
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTerpolymerizations of cyclohexene oxide, CO2, and isocyanates or isothiocyanates for the synthesis of poly(carbonate–urethane)s or poly(carbonate–thioimidocarbonate)s†
Koichi
Nakaoka
,
Satoshi
Muranaka
,
Io
Yamamoto
and
Tadashi
Ema
 *
*
Division of Applied Chemistry, Graduate School of Natural Science and Technology, Okayama University, Tsushima, Okayama 700-8530, Japan. E-mail: ema@cc.okayama-u.ac.jp
First published on 31st January 2024
Abstract
Terpolymerization of cyclohexene oxide (CHO), CO2, and aryl isothiocyanates produced poly(carbonate–thioimidocarbonate)s with gradient character, while that of CHO, CO2, and aryl isocyanates furnished poly(carbonate–urethane)s with random sequences. The former underwent partial degradation upon acid treatment or UV irradiation, while the latter was stable under the same conditions.
Introduction
The ring-opening copolymerization (ROCOP) of epoxides and carbon dioxide (CO2) for the synthesis of aliphatic polycarbonates is a green and sustainable synthetic technology with 100% atom economy, and it has been intensively studied.1–3 On the other hand, the terpolymerization of epoxides, CO2, and comonomers, such as lactones,4 lactides,5 cyclic acid anhydrides,6 and heteroallenes,7,8 is an effective strategy for the development of new CO2-based polymers (Scheme 1a). The thermal, optical, mechanical, or degradation properties can be added or tuned by incorporating new polymer backbones derived from the comonomers at the expense of the CO2 content. The scope of comonomers and the tunability of the physical properties are important factors in the terpolymerizations. Heteroallenes used as comonomers have been limited to SO2 and COS,7,8 which are gases to be carefully used, while isocyanates or isothiocyanates have never been used to prepare CO2-based polymers despite the commercial availability, good reactivity, and tunability with substituents.9 The terpolymerizations of epoxides, CO2, and isocyanates or isothiocyanates may open up a new way for the development of novel CO2-based polymers.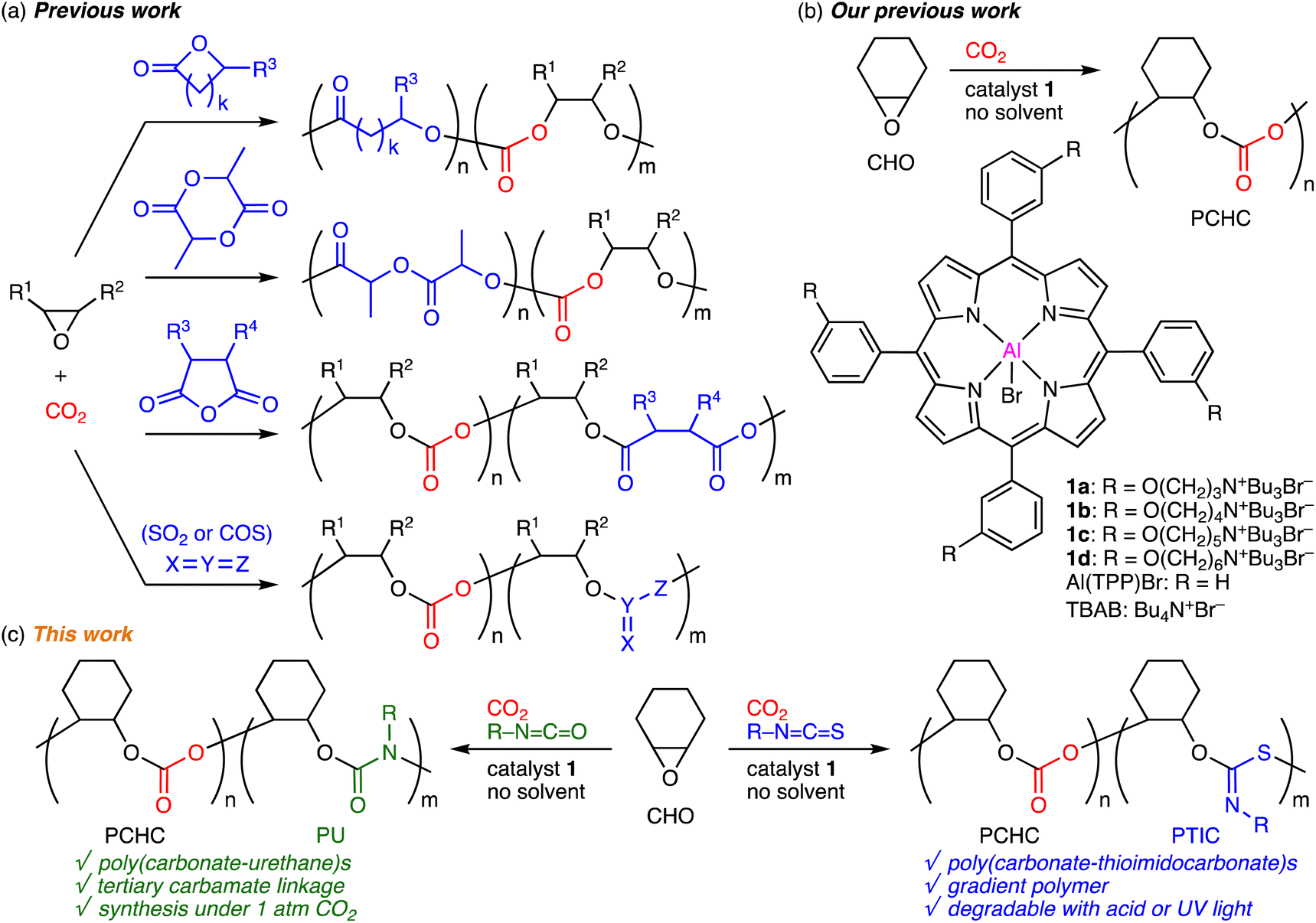 | ||
| Scheme 1 (a) Previous work on terpolymerization of epoxides, CO2, and comonomers. (b) Our previous work. (c) This work. | ||
In 2020, two groups independently achieved the first epoxide/isocyanate ROCOP (without CO2) to obtain new polyurethanes (PUs) with tertiary carbamate linkages,10a,b which are almost inaccessible via the conventional synthetic method with diisocyanates and diols. There are only several reports on this type of ROCOP partly because of the difficulty in using highly reactive isocyanates,10 which readily trimerize into isocyanurates.11 On the other hand, the epoxide/isothiocyanate ROCOP for the synthesis of poly(thioimidocarbonate)s (PTICs) was reported by three groups independently in 2021, and strong bases were used to activate isothiocyanates with poor reactivity as compared with isocyanates.12 These pioneering works suggest that the terpolymerizations of epoxides, CO2, and isocyanates or isothiocyanates might be difficult to achieve especially with a single catalyst because the ideal reaction conditions for the epoxide/CO2 ROCOPs are quite different from those for the epoxide/iso(thio)cyanate ROCOPs.
Previously, bifunctional MgII or ZnII porphyrin catalysts showed high activity for the synthesis of cyclic carbonates from epoxides and CO2,13 while bifunctional AlIII porphyrin catalysts 1 promoted the ROCOP of epoxides and CO2 efficiently to produce polycarbonates (Scheme 1b).14,15 The cooperative actions of the metal center and the quaternary ammonium halides led to the high activity and selectivity in both cases.16 More recently, we have also succeeded in the selective conversions of oxetanes and CO2 into trimethylene carbonates or poly(trimethylene carbonate)s with 1d.17 We envisioned that isocyanates or isothiocyanates could be used as comonomers in our catalytic system. Here we report the terpolymerizations of epoxide, CO2, and isocyanates or isothiocyanates for the first time (Scheme 1c). The terpolymerization of cyclohexene oxide (CHO), CO2 (2 MPa), and aryl isothiocyanates produced poly(carbonate–thioimidocarbonate)s with gradient character, while that of CHO, CO2 (1 atm), and aryl isocyanates afforded poly(carbonate–urethane)s with random sequences. The former underwent partial degradation upon acid treatment or UV irradiation to give polycarbonates, while the latter was stable under the same conditions.
Results and discussion
We investigated the terpolymerization of CHO, CO2 (2 MPa), and phenyl isothiocyanate (2a) with 1b, which showed the highest activity for the ROCOP of CHO and CO2.15 As a result, terpolymer 3a containing the poly(cyclohexene carbonate) (PCHC) and PTIC units were successfully obtained at 90 °C (Table 1, entry 1). 3a was isolated by reprecipitation (chloroform/methanol) and characterized by 1H NMR spectroscopy; the broad signals for the methine groups of the PCHC units appeared at 4.6 ppm, and those for the PTIC units were observed at 6.6–7.4 ppm (Fig. S6a, ESI†). Size-exclusion chromatography (SEC) indicated that 3a had high molecular weights with a bimodal distribution (Fig. S6c, ESI†). Bimodal molecular weight distributions are often observed for copolymerizations of epoxides and CO2, and higher-molecular-weight polymers are twice as large as lower-molecular-weight polymers.14 In the present terpolymerization, interestingly, the former was more than twice as large as the latter. The IR spectrum showed two strong absorptions at 1624 and 1759 cm−1 corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and CO stretching vibrations, respectively (Fig. S6d, ESI†). The MALDI-TOF mass spectrum of 3a showed two m/z intervals of 142 and 233 corresponding to the PCHC and PTIC units, respectively (Fig. S8, ESI†). The formation of terpolymers rather than blends was also confirmed by diffusion-ordered spectroscopy (DOSY) (Fig. S7, ESI†). Furthermore, we also synthesized a model compound to confirm the structure of 3a, and the NMR and IR spectra showed good similarities between 3a and the model compound (Fig. S13, ESI†).
N and CO stretching vibrations, respectively (Fig. S6d, ESI†). The MALDI-TOF mass spectrum of 3a showed two m/z intervals of 142 and 233 corresponding to the PCHC and PTIC units, respectively (Fig. S8, ESI†). The formation of terpolymers rather than blends was also confirmed by diffusion-ordered spectroscopy (DOSY) (Fig. S7, ESI†). Furthermore, we also synthesized a model compound to confirm the structure of 3a, and the NMR and IR spectra showed good similarities between 3a and the model compound (Fig. S13, ESI†).
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | 2 | S/Cb | Conv.c (%) | TONc | 3 | 4 | |||
| CHO | 2 | n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :md :md |
M
n
(kg mol−1) |
PDIe | Yieldc (%) | ||||
| a Reaction conditions: CHO (12.5 mmol), 2 (3.1 mmol), 1b (quantity indicated above), CO2 (2.0 MPa), 90 °C, 24 h, in an autoclave. b Ratio of CHO to 1b. c Determined by 1H NMR analysis of the crude reaction mixture. TON for the formation of 3. The yields of 4 were calculated based on 2. d Determined by 1H NMR analysis of the purified polymer. e Determined by SEC analysis of the purified polymer using THF as an eluent and polystyrene as a molecular-weight standard. Peaks had bimodal shapes. f CO2 (1.0 MPa). g CO2 (0.5 MPa). | |||||||||
| 1 | 2a | 6250 | 94 | 75 | 5400 | 4:1 |
83/34 | 1.1/1.1 | 6 |
| 2 | 2a | 20000 |
93 | 46 | 12400 |
5:1 |
164/63 | 1.0/1.3 | 3 |
| 3f | 2a | 20000 |
83 | 71 | 15600 |
3:1 |
167/55 | 1.1/1.3 | 7 |
| 4g | 2a | 20000 |
80 | 85 | 9000 | 2:1 |
85/22 | 1.1/1.4 | 10 |
| 5 | 2a | 40000 |
87 | 30 | 31600 |
8:1 |
183/57 | 1.1/1.5 | 3 |
| 6 | 2b | 40000 |
89 | 39 | 29600 |
9:1 |
121/41 | 1.1/1.4 | 0 |
| 7 | 2c | 40000 |
90 | 71 | 27600 |
3:1 |
74/25 | 1.1/1.3 | 0 |
| 8 | 2d | 40000 |
91 | 77 | 23500 |
4:1 |
132/45 | 1.1/1.4 | 0 |
| 9 | 2e | 40000 |
80 | >99 | 33100 |
2:1 |
164/59 | 1.1/1.2 | 0 |
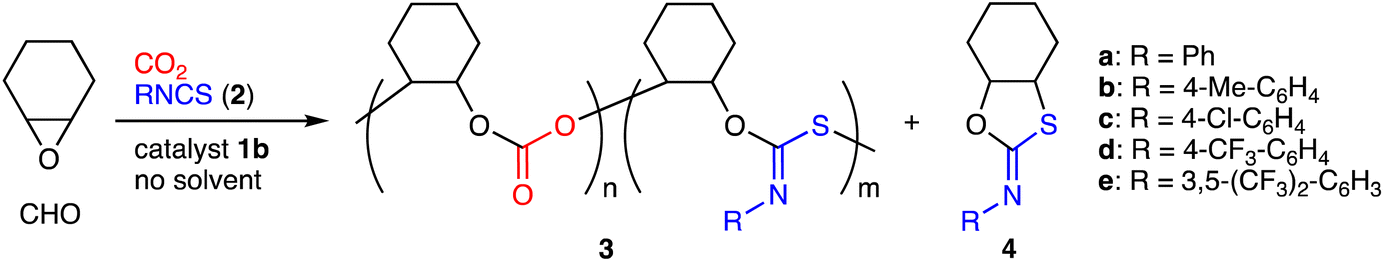
The reaction conditions were optimized. Higher temperature promoted the formation of byproduct 4a, while lower temperature resulted in a lower conversion of 2a; 90 °C was optimal (Table S1, entries 1–3, ESI†). When the molar ratio of CHO to 2a was set to 4:1, both the molecular weight of the polymer and the conversion of 2a were high (entries 2, 4–6). When the amount of catalyst 1b decreased from 0.016 to 0.0025 mol% (S/C = 40000), turnover numbers (TONs) increased from 5400 to 31600 to give high-molecular-weight polymers, and both the conversion of 2a and the PTIC content (n:m) decreased (Table 1, entries 1, 2 and 5). Interestingly, lower CO2 pressure led to a higher PTIC content (entries 2–4). These results suggest that although the formation of the PTIC units is slower than that of the PCHC units, the former becomes favorable as CO2 is consumed. This trend is consistent with the electrophilicity parameters reported for heteroallenes; isothiocyanates are less electrophilic than CO2.18 The linker length of catalysts 1 had a significant effect on the catalytic activity, and 1b exhibited the best result (Table S2, entries 1–4, ESI†). We consider that catalysts 1c and 1d have longer linkers that may hinder the polymer elongation owing to steric bulkiness, while catalyst 1a has shorter linkers that cannot assist well the ring-opening of CHO and/or the insertion of CO2 or 2a. In sharp contrast, a binary catalytic system composed of Al(TPP)Br and tetrabutylammonium bromide (TBAB) showed little or no activity for the PTIC formation under otherwise the same conditions (entry 5), which clearly demonstrates the importance of cooperative catalysis with bifunctional catalyst 1b.13–17
The scope of aryl isothiocyanates 2 was explored under the optimized conditions with 1b. Isothiocyanate 2b with the methyl group at the para position was modestly incorporated to form terpolymer 3b (Table 1, entry 6). In contrast, isothiocyanates 2c–e with electron-withdrawing groups showed much higher reactivity, and terpolymers 3c–e with higher PTIC contents were successfully obtained (entries 7–9).
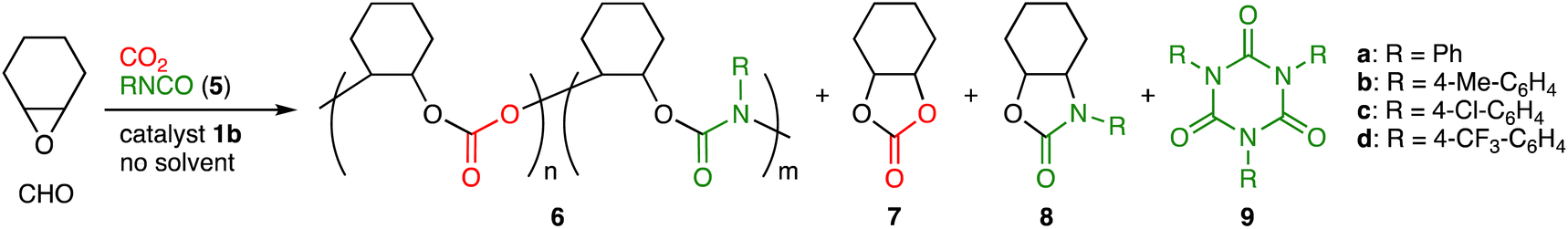
We next examined the reactivity of aryl isocyanates 5. In view of the facile conversion of 5a into isocyanurate 9a, we employed a syringe-pump for the slow dropwise addition of 5avia syringe under atmospheric CO2 pressure (balloon), which allowed us to optimize the reaction conditions for the terpolymerization of CHO, CO2, and 5a (Table S3, ESI†). Terpolymer 6a containing both the PCHC and PU units was obtained most efficiently with a catalyst loading of 0.016 mol% (S/C = 6250) (entries 1–3). The formation of byproduct 9a was minimal at 90 °C, although the formation of cyclohexene carbonate (7) and 2-oxazolidone 8a was suppressed at 80–100 °C (entries 2, 4 and 5). A faster dropwise addition of 5a resulted in the formation of a significant amount of 9a (entry 6). The use of 10 equiv. of CHO relative to 5a was better than that of 5 or 15 equiv. of CHO (entries 2, 7 and 8). The linker length of 1 had a crucial effect on the catalytic activity; 1b was the best catalyst (Table S4, entries 1–4, ESI†). In sharp contrast, a binary catalytic system composed of Al(TPP)Br and TBAB showed poor polymerization activity (entry 5), which demonstrates the advantage of bifunctional catalyst 1b.
Pure terpolymer 6a was obtained by reprecipitation (chloroform/methanol), and 6a was characterized by 1H NMR spectroscopy (Fig. S20a, ESI†). In addition to the broad signals at 4.6 ppm for the methine groups of the PCHC units, broad signals appeared at 6.9–7.5 ppm, which clearly indicates the incorporation of 5a. The 13C NMR spectrum showed the signals for the carbonyl groups of the PCHC and PU units at 153–154 ppm (Fig. S20b, ESI†). The IR spectrum showed two peaks for the CO stretching vibrations of the PU and PCHC units at 1707 and 1749 cm−1, respectively (Fig. S20d, ESI†). The structure of 6a was also analyzed by MALDI-TOF mass spectroscopy (Fig. S22a, ESI†); m/z intervals of 142 and 217 corresponding to the PCHC and PU units, respectively, were observed. DOSY also supported the formation of 6a (Fig. S21, ESI†). The analysis of hydrolysis products as well as the comparison of the NMR spectra between 6a and a model compound also supported the existence of the PCHC and PU units (Fig. S26 and S27, ESI†). Because 6a was prepared by the dropwise addition of 5a and because no systematic changes were seen in the ratios between the PCHC and PU units of 6a over the reaction time (data not shown), we consider that 6a had random sequences.
The scope of aryl isocyanates 5 was examined (Table 2). Isocyanate 5b with the methyl group at the para position was incorporated into terpolymer 6b accompanied with 9b. 4-Chlorophenyl isocyanate (5c) was successfully used to make 6c, while 4-(trifluoromethyl)phenyl isocyanate (5d) led to the formation of 6d with a larger molecular weight; byproduct 8d seems to result from the backbiting of the highly electrophilic carbonyl group of the PU unit by the adjacent terminal alkoxide ion. We also challenged the quaterpolymerizations of CHO, CO2, 2c or 2d, and 5c (Scheme 2 and Fig. S28–S33, ESI†). A mixture of 5c and CHO was added dropwise with the syringe-pump to a mixture of CHO, 2c or 2d, and 1b under CO2 (1 atm); as a result, quaterpolymers 10a with a Mn of 4.7 kg mol−1 and 10b with a Mn of 4.5 kg mol−1 were produced with the quantitative conversions of the comonomers (2c, 2d, and 5c). NMR, DOSY, IR, and mass spectra of the purified polymers indicated that the PCHC, PTIC, and PU units were contained in the polymer chains. When alkyl isocyanates were used instead of aryl isocyanates, the corresponding terpolymers 6 could not be obtained efficiently (data not shown). This is partly due to the rapid formation of the corresponding isocyanurates 9.
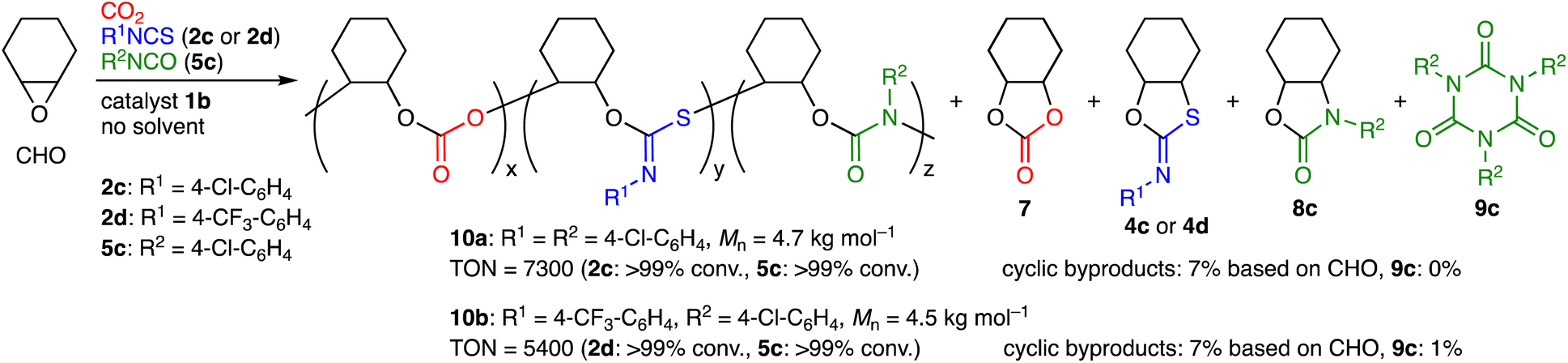 | ||
| Scheme 2 Quaterpolymerizations of CHO, CO2, 2c or 2d, and 5c. | ||
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | 5 | Conv.b (%) | TONb | 6 | Byproductb (%) | |||||
| CHO | 5 | n:mc |
M
n
(kg mol−1) |
PDId | 7 | 8 | 9 | |||
| a A mixture of CHO (1.2 mmol) and 5 (1.2 mmol) was added dropwise to a mixture of CHO (11.2 mmol) and 1b (S/C = 6250 for CHO, 0.016 mol%) at 15 μL h−1 with a syringe-pump under CO2 (1 atm, balloon) at 90 °C, and the mixture was then stirred at 90 °C for 2 h. b Determined by 1H NMR analysis of the crude reaction mixture. TON for the formation of 6. The yields of 7 were calculated based on CHO, and those of 8 and 9 were calculated based on 5. c Determined by 1H NMR analysis of the purified polymer. d Determined by SEC analysis of the purified polymer using THF as an eluent and polystyrene as a molecular-weight standard. e Addition at 24 μL h−1. | ||||||||||
| 1 | 5a | 81 | >99 | 3200 | 6:1 |
7.1 | 1.4 | 3 | 0 | 8 |
| 2 | 5b | 85 | >99 | 1900 | 2:1 |
4.2 | 1.4 | 1 | 0 | 29 |
| 3e | 5c | 79 | >99 | 3100 | 3:1 |
7.4 | 1.5 | 3 | 0 | 2 |
| 4e | 5d | 80 | >99 | 4200 | 9:1 |
14 | 1.3 | 4 | 8 | 12 |
Recently, degradable polymers have attracted much attention from the viewpoint of the promotion of chemical recycling and the mitigation of plastic pollution.19 Although sulfur-containing polymers are known to be susceptible to UV light or chemicals,19a there are no reports on the degradability of PTICs to our knowledge. We envisioned that terpolymers 3 containing the PTIC units might be degradable upon acid treatment or UV light irradiation. To our delight, 3 did undergo partial degradation by acid exposure or UV irradiation (Fig. 1a–c). Reprecipitation (chloroform/methanol) of the degradation mixtures and spectroscopic characterizations indicated that pure PCHCs were formed by the selective degradation of the PTIC linkages in both cases (Fig. S35 and S39, ESI†). Interestingly, Fig. 1b and c strongly suggests that terpolymers 3 have gradient character in composition (ratio of the PTIC to PCHC units);203c–e with electron-withdrawing groups showed larger decrease in molecular weight upon acid treatment or UV light irradiation than 3a and 3b. Accordingly, kinetic studies indicated that 2d was converted into the terpolymers faster than 2a (Fig. S14, ESI†). For comparison, block copolymer PCHC-b-PTIC (3e′) was synthesized in a one-pot two-step manner, which was confirmed to exhibit the selective degradation of the PTIC moiety (Fig. 1d, e and S42–S44, ESI†), while copolymer PTIC that was synthesized from CHO and 2e and purified in a similar manner was completely degraded (Fig. S49, ESI†). In sharp contrast, both PCHC and terpolymer 6a showed little or no degradability under the same conditions, which indicates that the PCHC and PU linkages are more robust (Fig. S37 and S41, ESI†).
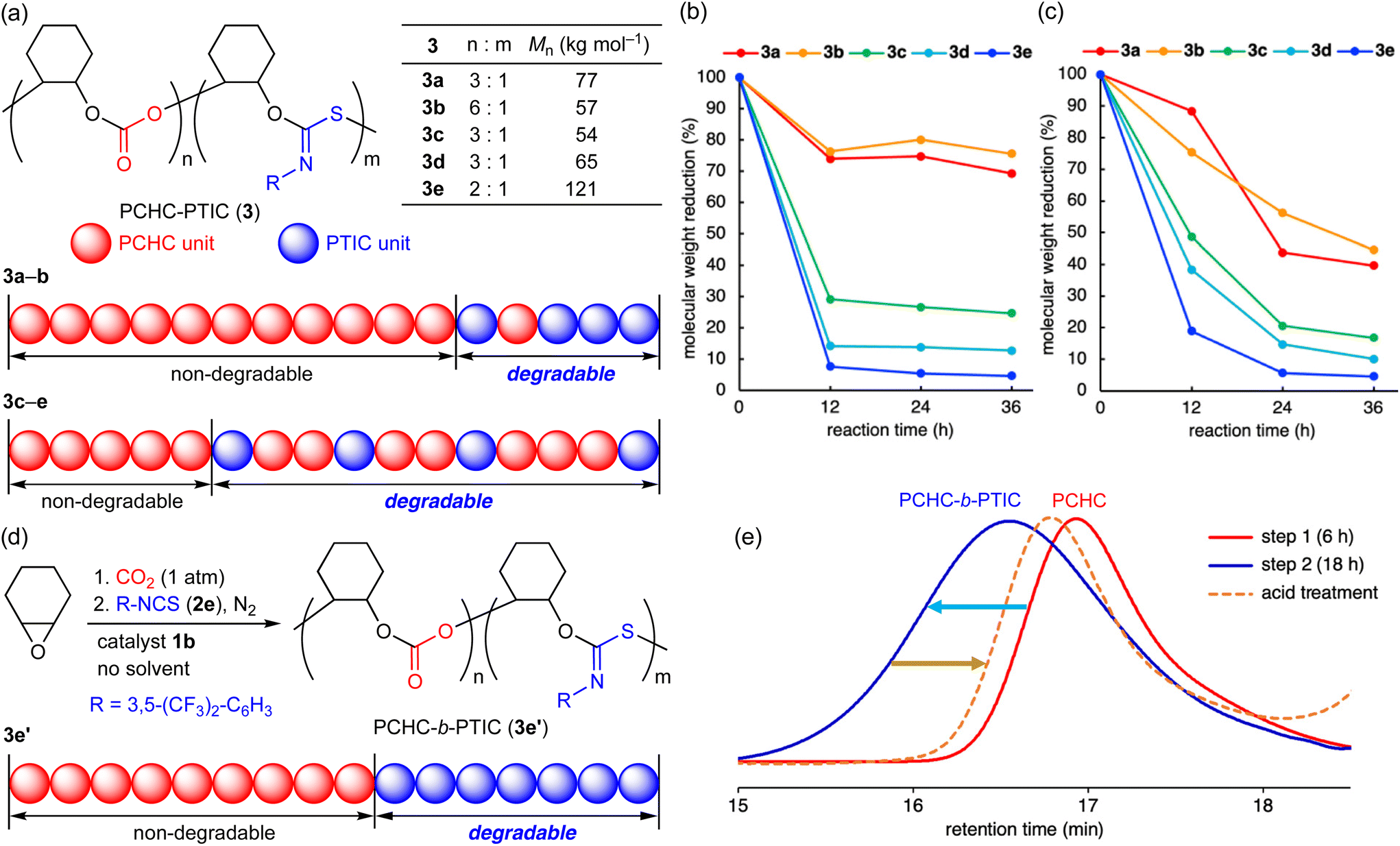 | ||
| Fig. 1 (a) Illustration of gradient character of 3. The molecular weights of 3 are average values for two peaks in SEC charts. Degradation of 3 upon (b) acid treatment and (c) UV irradiation. (d) One-pot two-step synthesis of block copolymer 3e′. (e) Synthesis and partial degradation of 3e’ monitored by SEC. | ||
Plausible catalytic cycles for the 1b-catalyzed polymerizations of CHO, CO2, and isothiocyanates/isocyanates are shown in Scheme 3. The nucleophilic attack of the counter anion on CHO activated by the Al center of 1b makes a new PCHC/PTIC/PU linkage, generating the alkoxide intermediate shown in the center. The subsequent insertion of CO2/isothiocyanate/isocyanate into the Al–O bond gives the carbonate/thioimidocarbonate/imidocarbonate anion, which then forms an ion pair with the quaternary ammonium ion of 1b upon CHO coordination. The S and N atoms of the thioimidocarbonate and carbamate anions, respectively, are more nucleophilic because the negatively charged atom with less electronegativity is more labile, which is the key determinant for the selective formation of the PTIC or PU linkage. In the PTIC cycle, the reaction of the alkoxide anion with isothiocyanate 2 is considered to be the rate-determining step, judging from the substituent effect of 2 on the ratio of the PTIC to PCHC units (Table 1), degradation behaviors with gradient character (Fig. 1), and kinetic studies (Fig. S14, ESI†). In the PU cycle, the PU linkage formation may be the rate-determining step, although the situation is complicated owing to the side reactions such as the formation of 9. Cyclic byproducts 7, 4, and 8 are formed by the backbiting of the terminal alkoxide ions just after the construction of the PCHC, PTIC, and PU linkages, respectively.
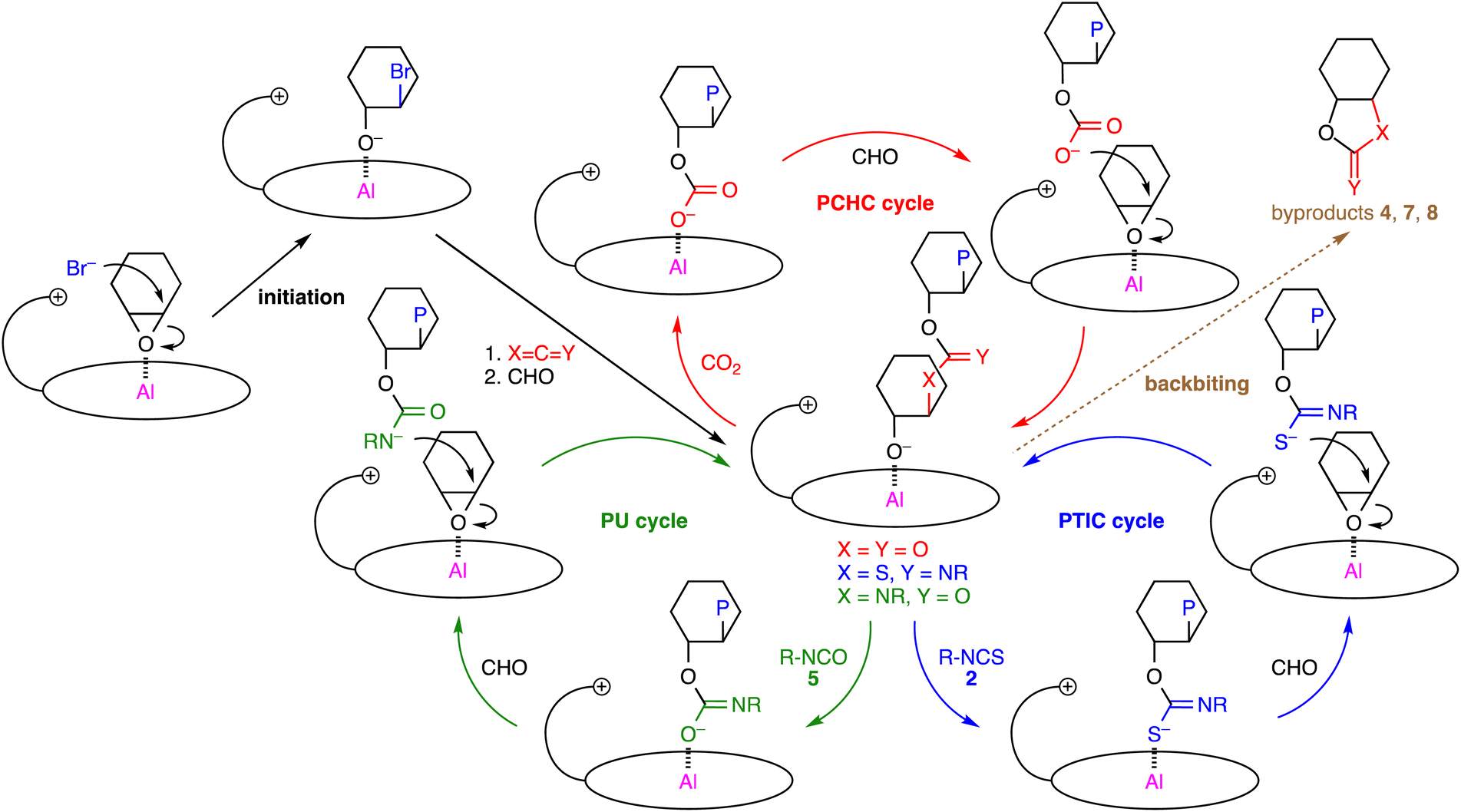 | ||
| Scheme 3 Plausible catalytic cycles, where P simply represents a part of a polymer chain that may differ in each step. | ||
The molecular weights of terpolymers 6 and quaterpolymers 10 synthesized at 1 atm CO2 pressure (Table 2 and Scheme 2) were much smaller than those of terpolymers 3 synthesized at 2.0 MPa CO2 pressure (Table 1). In the synthesis of 6 and 10, isocyanates 5 were added dropwise to minimize the formation of byproducts 9. It is likely that 6 and 10 growing at 1 atm CO2 pressure have the alkoxide anions at the ends, which may experience backbiting and protonation. In contrast, 3 growing at the high CO2 pressure can undergo the rapid addition of CO2 to the alkoxide anions to form the carbonate anions, which leads to the smooth ring-opening of CHO rather than backbiting or protonation. If 5 could be added dropwise at the high CO2 pressure, 6 and 10 with higher molecular weights would be obtained.
Conclusions
We have achieved terpolymerizations of epoxide, CO2, and isocyanates or isothiocyanates to synthesize new terpolymers for the first time. These terpolymerizations are fascinating because the reactions can proceed with 100% atom economy if no cyclic byproducts are formed. Herein, CHO was used as an epoxide because of the excellent physical properties of the resulting polycarbonates,15 while aryl iso(thio)cyanates were used because of the controllable reactivities. The terpolymerization of CHO, CO2, and isothiocyanates produced poly(carbonate–thioimidocarbonate)s showing degradability for acids and UV light. The ratio of the PTIC to PCHC units in the terpolymers could be controlled by the CO2 pressure, and the terpolymers had gradient character in composition. A block copolymer, poly(carbonate-b-thioimidocarbonate), was also synthesized in a one-pot two-step manner. On the other hand, the terpolymerization of CHO, CO2, and isocyanates yielded poly(carbonate–urethane)s; the slow addition of isocyanates under atmospheric CO2 pressure suppressed the formation of cyclic byproducts and enabled the formation of terpolymers with random sequences. It should be noted that all the terpolymerizations were catalyzed by a single catalyst, bifunctional AlIII porphyrin 1b, which demonstrates that cooperative catalysis with the metal center and the quaternary ammonium salts is effective for the conversions of the monomers with different reactivities into the terpolymers. The results reported here will be useful for the design and creation of environmentally benign polymers. Further studies on the application of the polymers and the creation of new CO2-based polymers are currently underway in our group.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by The Iwatani Naoji Foundation. We thank Ms. Tomoko Amimoto (Natural Science Center for Basic Research and Development (N-BARD), Hiroshima University) for the measurement of mass spectra of polymers.References
- Recent reviews: (a) Y. Wang and D. J. Darensbourg, Coord. Chem. Rev., 2018, 372, 85–100 CrossRef CAS; (b) C. M. Kozak, K. Ambrose and T. S. Anderson, Coord. Chem. Rev., 2018, 376, 565–587 CrossRef CAS; (c) M. Scharfenberg, J. Hilf and H. Frey, Adv. Funct. Mater., 2018, 28, 1704302 CrossRef; (d) A. J. Kamphuis, F. Picchioni and P. P. Pescarmona, Green Chem., 2019, 21, 406–448 RSC; (e) V. Paradiso, V. Capaccio, D. H. Lamparelli and C. Capacchione, Catalysts, 2020, 10, 825 CrossRef CAS; (f) J. Huang, J. C. Worch, A. P. Dove and O. Coulembier, ChemSusChem, 2020, 13, 469–487 CrossRef CAS PubMed; (g) G. A. Bhat and D. J. Darensbourg, Green Chem., 2022, 24, 5007–5034 RSC; (h) C. A. L. Lidston, S. M. Severson, B. A. Abel and G. W. Coates, ACS Catal., 2022, 12, 11037–11070 CrossRef CAS.
- Recent examples: (a) M. Reiter, S. Vagin, A. Kronast, C. Jandl and B. Rieger, Chem. Sci., 2017, 8, 1876–1882 RSC; (b) Z. Huang, Y. Wang, N. Zhang, L. Zhang and D. J. Darensbourg, Macromolecules, 2018, 51, 9122–9130 CrossRef CAS; (c) H. Nagae, R. Aoki, S. Akutagawa, J. Kleemann, R. Tagawa, T. Schindler, G. Choi, T. P. Spaniol, H. Tsurugi, J. Okuda and K. Mashima, Angew. Chem., Int. Ed., 2018, 57, 2492–2496 CrossRef CAS PubMed; (d) R. Duan, C. Hu, Z. Sun, H. Zhang, X. Pang and X. Chen, Green Chem., 2019, 21, 4723–4731 RSC; (e) N. G. Patil, S. K. Boopathi, P. Alagi, N. Hadjichristidis, Y. Gnanou and X. Feng, Macromolecules, 2019, 52, 2431–2438 CrossRef CAS; (f) H. Cao, Y. Qin, C. Zhuo, X. Wang and F. Wang, ACS Catal., 2019, 9, 8669–8676 CrossRef CAS; (g) H. Asaba, T. Iwasaki, M. Hatazawa, J. Deng, H. Nagae, K. Mashima and K. Nozaki, Inorg. Chem., 2020, 59, 7928–7933 CrossRef CAS PubMed; (h) W. Lindeboom, D. A. X. Fraser, C. B. Durr and C. K. Williams, Chem. – Eur. J., 2021, 27, 12224–12231 CrossRef CAS PubMed; (i) G.-W. Yang, C.-K. Xu, R. Xie, Y.-Y. Zhang, C. Lu, H. Qi, L. Yang, Y. Wang and G.-P. Wu, Nat. Synth., 2022, 1, 892–901 CrossRef; (j) Y.-N. Li, H.-H. Yang and X.-B. Lu, J. Polym. Sci., 2022, 60, 2078–2085 CrossRef CAS; (k) R. Zhang, Q. Kuang, H. Cao, S. Liu, X. Chen, X. Wang and F. Wang, CCS Chem., 2023, 5, 750–760 CrossRef CAS.
- S. Inoue, H. Koinuma and T. Tsuruta, J. Polym. Sci., Part B: Polym. Lett., 1969, 7, 287–292 CrossRef CAS.
- (a) C. Romain and C. K. Williams, Angew. Chem., Int. Ed., 2014, 53, 1607–1610 CrossRef CAS PubMed; (b) S. Paul, C. Romain, J. Shaw and C. K. Williams, Macromolecules, 2015, 48, 6047–6056 CrossRef CAS; (c) Y. Xu, S. Wang, L. Lin, M. Xiao and Y. Meng, Polym. Chem., 2015, 6, 1533–1540 RSC; (d) Y. Li, J. Hong, R. Wei, Y. Zhang, Z. Tong, X. Zhang, B. Du, J. Xu and Z. Fan, Chem. Sci., 2015, 6, 1530–1536 RSC; (e) S. Kernbichl, M. Reiter, F. Adams, S. Vagin and B. Rieger, J. Am. Chem. Soc., 2017, 139, 6787–6790 CrossRef CAS PubMed; (f) S. Kernbichl, M. Reiter, J. Mock and B. Rieger, Macromolecules, 2019, 52, 8476–8483 CrossRef CAS; (g) G. S. Sulley, G. L. Gregory, T. T. D. Chen, L. P. Carrodeguas, G. Trott, A. Santmarti, K.-Y. Lee, N. J. Terrill and C. K. Williams, J. Am. Chem. Soc., 2020, 142, 4367–4378 CrossRef CAS PubMed; (h) Z. Yang, C. Hu, F. Cui, X. Pang, Y. Huang, Y. Zhou and X. Chen, Angew. Chem., Int. Ed., 2022, 61, e202117533 CrossRef CAS PubMed; (i) Y. Zhou, Z. Gao, C. Hu, S. Meng, R. Duan, Z. Sun and X. Pang, Macromolecules, 2022, 55, 9951–9959 CrossRef CAS.
- (a) M. Kröger, C. Folli, O. Walter and M. Döring, Adv. Synth. Catal., 2006, 348, 1908–1918 CrossRef; (b) S. Liu, J. Wang, K. Huang, Y. Liu and W. Wu, Polym. Bull., 2011, 66, 327–340 CrossRef CAS; (c) L. Gu, Y. Qin, Y. Gao, X. Wang and F. Wang, Chin. J. Chem., 2012, 30, 2121–2125 CrossRef CAS; (d) L. Tang, W. Luo, M. Xiao, S. Wang and Y. Meng, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1734–1741 CrossRef CAS; (e) D. Xie, Z. Yang, L. Wu, C. Zhang and M. H. Chisholm, Polym. Int., 2018, 67, 883–893 CrossRef CAS; (f) C. Hu, R. Duan, S. Yang, X. Pang and X. Chen, Macromolecules, 2018, 51, 4699–4704 CrossRef CAS; (g) X. Li, C. Hu, X. Pang, R. Duan and X. Chen, Catal. Sci. Technol., 2018, 8, 6452–6457 RSC; (h) R. Duan, C. Hu, Z. Sun, H. Zhang, X. Pang and X. Chen, Green Chem., 2019, 21, 4723–4731 RSC; (i) N. Liu, C. Gu, M. Chen, J. Zhang, W. Yang, A. Zhan, K. Zhang, Q. Lin and L. Zhu, ChemistrySelect, 2020, 5, 2388–2394 CrossRef CAS; (j) Y. Chen, W. Wang, D. Xie, L. Wu and C. Zhang, J. Polym. Sci., 2021, 59, 1528–1539 CrossRef CAS; (k) X. Li, R.-L. Duan, C.-Y. Hu, X. Pang and M.-X. Deng, Polym. Chem., 2021, 12, 1700–1706 RSC; (l) X. Li, C.-Y. Hu, R.-L. Duan, Z.-Z. Liang, X. Pang and M.-X. Deng, Polym. Chem., 2021, 12, 3124–3131 RSC; (m) Y. Huang, C. Hu, X. Pang, Y. Zhou, R. Duan, Z. Sun and X. Chen, Angew. Chem., Int. Ed., 2022, 61, e202202660 CrossRef CAS PubMed.
- Recent reviews: (a) J. Liang, S. Ye, S. Wang, M. Xiao and Y. Meng, Polym. J., 2021, 53, 3–27 CrossRef; (b) D. H. Lamparelli and C. Capacchione, Catalysts, 2021, 11, 961 CrossRef CAS.
- Y. Zhi, Y. Miao, W. Zhao, J. Wang, Y. Zheng, H. Su, Q. Jia and S. Shan, Polymer, 2019, 165, 11–18 CrossRef CAS.
- T.-J. Yue, B.-H. Ren, W.-J. Zhang, X.-B. Lu, W.-M. Ren and D. J. Darensbourg, Angew. Chem., Int. Ed., 2021, 60, 4315–4321 CrossRef CAS PubMed.
- A. J. Plajer and C. K. Williams, Angew. Chem., Int. Ed., 2022, 61, e202104495 CrossRef CAS PubMed.
- (a) M. Jurrat, B. J. Pointer-Gleadhill, L. T. Ball, A. Chapman and L. Adriaenssens, J. Am. Chem. Soc., 2020, 142, 8136–8141 CrossRef CAS PubMed; (b) L. Song, W. Wei, M. A. Farooq and H. Xiong, ACS Macro Lett., 2020, 9, 1542–1546 CrossRef CAS PubMed; (c) M. Jia, N. Hadjichristidis, Y. Gnanou and X. Feng, Angew. Chem., Int. Ed., 2021, 60, 1593–1598 CrossRef CAS PubMed; (d) C. Hu, X. Chen, M. Niu, Q. Zhang, R. Duan and X. Pang, Macromolecules, 2022, 55, 652–657 CrossRef CAS; (e) Y.-Q. Teng, Y. Liu and X.-B. Lu, Macromolecules, 2022, 55, 9074–9080 CrossRef CAS.
- (a) Y. Taguchi, I. Shibuya, M. Yasumoto, T. Tsuchiya and K. Yonemoto, Bull. Chem. Soc. Jpn., 1990, 63, 3486–3489 CrossRef CAS; (b) Y. Nambu and T. Endo, J. Org. Chem., 1993, 58, 1932–1934 CrossRef CAS.
- (a) C. Chen, Y. Gnanou and X. Feng, Macromolecules, 2021, 54, 9474–9481 CrossRef CAS; (b) L. Song, M. Liu, D. You, W. Wei and H. Xiong, Macromolecules, 2021, 54, 10529–10536 CrossRef CAS; (c) T. Lai, P. Zhang, J. Zhao and G. Zhang, Macromolecules, 2021, 54, 11113–11125 CrossRef CAS; (d) X.-F. Zhu, X.-Y. Lu, C.-K. Xu, Y.-B. Fang, G.-W. Yang, W. Li, J. Wang and G.-P. Wu, Chin. J. Chem., 2023, 41, 3311–3318 CrossRef CAS.
- (a) T. Ema, Y. Miyazaki, S. Koyama, Y. Yano and T. Sakai, Chem. Commun., 2012, 48, 4489–4491 RSC; (b) T. Ema, Y. Miyazaki, J. Shimonishi, C. Maeda and J. Hasegawa, J. Am. Chem. Soc., 2014, 136, 15270–15279 CrossRef CAS PubMed; (c) C. Maeda, T. Taniguchi, K. Ogawa and T. Ema, Angew. Chem., Int. Ed., 2015, 54, 134–138 CrossRef CAS PubMed; (d) C. Maeda, J. Shimonishi, R. Miyazaki, J. Hasegawa and T. Ema, Chem. – Eur. J., 2016, 22, 6556–6563 CrossRef CAS PubMed; (e) C. Maeda, S. Sasaki and T. Ema, ChemCatChem, 2017, 9, 946–949 CrossRef CAS.
- J. Deng, M. Ratanasak, Y. Sako, H. Tokuda, C. Maeda, J. Hasegawa, K. Nozaki and T. Ema, Chem. Sci., 2020, 11, 5669–5675 RSC.
- C. Maeda, K. Kawabata, K. Niki, Y. Sako, T. Okihara and T. Ema, Polym. Chem., 2023, 14, 4338–4343 RSC.
- (a) J. Hasegawa, R. Miyazaki, C. Maeda and T. Ema, Chem. Rec., 2016, 16, 2260–2267 CrossRef CAS PubMed; (b) T. Ema, Bull. Chem. Soc. Jpn., 2023, 96, 693–701 CrossRef CAS.
- C. Maeda, H. Inoue, A. Ichiki, T. Okihara and T. Ema, ACS Catal., 2022, 12, 13042–13049 CrossRef CAS.
- Z. Li, R. J. Mayer, A. R. Ofial and H. Mayr, J. Am. Chem. Soc., 2020, 142, 8383–8402 CrossRef CAS PubMed.
- Recent reviews: (a) T.-J. Yue, L.-Y. Wang and W.-M. Ren, Polym. Chem., 2021, 12, 6650–6666 RSC; (b) A. Uva, A. Lin, J. Babi and H. Tran, J. Chem. Technol. Biotechnol., 2022, 97, 801–809 CrossRef CAS; (c) Q. Carboué, S. Fadlallah, M. Lopez and F. Allais, Macromol. Rapid Commun., 2022, 43, 2200254 CrossRef PubMed; (d) M. Dirauf, I. Muljajew, C. Weber and U. S. Schubert, Prog. Polym. Sci., 2022, 129, 101547 CrossRef CAS; (e) C. Hu, X. Pang and X. Chen, Macromolecules, 2022, 55, 1879–1893 CrossRef CAS.
- Selected review: M. M. Alam, K. S. Jack, D. J. T. Hill, A. K. Whittaker and H. Peng, Eur. Polym. J., 2019, 116, 394–414 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and compound characterizations. See DOI: https://doi.org/10.1039/d3py01165h |
| This journal is © The Royal Society of Chemistry 2024 |