. 5c and d). The control system has a milder temperature response until ∼160 °C where it then increases more aggressively which sensibly coincides with the large increases in T2 (relaxation time) for the rigid and mobile phases. It is important to note that the intra-system phase fraction comparison must be weighted by the T2 relaxation times for each system when doing a comparative analysis. For instance, that the mobile phase is higher for the catalyst containing system above ∼90 °C (Fig. 5d) is at face a bit misleading as, while that is relatively true, the catalyst mobile phase is overall more restricted according to the T2 relaxometry. It does, however, suggest that there is better mixing/interfacing of the lignin and rubber phases in the catalyst system and that the control lignin/ENR (without a catalyst) developed poorer lignin/rubber compatibility in the system during melt-mixing. The lack of catalyst failed to enable increased interface fraction during synthesis itself. The lack of network in the structure for control makes the lignin phase more mobile than the vitrimer sample above the Tg of the lignin. The latter undergoes catalytic TER above 125 °C which helps stabilize the rigid phase. Fig. 5b showing relatively low relaxation time for mobile phase also suggests mobile phase is under the influence of lignin due to better mixing via reaction during synthesis of material and even at high temperature the transesterification keeps the mobility of soft segments lower than that in control. It is notable that the phase behavior plateaus around the predicted Tv from rheology for the vitrimer system. It is possible that the bond switching events provide transient stabilization of the system as it competes with thermal induced flow which is random. It is also possible that this is simple a reflection of better network density.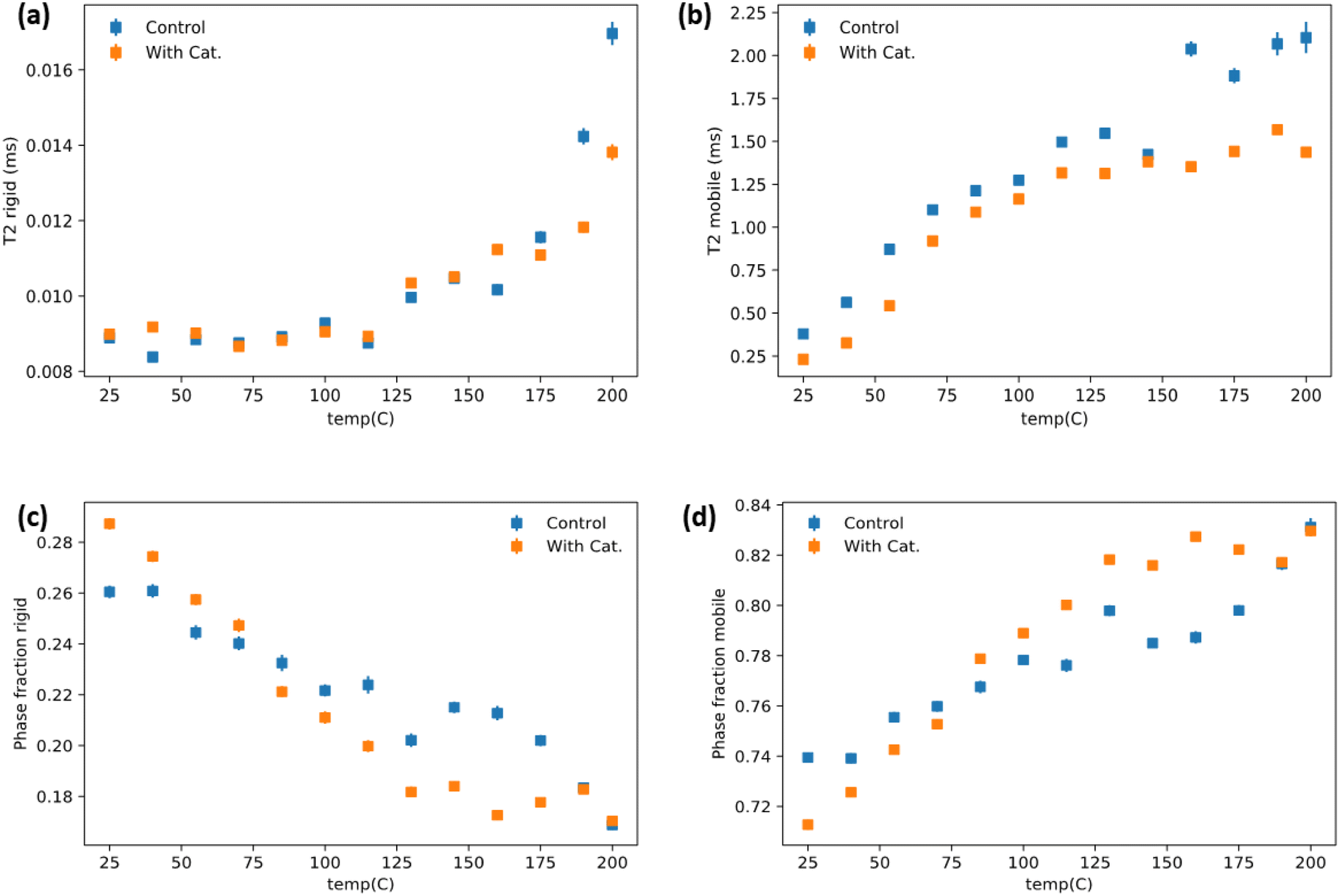 | ||
| Fig. 5 T 2 relaxation time spectra of (a) rigid lignin and (b) soft ENR phases obtained from 60 phr lignin loaded vitrimer and its catalyst free control, as a function of temperature by time domain 1H free induction decays. Phase fractions determined by 1H time domain NMR for the pure vitrimer and its corresponding control at various temperatures for (c) rigid lignin and (d) soft ENR fractions. As shown, the onset of mobility restriction in the mobile phase may be indicative of extensive bond exchange starts at ∼125 °C. | ||
Mechanical property analysis
Mechanical properties play a crucial role in defining the suitable applications of a vitrimeric material. Tensile testing was used to examine the mechanical properties of all the lignin-ENR vitrimers. The results are summarized in Table 1 and Fig. 6. The incorporation of higher mass fractions of highly branched aromatic lignin in the reaction mixture with inherently flexible ENR-25 segments markedly increased the tensile strength and toughness of the vitrimer. Of all compositions evaluated, 60 phr ACN-lignin/ENR25 exhibited the highest tensile strength and toughness of 9.45 MPa and 77.5 MJ m−3 respectively, with an elongation at break at ∼1350% (Fig. 6a and b). The gradual increase in the tensile strength and toughness of the vitrimer upon increasing the lignin mass fraction can be attributed to the increase in the rigidity and crosslinking density of the vitrimer. Moreover, the innate three-dimensional phenylpropanoid backbone of lignin containing multifunctional groups offers reinforcing capabilities to the rubber matrix.27,51 This was further evidenced by a systematic increase in the storage modulus of the vitrimer on increased lignin loading. At the onset (40 °C) of the rubbery plateau range (40 °C to 180 °C), as depicted in Fig. 6c, the 60 phr ACN-lignin/ENR25 vitrimer demonstrated a higher storage modulus (10.8 MPa) than the 50 phr (5.4 MPa) and 40 phr (4.8 MPa) vitrimer samples. Furthermore, the presence of secondary alcohols, ethers, and esters upon crosslinking are responsible for overall balance between hydrogen bonding acceptor and donor species in the material at any given time. It is well known that the dynamic character of hydrogen bonding often assists in dissipating energy during force loading, thus contributing to increased toughness of the material, as demonstrated for various elastomeric and hydrogel systems.52–56 The stress relaxation behavior induced by TERs at elevated temperatures is instrumental in imparting various sought-after properties like self-healing, shape-recovery, and reprocessability. The reprocessed mechanically failed vitrimer samples demonstrated almost identical tensile properties as the original specimen even after two reprocessing cycles (Fig. 6d and e). The elastic recovery of the original and reprocessed vitrimer samples was investigated by applying a cyclic tension-retraction test for both original and reprocessed 60 phr vitrimer samples. Both original and reprocessed vitrimer samples exhibited exceptional elastic recovery with little energy dissipated (low hysteresis) during cyclic loading (Fig. 6f). The strong covalent interaction between rubber with low elastic modulus and lignin reinforcement with a high elastic modulus resulted in a material with high toughness and low hysteresis loss.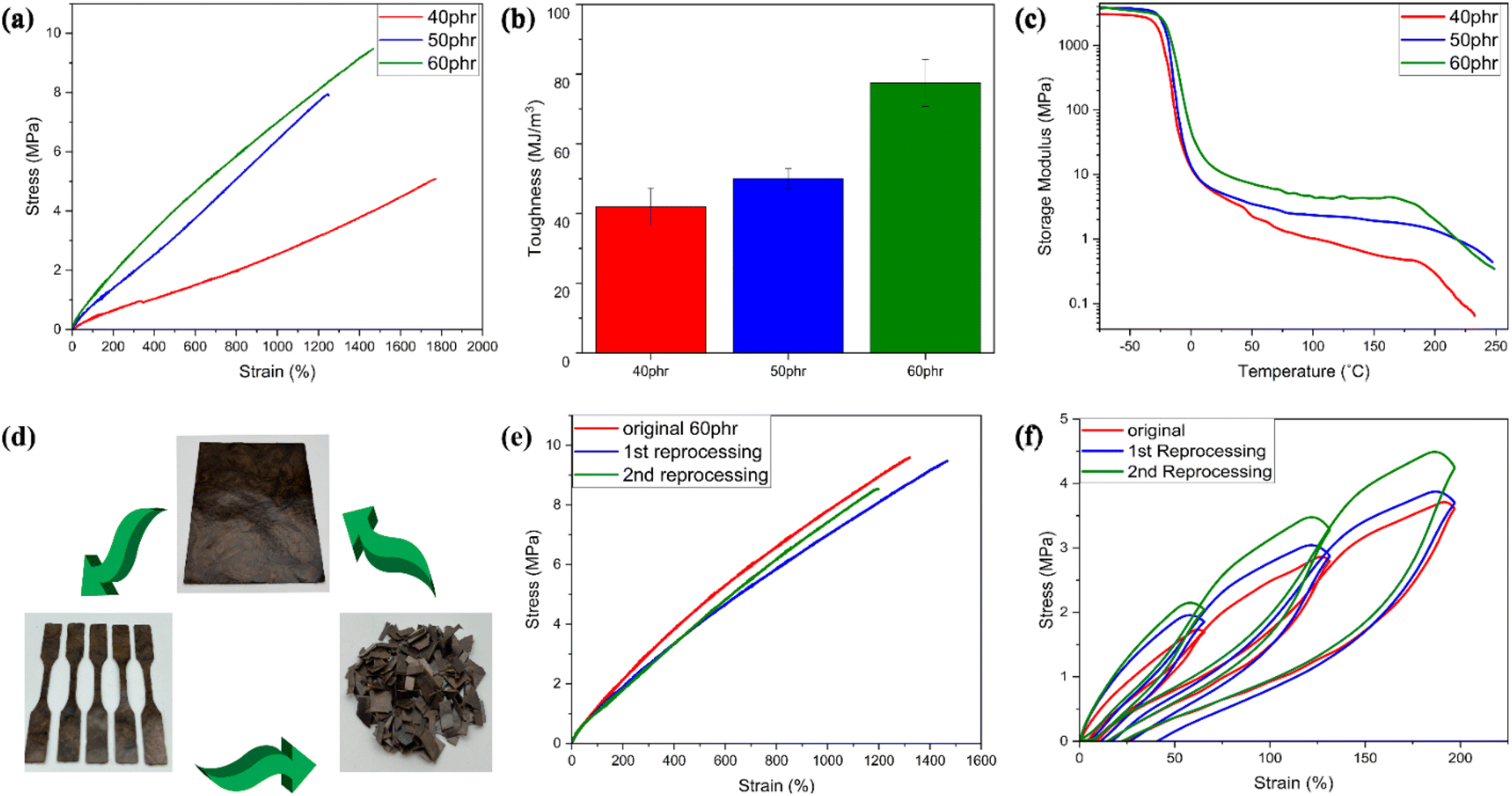 | ||
| Fig. 6 Mechanical characterization of ACN-lignin/ENR25 vitrimer: (a) stress–strain curves for ACN-lignin/ENR25 vitrimer with different lignin loadings. (b) Tensile toughness, proportionate to the area under the stress–strain curve. (c) Storage modulus of ACN-lignin/ENR25 vitrimer with different lignin loading as a function of temperature. (d) Schematic representation of reprocessability of vitrimer samples. (e) Stress–strain curves for 60 phr vitrimer sample for different reprocessing cycles at room temperature. (f) Extension–retraction stress–strain curves for original and reprocessed 60 phr vitrimer samples with different loading cycles. | ||
Conclusion
In summary, we demonstrated the synthesis of a TER-based vitrimer by using 100% renewable biomass starting materials. The vitrimer was synthesized by reacting functional groups of ACN-lignin and ENR25. The ACN fractionation of lignin received from pulp manufacturers resulted in a lignin with a narrower polydispersity and improved reactivity due to significant reduction in methoxy groups and around five-fold increase in the DS of carboxylic acid groups after extraction. A substantial increase in the Tg, modulus, and tensile properties was observed upon increasing lignin content in the reaction mixture. The ester cross-linkages undergo dynamic exchange with nucleophilic hydroxyl and carboxylic acid groups from lignin at elevated temperatures, which allows the topological rearrangement of the crosslinked matrix, thereby enabling material to show thermal stress relaxation behavior. The rate of relaxation decreased with lower lignin content, which was evidenced by increase in relaxation times and activation energy for productive exchange. The material was observed to retain its tensile properties after multiple reprocessing cycles, suggesting its potential utilization in circular applications. By taking advantage of sustainable and affordable feedstocks, this approach introduces a facile method to synthesize fully bio-based vitrimeric elastomers for potential applications in both industrial development and frontier research.Notes
This manuscript has been authored in part by UT-Battelle, LLC, under contract DE-AC0500OR22725 with the US Department of Energy (DOE). The US government retains and the publisher, by accepting the article for publication, acknowledges that the US government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for US government purpose. DOE will provide public access to these results of federally sponsored research in accordance with the DOE public access plan (https://energy.gov/downloads/doe-public-access-plan).Conflicts of interest
The authors declare the following financial interests which may be considered as potential competing interests: Naskar AK, Seo J, Rohewal SS, Toomey M, Kearney LT, Damron JT has pending US Patent Application (No. 18/532,599) titled “RECYCLABLE EPOXY-ANHYDRIDE POLYMER” assigned to UT Battelle, LLC.Acknowledgements
This work was supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, Materials Sciences and Engineering Division [FWP# ERKCK60], under contract DE-AC05-00OR22725 with UT-Battelle, LLC. S. S. R. acknowledges doctoral research scholarship through the U.S. Department of Energy, Office of Energy Efficiency and Renewable Energy (EERE), Vehicle Technologies Office (VTO).References
- A. I. Osman, M. Hefny, M. I. A. Abdel Maksoud, A. M. Elgarahy and D. W. Rooney, Environ. Chem. Lett., 2021, 19, 797–849 CrossRef CAS.
- H. Kaur and C.-C. Chen, Fluid Phase Equilib., 2020, 505, 112339 CrossRef CAS.
- R. S. El-Emam and H. Özcan, J. Cleaner Prod., 2019, 220, 593–609 CrossRef CAS.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y. X. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef CAS PubMed.
- H. Kaur, S. Abedi and C.-C. Chen, J. Chem. Eng. Data, 2022, 67, 1932–1950 CrossRef CAS.
- L. T. J. Korley, T. H. Epps Iii, B. A. Helms and A. J. Ryan, Science, 2021, 373, 66–69 CrossRef CAS PubMed.
- X.-L. Zhao, P.-X. Tian, Y.-D. Li and J.-B. Zeng, Green Chem., 2022, 24, 4363–4387 RSC.
- A. K. Mohanty, M. Misra and L. T. Drzal, J. Polym. Environ., 2002, 10, 19–26 CrossRef CAS.
- P. F. Cao, B. Li, T. Hong, J. Townsend, Z. Qiang, K. Xing, K. D. Vogiatzis, Y. Wang, J. W. Mays and A. P. Sokolov, Adv. Funct. Mater., 2018, 28, 1800741 CrossRef.
- M. Chen, L. Zhou, Y. Wu, X. Zhao and Y. Zhang, ACS Macro Lett., 2019, 8, 255–260 CrossRef CAS PubMed.
- Z. Tadmor, J. Appl. Polym. Sci., 1974, 18, 1753–1772 CrossRef CAS.
- L. Imbernon and S. Norvez, Eur. Polym. J., 2016, 82, 347–376 CrossRef CAS.
- B. T. Michal, C. A. Jaye, E. J. Spencer and S. J. Rowan, ACS Macro Lett., 2013, 2, 694–699 CrossRef CAS PubMed.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS PubMed.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef PubMed.
- Y. Jin, Z. Lei, P. Taynton, S. Huang and W. Zhang, Matter, 2019, 1, 1456–1493 CrossRef.
- F. I. Altuna, V. Pettarin and R. J. J. Williams, Green Chem., 2013, 15, 3360–3366 RSC.
- S. Dhers, G. Vantomme and L. Avérous, Green Chem., 2019, 21, 1596–1601 RSC.
- I. Choudhury and S. Hashmi, Encyclopedia of renewable and sustainable materials, Elsevier, 2020 Search PubMed.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna and M. Keller, et al. , Science, 2014, 344, 1246843 CrossRef PubMed.
- P. Phanthong, P. Reubroycharoen, X. Hao, G. Xu, A. Abudula and G. Guan, Carbon Resour. Convers., 2018, 1, 32–43 CrossRef.
- S. Zhang, T. Liu, C. Hao, L. Wang, J. Han, H. Liu and J. Zhang, Green Chem., 2018, 20, 2995–3000 RSC.
- A. Moreno, M. Morsali and M. H. Sipponen, ACS Appl. Mater. Interfaces, 2021, 13, 57952–57961 CrossRef CAS.
- D. Stewart, Ind. Crops Prod., 2008, 27, 202–207 CrossRef CAS.
- A. Naseem, S. Tabasum, K. M. Zia, M. Zuber, M. Ali and A. Noreen, Int. J. Biol. Macromol., 2016, 93, 296–313 CrossRef CAS PubMed.
- N. Kanbargi, M. Goswami, L. Collins, L. T. Kearney, C. C. Bowland, K. Kim, K. Rajan, N. Labbe and A. K. Naskar, ACS Appl. Polym. Mater., 2021, 3, 2911–2920 CrossRef CAS.
- J. Becker and C. Wittmann, Biotechnol. Adv., 2019, 37, 107360 CrossRef CAS PubMed.
- K. Sanderson, Nature, 2011, 474, S12–S14 CrossRef CAS PubMed.
- W. O. S. Doherty, P. Mousavioun and C. M. Fellows, Ind. Crops Prod., 2011, 33, 259–276 CrossRef CAS.
- T. Saito, R. H. Brown, M. A. Hunt, D. L. Pickel, J. M. Pickel, J. M. Messman, F. S. Baker, M. Keller and A. K. Naskar, Green Chem., 2012, 14, 3295–3303 RSC.
- J. H. Lora and W. G. Glasser, J. Polym. Environ., 2002, 10, 39–48 CrossRef CAS.
- M. Cui, N. A. Nguyen, P. V. Bonnesen, D. Uhrig, J. K. Keum and A. K. Naskar, ACS Macro Lett., 2018, 7, 1328–1332 CrossRef CAS PubMed.
- E. C. Corker, U. V. Mentzel, J. Mielby, A. Riisager and R. Fehrmann, Green Chem., 2013, 15, 928–933 RSC.
- M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, ACS Macro Lett., 2012, 1, 789–792 CrossRef CAS PubMed.
- L. Matějka, S. Pokomý and K. Dušek, Polym. Bull., 1982, 7, 123–128 CrossRef.
- A. K. Biswal, A. Nandi, H. Wang and A. Vashisth, Compos. Sci. Technol., 2023, 242, 110202 CrossRef CAS.
- L. Shechter and J. Wynstra, Ind. Eng. Chem., 1956, 48, 86–93 CrossRef CAS.
- P. N. Dave and A. Gor, Handbook of nanomaterials for industrial applications, 2018, pp. 36–66 Search PubMed.
- Y. Xu, K. Odelius and M. Hakkarainen, ACS Sustainable Chem. Eng., 2019, 7, 13456–13463 CrossRef CAS.
- C. A. Ruslimie, M. Y. Norhanifah, M. R. Fatimah Rubaizah and M. Asrul, CAMS-2015: 3rd International Conference on Chemical, Agricultural and Medical Sciences, 2015, 10–11 Search PubMed.
- T. Liu, C. Hao, L. Wang, Y. Li, W. Liu, J. Xin and J. Zhang, Macromolecules, 2017, 50, 8588–8597 CrossRef CAS.
- Y. Li, T. Liu, S. Zhang, L. Shao, M. Fei, H. Yu and J. Zhang, Green Chem., 2020, 22, 870–881 RSC.
- S. Wang, S. Ma, Q. Li, W. Yuan, B. Wang and J. Zhu, Macromolecules, 2018, 51, 8001–8012 CrossRef CAS.
- S. Salaeh, A. Das, S. Wießner and M. Stapor, Eur. Polym. J., 2021, 151, 110452 CrossRef CAS.
- C. Taplan, M. Guerre, J. M. Winne and F. E. Du Prez, Mater. Horiz., 2020, 7, 104–110 RSC.
- K. Pahnke, J. Brandt, G. Gryn'ova, P. Lindner, R. Schweins, F. G. Schmidt, A. Lederer, M. L. Coote and C. Barner-Kowollik, Chem. Sci., 2015, 6, 1061–1074 RSC.
- C. He, P. R. Christensen, T. J. Seguin, E. A. Dailing, B. M. Wood, R. K. Walde, K. A. Persson, T. P. Russell and B. A. Helms, Angew. Chem., Int. Ed., 2020, 59, 735–739 CrossRef CAS PubMed.
- P. Maity, S. V. Kasisomayajula, V. Parameswaran, S. Basu and N. Gupta, IEEE Trans. Dielectr. Electr. Insul., 2008, 15, 63–72 CrossRef CAS.
- Y. Horikawa, S. Hirano, A. Mihashi, Y. Kobayashi, S. Zhai and J. Sugiyama, Appl. Biochem. Biotechnol., 2019, 188, 1066–1076 CrossRef CAS PubMed.
- N. A. Mohamad Aini, N. Othman, M. H. Hussin, K. Sahakaro and N. Hayeemasae, Front. Mater., 2020, 6, 329 CrossRef.
- J.-Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef CAS PubMed.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. B. Ihsan, T. Akasaki, K. Sato, M. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932–937 CrossRef CAS PubMed.
- X. Zhao, Soft Matter, 2014, 10, 672–687 RSC.
- T. Shen, Z. Song, S. Cai and F. J. Vernerey, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2105974118 CrossRef CAS PubMed.
- X. Shi, Q. Ge, H. Lu and K. Yu, Soft Matter, 2021, 17, 2104–2119 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py01177a |
| This journal is © The Royal Society of Chemistry 2024 |