
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComparative study on the effects of the inclusion complexes of non-steroidal anti-inflammatory drugs with 2-hydroxypropyl-β-cyclodextrins on dissociation rates and supersaturation†
Yukiko
Oshite‡
,
Ayako
Wada-Hirai‡
,
Risa
Ichii
,
Chihiro
Kuroda
,
Kanji
Hasegawa
,
Ryosuke
Hiroshige
,
Hideshi
Yokoyama
,
Tomohiro
Tsuchida
 and
Satoru
Goto
*
and
Satoru
Goto
*
Faculty of Pharmaceutical Sciences, Tokyo University of Science, 2641 Yamazaki, Noda, Chiba 278-8510, Japan. E-mail: s.510@rs.tus.ac.jp
First published on 19th February 2024
Abstract
Active pharmaceutical ingredient (API) complexes with cyclodextrins (CDs) and their derivatives are widely formulated. Previously, we reported on the supersaturation effect and its benefits for CD inclusion complexes without polymers. The degree of amorphization and percentage of remaining crystals were determined using X-ray powder diffraction (XRPD) and differential scanning calorimetry (DSC) techniques. However, these properties clash with the stoichiometry of the solution according to the phase solubility diagram. In this study, the complexation contents of the prepared mixtures of indomethacin, piroxicam, diclofenac, and loxoprofen sodium with the 2-hydroxypropylated derivative of CD (HP-β-CD) were comparatively analyzed using dissolution curves. XRPD and DSC measurements indicated that equimolar mixtures were favorable for the interaction between these APIs and HP-β-CD. Enhancing the API solubility of HP-β-CD can be achieved through dissolution experiments. Mixtures of indomethacin with HP-β-CD consisted of an equimolar complex and corresponding remains. If the remaining component was HP-β-CD, then a gradual release of the equimolar complex was induced, and the release of diclofenac indicated similar dissolution behaviors. In contrast, the mixtures of indomethacin and diclofenac at molar ratios of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:1 showed immediate supersaturation and a gradual decrease in the equilibrium concentration. These results indicate that the unbound HP-β-CD in the mixture acts as a matrix for controlled release.
:1 and 1:1 showed immediate supersaturation and a gradual decrease in the equilibrium concentration. These results indicate that the unbound HP-β-CD in the mixture acts as a matrix for controlled release.
1. Introduction
In drug development, hydrophobicity and biodegradability affect many pharmacologically active resources, reducing the viability of potential new drug candidates. Therefore, enhancing the oral bioavailability of active pharmaceutical ingredients (API) is important. Various sutable methods have been developed for micronizing dispersoid particles, annexing surfactants, and modifying them as prodrugs.1 We focus on cyclodextrin (CD) analogs, which are expected to improve the aqueous solubility, dissolution rate, stability, and efficiency (bioavailability) of APIs.1 This allows for the conversion of crystalline APIs into solid solutions or solid dispersions, prevents interaction between APIs and additives, eliminates unpleasant tastes and odors, and reduces gastrointestinal irritation.2–11CD application exploits its ability to capture an insoluble API by encapsulating the API in CD's hydrophobic central cavity or by adsorbing it to CD's hydrophilic outer surface.6–9 As more CDs are analyzed, various API–CD structures have been proposed, including self-association and non-inclusion structures.6 The corresponding properties, especially stoichiometry, of a solid are not necessarily predictive of the functional features of API–CD complexes in solution. This may be thermodynamically reasonable because the atmosphere for API–CD complexes consists of protic or aprotic solvents or a crystalline lattice. Based on this, we propose that the beneficial or unfavorable potential of the solution/dispersion of API–CD complexes and their aggregates is based on crystal or amorphous solid analysis.
β-CD is a cyclic heptamer composed of glucopyranoses with α-1,4-glycosidic linkages.7–10 Because β-CD is readily available and has a cavity size suitable for the width of various APIs, it has been widely used in the early stages of drug development.8 However, its notably low aqueous solubility (18.5 mg mL−1) and nephrotoxicity has limited its use, especially in parenteral drug delivery.8,9 The partially 2-hydroxypropylated derivative of β-CD (HP-β-CD) (Chart 1) has a greater aqueous solubility (>600 mg mL−1) than β-CD as well as higher resistance to chemical degradation and photodegradation;7,10 this derivative is considered to have higher solubility and non-toxicity at low to moderate oral and intravenous doses8 and has generally been verified to be safe when administered parenterally in animals and humans.1,6 It is clinically utilized in APIs delivered via oral, intravenous, and suppository administration routes in the US, Belgium, Switzerland, and other countries.11 One drawback is that HP-β-CD consists of various degrees of hydroxypropyl substituents, which causes unevenness and diversity of physicochemical properties.
 | ||
| Chart 1 Chemical structures of non-steroidal anti-inflammatory drugs (NSAIDs) and cyclodextrin. | ||
CD in complex with piroxicam (PRX) (Chart 1) was developed as a pioneering drug. PRX is a nonsteroidal anti-inflammatory drug (NSAID) used to treat rheumatism, osteoarthritis, and primary dysmenorrhea; however, its oral administration causes adverse gastrointestinal (GI) effects.12 Reducing the GI adverse effect,13,14 β-CD applications for PRX (PRX/β-CD) were formulated at a molar ratio of 1 of PRX and 2.5 of CD,2,15,16 then further improved as the HP-β-CD complex (PRX/HP-β-CD). PRX has the advantage of inclusion because of its fluorescence and binding quenching.17 The PRX/HP-β-CD complex demonstrated high stability during photodegradation.18 The addition of hydroxypropylmethylcellulose (HPMC) to PRX/HP-β-CD improved the dissolution rate.19 Regarding conventional preparations of PRX/HP-β-CD, freeze-drying and fluid-bed coating techniques were developed to provide amorphous products significantly without crystalline signals on X-ray powder diffraction (XRPD) measurements.20,21 In the cold stress-induced gastric ulcer induced by PRX/HP-β-CD and the indomethacin (INM) (Chart 1) complex with HP-β-CD (INM/HP-β-CD), these complexes protected a slight level as non-administered groups of NSAIDs.22 The administration and analyses of INM and PRX inclusion complexes were performed, which relieved adverse effects.
Diclofenac (DCF) (Chart 1) is one of the most potent NSAIDs with an analgesic effect, alleviating neuropathic pain by inhibiting cyclooxygenase (COX) and is the leading choice in the support of narcotic analgesics for cancer. Currently, DCF is clinically prescribed with ibuprofen and loxoprofen (LXP) (Chart 1).23 Comparing the sodium salts of LXP and DCF, the period to reach the maximal blood concentration of LXP-Na was less than 1 h, whereas DCF-Na required 2.5 hours.24 Because the 1-octanol/water partition coefficients (logP) of LXP-acid and DCF-acid are 3.35 and 4.51, respectively, the hydrophobicity of DCF could delay its effect. The adverse effect that is induced by inhibiting COX isozyme COX-1 is potent in LXP, ibuprofen, INM, and ketoprofen, although that of DCF is not severe. The DCF inhibitory selectivity of another isozyme, COX-2, for COX-1 is approximately +1/2, which is as potent as that of celecoxib.25 Regarding cardiovascular and musculoskeletal disorders associated with COX-2 inhibition, the most frequently used DCF receives the most complaints in terms of NSAIDs after ibuprofen.26,27
Regarding renal impairment, LXP-Na showed no administration dependence, DCF-Na showed a significant correlation. This is caused by renal ischemia because DCF is harmful to renal function.28 DCF has individual advantages and disadvantages similar to other NSAIDs; therefore, this alone does not indicate that the more potent activity of DCF results in more predominant prescriptions than LXP. To accommodate the clinical data of the patients, DCF, LXP, celecoxib, ibuprofen, and other NSAIDs were chosen, considering the performance of the API and its combinations with other medicinal ingredients. Therefore, the aim of the development and research on DCF complexes with CDs (DCF/CDs) differs somewhat from that of PRX complexes with CDs. The deliberate planning of the efficient work of DCF should be addressed rather than protecting against GI disorders. Scrutinizing the dissolution profiles of various DCF formulations would be valuable for easing the renal burden.
A comparative study for the HP-β-CD inclusion complexes of DCF and conventional INM and PRX was published, focused on their phase-solubility diagrams and the time course of degradations of APIs.29,30 The stability constants of the acidic API and CD-inclusion complex (i.e., an association constant) decreased as the pH increased. The corresponding US patent in 1997 stated that the equimolar DCF complex with β-CD was the main composition, whereas the US patent in 2005 claimed that DCF and HP-β-CD were found at a molar ratio of 1:1.5–2.5, most preferably 1:2.31,32 The properties, especially the stoichiometry, of the CD inclusion complex in the solid state can be obtained using XRPD and thermal analyses. However, solid stoichiometry would not be adequate to explain the dissolution behavior in solution as the CD inclusion complex as a solute has quantitative features different from those of solid stoichiometry. The saturated concentration of the equimolar DCF complex with the HP-β-CD (DCF/HP-β-CD) (3.48 mM at pH 3.0) was enhanced to 7.5-fold compared to that of DCF-Na (0.47 mM).33
To examine the permeability across the silicon membrane, a DCF/HP-β-CD complex at a molar ratio of 1:2 was used to sustain a supersaturated solution and was found to maximize the permeation rate, whereas excess amounts (1:10 and 1:20) of HP-β-CD decreased the flux.33 Cross peaks obtained on the rotating frame nuclear Overhauser effect spectroscopy (ROESY) of the proton-nuclear magnetic resonance (1H-NMR) experiments supposed the 2,6-dichloroanilino and benzoic acid moieties of DCF as two binding sites, indicating that the DCF/HP-β-CD complex had a preferable molar ratio of 1:2.34 Contrarily, since the phase-solubility diagrams of DCF (pKa = 4.15) illustrated the linear correlations to the concentration of HP-β-CD at pH of 6.4, 6.8, and 7.7, their stoichiometric ratio was assumed to be 1:1 rather than 1:2.34,35
In our previous study, the concentrations of INM were measured at concentrations of more than 0.10 M of HP-β-CD (up to 0.30 mM), and the phase solubility diagram of INM to the concentration of HP-β-CD was verified as a parabolic curve.35,36 According to the Higuchi and Conner model,35 the results proved that the complex of INM and HP-β-CD has a 1:2 stoichiometry in solution. The final decision requires considerable evidence. In the solid state of INM, its mixture with less than 1/10 of HP-β-CD prompted phase transition from the most stable (in the sense of the neat condition at the standard temperature) to the metastable polymorphs.36 The most stable form of nifedipine (NIF), identically, had transformed to its metastable form by preparing the NIF mixture with HP-β-CD at a molar ratio of 9:1.37 The HCl salt of nicardipine (NIC), which is a cationic derivative of neutral NIF, cannot form an inclusion complex with CD.37 The equimolar NIC–HCl mixture with HP-β-CD demonstrated supersaturation in the dissolution test with a fluid at pH 1.2.37,38 At that time, it was concluded that the supersaturation of the NIC–HCl mixture with HP-β-CD would occur because of the positive charge of NIC.
However, the CD complex showed obvious supersaturation despite the fact that INM is a non-charged form of INM at pH 1.2.35,36 The anionic form of INM (pKa = 4.5) at pH 1.2 would never occupy a large part. Therefore, it was not possible to conclude that any ionic APIs directly caused the supersaturation of the API complex with the HP-β-CD. The INM/HP-β-CD complex showed supersaturation at pH 1.2, indicating its ability to induce a GI disorder. This property is unfavorable for INM, with an inhibitory effect selective for COX-1, although our goal differs from such pharmaceutical improvements. The “parachute” effect, which was accomplished in the CD complexation of INM and NIC–HCl, may be affected by the physicochemical properties in crystal or amorphous solids, dispatching certain signals to the experimental instruments.36,39,40 The regulation of the parachute effect, which achieves APIs with poor water solubility supersaturation, as well as other benefits delivered by CD inclusion, would improve the adsorption pathways and renal excretions of APIs, thereby expanding their applications. In this context, the practical and effective stoichiometry of NSAIDs is important; therefore, in the present study, we re-investigated the aqueous solubility and dissolution rate.
2. Experimental section
2.1. Materials
DCF acid was purchased from Tokyo Chemical Industry (Tokyo, Japan), and PRX, INM, and HP-β-CD were from Wako Pure Chemicals (Osaka, Japan). LXP sodium dihydrate was obtained from Teikoku Seiyaku Co. (Kagawa, Japan). The molecular weight and the degree of substitution of HP-β-CD were approximately 1400 and 6.32 ± 0.74, respectively.1,2 All the other commercially available materials and solvents were of analytical grade.Two mixing methods were used to produce the APIs/HP-β-CD mixtures: physical mixing (PM) and solvent mixing (SM). The PM method involved kneading the mixed API and HP-β-CD with an agate mortar and pestle. Neat API and plain HP-β-CD samples were used after grinding, similar to the PM preparation. The PM-prepared mixtures were prepared as equimolar mixtures of API and HP-β-CD.
To prepare the API/HP-β-CD mixture,54,55 a solution with a mass corresponding to the final 2–3 mmol kg−1 API was dissolved in 80 mL ethanol (Milli-Q water for LXP-Na). This was then mixed with aqueous solutions in the relative triple, double, equivalent, a half, and one-third molality of HP-β-CD, preparing the APIs/HP-β-CD mixtures with molar ratios of 1:3 1:2, 1:1, 2:1, and 3:1, respectively. Neat API and pure HP-β-CD were prepared as references with 1:0 and 0:1 labels, respectively, for SM preparation. Each SM-prepared sample was mounted on a rotary evaporator (Eyela, Tokyo, Japan) under a vacuum of 1.3 kPa using an oil rotary vacuum pump (G-20 DA; ULVAC, Inc., Kanagawa, Japan). The mixture was stirred continuously at 100 rpm in a water bath (BM-82; Yamato, Tokyo, Japan) at 313 K.
Furthermore, the PM- and SM-prepared samples were dried and stored in a reduced-pressure desiccator (60 kPa) containing fresh silica gel beads at a standard temperature for at least 24 hours to remove as much excess moisture from the obtained powder as possible. On-demand mixtures with mole fractions in intervals of 10% were prepared similarly.
2.2. XRPD diffractometry of API and API mixture with HP-β-CD
XRPD pattern measurements were performed using RINT 2000 (Rigaku Co., Tokyo, Japan) with a Cu Kα radiation source at a voltage of 40 kV and a current of 40 mA. The X-ray was filtered by the Ni filter to produce monochromatic radiation and progressed using the parallel-beam method in the 2θ range from 5 to 40 degrees at a scanning rate of 0.02 degrees steps. The spectra are presented as the average of five scans, and the scanning sequences were conducted at least in triplicate. The samples were crushed in an agate mortar and pestle, and the powders were mixed.To identify the polymorph, the diffractograms of the NSAID single-crystal structures were compared with those of the references. The reproduced references were converted from 3D coordinates using the Reflex Module of Powder Diffraction on Biovia/Accelrys Materials Studio 2022 (Dassault Systems), and the Miller indices of conspicuous peaks were calculated. The 3D crystalline coordinates were retrieved from the Cambridge Crystallographic Data Centre (CCDC). The most stable and metastable forms of INM and PRX were derived from the CCDC reference codes INDMET (1972), INDMET04 (2011), and BIYSEH (1982), respectively. For DCF, the most stable HD2-form (SIKLIH, C2/2), metastable HD1-form (SIKLIH02, P21/c), and metastable HD3-form (SIKLIH04, Pcan) forms were retrieved. Because no crystalline entries had been retrieved for dihydrated loxoprofen sodium salt, the observed signals were verified to the 2θ angles of 7.5, 11.1, 22.2, 30.0, and 33.8 degrees.46,47
2.3. Thermal analysis of neat API and API mixture with HP-β-CD
Differential scanning calorimetry (DSC) was performed using a DSC8230 instrument (Rigaku Co., Tokyo, Japan). Samples with a total mass amount of 5.0 mg were placed in an aluminum pan and sealed. The temperature was scanned from 303 K to 453 K at a rate of 5.0 or 10.0 K min−1 under a nitrogen gas flow of 30 mL min−1 (scanning range of 303–463 K for DCF). Tm was adopted as the melting start temperature obtained from the intersection of the extension of the baseline and the extension of the point of maximum slope of the peak using the Thermo Plus 2 software (Rigaku Co., Tokyo, Japan). If the resulting thermogram had a simple endothermic peak, the area enclosed by the endothermic curve and baseline was converted to the total melting enthalpy (ΔfusH) for a mass amount of the component through compensation with an instrumental coefficient. The total melting entropy (ΔfusS) for a mass amount of component was simultaneously approximated by the quotient of the ΔfusH divided by Tm, according to the classical definition of Clausius.2.4. HPLC measurement of API concentration in the sample solution
The sample solution was filtrated with a membrane filter (Minisart RC 4 with 0.22 μm pore size; Sartorius, Göttingen, Germany). The fractionation of the API in the filtrate was performed using HPLC (Shimadzu Co., Kyoto, Japan) with a mobile phase of acetonitrile–tetrahydrofuran–acetic acid–ultrapure water with the volume ratio of 50:5:0.2:44.8 or that of acetonitrile–ultrapure water with 50:50 at a flow rate of 1 mL min−1, mounting a reversed-phase column (Capcell Pak C18; Shiseido; 5 μm, 4.6 mm ∅ × 250 mm) at a temperature of 313 K. The amounts of INM, PRX, DCF, and LXP were determined by monitoring the absorbance at wavelengths of 320, 357, 348, and 223 nm, respectively.
2.5. Phase-solubility isotherm diagram of API to a concentration of HP-β-CD
The buffer solutions were as follows: 10 mM sodium citrate/HCl buffer (pH 1.2) or 25 mM KH2PO4/Na2HPO4 buffer (pH 6.8), which were prepared in compliance with solution media no. 2 for the dissolution test in the Japanese Pharmacopeia implemented international harmonization. HP-β-CD was added to the buffer solution to obtain a stock solution. The API was added to the solutions that were mixed with various concentrations of the HP-β-CD stock and buffer solutions and shaken in a water bath at 310 K. The concentration of the API in the supernatant was determined using HPLC.The phase solubility diagram consisted of the equilibrium concentrations of a guest API on the ordinate and the total concentration of the host CD on the abscissa.35 If a straight line and a parabolic curve could be approximated on the phase solubility diagram, then they were classified as AL- and AP-types according to the Higuchi and Connors catalogs, respectively.7,13,35 For the linear correlation corresponding to the AL-type, the regression analysis of the experimental dataset using eqn (1) provided the stability constant K1:1 for equimolar APD–CD complexation
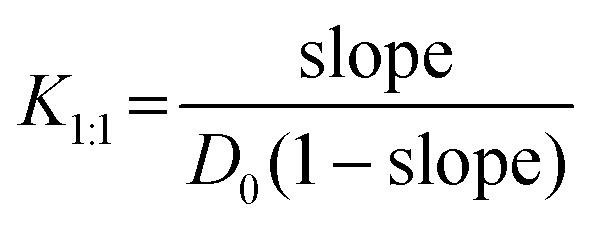 | (1) |
Next, the parabolic curve corresponding to the AP-type indicates that the complexes are associated with the stoichiometry of equimolar guest/host and that of single-guest/double-hosts (or further convoluted proportions).7,13,35 Multiple regression analysis of the measured dataset to the total concentration of the host ([CD]) and its square ([CD]2) with eqn (2) was used to obtain the stability constants for a combination of equimolar complex K1:1 and double-host associating complex K1:27,13,35
| Dt = K1:1K1:2D0[CD]2 + K1:1D0[CD] + D0 | (2) |
Occasionally, phase solubility diagrams may exhibit a hyperbolic curve, referred to as a saturation curve. This curve was classified as an AN-type on the Higuchi and Connors catalog.7,13,35 The curve can be described using eqn (3) for the Langmuir adsorption isotherm.41,42 To process with linear regression analysis of ordinate ([CD]/γ) referred to the abscissa ([CD]), eqn (3) is converted to Hanes and Woolf's expression as eqn (3′)
 | (3) |
 | (3′) |
In the rare cases mentioned by Szejtli,13 the ascending region in the lower concentration of the host is followed by a plateau region, the pattern of which is similar to the saturation curve of the AN-type, whereas it terminates at any host concentration threshold. This was named as the BS-type in Higuchi and Connors’ catalog,7,13,35 and for a concentration higher than the threshold, the guest decreases along a hyperbola, most likely according to the solubility product KSP.42 To analyze this type of pattern, we believe that a hypothesis that has not yet been discussed is needed.42 At the concentration of API in the plateau region, it is postulated that a soluble complex of API and CD is enlarged by the adhesion of the excess CD, like a snowball. At the threshold concentration of CD, the scale of the aggregated dispersoids transcends an upper limit, and the aggregation induces subsequent sedimentation.43 Thus, it is rational to consider that the solubility product is a property of the poorly soluble aggregated dispersoids containing a tolerable amount of API and an excess amount of CD. If the hypothesis is valid, then we would find an opportunity (trigger) to turn the soluble complexes into poorly soluble aggregated dispersoids.
It seems that the γ-CD, whose interior cavity is larger (427 Å3) than that of β-CD (262 Å3),13 frequently tends to provide the BS-type inclusion. In contrast, the solubility of free-CD (18 g L−1) is particularly low compared to that of the γ-CD (232 g L−1).13 Here, the specific density of the β-CD molecule (with fixed H2O molecules) is likely to be too high to disperse itself in solution, whereas it can be supposed that the γ-CD is 13-fold floating power relative to the β-CD. Although the aqueous solubilities of the methylated (>500 g L−1) and 2-hydroxypropylated (>330 g L−1) derivatives of CD are remarkably high (over 18-fold higher that of plain-CD), these modified CDs were derived by alkylating their hydroxyl groups. Their appearance increased their hydrophobicity because of the increased number of hydrocarbons. However, hydrogen bonding pairs between the 2- and 3-hydroxyl groups and those between the 6-hydroxyl groups contracted the molecular interior owing to their potent electrostatic forces. In contrast, the hydrogen bonding pairs of the substituted derivatives were disassociated, relaxing their molecular interiors to reduce the specific density. These substituted derivatives acquired a higher floating power than that of plain-CD. Investigating the 3D crystalline structures surveyed from the CCDC, the density of β-CD (containing 12 bound water molecules) and γ-CD (containing 14 bound water molecules) was 1.462 and 1.405, respectively. Although the density of the hydrated crystalline molecules with their packing structures did not coincide with the density of the solute in the liquid phase, it is expected that the compactness of CD is higher than the density of CD based on the supporting evidence. Although proof has been furnished neither experimentally nor computationally, it was supposed that the aggregation dispersoids containing excess CDs also became too high to be dispersed. This suggests that the intermolecular interaction between the API and CD causes the solubility of the complex to increase or decrease, respectively. As mentioned above, an attempt was made to classify the phase solubility diagram as the BS-type was divided to estimate the components of a stoichiometric inclusion complex and to discriminate non-stoichiometric aggregation dispersoids (snowball-like particles) as soluble and insoluble.43
2.6. The apparent solubility and dissociation rates of APIs/HP-β-CD
The mixture of API with HP-β-CD (or neat API) was added to 5 mL 10 mM sodium citrate/HCl buffer (pH 1.2) or 25 mM KH2PO4/Na2HPO4 buffer (pH 6.8) and shaken in a water bath at 310 K. The concentration of the API in the supernatant was determined using HPLC. The dissolution process of a neat API or guest API solid mixture with a host CD was assumed to correspond to a linear correlation or a saturation curve. The PM- and SM-prepared samples of the neat API and the API mixture with CD were kneaded beforehand with an agate mortar and pestle in the prescribed manner to align their particle size (no sieving). In this procedure, we could not avoid contaminating the superfine powders. An adequate amount of sample was added to the dissolved solution under the conditions described above. After an appropriate incubation time, t, the concentration of API C was measured. As the adhering powders were immediately dissolved in the dissolving solution at t = 0, their concentration was treated as the initial concentration, C = C0. If a sink condition is applicable because of the higher solubility of the API, then the API concentration-time diagram requires regression analysis as a linear correlation. If the dissociation curve traces a saturation curve, then non-linearly curve fitting with the Noyes–Whitney integral expression (eqn (4)) was applied using the Solver module of Microsoft Excel 2016 with the implemented GRG nonlinear option39,42| C = Ceq − (Ceq − C0)exp(−kSt) | (4) |
2.7. Kinetics of consecutively invertible step model for the supersaturating process
If the dissolution curve contained a supersaturating process, the recrystallization solid was considered to be different from the dissolution solid. The dissolution process of a guest API solid mixture with a host CD is assumed to correspond to a series of consecutive invertible steps.39–41 An API solid mixture with CD (X, as a mixture containing an inclusion complex) is dissolved in the buffer, in which the dissolution and precipitation rate constants are represented as k+1 and k−1, respectively. The APD–CD complex in the buffer (Y as a solute) continuously equilibrates with free API and CD (Y′), in which the dissociation and association rate constants are expressed as ka and kb, respectively. Finally, the free API equilibrates to a stable crystal (Z, as a pure or hydrated crystal) in which the precipitation and dissolution rate constants are k+2 and k−2, respectively. | (5) |
Because the concentrations of Y and Y′ are difficult to determine, we considered the exchange between Y and Y′ as a rapid balance step. This immeasurable process (Y to Y′) could be conjugated with the initiation step from X to Y and the termination step from Y′ to Z at both edges of the entire process. The differential equations for a consecutive invertible process are described in eqn (6).
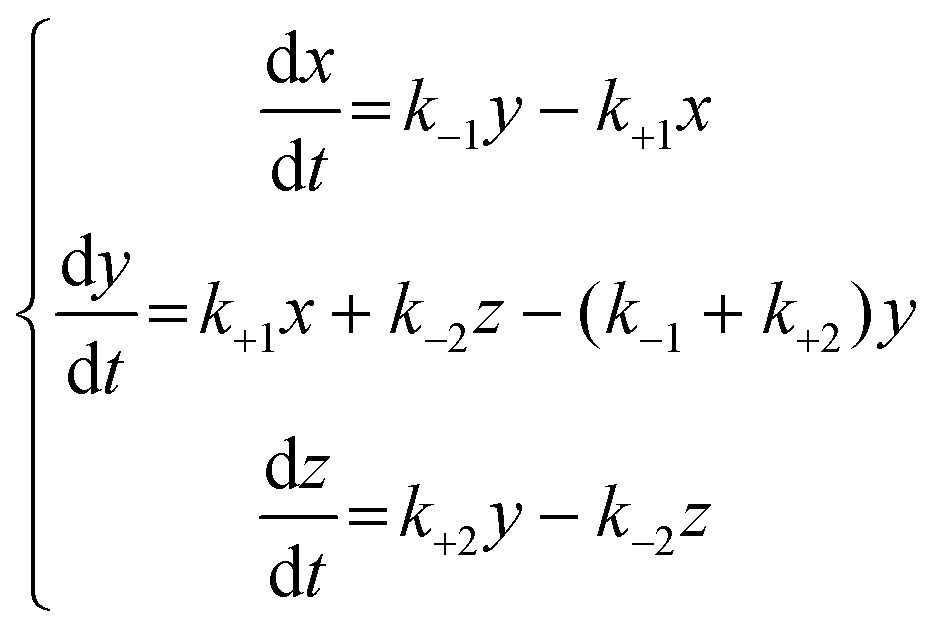 | (6) |
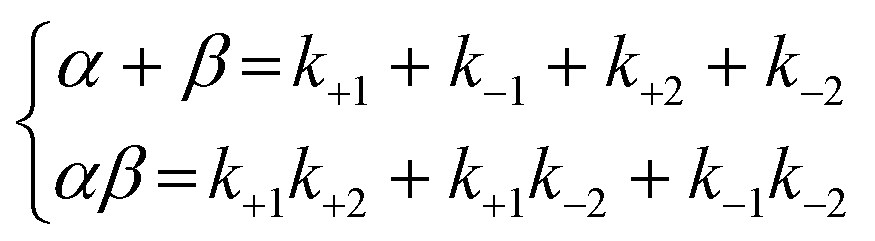 | (7) |
As the integral solutions of the given differential equations, the concentrations of x, y, and z are described with eqn (8)–(10) of the rate constants k1, k−1, k+2, k−2, and variables α and β.
 | (8) |
 | (9) |
| y = 1 − x − z | (10) |
To transform the mole fractions into molarities (mol L−1), x, y, and z were multiplied by coefficient A0. The rate constants k1, k−1, k+2, and k−2 for APD–CD were calculated by nonlinearly fitting the measured dissolution profiles to eqn (8)–(10), in which nonlinear fitting was performed using the solver module of Microsoft Excel 2016 with the implemented GRG nonlinear option.
2.8. Determination of the viscosity of the HC-β-CD aqueous solution
The sample solution was prepared using HP-β-CD dissolved in 50 mM KH2PO4/Na2HPO4 buffer (pH 6.8). An Ostwald viscometer was immersed in a water bath adjusted to the standard temperature of 298 K for thermal equilibration. Then, 8 mL sample liquid was used for viscosity measurement. After the sample liquid was pulled into the upper reservoir by suction, it was drained back into the lower reservoir. The time required for a predetermined volume of the sample liquid to drain due to gravity was measured. The equipment was calibrated using ultrapure water as the reference. To determine the viscosity of the DCF mixtures with HP-β-CD, an adequate amount of DCF sodium (pKa 4.15 for its conjugated acid) was added to the HP-β-CD solution at pH 6.8.2.9. Energetics for the stability of the equimolar DCF/HP-β-CD complex in water
The initial structures of the DCF complex with the HP-β-CD and their unbound forms were retrieved from the CCDC database as well as possible and arranged as either the 2,6-dichloroanilino group or propionic acid group of DCF, either intruding inside the C2–C3 rim (bottom interface of the truncated cone), intruding inside the C6 rim (upper interface of the truncated cone), or located outside (middle point between the bottom and upper faces beyond the periodic cells). The structures and conformations were geometrically optimized using the COMPASS III force field for molecular mechanics. Their amorphous cell modules with periodic structures consisted of a complex embedded in 500 spontaneously-generated water molecules. The molecular dynamics (MD) processing steps were performed at an interval of 1 fs. In the stabilizing process for 600 ps, the cell density gradually equilibrated under NPT conditions (constant number of molecules, stabilized pressure, and stabilized temperature). The thermal fluctuations were sustained for 1000 ps at 298 K. Modeling and MD simulations were performed using Biovia/Accelrys Materials Studio 2022 (Dassault Systems) in Windows (Intel Core-i7 11700 processor and 16 GB of PC4-25600/DDR4-3200 main memory).The Boltzmann distribution Pi was computed using eqn (11)
 | (11) |
3. Results and discussion
3.1. INM, DCF, and LXP formed a mixture with HP-β-CD with a XRPD halo pattern, while PRX showed XRPD signals in its mixture with HP-β-CD
First, we produced the inclusion complexes of NSAIDs/HP-β-CD: the SM- and PM-prepared mixtures of NSAIDs with HP-β-CD at the designated ratios were obtained. Then, the prepared mixtures were verified using the XRPD measurements. The diffractograms for the neat NSAID, SM-prepared NSAID mixture with HP-β-CD, and plain HP-β-CD are shown in Fig. S1, S2, S3, and S4,† respectively. The diffractogram of the neat INM contained the verifiable signals at 2θ angles of 11.66, 16.72, 19.64, 21.88, 26.66, and 29.40 degrees in Fig. S1(A),† illustrating the most stable polymorph γ-form of INM.36 The SM-prepared INM demonstrated the XRPD pattern of an ordinary metastable polymorph α-form with the signals at 2θ angles of 8.42, 14.54, 18.52, and 22.12 degrees.36 The observed angles for the γ- and α-forms of INM were reproduced with the diffractograms simulated with the 3D crystallographic structures, as shown in Fig. S1(B).† The SM-prepared equimolar INM mixture with HP-β-CD showed a halo pattern with two broad peaks that resembled that of the plain HP-β-CD, indicating that these INM/HP-β-CD complexes were significantly identical to the previous report.36 The prepared PM mixture was combined with the signals of the INM form and the halo pattern of HP-β-CD.The diffractogram of neat PRX contained the distinctive diffraction signals at 2θ angles of 8.74, 14.62, 17.80, 22.50, and 27.49 degrees, as shown in Fig. S2(A),† leading them to be identified as a cubic crystal (I form) of PRX.18,20,21 The signals were supported by the 3D crystalline structure of PRX, as shown in Fig. S2(B).† For the SM-prepared equimolar PRX mixture with HP-β-CD, the diffractogram shows distinctive diffraction signals corresponding to the cubic crystal overlapping the halo pattern of HP-β-CD. Similar results were observed in the diffractograms of PM-prepared equimolar mixtures. These mixtures were considered to contain the PRX/HP-β-CD complex and partially retain the crystalline PRX, which is consistent with a previous report.18 If an amorphous structure consisting of the PRX/HP-β-CD complex without crystals was accomplished, further improved techniques would be required.20,21
The diffractogram for neat DCF acid consisted of distinctive diffraction signals at 2θ of 10.66, 15.26, 18.86, 20.56, 24.42, and 28.54 degrees, as shown in Fig. S3(A),† corresponding to the published patterns.31,44,45 These signals were verified using the 3D crystalline structure of DCF (Fig. S3(B)†-HD2). Halo patterns were obtained for DCF mixtures. The XRPD patterns shown in Fig. S4† were obtained for neat LXP sodium salt dihydrate (LXP-Na), its mixtures, and plain HP-β-CD. That of neat LXP-Na was similar to the signals equivalent to the reported angles,46,47 but no 3D crystallographic structure of the LPX-Na dihydrate was observed. The halo patterns of the mixtures were similar to those of plain HP-β-CD. These results indicate the successful synthesis of the DCF/HP-β-CD and LXP/HP-β-CD complexes and destroyed the crystals.
3.2. Polymorphism of INM and partial crystals of PRX were observed in the HP-β-CD mixture
Second, we confirmed the physicochemical and material scientific details of the APD–CD complex in the solid state, and the related samples were examined for thermal analyses using differential scanning calorimetry (DSC) to verify the state of the INM/HP-β-CD complex. Fig. 1(A) shows the obtained thermograms of SM- and PM-treated INM, equimolar INM mixtures with HP-β-CD, and unbound HP-β-CD. The thermogram of the SM-treated pure INM contained an endothermic peak at 428 K, which disappeared in that of the PM-treated pure INM, namely, neat INM, with a melting point of 435 K.36 The correspondence of this signal to the melting of the α-form was supported by the XRPD diffractogram in Fig. S1(C).† During DSC scanning, the heating process transformed into the γ-form along with the melting of the α-form.36 For the PM-prepared INM mixture with HP-β-CD, after broad heat absorption at about 360 K, a sharp endothermic peak was observed at temperatures below 435 K. The second peak corresponds to the form of INM, according to the XRPD pattern in Fig. S1(C).† For the SM-prepared mixture, we found only a broad heat absorption at approximately 340 K. Its diffractogram suggested a similar amorphous state of the plain HP-β-CD, indicating the absence of the INM crystal.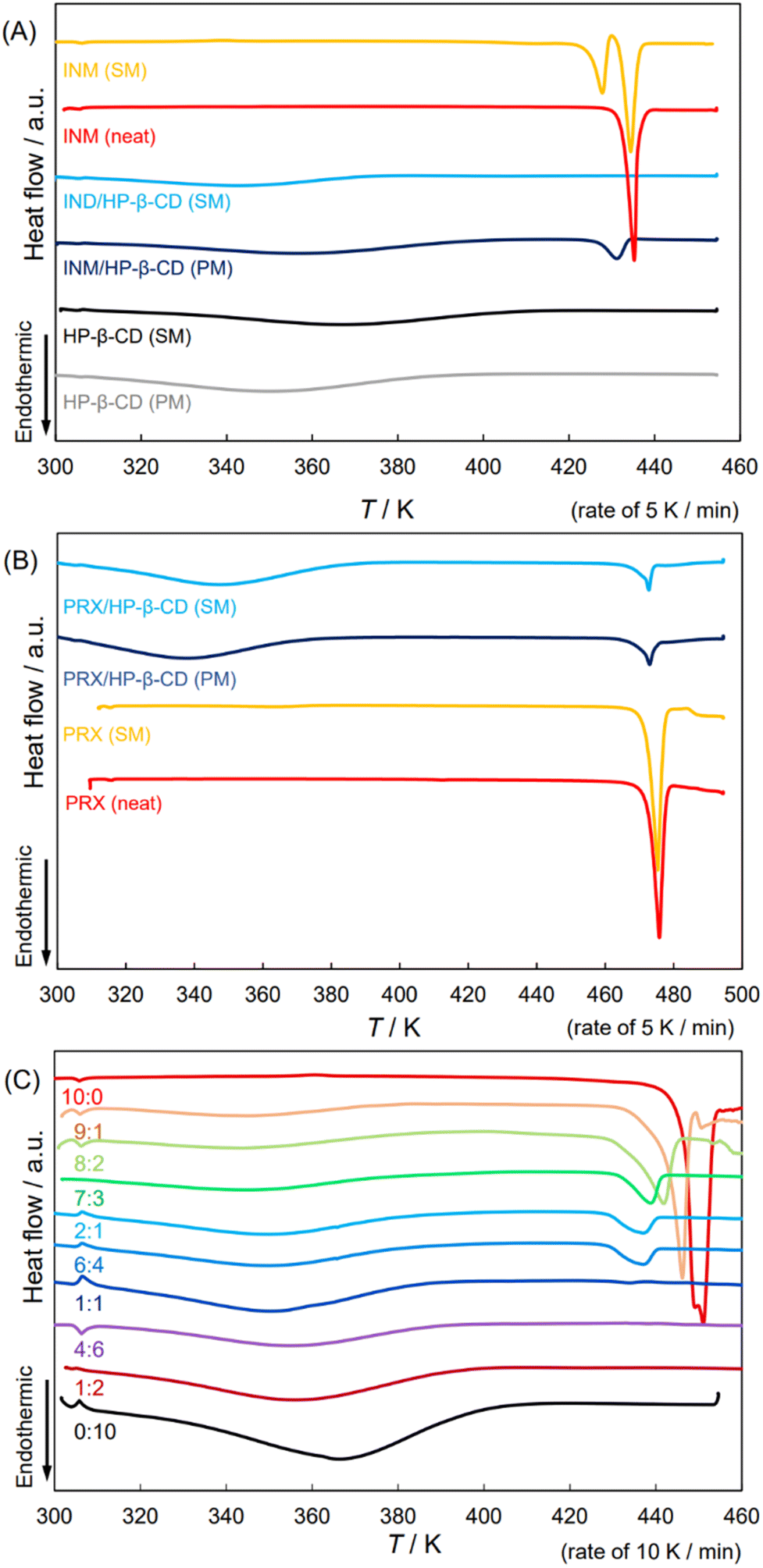 | ||
| Fig. 1 DSC thermograms of neat/SM-treated APIs; the SM-/PM-prepared equimolar mixtures of APIs and HP-β-CD and those of HP-β-CD. (A) INM, (B) PRX, and (C) DCF. | ||
Fig. 1(B) shows the thermal analyses of PRX mixtures with HP-β-CD. Similar to the XRPD diffractograms in Fig. S2(C),† the DSC patterns of PM- and SM-prepared PRX, and their mixtures with HP-β-CD indicated that all these samples contained more or less stable crystals of PRX. The observed endothermal peaks were detected at a temperature of 470 K.18,20,21 These mixtures might contain the PRX/HP-β-CD complex but partially remained in the crystalline PRX form, which was coincident with the report.18 Mixtures showed broad absorption at about 340 K, a slightly lower temperature than the absorption in the plain HP-β-CD. Although this was not sufficient for confirmation, the mixtures were thermodynamically anticipated to be in a slightly more stable state because of the significant reduction in their endothermic peaks.18
Fig. S5† shows the ATR-FTIR spectra of the SM-prepared PRX mixtures with HP-β-CD at the ratios of 2:1, 1:1, and 1:2 at the standard temperature. In the plain HP-β-CD spectrum, we found a broad band of OH stretching vibrations at 3322 cm−1, signals of C–H stretching vibrations at 2963 and 2918 cm−1, and a complex band of asymmetric bending vibrations around the ether bonds at 1016 cm−1.18 In the neat PRX spectrum, the sharp peak at 3332 cm−1 indicated the OH stretching vibration.18 Although the NH vibration at about 3390 cm−1 was not recognized, the high-intensity absorption of the NH deformation vibration appeared at 1523 cm−1, and the peaks at 1628, 1561, and 1434 cm−1 were assigned to pyridine and conjugated benzene rings at the temperature.18 The bands at 1348 and 1179 cm−1 were related to the asymmetric and symmetric bending vibrations of the SO2 group, respectively.18 Aromatic C–H bending vibrations in the planar C–H groups of the aromatic structures were observed at 876, 828, and 771 cm−1.18 We attempted to extract the spectral pattern specific to the PRX/HP-β-CD complex, comparing the FTIR spectra of the PRX mixtures with HP-β-CD, neat PRX, and plain HP-β-CD due to the singular value decomposition (SVD) procedure (see Fig. S5† for details). The analyses indicated that neither the peak appearance nor the peak shift was significant.
According to the thermal analysis, XRPD patterns, and FTIR spectroscopy, the SM- and PM-prepared PRX mixtures with HP-β-CD seldom contained observable evidence of the intermolecular interacting complex formation of PRX and HP-β-CD.
3.3. DCF remained HD2 polymorph both in neat form and in its mixture with HP-β-CD
In our previous study, the crystal signals of the INM mixture with HP-β-CD in the XRPD diffractograms disappeared at molar ratios less than 7:3.36 This study examined the behavior of DCF mixtures with HP-β-CD. Fig. 1(C) shows DSC thermograms of the DCF mixtures by SM-preparation at mole fractions in intervals of 1/10 (containing 2:1 and 1:2 mixtures). The diffractograms at 10:0, 1:1, and 0:10 ratios are shown in Fig. S3(A).† Identical signals that appeared in the XRPD thermogram of neat DCF were observed in the patterns with molar ratios of 9:1 and 8:2. The DSC endothermic peak beginning at the melting point of 451 K was assigned to the most stable polymorph of DCF, and neat DCF additionally exhibited a shoulder at 445 K at a heating rate of 10 K min−1. If the heating rate decreased (5 K min−1), the shoulder dissipated (Fig. S6(A) and S6(B)†). The DCF mixtures with small amounts of HP-β-CD at molar ratios of 9:1 and 8:2 contained endothermic DSC peaks at temperatures lower than the melting point. As described above, the XRPD diffraction peaks of the mixtures were invariable.
Lai and his colleagues44 reported two crystalline forms: “DCF1” (precipitated by neutralization of aqueous solution) with its endothermal peak at 449 K, and “DCF2” (recrystallized from chloroform) with its endothermal peak at 454 K (these peak positions that would be differently defined correspond to 445 and 451 K, respectively). They, however, claimed that the 2θ angles of signals in their diffractograms coincided with each other.44 A similar trend was observed in the present study. From the perspective of the DSC patterns, lower mole fractions and XRPD halo patterns were observed at molar ratios of 7:3 or less, and endothermic peaks were obtained at a temperature of approximately 430 K at molar ratios of 7:3, 2:1, and 6:4. This was a distinctive feature of the DCF mixtures compared to the thermal analysis of the INM mixtures. In the CCDC database, DCF polymorphisms were entered into HD1, HD2, and HD3 forms. As shown in Fig. S3,† we obtained significant diffractograms in agreement with those of the HD2 form. This XRPD pattern was consistent with the diffractogram reproduced according to the 3D crystalline structure of the HD2 form but was different from the diffractograms reproduced from those of the HD1 and HD3 forms (Fig. S3(B)†). States with the same diffractogram and different melting temperatures indicate the possibility of different assemblies composed of a common crystal lattice. The heat capacity in the clusters of microcrystal fragments may be less than that of the aligned crystals, which can be imaged as eutectic mixtures or solid dispersions consisting of microcrystals of the same material. During a phase transition, the entropy change is given by the integral of the heat capacity with respect to the temperature logarithm. The higher the heat capacity of the solid, the lower its melting entropy.
We examined the recrystallization of pure DCF using diethyl ether, acetone, chloroform, ethanol, 2-propanol, and acetonitrile. Fig. S6(B)† shows the DSC thermograms of the neat and recrystallized DCF samples. Their melting points and enthalpies were analyzed, and their melting entropies were calculated, as shown in Table 1. The melting temperatures of the samples treated with 2-propanol (446.4 K) and acetonitrile (449.8 K) deviated from the range of their average ± standard deviation (SD) (447.2–449.6 K), and the entropy changes (41.6 J K−1 mol−1) of the sample treated with 2-propanol also varied from their average ± standard deviation (SD) (49.1–62.0 J K−1 mol−1). However, the signals in the XRPD diffractograms of neat DCF and the sample treated with 2-propanol coincided significantly. Adding a small amount of HP-β-CD likely had an effect on DCF recrystallization, similar to recrystallization with 2-propanol. This may have introduced a sort of disorder into the crystalline regularity. In other words, the transformation to the form of INM was experimentally verified,36 whereas the shoulder of DCF at 445 K (Fig. S6(B)†) provided no evidence of the induction of a crystallographically apparent metastable polymorphism.
| T ini | T peak | T end | ΔH (J g−1) | ΔH (kJ mol−1) | ΔS (J K−1 mol−1) | |
|---|---|---|---|---|---|---|
| Neat | 449.6 | 451.7 | 454.2 | −89.4 | 26.5 | 58.9 |
| Diethyl ether | 449.1 | 451.7 | 454.1 | −88.9 | 26.3 | 58.6 |
| Acetone | 448.3 | 451.0 | 453.6 | −88.7 | 26.3 | 58.6 |
| Chloroform | 447.8 | 450.7 | 453.4 | −91.2 | 27.0 | 60.3 |
| Ethanol | 447.7 | 450.7 | 452.8 | −82.6 | 24.5 | 54.6 |
| 2-Propanol | 446.4 | 449.6 | 451.6 | −62.6 | 18.5 | 41.6 |
| Acetonitrile | 449.8 | 453.3 | 456.0 | −85.3 | 25.3 | 56.2 |
3.4. INM and PRX produced dual complexes with single HP-β-CD in solution
Third, we determined the mole fraction of the inclusion complex and the stability constant, and the phase solubility diagrams of INM and PRX were obtained in the solution at HP-β-CD concentrations of 0–300 mM at pH 1.2. The phase-solubility diagrams were AL-type for both INM/HP-β-CD and PRX/HP-β-CD at low concentrations of HP-β-CD (up to 50 mM).20,48 Here, the solubilities of INM and PRX increased almost linearly along a tangent line at lower HP-β-CD concentrations, whereas it involved a gradually increasing deviation from the tangent line at HP-β-CD concentrations of 100–300 mM (Fig. S7†). The itraconazole mixture with HP-β-CD showed a similar trend (10% w/v corresponded to 70 mM).49 INM and PRX, whose solubility was confirmed to be saturated after 2 days (INM) and 5 days (PRX), were analyzed by applying both a linear regression analysis assuming the AL-type and a multiple regression analysis assuming the AP-type, as shown in Table S1.† The coefficients of determination, namely, the squares of the regression coefficients, for INM as AL-type (eqn (1)) and AP-type (eqn (2)), were calculated to be 0.9688 and 0.9923, respectively. The latter was high, indicating that the diagram of INM solubility as a function of the concentration of HP-β-CD traced a parabolic curve corresponding to the AP-type. Stoichiometry was, therefore, determined as 1:2, and its stability constants of K1:1 = 1490 M−1 and K1:2 = 3.18 M−1 at pH 1.2. Backensfeld et al. reported that K1:1 was 1580 M−1 at pH 4.9 (phosphate buffer) and gradually decreased with an increasing pH.48 Although it seemed that the obtained K1:1 value was lower, their group did not examine solubility tests under such low pH conditions, and the effects of the discrepancy in buffer components are unexpected, according to the Henderson–Hasselbalch theory. Similarly, the coefficients of determination for PRX were 0.9855 for the AL-type and 0.9992 for the AP-type (Table S1†). Hence, the stoichiometry of PRX and HP-β-CD was determined to be 1:2, and the stability constants were K1:1 = 136 and K1:2 = 1.99 at pH 1.2. The K1:1 value of PRX/HP-β-CD was relatively small. Jug and colleagues reported K1:1 = 105.1 M−1 in an aqueous solution of HP-β-CD (i.e., not controlled pH).19 It could be assumed that the solubility of neutral PRX would slightly differ between distilled water and the buffered solution at pH 1.2 because of the low dissociation (and the relatively low contribution of coulombic interaction). These findings suggest that the K1:1 value obtained in the present study is acceptable. For DCF examined in the concentration range of 0–120 mM of HP-β-CD, the coefficients of determination were 0.9466 as AL-type and 0.9999 as AP-type (Table S1†), indicating that the stoichiometry of DCF and HP-β-CD was determined to be 1:2 and its stability constants were K1:1 = 3.18 and K1:2 = 113 at pH 1.2.
3.5. DCF formed an equimolar complex with a low concentration of HP-β-CD but unstable aggregations with a high concentration of HP-β-CD
Compared to INM and PRX, the GI adverse effects of DCF were not as severe, wherein the masking effect of CDs is not a desired emergent obligation. The phase-solubility isotherm diagram for DCF was examined at pH 6.8, which is close to intestinal or related physiological conditions. The equilibrium concentrations of DCF in the HP-β-CD aqueous solutions of various concentrations (pH 6.8) were surveyed for three days. However, the dissolution curves at concentrations of 120 mM and higher contained larger experimental errors, sometimes more than half of the determined values (Fig. S7(A)–(C)†). The viscosities of the HP-β-CD solutions at high concentrations were examined (Fig. S7(D)†). In the viscosity-concentration diagram, a linear increase in the viscosity appeared at HP-β-CD concentration until 120 mM (i.e., 17 w/v%), after which a positive deviation from linearity was observed. This upward curvature was observed at higher HP-β-CD concentrations (until 300 mM; i.e., 42 w/v%). For INM and PRX at pH 1.2, satisfactory parabolic correlations were observed in the 100–300 mM concentration range. Contrarily, for DCF at pH 6.8, it was conceived that the given complex DCF/HP-β-CD attracted each other with electrostatic interactions, enfolding the metal cations of the buffer components because DCF (pKa = 4.15) is almost dissociated and negatively charged. This could be the reason why the solutions were insufficiently agitated, and their deviations in concentrations involved diverse measurements.To determine the equilibrium concentration of DCF at pH 6.8, the dissolution curves for 10–14 days were traced. Fig. 2 shows the phase-solubility diagram; the ascending region at a low concentration of HP-β-CD was followed by an attenuation of the upward slope at concentrations over 60 mM. Scavonae and colleagues published the phase-solubility diagram of DCF and HP-β-CD in their review, which showed a linear correlation until the abscissa of the 150 mmol HP-β-CD per g solution, while the curve seemed to saturate after arriving at the ordinate of 0.23 mmol DCF per g solution.50 Although the provenance of their diagram is not presented, its trend is similar to that reported herein. Here, we obtained the value of K1:1 = 33.21 (pH 6.8) from a linear correlation up to 60 mM, which is consistent with the reported value of K1:1 = 57 M−1 (at pH 6.8, 1053 M−1 at pH 7.7) according to the linear correlation on the phase-solubility diagram presented by Bodley et al.34 Although our present study illustrated a saturation curve, the diagram was indeterminate at higher concentrations. Assuming that the excessive viscosity provided resistance to agitation under these conditions, the viscous solution would endlessly suspend poorly-soluble aggregated dispersoids, and sedimentation would also be disturbed.
 | ||
| Fig. 2 Phase-solubility diagram of IND at pH 1.2 (A), PRX at pH 1.2 (B), and DCF at pH 6.8 (C). INM solubility was measured after 1 (open squares), 2 (open triangles), and 4 days (open diamonds), and PRX solubility after 5 days (closed circles). Linear regression analysis of DCF solubility is represented as the equation and the R2 value. The inset shows the dissolution curves of DCF at pH 6.8. Curve fitting was computed using eqn (4). | ||
As Szejtli described, “it can point either to an increase of the host ratio within the complex or a change in the solute–solvent interaction (hydration and ionization of the guest) or both” in AN-type.13 The difference between AN- and BS-types can be ambiguous and depends on whether precipitation or sedimentation is confirmed at high concentrations.42 In addition, the saturation and plateau are difficult to distinguish from this observation. In this study, the HP-β-CD complex containing neutralized DCF was found to be negatively charged. Hence, the individual complexes were potently repulsive to each other and showed attractive interactions for the metal cations in the buffer components. Regardless of whether the classification was formally AN- or BS-types, the solution/suspension was inhomogeneous, and its concentration could not be measured with adequate accuracy after incubation for 72 hours. Messner et al. reported that the self-assembly of cyclodextrins depends on guest molecules.52 Ibuprofen and dextromethorphan flocculated at high concentrations of HP-β-CD (40 and 90 mM, respectively), forming impermeable aggregations on the cut-off membrane with a molecular weight of 100 kDa, corresponding to more than 60 units of APD–CD complexes. DCF may be dispersed because of the aggregated assembly of DCF/HP-β-CD, although the size transitions of the assembled particles in the phase-solubility diagram are being investigated by our research group.53 Under 60 mM of HP-β-CD, the stoichiometry of DCF/HP-β-CD was equimolar.
3.6. Both premade INM/HP-β-CD and makeshift PRX/HP-β-CD complexes enabled the parachute effect
The XRPD measurements and the DSC thermal analyses suggested that the INM gave rise to an amorphous mixture with HP-β-CD at a molar ratio of 2:1 (Fig. S1†), whereas PRX at any ratio produced amorphous patterns that partially contained crystals (Fig. S2†). As shown in Fig. 2(A) and (B), the phase solubility experiments at pH 1.2 indicated that INM and PRX demonstrated equivalent stoichiometry; however, the stability constant of PRX/HP-β-CD was particularly less than those of INM/HP-β-CD and DCF/HP-β-CD. The dissolution curves of the neat INM, neat PRX, and their mixtures with the HP-β-CD are described in section 3.6 to consider the influence of their properties on the dissolution curves of the mixtures. In our previous study, the SM-prepared INM mixture with HP-β-CD at a molar ratio of 1:2 dissolved monotonically, whereas the complexes at molar ratios of 2:1 and 1:1 rapidly dissolved to show supersaturation and gradually decreased until equilibrium was reached.36
Fig. 3(A1)–(A3) show the dissolution curves of the SM- and PM-prepared INM mixtures with HP-β-CD and neat INM at pH 1.2. Fig. 3(A1) illustrates the curves in a short period of 0–4 hours, while Fig. 3(A2) presents the same combination of their equilibrated curves in a long period of 0–432 hours. The inset in Fig. 3(A3) demonstrates that the dissolution curves of the γ- and α-crystal of INM were simple saturation curves whose equilibrium concentrations were not significantly different (dissolution rate constant α = 2.03 h−1 and recrystallization rate constant β = 6.10 h−1 for γ-form, α = 44.4 × 10−3 h−1 and β = 4.42 h−1 for α-form). A small amount of supersaturation was observed in the amorphous mixture, however this attenuated to equilibrium concentration after 12 hours. In Fig. 3(A1) and (A2), the distribution curve of the PM-prepared equimolar INM mixture could be approximated by a typical Noyes–Whitney curve with an equilibrium concentration of approximately three times that of neat INM (α = 22.8 h−1 and β = 226 h−1). For the SM-prepared equimolar INM mixture, the concentrations of the supersaturating INM (at 10 min) and the equilibrium INM (at 2 hours and more) were about 250-fold and 60-fold to that of neat INM, respectively (α = 1.77 h−1 and β = 12.3 h−1). The SM-prepared INM mixture with a molar ratio of 2:1 showed similar behavior to the equimolar mixture (α = 1.84 h−1 and β = 10.8 h−1). In contrast, for the SM-prepared INM mixture with HP-β-CD at a molar ratio of 1:2, supersaturation was observed after 4 hours, and its peak height was 40% of that of the equimolar mixture (α = 25.6 × 10−3 h−1, β = 0.667 h−1). These results indicate that the premade mixture containing an excess HP-β-CD succeeded to implement a rapid (1–2 h) parachute effect at a ten-fold amount compared to neat INM.36,39
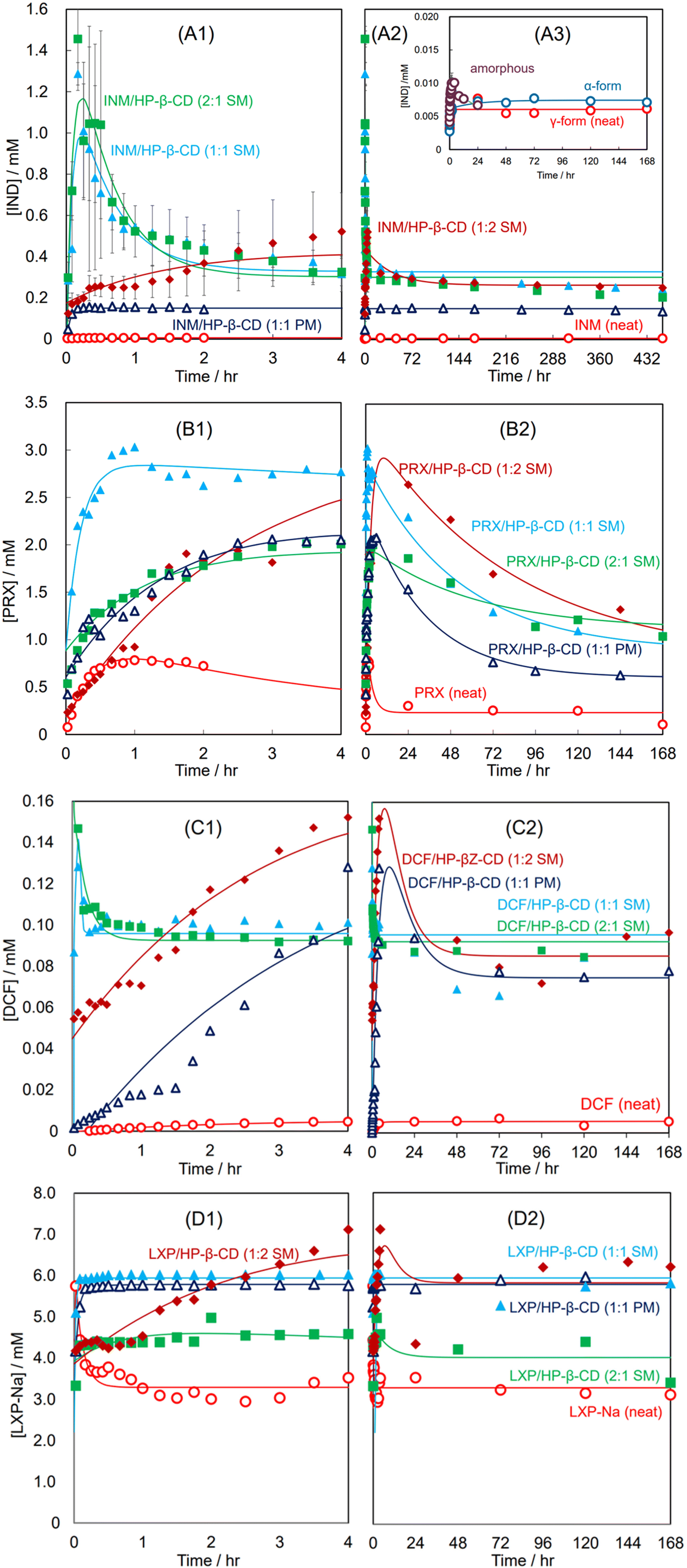 | ||
| Fig. 3 Dissolution curves of neat APIs and their SM-/PM-prepared mixtures with HP-β-CD at various molar ratios at pH 1.2: INM and its mixtures in 4 hours (A1) and in long periods (A2); γ-, α-forms, and amorphous INM in 168 hours in inset (A3); PRX and its mixtures in four hours (B1) and in long periods (B2). Curve fitting was performed using eqn (8)–(10). DCF and its mixtures in four hours (C1) and in long periods (C2); LXP and its mixtures in four hours (D1) and in long periods (D2). | ||
Fig. 3(B1) and (B2) show the dissolution curves of the SM- and PM-prepared PRX mixtures with HP-β-CD and neat PRX at pH 1.2. Fig. 3(B1) illustrates the incremental curves for a short period of 0–4 hours, whereas Fig. 3(B2) presents the same combination of decreasing curves over a long period of 0–168 hours. The neat PRX (anhydrate) dissolved at a concentration of 0.8 mM in the first hour; however, after supersaturation, the concentration gradually decreased to less than 0.3 mM for 24 hours (α = 0.316 h−1 and β = 2.72 h−1). The precipitation of the thermodynamically more stable hydrate crystal compared to the anhydrate one reduced the concentration in the solution and provided supersaturation, as described in our previous study.39 The SM-prepared equimolar PRX mixture with HP-β-CD immediately dissolved and remained at a concentration 11 times higher than that equilibrated with the PRX hydrate. After 20 hours, the concentration slowly decreased (α = 19.3 × 10−3 h−1 and β = 4.71 h−1). The concentration of the SM-prepared PRX mixture with a molar ratio of 1:2 increased slightly, reaching a concentration as high as that of the equimolar mixture. After 20 hours, although the amount of PRX in the solution was reduced, it remained at a higher concentration than the equimolar mixture at all times (α = 16.8 × 10−3 h−1 and β = 0.850 h−1). For the SM-prepared mixture with a molar ratio of 2:1, the maximum concentration was observed at 3/4 that of the equimolar mixture (α = 12.3 × 10−3 h−1 and β = 0.374 h−1). The dissolution curve of the PM-prepared equimolar mixture was similar to the SM-prepared 2:1 mixture for the first 4 hours, but its concentration decreased to the lowest level within 72 hours (α = 30.3 × 10−3 h−1 and β = 0.673 h−1).
As a result, the PRX mixture, in which amorphous inclusion complex formation was insufficient in the solid state, formed a makeshift complex of PRX/HP-β-CD in solution and succeeded in implementing a persistent parachute effect. These findings indicate that neither the dissolution performance nor the parachute effect require a halo pattern in the XRPD diffractogram.
3.7. The dissolution performance and parachute effect of DCF/HP-β-CD and LXP-Na/HP-β-CD complexes were indiscriminately targeted by the SM- and PM-preparations
The XRPD measurements and DSC thermal analyses suggested that DCF and LXP-Na efficiently produced amorphous mixtures with HP-β-CD at molar ratios of 1:1 and 2:1, respectively (Fig. S3 and S4†). Although the DCF solubility in the HP-β-CD solution at pH 6.8 traced a typical saturation curve dependent on the concentration of the HP-β-CD, it became difficult to determine the high concentration of HP-β-CD (more than 120 mM) due to interference with viscosity via a solution for the sedimentation of the insoluble aggregation. The dissolution performance and parachute effects of DCF/HP-β-CD and LXP-Na/HP-β-CD complexes are described in section 3.7. The aqueous solubility of neat DCF was as poor as that of INM, although that of LXP-Na was over 3 mM, for which some improvement was expected through the inclusion-complexation of LXP-Na/HP-β-CD.
Fig. 3(C1) and (C2) show the dissolution curves of the SM-prepared DCF mixtures with HP-β-CD at molar ratios of 2:1, 1:1, and 1:2, and that of the PM-prepared equimolar mixture at pH 1.2. Fig. 3(C1) illustrates the curves for a short period of 0–4 hours, whereas Fig. 3(C2) presents the same combination of equilibrated curves over a long period of 0–168 hours. For the neat DCF, a simple Noyes–Whitney type saturation curve was obtained with an equilibrium concentration of 0.0055 mM (α = 10.3 × 10−3 h−1 and β = 0.490 h−1). For the PM-prepared equimolar mixture, the DCF concentrations gradually increased until its supersaturation at about 0.13 mM after 10 hours and gradually decreased toward a concentration of 0.075 mM (α = 87.2 × 10−3 h−1 and β = 0.213 h−1). For the SM-prepared DCF mixtures at a molar ratio of 1:2, the supersaturated dissolution curve progressed parallelly to the PM-prepared equimolar mixture (α = 0.115 h−1 and β = 0.231 h−1). Its peak concentration of supersaturation reached a further high concentration of 0.155 mM (28-fold higher than the equilibrated concentration of the neat DCF). In contrast, the SM-prepared mixtures at molar ratios of 2:1 and 1:1 presented immediate supersaturation of 25-fold times the equilibrated concentration of the neat DCF. Subsequently, it readily decreased down to a concentration of 17-fold that of the neat DCF and equilibrated (α = 5.75 h−1 and β = 5938 h−1 for the 2:1 mixture and α = 36.6 h−1 and β = 121 h−1 for the equimolar mixture). These DCF results were qualitatively similar to those of the corresponding INM mixtures; however, the equilibrated concentrations after the supersaturation of the 2:1 and 1:1 mixtures remained higher. Furthermore, the equilibrium concentration of DCF in the PM- and SM-containing mixtures was maintained at relatively high levels. The mixtures containing HP-β-CD effectively succeeded in implementing a persistent parachute effect for 24 hours. The supersaturation of GI-adverse INM/HP-β-CD at an acidic pH is disadvantageous, while DCF is not a concern, for which high performance is expected.
Fig. 3(D1) and (D2) show the dissolution curves at pH 1.2 for the SM-prepared LXP-Na mixtures with HP-β-CD at various molar ratios and the PM-prepared equimolar mixture. The dissolution curve of the neat LXP-Na rapidly rose to 6 mM and immediately converged to 3 mM, maintaining α = 8.42 h−1 and β = 753 h−1. Similarly to neat LXP-Na, its PM- and SM-prepared equimolar mixtures immediately dissolved, while their equilibrium concentration remained at 6 mM, that is approximately twice that of neat LXP-Na, for 168 hours in the manner of Noyes–Whitney saturation curve (α = 2.77 h−1 and β = 20.7 h−1 for PM and α = 23.7 h−1 and β = 59.3 h−1 for PM). The SM-prepared mixture with a molar ratio of 2:1 was immediately dissolved to a concentration of 4.5 mM. Our results indicate that its LXP concentration reached a slightly lower concentration over a long period (α = 0.124 h−1 and β = 1.49 h−1). The SM-prepared mixture with a molar ratio of 1:2 demonstrated a persistent parachute effect. According to the non-linear fitting approximation, the supersaturation curve peaked at 8 hours and transferred to a stationary phase after 48 hours. However, the concentration at the top of the peak was 7 mM, which was at most twice; therefore, the pharmaceutical advantage of an inclusion complex with cyclodextrins seems poor for this quick-acting drug, as described above.
3.8. The regulating factors of API dissolution behaviors included API solubility, as well as the compositions and states of the solid matrix
INM, DCF, and LXP-Na prioritize being complexed with the equimolar HP-β-CD, and their equimolar mixtures are enclosed by the unbound HP-β-CD acting as a solid matrix. Here, the matrix is generally denoted as a polymer, providing the composite component with its shape and surface quality. In the science of physical pharmacy, the matrix is equipped with features to disperse APIs in itself as a solid and to assist in the release of APIs at a steady rate because of the collapse of the matrix. Using the unbound HP-β-CD matrix, we anticipated that the NSAID/HP-β-CD complexes would be gradually released from the solids. Indeed, the dissolution curves of the mixtures at the molar ratio of 1:2, namely, a sufficient amount of the unbound HP-β-CD, were gradually elevated. Meanwhile, the major components of the NSAID/HP-β-CD complexes at molar ratios of 2:1, namely, with less or no unbound HP-β-CD, and 1:1, were immediately released. The latter was considered to be due to the fact that the excess amount of solid in the mixtures did not contain a sufficient volume of the actual matrix, and the isolated NSAIDs would remain in the solid because the solution was saturated with the NSAIDs. In particular, the INM and DCF mixtures at molar ratios of 2:1 and 1:1 were immediately released and caused conspicuous supersaturation, subsequently slowing down to the equilibrium concentration due to the recrystallization of INM and DCF (or their hydrates).36 The equilibrium concentrations of their mixtures at molar ratios of 2:1 and 1:1 were not significantly different.36
The molar ratio of stable clathrates has a gap between the amorphous solid and the aqueous solution. The process of water seeping into the amorphous solid and dissolving it quickly leads to an apparent supersaturation state. However, as it slowly settles into an equilibrium state in solution to stabilize by containing equimolar amounts of indomethacin and diclofenac in cyclodextrin, the stability constant settles to a much smaller ratio. These results suggest that the ratio deemed appropriate in solid-phase analysis may not necessarily align with the stability constant in the aqueous solution.
The SM-prepared LXP-Na mixture at a molar ratio of 2:1 showed little advantage in solubility compared to neat LXP-Na; therefore, its dissolution curve was insignificantly different from that of neat LXP-Na.
For the PRX mixtures, the percentage of the complex content appeared to be relatively low, and it is likely that the soluble PRX/HP-β-CD complex was accompanied by a large amount of unbound HP-β-CD. As described above, the phase solubility experiments suggested a small stability constant for the PRX/HP-β-CD complex, which may explain why the proportion of the PRX/HP-β-CD complex to the unbound PRX decreased during SM preparation. The degree of dissociation is assumed to depend on the stability constants.8,51 The difference between the dissolution curves of the PRX and DCF mixtures was caused by the complexation efficiency of the solid mixtures. Hence, an imbalance of the molar ratios (2:1 and 1:2) would reduce the release of PRX complex since the PRX mixtures contained a lower amount of the PRX/HP-β-CD complex and a lot of matrices.
3.9. The DCF and HP-β-CD mixture contains as large a part of DCF/HP-β-CD inclusion-complex as possible
Fig. 4 shows the dissolution curves at pH 6.8 of the SM-prepared DCF mixtures with HP-β-CD at molar ratios of 1:1 and 1:2 and that of the PM-prepared equimolar mixture. The concentrations of DCF in the SM-prepared mixture at a molar ratio of 1:2, the PM-prepared equimolar mixture, and the neat DCF were immediately saturated at neutral pH (a dissolution kinetic constant kS = 9.57 × 105 d−1, a saturation concentration Cs = 7.32 mM for SM-prepared 1:2 mixture and kS = 10.9 d−1, Cs = 6.16 mM for the PM-prepared 1:1 mixture). Because the mixture consisted of DCF without sodium, the dissolution process involved neutralization with metal cations in the buffer. Contrarily, it can be explained that the DCF acid was extracted from the solid matrix by the electrostatic attraction of the metal cations. This predominantly influenced the dissolution rates of the SM-prepared DCF mixture at a molar ratio of 1:2 and the PM-prepared equimolar mixture. In contrast, the dissolution curve of the SM-prepared equimolar DCF mixture lost the supersaturation and gradually saturated to the equilibrium concentration of the mixture at a molar ratio of 1:2 (kS = 1.71 d−1 and Cs = 7.00 mM).
 | ||
| Fig. 4 Dissolution curves of neat DCF-acid, neat DCF-Na, their PM-prepared equimolar mixture with HP-β-CD, and the SM-prepared mixtures at various molar ratios at pH 6.8. Curve fitting was performed using eqn (4) and (8)–(10). | ||
We assumed that DCF moderately (partially) formed a DCF/HP-β-CD complex in the SM-prepared mixtures. The mixture consisted of free DCF and unbound HP-β-CD in addition to the DCF/HP-β-CD complexes at molar ratios of 1:1 and 1:2. Thus, the equimolar mixture contained less unbound HP-β-CD matrix than that with a molar ratio of 1:2. Therefore, the free DCF and the DCF/HP-β-CD complex in these mixtures are enclosed with less amount of matrix of the unbound HP-β-CD. The dissolution rate of the equimolar mixture was assumed to be higher than that at a molar ratio of 1:2. However, we obtained the opposite results. The experimental results supported an equimolar mixture predominantly consisting of the DCF/HP-β-CD complex. Because the DCF/HP-β-CD complex at a molar ratio of 1:2 has more aqueous solubility than the equimolar complex, the latter dissolves quickly, while the former dissolves more slowly. In conclusion, the dissolution patterns of equimolar DCF mixtures with HP-β-CD illustrated that equimolar complexes comprised a large proportion of the mixtures, and the DCF mixture at a molar ratio of 2:1 contained an equimolar complex and free DCF.
3.10. Energetics for the stability of the DCF/HP-β-CD complex in water
The solution pH transformed dissolution curves of the mixtures of the DCF acid with HP-β-CD and the supersaturation. In this process, DCF is extracted from the solid matrix by the coulombic attraction of the metal cations in the solution. MD simulations were performed to evaluate the effect of the acid dissociation of DCF on the inclusion to maintain the DCF/HP-β-CD complex in the solution. In the force field calculations, the systems containing the DCF anion without sodium cations or cationic sparkles (pseudo-atoms) were more stabilized during the computations of the structural optimizations and dynamics simulations than the systems with cations. In the 3D structure of HP-β-CD, the cyclic backbone with the α-1,4-glycosidic bonds among the seven glucose units shapes the side wall of a truncated cone, in which C3–H and C5–H protrude into the interior cavity. The hydroxyl groups of C2 and C3 were located at the larger rim (bottom interface) of the truncated cone, and the free or 2-hydroxypropylated hydroxyl group of C6 was positioned at the lower rim (upper interface) of the truncated cone. The results of the MD simulations indicate that either DCF acid or anion intruded across the C2–C3 rim interface and was more energetically favorable than their intrusions across the C6 rim interface, approaching the interior cavity of the HP-β-CD.
Fig. 5 shows the energy profiles of the MD trajectory of the water-filled cells for the systems of DCF acid and DCF anion associated and dissociated with HP-β-CD during the equilibrated thermal fluctuation from 600 to 1000 ps. The most stable complex of DCF acid with their 2,6-dichloroanilino group intruded across the C6 rim of HP-β-CD was equilibrated at a steric energy level of E0 = −276223.15 kJ mol−1. The system of the DCF acid dissociated with the HP-β-CD and the system of the propionic acid intruded across the C6 rim of HP-β-CD had energy differences ΔEi of 2364.94 and 4443.33 kJ mol−1 to the most stable system, respectively. The most stable complex of the DCF anion with their 2,6-dichloroanilino group intruded across the C6 rim of HP-β-CD was equilibrated at E0 = −274667.71 kJ mol−1. The system of the DCF anion with its propionate anion group intruded across the C6 rim of HP-β-CD, and the system of the DCF anion dissociated with the HP-β-CD and had ΔEi of 531.20 and 1424.87 kJ mol−1 to the most stable system, respectively.
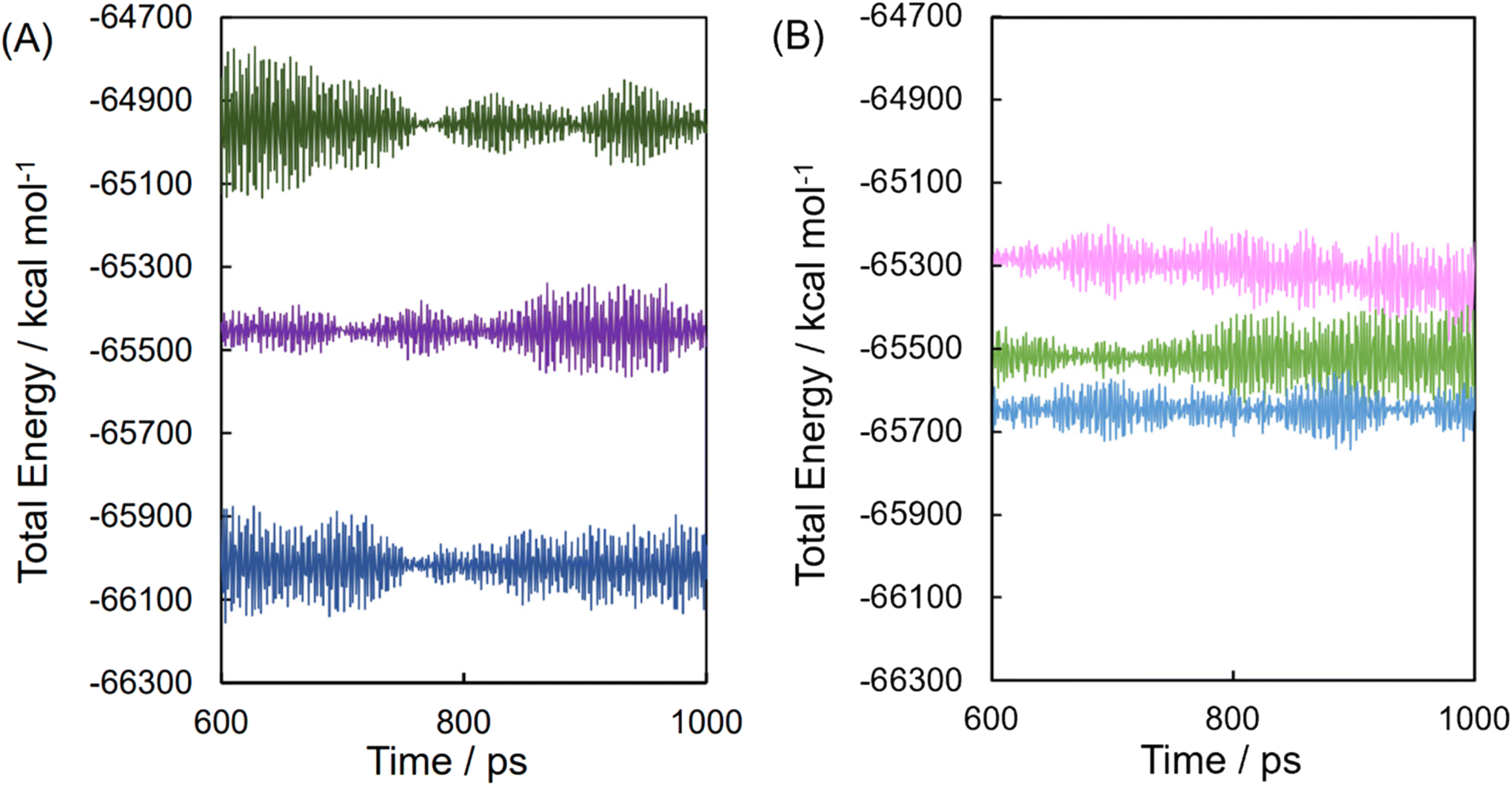 | ||
| Fig. 5 Molecular dynamic trajectory of DCF acid (A) or DCF anion (B) mixture with equimolar HP-β-CD after the equilibrium process. DCF acid approached the 6-hydroxylmethyl group side (dark green) and 2-/3-hydroxyl groups side (blue) and was isolated from HP-β-CD (violet), whereas the DCF anion approached the 6-hydroxylmethyl group side (green) and 2-/3-hydroxyl groups side (sky blue) and was isolated from HP-β-CD (pink). | ||
Although the steric energy levels of the most stable systems for DCF acid and DCF anion could not be compared because the systems contained different components, the energy differences of the metastable complexes were similar in the system for the DCF anion. This suggests that the anionic complex maintains energetic stability according to conformations containing similar Boltzmann distributions. Structural/conformational diversity (disorder) provides entropic superiority for dispersing solutes in solution. From the perspective of chemical potentials, it was considered that the anionic complex was predominant at pH 6.8; however, it was more stable than the neutral complex at pH 1.2, in addition to these complexes in solution.
4. Conclusion
The inclusion complex of APD–CDs derives solubilization and induces the “parachute” effect, thereby facilitating drug administration and release control. XRPD crystallography and the DSC thermal analysis based on the mole fraction of API to HP-β-CD indicate that the consumption of the API crystal was due to the complexation with HP-β-CD and provides the optimum ratio for the complexation of the API with HP-β-CD. The properties of the solid were not necessarily parallel to the stoichiometry obtained from the phase solubility diagram measured in solution. These findings demonstrated that the efficient solubility, dissolution rate, and optimum molar ratio of the APIs/HP-β-CD complexes were regulated by the stability constants, stoichiometry of complexations, and bulk phase in the solid matrix. As the mixture of DCF with HP-β-CD has properties similar to those of INM, we observed not only highly equilibrated concentration but also immediate supersaturation, specifically in the acidic solution. The proportion of the PRX/HP-β-CD complex affected the dissolution curve of delayed release and equilibrium solubility. Stoichiometry and stability constants do not guarantee improvements in solubility and dissolution rates. The apparent intensity of the complex/complex, complex/API, and API/API interactions can differ in the solid state and solution. Furthermore, the solubility and dissolution rates are influenced by many complicated factors, such as the equilibration of solutes/dispersoids with multiple solid states, the difference in solubility of the API and complex, and the balance in the chemical potential from the concentrations of solutes/dispersoids.Author contributions
Y. Oshite and W.-H. Ayako: writing, investigation, and visualization. R. Ichii: investigation for Fig. 3. C. Kuroda: investigation for the XRPD simulations and the SVD analysis for FTIR spectra. K. Hasegawa: investigation for the thermal analyses of recrystallized diclofenac. R. Hiroshuge, H. Yokoyama, T. Tsuchida: review and editing. S. Goto: supervision.Conflicts of interest
There are no conflicts to declare.References
- G. Tiwari, R. Tiwari and A. K. Rai, J. Pharm. BioAllied Sci., 2010, 2, 72–79, DOI:10.4103/0975-7406.67003.
- R. A. Rajewski and V. J. Stella, J. Pharm. Sci., 1996, 85, 1142–1169, DOI:10.1021/js960075u.
- D. Prasad, H. Chauhan and E. Atef, J. Pharm. Sci., 2014, 103, 3511–3523, DOI:10.1002/jps.24137.
- T. Lofsson and M. E. Brewster, J. Pharm. Sci., 1996, 85, 1017–1025, DOI:10.1021/js950534b.
- A. R. Hedges, Chem. Rev., 1998, 98, 2035–2044, DOI:10.1021/cr970014w.
- T. Loftsson, M. Masson and M. E. Brewster, J. Pharm. Sci., 2004, 93, 1091–1099, DOI:10.1002/jps.20047.
- K. Uekama, F. Hirayama and T. Irie, Chem. Rev., 1998, 98, 2045–2076, DOI:10.1021/cr970025p.
- R. Challa, A. Ahuja, J. Ali and R. K. Khar, AAPS PharmSciTech, 2005, 6, E329–E357, DOI:10.1208/pt060243.
- M. E. Brewster and T. Loftsson, Adv. Drug Delivery Rev., 2007, 59, 645–666, DOI:10.1016/j.addr.2007.05.012.
- S. Scalia, S. Villani and A. Casolari, J. Pharmacol., 1999, 51, 1367–1374, DOI:10.1211/0022357991777182.
- G. Moster and D. O. Thompson, Complexation and Cyclodextrins, in Encyclopedia of Pharmaceutical Technology, ed. J. Swarbrick and J. C. Boylan, Marcell Dekker, New York, 2nd edn, 2002, pp. 531–558 Search PubMed.
- C. Scarpignate, Piroxicam-β-cyclodextrin: A GI safer piroxicam, Curr. Med. Chem., 2013, 20, 2415–2437, DOI:10.2174/09298673113209990115.
- J. Szejtli, Res. Rev., 1994, 14, 353–386, DOI:10.1002/med.2610140304.
- V. J. Stella and R. A. Rajewski, Pharm. Res., 1997, 14, 556–567, DOI:10.0123/a:1012136608249.
- B. G. Woodcock, D. Acerbi, P. G. Merz, S. Rietbrock and N. Rietbrock, Eur. J. Rheumatol. Inflammation, 1993, 12, 12–28 CAS , PMID: 7805705.
- C. A. Bersani-Amado, S. F. Taniguchi, L. S. Sudo, E. Kimura and S. Oga, Gen. Pharmacol., 1995, 26, 809–813, DOI:10.1016/0306-3623(94)00204-Z.
- X. Guo, Y. Yang, G. Zhao, G. Zhang, J. Chao and S. Shuang, Spectrochim. Acta, Part A, 2003, 59, 3379–3386, DOI:10.1016/s1386-1425(03)00164-1.
- V. D. Nikolić, S. S. Ilić-Stojanović, L. B. Nikolić, M. D. Cakić, A. S. Zdravković, A. J. Kapor and M. M. Popsavin, Hem. Ind., 2014, 68, 107–116, DOI:10.2298/HEMIND130306034N.
- M. Jug, M. Bećirević-Laćan, B. Cetina-Čižmek and M. Horvat, Pharmazie, 2004, 59, 686–691 CAS , PMID: 15497750.
- M. Jug and M. Bećirević-Laćan, Eur. J. Pharm. Sci., 2004, 21, 251–260, DOI:10.1016/j.ejps.2003.10.029.
- X. Zhang, D. Wu, J. Lai, Y. Lu, Z. Yin and W. Wu, J. Pharm. Sci., 2009, 98, 665–675, DOI:10.1002/jps.21453.
- I. A. Alsarra, M. O. Ahmed, F. K. Alanazi, K. E. H. ElTahir, A. M. Alsheikh and S. H. Neau, Int. J. Med. Sci., 2010, 7, 232–239, DOI:10.7150/ijms.7.232.
- P. G. Conaghan, Rheumatol. Int., 2012, 32, 1491–1502, DOI:10.1007/s00296-011-2263-6.
- T. Mizuno, K. Ito, Y. Miyagawa, K. Ishikawa, Y. Suzuki, M. Mizuno, Y. Ito, Y. Funahashi, R. Hattori, M. Gotoh, K. Yamada and Y. Noda, Jpn. J. Clin. Oncol., 2012, 42, 1073–1078, DOI:10.1093/jjco/hys145.
- C. Scarpignate, Curr. Med. Chem., 2013, 20, 2415–2437, DOI:10.2174/09298673113209990115.
- P. McGettigan and D. Henry, PLoS Med., 2013, 10, e1001388, DOI:10.1371/journal.pmed.1001388.
- O. A. Teslim, M. K. Vyvienne, O. M. Olatokunbo, A. J. Oluwafisayo, N. B. Mlenzana, M. Shamila, T. Nesto and M. Grace, Am. J. Health Res., 2014, 2, 106–112, DOI:10.11648/j.ajhr.20140204.11.
- I. Nourwali, A. Namnakani, M. Almutairi, A. Alaufi, Y. Aljohani and S. Kassim, Dent. J., 2020, 8, 2, DOI:10.3390/dj8010002.
- T. Backensfeld, B. W. Müller and K. Kolter, Int. J. Pharm., 1991, 74, 85–93, DOI:10.1016/0378-5173(91)90225-D.
- O. Reer, T. K. Bock and B. W. Müller, Int. J. Pharm., 1994, 83, 1345–1349, DOI:10.1002/jps.2600830928.
- M. D. Bodley, M. R. Caira, L. A. Glintenkamp, V. J. Griffith, L. R. Nassimbeni, D. G. Murray, N. L. J. Penkler and M. C. B. Van Oudtshoorn, Inclusion complex of beta-cyclodextrin and diclofenac, its preparation and use, US005674854A, 1994.
- L. J. Penkler and B. P. Daisley, Stable injectable compositions, US2005/0238674A1, 2005.
- M. M. R. Dias, S. L. Raghavan, M. A. Pellett and J. Hadgraft, Int. J. Pharm., 2003, 263, 173–181, DOI:10.1016/s0378-5173(03)00366-1.
- M. D. Bodley, L. J. Penkler, L. A. Glintenkamp and M. C. Van Oudtshoorn, Pharmaceutical composition, containing diclofenac and 2-hydroxypropyl-beta-cyclodextrin, European Patent 0658347 A2, 1994.
- T. Higuchi and K. A. Connors, Phase Solubility Techniques, in Advanced Analytical Chemistry of Instrumentation, 1965, vol. 4, pp. 117–212 Search PubMed.
- A. Wada-Hirai, S. Shimizu, R. Ichii, C. Tsunoda, R. Hiroshige, M. Fujita, Y.-P. Li, Y. Shimada, Y. Otsuka and S. Goto, J. Pharm. Sci., 2021, 110, 3623–3630, DOI:10.1016/j.xphs.2021.07.002.
- Y.-P. Li, S. Goto, Y. Shimada, K. Komatsu, Y. Yokoyama, H. Terada and K. Makino, J. Phys. Chem. Biophys., 2015, 5, 1000187, DOI:10.4172/2161-0398.1000187.
- Y.-P. Li, S. Goto, Y. Shimada and K. Makino, J. Pharm. Sci. Technol., Jpn., 2016, 76, 267–273, DOI:10.14843/jpstj.76.267.
- M. Fujita, S. Goto, H. Chatani, Y. Otsuka, Y. Shimada, H. Terada and K. Inoo, RSC Adv., 2020, 10, 1572–1579, 10.1039/C9RA09952B.
- R. Hiroshige, S. Goto, R. Ichii, S. Shimizu, A. Wada-Hirai, Y.-P. Li, Y. Shimada, Y. Otsuka, K. Makino and H. Takahashi, J. Inclusion Phenom. Macrocyclic Chem., 2022, 102, 327–338, DOI:10.1007/s10847-021-01122-1.
- R. Hiroshige, S. Goto, C. Tsunoda, R. Ichii, S. Shimizu, Y. Otsuka, K. Makino, H. Takahashi and H. Yokoyama, J. Inclusion Phenom. Macrocyclic Chem., 2022, 102, 791–800, DOI:10.1007/s10847-022-01160-3.
- S. Shimizu, A. Wada-Hirai, Y.-P. Li, Y. Shimada, Y. Otsuka and S. Goto, J. Pharm. Sci., 2020, 109, 2206–2212, DOI:10.1016/j.xphs.2020.03.029.
- C. Schönbeck, K. Gaardahl and B. Houston, Mol. Pharm., 2019, 16, 648–654, DOI:10.1021/acs.molpharmaceut.8b00953.
- F. Lai, C. Sinico, G. Ennas, F. Marongiu, G. Marongiu and A. M. Fadda, Int. J. Pharm., 2009, 373, 124–132, DOI:10.1016/j.ijpharm.2009.01.024.
- S. Li, Y. Tang, X. Zhang, Y. Dou and X. Shen, J. Inclusion Phenom. Macrocyclic Chem., 2019, 94, 1–10, DOI:10.1007/s10847-019-00910-0.
- M. Farooq, M. H. Shoaib, R. I. Yousuf, F. Qazi and M. Hanif, Polym. Bull., 2019, 76, 2537–2558, DOI:10.1007/s00289-018-2510-z.
- T. Fujito, T. Oshima, K. Higashi, K. Ueda, M. Ito, H. Masu, S. Noguchi and K. Moribe, Cryst. Growth Des., 2022, 22, 1094–1103, DOI:10.1021/acs.cgd.1c01050.
- T. Backensfeld, B. W. Müller, M. Wiese and J. K. Seydel, Pharm. Res., 1990, 7, 484–490, DOI:10.1023/A:1015860531565.
- T. Loftsson, J. Pharm. Pharmacol., 2010, 62, 1607–1621, DOI:10.1111/j.2042-7158.2010.01030.x.
- C. Scavone, A. Colomba, S. Fiorentino, D. Cimmaruta, R. Cenami, M. Torella, T. Fossati and F. Rossi, Drug R. D., 2016, 16, 129–140, DOI:10.1007/s40268-016-0123-2.
- N. Ono, F. Hirayama, H. Arima and K. Uekama, Eur. J. Pharm. Sci., 1999, 8, 133–139, DOI:10.1016/s0928-0987(99)00002-0.
- M. Messner, S. V. Kurkov, R. Flavià-Piera, M. E. Brewster and T. Liftsson, Int. J. Pharm., 2011, 408, 235–247, DOI:10.1016/j.ijpharm.2011.02.008.
- H. Kataoka, Y. Sakaki, K. Komatsu, Y. Shimada and S. Goto, J. Pharm. Sci., 2017, 106, 3016–3021, DOI:10.1016/j.xphs.2017.04.010.
- C. Tsunoda, S. Goto, R. Hiroshige, T. Kasai, Y. Okumura and H. Yokoyama, Int. J. Pharm., 2023, 638, 122913, DOI:10.1016/j.ijpharm.2023.122913.
- C. Tsunoda, K. Hasegawa, R. Hiroshige, T. Kasai, H. Yokoyama and S. Goto, Mol. Pharm., 2023, 20(10), 5032–5042 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3pm00039g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |