
A facile access to aliphatic trifluoromethyl ketones via photocatalyzed cross-coupling of bromotrifluoroacetone and alkenes†
Satoshi
Mizuta
 *a,
Tomoko
Yamaguchi
a,
Masaharu
Iwasaki
bcde and
Takeshi
Ishikawa
*f
*a,
Tomoko
Yamaguchi
a,
Masaharu
Iwasaki
bcde and
Takeshi
Ishikawa
*f
aCenter for Bioinformatics and Molecular Medicine, Graduate School of Biomedical Sciences, Nagasaki University, 1-14 Bunkyo, Nagasaki, 852-8521, Japan. E-mail: s-miuzta@nagasaki-u.ac.jp
bLaboratory of Emerging Viral Diseases, International Research Center for Infectious Diseases, Research Institute for Microbial Diseases, Osaka University, 3-1 Yamadaoka, Suita, Osaka 565-0871, Japan
cCenter for Infectious Disease Education and Research, Osaka University, 2-8 Yamadaoka, Suita, Osaka 565-0871, Japan
dCenter for Advanced Modalities and Drug Delivery System, Osaka University, 2-8 Yamadaoka, Suita, Osaka 565-0871, Japan
eRNA Frontier Science Division, Institute for Open and Transdisciplinary Research Initiatives, Osaka University, Suita, Osaka, Japan
fDepartment of Chemistry, Biotechnology, and Chemical Engineering, Graduate School of Science and Engineering, Kagoshima University, 1-21-40 Korimoto, Kagoshima 890-0065, Japan. E-mail: ishi@cb.kagoshima-u.ac.jp
First published on 11th September 2024
Abstract
Biological molecules incorporating trifluoromethyl ketones (TFMKs) have emerged as reversible covalent inhibitors, aiding in the management and treatment of inflammatory diseases, cancer, and respiratory conditions. TFMKs, renowned for their versatile binding properties and adaptability, are pivotal in the rational design of novel drugs for diverse diseases. The photocatalytic insertion of alkenes, abundant feedstocks, into the α-carbon of trifluoromethylacetone represents a highly effective and atom-economical method for synthesizing valuable TFMKs. However, these processes typically necessitate high-energy photoirradiation (λ > 300 nm, Hg lamp) and stoichiometric oxidants to generate the acetonyl radical from acetone. In our study, we demonstrate the visible-light photocatalytic radical addition into olefins using bromotrifluoroacetone as the trifluoroacetonyl radical precursor under mild conditions. Aliphatic trifluoromethyl ketones or the corresponding bromo-substituted products can be obtained by selecting an appropriate photocatalyst and solvent. Comprehensive experimental investigations, including cyclic voltammetry, Stern–Volmer quenching studies, and kinetic isotope effects, corroborate the synthesis of trifluoroacetonyl radical species from bromotrifluoroacetone under photoredox conditions. Further, we demonstrate the efficient synthesis of an oseltamivir derivative bearing a trifluoromethylketone moiety, which shows promising biological activity. Hence, this methodology will streamline the direct introduction of trifluoromethyl ketone into biological target molecules during drug discovery.
Introduction
Trifluoromethyl ketones (TFMKs) are valuable synthetic intermediates1 and have been extensively used in pharmaceuticals and biological targets. The carbonyl group of TFMK is susceptible to nucleophilic attack owing to the adjacent trifluoromethyl group exhibiting a strong electron-withdrawing property.2 Biological molecules with TFMK functional groups in aqueous solutions comprise gem-diol in equilibrium, targeting enzymatic catalytic sites or non-catalytic residues to form reversible covalent bonds (Fig. 1A). As the equilibria composition acts as potential enzyme inhibitors, numerous biologically active TFMKs have been developed as reversible covalent inhibitors toward phospholipase A2,3 histone deacetylase,4 human leukocyte elastase,5 and more6 (Fig. 1B). Consequently, TFMKs may serve as potentially privileged motifs in the rational drug design of reversible covalent inhibitors.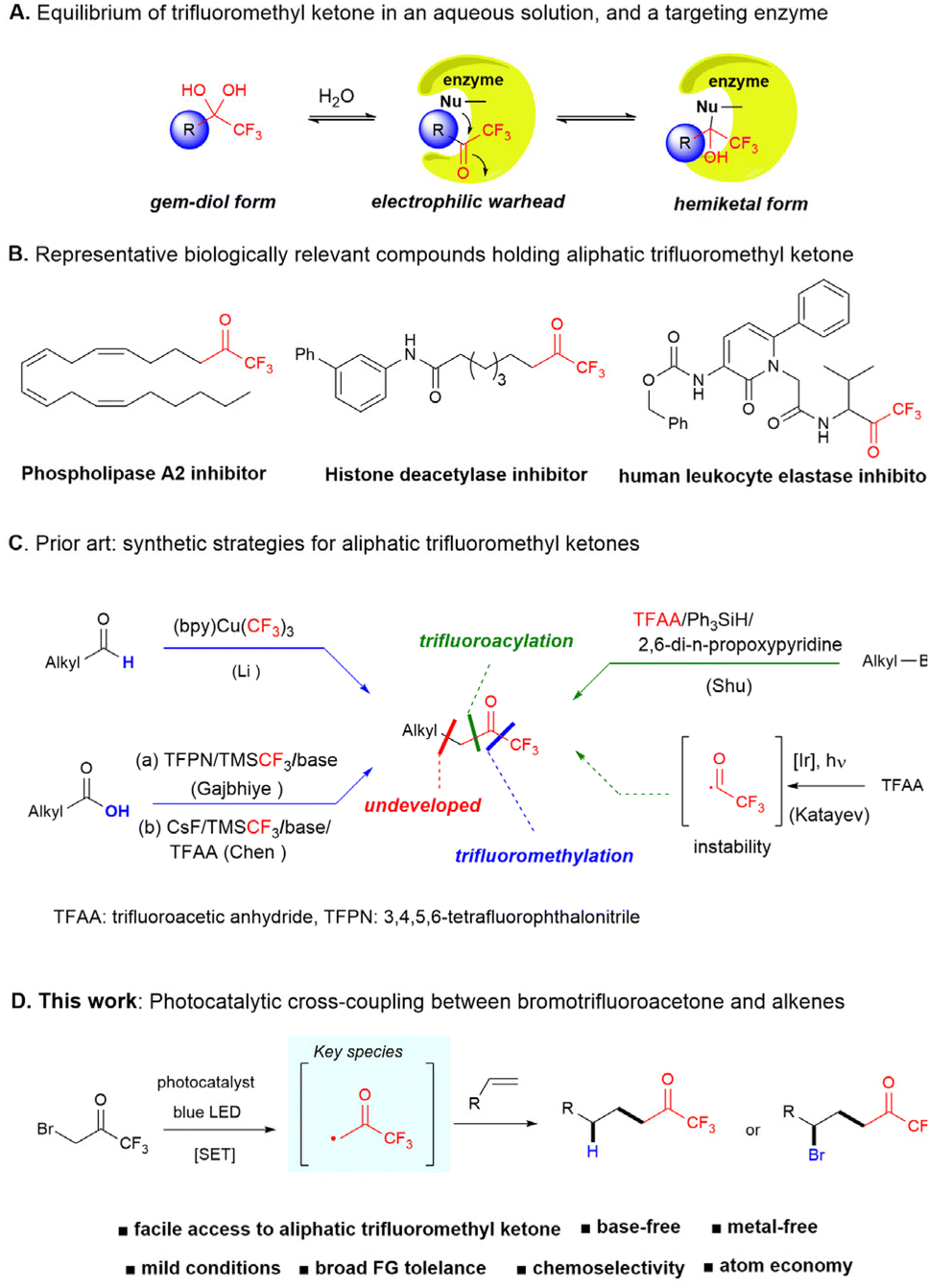 | ||
| Fig. 1 Efficient approaches for synthesizing aliphatic trifluoromethyl ketones. | ||
Traditional methods to access TFMKs predominantly include the stoichiometric trifluoromethylation of carbonyl compounds;1,7 however, these reactions are limited by a narrow substrate scope and low regioselectivity. An alternative approach, the oxidation of α-trifluoromethyl alcohol, requires large quantities of oxidants and multiple steps.8 Two strategies using trifluoromethylation and trifluoroacetylation reactions have been primarily developed, as illustrated in recent reports (Fig. 1C). For instance, the oxidative trifluoromethylation of an aldehyde with (bpy)Cu(CF3)2 occurs in the presence of silane; however, it requires stoichiometric metal species.9 Further, deoxytrifluoromethylation using carboxylic acids has been reported.10 Recently, several remarkable catalytic methods have been reported to access TFMKs. Katayev et al. reported trifluoroacetylation of alkenes using trifluoracetic anhydride (TFAA) and Ir photocatalysts via the generation of a trifluoroacetyl radical, yielding the corresponding CF3-enones.11 However, owing to its instability, the trifluoroacetyl radical collapsed rapidly into carbon monoxide and the CF3 radical. Further, the hydrotrifluoroacetylation of electron-poor olefins, mediated by the decarboxylation of masked acyl reagents via photocatalysis has been successfully demonstrated; this method effectively generates TFMKs from the resulting products.12 To the best of my knowledge, the most noteworthy recent advance in this area is the report by Shu of the radical alkylation between alkyl bromides and TFAA using an Ir photocatalyst in the presence of silane and 2,6-dipropoxypyridine, thus, providing access to various aliphatic TFMKs.13
We envisaged that the insertion of alkenes, which are abundant feedstocks, into the α-carbon of trifluoromethylacetone via photocatalysis14 can be a powerful tool for synthetic transformations; this approach offers a highly effective and atom-economical strategy for preparing structurally valuable TFMKs. A C–C bond coupling between olefins and photoactivated acetone resulted in the formation of methyl ketone.15 Li et al. reported silver-catalyzed carbofluorination of alkenes with acetone via atom transfer radical addition (ATRA) reactions in aqueous solutions.16 However, these processes require high energy photoirradiation (λ > 300 nm, Hg lamp) and stoichiometric oxidants to generate the acetonyl radical from acetone. Although photoredox catalytic radical addition of the common α-bromo acetophenone to unactivated olefins has been achieved,17 the alkylation of α-bromo alkyl ketone derived radicals in this area is less studied. In this study, we demonstrate the visible-light photocatalytic radical addition into olefins using bromotrifluoroacetone as the trifluoroacetonyl radical precursor (Fig. 1D). Further, we propose that capturing a putative carbon radical with hydrogen- or bromine radicals results in hydrogenation and bromination products, respectively. This reaction facilitates a direct and modular synthesis of various aliphatic TFMKs, including pharmaceutical ingredients, offering several advantages such as tolerance for diverse functional groups, absence of metals and bases, and broad applicability.
Results and discussion
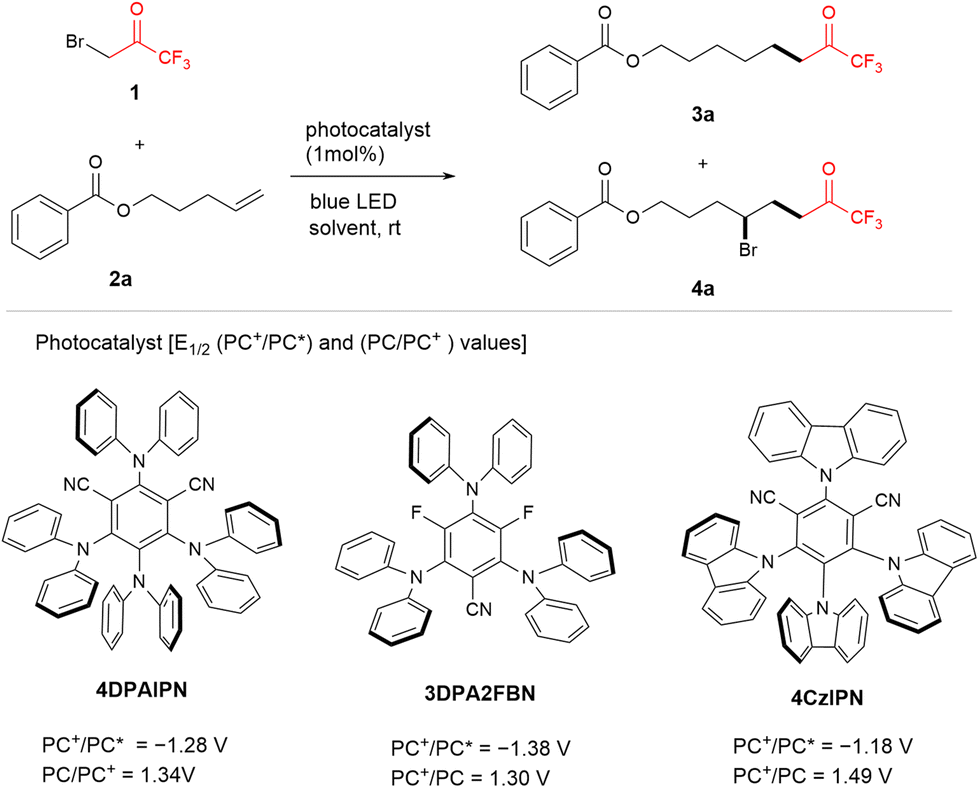
A readily available reagent, bromotrifluoroacetone (1), has been widely used for synthesizing heteroarene with substituted trifluoromethyl groups;18 however, studies on its utilization as a radical precursor of 1 are limited. We optimize the reaction parameters using pent-4-en-1-yl benzoate (2a) as a reaction partner and photocatalyst under irradiation from blue LEDs at 470 nm and room temperature (Table 1). The reaction of 1 using 2,4,5,6-tetrakis(diphenylamino)isophthalonitrile (4DPAIPN) [E1/2(PC+/PC*) = −1.28 V vs. SCE]14b,19 as the photocatalyst in 1,4-dioxane affords a mixture of product 3a and corresponding carbobrominated product 4a in 33% and 11% 19F NMR yields, respectively (entry 1). Further, the effect of the solvent on the photo-catalyzed cross-coupling between bromotrifluoroacetone (1) and substrate 2a (entries 2–5) was analyzed. Tetrahydrofuran (THF) is an excellent solvent, yielding 3a as the primary product (entry 4). In contrast, a carbobromination reaction of 2a with 1 is observed in the MeCN solvent, yielding the product 4a with high chemoselectivity (entry 5). Further, screening of photocatalysts including 3DPA2FBN [E1/2(PC+/PC*) = −1.38 V vs. SCE],20 4CzIPN [E1/2(PC+/PC*) = −1.04 V vs. SCE],14b,19 and [Ir[dF(CF3)ppy]2(dtbbpy)]PF6 [E1/2(Ir+/Ir*) = −0.89 V vs. SCE]14b,19 was performed using THF as a solvent under optimized conditions. A notable achievement is obtained with 3DPA2FBN, resulting in a 75% isolated yield of product 3a (entry 7). A similar result is obtained with 4CzIPN (entry 8); however, the Ir catalyst exhibits less reactivity and chemoselectivity (entry 9). Control experiments reveal that photocatalyst and light are required for this transformation (entry 10). We further optimized the photocatalysts for chemoselective formation of the target product 4a using MeCN as a solvent (entries 11–14). Among photocatalysts, 3DPA2FBN offers the most promising chemoselectivity and 72% isolated yield of 4a by extending the irradiation time to 8 h (entry 11).|
|
|||
|---|---|---|---|
| Entry | Photocatalyst | Solvent | Yield (isolated) (3a/4a)%a |
a Yields determined by 19F NMR spectroscopy using benzotrifluoride as an internal standard.
b Reaction conditions: 1 (0.45 mmol), 2a (0.3 mmol), photocatalyst (1 mol%), solvent (500 μL), 12 W blue LEDs, room temperature (rt), 3 h.
c ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1 (0.6 mmol).
d For 8 h. PC = photocatalyst, MTBE = tert-butyl methyl ether. 1 (0.6 mmol).
d For 8 h. PC = photocatalyst, MTBE = tert-butyl methyl ether.
|
|||
| 1b | 4DPAIPN | 1,4-Dioxane | 33/11 |
| 2b | 4DPAIPN | MTBE | 22/6 |
| 3b | 4DPAIPN | MeOH | 0/0 |
| 4b | 4DPAIPN | THF | 48/5 |
| 5b | 4DPAIPN | MeCN | 0/63 (63) |
| 6c | 4DPAIPN | THF | 60/13 |
| 7c | 3DPA2FBN | THF | 77 (75)/12 |
| 8c | 4CzIPN | THF | 72/12 |
| 9c | Ir[(dFCF3)ppy]2(dtbbpy)PF6 | THF | 39/13 |
| 10c | No PC/no light | THF | 0/0 |
| 11c,d | 4DPAIPN | MeCN | 0/75 (72) |
| 12c,d | 3DPA2FBN | MeCN | 4/49 |
| 13c,d | 4CzIPN | MeCN | 13/57 |
| 14c,d | Ir[(dFCF3)ppy]2(dtbbpy)PF6 | MeCN | 5/70 |
We determined the substrate scope of this transformation using the optimized reaction conditions (Table 1, entry 7; Fig. 2). This photocatalytic process tolerates a variety of functional groups, including sensitive ones such as ester, alcohol, acid, and boronate ester in unactivated alkenes, affording the corresponding target products (3a–l) in moderate to high yields. The photo-catalyzed functionalization of 2a was performed on a 2.4 mmol scale by increasing the irradiation time, yielding the product 3a in 66% yield. Alkene substrates (2m–o) with various substitute patterns, including bromine and ether group, undergo the cross-coupling reactions, yielding alkyl TFMKs (3m–o) as major products and bromo-substituted products in relatively low ratios (4m–o). These reaction conditions affect the reactivity and chemoselectivity of alkene substrates containing the amide moiety. When 4CzIPN is used as a photocatalyst instead of 3DPA2FBN, the substrate 2p converts into hydro- and bromo-products with 59% yield in a 1:1 ratio. Under identical conditions, the related substrate 2q containing a phthalimide group chemoselectively transforms into the corresponding TFMK 3q in 64% yield. Further experiments display good functional group compatibility with 1,2-disubstituted alkenes (2r and 2s) and cyclooctene (2t), affording the desired products (3r–u) in moderate yields. This photocatalytic reaction using 3DPA2FBN and 4CzIPN applies to biologically relevant ibuprofen 2u, naproxen 2v, and estrone derivatives 2w, affording the desired products (3u–w) in 91%, 66%, and 36% yields, respectively.
 | ||
| Fig. 2 Substrate scope of alkene partners. Isolated yield is given for each compound. Reaction conditions: 1 (2.0 eq.), 2 (0.3 or 0.50 mmol), photocatalyst (1 mol%), THF, rt, 3 h. aRun on 2.4 mmol scale (isolated yield). Reaction was performed with 4CzIPN (1 mol%) at rt for b8 h and c22 h. | ||
Next, we investigated the substrate scope of carbobromination of 1 under the optimized reaction conditions (Table 1, entry 11; Fig. 3). The reaction tolerates a broad array of functional groups, including ester (4a–f), alcohol (4g), aryl (4h), ether (4j), acid (4k), ether (4o), and amides (4p, 4y). Their desired products are obtained in moderate to high yields; however, the yield of the product (4q) containing a phthalimide moiety is low. This protocol with 2a is scaled up to 2.4 mmol, which affords the product 4a in 85% yield. Cycloalkenes (2t and 2aa) and 1,2-disubstituted alkenes (2s, and 2z) are compatible, resulting in the desired products with moderate to high yields. Finally, tetraphenylethylene (TPE) derivative 2ab is tolerated, affording the corresponding product 4ab in 56% yield.
 | ||
| Fig. 3 Substrate scope of photocatalyzed carbobromination of alkene partners. The isolated yield is given for each compound. Reaction conditions: 1 (2.0 eq.), 2 (0.3 mmol), photocatalyst (1 mol%), MeCN, rt, 8 h. aRun on 2.4 mmol scale (isolated yield). bReaction was performed at rt for 20 h. | ||
Mechanistic experiments were conducted to gain insight into the mechanism of this transformation under standard conditions. The reduction of bromotrifluoroacetone 1via 3DPA2FBN in THF-d8 under blue LED irradiation affords the product [D]5 in 36% 19F NMR yield (Fig. 4A (1)). The reaction of 2a in THF-d8 leads to the formation of product 3a in 54% yield with 53% deuterium incorporation (Fig. 4A (2)). Hence, trifluoroacetonyl radical is synthesized from 1via a single electron transfer followed by hydrogen trapping using THF. Moreover, (2,2,6,6-tetramethylpiperidin-1-yl)oxyl TEMPO, a radical scavenger, completely impedes the basic conditions (Fig. 4A (3)). When the photocatalytic reaction of 2,2-diallyl malonate 6 is performed under standard conditions, two cyclized products, 7a and 7b, are obtained in 26% yield, as confirmed by NMR and electrospray ionization mass spectrometry (ESI-MS) analyses (Fig. 4A (4)).
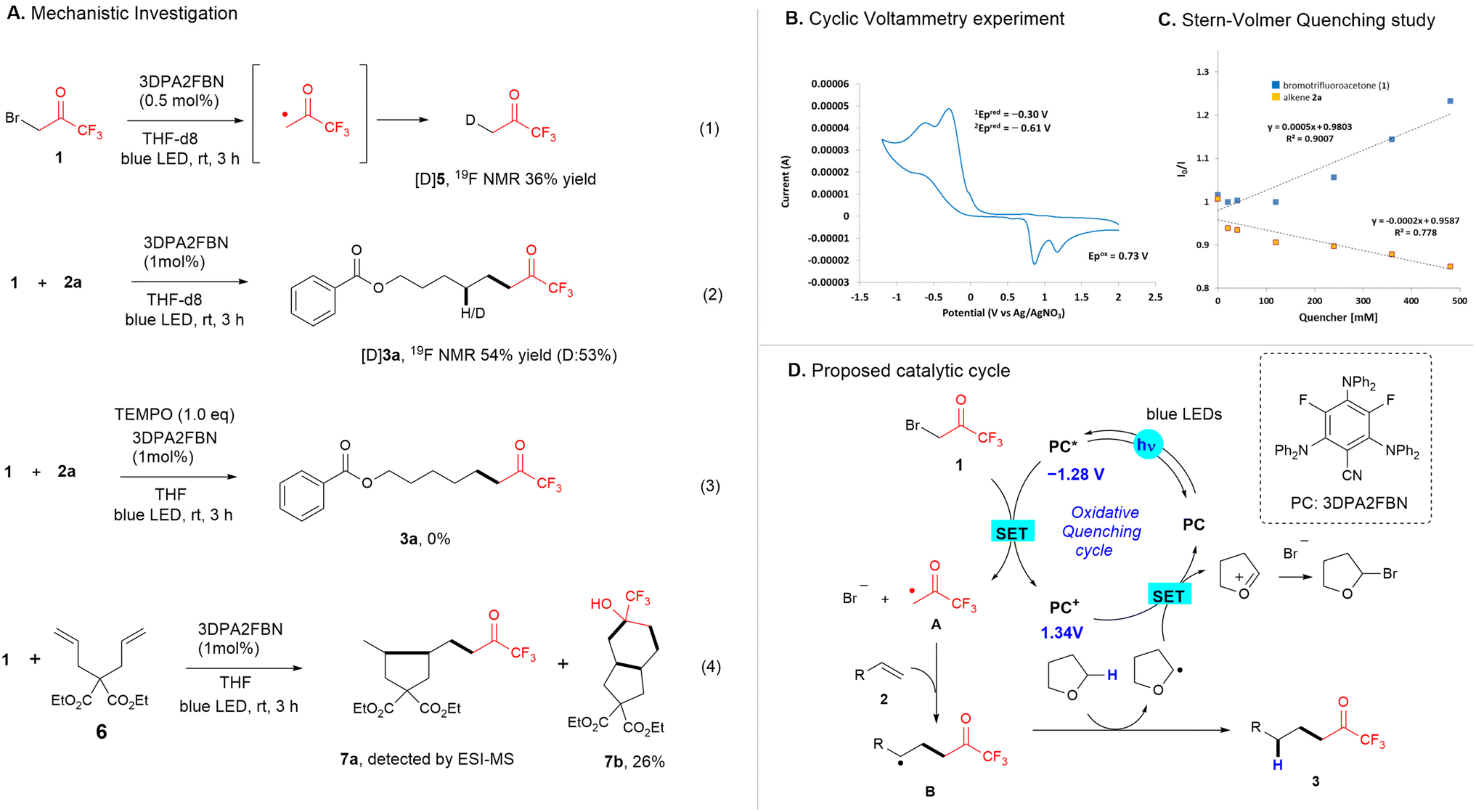 | ||
| Fig. 4 (A) Mechanistic experiments. (B) Cyclic voltammetry experiments. (C) Stern–Volmer quenching of 3DPA2FBN (PC*) by 1. (D) Proposed reaction pathway. | ||
Further, cyclic voltammetry (CV) was performed to understand the redox properties of 1 (Fig. 4B). Two higher reduction potentials (Ered) at −0.30 and −0.60 V vs. Ag/AgNO3 compared with those of phenacyl bromide as a known radical precursor21 (Ered = −0.95 and −1.28 V vs. Ag/AgNO3, Fig. S12†) and a high oxidation potential (Eox) at 0.73 V vs. Ag/AgNO3 in MeCN are observed. The two reduction peaks are attributed to the readily initial reduction of the C–Br bond owing to the trifluoromethyl ketone and a second reduction of mesolytically cleaved carbon radical for ˙CH2COCF3/−CH2COCF3.22 The oxidation potential suggests oxidation of the resultant bromide anion to the corresponding bromine radical. Moreover, Stern–Volmer quenching studies reveal an efficient quenching of the excited T1-state of photocatalyst (PC*) by 1 (Fig. 4C). Hence, we propose that the catalytic cycle is initiated via a single electron reduction of 1 by PC*, affording a trifluoroacetonyl radical.
Based on these data and previous reports,23 a plausible mechanistic pathway for this transformation is elucidated (Fig. 4D). The catalytic cycle on the oxidative quenching cycle is initiated by a single electron transfer (SET) reduction of 1 (Ered = −0.30 V) using the excited state of catalysts, i.e., 3DPA2FBN [E1/2(PC+/PC*) = −1.38 V vs. SCE],20 leading to a trifluoroacetonyl radical A, a bromide ion, and the corresponding oxidized photocatalysts. Subsequently, the radical addition of A to alkenes leads to a putative carbon radical intermediate B, followed by hydrogen atom abstraction from THF to give the product 3. PC+ [E1/2(PC+/PC) = 1.30 V vs. SCE] oxidizes the THF α-radical,20 which restores the ground state of PC, producing an oxonium ion that could be trapped by a bromide ion affording α-bromo-THF.
Then, we performed control experiments to elucidate the mechanism of carbobromination. The Stern–Volmer quenching studies reveal the higher ability of 1 to quench the excited state of 4DPAIPN in MeCN compared with that of 3DPA2FBN in THF (Fig. 5A). The presence of TEMPO under the basic conditions of carbobromination leads to a low yield of 16% for 4a (Fig. 5B (1)). The reaction of 2,2-diallyl malonate 6 using 4DPAIPN in MeCN results in the formation of the cyclized product 7b along with a trace quantity of bromo-substituted product 8a, and its exact mass was determined by high-resolution mass spectrometry (HRMS) analysis (Fig. 5B (2)). Hence, we propose the ATRA reaction24 as a plausible mechanistic pathway to bromo-substituted product 4 (Fig. 5C). The initial SET of 1 by the excited state of 4DPAIPN [E1/2(PC+/PC*) = −1.28 V]14b,19 leads to C–Br bond cleavage, inducing the formation of radical A and PC+ [E1/2(PC+/PC) = 1.34 V].14b,19 Subsequent radical addition of A to alkene results in the formation of carbon-centered radical B. The oxidation of B by PC+ under standard conditions releases the ground state of PC and the carbocation intermediate C that undergoes nucleophilic bromine substitution. Alternatively, a halogen-atom transfer (XAT) reaction of 1 as a bromine source to lead the radical chain propagation could be a proposed pathway for affording the product 4.
 | ||
| Fig. 5 Mechanistic studies (A) Stern–Volmer quenching of 4DPAIPN (PC*) by 1. (B) Control experiments. (C) Proposed reaction mechanism. | ||
We accomplished a series of synthetic transformations of 4a on biologically valuable compounds (Fig. 6A). The efficient intramolecular cyclopropanation of 4a with cesium carbonate provides the product 9a with an 80% yield. The heterocyclization of o-phenylenediamine leads to a biologically relevant compound 9b. Additionally, the transformation of 4a using a nucleophilic reaction partner, NaN3, leads to the azide product 9c in 55% yield. Finally, the antiviral drug oseltamivir derivative 2ac exhibiting multiple functional groups is used for this transformation with chloroform as a co-solvent owing to its poor solubility, affording 10 in 31% yield (Fig. 6B). Condensations of oseltamivir phosphate and acid 3i gave a TFMK folding oseltamivir derivative (11) in 62% yield.
 | ||
| Fig. 6 (A) Synthetic transformation of TFMK 4a. (B) Synthesis of TFMK-folding oseltamivir derivatives (10 and 11). (C) Profile of anti-influenza activity for 2ac, 10, 11 and oseltamivir. CV staining determined a 50% effective concentration (EC50) against the A/WSN/33 virus. EC50 values were calculated from two independent experiments. Compound 10 exhibited promising antiviral activity (EC50 = 0.46 ± 0.09 μM) against the A/WSN/33 virus compared with that of oseltamivir phosphate (EC50 = 0.43 ± 0.15). Substrate 2ac and compound 11 did not exhibit antiviral activity at 10 μM concentration. (D) Crystal structure (PDB code: 6HP0) of NA (gray) and the binding structure of 10 (blue) from docking simulations. Ary293, ILE427, and Lys432 in the cavity of NA are illustrated with some key NA residues (pink). | ||
Influenza viruses cause acute respiratory illnesses in humans during seasonal epidemics worldwide, resulting in hundreds of thousands of deaths annually.25 Influenza-A viruses (IAVs) possess a surface glycoprotein, neuraminidase (NA), an exosialidase that cleaves the glycosidic linkages between the sialic acid and the host glycoconjugate.26 Its activity facilitates the viral movement that releases the progeny viruses from the host cells. As a selective neuraminidase inhibitor, oseltamivir has been used worldwide to prevent the spread of influenza infections.27 The TFMK-holding oseltamivir derivative with a bromo atom (10) and that without the bromo atom (11) are assayed in vitro against the A/WSN/33 virus (Fig. 6C). Compound 10 exhibits potent antiviral activity equivalent to that of the parent compound oseltamivir, with a half-effective concentration (EC50) of 0.46 ± 0.09 μM. In comparison, the inhibitory effect of alkene substrate 2ac and compound 11 could not be observed even at 10 μM. The oseltamivir structure comprises the ethyl ester prodrug of oseltamivir carboxylate, which can convert to its active form by esterases.28 These results suggest that the bromo atom and TFMK of the compound 10 contribute to the inhibition of the neuraminidase enzymatic activity. Next, we performed the docking simulation of the acid form of 10 with the crystal structure of neuraminidase from H1N1 influenza virus (PDB code: 6HP0)29 using AutoDockVina (Fig. 6D). The binding mode of the most reasonable structure obtained from the docking calculation revealed a hydrogen bonding interaction between the acid moiety and Arg293. Additionally, we observe that the TFMK moiety is located in the cavity of a nucleophilic amino acid Lys432 and a hydrophobic amino acid ILE427. Bromo substitute on the alkyl chain of compound 10 might assist the TFMK moiety to orient toward the cavity. These computational findings agree with the inhibitory activity observed experimentally.
Conclusions
In this study, we have demonstrated a metal- and base-free, visible light photocatalyzed cross-coupling between bromotrifluoroacetone and unactivated alkenes, providing direct access to aliphatic TFMKs, including the biologically relevant derivatives. The protocol is carried out under mild conditions with the appropriate choice of photocatalyst and solvent, providing chemoselectively aliphatic TFMKs and the corresponding bromo-substituted products, exhibiting remarkably broad functional group tolerance. This methodology successfully facilitated the synthesis of an oseltamivir derivative featuring an aliphatic TFMK exhibiting potential anti-influenza activity. Hence, the photocatalyzed radical addition of bromotrifluoroacetone offers a streamlined approach for synthesizing TFMK derivatives, which can be pivotal in the design of reversible covalent enzyme inhibitors for drug development. We are currently investigating the applications of our methodology toward the synthesis of biological targets incorporating TFMKs, and photoredox-catalyzed C–H alkylation of arenes.Author contributions
The synthesis of aliphatic trifluoromethyl ketones was carried out by S. M. The docking studies were carried out by T. I. The biological data were evaluated by T. Y. and M. I. The characterization and data curation were made by S. M. and T. I. S. M., T. I. and M. I. were responsible for financial resource. The manuscript was written through contributions of S. M. and T. I. All authors have given approval to the final version of the manuscript.Data availability
Experimental procedures and characterization data for all the products can be found in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported financially by the Japan Society for the Promotion of Science (JSPS) KAKENHI (grant numbers 21K07704 (S. M.) and 23K28190 (T. I.)), and AMED (grant numbers JP23fk0108657 and JP24fk0108657 (S. M. and M. I.)).References
- (a) Y. Dongmei, Z. Yuhan, C. Qing, Z. Yilong and Q. Jingping, Bioactivities and Synthesis of Trifluoromethyl Alkyl Ketones, Prog. Chem., 2014, 26, 976–986 Search PubMed; (b) C. B. Kelly, M. A. Mercadante and N. E. Leadbeater, Trifluoromethyl Ketones: Properties, Preparation, and Application, Chem. Commun., 2013, 49, 11133–11148 RSC; (c) J.-P. Bégué and D. Bonnet-Delpon, Preparation of Trifluoromethyl Ketones and Related Fluorinated Ketones, Tetrahedron, 1991, 47, 3207–3258 CrossRef.
- (a) I. Columbus, L. Ghindes-Azaria, I. M. Herzog, E. Blum, G. Parvari, Y. Eichen, Y. Cohen, E. Gershonov, E. Drug, S. Saphier, S. Elias, B. Smolkin and Y. Zafrani, Species-Specific Lipophilicities of Fluorinated Diketones in Complex Equilibria Systems and Their Potential as Multifaceted Reversible Covalent Warheads, Commun. Chem., 2023, 6, 197 CrossRef CAS PubMed; (b) R. A. Smith, L. J. Copp, S. L. Donnelly, R. W. Spencer and A. Krantz, Inhibition of Cathepsin B by Peptidyl Aldehydes and Ketones: Slow-Binding Behavior of a Trifluoromethyl Ketone, Biochemistry, 1988, 27, 6568–6573 CrossRef CAS PubMed.
- (a) C. Baskakis, V. Magrioti, N. Cotton, D. Stephens, V. Constantinou-Kokotou, E. A. Dennis and G. Kokotos, Synthesis of Polyfluoro Ketones for Selective Inhibition of Human Phospholipase A2 Enzymes, J. Med. Chem., 2008, 51, 8027–8037 CrossRef CAS PubMed; (b) I. P. Street, H.-K. Lin, F. Laliberte, F. Ghomashchi, Z. Wang, H. Perrier, N. M. Tremblay, Z. Huang, P. K. Weech and M. H. Gelb, Slow- and Tight-Binding Inhibitors of the 85 kDa Human Phospholipase A2, Biochemistry, 1993, 32, 5935–5940 CrossRef CAS PubMed.
- (a) A. S. Madsen and C. A. Olsen, A Potent Trifuormethyl Ketone Histone Deacetylase Inhibitor Exhibits Class-Dependent Mechanism of Action, MedChemComm, 2016, 7, 464–470 RSC; (b) C.-J. Gong, A.-H. Gao, Y.-M. Zhang, M.-B. Su, F. Chen, L. Sheng, Y.-B. Zhou, J.-Y. Li and F.-J. Na, Design, Synthesis and Biological Evaluation of Bisthiazole-Based Trifluoromethyl Ketone Derivatives as Potent HDAC Inhibitors with Improved Cellular Efficacy, Eur. J. Med. Chem., 2016, 112, 81–90 CrossRef CAS PubMed; (c) J. Cai, H. Wei, K. H. Hong, X. Wu, X. Zong, M. Cao, P. Wang, L. Li, C. Sun, B. Chen, G. Zhou, J. Chen and M. Ji, Discovery, Bioactivity and Docking Simulation of Vorinostat Analogues Containing 1,2,4-Oxadiazole Moiety as Potent Histone Deacetylase Inhibitors and Antitumor Agents, Bioorg. Med. Chem., 2015, 23, 3457–3471 CrossRef CAS PubMed.
- (a) C. A. Veale, P. R. Bernstein, F. J. Brown, C. Bryant, J. R. Damewood, R. Earley, S. W. Feeney, P. D. Edwards, B. Gomes, J. M. Hulsizer, B. J. Kosmider, R. D. Krell, G. Moore, T. W. Salcedo, A. Shaw, D. S. Silberstein, G. B. Steelman, M. Stein, A. Stimpler, R. M. Tohmas, E. P. Vacek, D. J. Wolanin and S. Woolson, Orally Active Trifluoromethyl Ketone Inhibitors of Human Leukocyte Elastase, J. Med. Chem., 1997, 40(20), 3173–3181 CrossRef CAS PubMed; (b) P. R. Bernstein, B. C. Gomes, B. J. Kosmider, E. P. Vacek and J. C. Williams, Nonpeptidic Inhibitors of Human Leukocyte Elastase. 6. Design of a Potent, Intratracheally Active, Pyridone-Based Trifluoromethyl Ketone, J. Med. Chem., 1995, 38(1), 212–215 CrossRef CAS PubMed.
- (a) Y.-M. Shao, W.-B. Yang, T.-H. Kuo, K.-C. Tsai, C.-H. Lin, A.-S. Yang, P.-H. Liang and C.-H. Wong, Design, Synthesis, and Evaluation of Trifluoromethyl Ketones as Inhibitors of SARS-CoV 3CL Protease, Bioorg. Med. Chem., 2008, 16, 4652–4660 CrossRef CAS PubMed; (b) A. C. Gibbs, R. Steele, G. Liu, B. A. Tounge and G. T. Montelione, Inhibitor Bound Dengue NS2B-NS3pro Reveals Multiple Dynamic Binding Modes, Biochemistry, 2018, 57, 1591–1602 CrossRef CAS PubMed.
- (a) X. Ispizua-Rodriguez, S. B. Munoz, V. Krishnamurti, T. Mathew and G. K. S. Prakash, Direct Synthesis of Tri/Difluoromethyl Ketones from Carboxylic Acids by Cross-Coupling with Acyloxyphosphonium Ions, Chem. – Eur. J., 2021, 27, 15908–15913 CrossRef CAS PubMed; (b) W. Wang, W. Xiong, J. Wang, Q.-A. Wang and W. Yang, Brønsted Acid-Catalyzed Asymmetric Friedel–Crafts Alkylation of Indoles with Benzothiazole-Bearing Trifluoromethyl Ketone Hydrates, J. Org. Chem., 2020, 85, 4398–4407 CrossRef CAS PubMed; (c) A. Sanz-Marco, G. Blay, M. C. Munoz and J. R. Pedro, Catalytic Enantioselective Conjugate Alkynylation of α,β-Unsaturated 1,1,1-Trifluoromethyl Ketones with Terminal Alkynes, Chem. – Eur. J., 2016, 22, 10057–10064 CrossRef CAS PubMed; (d) L. Anthore and S. Z. Zard, A Convergent Radical Based Route to Trifluoromethyl Ketones and to α,β-Unsaturated Trifluoromethyl Ketones, Org. Lett., 2015, 17, 3058–3061 CrossRef CAS PubMed; (e) Y. Zhou, D. Yang, G. Luo, Y. Zhao, Y. Luo, N. Xue and J. Qu, Highly Selective Trifluoroacetic Ester/Ketone Metathesis: An Efficient Approach to Trifluoromethyl Ketones and Esters, Tetrahedron, 2014, 70, 4668–4674 CrossRef CAS.
- (a) M. Kirihara, K. Suzuki, K. Nakakura, K. Saito, R. Nakamura, K. Tujimoto, Y. Sakamoto, Y. Kikkawa, H. Shimazu and Y. Kimura, Oxidation of Fluoroalkyl Alcohols Using Sodium Hypochlorite Pentahydrate [1], J. Fluor. Chem., 2021, 243, 109719 CrossRef CAS; (b) Y. Kadoh, M. Tashiro, K. Oisaki and M. Kanai, Organocatalytic Aerobic Oxidation of α-Fluoroalkyl Alcohols to Fluoroalkyl Ketones at Room Temperature, Adv. Synth. Catal., 2015, 357, 2193–2198 CrossRef CAS.
- P. Zhan, H. Shen, L. Zhu, W. Cao and C. Li, Radical C(sp2)–H Trifluoromethylation of Aldehydes in Aqueous Solution, Org. Lett., 2018, 20, 7062–7065 CrossRef PubMed.
- (a) M. S. Said, N. S. Khonde, P. Kumar and J. M. Gajbhiye, Electron-Deficient Fluoroarene-Mediated Synthesis of Trifluoromethyl Ketones from Carboxylic Acids, Org. Lett., 2023, 25, 1094–1098 CrossRef CAS PubMed; (b) X. Liu, L. Liu, T. Huang, J. Zhang, Z. Tang, C. Li and T. Chen, Trifluoromethylation of Benzoic Acids: An Access to Aryl Trifluoromethyl Ketones, Org. Lett., 2021, 23, 4930–4934 CrossRef CAS PubMed.
- K. Zhang, D. Rombach, N. Y. Nötel, G. Jeschke and D. Katayev, Radical Trifluoromethylation of Alkenes Triggered by a Visible-Light-Promoted C–O Bond Fragmentation of Trifluoroacetic Anhydride, Angew. Chem., Int. Ed., 2021, 60, 22487–22495 CrossRef CAS PubMed.
- (a) Z.-P. Ye, M. Guo, Y.-Q. Ye, C.-P. Yuan, H.-L. Wang, J.-S. Yang, H.-B. Chen, H.-Y. Xiang, K. Chen and H. Yang, Iodine(III)-Mediated Trifluoroacetylation of a C(sp2)–H or C(sp)–H Bond with Masked Trifluoroacyl Reagents, Org. Lett., 2024, 26, 5196–5201 CrossRef CAS PubMed; (b) S. Han, K. L. Samony, R. N. Nabi, C. A. Bache and D. K. Kim, Hydrotrifluoroacetylation of Alkenes via Designer Masked Acyl Reagents, J. Am. Chem. Soc., 2023, 145(21), 11530–11536 CrossRef CAS PubMed.
- H.-W. Du, Y.-D. Dan, X.-W. Zeng and W. Shu, Access to Trifluoromethylketones from Alkyl Bromides and Trifluoroacetic Anhydride by Photocatalysis, Angew. Chem., Int. Ed., 2023, 62, e202308732 CrossRef CAS PubMed.
- State-of-the-art reviews on photocatalysis: see: (a) L. Candish, K. D. Collins, G. C. Cook, J. J. Douglas, A. Gómez-Suárez, A. Jolit and S. Keess, Photocatalysis in the Life Science Industry, Chem. Rev., 2022, 122, 2907–2980 CrossRef CAS PubMed; (b) J. D. Bell and J. A. Murphy, Recent Advances in Visible Light-Activated Radical Coupling Reactions Triggered by (i) Ruthenium, (ii) Iridium and (iii) Organic Photoredox Agents, Chem. Soc. Rev., 2021, 50, 9540–9685 RSC.
- (a) Y. Shiraishi, D. Tsukamoto and T. Hirai, Highly Efficient Methyl Ketone Synthesis by Water-Assisted C–C Coupling between Olefins and Photoactivated Acetone, Org. Lett., 2008, 10, 3117–3120 CrossRef CAS PubMed; (b) W.-S. Chung and C.-C. Ho, Photochemistry of Acetone in the Presence of Exocyclic Olefins: An Unexpected Competition between the Photo-Conia and Paternò-Büchi Reactions, Chem. Commun., 1997, 317–318 RSC.
- L. Zhu, H. Chen, Z. Wang and C. Li, Formal Fluorine Atom Transfer Radical Addition: Silver-Catalyzed Carbofluorination of Unactivated Alkenes with Ketones in aqueous Solution, Org. Chem. Front., 2014, 1, 1299–1305 RSC.
- (a) X.-H. Huang, F.-L. Liu, T.-F. Fu, X. Hu, Y.-Y. Wang, B. Liu, M.-Y. Teng and G.-L. Huang, Visible Light-Induced Radical Cascade Acylmethylation/Cyclizatin of 2-(Allyloxy)arylaldehydes with α-Bromo Ketones: Access to Cyclic 1,5-Dicarbonyl-Containing Chroman-4-one Skeletons, Org. Biomol. Chem., 2023, 21, 6772–6777 RSC; (b) D. M. Fischer, M. Freis, W. M. Amberg, H. Lindner and E. M. Carreira, Organophotocatalytic Carbo-Heterofunctionalization of Unactivated Olefins with Pendant Nucleophiles, Chem. Sci., 2023, 14, 7256–7261 RSC; (c) H.-L. Huang, J. Xu, Y.-X. Fan, Q.-Q. Su, J.-Y. Du, R.-F. Zhang, Y. I. Wang, H. Hu and F. Gao, Visible-Light-Induced Difunctionalization of Alkenyl Ketones with α-Carbonyl Alkyl Bromide: Concomitant Installation of C–C Bonds, J. Org. Chem., 2022, 87, 14093–14102 CrossRef CAS PubMed.
- For selected recently published literature. see: (a) A. Ali Adnan, A. Mohammad, T. Ahmed, H. Muhammad, A. Tashfeen and W. Simon, Synthesis of Substituted Arylidene Hydrazinyl Trifluoromethyl Thiazole Derivatives and Their Antibacterial Studies Using Different Genes Expression, ChemistrySelect, 2024, 9, e202401206 CrossRef; (b) A. K. Ghosh, J. L. Mishevich, S. Kovela, R. Shaktah, A. K. Ghosh, M. Johnson, Y.-F. Wang, A. Wong-Sam, J. Agniswamy, M. Amano, Y. Takamatsu, S.-I. Hattori, I. T. Weber and H. Mitsuya, Exploration of Imatinib and Nilotinib-Derived Templates as the P2-Ligand for HIV-1 Protease Inhibitors: Design, Synthesis, Protein X-Ray Structural Studies, and Biological Evaluation, Eur. J. Med. Chem., 2023, 255, 115385 CrossRef CAS PubMed; (c) D. Qingsong, G. Jun, Z. Huan, Z. Youlai and M. Xiangtai, Sustainable Access to Benzothiophene Derivatives Bearing a Trifluoromethyl Group via a Three-Component Domino Reaction in Water, Org. Biomol. Chem., 2022, 20, 7424–7428 RSC.
- (a) P. P. Singh and V. Srivastava, Recent Advances in Using 4DPAIPN in Photocatalytic Transformations, Org. Biomol. Chem., 2021, 19, 313–321 RSC; (b) J. Luo and J. Zhang, Donor-Acceptor Fluorophores for Visible-Light-Promoted Organic Synthesis: Photoredox/Ni Dual Catalytic C(sp3)–C(sp2) Cross-Coupling, ACS Catal., 2016, 6, 873–877 CrossRef CAS; (c) G. J. Choi, Q. Zhu, D. C. Miller, C. J. Gu and R. R. Knowles, Catalytic Alkylation of Remote C-H Bonds Enabled by Proton-Coupled Electron Transfer, Nature, 2016, 539, 268–271 CrossRef CAS PubMed.
- E. Speckmeier, T. G. Fischer and K. Zeitler, A Toolbox Approach to Construct Broadly Applicable Metal-Free Catalysts for Photoredox Chemistry: Deliberate Tuning of Redox Potentials and Importance of Halogens in Donor-Acceptor Cyanoarenes, J. Am. Chem. Soc., 2018, 140, 15353–15365 CrossRef CAS PubMed.
- (a) N. S. Dange, A. H. Jatoi, F. Rovert and Y. Landais, Visible-Light-Mediated Addition of Phenacyl Bromides onto Cyclopropens, Org. Lett., 2017, 19, 3652–3655 CrossRef CAS PubMed; (b) D. A. Nicewicz and W. C. MacMillan, Merging Photoredox Catalysis with Organocatalysis: The Direct Asymmetric Alkylation of Aldehydes, Nature, 2008, 322, 77–80 CAS.
- (a) D. D. M. Wayner, D. J. McPhee and D. Griller, Oxidation and Reduction Potentials of Transient Free Radicals, J. Am. Chem. Soc., 1998, 110, 132–137 CrossRef; (b) N. Bortolamei, A. A. Isse and A. Gennaro, Estimation of Standard Reduction Potentials of Alkyl Radicals Involved in Atom-Transfer Radical Polymerization, Electrochim. Acta, 2010, 55, 8312–8318 CrossRef CAS.
- For selected examples, see: (a) J. Zhan, J. Chen, J. Chen, Y. Luo and Y. Xia, Tetrahedron Lett., 2022, 98, 153835 CrossRef; (b) F. Chen, X.-H. Xu and F.-L. Qing, Photoredox-Catalyzed Addition of Dibromofluoromethane to Alkenes: Direct Synthesis of 1-Bromo-1-Fluoroalkanes, Org. Lett., 2021, 23, 2364–2369 CrossRef CAS PubMed; (c) Y. Ouyang, X.-H. Xu and F.-L. Qing, Hydrotrifluoromethylthiolation of Unactivated Alkenes and Alkynes with Trifluoromethanesulfonic Anhydride through Deoxygenative Reduction and Photoredox Radical Processes, Angew. Chem., Int. Ed., 2019, 58, 18508–18512 CrossRef CAS PubMed; (d) Y. Wan, T. Shang, Z. Lu and G. Zhu, Photocatalytic 1,1-Hydrofluoroalkylation of Alkynes with a Concurrent Vicinal Acylation: An Access to Fluoroalkylated Cyclic Ketones, Org. Lett., 2019, 21, 4187–4191 CrossRef CAS PubMed; (e) Y.-Y. Ren, X. Zheng and X. Zhang, Bromotrifluoromethane: A Useful Reagent for Hydrotrifluoromethylation of Alkenes and Alkynes, Synlett, 2018, 29, 1028–1032 CrossRef CAS; (f) Q.-Y. Lin, X.-H. Xu, K. Zhang and F.-L. Qing, Visible-Light-Induced Hydrodifluoromethylation of Alkenes with a Bromodifluoromethylphosphonium Bromide, Angew. Chem., Int. Ed., 2016, 55, 1479–1483 CrossRef CAS PubMed; (g) W. Yu, X.-H. Xu and F.-L. Qing, Photoredox Catalysis Mediated Application of Methyl Fluorosulfonyldifluoroacetate as the CF2CO2R Radical Source, Org. Lett., 2016, 18, 5130–5133 CrossRef CAS PubMed; (h) B. Yang, X.-H. Xu and F.-L. Qing, Synthesis of Difluoroalkylated Arenes by Hydroaryldifluoromethylation of Alkenes with α,α-Difluoroarylacetic Acids under Photoredox Catalysis, Org. Lett., 2016, 18, 5956–5959 CrossRef CAS PubMed.
- For selected examples, see: (a) T. Rawner, E. Lutsker, C. A. Kaiser and O. Reiser, The Different Faces of Photoredox Catalysts: Visible-Light-Mediated Atom Transfer Radical Addition (ATRA) Reactions of Perfluoroalkyl Iodides with Styrenes and Phenylacetylenes, ACS Catal., 2018, 8, 3950–3956 CrossRef CAS; (b) D. P. Tiwari, S. Dabral, J. Wen, J. Wiesenthal, S. Terhorst and C. Bolm, Organic Dye-Catalyzed Atom Transfer Radical Addition–Elimination (ATRE) Reaction for the Synthesis of Perfluoroalkylated Alkenes, Org. Lett., 2017, 19, 4295–4298 CrossRef CAS PubMed; (c) Ł. Woźniak, J. J. Murphy and P. Melchiorre, Photo-Organocatalytic Enantioselective Perfluoroalkylation of β-Ketoesters, J. Am. Chem. Soc., 2015, 137, 5678–5681 CrossRef PubMed; (d) N. Iqbal, J. Jung, S. Park and E. J. Cho, Controlled Trifluoromethylation Reactions of Alkynes through Visible-Light Photoredox Catalysis, Angew. Chem., Int. Ed., 2014, 53, 539–542 CrossRef CAS PubMed; (e) C.-J. Wallentin, J. D. Nguyen, P. Finkbeiner and C. R. J. Stephenson, Visible Light-Mediated Atom Transfer Radical Addition via Oxidative and Reductive Quenching of Photocatalysts, J. Am. Chem. Soc., 2012, 134, 8875–8884 CrossRef CAS PubMed.
- (a) C. Paules and K. Subbarao, Influenza, Lancet, 2017, 390, 697–708 CrossRef PubMed; (b) J. Dunning, J. K. Baillie, B. Cao and F. G. Hayden, Antiviral Combinations for Severe Influenza, Lancet Infect. Dis., 2014, 14, 1259–1270 CrossRef CAS PubMed.
- E. D. Clercq, Antiviral Agents Active against Influenza A Viruses, Nat. Rev. Drug Discovery, 2006, 5, 1015–1025 CrossRef PubMed.
- J. L. McAuley, B. P. Gilbertson, S. Trifkovic, L. E. Brown and J. L. McKimm-Breschkin, Influenza Virus Neuraminidase Structure and Functions, Front. Microbiol., 2019, 10, 39 CrossRef PubMed.
- (a) B. E. Davies, Pharmacokinetics of Oseltamivir: An Oral Antiviral for the Treatment and Prophylaxis of Influenza in Diverse Populations, J. Antimicrob. Chemother., 2010, 65(Suppl 2), ii5–ii10 CAS.
- V. Zima, C. B. Albiñana, K. Rojíková, J. Pokorná, O. Pachl, P. Řezáčová, J. Hudlicky, V. Navrátil, P. Majer, J. Konvalinka, M. Kožíšek and A. Machara, Investigation of Flexibility of Neuraminidase 150-loop using Tamiflu derivatives in influenza A viruses H1N1 and H5N1, Bioorg. Med. Chem., 2019, 27, 2935–2947 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob01247j |
| This journal is © The Royal Society of Chemistry 2024 |