
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A zuranolone nanocrystal formulation enables solubility-independent in vivo study of pentylenetetrazol-induced seizures in a rat model
Stanislav
Chvíla
 a,
Hana
Kubová
c,
Pavel
Mareš
c,
Eva
Kudová
*b and
František
Štěpánek
*a
a,
Hana
Kubová
c,
Pavel
Mareš
c,
Eva
Kudová
*b and
František
Štěpánek
*a
aDepartment of Chemical Engineering, University of Chemistry and Technology Prague, Technická 3, 166 28 Prague 6, Czech Republic. E-mail: stepanef@vscht.cz
bInstitute of Organic Chemistry and Biochemistry, Czech Academy of Sciences, Flemingovo nám. 542, 160 00 Prague 6, Czech Republic. E-mail: kudova@uochb.cas.cz
cInstitute of Physiology, Academy of Sciences of the Czech Republic, Videnska 1083, 142 20 Prague 4, Czech Republic
First published on 5th February 2024
Abstract
Neuroactive steroids are a promising class of substances with many potential therapeutic applications, but their preclinical evaluation is challenging due to very low aqueous solubility. A common practice is to “solubilise” such drugs using water-miscible solvents, but this approach has drawbacks: the drug can precipitate uncontrollably after injection, the solvent can artificially increase membrane permeability, and such formulations are not directly transferrable to humans. It would be beneficial to use the same physical form of the drug during preclinical and clinical studies. This work reports an approach based on an aqueous suspension of phospholipid-coated nanocrystals of zuranolone, chosen as a representative of poorly soluble neuroactive steroid drugs. The wet stirred media milling method was used for creating a nanosuspension with a mean particle size of d1,0 = 114 ± 39 nm, colloidally stable in PBS over 24 months at a concentration up to 100 mg mL−1. The applicability of the nanosuspension was demonstrated in a study of pentylenetetrazol-induced seizures in developing rats as a model of human generalized tonic–clonic seizures. The incidence and severity of seizures were assessed for the zuranolone nanosuspension and compared to an established dosage as a cyclodextrin complex. The incidence of generalized seizures with or without the tonic phase was found to be lower in P12 rats receiving zuranolone in doses of 0.5 and 1 mg kg−1 in the nanocrystal formulation than in those receiving the cyclodextrin solution. In contrast, both formulations significantly decreased seizure severity in P25 rats at a dose of 1 mg kg−1. Crucially, the nanocrystal formulation enabled the creation of a concentration series independent of the thermodynamic solubility of the drug. A constant volume appropriate to the body size of the young rats could therefore be injected during the in vivo study.
1. Introduction
Neurosteroids are endogenous molecules produced in the nervous system from cholesterol.1 The synthetic analogues of neurosteroids – neuroactive steroids – are rationally designed and synthesized with modified physicochemical or biological properties.2,3 Neurosteroids modulate neurotransmission through ligand-gated or voltage-gated ion channels, G protein-coupled receptors (GPCRs) or other mechanisms.4 Their physiological and pathological implications are related to the modulation of γ-aminobutyric acid type A receptors (GABAARs).5–7 Abnormalities in the operation of the GABAergic system have been closely linked to many central nervous system (CNS) diseases.8 Therefore, neurosteroids have been explored as potential candidates for treatment of neurodevelopmental and neuropsychiatric disorders, including but not limited to epilepsy, depression, schizophrenia, and traumatic brain injury.9 However, the physico-chemical properties of neurosteroids and neuroactive steroids, especially low aqueous solubility, make their pharmaceutical application challenging.Examples of successfully developed neurosteroid-based drugs include ganaxolone (ZTALMY®; Marinus Pharmaceuticals), which received approval in 2022 for the treatment of seizures associated with cyclin-dependent kinase-like 5 deficiency disorder in patients 2 years of age and older. Ganaxolone is available as an orally administered suspension.10 Additionally, allopregnanolone was approved in 2019 for the treatment of moderate to severe postpartum depression as a 60 h infusion with sulfobutylether-β-cyclodextrin due to its low oral bioavailability.11 Strategies for improving the oral bioavailability of allopregnanolone include prodrug formation (LYT-300, PureTech Health)12 or synthetic modification. Zuranolone (SAGE-217, Sage Therapeutics)13 is a new drug obtained by allopregnanolone substitution with a pyrazole moiety.14 It was approved for the treatment of post-partum depression in August 2023. Zuranolone has been tested in clinical trials also for major depressive disorder (MDD), bipolar depression, and even Parkinson's disease.13,15–17 Interestingly, a recently concluded Phase III study has shown that significant improvement of MDD symptoms has been achieved for an oral dose of 30 mg but not for 20 mg.18 Existing data demonstrate efficacy mostly at approximately the two-week mark, with the desired effect slowly tapering off.19,20
As evidenced by the above-mentioned studies, neurosteroid-based drugs can exhibit non-trivial dose–response relationships and complex temporal effects. It is therefore important to investigate this class of substances more thoroughly both in vitro and in vivo. The most common approach for conducting bioactivity studies with poorly soluble compounds is their “solubilisation” e.g. by forming a complex with cyclodextrins or by dissolution in a DMSO-containing buffer.21 This approach, however, does not guarantee consistency with later-used vehicles for experimental animal models and eventually formulations for human use. For example, DMSO exhibits a range of pharmacological effects22,23 such as phospholipid membrane disruption, which artificially increases drug permeation.24–27 Solubilisation vehicles also carry the risk of uncontrolled precipitation when the solution is injected into an aqueous environment, resulting in poor experimental reproducibility.28 Finally, the highest achievable drug concentration is still limited even when using a “solubilisation” vehicle, which can make dose–response studies challenging in small animal models where a certain maximum injectable volume cannot be exceeded. Therefore, it would be ideal to possess an aqueous vehicle in which an arbitrary concentration series of a poorly soluble drug could be readily prepared, and which could be directly transferable from pre-clinical to clinical studies without having to fundamentally change the chemical composition or physical form. Aqueous suspensions of drug nanocrystals represent an emerging formulation approach enabling just that.
Besides enabling the creation of solubility-independent concentration series, drug nanocrystal formulations offer a possible way of increasing the bioavailability and dissolution rate29 of poorly soluble substances thanks to a very high surface-to-volume ratio.30 It was recently shown that phospholipid-stabilised nanocrystals of curcumin exhibited excellent colloidal stability in a range of biorelevant media and their anti-proliferative activity on a carcinoma cell line was superior to that of first-line cytostatic drugs.28 A well-documented method for obtaining drug nanocrystals is wet stirred media milling31,32 whose main advantage is robust scalability, good control over particle size, and ability to work under sterile conditions. A comprehensive study on the wet stirred media milling method using a set of 10 poorly soluble FDA-approved active substances was recently conducted, demonstrating that a suspension of phospholipid-stabilised nanocrystals can be prepared universally.61 Apart from water, drug nanocrystal formulations contain two main components: the drug itself and a stabiliser system whose role is to ensure colloidal stability of the aqueous suspension during storage, in cell culture media, and after in vivo application.28 Stabilisers typically comprise polymers, surfactants or their combination to provide steric33 and/or electrostatic34 repulsion between the nanocrystal surfaces. In the present work, a new approach based on phospholipid stabilization of nanocrystals was employed. The phospholipid coating of drug nanocrystals was shown to provide excellent colloidal stability both for individual nanocrystals35,36 and composite vesicles.37–39 In the present work, a phospholipid-stabilised nanocrystal formulation of the neurosteroid drug zuranolone has been developed and its efficacy was evaluated in an experimental animal model of pentetrazol-induced seizures in young rats40 using a hydroxypropyl-β-cyclodextrin inclusion complex as a reference. For the sake of the present work, zuranolone was chosen as a model drug representing a class of poorly soluble neuroactive steroids. During preclinical development of these substances, it is common to conduct in vivo bioactivity studies during which it is preferred to administer the investigated substance by a parenteral route to ensure accuracy and consistency of dosing. Therefore, even if the final marketed product is administered orally, there is a need for a solubility-independent formulation approach suitable for injection. The aim of this work is to demonstrate such an approach.
2. Materials and methods
2.1. Materials
Zuranolone (ChemShuttle, Hayward, CA, USA, catalogue number 186443, CAS 1632051-40-1) for this study was kindly provided by the Neurosteroids group at the Institute of Organic Chemistry and Biochemistry (Prague, Czech Republic). Sodium hydroxide (p.a.) and phosphate buffered saline (PBS) tablets were purchased from Sigma-Aldrich. Dipalmitoylphosphatidylcholine (DPPC), sodium dipalmitoylphosphoglycerole (DPPG) and N-(carbonyl-methoxypolyethylenglycol 2000)-1,2-dimyristoyl-sn-glycero-3-phosphoethanol-amine sodium salt (MPEG-2000-DMPE) were purchased from Corden Pharma (Switzerland). Deionized water (0.07 μS cm−1) used for all reactions and treatment processes was prepared using an Aqual 25 generator. All chemicals were used as received.2.2. Preparation of nanocrystal formulation
An aqueous suspension of lipid-coated zuranolone nanocrystals was produced through a batch wet stirred media milling process as follows: 60 mg of zuranolone, 30.2 mg of DPPC, 3.6 mg of DPPG, and 6.2 mg of MPEG-2000-DMPE were placed in a 25 mL amber glass vial with 5 g of 0.4–0.5 mm ZrO2 milling beads and 1 mL of water. Milling was carried out at room temperature for 24 h at 600 rpm using a cross-magnetic stirrer (8 × 20 mm). Afterwards, the suspension was worked up by the addition of 2 mL of deionized water, stirred for 2 minutes to achieve homogenization and subsequently removed from the milling beads using a pipette, obtaining a stock nanosuspension with a zuranolone concentration of approximately 20 mg mL−1. This suspension of nanocrystals was stored in darkness at 4 to 8 °C for up to 24 months to determine its long-term stability. A drug-free vehicle to be used as a control during in vivo experiments (i.e. a solution of phospholipids in water) was prepared using the same procedure, except without zuranolone.2.3. Preparation of a cyclodextrin complex
A common formulation approach used during pre-clinical evaluation of poorly soluble drugs including neuroactive steroids is the preparation of cyclodextrin solutions. For this reason, a cyclodextrin (CDX) solution of zuranolone was used as a reference and compared with the lipid-coated nanocrystal suspension. The solution was prepared as follows: 30 mg of zuranolone was dissolved in a solution of 3 g of (2-hydroxypropyl)-β-cyclodextrin (CDX, Sigma-Aldrich) and 157 mg of citric acid (3-hydroxypenta-1,3,5-tricarboxylic acid, Sigma-Aldrich) in 30 mL of distilled water. The pH was adjusted to 7.4 with sodium hydroxide (NaOH, Sigma-Aldrich). A drug-free vehicle to be used as a control during in vivo experiments was prepared using the same procedure, except without zuranolone.2.4. Characterization of raw zuranolone
The morphology of crude zuranolone crystals before milling was examined by scanning electron microscopy (SEM, Jeol JCM-5700). Their degree of crystallinity was determined by X-ray powder diffraction (XRPD). The XRPD data were collected at room temperature using a PANalytical X'Pert PRO powder diffractometer equipped with a PIXcellD_1D detector over the angular range of 5–50° (2θ) with a step size of 0.039° (2θ). Data evaluation was performed using the software package HighScore Plus 3.0e.2.5. Characterization of zuranolone nanocrystal suspensions
The particle size distribution of freshly prepared and aged nanosuspensions (1 and 2 years of storage) was determined by dynamic light scattering (DLS) utilizing a NANO-flex fibre optic instrument (Microtrac, Germany). Prior to each measurement, 50 μL of the nanosuspension was diluted with 950 μL of deionized water in an Eppendorf vial, briefly shaken to homogenise the mixture and measured in a sequence of five 60-second DLS measurements. The number- and volume-based particle size distributions were used for calculating the mean values of particle size and their standard deviations. The morphology of zuranolone nanocrystals after milling was analysed by transmission electron microscopy (TEM, Jeol JEM-1010) using an acceleration voltage of 80 kV. Before deposition on the TEM grids, the nanosuspensions were diluted 20 times to avoid particle overlap.The crystallinity of the nanosuspension was determined by XRPD using the same instrument and measurement conditions as those used for raw zuranolone. Before XRPD analysis, the nanocrystal suspension was freeze-dried as follows: 750 μL of the zuranolone nanosuspension was placed in a 2 mL cryogenic vial, closed without airtight sealing and shock-frozen in liquid nitrogen for 150 seconds. The vial was then transferred into a round-bottom flask and lyophilised for 48 hours. The lyophilised material was stored in darkness at 4 to 8 °C before XRPD measurement.
2.6. Determining the concentration of zuranolone nanosuspensions
To determine that zuranolone was not degraded during the wet milling process, the actual concentration of zuranolone in the nanosuspension was measured via high-performance liquid chromatography (HPLC) as follows: 20 μL of the nanosuspension was diluted in 25 mL of isopropanol and subjected to liquid chromatography using an Agilent 1100 HPLC device. A Symmetry® C18 100 Å, 3.5 μm, 4.6 × 75 mm column was used as the stationary phase. The mobile phase was a mixture of ammonium phosphate buffer (pH 2.5) (A) and acetonitrile/water (B), detailed in Table 1. The flow rate was set to 1 mL min−1, the detection wavelength to 220 nm, the column temperature to 40 °C, and the injection volume was 15 μL. The run time was 11 minutes, with the main peak at 6.7 min. The dilution step was carried out four times to account for any pipetting errors.| Time [min] | Perc. phase B | Flow [mL min−1] |
|---|---|---|
| 0.0 | 10.0 | 1.0 |
| 0.4 | 10.0 | 1.0 |
| 4.5 | 80.0 | 1.0 |
| 9.0 | 80.0 | 1.0 |
| 11.0 | 10.0 | 1.0 |
2.7. In vivo study
Experiments were performed on male Wistar albino rats (n = 98, Institute of Physiology of the Czech Academy of Sciences) on two postnatal days (P12 and P25). The day of birth was defined as day 0. Rats were housed in a controlled environment (temperature 22 ± 1 °C, humidity 50–60%, lights on 6 am–6 pm) with free access to food and water. During experiments with pups, the temperature in Plexiglas cages was maintained at 32 ± 2 °C using an electric heating pad connected to a digital thermometer to compensate for immature thermoregulatory functioning at this age.41All procedures involving animals and their care were conducted according to the ARRIVE guidelines42 in compliance with national (Act No. 246/1992 Coll.) and international laws and policies (EU Directive 2010/63/EU for animal experiments and the National Institutes of Health Guide for the Care and Use of Laboratory Animals, NIH Publications No. 8023, revised 1978). The experimental protocol was approved on July 13, 2022, by the Ethical Committee of the Czech Academy of Sciences (Approval No. 35/2022).
The nanocrystal suspension of zuranolone, the cyclodextrin (CDX) solution of zuranolone, or drug-free vehicles were administered intraperitoneally. The zuranolone concentration in both the CDX solution and the nanocrystal suspension was 1 mg mL−1. To study its anticonvulsant action, zuranolone dissolved in CDX was administered in doses of 0.25, 0.5 and 1 mg kg−1. The nanocrystal formulation was administered in doses of 0.5 and 1 mg kg−1; the dose of 0.25 mg kg−1 was administered to the group of 25-day-old rats. Controls were injected with each vehicle of the corresponding volume.
The anticonvulsant effect of zuranolone was tested in a model of pentylenetetrazol (PTZ)-induced seizures described before.43 Pentylenetetrazol (Sigma-Aldrich) dissolved in distilled water at a concentration of 50 mg mL−1 was administered subcutaneously at a dose of 100 mg kg−1, 20 min after the administration of zuranolone. Seizure severity and incidence of generalized seizures (GS) starting with a loss of righting reflexes with (GTCS) or without (GCS) the tonic phase44 were registered and statistically evaluated. Data acquisition and analysis were done blinded to the treatment. Data were analyzed using GraphPad Prism 8 (GraphPad Software, United States) software. Using the D'Agostino–Pearson normality test, all data sets were first analysed to determine whether the values were derived from a Gaussian distribution. The Kruskal–Wallis test was used to compare the effects of zuranolone in either formula on the severity of PTZ-induced seizures. The incidence of GTCS, the most severe type of seizure induced with PTZ, was statistically compared by means of Fisher's test. In the text, data are presented as means with SD. The level of statistical significance was set as p = 0.05.
3. Results and discussion
3.1. Physico-chemical properties of zuranolone nanocrystal suspensions
Raw zuranolone consisted of clusters of rectangular cuboid crystals ranging in size from 1 to 100 μm (Fig. 1A). After wet stirred media milling, the nanocrystals retained the rectangular cuboid morphology, ranging in size between approximately 100 and 500 nm (Fig. 1B). The particle size distribution of zuranolone nanocrystals measured by DLS (Fig. 2A) confirmed the qualitative TEM observation and provided a number-mean particle size of d1,0 = 114 ± 39 nm for the freshly milled nanosuspensions, with a polydispersity index (PDI) of 0.311. The nanosuspension revealed remarkably good colloidal stability over time: even after 24 months of storage, no particle aggregation, sedimentation, or phase separation was observed, and the particle size distribution remained practically unchanged (Fig. 2A). The number-mean particle size after 24 months was d1,0 = 122 ± 41 nm with a PDI of 0.388. The 10th, 50th and 90th percentiles of the particle size distribution (d10, d50 and d90) for the nanocrystal suspensions determined for both timeframes are summarised in Table 2. | ||
| Fig. 1 (A) SEM micrograph of raw crystalline zuranolone. (B) TEM micrographs of zuranolone nanocrystals produced by wet milling, shown at two different magnifications to reveal the uniformity of the crystal size and morphology. | ||
 | ||
| Fig. 2 (A) Number-weighted particle size distributions of zuranolone nanosuspensions measured by DLS immediately after milling and dilution (orange line), and after 24 months of storage in darkness at 4–8 °C (blue line). (B) Concentration series of zuranolone nanocrystal suspensions (the values are concentrations in mg mL−1). | ||
| Time | d 10 [nm] | d 50 [nm] | d 90 [nm] |
|---|---|---|---|
| Immediate | 51.1 | 68.8 | 168.2 |
| 24 months | 67.0 | 82.4 | 195.1 |
Visually, the nanosuspension was a homogeneous milky liquid of water-like viscosity (Fig. 2B). The main advantage of the nanocrystal suspension is that a broad range of concentrations can be created regardless of the thermodynamic solubility of zuranolone. Zuranolone is practically insoluble in water, and even its solubility in organic solvents is not very high: 5 mg mL−1 in ethanol and 82 mg mL−1 in DMSO.45 On the other hand, it was possible to prepare a 100 mg mL−1 aqueous nanocrystal suspension with relative ease (Fig. 2B). The nanocrystal suspension can be handled in the same way as aqueous solutions: it can be diluted arbitrarily, pipetted to well-plates containing a cell culture, or injected into animals.
The analytically determined zuranolone content in the freshly prepared nanocrystal suspension for the in vivo study (prepared as described in section 2.2) was 1.02 ± 0.03 mg mL−1, indicating that no chemical degradation or decomposition took place during the milling process. Analysis by XRPD (Fig. 3 and Table 3) revealed that the nanocrystals retained the original crystalline form of zuranolone, implying that the mechanical stresses endured during the wet milling step did not cause amorphization or polymorphic transformation. Likewise, no polymorphic transformation occurred during storage, as identical characteristic XRPD peaks were found in the aged sample.
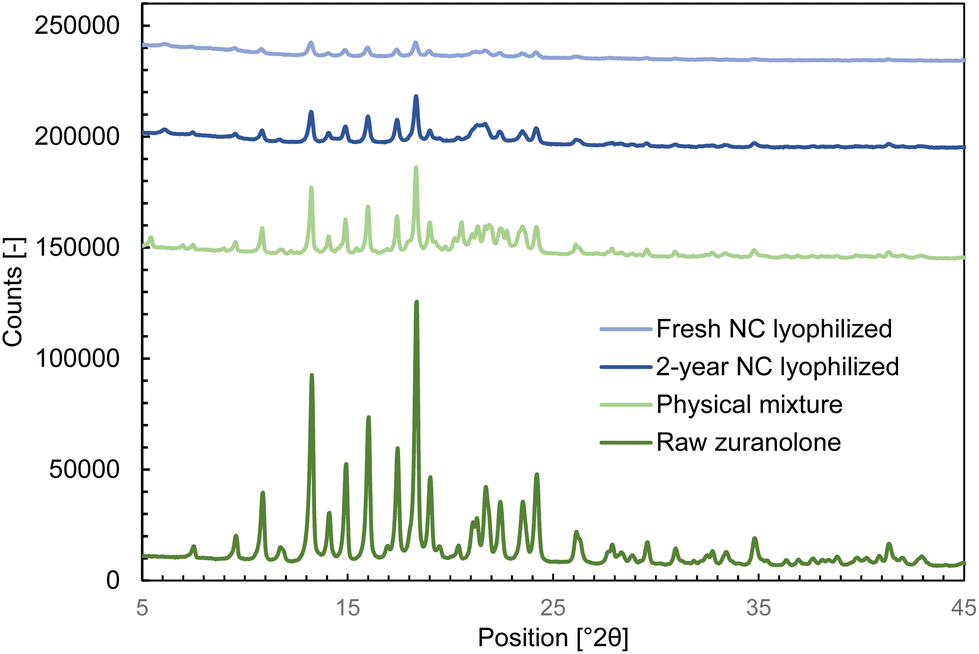 | ||
| Fig. 3 XRPD diffractograms of a freeze-dried freshly prepared zuranolone nanocrystal suspension, freeze-dried nanocrystal suspension after 24 months of storage, physical mixture of non-milled crystalline zuranolone with phospholipids at the same ratio as in the nanosuspension, and pure non-milled crystalline zuranolone. | ||
| Lyophilized nanocrystals | Zuranolone Form A46 |
|---|---|
| 9.6 | 9.3 to 9.7 |
| 10.9 | 10.6 to 11.0 |
| 13.3 | 13.0 to 13.4 |
| 14.9 | 14.7 to 15.1 |
| 16.0 | 15.8 to 16.2 |
| 18.4 | 18.1 to 18.5 |
| 19.0 | 18.7 to 19.1 |
| 21.1 to 21.3 | 20.9 to 21.3 |
| 21.7 | 21.4 to 21.8 |
| 23.5 | 23.3 to 23.7 |
3.2. In vivo efficacy of the zuranolone nanocrystal formulation
It has been widely recognized that the developing brain is peculiarly vulnerable to various hazards, including seizures. As illustrated using ganaxolone,10 neurosteroids possess great potential for the treatment of paediatric types of seizures. Therefore, a model of PTZ-induced seizures in developing brains of 12- and 25-day-old animals has been used in the present work for the evaluation of the efficacy of zuranolone. The animal model of PTZ-induced seizures in developing rats is a relevant model of human generalized tonic–clonic seizures and has led to the identification of most of the currently approved drugs.47 For the extrapolation of experimental data to human studies, the correlation between the age of animals and that of humans has already been described in the literature. In brief, 12- and 25-day-old rats correspond to early postnatal and preschool or early school age in infants and children, respectively.48–51The setting of the experiment was established as follows. The animals were administered the nanocrystal formulation of zuranolone or zuranolone in CDX solution (for dose, see the Materials and methods section). After 20 minutes of pre-treatment, the animals were administered pentylenetetrazol (100 mg kg−1s.c.). The rats were placed individually in plexiglass boxes and were then observed for 30 min after PTZ injection. The incidence and severity of seizures were assessed. The results are summarized in Fig. 4. As revealed by the Kruskal Wallis test (H = 28.30), in P12 rats the seizure severity decreased after pretreatment with either dose of zuranolone in the nanocrystal formulation (p = 0.014 and p = 0.0011, respectively). Zuranolone dissolved in CDX did not significantly affect the severity of seizures in P12 rats, expressed as a score (for details, see ref. 44). The incidence of generalized seizures (GS) with or without the tonic phase was lower in animals receiving zuranolone in doses of 0.5 and 1 mg kg−1 in the nanocrystal formulation (Fisher's test: p = 0.0125 and p = 0.009, respectively) than those receiving zuranolone dissolved in CDX.
 | ||
| Fig. 4 Effects of zuranolone in a nanocrystal formulation (white columns) and CDX solution (blue columns) on the severity of seizures and incidence of generalized seizures with or without the tonic phase (GS) induced with pentylenetetrazol (PTZ) administered subcutaneously in a dose of 100 mg kg−1 in 12-day (P12, on the left) and 25-day (P25, on the right) old rats. In P12 rats, seizure severity (on the top) expressed as a score was significantly suppressed by the nanocrystal formulation of zuranolone in doses of 0.5 and 1 mg kg−1, administered 20 min before the injection of PTZ, whereas zuranolone dissolved in CDX was ineffective. The protective effect was driven mostly by the decreased incidence of generalized seizures (GS on the bottom). In P25 rats, zuranolone in a dose of 0.5 mg kg−1 significantly decreased seizure severity in both formulations. Both formulations equally suppressed the incidence of GS in doses of 0.5 and 1 mg kg−1. *p < 0.05 compared to corresponding controls, #p < 0.05 comparison of corresponding doses in different formulations. | ||
In contrast, in P25 rats, both formulations of zuranolone in a dose of 1 mg kg−1 significantly decreased seizure severity (Kruskal Wallis test, H = 28.80; p = 0.0241 and 0.0003, respectively). Zuranolone in doses of 0.5 and 1 mg kg−1 decreased the incidence of GS in both formulations (Fisher's test: nanocrystal formulation of zuranolone p = 0.001 and p = 0.0256, respectively; zuranolone in CDX p = 0.0047 and p = 0.0047, respectively). The significant effect on the incidence of GS in P12 animals after the administration of a lower dose of the nanosuspension than that of the CDX solution might be caused by higher concentration levels of zuranolone in the brain. A possible difference in brain concentrations could have various causes such as improved penetration into the brain, reduced efflux, or changes at the level of metabolism.
The definitions of “developing” and “adult” animals are not related only to the sexual maturity of the rodents. Many systems in developing rats are immature and it takes weeks or months for these systems to develop to full maturity. For example, changes in the expression of major drug efflux transporters (e.g. adenosine triphosphate binding cassette (ABC) transporters) and related enzymes (CYPP450, glutathione-s-transferase, sulfotransferase, etc.) at the blood–brain and blood–cerebrospinal-fluid barriers may significantly affect the permeability of drugs into the central nervous system.52,53 The ABC transporters regulate the entry of lipidic molecules across cellular barriers to reduce the entry of compounds from the blood into the brain by active efflux.54 The current literature demonstrates an increase in transcript expression of efflux transporters and related enzymes during development.55,56 Additionally, the expression and function of transporters are regulated by a variety of factors, including age, dose and length of the treatment,55,57 nuclear receptors,58 xenobiotics/drugs59 and a variety of inflammatory molecules.60
Similarly to changes in the expression of drug transporters, changes in the developmental expression patterns of GABAA receptor subunits should be considered. All hypotheses suggested are heavily limited by insufficient data available in the current literature related to CNS research. While the verification of these hypotheses falls beyond the scope of the present work, our study provides a general framework for the administration of poorly soluble substances to young rats in the form of a nanosuspension formulation, which is an enabling tool for further research on neuroactive steroids in the developing brain.
4. Conclusion
Suspensions of phospholipid-stabilized nanocrystals of zuranolone, a poorly soluble neuroactive steroid drug, have been successfully and repeatedly prepared by wet stirred media milling. These nanocrystal suspensions retained their particle size distribution, colloidal stability, and crystallinity over 24 months of storage in darkness at low temperatures. A broad range of zuranolone concentrations in the nanocrystal suspension could be achieved independently of its thermodynamic solubility. Specifically, a colloidally stable aqueous nanocrystal suspension with a concentration of 100 mg mL−1 could be prepared with ease, exceeding even the solubility of zuranolone in pure DMSO (82 mg mL−1). The nanocrystal suspension can be handled in the same way as aqueous solutions (arbitrary dilution series, pipetting, and application by injection).To evaluate the applicability of the nanocrystal formulation, a comparative in vivo study of pentylenetetrazol-induced seizures in developing brains of 12- and 25-day-old rats was carried out. The efficacy of zuranolone nanocrystal suspensions was compared with that of an existing cyclodextrin inclusion complex of zuranolone. After the administration of the zuranolone nanocrystal formulation (1, 0.5 or 0.25 mg kg−1, i.p.) and 20 minutes of pre-treatment, seizures were induced by dosing pentylenetetrazol (100 mg kg−1, s.c.) and the animals were observed. The severity of induced seizures was greatly diminished in 12-day rats, while administration of zuranolone in cyclodextrin solution showed no such effect; moreover, the incidence of generalized seizures was also decreased only in the case of administration of the nanocrystalline formulation. Conversely, in 25-day rats, the seizure severity and incidence were decreased after the administration of either tested formulation.
In summary, the phospholipid-stabilised nanocrystal suspension was found to have two main advantages: (i) creation of concentration series independent of the thermodynamic solubility of the drug, enabling dose–response studies to be conducted over any concentration range without having to change the administered volume of the suspension. This is particularly important for studies in developing animals where the administered volume is limited due to body weight. (ii) Owing to its excellent colloidal stability and rapid cell uptake, as shown in ref. 28, the nanocrystal formulation may lead to a higher in vivo efficacy.
It was recently shown from a pool of 10 different APIs61 that phospholipid-stabilised nanocrystal suspensions can be prepared from a diverse range of compounds and applied to in vitro cell uptake, cytotoxicity and efficacy studies.28 The present work extends this knowledge by demonstrating the applicability of nanocrystal suspensions also to in vivo studies. The range of compounds processable into the nanocrystalline form has hereby been extended to zuranolone, chosen as a representative of neuroactive steroids that exhibit extremely low aqueous solubility. Due to its composition (only API, well-tolerated common lipids, and water), the same nanocrystal formulation can in principle be translated from preclinical to clinical development without the need for fundamental modification, reducing both time before first-in-human studies and the risk of formulation-related differences in the study outcomes.
Abbreviations
| FIH | First-in-human |
| API | Active pharmaceutical ingredient |
| PTZ | Pentylenetetrazol |
| CDX | (2-Hydroxypropyl)-β-cyclodextrin |
| GABAARs | γ-Aminobutyric acid type A receptors |
| GPCRs | G protein-coupled receptors |
| CDKL5 | Cyclin-dependent kinase-like 5 |
| MDD | Major depressive disorder |
| DMSO | Dimethyl sulfoxide |
| CNS | Central neural system |
| PBS | Phosphate buffered saline |
| DPPC | Dipalmitoylphosphatidylcholine |
| DPPG | Dipalmitoylphosphoglycerol |
| MPEG-2000-DMPE | N-(Carbonyl-methoxypolyethylenglycol 2000)-1,2-dimyristoyl-sn-glycero-3-phosphoethanol-amine sodium salt |
| SEM | Scanning electron microscopy |
| XRPD | X-ray powder diffraction |
| TEM | Transmission electron microscopy |
| NIH | National Institutes of Health |
| GS | Generalized seizures |
| SD | Standard deviation |
| GTCS | Generalized seizures starting with a loss of righting reflexes |
| GTS | Generalized seizures without a loss of righting reflexes |
| DLS | Dynamic light scattering |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Erik Sonntag for help with HPLC. F. Š. would like to acknowledge the financial support from the Czech Science Foundation (project no. 19-26127X). S. Ch. would like to acknowledge the support from The Pharmaceutical Applied Research Centre (The PARC). E. K. was supported by the project National Institute for Research of Metabolic and Cardiovascular Diseases (Programme EXCELES, ID Project No. LX22NPO5104) – Funded by the European Union – Next Generation EU, by the Czech Academy of Sciences institutional support RVO:61388963, and by the European Regional Development Fund–ERDF/ESF Project “PharmaBrain”, No. CZ.02.1.01/0.0/0.0/16_025/0007444. H. K. would like to acknowledge the Czech Academy of Sciences institutional support RVO:67985823.References
- E. E. Baulieu, Neurosteroids: A Novel Function of the Brain, Psychoneuroendocrinology, 1998, 23, 963–987 CrossRef CAS PubMed.
- R. C. Melcangi, L. M. Garcia-Segura and A. G. Mensah-Nyagan, Neuroactive steroids: State of the art and new perspectives, Cell. Mol. Life Sci., 2007, 65, 777 CrossRef PubMed.
- R. C. Melcangi and G. C. Panzica, Neuroactive steroids: Old players in a new game, Neuroscience, 2006, 138, 733–739 CrossRef CAS PubMed.
- E. Kudova, Rapid effects of neurosteroids on neuronal plasticity and their physiological and pathological implications, Neurosci. Lett., 2021, 750, 135771 CrossRef CAS PubMed.
- M. Chebib and G. A. R. Johnston, The ‘ABC’ of Gaba Receptors: A Brief Review, Clin. Exp. Pharmacol. Physiol., 1999, 26, 937–940 CrossRef CAS PubMed.
- H. Möhler, GABAA Receptors in Central Nervous System Disease: Anxiety, Epilepsy, and Insomnia, J. Recept. Signal Transduction, 2006, 26, 731–740 CrossRef PubMed.
- G. Akk, et al., Mechanisms of neurosteroid interactions with GABAA receptors, Pharmacol. Ther., 2007, 116, 35–57 CrossRef CAS PubMed.
- A. Ghit, D. Assal, A. S. Al-Shami and D. E. E. Hussein, GABAA receptors: structure, function, pharmacology, and related disorders, J. Genet. Eng. Biotechnol., 2021, 19, 123 CrossRef PubMed.
- D. Belelli and J. J. Lambert, Neurosteroids: endogenous regulators of the GABAA receptor, Nat. Rev. Neurosci., 2005, 6, 565–575 CrossRef CAS PubMed.
- Marinus Pharmaceuticals Inc. ZTALMY® (ganaxolone) oral suspension, CXX [pending controlled substance scheduling]: US prescribing information, 2022.
- L. J. Scott, Brexanolone: First Global Approval, Drugs, 2019, 79, 779–783 CrossRef CAS PubMed.
- D. K. Bonner, et al., US2022395513A1 Lipid Prodrugs of Neurosteroids, 2023.
- G. Martinez Botella, et al., Neuroactive Steroids. 2. 3α-Hydroxy-3β-methyl-21-(4-cyano-1H-pyrazol-1′-yl)-19-nor-5β-pregnan-20-one (SAGE-217): A Clinical Next Generation Neuroactive Steroid Positive Allosteric Modulator of the (γ-Aminobutyric Acid)A Receptor, J. Med. Chem., 2017, 60, 7810–7819 CrossRef CAS PubMed.
- K. Singh, R. Pal, S. A. Khan, B. Kumar and M. J. Akhtar, Insights into the structure activity relationship of nitrogen-containing heterocyclics for the development of antidepressant compounds: An updated review, J. Mol. Struct., 2021, 1237, 130369 CrossRef CAS.
- A. L. Althaus, et al., Preclinical characterization of zuranolone (SAGE-217), a selective neuroactive steroid GABAA receptor positive allosteric modulator, Neuropharmacology, 2020, 181, 108333 CrossRef CAS PubMed.
- A. Arnaud, et al., Number Needed to Treat and Number Needed to Harm analysis of the zuranolone phase 2 clinical trial results in major depressive disorder, J. Affective Disord., 2021, 285, 112–119 CrossRef CAS PubMed.
- A. Bullock, et al., Zuranolone as an oral adjunct to treatment of Parkinsonian tremor: A phase 2, open-label study, J. Neurol. Sci., 2021, 421, 117277 CrossRef CAS PubMed.
- A. H. Clayton, et al., Zuranolone in Major Depressive Disorder: Results From MOUNTAIN-A Phase 3, Multicenter, Double-Blind, Randomized, Placebo-Controlled Trial, J. Clin. Psychiatry, 2023, 84(2), 1–11 Search PubMed.
- F. ten Doesschate, J. A. van Waarde and G. A. van Wingen, Non-superiority of zuranolone (SAGE-217) at the longer-term, J. Affective Disord., 2021, 291, 329–330 CrossRef CAS PubMed.
- F. Ten Doesschate, J. A. van Waarde and G. A. van Wingen, Still no evidence for the efficacy of zuranolone beyond two weeks: Response to Arnaud and Bonthapally, J. Affective Disord., 2022, 313, 149–150 CrossRef CAS PubMed.
- M. Timm, L. Saaby, L. Moesby and E. W. Hansen, Considerations regarding use of solvents in in vitro cell based assays, Cytotechnology, 2013, 65, 887–894 CrossRef CAS PubMed.
- M. Colucci, et al., New insights of dimethyl sulphoxide effects (DMSO) on experimental in vivo models of nociception and inflammation, Pharmacol. Res., 2008, 57, 419–425 CrossRef CAS PubMed.
- M. Verheijen, et al., DMSO induces drastic changes in human cellular processes and epigenetic landscape in vitro, Sci. Rep., 2019, 9, 4641 CrossRef CAS PubMed.
- J. Hadgraft and M. E. Lane, Skin permeation: The years of enlightenment, Int. J. Pharm., 2005, 305, 2–12 CrossRef CAS PubMed.
- T. J. Anchordoguy, J. F. Carpenter, J. H. Crowe and L. M. Crowe, Temperature-dependent perturbation of phospholipid bilayers by dimethylsulfoxide, Biochim. Biophys. Acta, Biomembr., 1992, 1104, 117–122 CrossRef CAS PubMed.
- Z. T. Chowhan and R. Pritchard, Effect of Surfactants on Percutaneous Absorption of Naproxen I: Comparisons of Rabbit, Rat, and Human Excised Skin, J. Pharm. Sci., 1978, 67, 1272–1274 CrossRef CAS PubMed.
- B. Gironi, et al., Effect of DMSO on the Mechanical and Structural Properties of Model and Biological Membranes, Biophys. J., 2020, 119, 274–286 CrossRef CAS PubMed.
- D. Lizoňová, et al., Surface stabilization determines macrophage uptake, cytotoxicity, and bioactivity of curcumin nanocrystals, Int. J. Pharm., 2022, 626, 122133 CrossRef PubMed.
- M. Jarvis, V. Krishnan and S. Mitragotri, Nanocrystals: A perspective on translational research and clinical studies, Bioeng. Transl. Med., 2019, 4, 5–16 CrossRef PubMed.
- A. A. Noyes and W. R. Whitney, The Rate of Solution of Solid Substances in their Own Solutions, J. Am. Chem. Soc., 1897, 19, 930–934 CrossRef.
- L. Peltonen, Design Space and QbD Approach for Production of Drug Nanocrystals by Wet Media Milling Techniques, Pharmaceutics, 2018, 10(3), 104–120 CrossRef CAS PubMed.
- M. Li, M. Azad, R. Davé and E. Bilgili, Nanomilling of Drugs for Bioavailability Enhancement: A Holistic Formulation-Process Perspective, Pharmaceutics, 2016, 8(2), 17–51 CrossRef PubMed.
- J. Li, Z. Wang, H. Zhang, J. Gao and A. Zheng, Progress in the development of stabilization strategies for nanocrystal preparations, Drug Delivery, 2021, 28, 19–36 CrossRef CAS PubMed.
- C. Schneider, M. Hanisch, B. Wedel, A. Jusufi and M. Ballauff, Experimental study of electrostatically stabilized colloidal particles: Colloidal stability and charge reversal, J. Colloid Interface Sci., 2011, 358, 62–67 CrossRef CAS PubMed.
- L. Vidlářová, G. B. Romero, J. Hanuš, F. Štěpánek and R. H. Müller, Nanocrystals for dermal penetration enhancement – Effect of concentration and underlying mechanisms using curcumin as model, Eur. J. Pharm. Biopharm., 2016, 104, 216–225 CrossRef PubMed.
- K. Peters, et al., Preparation of a clofazimine nanosuspension for intravenous use and evaluation of its therapeutic efficacy in murine Mycobacterium avium infection, J. Antimicrob. Chemother., 2000, 45, 77–83 CrossRef CAS PubMed.
- Y. Barenholz, (Chezy). Doxil®—The first FDA-approved nano-drug: Lessons learned, J. Controlled Release, 2012, 160, 117–134 CrossRef CAS PubMed.
- Y. Xiao, et al., PEGylation and surface functionalization of liposomes containing drug nanocrystals for cell-targeted delivery, Colloids Surf., B, 2019, 182, 110362 CrossRef CAS PubMed.
- T. Li, et al., Controlling the size and shape of liposomal ciprofloxacin nanocrystals by varying the lipid bilayer composition and drug to lipid ratio, J. Colloid Interface Sci., 2019, 555, 361–372 CrossRef CAS PubMed.
- L. Velisek, et al., Pentylenetetrazol-induced seizures in rats: an ontogenetic study, Naunyn-Schmiedeberg's Arch. Pharmacol., 1992, 346, 588–591 CrossRef CAS PubMed.
- P. Conklin and F. W. Heggeness, Maturation of tempeature homeostasis in the rat, Am. J. Physiol., 1971, 220, 333–336 CrossRef CAS PubMed.
- NC3Rs. ARRIVE guidelines. https://www.nc3rs.org.uk/arrive-guidelines.
- E. Kudova, et al., The Neuroactive Steroid Pregnanolone Glutamate: Anticonvulsant Effect, Metabolites and Its Effect on Neurosteroid Levels in Developing Rat Brains, Pharmaceuticals, 2021, 15(1), 49–67 CrossRef PubMed.
- M. Pohl and P. Mares, Effects of flunarizine on Metrazol-induced seizures in developing rats, Epilepsy Res., 1987, 1, 302–305 CrossRef CAS PubMed.
- Selleck Chem. Zuranolone (SAGE-217). https://www.selleckchem.com/products/zuranolone-sage-217.html.
- P. S. Watson, et al., A crystalline 19-nor C3, 3-disubstituted C21-N-pyrazolyl steroid, 2017 Search PubMed.
- W. Löscher, Critical review of current animal models of seizures and epilepsy used in the discovery and development of new antiepileptic drugs, Seizure, 2011, 20, 359–368 CrossRef PubMed.
- J. Dobbing and J. L. Smart, Vulnerability of developing brain and behaviour, Br. Med. Bull., 1974, 30, 164–168 CrossRef CAS PubMed.
- P. Mares, A. Zouhar and G. Brozek, Ontogenetic development of electrocorticogram in the rat, Act. Nerv. Super., 1979, 21, 218–225 CAS.
- B. E. Piacsek, N. J. Statham and M. P. Goodspeed, Sexual maturation of male rats in continuous light, Am. J. Physiol., 1978, 234, E262–E266 CAS.
- R. J. Ellingson and G. H. Rose, Ontogenesis of the electroencephalogram, in Developmental Neurobiology, ed. W. A. Himwich and C. Charles, 1970, pp. 441–474 Search PubMed.
- M. E. Eng, G. E. Imperio, E. Bloise and S. G. Matthews, ATP-binding cassette (ABC) drug transporters in the developing blood–brain barrier: role in fetal brain protection, Cell. Mol. Life Sci., 2022, 79, 415 CrossRef CAS PubMed.
- N. Strazielle and J.-F. Ghersi-Egea, Efflux transporters in blood-brain interfaces of the developing brain, Front. Neurosci., 2015, 9, 21 Search PubMed.
- J. Neumann, D. Rose-Sperling and U. A. Hellmich, Diverse relations between ABC transporters and lipids: An overview, Biochim. Biophys. Acta, Biomembr., 2017, 1859, 605–618 CrossRef CAS PubMed.
- L. M. Koehn, et al., Efflux transporters in rat placenta and developing brain: transcriptomic and functional response to paracetamol, Sci. Rep., 2021, 11, 19878 CrossRef CAS PubMed.
- C. J. Ek, et al., Efflux mechanisms at the developing brain barriers: ABC-transporters in the fetal and postnatal rat, Toxicol. Lett., 2010, 197, 51–59 CrossRef CAS.
- W. Zhang, et al., Differential Expression of ABC Transporter Genes in Brain Vessels vs. Peripheral Tissues and Vessels from Human, Mouse and Rat, Pharmaceutics, 2023, 15, 5 CAS.
- B. Bauer, A. M. S. Hartz, G. Fricker and D. S. Miller, Pregnane X Receptor Up-Regulation of P-Glycoprotein Expression and Transport Function at the Blood-Brain Barrier, Mol. Pharmacol., 2004, 66, 413–419 CAS.
- V. S. Narang, et al., Dexamethasone increases expression and activity of multidrug resistance transporters at the rat blood-brain barrier, Am. J. Physiol.: Cell Physiol., 2008, 295, C440–C450 CrossRef CAS PubMed.
- M. Iqbal, et al., Pro-Inflammatory Cytokine Regulation of P-glycoprotein in the Developing Blood-Brain Barrier, PLoS One, 2012, 7, e43022 CrossRef CAS PubMed.
- F. Hládek, S. Chvíla, O. Navrátil, M. Balouch and F. Štěpánek, Systematic Investigation of Wet-Milling Kinetics and Colloidal Stability of Pharmaceutical Nanocrystals, Cryst. Growth Des., 2022, 22, 6928–6940 CrossRef.
| This journal is © The Royal Society of Chemistry 2024 |