
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct C–H functionalisation of azoles via Minisci reactions
Ai-Lan
Lee
 *a,
David T.
Mooney
b and
Heather
McKee
b
*a,
David T.
Mooney
b and
Heather
McKee
b
aEaStCHEM School of Chemistry, University of Edinburgh, David Brewster Road, Edinburgh, EH9 3FJ, UK. E-mail: AiLan.Lee@ed.ac.uk
bInstitute of Chemical Sciences, School of Engineering and Physical Sciences, Heriot-Watt University, Edinburgh, EH14 4AS, UK
First published on 25th October 2024
Abstract
Azoles have widespread applications in medicinal chemistry; for example, thiazoles, imidazoles, benzimidazoles, isoxazoles, tetrazoles and triazoles appear in the top 25 most frequently used N-heterocycles in FDA-approved drugs. Efficient routes for the late-stage C–H functionalisation of azole cores would therefore be highly desirable. The Minisci reaction, a nucleophilic radical addition reaction onto N-heterocyclic bases, is a direct C–H functionalisation reaction that has the potential to be a powerful method for C–H functionalisations of azole scaffolds. However, azoles have not been as widely studied as substrates for modern Minisci-type reactions as they are often more electron-rich and thus more challenging substrates compared to electron-poor 6-membered N-heterocycles such as quinolines, pyrazines and pyridines typically used in Minisci reactions. Nevertheless, with the prevalence of azole scaffolds in drug design, the Minisci reaction has the potential to be a transformative tool for late-stage C–H functionalisations to efficiently access decorated azole motifs. This review thus aims to give an overview of the C–H functionalisation of azoles via Minisci-type reactions, highlighting recent progress, existing limitations and potential areas for growth.
 Ai-Lan Lee | Ai-Lan obtained her MSci (Hons) (2000) and PhD (2004) from the University of Cambridge, under the supervision of Prof. Steven V. Ley. Following a Lindemann Trust Fellowship (2004–2005) at Boston College with Prof. Amir Hoveyda, Ai-Lan was appointed as a fixed-term Lecturer at the University of Edinburgh (2006), carrying out research with Prof. David Leigh. She was a Lecturer (2007–2013) and Associate Professor, Reader (2013–2024) at Heriot-Watt University before taking up her current position as Chair in Organic Chemistry at the University of Edinburgh (2024). Her research interests include decarboxylative radical reactions and the development of new gold-, palladium- and photo-catalysed reactions. |
 David T. Mooney | David T. Mooney received his MSci (Hons) in Chemistry with Medicinal Chemistry from the University of Glasgow in 2020. He also completed a placement year in industry at AstraZeneca (Macclesfield) before returning to Glasgow to complete a Master's project in the France group, working on the development of anti-viral compounds for targeting Ebola. He is currently pursuing his doctoral studies at Heriot-Watt University under the supervision of Ai-Lan Lee and Samuel Drane (AstraZeneca), funded by an EPSRC/AstraZeneca Industrial CASE Award. His research interests focus on the development of direct C–H amidations of N-heterocycles via decarboxylative radical formation. |
 Heather McKee | Heather received her MChem (Hons) in Chemistry from Heriot-Watt University in 2023, undertaking her MChem project under the supervision of Prof. Ai-Lan Lee, developing a light-mediated methodology for the direct C–H amidation of 1,3-azoles. A summer placement at the Institute of Cancer and Genetics, University of Edinburgh, fuelled her interest in medicinal chemistry further, and since graduating, Heather has entered the pharmaceutical industry and is a Development Chemist at Veranova (Macfarlan Smith). |
1. Introduction
Azoles are a subclass of heterocycles with modern applications in various fields, including supramolecular materials, coordination and organometallic chemistry, and the agrochemical industry.1 One of the main applications of azoles, however, lies within the field of medicinal chemistry (see Fig. 1);2 therefore establishing an efficient route to the late-stage functionalisation3 of azoles would be highly desirable. Methods for direct C–H functionalisation of azoles would be ideal, as it allows for shorter, more efficient pathways and thus more cost-effective protocols.4 The Minisci reaction is one such direct C–H functionalisation reaction which has sparked a resurgence of interest, given that it facilitates the nucleophilic attack of a carbon radical species (e.g., I–III, Scheme 1) onto N-heterocyclic bases.5 | ||
| Fig. 1 Examples of pharmaceutical compounds with azole cores. | ||
 | ||
| Scheme 1 Minisci-type C–H functionalisations of azoles. | ||
So far, however, azoles have not been as widely studied as substrates for modern Minisci-type reactions, despite their aforementioned prevalence in medicinal chemistry. This is likely because azoles are more electron-rich and thus more challenging substrates compared to more typically studied electron-poor 6-membered N-heterocycles such as quinolines, pyrazines and pyridines.6 Azoles are thus thought to be less reactive towards nucleophilic radical additions required in a typical Minisci-type reaction. Indeed, azole drug compounds are notably absent from a recent review summarising the list of pharmaceutical compounds that can undergo Minisci-type reactions.5f Nevertheless, the direct Minisci C–H functionalisation of azoles could potentially be a very powerful method for efficiently accessing decorated azole motifs. Therefore, the purpose of this review is to give an overview of Minisci-type C–H functionalisations of azoles (Scheme 1), with particular emphasis on recent advances, existing limitations and areas for further development.
2. Azoles
2.1 Background and application
Azoles are classified as five-membered heterocyclic aromatic compounds which contain nitrogen and at least one other heteroatom and are common scaffolds in drug design.2a For example, imidazoles, thiazoles, tetrazoles, benzimidazoles, pyrazoles, 1,2,4-triazoles and isoxazoles are included in the top 25 most frequently used N-heterocycles in U.S. FDA-approved drugs.2b,c,7 Thiazoles, imidazoles, tetrazoles and benzimidazoles also constitute four out of the top five most common five-membered aromatic nitrogen heterocycles in U.S. FDA approved drugs.2b,c In addition to the azoles listed above, 2,4,-dihydro-1,2,4-triazol-3-one, 1,3,4-thiadiazoles, indazoles, benzoindazoles, 1,2,3-triazoles and benzothiazoles also appear in the top 100 most frequently used ring systems in small-molecule drugs.2c2.2 Routes to the functionalisation of azoles
Due to their importance as scaffolds in medicinal chemistry, methods for functionalising unactivated azole cores are crucial for transforming azole cores into pharmaceutically relevant compounds. Such functionalisations have classically proceeded via indirect methods (1 → 2 → 3, Scheme 2A), whereby the installation of a functional group (X in 2) is required.8 For example, acylation of (benzo)thiazole motifs has been achieved through the use of Grignard reagents9 and alkylations of imidazoles have been conducted via directed lithiations.10 Nevertheless, there is clear inefficiency in synthetic pathways that require multiple steps and non-atom economical pre-functionalisations, and the use of strongly basic reagents such as Grignard reagents and lithiations can preclude late-stage functionalisations. | ||
| Scheme 2 General indirect (A) and direct (B) methods for azole functionalisation. | ||
In contrast, direct C–H functionalisations (1 → 3, Scheme 2B) such as the Minisci-type reactions, occur in one step and can potentially be utilised for late-stage functionalisations.5a–c,f A brief overview of the classical Minisci reaction will be given in the next section (Section 3) before the review will focus on its main topic, the Minisci-type reactions of azoles (Section 4).
3. Classical Minisci reaction
In 1971, Minisci and co-workers demonstrated that alkyl carboxylic acids 5 can be used as cheap and readily available alkyl radical I precursors11 for the C–H alkylation of N-heterocyclic bases 4, using AgNO3, (NH4)2S2O8 as an oxidant, and sulfuric acid to activate the N-heterocyclic base substrates (Scheme 3A).12 C–H acylations soon followed, using α-keto acids136 as acyl radical II precursors (Scheme 3B),14 although acylation can alternatively proceed via aldehydes 11 using iron(II) sulfate, tert-butylhydroperoxide, H2SO4 and acetic acid (Scheme 3D).14,15 The carboxyamidation equivalent was not developed until the 1990s using oxamic acids 716 as carbamoyl radical III precursors (Scheme 3C).17 An earlier carboxyamidation method mediated by iron(II) sulfate and hydroxylamine O-sulfonic acid (HAS) in the presence of H2SO4 is less versatile, as it requires solvent quantities of formamide 12 as the radical precursor (Scheme 3E).18 N-heterocycles 4 typically used in these early studies were pyridines, (iso)quinolines and pyrazines, although a study on one azole (benzothiazole) was carried out under the iron(II) sulfate-mediated conditions shown in Scheme 3D.19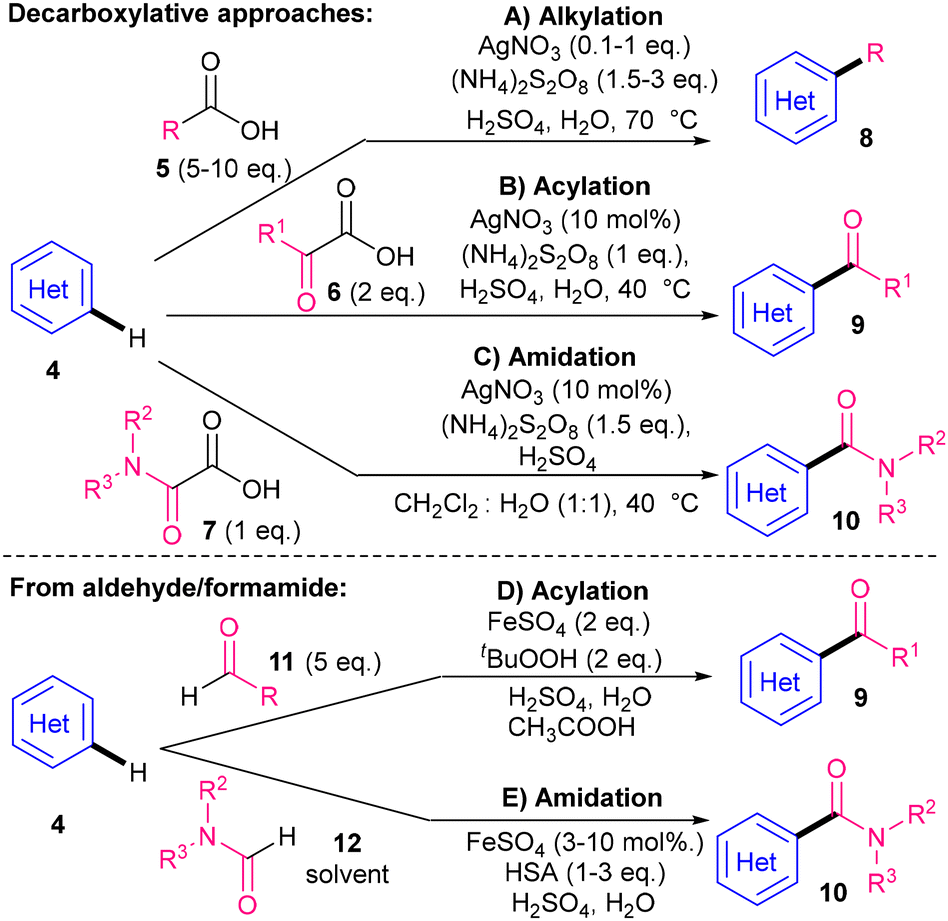 | ||
| Scheme 3 Minisci and co-workers’ seminal results. | ||
There are, however, several apparent sustainability concerns with the classical Minisci conditions, including the reliance on substoichiometric amounts of expensive and non-sustainable silver,20 the large excess of radical precursors (for 5, 11 and 12), some high temperatures and strong acid activators, which can limit the substrate scope.21 Thus, many modern Minisci-type reactions have been developed in an attempt to overcome some of these limitations,5a and milder reaction conditions are also preferable for applications in late-stage C–H functionalisations.
The prevalence of azoles in pharmaceutical drugs is the reason for the focus on azoles in this review (Section 4).7 This review will focus on publications published since 201222 and will cover only Minisci-type reactions of azoles (i.e., nucleophilic radical additions onto azoles). Other methodologies for C–H functionalisation of azoles, such as transition metal cross-couplings,23 as well as tri- and di-fluoromethylations are outside the scope of this review.24
4. Minisci reactions of azoles
Recent developments in various Minisci-type reactions initially focused on electron-poor six-membered N-heterocycles, typically incorporating only one or two azole moieties (if any) into the substrate scope.5a Optimisation studies were not typically carried out on azole substrates. It is only recently that more thorough investigations into Minisci reactions of azoles have been carried out, and more emphasis will be placed on these publications. This section will be divided into three major sub-sections: Minisci-type alkylations (Section 4.1), acylations (Section 4.2) and amidations (Section 4.3).4.1 Minisci alkylation of azoles
Minisci alkylations are by far the most studied of the three classes of functionalisations mentioned above, and many groups who developed Minisci-type alkylation protocols may have included one azole (usually benzothiazole) in their substrate scope studies. This review will only cover selected seminal examples of those and will place more emphasis on Minisci alkylation studies that specifically focused on azole substrates. | ||
| Scheme 4 Silver-catalysed alkylation of azoles.25 | ||
An extension of this methodology was reported by Li's group in 2017, using cycloalkanols 14 as radical precursors (Scheme 5).27 Their methodology was applicable to benzothiazoles, thiazoles and benzoxazole substrates. Hydrogen atom transfer (HAT) with cycloalkanol 14 followed by fragmentation of the subsequent alkoxy radical IV gives a primary C-centred radical nucleophile V, which adds to the azole substrates in the usual Minisci fashion (Scheme 5).
 | ||
| Scheme 5 Silver-catalysed alkylation of azoles using cycloalkanols.27 | ||
In 2017, iron catalysis was used to access primary, secondary and tertiary alkylations of azoles (Scheme 6).28 Bao's group used diacyl peroxides 15 as acid precursors for primary and secondary alkylations whereas alkyl tert-butyl peresters 16 were employed for secondary and tertiary alkylations (e.g.13a, Scheme 6). The main azole investigated was benzothiazole, although two benzoxazoles (e.g.13m), one N-Me-benzimidazole (13n) and one thiazole (13o) also reacted well. Advantages of this protocol are the use of a cheaper and more sustainable iron catalyst, good yields and a broad substrate scope. However, 15 and 16 are less accessible than their carboxylic acid counterparts 5 and the use of peroxides with heating may also pose safety concerns, especially on larger scales.
 | ||
| Scheme 6 Iron-catalysed alkylation of azoles.28 | ||
In 2020, the alkylation of 2H-indazoles 16 using alkyl-DHP reagents 18 as radical precursors was reported by the groups of Du and Tan (Scheme 7).29 Their conditions were compatible with secondary alkylations (19a–c) and one example of a tertiary alkylation was also shown (19d). The short reaction times (2 h) are a highlight, although the use of alkyl-DHP 18 is less atom economical and not as readily available as using 5 directly (e.g.Scheme 4). The silver nitrate loading was relatively high at 40 mol%, and TFA was required to activate the indazole substrates.
 | ||
| Scheme 7 Silver-catalysed acylation of 2H-indazoles.29 | ||
Very recently, Ronchi and Bellina and co-workers disclosed a direct decarboxylative method for the alkylation of azoles using acids 5 (Scheme 8).6 Unlike the work described in Scheme 4,25 this work focused primarily on (benz)imidazoles, using sub-stoichiometric silver nitrate (60 mol%), ammonium persulfate as the oxidant, and crucially, various acid additives were screened to reveal TFA as optimal for activating the azole substrates.6 While tertiary (13p–q) and secondary (13r–s) alkylations proceeded smoothly, primary alkylations were less successful (13t–u, 0–10%). The highlight of this work is that it is one of the very few examples to investigate azole substrates with three heteroatoms: oxadiazole (20a, 53%), thiadiazole (dialkylated, 20b, 75%) and 1,2,4-triazole (dialkylated, 20c, 34%). Crucially, two benzimidazole drug precursors were also successfully formed in one step from benzimidazole (13x, 73% and 13y, 38%), showcasing the potential of the Minisci reaction for synthesis of azole drug molecules. Nevertheless, the conditions are still relatively demanding, using 60 mol% of the expensive and non-sustainable silver catalyst,20 70 °C and an acid additive.
 | ||
| Scheme 8 Silver-mediated alkylation of azoles and its application to the synthesis of drug precursors.6 | ||
4.1.2.1 Transition metal photocatalysed alkylations. In 2016, Barriault and co-workers described a gold(I)-catalysed alkylation of N-heterocycles using alkyl bromides 21 (Scheme 9).30,31 The study included 3 azoles: benzothiazole was the most reactive (13g, 90%), followed by benzimidazole (13r, 13z, 75–80%) and benzoxazole (13aa, 55%). The methodology was pioneering; however, drawbacks include the use of an expensive gold catalyst and high energy UV irradiation.32 Most of the subsequent light-mediated methodologies have moved towards visible light irradiation.
 | ||
| Scheme 9 Gold-photocatalysed alkylation (4 azole examples).30 | ||
For example, in 2016 a Ru(II)-catalysed alkylation of N-heterocycles was reported by the groups of Liu and Chen, using primary and secondary alkyl boronic acids 22 as radical precursors (Scheme 10).33 The key advancement here is the use of visible light irradiation, which does not require specialized setup and is more functional group tolerant compared to UVA irradiation.32 The authors incorporated three benzimidazoles (13z, 13ab–ac) and two benzothiazoles (13ad and 13g) into the substrate scope, forming the desired products in good yields (72–90%). Advantages include good yields and low catalyst loading, although ruthenium has toxicity, cost and sustainability implications.20 The use of costly 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) solvent also poses environmental and toxicity concerns.
 | ||
| Scheme 10 Ru-photocatalysed alkylation (5 azole examples).33 | ||
Ir-photocatalysed alkylations using carboxylic acids 5 by the group of Glorius (Scheme 11A)34 have the advantages of wide availability, stability, non-toxicity and low cost of 5 compared to many other radical precursors.11 The heteroaromatic scope included a benzimidazole (13r, 46%) and benzothiazole (13g, 29%). Limitations, however, include the use of a large excess of radical precursor 4 (10 eq.), as well as the use of an expensive and non-sustainable20 Ir photocatalyst (£858.50 per g).35 In 2019, Jin and co-workers developed a cheaper iron-catalysed Minisci alkylation, utilising the ligand-to-metal charge transfer (LMCT) pathway of in situ formed iron carboxylate complexes (Scheme 11B).36
 | ||
| Scheme 11 (A) Ir- and (B) Fe-photocatalysed alkylation of N-heterocycles (2 and 7 azole examples).34,36 | ||
An Ir-photocatalysed methodology using radical precursors 23 and tert-butyl peracetate (tPBA) as the oxidant was revealed by Wang's group in 2018 (Scheme 12).37 Although the reaction was initially developed to achieve α-aminoalkylations (e.g.13ae–af), it was also found to work on other H donors 23, such as ethers (e.g.13ag, 46%), aldehydes (to form acylation product 24a, 95%), formamides (to form amidation product 25a, 30%), p-xylene (13ah, 43%), and alkanes (e.g.13g, 13e 56–60%). The main azole studied was benzothiazole (32 examples), although one benzimidazole also showed a decent yield (13ai, 54%). The key advantages are the atom-economical nature of 23, the good substrate scope and mild conditions. Nevertheless, as the key radical R˙ is formed via HAT, the reaction works best for stabilised R˙ (α-to N/O, or benzylic). Unstabilised alkyls require a large excess of 23 (13g, 13e) and can result in regioisomers (e.g.13e) unless symmetrical cyclic alkanes are utilised. The reaction also requires the use of expensive and non-sustainable20 Ir photocatalysts, TFA as an acid activator and tert-butyl peracetate (tPBA) as the oxidant.
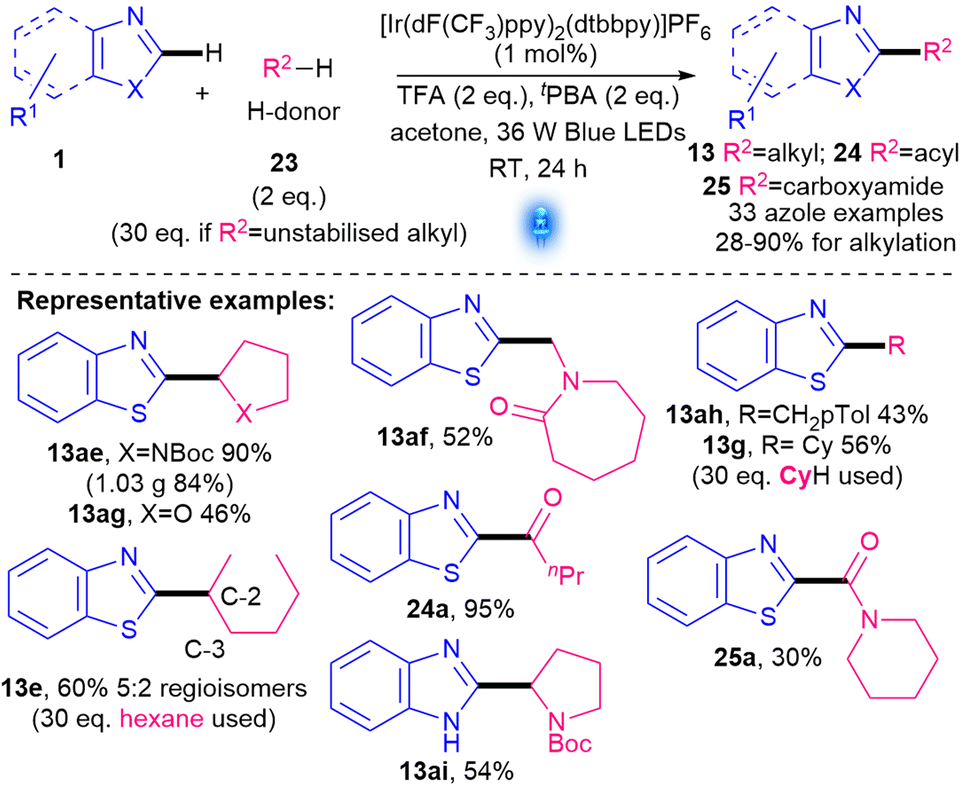 | ||
| Scheme 12 Ir-photocatalysed alkylation of azoles.37 | ||
In the same year, a Ru-photocatalysed methodology using N-hydroxyphthalimide esters (NHP-esters) 26 as radical precursors was developed by Opatz and co-workers (Scheme 13).38 A highlight of their protocol is that further external oxidant is not required. Alkylation of benzothiazole proceeded smoothly (13g, 13aj–ak); however, the reaction with Boc-protected imidazole yielded no desired product (13al). Only 1.5 eq. of NHP-ester 26 were required since 26 is more activated towards decarboxylation than acids 5. However, NHP-esters 26 are less atom-economical and less accessible than 5 or 23. Nevertheless, the yields with benzothiazole are good. Other potential limitations include the use of an expensive and non-sustainable Ru catalyst as well as extended reaction times (48 h).
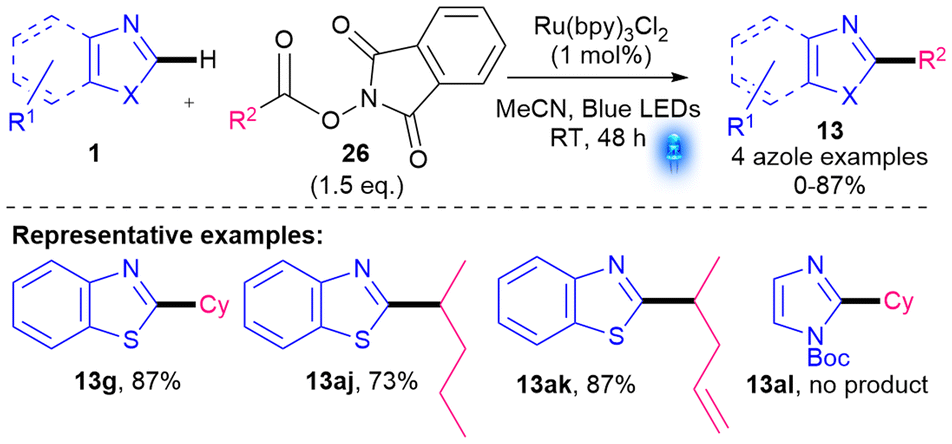 | ||
| Scheme 13 Ru-catalysed alkylation using NHP-esters (4 azole examples).38 | ||
An alternative method which avoids the use of a stoichiometric oxidant was revealed by Li's group in 2019 (Scheme 14).39 A cobalt co-catalyst [Co(dmgH)2Py2]PF6 is used instead, and the base, nBu4NOAc, is only needed in sub-stoichiometric amounts. The authors demonstrated perhaps the best 1,3-azole scope so far, with the whole range of 1,3-azoles: benzothiazole, thiazole, benzoxazole, oxazole, benzimidazole and imidazole all successfully C–H alkylated. Their procedure is best for tertiary alkylation (13am–at) and poor for unstabilised primary and secondary alkylations (13au). Primary and secondary alkylations where the radical is stabilised (α-to O/N, e.g.13ag, 13av) worked well. Advantages of this methodology are the removal of a stoichiometric oxidant and the excellent 1,3-azole substrate scope. Nevertheless, the use of [Co(dmgH)2Py2]PF6 introduces sustainability issues related to cobalt, along with the sustainability and cost issues already associated with Ir.20
 | ||
| Scheme 14 Ir-photocatalysed alkylation of azoles with a Co co-catalyst.39a | ||
In 2023, chlorine radicals were used as an HAT reagent for alkanes 23 (Scheme 15).40 The reaction developed by Jian, Tong and co-workers is catalysed by inexpensive FeCl3, with either LiCl or seawater as an additive (and an extra Cl source in addition to FeCl3), although yields are generally better with LiCl. As part of their substrate scope, eight different benzothiazoles were studied with three different alkanes 23. While benzothiazole itself gave a decent 67% yield of 13g, substituents on the benzothiazole core appear to cause a drop in yields (13aw-ax). Advantages of this method are the use of atom-economical alkanes 23 and a cheap FeCl3 photocatalyst. Disadvantages, however, include the requirement of 20 eq. of radical precursor 23, the use of symmetrical alkanes to avoid regioselectivity issues and generally moderate yields.
 | ||
| Scheme 15 Fe-photocatalysed alkylation of azoles.40 | ||
4.1.2.2 Organophotocatalysed alkylations. With the drive for greener chemistry, many research groups subsequently moved to organophotocatalysis instead of transition metal-based photocatalysis.
A noble-metal-free photocatalytic Minisci alkylation protocol for benzothiazoles was revealed by Jian and Tong's groups in 2022 (Scheme 16).41 Readily available alcohols 30 are utilised as the radical precursors, with a cheap organic dye, Eosin Y 27 (Fig. 2), as the photocatalyst and Fe2(SO4)3 as the Lewis acid. One of the interesting aspects of their protocol is that the reaction can be switched from alkylation if performed under argon to acylation if performed under air (Scheme 16A). Under argon, the reactive radical species is VI whereas in air, VI undergoes oxidation and HAT to give acyl radical VII (Scheme 16B). In contrast to examples using 5 as the radical precursor, their protocol works best for primary alkylations (13bb–bf, 13ad, 13i), is moderate for secondary alkylations (13bg) and does not work for tertiary alkylations (13a). The procedure is therefore very complementary to procedures using 5. Nevertheless, one drawback is the need for a large excess of the alcohol reactant 30 (31–85 eq.).
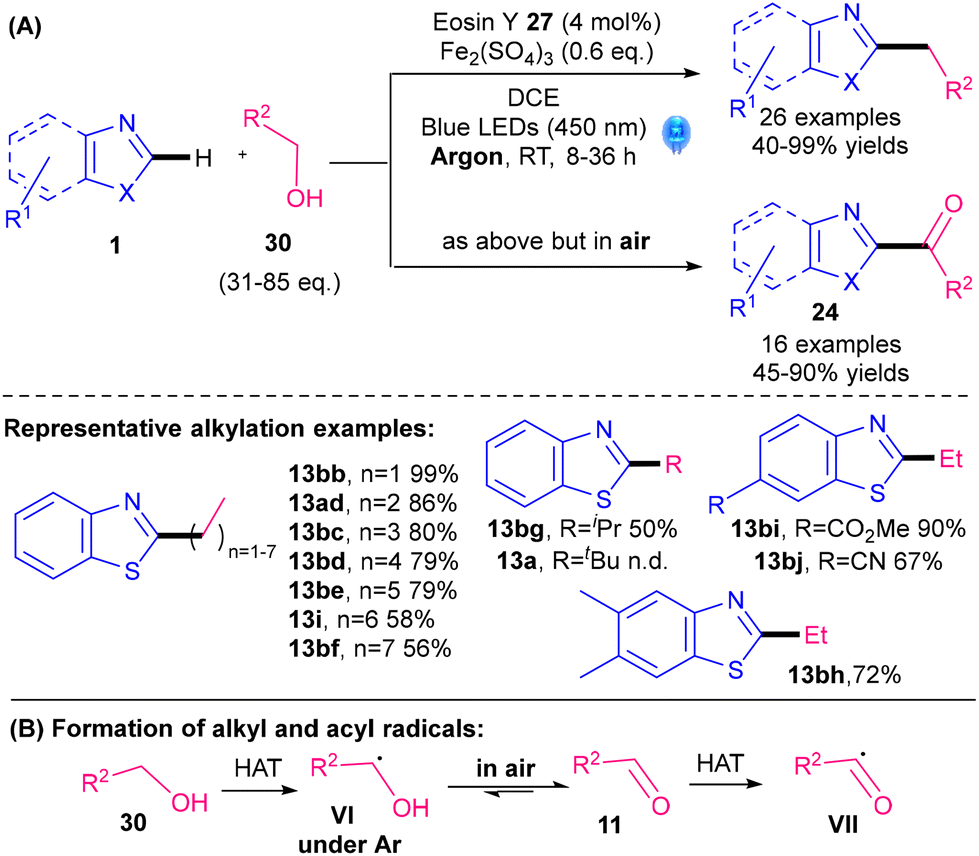 | ||
| Scheme 16 Noble-metal-free photocatalytic alkylation of benzothiazoles.41 | ||
 | ||
| Fig. 2 Examples of organophotocatalysts. | ||
Sherwood's group utilised 4-CzIPN 28 for alkylations, proceeding via the in situ formation of N-acyloxyphthalimide (NAPs) from the reaction of 5 with N-hydroxyphthalimide (Scheme 17).42 Four azoles were studied as part of their substrate scope: alkylation of benzimidazole (13z, 39%) and benzothiazole (13g, 31%) were low yielding, and only trace amounts of alkylated benzoxazole (13aa) and 2H-indazoles (33a) were formed. Advantages include the use of an organophotocatalyst instead of a metal-catalyst and only 1.5 eq. of the readily available acids 5. The formation of activated NAPs in situ, however, reduces the atom economy and requires the use of toxic reagents including DIC and DMAP.
 | ||
| Scheme 17 4-CzIPN-Photocatalysed alkylation (4 azole examples).42 | ||
Ji, Zhao, Huang and co-workers later utilised 4-CzIPN 28 in the aerobic photocatalytic alkylation of N-heterocycles, via the decarbonylation of aldehyde precursors 11 (Scheme 18).43 Only one azole (1a) was investigated, giving 13a in a moderate 47% yield.
 | ||
| Scheme 18 4-CzIPN-Photocatalysed alkylation (1 azole example).43a | ||
Despite 4-CzIPN 28 being a step in the right direction in terms of sustainability, it is currently very expensive, costing approximately £3596 per g.35 Various research groups subsequently utilised the much cheaper (£62 per g)35 Fukuzumi photocatalyst [Acr+-Mes][ClO4−] (29, Fig. 2). Molander and co-workers utilised alkyltrifluoroborates 31 as radical precursors using photocatalyst 29 and K2S2O8 as the oxidant (Scheme 19), obtaining yields ranging from <5% for benzimidazole to 80% for 1H-indazole (13a, 13q, 33b).44 A very appealing feature is that the reaction relies on only one equivalent of 31, albeit they are relatively expensive radical precursors compared to the more readily available acids 5 and aldehydes 11.
 | ||
| Scheme 19 Use of alkyltrifluoroborates for alkylations (3 azole examples).44 | ||
An alternative approach by Frenette's group makes use of acid precursors 5 in the [Acr+-Mes][ClO4−]-catalysed alkylations with a hypervalent iodine reagent PhI(OCOCF3)2 as the oxidant (Scheme 20).45 The scope included benzimidazoles and benzothiazoles, furnishing 13 in moderate yields (13x, 13s and 13bg, 44–69%). An advantage of the approach is that it uses only 1 mol% of photocatalyst. The use of acids 5 is also highly advantageous in terms of commercial availability, cost and atom economy,11 especially since the equivalents of 5 are greatly reduced compared to previous methods.34
 | ||
| Scheme 20 Organophotocatalysed alkylation (3 azole examples).45 | ||
In 2019, Li, Wang and co-workers also utilised [Acr+-Mes][ClO4−] 29 and acid precursors 5, but this time the study was focused solely on benzothiazoles and the protocol used sustainable air as the oxidant (Scheme 21).46 Alkylations proceeded in moderate to good yields (48–92%). The ambient reaction temperature and availability of reagents are highlights; however, the reaction time is relatively lengthy at 36 h. Tertiary carboxylic acids gave the respective alkylated products in excellent yields (13a, 13am), secondary carboxylic acids in moderate yields (13bk, 13aj, 13g), while primary carboxylic acids did not react, reflecting the relative stabilities of the corresponding alkyl radicals.
 | ||
| Scheme 21 Alkylation of benzothiazoles using air as an oxidant.46 | ||
While 28 and 29 are well-known organophotocatalysts, other cheaper organophotocatalysts can also be utilised. In 2020, Chu, Sun and co-workers used the anthocyanidin pigment, cyanidin 32, as a photocatalyst (Scheme 22).47 Benzothiazoles seem to react better than benzoxazoles, which in turn perform better than benzimidazole (13bk 65% vs.13bo 50% vs.13z 36%). In terms of the alkylation scope, tertiary (13a) and secondary (13bk) alkylations were possible, but primary alkylation was not successful.
 | ||
| Scheme 22 Cyanidin-catalysed alkylation of (benzo)azoles.47 | ||
In 2019, Hajra's group reported on the etherification of 2H-indazoles 16 using a Minisci-like reaction (Scheme 23).48 Advantages include the use of the cheap organic dye rose bengal as the catalyst; however, limitations include the large excess of ether precursor 33 required.
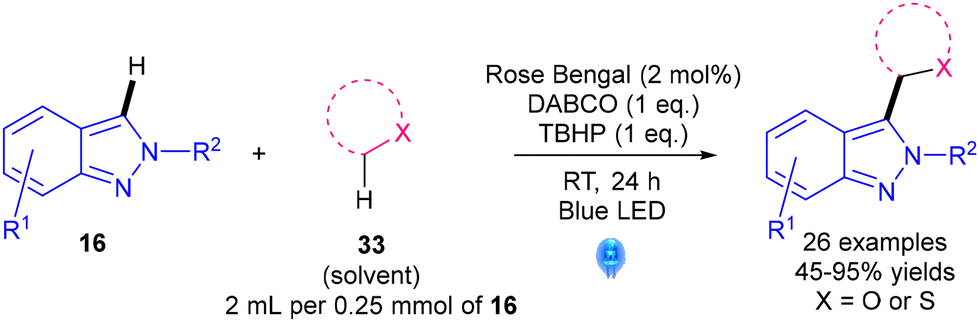 | ||
| Scheme 23 Rose bengal-photocatalysed etherification of 2H-indazoles.48 | ||
Very recently, a visible-light methodology that uses neutral 9-arylacridinium pre-catalyst 34 in the presence of TFA and pyridinium N-oxide to alkylate N-heterocycles, including 2 azoles, was revealed by Bosque and Gonzalez-Gomez's groups (Scheme 24).49 The authors highlight that the methodology is free of chemical oxidants, metals or chlorinated solvents. Nevertheless, a perfluorinated solvent (HFIP) is required, and the reaction times are relatively long at 48 h.
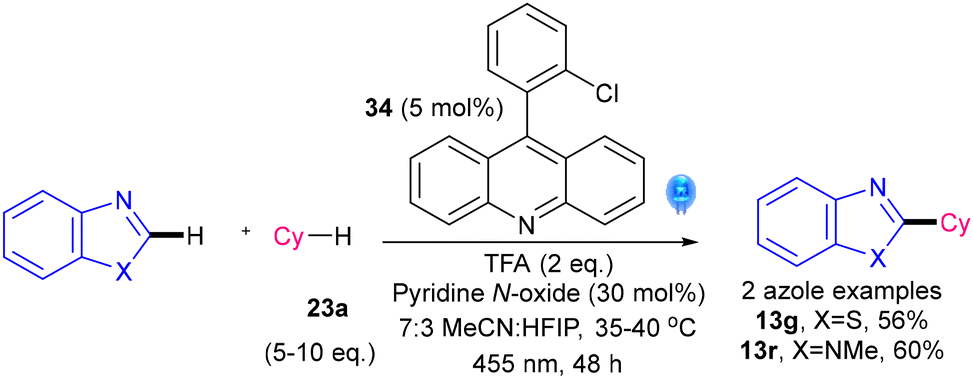 | ||
| Scheme 24 Alkylations using a neutral 9-arylacridinium catalyst (2 azole examples).49 | ||
4.1.2.3 Photocatalyst-free light-mediated alkylations. In 2017, a UV light-mediated approach to functionalise various heteroaromatics, which included one azole alkylation, was reported by C.-J. Li's group (Scheme 25A).50 Advantages include the discovery of an air- and water-stable sulfone compound 35 that can be cleaved under light irradiation without a photocatalyst. Disadvantages include the use of UV light and the relatively poor atom economy of 35.
 | ||
| Scheme 25 Photocatalyst-free (A) UV light-mediated alkylation (1 azole example)50 and (B) PhI(OCOCF3)2-mediated alkylation (1 azole example).51 | ||
A year later, a photocatalyst-free visible light-mediated procedure was developed using 2 eq. of PhI(OCOCF3)2 (Scheme 25B).51 Yang and Zhang's groups investigated only one azole (benzothiazole) as part of their substrate scope, and although the yield of 13g was good (75%), the reaction time was much longer compared to other N-heterocyclic bases screened (48 h vs. 12 h).
In 2021, Tan and Du's groups devised a procedure focused specifically on benzothiazoles (Scheme 26).52 The procedure requires only 2 eq. of DHP 18a and Na2S2O8, and it proceeds at RT in only 2 h. A Lewis acid additive was found to be essential. Tertiary (13a) and secondary (13g) alkylations were successful, but not primary alkylations (13bs). Although only 2 eq. of 18a are required, a downside is that this radical precursor is neither atom-economical nor commercially available.
 | ||
| Scheme 26 Photocatalyst-free alkylation of benzothiazoles.52 | ||
An alkylation of benzothiazoles with alcohols, ethers, lactams, amides and alkanes 36 using combined self-photoredox catalysis and HAT was reported by J. Li's group in 2022 (Scheme 27).53 The authors consider the reaction self-catalysed due to the photosensitivity of benzothiazoles. As expected, benzimidazoles and benzoxazoles are not suitable substrates as they are not photosensitive under visible light. Thiazole and 4,5-dimethylthiazole also failed to react. The advantages of this procedure are that it is metal- and photosensitiser-free; however, 36, although atom economical in nature, has to be used in large excess. The self-catalysed nature of the procedure is a highlight, although it necessarily limits the procedure to azoles that are photosensitive under visible light (i.e. benzothiazoles).
 | ||
| Scheme 27 “Self-catalysed” alkylation of benzothiazoles.53 | ||
In 2019, the groups of Chen and Wang very elegantly showed the utility of visible light-mediated Minisci-type alkylation of azoles in their histidine-specific peptide modification (Scheme 28).54 Since histidine carries an imidazole side chain, it allows for highly chemoselective alkylations onto the imidazole of histidine in native peptides/proteins 37. This overcomes the previous limitations of histidine modifications, which relied on N-substitution reactions of the imidazole, thereby inherently suffering from interferences with cysteine and lysine residues. Additionally, the reaction is transition-metal-free and exhibits a broad scope for peptides 37 and DHP 18a. DHP 18a is thought to act as both the alkylating reagent and the oxidant. Their report very elegantly showcases the application of the Minisci reaction of azoles in chemical biology.
 | ||
| Scheme 28 Minisci alkylation in histidine-specific peptide modification.54 | ||
Despite the utility of light-mediated protocols for enabling milder reactions, one potential drawback is that batch scale-up can be problematic due to less permeating light irradiation.55 Although continuous flow can sometimes address this issue,55 the option of cheap and operationally simple procedures is also in demand. As such, several metal- and light-free Minisci alkylation procedures have been developed and will be the topic of the next section.
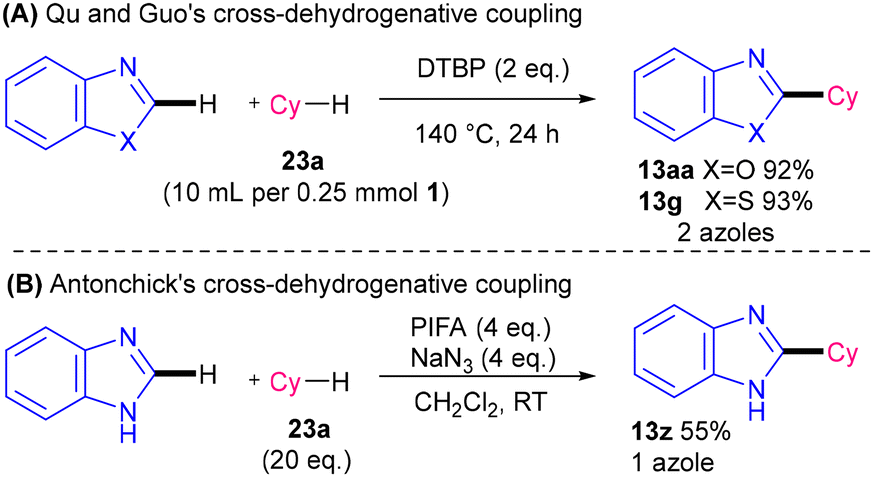 | ||
| Scheme 29 (A) DTBP-56 and (B) PIFA-mediated58 metal-free alkylation of N-heterocycles (2 azole and 1 azole examples respectively). | ||
In 2013, hypervalent iodine PIFA was utilised in conjunction with NaN3 for a much milder cross-dehydrogenative coupling (Scheme 29B).58 Only one azole (benzimidazole) was included as part of Antonchick and co-workers’ substrate scope (55%, 13z). The reaction is mild, transition metal-free, and uses atom economical alkanes 23. However, a large excess (20 eq.) of 23 is required and the reaction requires the use of toxic NaN3.
A more azole specific study was carried out by Cai and co-workers in 2017, using oxidants DTBP or dicumyl peroxide (DCP) at 120 °C in a metal-free procedure (Scheme 30).59 Alkylations with CH2Ar and Cy were carried out using DTBP as the oxidant and methylarene or cyclohexyl respectively as the alkyl radical precursor, whereas methylation was carried out using DCP as the oxidant and acetic acid as the Me radical precursor (Scheme 31). The methylation was compatible with benzothiazole (13bs), benzoxazole (13bt) and benzimidazole (13bu) whereas the DTBP method was shown to be compatible with benzothiazole (13bv), benzoxazole (13aa) and thiazole (13h) but not with benzimidazole (13bw). Advantages of this method include a good azole substrate scope and the use of methylarenes and cycloalkanes as radical precursors, although they have to be used in solvent quantities. The alkylation scope is also limited to –Me, –Cy and –CH2Ar groups and the use of peroxides at elevated temperatures may pose safety risks at larger scales.
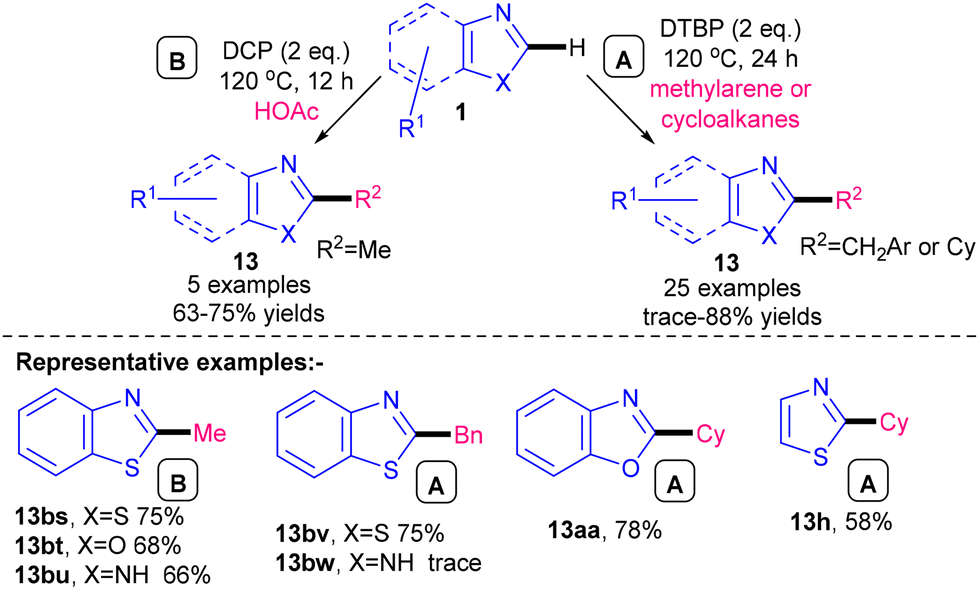 | ||
| Scheme 30 DTBP- and DCP-mediated metal-free alkylation of azoles.59 | ||
 | ||
| Scheme 31 Molecular oxygen-mediated alkylation (5 azole examples).60 | ||
In 2017, a molecular oxygen-mediated alkylation with boronic acids was developed by Liu and co-workers and 3 azoles were included in their substrate scope (Scheme 31).60 The advantage of this procedure is the metal-free aspect and the use of O2 as an oxidant; however, elevated temperatures (110 °C) are required, as well as 5 eq. of alkylboronic acids 22, which are not as cheap or readily available as 5 or 23.
In 2018, our research group devised a much milder (40 °C) metal- and light-free procedure for the alkylation of N-heterocycles (Scheme 32A).61 Whilst good to excellent yields were obtained across a variety of N-heterocycles (50–91%), a much lower yield (25%) was presented for benzothiazole (13g). In 2019, Wang's group also revealed metal-free Minisci alkylations using (NH4)2S2O8 in DMSO, using α-ketoacids62 or organosilanes63 as radical precursors, which included 1–2 benzothiazoles in their substrate scope. Weng's group revealed a persulfate-mediated hydroxyalkylation of benzothiazoles in 2019, using alcohols 30 as radical precursors (Scheme 32B).64 Ethers 39 were also suitable radical precursors (Scheme 32C). While alcohols and ethers are readily available, one disadvantage is that these radical precursors are used in large excess as solvents.
 | ||
| Scheme 32 Persulfate-mediated metal-free alkylations using: (A) carboxylic acids,61 (B) alcohols,64 (C) ethers64 and (D) amides.65 | ||
It should be noted that in 2020 a similar reaction was reported using Selectfluor with light66 and in 2024 Mantry and Gandeepan developed a related visible light-induced PhI(OAc)2 mediated reaction.67 An amidoalkylation of benzothiazoles was developed by Huang and Zhu's groups in 2016, again with the disadvantage that the radical precursor 40 is used as a solvent in large excess (Scheme 32D).65 In 2019, Weng and co-workers used Eosin Y and visible light irradiation with K2S2O8 to carry out a very similar reaction, but at room temperature.68
In 2022, a metal-free alkylation of 2H-indazoles 16 using aldehydes 11 as radical precursors and mediated by DTBP at 120 °C was revealed by Lin's group (Scheme 33).69 Alkylation occurred when R2 = alkyl and acylation occurred when R2 = aryl (see Section 4.2.3), likely due to the reduced propensity for R2CO˙ to decarbonylate when R2 = aryl. While the reaction is metal-free, heating a peroxide to 120 °C and the use of toxic chlorobenzene may pose safety issues on larger scales.
 | ||
| Scheme 33 DTBP-mediated metal-free alkylation of 2H-indazoles.69 | ||
 | ||
| Scheme 34 Electrophotocatalytic alkylations (A) from carboxylic acids (1 azole example)71 and (B) from alkanes (3 azole examples).73 | ||
Xu's group followed up with an electrophotocatalytic methodology using atom economical alkanes 23 as radical precursors, employing chlorine radicals (generated from HCl) as a simple but effective HAT reagent for 23 (Scheme 34B).73,74 The reaction worked well for benzothiazoles (13g, 13br) but not for benzoxazole (13aa). As with many procedures using alkanes 23, a large excess of 23 is required.
In 2021, an electrophotocatalytic Minisci alkylation of benzothiazoles using alkanes 23 as radical precursors was revealed by Ravelli's group (Scheme 35).75 A stoichiometric oxidant is not required and TBADT was utilised in a threefold role: as the HAT photocatalyst to activate the sp3 C–H bond of 23, as a photoredox catalyst and as an electrocatalyst.75 Various benzothiazole cores were successfully alkylated (13be, 13bx–ca). While symmetrical alkanes 23 are usually relied on to avoid regioselectivity issues, Ravelli's results are notable for their selective functionalisation of isocapronitrile (13bz) and cyclopentanone (13ca) in good yields.
 | ||
| Scheme 35 Electrophotocatalytic alkylation of benzothiazoles.75 | ||
More recently in 2022, an electrochemical Minisci-type alkylation using alkyl iodides 41 was disclosed by the groups of Fernández-Salas and Alemán (Scheme 36).76 Two azoles were investigated as part of their substrate scope studies: benzothiazole (13g) and benzimidazole (13r). Advantages include being free of stoichiometric chemical oxidants; however, alkyl iodides 41 are not as readily available or cheap as other commonly used radical precursors such as acids 5 or alkanes 23.
 | ||
| Scheme 36 Electrocatalytic alkylation (2 azole examples).76 | ||
Electrochemical and electrophotocatalytic Minisci reactions are clearly areas of recent development which allow the Minisci reaction to occur without stoichiometric chemical oxidants. The seminal reports were not necessarily developed for azole substrates, so the yields for the 1–2 azoles tested were not always good. Nevertheless, recent reports have started to focus more on azole substrates75 and there will undoubtedly be further future developments in this area.
4.2 Minisci acylation of azoles
Minisci-type acylations of N-heterocycles are the second most studied reaction after alkylations. This section gives an overview of some of the methods available for Minisci acylation of azoles.In 2018, Oh and co-workers reported a protocol for the acylation of 2-H indazoles 16 using α-keto acids13,776 as precursors for acyl radicals via silver- and persulfate-mediated decarboxylation (Scheme 37).78 The reaction proceeded at mild RT, forming 3-acyl-2H-indazoles 42 in 25–83%. The use of 20 mol% AgNO3, however, is a potential limitation in terms of sustainability and cost. The reaction was found to be compatible with phenyl-substituted α-keto acids 6 (R3 = Ar, 42a–b). Alkyl α-keto acids 6 (R3 = alkyl), however, required higher temperatures of 50 °C, yet still achieved only 40% yield (42c). Moving from an N-2 phenyl substituent (42a 81%) to an N-2 alkyl substituent (42d–e 25–38%) also causes a drop in yield.
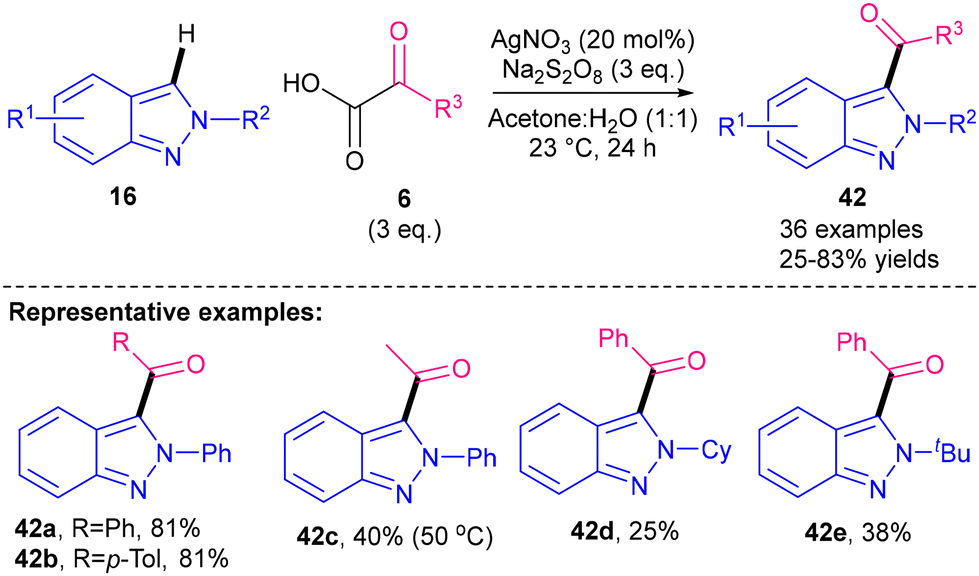 | ||
| Scheme 37 Silver-catalysed acylation of 2H-indazoles.78 | ||
An alternative iron(II) catalysed acylation of N-heterocycles making use of triethyl orthoformate 43 and tBuOOH 44 was reported in 2019 by Reddy's group (Scheme 38).79 Only one azole was investigated: benzothiazole 1a to yield 24b (42%). Triethylorthoformate 43 acts as both a substrate and solvent under their conditions.
 | ||
| Scheme 38 Fe-catalysed acylation of N-heterocycles (1 azole example).79 | ||
Also in 2019, an iron-catalysed acylation of N-heterocycles using α-keto acids 6 as the acyl radical precursor was reported by Zeng's group (Scheme 39).80 The authors were able to render the classical Minisci reaction silver-free by instead utilising a much cheaper and more abundant iron catalyst. However, the methodology was not very successful with azoles. Three azoles were investigated as part of the scope: a benzothiazole (19b, 20%) and two triazoles which were both unsuccessful (45a, 46a, Scheme 39).
 | ||
| Scheme 39 Fe-catalysed acylation (3 azole examples).80 | ||
In 2020, Du, Tan and co-workers reported the acylation of 2-H indazoles 16, using acyl-DHP 47 as radical precursors (Scheme 40).29 While the substrate scope was generally good, the reaction may not compare as favourably with the previously described methodology using α-keto acids 6 (Scheme 37),78 since non-commercial and less atom-economical acyl-DHP 47, higher AgNO3 catalyst loadings (40 mol%) and the addition of TFA (3 eq.) are needed. Nevertheless, an advantage is that the reaction is more time efficient (4 h vs. 24 h).
 | ||
| Scheme 40 Silver-catalysed acylation of 2H-indazoles.29 | ||
4.2.2.1 Photocatalysed acylations. In 2018, a Ru(bpy)3Cl2 photocatalysed acylation of N-heterocycles using K2S2O8 as the oxidant and TFA as an acid activator was reported by Shah's group (Scheme 41A).81 Interestingly, alkynes are used as the radical precursor via in situ generation of α-keto acid 6a. Only one azole was studied as part of the general substrate scope (24c).

A year later, Xu and co-workers published a photocatalysed Minisci-type reaction of N-heteroarenes.82 One example of an acylated benzothiazole (24c) was reported using acyl chloride 48 as the radical precursor (Scheme 41B). While this method is milder than the classical silver catalysed Minisci-type acylations, silver has now been replaced by a more expensive iridium catalyst.20 Other limitations include extended reaction times and the use of 10 eq. of acid chloride 48.
As discussed in Section 4.1.2, a noble-metal-free photocatalytic protocol was developed for benzothiazoles by Jian and Tong's groups (Scheme 16).41 The reaction can be switched from alkylation (under Ar) to acylation (under air, Scheme 42). One big advantage is that the acylation scope is complementary to the corresponding reactions using α-keto acids 6 or activated ester derivatives; the latter tends to work best with aromatic acyls and poorly or not at all with aliphatic acyls. The method in Scheme 42, in contrast, works well with aliphatic acyls (e.g.24ab,d–h). A drawback, however, is the need for a large excess of the alcohol reactant 30 (31–85 eq.).
 | ||
| Scheme 42 Noble-metal-free photocatalytic acylation of benzothiazoles.41 | ||
4.2.2.2 Photocatalyst-free light-mediated acylations. In 2018, a seminal persulfate- and visible light-mediated acylation of N-heterocycles was developed by Wencel-Delord's group, using α-keto acid 6 without the need for a photocatalyst (Scheme 43A).83 The heterocyclic scope included 1 azole: benzothiazole 1a (84% 24c). In the same year, Zhang's group independently reported a procedure using hypervalent iodine(III) reagent PhI(OCOCF3)2 (Scheme 43B, 50% 24c).51 Both methodologies have the advantage of not requiring a photocatalyst; however, PhI(OCOCF3)2 is less economical than persulfate.

In 2021, an alternative light-mediated route to acylation was devised by Tan and Du's groups, focusing on benzothiazoles (Scheme 44).52 The reaction uses DHP 47 as radical precursors, synthesised from their parent glyoxal hydrates. The optimised conditions rely on only 2 equiv. of DHP 47 and Na2S2O8 as the oxidant and a Lewis acid additive. One downside is the non-atom-economical nature of the DHP radical precursor 47 and the fact that it is not commercially available.
 | ||
| Scheme 44 Acylation of benzothiazoles using DHP.52 | ||
Very recently in 2024, a metal-, photocatalyst- and oxidant-free methodology for the acylation of 2H-indazoles 16 was reported by the groups of Cao, Li and Shen (Scheme 45).84 They propose that 16 absorbs visible light to form its excited state 16*, which then undergoes energy transfer to α-ketoacid 6, with the excited 6* subsequently homolyzing to form the key acyl radical II. The protocol may therefore be specific to visible light-absorbing 2H-indazole substrates 16. However, their UV-vis studies show that 16 does not absorb visible light, which somewhat contradicts the proposed mechanism. Nevertheless, advantages include not requiring metals, photocatalysts or oxidants, but the use of costly and toxic HFIP solvent could be a disadvantage.
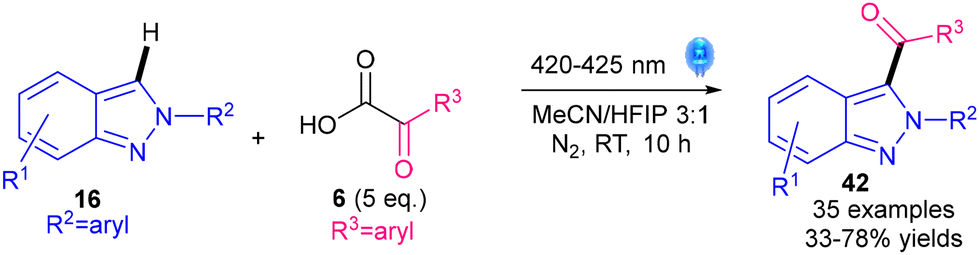 | ||
| Scheme 45 Photocatalyst- and oxidant-free acylation of 2H-indazoles.84 | ||
 | ||
| Scheme 46 PIFA-mediated acylation of benzothiazole.85 | ||
In 2014, a metal-free method for acylating benzothiazoles using H-dialkyl phosphonates 49 was disclosed by Chen and Qu's groups (Scheme 47).86 Benzothiazoles were successfully acylated (24b, 24q–s). The authors also investigated thiazole and benzoxazole, but both failed to produce any acylated products (24u, 24v). One of the key advantages of their method is that it has a good substrate scope for aliphatic acyls, particularly given that most Minisci acylation methodologies tend to focus on aroylations. Nevertheless, limitations include the acute toxicity of TBHP, the safety implications of heating TBHP to elevated temperatures (especially at larger scales) and the non-atom-economical nature of radical precursor 49.
 | ||
| Scheme 47 Metal-free acylation using H-dialkyl phosphonates.86 | ||
Our group developed a metal- and light-free method for direct C–H acylations of N-heterocycles in 2019,87 optimised for (iso)quinolines, where yields were up to 98%. Only one azole was investigated: benzothiazole 1a, which gave a poor yield of 29% for 24w (Scheme 48A). In 2020, Laha's group developed a persulfate-mediated methodology for the acylation of electron-rich pyrroles,88 and included one azole as part of their substrate scope study (24c, Scheme 48BI).

Two years later, Laha's group disclosed a procedure using glucose to activate the persulfate and break it down to the key sulfate radical anion at RT in water, thereby avoiding the need for high temperatures, UV light or metals (Scheme 48BII).89 Two azoles were investigated: benzothiazole worked well (24c) but indazole 16 failed to undergo acylation. This methodology looks promising as the reaction is mild and a green solvent is used. Nevertheless, more studies are needed on azole motifs to ascertain whether the methodology is applicable to other azoles apart from benzothiazole.
In 2022, a metal-free acylation of 2H-indazoles 16 using aldehydes 11 as radical precursors and mediated by DTBP at 120 °C was revealed by Lin's group (Scheme 49).69 Acylation occurred when R2 = aryl (and alkylation occurred when R2 = alkyl, see Section 4.1.3). Advantages include being metal-free, although the heating of a peroxide to 120 °C and the use of toxic chlorobenzene may pose safety issues on larger scales.
 | ||
| Scheme 49 Metal-free acylation of 2H-indazoles.69 | ||
4.3 Minisci amidations of azoles
Minisci amidations of azoles are far less studied than alkylations and acylations; however, some recent studies have significantly expanded the scope of azoles that can be successfully amidated via Minisci-type reactions.4.3.1.1 Photocatalysed amidations. In 2019, an amidation of N-heterocycles via a photocatalytic Minisci-type reaction using 4-CzIPN 28 as a photocatalyst was reported by Landais (Scheme 50).90 Although a wide substrate scope was demonstrated, only two azoles were studied to give 25b and 25c in moderate yields of 41% and 59% respectively. The reaction is metal-free, very mild and is complete in 12 h at RT. Nevertheless, the 4-CzIPN 28 catalyst is currently very expensive commercially (£3596 per g).35
 | ||
| Scheme 50 Photocatalysed azole amidation (2 azole examples).90 | ||
4.3.1.2 Photocatalyst-free light-mediated amidations. In 2016, a visible light-mediated Minisci-type amidation of azoles, using stoichiometric benzaldehyde as a photosensitiser, was published by Ji's group (Scheme 51).91 Benzothiazoles (25d–i, 6 examples) and benzimidazoles (25j–n, 5 examples) were studied in more detail but one thiazole (25p) was also shown to be compatible. An attempt to functionalise benzoxazoles, though, was not successful (25o). The reaction has the advantage of proceeding at a mild temperature (29 °C). One limitation, however, is that only functionalisation with primary amides was possible and formamide was used in solvent quantities.
 | ||
| Scheme 51 Light-mediated amidation using formamide.91 | ||
We recently developed one thermal methodology (Conditions A) and one photosensitiser-free, light-mediated methodology (Conditions B) for direct Minisci-type C–H amidation of 1,3-azoles (Scheme 52).92 The reaction was applicable to the four most important 1,3-azoles in medicinal chemistry: thiazoles (25t–y), benzothiazoles (25s), benzimidazoles (25r) and imidazoles (25q). Both conditions worked well, although Conditions A were more amenable to batch scale-up (25s, 81% g scale) and the milder and more efficient Conditions B were more functional group tolerant (25u). A wide variety of primary (25w), secondary (25x) and tertiary (25y) amides were successfully installed onto all four 1,3-azole classes investigated. It is of note that the reaction concentration had to be optimised separately for each azole class in order to achieve good yields. Late-stage C–H amidation of 1,3-azole-based drug molecules was successfully demonstrated (e.g.25z–25aa),92 highlighting the application of this methodology.
 | ||
| Scheme 52 Thermal- and light-mediated amidation of 1,3-azoles and its application to the C–H functionalisation of drug molecules.92 | ||
 | ||
| Scheme 53 Amidation of azoles using formamides.93 | ||
In 2021, a metal- and light-free protocol for the amidation of 2H-indazoles was reported by Bhat and Lee (Scheme 54).94 The thermally mediated reaction utilises oxamic acid 7 as the carbamoyl precursor and ammonium persulfate as the oxidant at 60 °C. Advantages include a short reaction time, the absence of metals and efficient scalability to a 1 g scale. Various electron-withdrawing and -donating substituents were tolerated on the N2-phenyl ring (56c–d) but only secondary amidations were reported (e.g.56e–g). In 2022, Zhang and Jiang developed a photocatalysed version of the same transformation, using 4-CzIPN 28 as the photocatalyst.95
 | ||
| Scheme 54 Amidation of 2H-indazoles.94 | ||
Li and co-workers published an amidation of N-heterocycles in 2021, this time via isocyanide (Scheme 55).96 Benzothiazole was included in their substrate scope (25ai), along with benzimidazole and benzoxazole, although the latter two were not suitable substrates (25aj–ak). The major drawback of this chemistry is the elevated temperature (120 °C), which will likely preclude it from being applicable to late-stage functionalisations.
 | ||
| Scheme 55 Amidation using isocyanide (3 azole examples).96 | ||
Our research group also developed metal- and light-free amidations of azoles which are already discussed in Scheme 52.92
 | ||
| Scheme 56 Electrophotocatalytic amidation (2 azole examples).71 | ||
5. Conclusion and outlook
Whilst the pivotal work of Minisci and co-workers laid the foundations, growing sustainability concerns as well as the drive towards milder reaction conditions have resulted in multiple developments in the Minisci reaction. For example, there has been a push to replace costly and non-sustainable silver20 with metal-free procedures. Many visible-light-mediated methodologies have also been developed to render Minisci-type reactions milder, with the initial use of expensive and non-sustainable20 Ir and Ru photocatalysts being subsequently overtaken by a variety of organophotocatalytic and photocatalyst-free protocols. Stoichiometric oxidant-free procedures have also been revealed, such as the use of electrophotocatalytic methods, although these are in their early stages of development. Nevertheless, it is clear that there is a consistent move towards developing metal-free, milder and more sustainable protocols for Minisci-type reactions.A review of the literature reveals 1,3-azoles (benzothiazoles, benzimidazoles, benzofurans, thiazoles, imidazoles and oxazoles) to be the most explored azole substrates for Minisci-type reactions (Fig. 3). Minisci-type C–H functionalisations of benzothiazoles, in particular, have been very well studied. Conversely, only one class of 1,2-azoles (indazoles) has been routinely investigated, leaving plenty of room for further developments with other 1,2-azole motifs. To the best of our knowledge, there are very few reports of successful Minisci-type C–H functionalisation of azoles with three heteroatoms. Promisingly, a handful of initial successful reactions with these motifs6 hint at potential future opportunities. Since isoxazoles, triazoles and thiadiazoles are important motifs in medicinal chemistry,2b,c,7 a future challenge would be the successful Minisci direct C–H functionalisations of these motifs under mild and sustainable conditions.
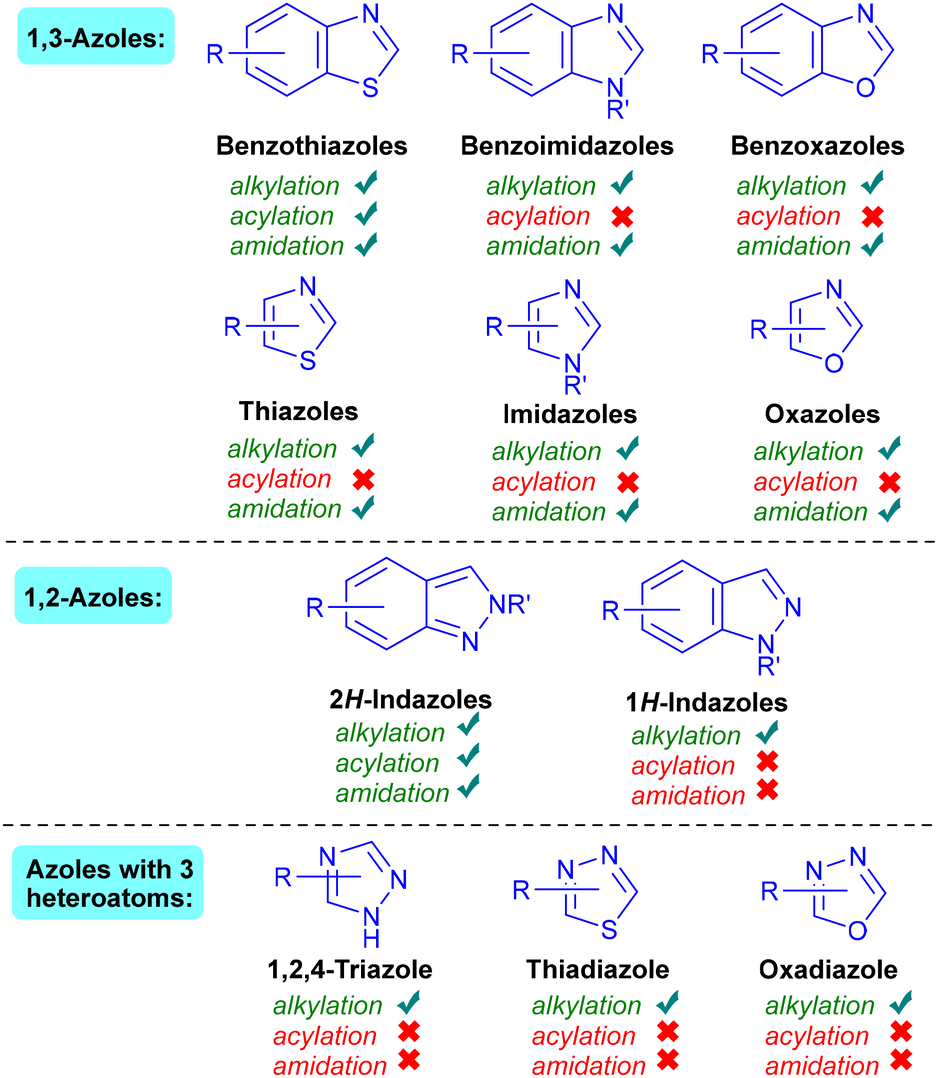 | ||
| Fig. 3 List of azoles that have been successfully C–H functionalised via the Minisci reaction. | ||
The vast majority of Minisci reactions of azoles focus on alkylations and acylations, with fewer publications focusing on amidations. Nevertheless, it is in fact acylations that currently exhibit the poorest azole substrate scope (Fig. 3), with successful reports on only benzothiazoles and indazoles. This is likely the result of acyl radicals being less nucleophilic than carbamoyl or alkyl radicals.94 As such, there is potential for further developments, particularly with respect to the lesser-studied azole cores such as triazoles, thiadiazoles and oxadiazoles, as well as acylations of azoles other than benzothiazoles and indazoles. It is also evident that successful Minisci C–H functionalisations of azoles often require specific optimisation for different azole cores and this should be taken into consideration.92
Recently, applications in chemical biology,54 synthesis of drug precursors6 and late-stage C–H functionalisations of drug molecules92 have emerged. Chen, Wang and co-workers’ histidine-specific peptide modification54 elegantly showcases the application of the Minisci reaction of azoles in chemical biology. Our late-stage C–H amidations on azole drug molecules also showcase the potential synthetic applications of this methodology.92 We envisage that further future applications of Minisci C–H functionalisations of azoles will emerge as more efficient, milder and more sustainable methodologies are being developed for azoles.
Data availability
Not applicable as this is a review article with no new data generated.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Engineering and Physical Sciences Research Council and AstraZeneca for financial support [Industrial CASE Ph.D. studentship to DTM; Grant code: EP/V519522/1].References
- (a) B. Schulze and U. S. Schubert, Chem. Soc. Rev., 2014, 43, 2522–2571 RSC; (b) A. Diaz-Ortiz, P. Prieto, J. R. Carrillo, R. Martin and I. Torres, Curr. Org. Chem., 2015, 19, 568–584 CrossRef CAS; (c) E. Peris, Chem. Rev., 2018, 118, 9988–10031 CrossRef CAS PubMed; (d) A. Tigreros and J. Portilla, RSC Adv., 2020, 10, 19693–19712 RSC; (e) G. Aromi, L. A. Barrios, O. Roubeau and P. Gamez, Coord. Chem. Rev., 2011, 255, 485–546 CrossRef CAS; (f) J. D. Crowley and D. A. McMorran, Top. Heterocycl. Chem., 2012, 28, 31–83 CAS.
- (a) A. K. Kabi, S. Sravani, R. Gujjarappa, A. Garg, N. Vodnala, U. Tyagi, D. Kaldhi, R. Velayutham, S. Gupta and C. C. Malakar, in Nanostructured Biomaterials: Basic Structures and Applications, ed. B. P. Swain, Springer Singapore, Singapore, 2022, pp. 79–99, DOI:10.1007/978-981-16-8399-2_4; (b) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed; (c) J. Shearer, J. L. Castro, A. D. G. Lawson, M. MacCoss and R. D. Taylor, J. Med. Chem., 2022, 65, 8699–8712 CrossRef CAS PubMed.
- T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546 RSC.
- J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS PubMed.
- (a) R. S. J. Proctor and R. J. Phipps, Angew. Chem., Int. Ed., 2019, 58, 13666–13699 CrossRef CAS PubMed; (b) M. A. J. Duncton, MedChemComm, 2011, 2, 1135–1161 RSC; (c) C. Punta and F. Minisci, Trends Heterocycl. Chem., 2008, 13, 1–68 CAS; (d) W. Wang and S. Wang, Curr. Org. Chem., 2021, 25, 894–934 CrossRef CAS; (e) A. C. Sun, R. C. McAtee, E. J. McClain and C. R. J. Stephenson, Synthesis, 2019, 1063–1072 CAS; (f) X. Zhang, S. Li, F. Qiu, H. T. Ang, J. Wu and P. Jia, Green Chem., 2024, 26, 3595–3626 RSC.
- E. Rosadoni, E. Bombonato, A. Del Vecchio, S. Guariento, P. Ronchi and F. Bellina, J. Org. Chem., 2023, 88, 14236–14241 CrossRef CAS PubMed.
- Purines are considered a separate class of heterocycles and are outside the scope of this review. For example, see: D. T. Mooney, P. R. Moore and A.-L. Lee, Org. Lett., 2022, 24, 8008–8013 CrossRef CAS PubMed and references cited therein.
- For example, the following review gives methods for forming 2-ketoaryl azole derivatives: N. Aljaar, M. M. Ibrahim, E. A. Younes, M. Al-Noaimi, K. A. Abu-Safieh, B. F. Ali, K. Kant, N. Al-Zaqri, R. Sengupta and C. C. Malakar, Asian J. Org. Chem., 2023, 12, e202300036 CrossRef CAS.
- T. Huang, X. Wu, Y. Yu, L. An and X. Yin, Tetrahedron Lett., 2019, 60, 1667–1670 CrossRef CAS.
- B. L. Eriksen, P. Vedsø, S. Morel and M. Begtrup, J. Org. Chem., 1998, 63, 12–16 CrossRef CAS PubMed.
- J. Schwarz and B. König, Green Chem., 2018, 20, 323–361 RSC.
- (a) F. Minisci, R. Bernardi, F. Bertini, R. Galli and M. Perchinummo, Tetrahedron, 1971, 27, 3575–3579 CrossRef CAS; (b) F. Minisci, F. Fontana and E. Vismara, J. Heterocycl. Chem., 1990, 27, 79–96 CrossRef CAS; (c) F. Minisci, E. Vismara, F. Fontana, G. Morini, M. Serravalle and C. Giordano, J. Org. Chem., 1987, 52, 730–736 CrossRef CAS.
- F. Penteado, E. F. Lopes, D. Alves, G. Perin, R. G. Jacob and E. J. Lenardão, Chem. Rev., 2019, 119, 7113–7278 CrossRef CAS PubMed.
- (a) T. Caronna, G. Fronza, F. Minisci and O. Porta, J. Chem. Soc. Perkin Trans. 2, 1972, 2035–2038, 10.1039/P29720002035; (b) T. Caronna, G. Fronza, F. Minisci, O. Porta and G. P. Gardini, J. Chem. Soc., Perkin Trans. 2, 1972, 1477–1481, 10.1039/P29720001477; (c) F. Fontana, F. Minisci, M. C. Nogueira Barbosa and E. Vismara, J. Org. Chem., 1991, 56, 2866–2869 CrossRef CAS.
- T. Caronna, G. P. Gardini and F. Minisci, J. Chem. Soc. D: Chem. Commun., 1969, 201–201, 10.1039/C29690000201.
- I. M. Ogbu, G. Kurtay, F. Robert and Y. Landais, Chem. Commun., 2022, 58, 7593–7607 RSC.
- F. Coppa, F. Fontana, E. Lazzarini and F. Minisci, Heterocycles, 1993, 36, 2687–2696 CrossRef CAS.
- A. Citterio, A. Gentile, F. Minisci, M. Serravalle and S. Ventura, J. Org. Chem., 1984, 49, 3364–3367 CrossRef CAS.
- T. Caronna, R. Galli, V. Malatesta and F. Minisci, J. Chem. Soc. C, 1971, 1747–1750, 10.1039/J39710001747.
- D. J. Cole-Hamilton, Chem. Int., 2019, 41, 23–28 CAS.
- M. Sharique, J. Majhi, R. K. Dhungana, L. M. Kammer, M. Krumb, A. Lipp, E. Romero and G. A. Molander, Chem. Sci., 2022, 13, 5701–5706 RSC.
- See review ref. 5b for examples before 2011.
- Note that there are some transition metal catalysed reactions where the exact mechanisms are not known, but there is evidence to show that they may involve radicals. These have not been included in the review. For example, see: (a) P. Ren, I. Salihu, R. Scopelliti and X. Hu, Org. Lett., 2012, 14, 1748–1751 CrossRef CAS PubMed; (b) X. Wu, J. W. T. See, K. Xu, H. Hirao, J. Roger, J.-C. Hierso and J. Zhou, Angew. Chem., Int. Ed., 2014, 53, 13573–13577 CrossRef CAS PubMed.
- The more electrophilic nature of the tri- and di-fluoromethyl radicals tends to make them diverge from Minisci-like reactivity. For example, see: (a) P. Ghosh, S. Mondal and A. Hajra, J. Org. Chem., 2018, 83, 13618–13623 CrossRef CAS PubMed; (b) A. G. O'Brien, A. Maruyama, Y. Inokuma, M. Fujita, P. S. Baran and D. G. Blackmond, Angew. Chem., Int. Ed., 2014, 53, 11868–11871 CrossRef PubMed; (c) Y. Fujiwara, J. A. Dixon, R. A. Rodriguez, R. D. Baxter, D. D. Dixon, M. R. Collins, D. G. Blackmond and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 1494–1497 CrossRef CAS PubMed.
- W.-M. Zhao, X.-L. Chen, J.-W. Yuan, L.-B. Qu, L.-K. Duan and Y.-F. Zhao, Chem. Commun., 2014, 50, 2018–2020 RSC.
- Molander developed a super-stoichiometric Mn(OAc)3-mediated Minisci-type alkylation which included 7 azoles (11–60%) that is outside the timescale of our review. G. A. Molander, V. Colombel and V. A. Braz, Org. Lett., 2011, 13, 1852–1855 CrossRef CAS PubMed.
- S.-C. Lu, H.-S. Li, S. Xu and G.-Y. Duan, Org. Biomol. Chem., 2017, 15, 324–327 RSC.
- K. R. Babu, N. Zhu and H. Bao, Org. Lett., 2017, 19, 46–49 CrossRef CAS PubMed.
- L. Liu, P. Jiang, Y. Liu, H. Du and J. Tan, Org. Chem. Front., 2020, 7, 2278–2283 RSC.
- T. McCallum and L. Barriault, Chem. Sci., 2016, 7, 4754–4758 RSC.
- For a related study using alkyl iodide, see: N. B. Bissonnette, M. J. Boyd, G. D. May, S. Giroux and P. Nuhant, J. Org. Chem., 2018, 83, 10933–10940 CrossRef CAS PubMed.
- T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed.
- G.-X. Li, C. A. Morales-Rivera, Y. Wang, F. Gao, G. He, P. Liu and G. Chen, Chem. Sci., 2016, 7, 6407–6412 RSC.
- R. A. Garza-Sanchez, A. Tlahuext-Aca, G. Tavakoli and F. Glorius, ACS Catal., 2017, 7, 4057–4061 CrossRef CAS.
- Price from Sigma-Aldrich on 6th Sept 2024, excluding taxes.
- Z. Li, X. Wang, S. Xia and J. Jin, Org. Lett., 2019, 21, 4259–4265 CrossRef CAS PubMed.
- J. Dong, Q. Xia, X. Lv, C. Yan, H. Song, Y. Liu and Q. Wang, Org. Lett., 2018, 20, 5661–5665 CrossRef CAS.
- L. M. Kammer, A. Rahman and T. Opatz, Molecules, 2018, 23, 764 CrossRef.
- (a) W.-F. Tian, C.-H. Hu, K.-H. He, X.-Y. He and Y. Li, Org. Lett., 2019, 21, 6930–6935 CrossRef CAS PubMed; (b) J. Dong, X. Lyu, Z. Wang, X. Wang, H. Song, Y. Liu and Q. Wang, Chem. Sci., 2019, 10, 976–982 RSC; (c) J. Dong, F. Yue, W. Xu, H. Song, Y. Liu and Q. Wang, Green Chem., 2020, 22, 5599–5604 RSC; (d) J. Wang, G.-X. Li, G. He and G. Chen, Asian J. Org. Chem., 2018, 7, 1307–1310 CrossRef CAS.
- Z.-T. Pan, L.-M. Shen, F. W. Dagnaw, J.-J. Zhong, J.-X. Jian and Q.-X. Tong, Chem. Commun., 2023, 59, 1637–1640 RSC.
- Z.-T. Pan, X.-K. Qi, Q. Xiao, X.-W. Liang, J.-J. Zhong, J.-X. Jian and Q.-X. Tong, Chem. Commun., 2022, 58, 8810–8813 RSC.
- T. C. Sherwood, N. Li, A. N. Yazdani and T. G. M. Dhar, J. Org. Chem., 2018, 83, 3000–3012 CrossRef CAS PubMed.
- (a) Z. Wang, X. Ji, J. Zhao and H. Huang, Green Chem., 2019, 21, 5512–5516 RSC See also: (b) S. Pillitteri, P. Ranjan, G. M. Ojeda-Carralero, L. Y. Vázquez Amaya, J. E. Alfonso-Ramos, E. V. Van der Eycken and U. K. Sharma, Org. Chem. Front., 2022, 9, 6958–6967 RSC.
- J. K. Matsui, D. N. Primer and G. A. Molander, Chem. Sci., 2017, 8, 3512–3522 RSC.
- J. Genovino, Y. Lian, Y. Zhang, T. O. Hope, A. Juneau, Y. Gagné, G. Ingle and M. Frenette, Org. Lett., 2018, 20, 3229–3232 CrossRef CAS PubMed.
- B. Wang, P. Li, T. Miao, L. Zou and L. Wang, Org. Biomol. Chem., 2019, 17, 115–121 RSC.
- R. Guo, M. Zuo, Q. Tian, C. Hou, S. Sun, W. Guo, H. Wu, W. Chu and Z. Sun, Chem. – Asian J., 2020, 15, 1976–1981 CrossRef CAS PubMed.
- M. Singsardar, S. Laru, S. Mondal and A. Hajra, J. Org. Chem., 2019, 84, 4543–4550 CrossRef CAS PubMed.
- L. Laze, B. Quevedo-Flores, I. Bosque and J. C. Gonzalez-Gomez, Org. Lett., 2023, 25, 8541–8546 CrossRef CAS PubMed.
- P. Liu, W. Liu and C.-J. Li, J. Am. Chem. Soc., 2017, 139, 14315–14321 CrossRef CAS PubMed.
- X.-Y. Zhang, W.-Z. Weng, H. Liang, H. Yang and B. Zhang, Org. Lett., 2018, 20, 4686–4690 CrossRef CAS PubMed.
- P. Jiang, L. Liu, J. Tan and H. Du, Org. Biomol. Chem., 2021, 19, 4487–4491 RSC.
- J. Zhou, C. Wang, L. Huang, C. Luo, S. Ye, N. Xu, Y. Zhu, L. Liu, Q. Ren, Z. Chen, S. Song and J. Li, Green Chem., 2022, 24, 4606–4613 RSC.
- X. Chen, F. Ye, X. Luo, X. Liu, J. Zhao, S. Wang, Q. Zhou, G. Chen and P. Wang, J. Am. Chem. Soc., 2019, 141, 18230–18237 CrossRef CAS PubMed.
- D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276–10341 CrossRef PubMed.
- R. Xia, H.-Y. Niu, G.-R. Qu and H.-M. Guo, Org. Lett., 2012, 14, 5546–5549 CrossRef CAS PubMed.
- T. He, L. Yu, L. Zhang, L. Wang and M. Wang, Org. Lett., 2011, 13, 5016–5019 CrossRef CAS PubMed.
- A. P. Antonchick and L. Burgmann, Angew. Chem., Int. Ed., 2013, 52, 3267–3271 CrossRef CAS PubMed.
- Z.-L. Li, L.-K. Jin and C. Cai, Org. Chem. Front., 2017, 4, 2039–2043 RSC.
- L. Zhang and Z.-Q. Liu, Org. Lett., 2017, 19, 6594–6597 CrossRef CAS PubMed.
- D. R. Sutherland, M. Veguillas, C. L. Oates and A.-L. Lee, Org. Lett., 2018, 20, 6863–6867 CrossRef CAS PubMed.
- J. Dong, Z. Wang, X. Wang, H. Song, Y. Liu and Q. Wang, J. Org. Chem., 2019, 84, 7532–7540 CrossRef CAS PubMed.
- J. Dong, X. Wang, Z. Wang, H. Song, Y. Liu and Q. Wang, Org. Chem. Front., 2019, 6, 2902–2906 RSC.
- W.-X. Xu, X.-Q. Dai and J.-Q. Weng, ACS Omega, 2019, 4, 11285–11292 CrossRef CAS PubMed.
- J. Wang, J. Li, J. Huang and Q. Zhu, J. Org. Chem., 2016, 81, 3017–3022 CrossRef CAS PubMed.
- Y. Kong, W. Xu, X. Liu and J. Weng, Chin. Chem. Lett., 2020, 31, 3245–3249 CrossRef CAS.
- L. Mantry and P. Gandeepan, J. Org. Chem., 2024, 89, 6539–6544 CrossRef CAS PubMed.
- J.-Q. Weng, W.-X. Xu, X.-Q. Dai, J.-H. Zhang and X.-H. Liu, Tetrahedron Lett., 2019, 60, 390–396 CrossRef CAS.
- B. Wang, X. Zhong, H. Yao, R. Deng, Z. Yan, M. Gao and S. Lin, Asian J. Org. Chem., 2022, 11, e202200152 CrossRef CAS.
- H. Yan, Z.-W. Hou and H.-C. Xu, Angew. Chem., Int. Ed., 2019, 58, 4592–4595 CrossRef CAS PubMed.
- X.-L. Lai, X.-M. Shu, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2020, 59, 10626–10632 CrossRef CAS.
- M. C. Leech and K. Lam, Acc. Chem. Res., 2020, 53, 121–134 CrossRef CAS PubMed.
- P. Xu, P.-Y. Chen and H.-C. Xu, Angew. Chem., Int. Ed., 2020, 59, 14275–14280 CrossRef CAS PubMed.
- Z. Tan, X. He, K. Xu and C. Zeng, ChemSusChem, 2022, 15, e202102360 CrossRef CAS.
- L. Capaldo, L. L. Quadri, D. Merli and D. Ravelli, Chem. Commun., 2021, 57, 4424–4427 RSC.
- R. d. Río-Rodríguez, L. Fragoso-Jarillo, A. F. Garrido-Castro, M. C. Maestro, J. A. Fernández-Salas and J. Alemán, Chem. Sci., 2022, 13, 6512–6518 RSC.
- H. Zhou, Q. Liu, M. Hua, C. Wang, D. Chen and H. Fu, Heterocycles, 2018, 96, 1226 CrossRef.
- G. Bogonda, H. Y. Kim and K. Oh, Org. Lett., 2018, 20, 2711–2715 CrossRef CAS PubMed.
- A. Srinivasulu, B. Shantharjun, D. Vani, K. C. Ashalu, A. Mohd, J. Wencel-Delord, F. Colobert and K. R. Reddy, Eur. J. Org. Chem., 2019, 1815–1819 CrossRef CAS.
- X.-Z. Wang and C.-C. Zeng, Tetrahedron, 2019, 75, 1425–1430 CrossRef CAS.
- S. Sultan, M. A. Rizvi, J. Kumar and B. A. Shah, Chem. – Eur. J., 2018, 24, 10617–10620 CrossRef CAS PubMed.
- R. Chang, J. Fang, J.-Q. Chen, D. Liu, G.-Q. Xu and P.-F. Xu, ACS Omega, 2019, 4, 14021–14031 CrossRef CAS PubMed.
- L. Guillemard, F. Colobert and J. Wencel-Delord, Adv. Synth. Catal., 2018, 360, 4184–4190 CrossRef CAS.
- M. Niu, C. Yang, M. Leng, Q. Cao, M. Li and Z. Shen, J. Org. Chem., 2024, 89, 6159–6168 CrossRef CAS PubMed.
- K. Matcha and A. P. Antonchick, Angew. Chem., Int. Ed., 2013, 52, 2082–2086 CrossRef CAS PubMed.
- X.-L. Chen, X. Li, L.-B. Qu, Y.-C. Tang, W.-P. Mai, D.-H. Wei, W.-Z. Bi, L.-K. Duan, K. Sun, J.-Y. Chen, D.-D. Ke and Y.-F. Zhao, J. Org. Chem., 2014, 79, 8407–8416 CrossRef CAS PubMed.
- M. T. Westwood, C. J. C. Lamb, D. R. Sutherland and A.-L. Lee, Org. Lett., 2019, 21, 7119–7123 CrossRef CAS PubMed.
- J. K. Laha, M. Kaur Hunjan, S. Hegde and A. Gupta, Org. Lett., 2020, 22, 1442–1447 CrossRef CAS PubMed.
- J. K. Laha and M. K. Hunjan, Chem. Commun., 2021, 57, 8437–8440 RSC.
- A. H. Jatoi, G. G. Pawar, F. Robert and Y. Landais, Chem. Commun., 2019, 55, 466–469 RSC.
- Y. Zhang, K. B. Teuscher and H. Ji, Chem. Sci., 2016, 7, 2111–2118 RSC.
- D. Mooney, H. McKee, T. Batch, S. Drane, P. Moore and A.-L. Lee, Chem. Commun., 2024, 60, 10752–10755 RSC.
- T. He, H. Li, P. Li and L. Wang, Chem. Commun., 2011, 47, 8946–8948 RSC.
- V. S. Bhat and A. Lee, Eur. J. Org. Chem., 2021, 3382–3385 CrossRef CAS.
- C. Ma, L. Shang, H. Zhao, X. He, Q. Lv, D. Zhang and Y. Jiang, Front. Chem., 2022, 10, 1087834 CrossRef CAS PubMed.
- Z. Zhou, H. Ji, Q. Li, Q. Zhang and D. Li, Org. Biomol. Chem., 2021, 19, 2917–2922 RSC.
- H. He, C.-M. Pan, Z.-W. Hou, M. Sun and L. Wang, J. Org. Chem., 2024, 89, 7531–7540 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |