
Photooxidative C–C double bond cleavage of β-enaminocarbonyl compounds: toward selective N-formylation of amines†
Hayeon
You
,
Suk Hyun
Lim
* and
Dae Won
Cho
 *
*
Department of Chemistry, Yeungnam University, Gyeongsan, Gyeongbuk 38541, Republic of Korea. E-mail: dwcho00@yu.ac.kr; sukhyun@ynu.ac.kr
First published on 11th November 2024
Abstract
A photooxidative C–C double bond cleavage of electron-deficient β-enaminocarbonyl compounds possessing a silyl group at the α-position to the nitrogen atom using methylene blue (MB) as the photosensitizer was explored. Photochemically generated 1O2 was added across the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond with the aid of a tethered silyl group to cleave it and form N-formylamines. This reaction protocol exhibited compatibility with numerous β-enaminocarbonyl substrates, including those with various N-alkyl, N-benzyl and N-aryl substituents.
C bond with the aid of a tethered silyl group to cleave it and form N-formylamines. This reaction protocol exhibited compatibility with numerous β-enaminocarbonyl substrates, including those with various N-alkyl, N-benzyl and N-aryl substituents.
Oxidative cleavage reactions of C–C double bonds using oxygen-containing species are promising and versatile transformations for the synthesis of a diverse array of valuable carbonyl compounds, ranging from aldehydes and ketones to carboxylic acids and other complex molecules.1–9 Among the various substrates containing C–C double bonds, oxidative cleavage reactions of enamines utilizing singlet oxygen (1O2) have been among the most widely studied9–19 since the pioneering work of Foote and Lin.20–22 In the reactions of electron-rich enamines, including N-alkyl enamines12,13,15,20–22 and N-alkyl indoles,23–251O2 is readily added to the C–C double bond moieties to form initial dioxetane intermediates, which undergo C–C and O–O bond cleavage processes to yield carbonyl and amide fragments (Scheme 1a). In reactions using electron-deficient enamine derivatives that possess an electron-withdrawing group at the β-carbon position,14,16,171O2-promoted cleavage reactions can decompose the substrates into two carbonyl fragments. However, to promote oxidative cleavage of these substrates efficiently, harsh reaction conditions (i.e., extreme low temperature (−78 °C) and intense UV light source) as well as cautious workup processes are required16,17 (Scheme 1b). From our recent study, we observed that while highly electron-deficient enaminoesters typically disfavored oxidative C–C double bond cleavage reactions by 1O2 (Scheme 1b),19 the silyl group-tethered analogs were prone to undergo oxidative C–C double bond cleavage under mild reaction conditions (Scheme 1b). It is likely that the silyl group present within the enaminoester substrate plays a pivotal role in facilitating 1O2-promoted bond cleavage.19 These findings motivated us to further investigate silyl group-assisted C–C bond cleavage reactions of various electron-deficient enamine derivatives. Herein, we report the photooxidative 1O2-promoted C–C double bond cleavage reactions of β-enaminocarbonyl compounds (i.e., β-enaminoesters and β-enaminoketones) possessing a trimethylsilyl group at the α-position to the nitrogen atom and the involvement of the tethered silyl group in the cleavage process (Scheme 1c).
 | ||
| Scheme 1 1O2-promoted oxidative C–C double bond cleavage reactions of enamine derivatives. | ||
We initiated photooxidative C–C double bond cleavage reactions using the non-silyl and silyl group-containing β-enaminoesters 1a and 1b as the model substrates to screen the optimized reaction conditions. An oxygenated (O2-purged) β-enaminoester (0.38 mmol, 1 equiv.) and a photosensitizer (sens, 5 mol%), which can serve as an efficient 1O2 generator, were irradiated with a 20 W compact fluorescent lamp (CFL) for 48 h, and then the photoproducts were analyzed. As shown in Table 1, visible light irradiation of an MeCN solution of the β-enaminoester 1a and the methylene blue (MB) photosensitizer led to the formation of N-(trimethylsilyl)methyl-N-phenylformamide 2a (entry 1). Solvent screening revealed that product yields were significantly enhanced when either DMF or 5% H2O–MeCN (i.e., H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeCN = 5:95 (v/v)) was employed as the reaction medium (entries 4–6). As expected, other organic photosensitizers, such as rose bengal (RB) and eosin Y (EY), also facilitated the efficient conversion of substrate 1a into substrate 2a, although they were slightly less efficient than MB (entries 7 and 8). In contrast, visible light irradiation of the non-silyl-tethered β-enaminoester 1b26 in the presence of MB did not result in any oxidative C–C double bond cleavage reactions (entry 9). Control experiments indicated that a photosensitizer, oxygen, and visible light were essential for this photochemical C–C double bond cleavage transformation (entries 10–12).
:MeCN = 5:95 (v/v)) was employed as the reaction medium (entries 4–6). As expected, other organic photosensitizers, such as rose bengal (RB) and eosin Y (EY), also facilitated the efficient conversion of substrate 1a into substrate 2a, although they were slightly less efficient than MB (entries 7 and 8). In contrast, visible light irradiation of the non-silyl-tethered β-enaminoester 1b26 in the presence of MB did not result in any oxidative C–C double bond cleavage reactions (entry 9). Control experiments indicated that a photosensitizer, oxygen, and visible light were essential for this photochemical C–C double bond cleavage transformation (entries 10–12).
a
|
|
|||||
|---|---|---|---|---|---|
| Entry | Sub. | Sens. | Reaction conditions | Conver. (%) | Yieldb (%) |
| a Reaction conditions: oxygenated (O2-purged) solutions (80 mL) of β-enaminoesters (1a and 1b, 4.75 mM) and a photosensitizer (5 mol%) were irradiated using 20 W CFL for 48 h. b Isolated yields. c Not detected. d Photoreaction under deoxygenated (N2-purged) conditions. e Reaction in the dark (no light). | |||||
| 1 | 1a | MB | MeCN | 100 | 46 |
| 2 | 1a | MB | MeOH | 81 | 27 |
| 3 | 1a | MB | DMSO | 100 | 22 |
| 4 | 1a | MB | DMF | 100 | 62 |
| 5 | 1a | MB | 5% H2O–MeCN | 100 | 75 |
| 6 | 1a | MB | 5% H2O–DMF | 100 | 68 |
| 7 | 1a | RB | 5% H2O–MeCN | 100 | 67 |
| 8 | 1a | EY | 5% H2O–MeCN | 100 | 66 |
| 9 | 1b | MB | 5% H2O–MeCN | 0 | n.d.c |
| 10 | 1a | No sens. | MeCN | 0 | n.d.c |
| 11d | 1a | MB | 5% H2O–MeCN | 0 | n.d.c |
| 12e | 1a | MB | 5% H2O–MeCN | 0 | n.d.c |

With these optimized conditions in hand (entry 5 in Table 1), we explored the substrate scope of this photooxidative reaction. Initially, a range of electron-donating (e.g., Me and OMe) or electron-withdrawing (e.g., F, Cl, and CF3) group-substituted phenyl-containing β-enaminoesters, 1c–1o, were examined. As shown in Scheme 2, visible light illumination of a 5% H2O–MeCN solution of β-enaminoesters possessing an electron-donating group at the meta- (1d and 1g) or para-(1e and 1h) positions led to the formation of N-formamides 2d, 2g, 2e, and 2h in moderate yields. In the case of substrates bearing an electron-donating group at the ortho-position (1c and 1f), distinctively, the MB-sensitized photoreactions afforded the oxidized products 2c and 2f in much lower yields. Photooxidative reactions of the electron-withdrawing group-containing β-enaminoesters 1i–1o occurred to produce N-formamides 2i–2o, albeit with lower efficiency compared to their electron-donating group-substituted analogs.
 | ||
| Scheme 2 Photooxidative reactions of β-enaminoesters 1c–1o. | ||
Next, a variety of N-alkyl substituted β-enaminoesters 3a–3t were explored as substrates for the photooxidative reactions. The results, shown in Scheme 3, revealed that the photoreactions of N-acyclic alkyl (3a–3h), N-allyl (3i), N-cyclic alkyl (3j and 3k), N-arylmethyl (3l–3n), N-benzyl (3o–3v), and N-phenethyl (3w–3y) substituted β-enaminoesters furnished the corresponding N-formamides in moderate to good yields (52–86%) and, notably, these reactions took place much more efficiently than those of N-phenyl analogs, even in the case where the N-substituent had an electron withdrawing group (i.e., 3e–3h, 3r–3t, and 3x–3y). These observations revealed that the structural and electronic nature of N-alkyl substituents had little impact on the photooxidative C–C bond cleavage efficiency. As analogs of β-enaminoesters, N-phenyl- or N-benzyl substituted β-enaminoketones were also employed as substrates for the photooxidative cleavage reaction. Under the same photochemical reaction conditions, N-phenyl (5a–5c) and N-benzyl (5d–5f)-substituted β-enaminoketones were also decomposed to the corresponding N-formamides (Scheme 4).
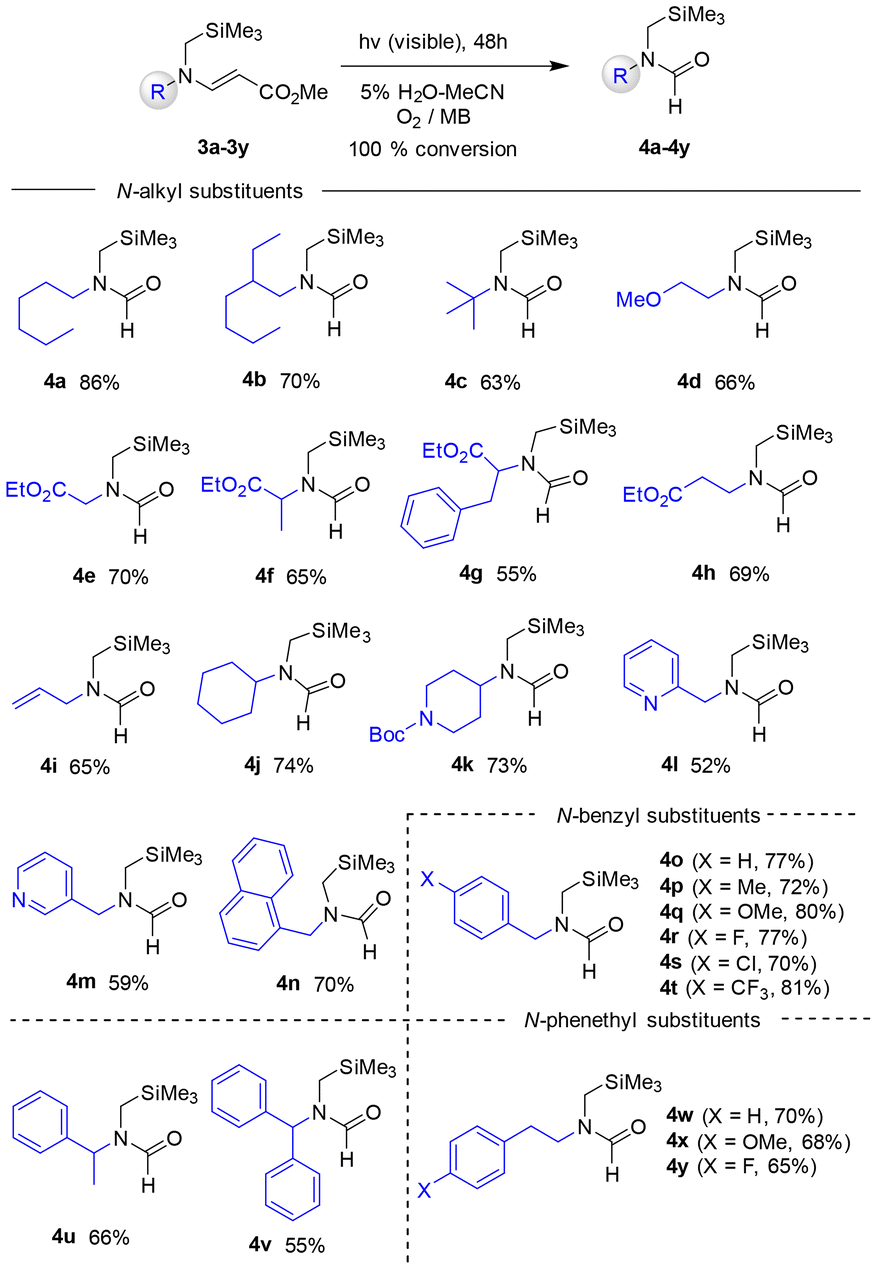 | ||
| Scheme 3 Photooxidative reactions of β-enaminoesters 3a–3y. | ||
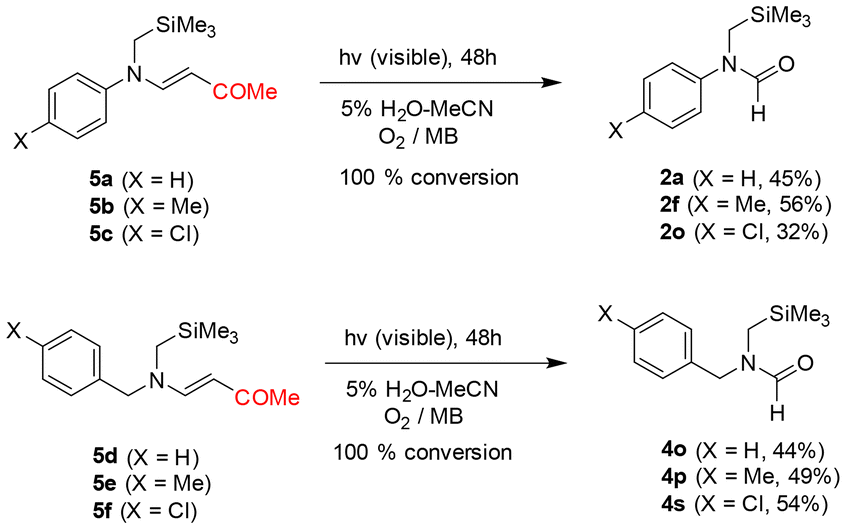 | ||
| Scheme 4 Photooxidative reactions of β-enaminoketones 5a–5f. | ||
To obtain an insight into the reaction mechanism associated with the photooxidative 1O2-promoted C–C double bond cleavage reactions of silyl group-containing β-enaminocarbonyl compounds, a series of experiments were performed. To clarify the involvement of singlet oxygen in the photoproduct formation process, MB-sensitized photoreactions of the β-enaminoester 1a were carried out in the presence of an efficient singlet oxygen quencher, DABCO, and the changes in both the percent conversion of 1a and the yield of the photoproduct 2a were monitored. The results showed that as the concentration of DABCO increased, significant decreases in both percent conversion of 1a and the yield of 2a were observed, and no photoproduct was observed when nearly 1 equiv. of DABCO (0.38 mmol, 4.75 mM) was added to the solution (Fig. 1). These observations are likely due to the depletion of generated 1O2 by DABCO through a physical quenching process.19,27–31
 | ||
| Fig. 1 Plots of percent conversion of 1a and product yields of 2a as a function of [DABCO] in MB-sensitized photooxidative reactions of β-enaminoester 1a. Reaction conditions: the oxygenated (O2-purged) 5% H2O–MeCN solutions (80 mL) of 1a (4.75 mM) containing MB (5 mol%) and varying concentrations of DABCO (0, 2.37, 4.75, 9.49, and 23.8 mM, respectively) were irradiated using a 20 W CFL for 48 h. | ||
To determine how the silyl group within the substrates is involved in the C–C bond cleavage, the sterically hindered silyl (i.e., SiMe2Ph)- and (trimethylsilyl)propyl group-substituted β-enaminoesters 6a–6c were prepared and their MB-sensitized photoreactions were compared with those of the less hindered silyl-substituted β-enaminoesters 1a, 3a, and 3p. As shown in Scheme 5, the photoproduct yields resulting from the reactions of the N-(dimethylphenylsilyl) group-substituted analogs 6a and 6b were lower than those from the reactions of the trimethylsilyl-substituted β-enaminoesters 1a and 3a. Importantly, no photoproduct was observed in the reaction using the N-(trimethylsilyl) propyl-tethered substrate 6c. These observations revealed that the exposure of silicon atoms to oxygen species (i.e., 1O2) and the spatial proximity of silicon to a nitrogen atom influence the photooxidative C–C double bond cleavage reaction.
 | ||
| Scheme 5 Photooxidative reactions of various silyl group-substituted β-enaminoesters. | ||
Based on the above observations and the results of earlier studies,19 a feasible mechanistic pathway responsible for the 1O2-promoted photooxidative C–C double bond cleavage reaction of electron-deficient β-enaminocarbonyl substrates was suggested (Scheme 6). Initially, a photochemically generated singlet excited state of MB (1MB*), via visible light absorption, undergoes efficient intersystem crossing (ISC) to form the triplet excited state of MB (3MB*).32–35 Among the diverse deactivation processes open to 3MB*, triplet energy transfer to molecular oxygen (3O2) occurs more efficiently, generating singlet oxygen (1O2). Considering that the features of the silyl group and the proximity of the silicon atom to a nitrogen atom play an essential role in the reactions, it is highly likely that the in situ-formed 1O2 is added to β-enaminocarbonyls 8 to initially produce the per-epoxide intermediate 9,36–41 which then subsequently rearranges to form the dioxetane intermediate 11. In particular, the six-membered cyclic conformation resulting from the interaction between the oxophilic silicon atom and the negatively charged oxygen atom seems to facilitate both the formation of per-epoxide 9 and its conversion to dioxetane 11. Finally, the generated dioxetane 11 undergoes either concerted or stepwise C–C and O–O bond cleavages to produce N-formamide 12.
 | ||
| Scheme 6 Suggested reaction mechanism. | ||
The involvement of the per-epoxide intermediate was supported by the MB-sensitized photoreaction of 1a in the presence of methyl phenyl sulfoxide (MeS(O)Ph), which can serve as a trapping agent for the per-epoxide.19,38,39 The results showed that as the concentration of sulfoxide 13 increased, the yield of 2a gradually decreased due to the quenching of the generated per-epoxide (Scheme 7).
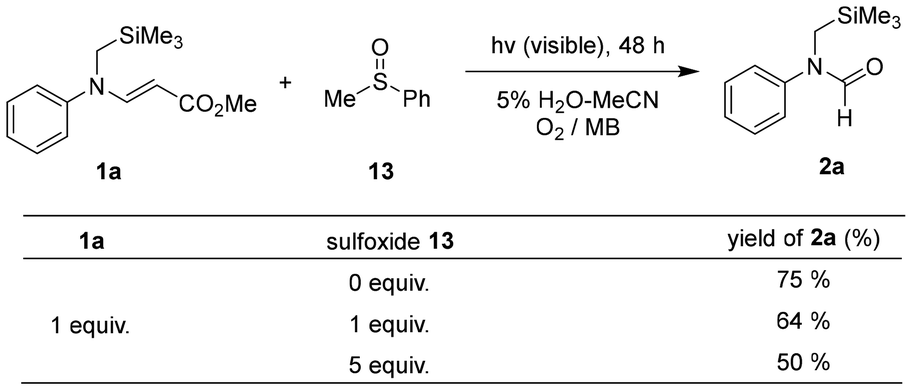 | ||
| Scheme 7 Photooxidative reactions of β-enaminoester 1a in the presence of methyl phenyl sulfoxide. | ||
Conclusions
We explored the photooxidative C–C double bond cleavage reactions of electron-deficient β-enaminocarbonyl compounds using the visible light photosensitizer MB. Photochemically generated 1O2, an in situ-formed oxidant, was added across the C–C double bond to cleave it. Importantly, the presence of a silyl group on the carbon atom next to the nitrogen atom played an essential role in the photooxidative C–C double bond cleavage reaction. This silyl assisted reaction protocol exhibited compatibility with a wide range of β-enaminocarbonyl substrates, including those with a series of N-alkyl, N-benzyl, and N-aryl substituents. We believe that this photochemical strategy will broaden our understanding of singlet oxygen-mediated oxidative processes.Author contributions
S. H. Lim and D. W. Cho conceptualized the project. H. You and S. H. Lim performed the experiments. D. W. Cho wrote the manuscript. All authors discussed and commented on the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by grants from the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science, and Technology (2021R1I1A304981914 to D. W. Cho, RS-2023-00211633 to S. H. Lim) and a Korea Basic Science Institute (National Research Facilities and Equipment Center) grant funded by the Ministry of Education (2019R1A6C1010046).References
- S. Caron, R. W. Dugger, S. G. Ruggeri, J. A. Ragan and D. H. B. Ripin, Chem. Rev., 2006, 106, 2943 CrossRef CAS.
- Z. Hassan, M. Stahlberger, N. Rosenbaum and S. Brase, Angew. Chem., Int. Ed., 2021, 60, 15138 CrossRef CAS PubMed.
- R. Lin, F. Chen and N. Jiao, Org. Lett., 2012, 14, 4158 CrossRef CAS PubMed.
- A. Rajagopalan, M. Lara and W. Kroutil, Adv. Synth. Catal., 2013, 355, 3321 CrossRef CAS.
- D. Xing, B. Guan, G. Cai, Z. Fang, L. Yang and Z. Shi, Org. Lett., 2006, 8, 693 CrossRef CAS PubMed.
- F. Mecozzi, J. J. Dong, D. Angelone, W. R. Browne and N. N. H. M. Eisink, Eur. J. Org. Chem., 2019, 7151 CrossRef CAS.
- P. Spannring, P. C. A. Bruijnincx, B. M. Weckhuysen and R. J. M. K. Gebbink, Catal. Sci. Technol., 2014, 4, 2182 RSC.
- R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981 CrossRef CAS.
- M. Adib, R. Pashazadeh, S. J. A. Gohari and F. Shahsavari, Synlett, 2017, 1481 CrossRef CAS.
- T. Poon, N. J. Turro, J. Chapman, P. Lakshminarasimhan, X. Lei, S. Jockusch, R. Franz, I. Washington, W. Adam and S. G. Bosio, Org. Lett., 2003, 5, 4951 CrossRef CAS PubMed.
- J. Sivaguru, H. Saito, T. Poon, T. Omonuwa, R. Franz, S. Jockusch, C. Hooper, Y. Inoue, W. Adam and N. J. Turro, Org. Lett., 2005, 7, 2089 CrossRef CAS PubMed.
- J. Li, S. Cai, J. Chen, Y. Zhao and D. Z. Wang, Synlett, 2014, 1626 Search PubMed.
- W. Ando, T. Saiki and T. Migita, J. Am. Chem. Soc., 1975, 97, 5028 CrossRef CAS.
- I. Erden, G. Ozer, C. Hoarau, W. Cao, J. Song, C. Gartner, I. Baumgardt and H. Butenschon, J. Org. Chem., 2008, 73, 6943 CrossRef CAS PubMed.
- H. H. Wasserman and S. Terao, Tetrahedron Lett., 1975, 16, 1735 CrossRef.
- H. H. Wasserman and J. L. Ives, J. Org. Chem., 1985, 50, 3573 CrossRef CAS.
- H. H. Wasserman and J. L. Ives, J. Org. Chem., 1978, 43, 3238 CrossRef CAS.
- N. H. Martin and C. W. Jefford, Helv. Chim. Acta, 1982, 65, 762 CrossRef CAS.
- S. H. Lim, M. Kim, K. Wee, D. H. Lim, Y. Kim and D. W. Cho, J. Org. Chem., 2023, 88, 172 CrossRef CAS PubMed.
- C. S. Foote, Acc. Chem. Res., 1968, 1, 104–110 CrossRef CAS.
- C. S. Foote and J. W. Lin, Tetrahedron Lett., 1968, 9, 3267 CrossRef.
- C. S. Foote, A. A. Dzakpasu and J. H.-P. Lin, Tetrahedron Lett., 1975, 16, 1247 CrossRef.
- I. Saito, M. Imuta, S. Matsugo and T. Matsuura, J. Am. Chem. Soc., 1975, 97, 7191 CrossRef CAS.
- I. Saito, M. Imuta and T. Matsuura, Chem. Lett., 1972, 1, 1173 CrossRef.
- I. Saito, S. Matsugo and T. Matsuura, J. Am. Chem. Soc., 1979, 101, 7332 CrossRef CAS.
- M. Zhao, X. Lian, Z. Ren, Y. Wang and Z. Guan, RSC Adv., 2014, 4, 62042 CAS.
- S. H. Lim, M. Kim, K. Wee and D. W. Cho, J. Org. Chem., 2023, 88, 12294 CrossRef CAS PubMed.
- B. M. Monroe, J. Phys. Chem., 1977, 81, 1861 CrossRef CAS.
- E. A. Ogryzlo and C. W. Tang, J. Am. Chem. Soc., 1970, 92, 5034 CrossRef CAS.
- C. Ouannes and T. Wilson, J. Am. Chem. Soc., 1968, 90, 6527 CrossRef CAS.
- A. P. Darmanyan, W. S. Jenks and P. Jardon, J. Phys. Chem. A, 1998, 102, 7420 CrossRef CAS.
- R. I. Patel, A. Sharma, S. Sharma and A. Sharma, Org. Chem. Front., 2021, 8, 1694 RSC.
- X. Yang, B. Chen, L. Zheng, L. Wu and C. Tung, Green Chem., 2014, 16, 1082 RSC.
- S. P. Pitre, C. D. McTiernan, H. Ismaili and J. C. Scaiano, ACS Catal., 2014, 4, 2530 CrossRef CAS.
- S. P. Pitre, C. D. McTiernan and J. C. Scaiano, Acc. Chem. Res., 2016, 49, 1320 CrossRef CAS PubMed.
- J. G. Luis, L. S. Andres and W. Q. Fletcher, Tetrahedron Lett., 1994, 35, 179 CrossRef CAS.
- A. G. Leach, K. N. Houk and C. S. Foote, J. Org. Chem., 2008, 73, 8511–8519 CrossRef CAS.
- A. P. Schaap, S. G. Recher, G. R. Faler and S. R. Villasenor, J. Am. Chem. Soc., 1983, 105, 1691 CrossRef CAS.
- T. H. W. Poon, K. Pringle and C. S. Foote, J. Am. Chem. Soc., 1995, 117, 7611 CrossRef CAS.
- A. A. Frimer, Chem. Rev., 1979, 79, 359–387 CrossRef CAS.
- M. Orfanopoulos, Photochem. Photobiol., 2021, 97, 1182 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob01688b |
| This journal is © The Royal Society of Chemistry 2025 |